

Abstract
The spinocerebellar ataxias are a genetically heterogeneous group of disorders with clinically overlapping phenotypes arising from Purkinje cell degeneration, cerebellar atrophy and varying degrees of degeneration of other grey matter regions. For 22 of the 32 subtypes, a genetic cause has been identified. While recurring themes are emerging, there is no clear correlation between the clinical phenotype or penetrance, the type of genetic defect or the category of the disease mechanism, or the neuronal types involved beyond Purkinje cells. These phenomena suggest that cerebellar Purkinje cells may be a uniquely vulnerable neuronal cell type, more susceptible to a wider variety of genetic/cellular insults than most other neuron types.
The autosomal dominant spinocerebellar ataxias (SCAs) are a genetically and clinically heterogeneous group of neurodegenerative disorders (tables 1 and 2). To date, 32 unique subtypes attributed to distinct genetic loci have been identified, comprising a collective global prevalence of ~4/100 000, with evidence of regional increases in prevalence of some SCAs due to the founder effect.1–4 All subtypes share the common end point of cerebellar and predominantly Purkinje cell degeneration.5 For 22 of the 32 subtypes, specific genetic defects have been identified (tables 3 and 4).5–31 The known genetic causes include CAG repeat expansions encoding expanded polyglutamine repeats in unrelated proteins,6–10,12,13,21 untranslated repeat expansions,14,15,17,31,28 and conventional mutations in critical genes (figure 1).11,16,18–20,23,25–27,30
Table 1.
The mixed spinocerebellar ataxias
| SCA | Phenotype | Age of onset: mean (range) |
Predominant geographical distribution5 |
|---|---|---|---|
| Mixed | |||
| SCA1 | CA/spasticity/ophthalmoplegia6 | 33 (4–74)7,38 | South Africa, India, Japan, Italy, Australia |
| SCA2 | CA/dystonia/parkinsonsism/arreflexia/loss of saccades39 | 32 (1–65)39,40 | USA, Spain, India, Mexico, Italy |
| SCA3 | CA/dystonia/spasticity/peripheral neuropathy/sleep disorders41 | 36 (5–70)10,41 | Most common worldwide |
| SCA4 | CA/motor and sensory neuropathy42 | 39 (19–72)43 | USA,44 Germany45 |
| SCA7 | RP/CA46 | 18–41 (0–70)13,46 | Finland, Mexico, South Africa, UK, Belgium, France, Germany, Japan |
| SCA8 | CA/EP/spasticity47 | 40 (1–73)47,48 | USA/Finland |
| SCA10 | CA/seizures49 | 36 (10–49)49,50 | Mexico, Brazil |
| SCA12 | CA/EP51 | 35 (8–55)51,52 | India |
| SCA13 | Variable18 | Childhood (0–45)18,53 | France, Philippines |
| SCA14 | CA/axial myoclonus/dystonia19 | 34 (5–70)19,54 | UK, France, The Netherlands, USA, Japan, Australia |
| SCA17 | Variable/EP/psychosis42 | 33 (3–55)55 | Japan, Portugal, USA |
| SCA18 | CA/sensorimotor neuropathy56 | 15 (12–25)56 | USA56 |
| SCA19/22 | Variable42 | 34 (10–46)22,57,58 | The Netherlands,57 China,58 France59 |
| SCA20 | CA/palatal tremor/dysphonia60 | 47 (19–64)60,61 | Australia |
| SCA21 | CA/cognitive deficits/EP62 | 18 (7–30)62 | France62 |
| SCA23 | CA/sensory neuropathy63 | 50 (43–56)63 | The Netherlands |
| SCA25 | CA/sensory neuropathy24 | ? (1–39)53 | France64 |
| SCA27 | CA/cognitive deficits26 | ? (15–20)26 | The Netherlands |
| SCA28 | CA/hyper-reflexia65 | 19 (12–36)27 | Italy, France, UK |
| SCA29 | Congenital non-progressive ataxia66 | Congenital66 | Canada,67 Australia66 |
| SCA32 | CA/cognitive deficits/azoospermia68 | Childhood to 60s | US68 |
| SCA35 | CA/torticollis30 | 44 (40–48)30 | China |
| SCA36 | CA/motor neuron disease31 | 53 (48–57)31 | Spain, Japan |
| SCA37 | CA/impaired vertical eye movements37 | 48 (38–64)37 | Spain37 |
Clinical phenotypes, average age of onset (if reported in the literature) and predominant geographical distribution.
CA, cerebellar ataxia; EP, extrapyramidal or Parkinsonian features; RP, retinopathy; SCA, spinocerebellar ataxia.
