

Summary
A20, a pivotal anti‐inflammatory protein, preserves immune homeostasis and regulates prolonged inflammation. A previous study has shown that A20 expression levels are down‐regulated in peripheral blood mononuclear cells (PBMCs) from patients with ankylosing spondylitis (AS). However, the precise role of A20 in reducing autoimmune disorders needs to be further elucidated. In this study, A20 expression was found to be preferentially reduced on circulating CD56bright natural killer (NK) cells in patients with AS, and its level was negatively correlated with that of proinflammatory cytokines. Further investigation demonstrated that A20 reduces interferon (IFN)‐γ and tumour necrosis factor (TNF)‐α production in CD56bright NK cells after stimulation with monokines or phorbol myristate acetate (PMA)/ionomycin(P/I). Furthermore, CD56bright NK cells isolated from AS patients promote TNF‐α secretion by autologous monocytes, and increasing the A20 expression level partially attenuates this process. More importantly, decreased A20 expression on circulating CD56bright NK cells is associated with worse disease status in patients with AS. Our findings reveal that A20 participates in the pathogenesis of AS by negatively regulating CD56bright NK cells and that its reduced expression contributes to a worsened disease status in patients with AS.
Keywords: A20, ankylosing spondylitis, CD56bright NK cells, disease activity, IFN‐γ, TNF‐α
Introduction
A20, encoded by TNFAIP3, is a pivotal anti‐inflammatory protein that inhibits nuclear factor (NF)‐κB activity 1, 2. It has been shown that A20 is tightly associated with the pathogenesis of several autoimmune diseases, including systemic lupus erythematosus (SLE), inflammatory bowel disease and rheumatoid arthritis (RA) 3, 4, 5, 6. Mice lacking A20 in dendritic cells (DCs) spontaneously develop lymphocyte‐dependent inflammatory bowel disease (IBD) and IBD‐associated seronegative ankylosing arthritis 2. A study using the genomewide association method suggested that TNFAIP3 is also associated with psoriasis and/or psoriatic arthritis 7. More importantly, clinical investigations have revealed that A20 mRNA expression in peripheral blood mononuclear cells (PBMCs) from patients with ankylosing spondylitis (AS) is down‐regulated and its expression level has been shown to be a strong predictive marker for AS, indicating that A20 plays a key role in the pathogenesis of AS 8. However, the clear association between A20 and AS needs to be further investigated.
Ankylosing spondylitis (AS) is characterized by inflammatory arthritis with erosive and osteoproliferative bone changes in the sacroiliac joints and vertebral bodies of the spine 8, 9. Several studies have shown that cytokines, particularly tumour necrosis factor (TNF)‐α and interferon (IFN)‐γ, play pivotal pathological roles in AS 10, 11. Interestingly, both TNF‐α and IFN‐γ can significantly activate the promoter of human leucocyte antigen (HLA)‐B27, indicating that they might contribute to the onset of AS 12. Recently, natural killer (NK) cells have been shown to play a key role in the pathogenesis of autoimmune diseases 13. Human NK cells are a heterogeneous population that can generally be distinguished as CD56dim and CD56bright subsets. The CD56dim subset comprises approximately 90% of the circulating NK cells. This subset is mainly involved in cytotoxicity responses. In contrast, the CD56bright subset accounts for 10% of NK cells in the peripheral blood, and primarily exerts immunoregulatory functions by producing certain cytokines, particularly IFN‐γ and TNF‐α 14, 15, 16. A20 has been shown to function as a proinflammatory cytokine inhibitor in several cell types 17, 18. However, it is still unclear whether A20 is essential for the functional performance of the CD56bright NK cell subset, and thus contributes to the pathogenesis of AS.
In this study, we identified that the expression of A20 on circulating NK cells is decreased preferentially on the CD56bright subset. Importantly, the surface A20 level is negatively correlated with serum IFN‐γ and TNF‐α. Moreover, A20 suppresses the secretion of IFN‐γ and TNF‐α in the process of cytokine‐induced CD56bright NK cell activation. Furthermore, by co‐culturing circulating CD14+ monocytes and the CD56bright NK cell subset, we identified that A20 is responsible for ameliorating the response of monocytes to monokines. Moreover, decreased A20 expression on circulating CD56 bright NK cells is associated with disease activity in patients with AS, suggesting that decreased A20 levels might initiate the activation of CD56 bright NK cells and thus contribute to the pathogenesis of AS. In summary, our findings suggest that reduced A20 expression on circulating CD56bright NK cells is a contributory factor in the pathogenesis of AS and will be a useful prognostic indicator and potential therapeutic target in the future.
Methods
Human subjects
Fifty patients with AS who were not receiving any treatment and who fulfilled the modified 1984 New York criteria for a diagnosis of AS 42 were enrolled from Zibo Central Hospital Affiliated to Shandong University. The 50 age‐ and sex‐matched healthy subjects were randomly recruited from Zibo Central Hospital Medical Center and confirmed to not have any autoimmune and inflammatory diseases. Each subject provided informed consent to join the study. All procedures involving study participants were approved by the Human Research Ethics Committee of Zibo Central Hospital Affiliated to Shandong University.
