

Summary
Despite improvements in treatment, coronary artery disease is still responsible for one‐third of all deaths globally, due predominantly to myocardial infarction (MI) and stroke. There is an important potential in developing new strategies for treatment of patients with these conditions. Inflammation, and in particular the actions of the complement system, has emerged as part of the pathogenesis in reperfusion injury in patients with MI. To further qualify this, we examined the association between the plasma levels of lectin pathway proteins and myocardial end‐points, left ventricular ejection fraction (LVEF) and infarct size in a cohort of patients with ST‐elevation myocardial infarction (STEMI). A blood sample was drawn the day after percutaneous coronary intervention from 73 patients with STEMI. The primary end‐points, LVEF and infarct size, were measured with magnetic resonance imaging 6–9 days after the infarct. Complement pattern‐recognition molecules of the lectin pathway (mannan‐binding lectin, H‐ficolin, L‐ficolin and M‐ficolin) were analysed along with soluble membrane attack complex (sMAC) and C‐reactive protein (CRP) in plasma with immunofluorometric assays <50%. CRP correlated negatively with LVEF, regression coefficient = –0·17 (P = 0·01). None of the lectin pathway proteins correlated to LVEF or infarct size, nor did soluble membrane attack complex (sMAC). There were no differences in plasma levels of these complement proteins when comparing patients with ejection fraction <50% to patients with ejection fraction <50%. Pattern‐recognition molecules of the lectin pathway and sMAC do not predict short‐term cardiac outcomes after MI.
Keywords: complement system, ischaemia/reperfusion injury, lectin pathway, myocardial infarction
Introduction
Coronary artery disease (CAD) is one of the leading causes of mortality and morbidity worldwide. Although there has been an improvement in prognosis, there is still a residual risk which demonstrates the scientific and therapeutic challenge we are facing in the search for improved treatment of CAD‐related diseases such as myocardial infarction (MI) 1, 2. The pathogenesis of acute MI is a complex multi‐factorial process, in which inflammation is an essential causative factor 3, 4. Inflammation normally arises in response to tissue damage caused by pathogens or irritants. The primary role of inflammation is defence, but paradoxically inflammation is suggested to worsening the outcome of MI during reperfusion. A possible explanation is that dying cells present patterns of neoepitopes, which is recognized by pattern‐recognition molecules and thus activate the complement system during reperfusion. Recently, the association between the lectin pathway and ischaemia/reperfusion (I/R) injury has generated scientific attention in a comprehensive review and calls for further investigation in clinical cohorts 5.
The lectin pathway is one of three pathways capable of activating the complement system, which integrates the innate and the adaptive immune systems. The lectin pathway is activated by five soluble pattern‐recognition molecules: mannan‐binding lectin (MBL), H‐ficolin, L‐ficolin, M‐ficolin and collectin‐LK 6, 7, 8, followed by activation of the MBL‐associated serine proteases (MASPs), and ultimately formation of the membrane attack complex (MAC). Collard et al. was the first to investigate the association between MBL and I/R injury in an animal model. They observed MBL deposition in ischaemic rat heart and furthermore illustrated that MBL was responsible for iC3b deposition after oxidative stress in an in‐vitro study 9. The research performed in murine I/R models is persuasive, and recently Clark et al. showed that mice treated with anti‐MASP‐2 antibody have smaller infarcts than their controls 10.
In clinical research, the association between the lectin pathway and myocardial ischaemia is controversial. On one hand, MBL is shown to be protective in the development of coronary artery disease, but on the other hand it is suggested to be harmful 11, 12. L‐ficolin and combinations of L‐ficolin, MBL and MAp44 are shown to be associated with left ventricular dilatation after MI 13. We showed that proteins involved in regulation of the lectin pathway, MAp44, MASP‐1 and MASP‐3, are found at higher concentrations in patients with acute MI compared to healthy controls, but these proteins had no association with infarct size 14. In contrast, two small clinical studies reported lower MASP‐2 level in patients with acute MI compared to healthy controls 15, 16.