Table 2.
The pure spinocerebellar ataxias
| SCA | Phenotype | Age of onset: mean (range) |
Predominant geographical distribution5 |
|---|---|---|---|
| Pure | |||
| SCA5 | Pure CA69 | 30 (10–68)69,70 | USA, Germany, France |
| SCA6 | Pure CA71 | 43–52 (19–71)71 | USA, Germany, Australia, Taiwan |
| SCA11 | Pure CA72 | 25 (15–43)16,72 | UK, France, Germany |
| SCA15/16 | Pure CA60 | 31 (7–66)60 | UK, France |
| SCA26 | Pure CA36 | 42 (26–60)36 | USA36 |
| SCA30 | Pure CA73 | 52 (5–76)73 | Australia73 |
| SCA31 | Pure CA42 | 52–62 (8–72)29,74 | Japan |
| SCA34 | Pure CA, erythrokeratodermia75 | Skin—childhood; ataxia—>4075 | Canada75 |
Clinical phenotypes, average age of onset (if reported in the literature) and predominant geographical distribution.
CA, cerebellar ataxia; SCA, spinocerebellar ataxia.
Table 3.
The mixed spinocerebellar ataxias
| SCA | Locus | Neuroradiological findings117 | Gene | Cause |
|---|---|---|---|---|
| Mixed | ||||
| SCA1 | 6p23 | OPCA38 | ATXN1 | PolyQ-encoding CAG repeat expansion |
| SCA2 | 12q24 | OPCA, spinal/cortical atrophy | ATXN2 | PolyQ-encoding CAG repeat expansion |
| SCA3 | 14q24.3-q31 | OPCA, enlarged 4th ventricle | ATXN3 | PolyQ-encoding CAG repeat expansion |
| SCA4 | 16q22.1 | CA | ||
| SCA7 | 3p21.1-p12 | OPCA | ATXN7 | PolyQ-encoding CAG repeat expansion |
| SCA8 | 13q21 | CA | ATXN8, ATXN8os | Non-coding CTG×CAG repeat |
| SCA10 | 22q13 | CA | ATXN10 | Non-coding pentanucleotide repeat |
| SCA12 | 5q31-q33 | CA+cerebral atrophy | PPP2R2B | Non-coding CAG expansion (5′UTR) |
| SCA13 | 19q13.3-q13.4 | OPCA | KCNC3 | Multiple missense mutations |
| SCA14 | 19q13.4 | CA (vermis) | PRKCG | Multiple missense mutations |
| SCA17 | 6q27 | CA±general atrophy | TBP | PolyQ-encoding (CAG or CAA repeat expansioi |
| SCA18 | 7q22-q32 | CA | ||
| SCA19/22 | 1p21-q21 | CA±cerebral atrophy | KCND3 | Multiple missense mutations |
| SCA20 | 11p13-q11 | CA | ||
| SCA21 | 7p21.3-p15.1 | CA | ||
| SCA23 | 20p13 | OPCA118 | PDYN | Multiple missense mutations |
| SCA25 | 2p21-p13 | CA | ||
| SCA27 | 13q34 | CA | FGF14 | Missense mutation F145S |
| SCA28 | 18p11 | CA27 | AFG3L2 | Multiple point mutations |
| SCA29 | 3p26 | Cerebellar hypoplasia66 | ||
| SCA32 | 7q32-q33 | CA68 | ||
| SCA35 | 20p13 | OPCA30 | TGM6 | Multiple point mutations |
| SCA36 | 20p13 | CA31 | NOP56 | Intronic hexanucleotide repeat expansion |
| SCA37 | 1p32 | CA37 | ||
Mapped locus, predominant neuroradiological findings, gene (if known) and mutation type (if known).
CA, cerebellar atrophy; OPCA, olivopontocerebellar atrophy; SCA, spinocerebellar ataxia.
Table 4.
The pure spinocerebellar ataxias
| SCA | Locus | Neuroradiological findings117 |
Gene | Cause |
|---|---|---|---|---|
| Pure | ||||
| SCA5 | 11q13 | CA | SPTBN2 | Missense or in-frame deletions |
| SCA6 | 19p13 | CA | CACNA1A | PolyQ-encoding expansion |
| SCA11 | 15q15.2 | CA | TTBK2 | Various mutations |
| SCA15/16 | 3p26-p25 | CA (vermis) | ITPR1 | Large genomic deletions |
| SCA26 | 19p13.3 | CA36 | eEF2 | Missense mutation P596H |
| SCA30 | 4q34.3-q35.1 | CA75 | ||
| SCA31 | 16q21 | CA | BEAN | Intronic pentanucleotide repeat expansion |
| SCA34 | 6q12.3-q16.2 | |||
Mapped locus, predominant neuroradiological findings, gene (if known), and mutation type (if known).