Flow cytometry analysis and cell sorting
PBMCs were isolated from peripheral blood and stained for CD3 (#300327; Biolegend, San Diego, CA, USA), CD16 (#302011; Biolegend) and CD56 (#304605; Biolegend) for the detection of NK cells. The circulating CD56bright subset and monocytes were sorted by a fluorescence activated cell sorter (FACS)‐Aria II flow cytometer (Becton Dickinson, San Jose, CA, USA) and their purity was always greater than 98% by flow cytometry. The isolated CD56bright subset was incubated in RPMI‐1640 supplemented with 10% fetal calf serum (FCS) (GIBCO, Carlsbad, ca, usa), 1% penicillin/streptomycin, 1% L‐glutamine (gibco) and 200 IU/ml recombinant IL‐2 (Sigma, St Louis, MO, USA) in 5% CO2 at 37°C. Data were analysed using FlowJo software (TreeStar Inc., Ashland, OR, USA. The results were expressed as the percentage of positive cells.
Staining for the expression of intracellular A20, IFN‐γ and TNF‐α
After surface staining, as described above, mononuclear cells were fixed using Inside Fix (Miltenyi Biotec, Auburn, CA, USA) containing 3·7% formaldehyde and incubated for 20 min at room temperature, washed twice using magnetic activated cell sorting (MACS) bovine serum albumin (BSA) solution (Miltenyi Biotec), and then stained with the respective antibody (A20, #ab197541, Abcam, Cambridge, MA, USA; IFN‐γ, #502509, Biolegend; TNF‐α, #502909, Biolegend) in Inside Perm for 30 min. Cells were washed, resuspended in MACS BSA solution and analysed on a FACSCalibur as described above.
Enzyme‐linked immunosorbent assay (ELISA)
Serum or culture supernatant were separated and prepared for analysis. The concentrations of IL‐1, IL‐6, TNF‐α and IFN‐γ (eBioscience) were analysed by ELISA, as indicated by the manufacturer. The intra‐ and interassay coefficients of variation (CV) were all <10% and two duplicate wells were prepared for each sample.
Detection of IFN‐γ and TNF‐α levels in CD56bright NK cells
Paired samples of CD56bright NK cells isolated from 10 active AS patients and 10 healthy controls were incubated in culture medium alone or in culture medium supplemented with IL‐12 (10 ng/ml) and IL‐18 (10 ng/ml) for 24 h at 37°C for the detection of IFN‐γ. For TNF‐α detection, the cells were stimulated with 1 μg/ml phorbol myristate acetate (PMA) and 0·5 μg/ml ionomycin for 5 h at 37°C. Finally, the cells were washed and stained with anti‐IFN‐γ or anti‐TNF‐α antibodies, and the cells were analysed on a FACSCalibur, as described above.
Co‐culture of circulating CD56bright NK cell subset and monocytes
The purified circulating CD56bright NK cell subset and autologous CD14+ monocytes were co‐cultured at a 1 : 1 ratio in complete medium with or without IL‐12 (10 ng/ml; Biolegend), IL‐15 (10–ng/ml; Biolegend) and IL‐18 (10 ng/ml; Biolegend) and incubated for 24 h at 37°C, according to previous research 21. After cell harvesting, monocytes were stained for the surface expression of CD14 (Biolegend) and intracellular expression of TNF‐α. To further clarify the functions of A20, CD56bright NK cells from AS patients over‐expressing A20 or empty vector were then co‐cultured with monocytes, as described above.
Western blot analysis
The cultured NK cells were collected and lysed in ice‐cold lysis buffer. Protein concentrations were measured by using a bicinchoninic acid Protein Assay Kit (Pierce, Rockford, IL, USA). The protein was separated by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis, and then electrotransferred onto polyvinylidene fluoride membranes (Millipore, Billerica, CA, USA). The following primary antibodies were used: anti‐A20 (1 : 1000 dilution, ab13597; Abcam) and anti‐glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (1 : 2000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Next, the membranes were incubated with a horseradish peroxidase (HRP)‐conjugated anti‐rabbit immunoglobulin (Ig)G antibody (1 : 5000 dilution; Cell Signaling Technology, Danvers, MA, USA) for 1 h, followed by enhanced chemiluminescence (Santa Cruz Biotechnology) and autoradiography.
Laboratory measurements
For all AS patients, the erythrocyte sedimentation rates (ESRs) were determined by the Westergren method. Plasma C‐reactive protein (CRP) levels were obtained by immunoturbidimetry measured with a Beckman Coulter biochemical analyser.
Statistical analysis
Statistical analysis was performed with GraphPad Prism version 7.0 (GraphPad Software Inc., San Diego, CA, USA). Student’s t‐test analysis was used for differences between two groups, while Spearman’s rank test was used to assess the association between two ranked variables. A P‐value <0·05 was considered statistically significant for all tests.
Results
A20 expression is preferentially decreased on circulating CD56bright subset in patients with AS
To preliminarily determine the effects of A20 on NK cells in patients with AS, its expression level on circulating total NK cells, CD56bright and CD56dim subsets was determined by flow cytometry for each patient and control. As shown in Fig. 1, we observed no significant difference in A20 expression in the total NK cells (Fig. 1a, 1) and CD56dim subset (Fig. 1c). However, A20 expression on the CD56bright subset was dramatically decreased compared to that of controls (Fig. 1d), indicating that A20 might participate in the pathogenesis of AS by driving CD56bright subset NK cells.