In this study, we aimed to investigate if markers of structural and functional cardiac damage [infarct size and left ventricular ejection fraction (LVEF)] in patients with STEMI were associated with plasma concentration of lectin pathway components. We therefore examined pattern‐recognition molecules which are central for initiation of the lectin pathway (MBL, M‐ficolin, L‐ficolin, H‐Ficolin) and the soluble membrane attack complex (sMAC) as an end‐point of complements activation.
Methods
Study population
This study includes STEMI patients originally enrolled into a prospective randomized study at Karolinska University Hospital, Sweden and admitted to the coronary care unit for primary percutaneous coronary intervention (PCI) 17. In brief, 89 patients met the inclusion criteria; chest pain >30 min and ≤6 h, ST elevation ≥0·1 mV (≥0·2 mV in V1–V3) in two contiguous electrocardiogram (ECG) leads or left bundle branch block, and a thrombolysis in myocardial infarction (TIMI) grade 0 flow in the infarct‐related artery. The patients were randomly allocated to either primary PCI (n = 45) or primary PCI and postconditioning (n = 44). In the postconditioning group, an angioplasty balloon was positioned upstream for the primary stent at a pressure of 2–4 atm within the first minute of reflow. The balloon was reinflated four times for 1 min, each separated by 1 min of reflow. Seven patients randomized to the primary PCI group and seven patients from the postconditioning group were excluded because of unwillingness, poor image quality, sudden death (n = 1), missing plasma samples or the protocol was not followed.
The study thus included 73 patients in total; 36 patients were allocated to the control group and 37 patients were allocated to the postconditioning group. The original study by Sörensson et al. 17, investigating the effect of postconditioning after PCI in STEMI, showed no difference in LVEF or infarct size between the two randomized groups. Based on this, and the fact that there were no differences in the complement proteins or C‐reactive protein between the control group and the postconditioning group (see Table 1 in the Result section), we pooled the two groups into one group of STEMI patients.
Table 1.
Mean plasma concentration of the complement proteins are divided into two groups: percutaneus coronary intervention (PCI) and PCI with postconditioning.
| PCI (mean ± s.e.) | PCI + postconditioning (mean ± s.e.) | P‐value | |
|---|---|---|---|
| MBL (µg/l) | 1847 ± 287 | 1855 ± 260 | 0·82* |
| M‐ficolin (µg/l) | 4166 ± 207 | 4700 ± 177 | 0·06 |
| L‐ficolin (µg/l) | 3294 ± 159 | 3293 ± 160 | 0·99 |
| H‐ficolin (µg/l) | 22 584 ± 648 | 23 090 ± 748 | 0·61 |
| sMAC (µg/l) | 136 ± 6 | 158 ± 15 | 0·69* |
| CRP (µg/l) | 17 ± 3 | 22 ± 3 | 0·17 |
MBL = mannan‐binding lectin; sMAC = soluble membrane attack complex; CRP = C‐reactive protein.
Student's t‐test was used to compare and test the levels between the groups. Mannan‐binding lectin (MBL) and soluble membrane attack complex (sMAC) were tested with Wilcoxon's rank sum test.
The original randomized study was approved by the local ethics committee at the Karolinska Institutet and conducted in accordance with the Declaration of Helsinki. Written informed consent was given by all patients included 17.
Ethylenediamine tetra‐acetic acid (EDTA)‐plasma was drawn the day after reperfusion and stored at –80°C for later analysis.
Clinical end‐points
Infarct size and LVEF were measured on days 6–9 after the infarct with magnetic resonance imaging (MRI), as previously described in the original study 17. In brief, the MRI was performed in the supine position with an eight‐channel cardiac coil. Gadolinium contrast (0·2 mmol/kg) was administrated prior to scanning the patient. The imaging protocol included Scout images, localization of the short axis and then covering the whole left ventricle with retrospectively gated cine steady‐state free precession images. Delayed‐enhancement images were acquired 15–20 min after injection of the contrast. The images were analysed with segmentation software (Segment version 1.8 R0857). Infarct size is expressed as the percentage of area at risk determined with bi‐plane left ventriculography before primary PCI at admission day.