CA, cerebellar atrophy; OPCA, olivopontocerebellar atrophy; SCA, spinocerebellar ataxia.
Figure 1.
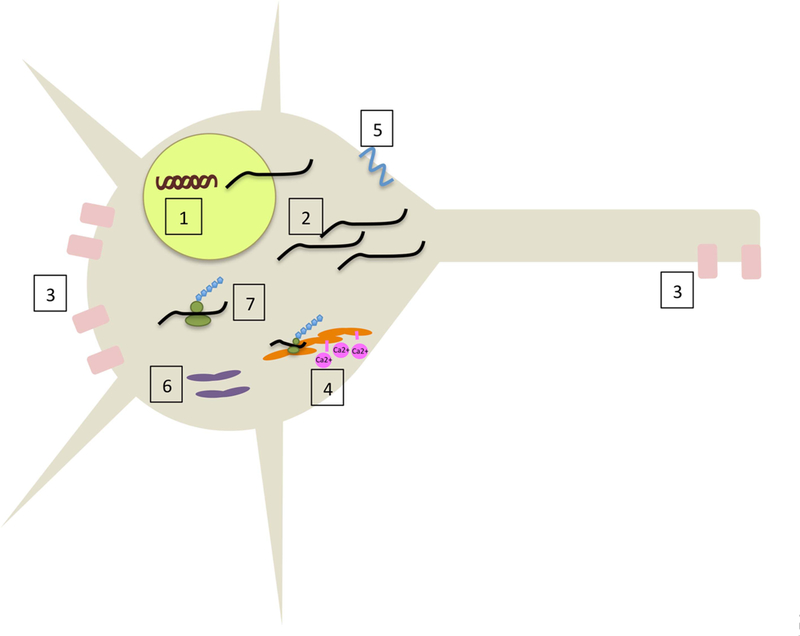
Overview of the spinocerebellar ataxia (SCA) disease mechanisms. 1: Transcriptionopathies (SCA1, 2, 3, 6, 7, 17). 2: Non-coding repeat expansions/RNA toxicity (SCA8, 10, 12, 31 36). 3: Voltage-gated potassium channel dysfunction (SCA13, 19/22). 4: ITPR1 loss (SCA15/16). 5: β3-Spectrin dysfunction (SCA5). 6: Mitochondrial dysfunction (SCA28). 7: Individual protein dysfunction (SCA11, 14, 23, 26, 27, 35).
CLINICAL FEATURES OF THE SCAS
The autosomal dominant SCAs have been clinically classified into three groups, autosomal dominant cerebellar ataxia (ADCA) types I, II and III.32 ADCAI represents mixed cerebellar ataxias, that is, involving other neurological symptoms in addition to cerebellar ataxia. ADCAII is specifically limited to cerebellar ataxias that include retinopathy, of which there is only one, SCA7. ADCAIII contains the pure cerebellar ataxias, in which cerebellar ataxia is the only or predominant neurological manifestation of disease. As more causative genes and disease mechanisms have been identified, no definitive correlation between phenotype and gene or mechanistic class has been delineated. Presently, although the division is not always homogenous within genotype, the autosomal dominant SCAs are typically classified as either mixed or pure. Key clinical features of the SCAs are summarised in tables 1 and 2.22,23,26,28,30,31,33–37
While patients with any of the mixed cerebellar ataxias may exhibit essentially pure cerebellar features, typically they develop additional neurological deficits, such as extrapyramidal symptoms, arreflexia, seizures, sensory deficits and cognitive deficits early in the course of the disease. These SCAs include SCA1–4, 7, 8, 10, 12–14, 17–21, 23, 25, 27–29, 32, 35 and 36.33–35 SCA7 is the mixed ataxia associated with progressive blindness due to a retinal rod-cone dystrophy that was originally identified as ADCA II. Genetic loci, neuroradiological findings and causative genes with mutation type (if known) of the mixed SCAs are listed in table 3.
The pure cerebellar ataxias are distinguished by exclusive cerebellar ataxia without other neurological symptoms, and include SCA5, 6, 11, 15/16, 26, 30, 31 and 34.33–35 However, rare reports for some of these predominantly pure cerebellar ataxias have noted involvement of other systems, resulting in pyramidal symptoms, peripheral neuropathy and movement disorders.76 Genetic loci, neuroradiological findings and causative genes with mutation type (if known) for the pure cerebellar ataxias are indicated in table 4.