Figure 1.
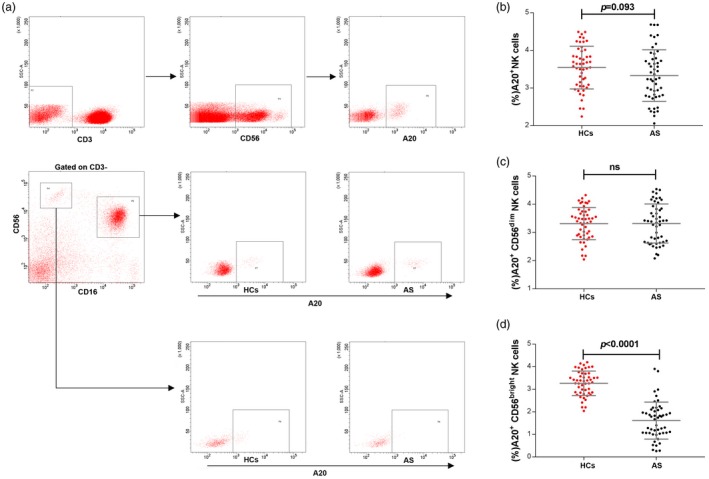
A20 expression on circulating natural killer (NK) cells in patients with ankylosing spondylitis (AS). (a) CD3 and CD56 staining was used to determine NK cells. (b) Graph comparing A20 expression on total NK cells from patients with AS (n = 50) and healthy controls (HCs, n = 50). (c) Graph comparing A20 expression on CD56dim NK cells from patients with AS and healthy controls. (d) Graph comparing A20 expression on CD56bright NK cells from patients with AS and healthy controls. Cumulative data are shown as the mean ± standard deviation (s.d.). A P‐value <0·05 was considered statistically significant for the test; n.s. = not significant by unpaired t‐test.
A20 expression level on the CD56bright subset is inversely correlated with serum TNF‐α and IFN‐γ
Because CD56bright NK cells are characterized as ‘immunoregulatory cells’ based on their ability to secrete cytokines, and A20 serves as a proinflammatory cytokine inhibitor 17, we assumed that reduced A20 expression on the circulating CD56bright subset might result in increased proinflammatory cytokine secretion in patients with AS. Therefore, we analysed the correlations between A20 levels and the serum proinflammatory cytokines, including TNF‐α, IFN‐γ, IL‐1β and IL‐6. Interestingly, A20 expression on the CD56bright subset was negatively correlated with IFN‐γ and TNF‐α, but not IL‐1 and IL‐6 (Fig. 2a–d), although the latter had a negative correlation tendency.
Figure 2.
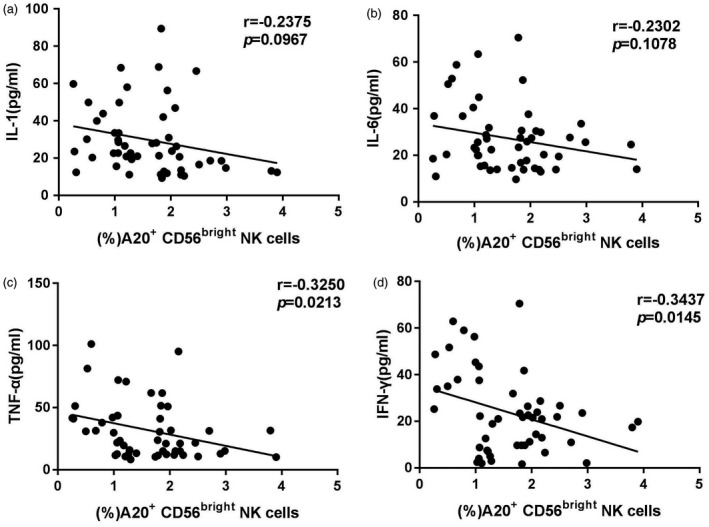
Correlation analysis of A20 expression on the circulating CD56bright natural killer (NK) subset with interleukin (IL)‐1, IL‐6, tumour necrosis factor (TNF)‐α and interferon (IFN)‐γ in patients with ankylosing spondylitis (AS). (a,b) A negative tendency but no significant correlations were found between A20 expression on the CD56bright subset and serum IL‐1 or IL‐6 in patients with AS. (c,d) Significant negative correlations between A20 expression on the CD56bright subset and serum TNF‐α or IFN‐γ in patients with AS. Statistical confidence was analysed by Spearman’s rank test. A P‐value <0·05 was considered statistically significant for the test.