Analysis of plasma proteins
MBL concentration was measured using our in‐house time‐resolved immunofluorometric assay (TRIFMA) assay based on ligand recognition, as described elsewhere 18. The lower level of detection was 5 µg/l, and the intra‐ and interassay coefficients of variance (CV) were both below 10%.
M‐ficolin 19 and H‐ficolin 20 were estimated with an in‐house TRIFMA. The interassay coefficient of variance was 13% for M‐ficolin and 16% for H‐ficolin. Intra‐assay imprecision was below 10% in both assays. L‐ficolin was measured using an enzyme‐linked immunosorbent assay (ELISA) from Hycult Biotech (Uden, the Netherlands), according to the manufacturer's instructions. The intra‐ and interassay CVs are below 6 and 1%, respectively. Plasma sMAC was determined by an in‐house TRIFMA, described in detail elsewhere 21. The limit of detection for the sMAC assay was 1 µg/l. The intra‐ and interassay CVs were below 5 and 12%, respectively.
Levels of C‐reactive protein (CRP) were quantified by commercially available monoclonal antibodies (Biotechne, R&D Systems, Minneapolis, MN, USA) using an in‐house assay for high sensitivity CRP. Briefly, wells were coated with 0·1 µg anti‐CRP antibody (MAB17071; R&D Systems) in 100 µl phosphate‐buffered saline (PBS) overnight at 4°C. Residual protein‐binding sites were blocked with PBS, 1% Tween 20 (Tw) for 1 h at room temperature and washed in PBS containing 0·05% Tween 20 (PBS/Tw). Recombinant human CRP (NIBSC 85/506) was used as standard and samples were diluted 200‐ or 1000‐fold in assay buffer (PBS, 1% Tw, 0·1% human serum albumin and 25 µM EDTA) and incubated overnight at 4°C. Bound CRP was determined by incubation with 25 ng biotinylated anti‐CRP antibody (BAM17072; R&D Systems) in 100 µl assay buffer for 2 h at room temperature. The wells were washed and subsequently incubated with 10 ng Eu3+‐labelled streptavidin (Perkin Elmer, Waltham, MA, USA) in 100 µl assay buffer for 1 h at room temperature. After washing, bound Eu3+ was detected by the addition of 200 µl of enhancement solution (Perkin Elmer), 5 min of vigorous shaking and reading the time‐resolved fluorescence on a DELFIA fluorometer (Victor3; Perkin Elmer). The limit of detection was 0·05 µg/l and the intra‐ and interassay CV were below 5 and 6%, respectively.
Statistical analysis
Assumptions of normally distributed data were checked with q–q plots and histograms. If data were not normally distributed, log‐transformation was performed before statistical analysis. Results are reported as mean ± standard deviation (s.d.) or median (25th percentile; 75th percentile), as appropriate. Pearson's correlation was used to test the relationship of plasma levels of sMAC, ficolins and CRP with LVEF and infarct size. For the association between MBL and the cardiac outcomes we used the Spearman's rank correlation test due to the non‐normal distributed MBL plasma levels. A t‐test was used to test the difference between two groups, but due to unequal variance between groups when measuring H‐ficolin and sMAC, we used Wilcoxon's rank‐sum test to test our hypothesis.
The statistical analysis was performed by stata version 15.0 for Mac OS, and figures are made in GraphPad Prism version 8.0.1.
Results
Levels of plasma CRP did not vary between the two groups defined in the original study: 22·7 µg/l [95% confidence interval (CI) = 16·9, 28·5] for the PCI and postconditioning group versus 17·4 µg/l (95% CI = 12·1, 22·6) in the PCI group, P = 0·17, nor did the two randomized groups differ when looking at the lectin pathway proteins; see Table 1.