Decreased A20 expression drives CD56bright NK cells to produce more IFN‐γ and TNF‐α
Because A20 expression on the CD56bright subset is inversely correlated with serum TNF‐α and IFN‐γ, we isolated the CD56bright subset from 10 active AS patients and 10 healthy controls, and then stimulated the cells with monokines or PMA/ionomycin(P/I) to observe the IFN‐γ and TNF‐α secretion levels, respectively. Compared to controls, as shown in Fig. 3a, the expression frequency of IFN‐γ on the CD56bright subset treated with IL‐12+IL‐18 was dramatically increased in patients with AS (left panel), and the elevated IFN‐γ level in the culture supernatant was observed in patients with AS (right panel). Similar results were observed for the TNF‐α level (Fig. 3B). Furthermore, we transfected the A20 lentiviral vector into CD56bright NK cells, and then repeated the experiment. The efficiency of A20 over‐expression was determined by Western blot analysis (Fig. 3c) and flow cytometry method (Fig. 3d). Unsurprisingly, the IFN‐γ and TNF‐α levels on the CD56bright NK cells were dramatically lower than the controls (Fig. 3e,f). Together, these results indicate that CD56bright NK cells secrete abundant levels of IFN‐γ and TNF‐α due primarily to decreased A20 expression.
Figure 3.
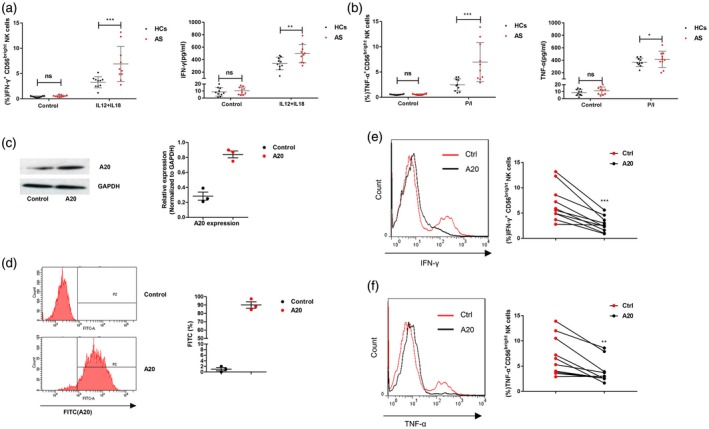
Circulating CD56bright subset natural killer (NK) cells from patients with ankylosing spondylitis (AS) produce abundant amounts of interferon (IFN)‐γ and tumour necrosis factor (TNF)‐α after stimulation. (a) Left panel: percentage of IFN‐γ+ CD56bright subset NK cells isolated from patients with AS (n = 10) or healthy controls (n = 10) after IL‐12+IL‐18 stimulation. The control group cells were treated with medium. Right panel: IFN‐γ concentration in the culture supernatant. (b) Left panel: percentage of TNF‐α+ CD56bright subset NK cells isolated from patients with AS (n = 10) or healthy controls (n = 10) after phorbol myristate acetate (PMA)+ionomycin (P/I) stimulation. The control group cells were treated with medium. Right panel: TNF‐α concentration in the culture supernatant. The efficiency of A20 over‐expression was determined by Western blot analysis (c) and flow cytometry (d) (the experiments were repeated three times). (e) Percentage of IFN‐γ+ CD56bright subset NK cells transfected with A20 lentiviral vector (A20) or empty vector (Ctrl) after treatment with IL‐12+IL‐18. (f) Percentage of TNF‐α+ CD56bright subset NK cells transfected with A20 or Ctrl after treatment with P/I. Values represent the mean ± standard deviation (s.d.); for all panels, *P < 0·05, **P < 0·01 and ***P < 0·001; n.s. = not significant by unpaired t‐test.
A20 can attenuate mononuclear cell production of TNF‐α that is stimulated by the CD56bright NK cell subset
TNF‐α plays a pivotal role in the pathogenesis of AS and the administration of TNF‐α antagonists, including etanercept or infliximab, has provided effective results in clinical practice 19, 20. A previous study demonstrated that NK cells, particularly the CD56bright subset, can stimulate monocytes to produce TNF‐α partly due to the elevated IFN‐γ levels 21. Thus, we speculated that the CD56bright NK cell subset from patients with AS can regulate monocytes to produce more TNF‐α compared to that from healthy controls. Therefore, we freshly isolated the circulating CD56bright subset NK cells and co‐cultured them with autologous CD14+ monocytes at a ratio of 1 : 1. After co‐culturing for 24 h, the CD56bright NK cells isolated from AS patients triggered monocytes to produce more TNF‐α than those from healthy controls. As shown in Fig. 4a,b, the levels of TNF‐α were increased both in CD14+ monocytes and the culture supernatant in response to stimulation compared to the controls. Furthermore, we transfected the A20 lentiviral vector into CD56bright NK cells and then co‐cultured them with monocytes, the elevation in TNF‐α levels was partly abrogated (Fig. 4c). These results indicate that decreased A20 levels not only stimulate CD56bright NK cells to produce more TNF‐α, but also stimulate monocytes to secrete a large amount of TNF‐α. A study has illustrated that CD56bright NK cells activate monocytes to produce increased levels of TNF‐α due partly to the elevated IFN‐γ secreted by CD56bright NK cells 21. Our results also demonstrate that decreased A20 levels induce the production of IFN‐γ in CD56bright NK cells (Fig. 3), which can partly explain why decreased A20 on the CD56bright subset promotes mononuclear cells to produce increased levels of TNF‐α. However, the precise underlying mechanisms need to be further investigated.
Figure 4.
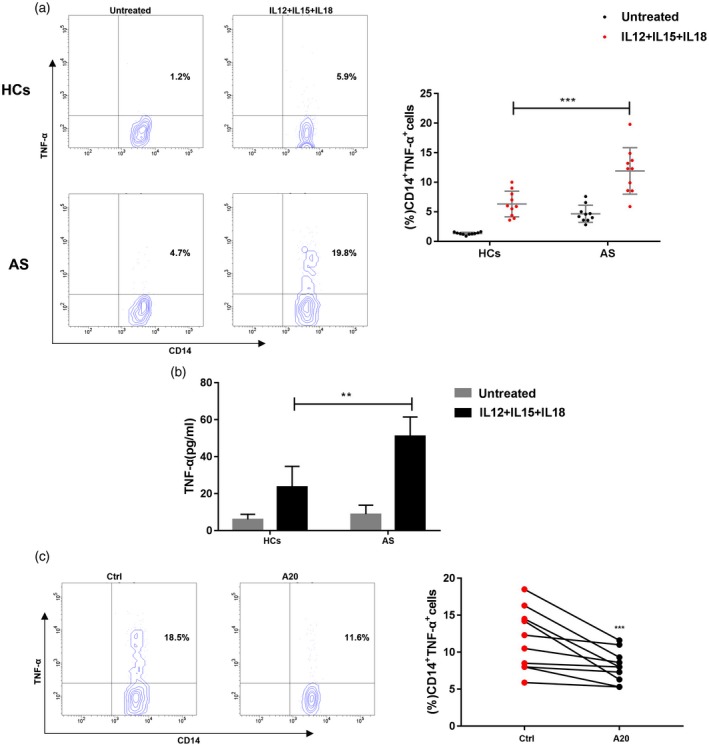
Circulating CD56bright subset from patients with ankylosing spondylitis (AS) stimulates tumour necrosis factor (TNF)‐α production by monocytes. CD56bright natural killer (NK) cell subsets and CD14+ monocytes were isolated from the blood of patients with AS (n = 10) and healthy controls (n = 10) by flow cytometry sorting and co‐cultured at a 1 : 1 ratio in complete medium in the absence (untreated) or the presence of interleukin (IL)‐12, IL‐15 and IL‐18 (IL‐12+IL‐15+IL‐18). (a) Percentage of TNF‐α+ and CD14+ monocytes isolated from patients with AS or healthy controls after IL‐12+IL‐15+IL‐18 stimulation. (b) TNF‐α concentration in the culture supernatant. (c) Percentage of TNF‐α+ and CD14+ monocytes measured by intracellular flow cytometry after transfection with A20 lentiviral vector (A20) or control vector (Ctrl). Values represent the mean ± standard deviation (s.d.); **P < 0·01 and ***P < 0·001.
Decreased A20 expression on circulating CD56bright NK cells is associated with disease activity in patients with AS
Our above results preliminarily demonstrated that decreased A20 expression on CD56bright subset isolated from AS patients regulates cells to produce elevated levels of IFN‐γ and TNF‐α, and thus contributes to the pathogenesis of AS. In this study, we used the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) and AS Disease Activity Score (ASDAS, CRP‐based) to assess disease activity. A20 expression on the surface of CD56bright subset cells was negatively correlated with the BASDAI and ASDAS (Fig. 5). ASDAS and BASDAI both reflect systemic inflammatory disease activity. Therefore, our results suggest that A20 expression on the CD56bright subset may serve as a biomarker of disease activity in the future.
Figure 5.
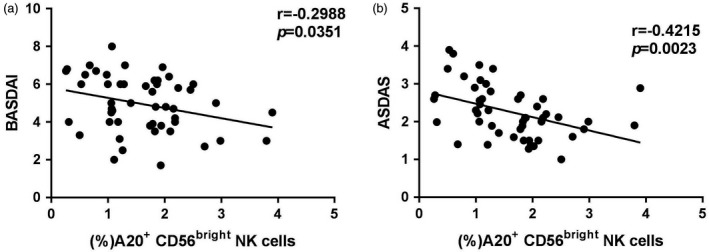
A20 on the circulating CD56bright subset is negatively associated with disease activity. (a) Negative correlations between A20 expression on the CD56bright subset and Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) in ankylosing spondylitis (AS) patients. (b) Significant negative correlations between A20 expression on the CD56bright subset and AS Disease Activity Score (ASDAS) in patients with AS. A P‐value <0·05 was considered statistically significant for the test.