Plasma CRP correlated negatively with LVEF, with a regression coefficient = –0·17, P = 0·01. This was expected, and indicates that higher CRP correspond to worsening and lowering of LVEF; see Fig. 1.
Figure 1.
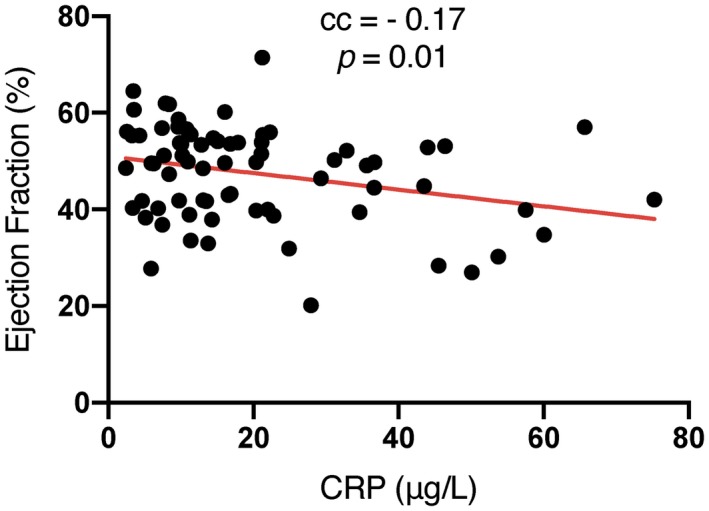
Left ventricular ejection fraction plotted with C‐reactive protein (CRP) on the x‐axis and the fitted linear model in red.
We found no association between LVEF or infarct size and plasma levels of the pattern‐recognition molecules from the lectin pathway or sMAC (Table 2). When stratifying the patients into the original randomized groups the cardiac outcomes remained without an association with complement factors (results not depicted).
Table 2.
Linear regression, correlation analyses.
| LVEF | Infarct size | |||
|---|---|---|---|---|
| Regression coefficient (95% CI) | P‐value | Regression coefficient (95% CI) | P‐value | |
| MBL (µg/l) | Spearman | P = 0·95 | Spearman | P = 0·71 |
| M‐ficolin (µg/l) | –1·88 (–10·8, 7·07) | P = 0·67 | –0·24 (–15·41, 14·92) | P = 0·97 |
| L‐ficolin (µg/l) | 0·00 (–0·00, 0·00) | P = 0·16 | –0·00 (–0·00, 0·00) | P = 0·74 |
| H‐ficolin (µg/l) | 0·00 (–0·00, 0·00) | P = 0·42 | –0·00 (–0·00, 0·00) | P = 0·94 |
| sMAC (µg/l) | 0·00 (–0·02, 0·02) | P = 0·83 | 0·02 (–0·02, 0·05) | P = 0·30 |
| CRP (µg/l) | –0·17 (–0·31, –0·04) | P = 0·01 | 0·14 (–0·10, 0·38) | P = 0·25 |
Pearson's correlation was applied to check if complement proteins correlated with left ventricular ejection fraction (LVEF) or infarct size. The regression coefficient was determined and the corresponding 95% confidence interval (CI). Spearman's rank correlation test is marked with ‘Spearman’.
In order to investigate if the lectin pathway alter cardiac function after MI, we divided patients into two groups; low ejection fraction (<50%) and normal ejection fraction (≥50%) in accordance with the American Heart Association 22. We found no differences in plasma levels of sMAC, MBL, M‐, L‐ and H‐ficolins or CRP when comparing patients with EF <50% (n = 40) with patients with EF ≥50% (n = 33); see Fig. 2 for details.
Figure 2.