Discussion
In this study, we investigated the correlation between circulating CD56bright NK cells and A20 in patients with AS. Accumulated studies have shown a correlation between autoimmune diseases and NK cells 22, 23. However, NK cells are a unique subset that play different roles in different autoimmune disease models. For instance, NK cells play a protective role in certain autoimmune diseases, whereas they play a pathogenic role in the onset of other autoimmune diseases 13. Furthermore, the proportion of CD56bright or CD56dim NK cells in the peripheral blood of patients with certain autoimmune diseases is similar to that in healthy subjects 24, 25, 26. In contrast, the circulating CD56bright subset is increased in primary Sjogren’s syndrome and is decreased in juvenile dermatomyositis and systemic sclerosis 27, 28, 29. The ubiquitin‐editing enzyme A20 is necessary for the development and functional performance of several immune cells, including B cells, T cells, DCs and macrophages, which play a crucial role in the pathogenesis of autoimmune diseases 30. However, the effect of A20 on the CD56bright subset of NK cells has not been elucidated. Here, we found that the A20 expression on the circulating CD56bright NK subset in patients with AS was dramatically decreased compared to that of controls. Subsequently, we analysed the correlations between A20 expression on the circulating CD56bright NK subset and serum proinflammatory cytokine levels. The results suggested that A20 expression on the CD56bright subset was negatively correlated with serum TNF‐α and IFN‐γ, but not IL‐1β and IL‐6. Therefore, our results demonstrate that the markedly decreased expression of A20 on the circulating CD56bright NK subset might contribute to the chronic invasive immune processes in AS, which initiates aberrant inflammation.
Generally, the CD56bright NK cell subset is considered to be immunoregulatory, and has a great capacity to secrete a large amount of cytokines in the target tissues in autoimmune diseases 31. Accumulated studies have demonstrated that CD56bright NK cells participate in the pathogenesis of numerous autoimmune diseases 32, 33, 34. For instance, the CD56bright NK cell subset is greatly expanded in the synovial fluid of patients with inflammatory arthritis, indicating that CD56bright NK cells maintain inflammatory conditions in autoimmune diseases 25. In this study, we isolated the circulating CD56bright subset of NK cells from 10 active AS patients and 10 healthy controls, and then stimulated them with IL‐18+IL‐12 or PMA/ionomycin. A striking observation in this study was that IFN‐γ and TNF‐α levels were dramatically elevated in patients with AS. To investigate this phenomenon, we transfected the A20 lentiviral vector into CD56bright NK cells and repeated the experiment. The results showed that both the supernatant levels and intracellular expression levels of IFN‐γ and TNF‐α were alleviated compared to those of the controls, suggesting that CD56bright NK cells secrete abundant amounts of IFN‐γ and TNF‐α due primarily to the decreased A20 expression.
Genetic and biochemical studies have suggested that A20 deficiency contributes to systemic inflammation and autoimmunity 35, 36. Strikingly, A20‐deficient haematopoietic stem cells (HSCs) displayed increased IFN‐γ signalling, and deletion of both IFN‐γ and A20 in haematopoietic cells resulted in a partial rescue of the HSC phenotype, suggesting that A20 is an important inhibitor of IFN‐γ secretion 17. In another study, A20‐deficient astrocytes exhibited stronger activation of NF‐κB in response to monokines, and were found to be responsible for accelerating experimental autoimmune encephalomyelitis (EAE) by suppressing NF‐κB and signal transducer and activator of transcription 1 (STAT‐1)‐dependent IFN‐γ and TNF‐α production 18. In this study, we demonstrated that a previously unknown down‐regulation of A20 on CD56bright NK cells contributes to elevated IFN‐γ and TNF‐α levels, and thus contributes to the pathogenesis of AS. However, IFN‐γ and TNF‐α are also produced by cell types other than CD56bright NK cells 37. Our study demonstrated that A20 expression on the CD56bright subset, the key source of proinflammatory cytokines in patients with AS, was negatively correlated with IFN‐γ and TNF‐α, indicating that the decreased A20 might increase the severity of disease, at least partly. Furthermore, monocytes or macrophages were demonstrated to be the predominant source of the proinflammatory cytokines, preferentially producing TNF‐α 38, 39, 40. A previous study also illustrated that NK cells, particularly the CD56bright subset, can stimulate monocytes to produce abundant amounts of TNF‐α 21. Hence, we reasoned that the CD56bright subset isolated from AS patients might drive monocytes to produce more TNF‐α. As shown in Fig. 4, the CD56bright NK cells from AS patients triggered monocytes to produce increased amounts of TNF‐α, and the elevation of TNF‐α levels was partly abrogated after transfection of A20 into CD56bright NK cells. Another study found that CD56bright NK cells activate monocytes to produce abundant levels of TNF‐α due partly to the elevated IFN‐γ secreted by CD56bright NK cells 21. Our results also demonstrate that decreased A20 levels drive the production of IFN‐γ in CD56bright NK cells, which can partly explain why decreased A20 on the CD56bright NK cell subset promotes mononuclear cells to produce increased amount of TNF‐α. However, the precise underlying mechanisms regarding A20 mediated proinflammatory cytokine production in cells need to be further investigated. Our results indicate that elevated levels of TNF‐α in patients with AS are probably induced by decreased A20 expression on CD56bright NK cells, at least in part.
More interestingly, our results demonstrated that decreased A20 expression on the CD56bright NK cell subset isolated from AS patients was associated with disease activity. As shown in Fig. 5, A20 expression on the CD56bright subset was negatively correlated with BASDAI and ASDAS. ASDAS and BASDAI both reflect systemic inflammatory disease activity 41. Compared with BASDAI, ASDAS is generally more strongly associated with inflammatory conditions in peripheral blood and might thus be a better measure of inflammatory activity than BASDAI in patients with AS 41. Our results also revealed that A20 expression on the CD56bright NK subset is associated more with ASDAS than BASDAI, indicating that A20 might be a good indicator of the underlying inflammatory processes in AS. Nevertheless, this case–control study lacks mechanistic investigation and research on the tissue situation. Therefore, the exact role of A20 in AS pathogenesis warrants further investigation.