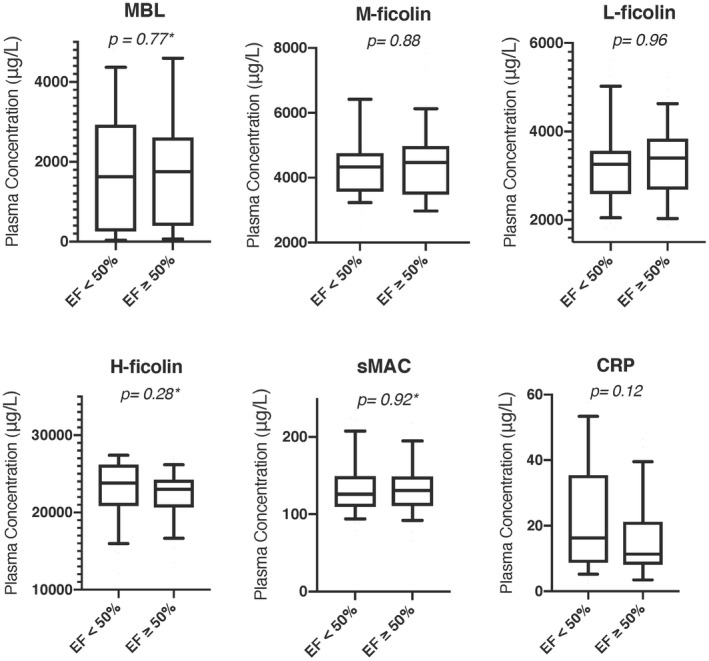
Plasma concentration of mannan‐binding lectin (MBL), M‐ficolin, L‐ficolin, H‐ficolin, soluble membrane attack complex (sMAC) and C‐reactive protein (CRP) given with 10th percentile, first quartile, median, third quartile and 90th percentile and divided into ejection fraction <50% and ejection fraction ≥50%.
Finally, we examined if the patients deficient in MBL (<100 ng/ml) differed in LVEF or infarct size from the patient with sufficient MBL. We found no differences between these two groups.
Discussion
In this study we show that the plasma level of CRP measured the day after PCI in STEMI patients is negatively associated with LVEF (Fig. 1). The present findings are in line with previous studies 23, 24, which have shown that increasing CRP levels are correlated with decreased left ventricular function; however, when stratifying the patients into LVEF above and below 50%, the disadvantages with having a high CRP level disappear in our study (Fig. 2).
Although inflammation is known to play an important role after MI, the exact inflammatory mechanism is not clear. Several studies using animal models support a role for the lectin pathway in the severity of MI after reperfusion 9, 10, 25, but only a limited number of human clinical studies have investigated this. Even though we found an association between inflammation (CRP) and LVEF, the pattern‐recognition molecules of the lectin pathway and sMAC, as a measure of downstream complement activity, was not associated with LVEF or infarct size following MI. However, our study cannot rule out local production of complement proteins and/or local capture of the lectins to neoepitopes in the heart. In theory, this may contribute to local tissue damage and will not necessarily be reflected in the circulation. The pathogenesis of apoptosis of cardiomyocytes following MI is highly complex, and is probably the net result of insufficient reperfusion, inflammatory response, reduced clearance and complement activation 26. As an example, pentraxins (e.g. CRP, PTX3) are also described as pattern‐recognition molecules and are reported to be capable of modulating the complement system both via the classical and lectin pathways, and may thus be involved in the inflammatory processes 27, 28. Due to the pleiotropic effects of PTX3, it is still debated if PTX3 is cardioprotective or a negative prognostic factor in cardiovascular diseases 29.
A potential association between lectin pathway proteins and LVEF or infarct size could, in theory, be obliterated by selection bias. As an example, the inclusion criteria require a TIMI grade at 0, which is defined as ‘no perfusion’ in the coronary artery involved. The STEMI patients are therefore a very homogeneous group, which narrows the variability of the two outcomes, LVEF and infarct size.
By determining sMAC levels, we assume that this reflects the activation of the entire complement activation. However, we do not know if MAC is deposited locally in the heart and not reflected by the soluble form.
No pharmaceutical drug targeting the complement system is approved in MI treatment. Panagiotou et al. 5 have reviewed clinical trials using the complement inhibitor C1 esterase inhibitor (C1INH) in human myocardial I/R injury 30, 31. C1INH is an obvious first choice in the search for treatment against IR injuries, as it has an additional effect by preventing cells to alter into procoagulatory and anti‐fibrolytic states 32. The most recent clinical study, a randomized clinical trial with 78 STEMI patients, showed a significantly better myocardial contractility in the group receiving plasma derived C1INH compared to placebo 33. Thus, pdC1INH is safe, with no adverse effects reported, and so far the clinical studies indicate that C1 inhibition is effective. These clinical trials have not been conducted with the state‐of‐the‐art revascularization, such as stents and anti‐thrombotic treatment, which is questioning if any additive effect of C1 inhibition remains when testing together with state‐of‐the‐art treatment. In the same trial, C3a and C4a plasma concentrations are increasing during and after surgery (coronary artery bypass grafting). These concentrations are reaching a maximum concentration approximately 2 h after surgery. This suggests that the complement system is mainly activated during and immediately after reperfusion.
The association between MBL and cardiac I/R injury is contentious. Personen et al. found higher serum levels of MBL and lower serum concentration of C3 in a cohort of patients with unstable angina pectoris or acute MI when compared with healthy controls 11. Our data confirmed this, with higher serum levels of MBL and lower sMAC in patients with STEMI as reported in healthy controls 21. Plasma MBL levels could be persistently higher in patients with high MI risk or it might be an acute immunological response due to cardiac ischaemia, thereby adversely causing enhanced thrombosis and complement‐mediated IR injury. Other studies suggest MBL as being protective in the development of acute coronary syndrome, as it lowers the atherosclerosis by eliminating infection and clearing cellular debris and apoptotic cells. Saevarsdottir et al. found that high MBL levels were associated with decreased risk of MI 12, and Vengen et al. found that individuals with MBL‐deficiency associated genotypes had twice the risk of developing a MI 34.
In the present study we did not focus on whether MBL or ficolin levels were higher or lower in patients with STEMI compared to healthy controls, but merely studied the impact of the lectin pathway on short‐term cardiac outcomes, LVEF and infarct size after ischaemia. Our study shows no correlation between levels of the initiating molecules of the lectin pathway, nor the final complement activation end‐product and the magnitude of the cardiac damage. This support our previous finding, that even though the lectin pathway components are elevated in patients with MI, they are not directly associated with the extent of cardiac damage 14.
The present findings must be carefully interpreted due to the cross‐sectional design and small number of patients. In particular, it is unclear whether the plasma levels of complement factors are stable and represent the level prior to the MI or if local inflammation have caused acute alteration in the complement system during MI. Furthermore, peak CRP levels would have been more appropriate to examine due to liver production latency.
In conclusion, this study demonstrates that structural and functional damage following ischaemia are not associated with physiological variations in plasma concentration of MBL or ficolins. In the acute phase, the plasma levels of the pattern‐recognition molecules of the lectin pathway (MBL and ficolins) or the end‐product of total complement activation (sMAC) do not appear to be of predictable prognostic value of short‐term cardiac outcomes after MI. Thus, it seems that once cardiac ischaemia has occurred, the patients with low plasma levels of either MBL or ficolins are not protected against the consequences of cardiac I/R compared to the patients with high plasma levels.
Future research must aim for randomized clinical trial using up‐to‐date revascularization and testing a specific lectin pathway inhibitor. Furthermore, to evaluate complement proteins in MI patients as a prognostic marker it is important to obtain bigger sample sizes and retrieve plasma samples during or immediately after the reperfusion.
Disclosures
The authors declare that there are no conflicts of interest concerning the publication of this paper.
Author contributions
P. S., L. M., T. K. H. and M.B. designed the study. The laboratory work was conducted by C. B. H. and the above‐mentioned laboratory technicians with supervision from S. T. and M. B. C. B. H. conducted the analysis of raw data and performed the statistics with supervision from J. A. Ø. and M. B. All authors contributed to writing this article.