In summary, as revealed in our study, A20 on the CD56bright NK cell subset plays a protective role in the process of AS by suppressing IFN‐γ and TNF‐α secretion. Furthermore, CD56bright cells isolated from 10 AS patients showed increased IFN‐γ and TNF‐α expression when stimulated and lentiviral transduction of A20 was associated with reduced IFN‐γ and TNF‐α expression in response to subsequent stimulation. A20 ameliorates the response of monocytes to monokines. More interestingly, decreased A20 expression on circulating CD56bright NK cells is associated with disease activity in AS patients, indicating that A20 should be considered a novel therapeutic target for AS.
Disclosures
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province (ZR2019MH099/ZR2015HM031/ZR2016HB25), the Natural Science Foundation of China (Grant nos 81600348/81600695) and the Medicine and Health Science Technology Development Projects of Shandong Province (Grant nos 2018WS534/2016WS0748).
Contributor Information
T. Li, Email: zbszxyyky@163.com
P. Zhao, Email: bzjzzpq@163.com.
References
- 1. Lee EG, Boone DL, Chai S et al Failure to regulate TNF‐induced NF‐kappaB and cell death responses in A20‐deficient mice. Science 2000; 289:2350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hammer GE, Turer EE, Taylor KE et al Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol 2011; 12:1184–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kool M, van Loo G, Waelput W et al The ubiquitin‐editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity 2011; 35:82–96. [DOI] [PubMed] [Google Scholar]
- 4. Billmann‐Born S, Till A, Arlt A et al Genome‐wide expression profiling identifies an impairment of negative feedback signals in the Crohn's disease‐associated NOD2 variant L1007fsinsC. J Immunol 2011; 186:4027–38. [DOI] [PubMed] [Google Scholar]
- 5. Matmati M, Jacques P, Maelfait J et al A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet 2011; 43:908–12. [DOI] [PubMed] [Google Scholar]
- 6. Hah YS, Lee YR, Jun JS et al A20 suppresses inflammatory responses and bone destruction in human fibroblast‐like synoviocytes and in mice with collagen‐induced arthritis. Arthritis Rheum 2010; 62:2313–21. [DOI] [PubMed] [Google Scholar]
- 7. Reveille JD. The genetic basis of spondyloarthritis. Ann Rheum Dis 2011; 70(Suppl 1):i44–50. [DOI] [PubMed] [Google Scholar]
- 8. Duan R, Leo P, Bradbury L, Brown MA, Thomas G. Gene expression profiling reveals a downregulation in immune‐associated genes in patients with AS. Ann Rheum Dis 2010; 69:1724–9. [DOI] [PubMed] [Google Scholar]
- 9. Bleil J, Maier R, Hempfing A, Sieper J, Appel H, Syrbe U. Granulation tissue eroding the subchondral bone also promotes new bone formation in ankylosing spondylitis. Arthritis Rheumatol 2016; 68:2456–65. [DOI] [PubMed] [Google Scholar]
- 10. Smith JA, Barnes MD, Hong D, DeLay ML, Inman RD, Colbert RA. Gene expression analysis of macrophages derived from ankylosing spondylitis patients reveals interferon‐gamma dysregulation. Arthritis Rheum 2008; 58:1640–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ciurea A, Scherer A, Exer P et al Rheumatologists of the Swiss Clinical Quality Management Program for Axial S. Tumor necrosis factor alpha inhibition in radiographic and nonradiographic axial spondyloarthritis: results from a large observational cohort. Arthritis Rheum 2013; 65:3096–106. [DOI] [PubMed] [Google Scholar]
- 12. Zhao L, Fong Y, Granfors K, Gu J, Yu D. Identification of cytokines that might enhance the promoter activity of HLA‐B27. J Rheumatol 2008; 35:862–8. [PubMed] [Google Scholar]
- 13. Gianchecchi E, Delfino DV, Fierabracci A. NK cells in autoimmune diseases: linking innate and adaptive immune responses. Autoimmun Rev 2018; 17:142–54. [DOI] [PubMed] [Google Scholar]
- 14. Yu J, Mao HC, Wei M et al CD94 surface density identifies a functional intermediary between the CD56bright and CD56dim human NK‐cell subsets. Blood 2010; 115:274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Timmons BW, Cieslak T. Human natural killer cell subsets and acute exercise: a brief review. Exercise Immunol Rev 2008; 14:8–23. [PubMed] [Google Scholar]
- 16. Cooper MA, Fehniger TA, Turner SC et al Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001; 97:3146–51. [DOI] [PubMed] [Google Scholar]
- 17. Nakagawa MM, Thummar K, Mandelbaum J, Pasqualucci L, Rathinam CV. Lack of the ubiquitin‐editing enzyme A20 results in loss of hematopoietic stem cell quiescence. J Exp Med 2015; 212:203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X, Deckert M, Xuan NT et al Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF‐kappaB‐ and STAT1‐dependent chemokine production in astrocytes. Acta Neuropathol 2013; 126:711–24. [DOI] [PubMed] [Google Scholar]