Acknowledgements
The authors thank Lisbeth Jensen and Annette G. Hansen at the Department of Microbiology and Immunology, Aarhus University, and Zahra Partovi Nasr at the Medical Research Laboratory, Aarhus University, for laboratory assistance. This study was supported by Aarhus University.
References
- 1. The top 10 causes of death [internet] . 2018. Available at: https://www.who.int/news-room/factsheets/detail/diabetes (accessed 27 May 2019).
- 2. Szummer K, Wallentin L, Lindhagen L et al Improved outcomes in patients with ST‐elevation myocardial infarction during the last 20 years are related to implementation of evidence‐based treatments: experiences from the SWEDEHEART registry 1995–2014. Eur Heart J 2017; 38:3056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol 2009; 54:2129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carter AM. Inflammation, thrombosis and acute coronary syndromes. Diabetes Vascular Dis Res 2005; 2:113–21. [DOI] [PubMed] [Google Scholar]
- 5. Panagiotou A, Trendelenburg M, Osthoff M. The lectin pathway of complement in myocardial ischemia/reperfusion injury‐review of its significance and the potential impact of therapeutic interference by C1 esterase inhibitor. Front Immunol 2018; 9:1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stover CM, Thiel S, Thelen M et al Two constituents of the initiation complex of the mannan‐binding lectin activation pathway of complement are encoded by a single structural gene. J Immunol 1999; 162:3481–90. [PubMed] [Google Scholar]
- 7. Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol 2003; 21:547–78. [DOI] [PubMed] [Google Scholar]
- 8. Henriksen ML, Brandt J, Andrieu JP et al Heteromeric complexes of native collectin kidney 1 and collectin liver 1 are found in the circulation with MASPs and activate the complement system. J Immunol 2013; 191:6117–27. [DOI] [PubMed] [Google Scholar]
- 9. Collard CD, Vakeva A, Morrissey MA et al Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol 2000; 156:1549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clark JE, Dudler T, Marber MS, Schwaeble W. Cardioprotection by an anti‐MASP‐2 antibody in a murine model of myocardial infarction. Open Heart 2018; 5:e000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pesonen E, Hallman M, Sarna S et al Mannose‐binding lectin as a risk factor for acute coronary syndromes. Ann Med 2009; 41:591–8. [DOI] [PubMed] [Google Scholar]
- 12. Saevarsdottir S, Oskarsson OO, Aspelund T et al Mannan binding lectin as an adjunct to risk assessment for myocardial infarction in individuals with enhanced risk. J Exp Med 2005; 201:117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schoos MM, Munthe‐Fog L, Skjoedt MO et al Association between lectin complement pathway initiators, C‐reactive protein and left ventricular remodeling in myocardial infarction‐a magnetic resonance study. Mol Immunol 2013; 54:408–14. [DOI] [PubMed] [Google Scholar]
- 14. Holt CB, Thiel S, Munk K, Ostergaard JA, Botker HE, Hansen TK. Association between endogenous complement inhibitor and myocardial salvage in patients with myocardial infarction. Eur Heart J Acute Cardiovasc Care 2014; 3:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frauenknecht V, Thiel S, Storm L et al Plasma levels of mannan‐binding lectin (MBL)‐associated serine proteases (MASPs) and MBL‐associated protein in cardio‐ and cerebrovascular diseases. Clin Exp Immunol 2013; 173:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang M, Hou YJ, Cavusoglu E et al MASP‐2 activation is involved in ischemia‐related necrotic myocardial injury in humans. Int J Cardiol 2013; 166:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sorensson P, Saleh N, Bouvier F et al Effect of postconditioning on infarct size in patients with ST elevation myocardial infarction. Heart 2010; 96:1710–5. [DOI] [PubMed] [Google Scholar]
- 18. Thiel S, Moller‐Kristensen M, Jensen L, Jensenius JC. Assays for the functional activity of the mannan‐binding lectin pathway of complement activation. Immunobiology 2002; 205:446–54. [DOI] [PubMed] [Google Scholar]
- 19. Wittenborn T, Thiel S, Jensen L, Nielsen HJ, Jensenius JC. Characteristics and biological variations of M‐ficolin, a pattern recognition molecule, in plasma. J Innate Immun 2010; 2:167–80. [DOI] [PubMed] [Google Scholar]
- 20. Krarup A, Sorensen UB, Matsushita M, Jensenius JC, Thiel S. Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan‐binding lectin, L‐ficolin, and H‐ficolin. Infect Immun 2005; 73:1052–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haahr‐Pedersen S, Bjerre M, Flyvbjerg A et al Level of complement activity predicts cardiac dysfunction after acute myocardial infarction treated with primary percutaneous coronary intervention. J Invasive Cardiol 2009; 21:13–9. [PubMed] [Google Scholar]
- 22. Ejection Fraction Heart Failure Measurements [internet]. 2017. Available at:: https://www.heart.org/en/health-topics/heart-failure (accessed 27 May 2019).