- 19. Clegg DO. Treatment of ankylosing spondylitis. J Rheumatol Suppl 2006; 78:24–31. [PubMed] [Google Scholar]
- 20. Zhao P, Wang L, Xiang X et al Increased expression of TIPE2 mRNA in PBMCs of patients with ankylosing spondylitis is negatively associated with the disease severity. Hum Immunol 2017; 78:232–7. [DOI] [PubMed] [Google Scholar]
- 21. Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF. CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol 2004; 173:6418–26. [DOI] [PubMed] [Google Scholar]
- 22. Schleinitz N, Vely F, Harle JR, Vivier E. Natural killer cells in human autoimmune diseases. Immunology 2010; 131:451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bielekova B, Catalfamo M, Reichert‐Scrivner S et al Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL‐2Ralpha‐targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci USA 2006; 103:5941–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pridgeon C, Lennon GP, Pazmany L, Thompson RN, Christmas SE, Moots RJ. Natural killer cells in the synovial fluid of rheumatoid arthritis patients exhibit a CD56bright, CD94bright, CD158negative phenotype. Rheumatology 2003; 42:870–8. [DOI] [PubMed] [Google Scholar]
- 25. Dalbeth N, Callan MF. A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum 2002; 46:1763–72. [DOI] [PubMed] [Google Scholar]
- 26. Daien CI, Gailhac S, Audo R et al High levels of natural killer cells are associated with response to tocilizumab in patients with severe rheumatoid arthritis. Rheumatology 2015; 54:601–8. [DOI] [PubMed] [Google Scholar]
- 27. Rusakiewicz S, Nocturne G, Lazure T et al NCR27/NKp30 contributes to pathogenesis in primary Sjogren’s syndrome. Sci Transl Med 2013; 5:195ra96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ernste FC, Crowson CS, de Padilla CL, Hein MS, Reed AM. Longitudinal peripheral blood lymphocyte subsets correlate with decreased disease activity in juvenile dermatomyositis. J Rheumatol 2013; 40:1200–11. [DOI] [PubMed] [Google Scholar]
- 29. Almeida I, Silva SV, Fonseca AR, Silva I, Vasconcelos C, Lima M. T and NK cell phenotypic abnormalities in systemic sclerosis: a cohort study and a comprehensive literature review. Clin Rev Allergy Immunol 2015; 49:347–69. [DOI] [PubMed] [Google Scholar]
- 30. Zhang M, Peng LL, Wang Y et al Roles of A20 in autoimmune diseases. Immunol Res 2016; 64:337–44. [DOI] [PubMed] [Google Scholar]
- 31. Conigliaro P, Scrivo R, Valesini G, Perricone R. Emerging role for NK cells in the pathogenesis of inflammatory arthropathies. Autoimmun Rev 2011; 10:577–81. [DOI] [PubMed] [Google Scholar]
- 32. Gross CC, Schulte‐Mecklenbeck A, Runzi A et al Impaired NK‐mediated regulation of T‐cell activity in multiple sclerosis is reconstituted by IL‐2 receptor modulation. Proc Natl Acad Sci USA 2016; 113:E2973–E2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laroni A, Armentani E, Kerlero de Rosbo N et al Dysregulation of regulatory CD56(bright) NK cells/T cells interactions in multiple sclerosis. J Autoimmun 2016; 72:8–18. [DOI] [PubMed] [Google Scholar]
- 34. Liu Y, Mu R, Gao YP et al A cytomegalovirus peptide‐specific antibody alters natural killer cell homeostasis and is shared in several autoimmune diseases. Cell Host Microbe 2016; 19:400–8. [DOI] [PubMed] [Google Scholar]
- 35. Catrysse L, Vereecke L, Beyaert R, Van GL. A20 in inflammation and autoimmunity. Trends Immunol 2014; 35:22–31. [DOI] [PubMed] [Google Scholar]
- 36. Zhou Q, Wang H, Schwartz DM et al Loss‐of‐function mutations inTNFAIP3leading to A20 haploinsufficiency cause an early onset autoinflammatory syndrome. Nat Genet 2016; 48:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun G, Yang S, Cao G et al Gammadelta T cells provide the early source of IFN‐gamma to aggravate lesions in spinal cord injury. J Exp Med 2018; 215:521–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee CH, Lam KSL. Obesity‐induced insulin resistance and macrophage infiltration of the adipose tissue: a vicious cycle. J Diabetes Invest 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Efimov GA, Kruglov AA, Khlopchatnikova ZV et al Cell‐type‐restricted anti‐cytokine therapy: TNF inhibition from one pathogenic source. Proc Natl Acad Sci USA 2016; 113:3006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parameswaran N, Patial S. Tumor necrosis factor‐alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr 2010; 20:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pedersen SJ, Sorensen IJ, Garnero P et al ASDAS, BASDAI and different treatment responses and their relation to biomarkers of inflammation, cartilage and bone turnover in patients with axial spondyloarthritis treated with TNFalpha inhibitors. Ann Rheum Dis 2011; 70:1375–81. [DOI] [PubMed] [Google Scholar]
- 42. van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum 1984; 27:361–8. [DOI] [PubMed] [Google Scholar]