- 23. Vanhaverbeke M, Veltman D, Pattyn N et al C‐reactive protein during and after myocardial infarction in relation to cardiac injury and left ventricular function at follow‐up. Clin Cardiol 2018; 41:1201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reindl M, Reinstadler SJ, Feistritzer HJ et al Relation of inflammatory markers with myocardial and microvascular injury in patients with reperfused ST‐elevation myocardial infarction. Eur Heart J Acute Cardiovasc Care 2017; 6:640–9. [DOI] [PubMed] [Google Scholar]
- 25. Jordan JE, Montalto MC, Stahl GL. Inhibition of mannose‐binding lectin reduces postischemic myocardial reperfusion injury. Circulation 2001; 104:1413–8. [DOI] [PubMed] [Google Scholar]
- 26. Griselli M, Herbert J, Hutchinson WL et al C‐reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med 1999; 190:1733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nauta AJ, Bottazzi B, Mantovani A et al Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur J Immunol 2003; 33:465–73. [DOI] [PubMed] [Google Scholar]
- 28. Gout E, Moriscot C, Doni A et al M‐ficolin interacts with the long pentraxin PTX3: a novel case of cross‐talk between soluble pattern‐recognition molecules. J Immunol 2011; 186:5815–22. [DOI] [PubMed] [Google Scholar]
- 29. Chu Y, Teng J, Feng P, Liu H, Wang F, Li X. Pentraxin‐3 in coronary artery disease: a meta‐analysis. Cytokine 2019; 119:197–201. [DOI] [PubMed] [Google Scholar]
- 30. Bauernschmitt R, Bohrer H, Hagl S. Rescue therapy with C1‐esterase inhibitor concentrate after emergency coronary surgery for failed PTCA. Intensive Care Med 1998; 24:635–8. [DOI] [PubMed] [Google Scholar]
- 31. de Zwaan C, Kleine AH, Diris JH et al Continuous 48‐h C1‐inhibitor treatment, following reperfusion therapy, in patients with acute myocardial infarction. Eur Heart J 2002; 23:1670–7. [DOI] [PubMed] [Google Scholar]
- 32. Zhang S, Shaw‐Boden J, Banz Y et al Effects of C1 inhibitor on endothelial cell activation in a rat hind limb ischemia‐reperfusion injury model. J Vasc Surg 2018; 68(Suppl 6):209S–221S.e2. [DOI] [PubMed] [Google Scholar]
- 33. Fattouch K, Bianco G, Speziale G et al Beneficial effects of C1 esterase inhibitor in ST‐elevation myocardial infarction in patients who underwent surgical reperfusion: a randomised double‐blind study. Eur J Cardiothorac Surg 2007; 32:326–32. [DOI] [PubMed] [Google Scholar]
- 34. Vengen IT, Madsen HO, Garred P, Platou C, Vatten L, Videm V. Mannose‐binding lectin deficiency is associated with myocardial infarction: the HUNT2 study in Norway. PLOS ONE 2012; 7:e42113. [DOI] [PMC free article] [PubMed] [Google Scholar]