GENETICS AND DISEASE MECHANISMS OF THE REPEAT EXPANSION-ASSOCIATED SCAS
Polyglutamine expansion ataxias
The mixed ataxias SCA1–3, 7 and 17, as well as the pure ataxia SCA6, are caused by expansions of CAG repeats encoding polyglutamine tracts within several unrelated genes and, as with other repeat expansion disorders, there is an inverse correlation between repeat length and age of onset.7,21,39,41,46,77 The expanded polyglutamine tracts impair native protein folding, function, DNA-protein or protein–protein interactions of the mutant protein, often leading to dysregulation of their function in transcription. The group is thus often considered part of a larger group of disorders called transcriptionopathies.34 Several of the polyglutamine ataxias are associated with the formation of cytoplasmic or intranuclear aggregates in affected tissue. While originally believed to be deleterious, the aggregates may actually represent protective sequestration of the misfolded protein.78
SCA1 is caused by an expanded CAG repeat in the gene, ATXN1, that encodes an N-terminal polyglutamine tract in the widely expressed ataxin-1 protein7 (normal range 6–39, pathological ~39–83).6,7 Borderline alleles also become pathological when interrupting CAT sequences normally present in the non-disease-associated CAG repeat sequence are absent and are unable to provide a stabilising effect during DNA replication and prevent repeat expansion.7
Ataxin-1 has been studied intensely, and a comprehensive review is beyond the scope of this summary. In short, ataxin-1 is a chromatin-binding factor that suppresses Notch signalling in the absence of Notch.79 Ataxin-1 is phosphorylated on a key residue, S776, that regulates nuclear translocation through its interaction with Capicua.6,80,81 The polyglutamine expansion increases complex formation with the RNA-binding RBM17 protein.82 Alterations in calcium homeostasis,83 nuclear transcription,81 and protein aggregation84 have all been implicated in the pathogenesis of SCA1.
SCA2 is caused by an expanded CAG repeat in the gene, ATXN2, resulting in an N-terminal polyglutamine tract in the widely expressed ataxin-2 protein (normal 13–31, pathological 32–79 repeats, superexpansions >100, incomplete penetrance 32–34).8,9,85 Repeat expansions in the ATXN2 gene of up to 500 repeats have been identified in patients presenting with a fatal infantile encephalopathy.86 Recent studies have also suggested that intermediate expansion lengths (27–33) also confer increased susceptibility to amyotrophic lateral sclerosis (ALS).87 Ataxin-2 is thought to be involved in RNA processing based on sequence homology to other RNA binding proteins.39 There is some evidence for the role of altered calcium homeostasis.88
SCA3 is caused by a CAG repeat expansion in the gene ATXN3 resulting in a C-terminal polyglutamine tract in the ubiquitously expressed ataxin-3 protein (normal 44 or less, pathological 52–86, incomplete penetrance 45–51).41 Small expansions (45–59) tend to manifest initially with predominantly neuropathic features and sleep disorders, developing ataxia later; modest expansions (≥60, average 73–76) present with an ataxia predominant syndrome; and large expansions (≥60, average 80) present with severe dystonia.41
The function of ataxin-3 has not been fully clarified,10 but it has demonstrated deubiquitinase activity, with a predilection for longer ubiquitin chains.41,89 Several mechanisms have been postulated for the molecular pathogenesis of SCA3, including toxic effects of a proteolytic fragment of the expanded ataxin-3 protein; altered protein-protein interactions leading to transcriptional dysregulation; or perturbation of axonal transport.41,90,91 Several groups have also implicated mitochondrial dysfunction,92,93 or altered calcium homeostasis.88
SCA6 is caused by expansion of a small CAG repeat in the CACNA1A gene. The expanded polyglutamine tract, the smallest in the SCA family, (normal 4–18 repeats, pathological 19–33 repeats),77 appears in two proteins encoded by this gene: within the C-terminus of some splice variants of the a1A subunit of the voltage-gated P/Q-type calcium channel, and within a separate transcription factor protein, a1ACT.12,94
The α1ACT transcription factor was recently discovered to be the product of an internal ribosome entry site within the CACNA1A gene, and is important for neural and Purkinje cell development. The polyglutamine expanded α1ACT loses transcription factor function, leading to cell death in cultured cells and cerebellar atrophy and clinical ataxia in transgenic mice.94
SCA7 is caused by an expanded CAG repeat in the gene, ATXN7, resulting in a polyglutamine tract in the ataxin-7 protein (normal 7–17, pathological 38–150).13,46 Longer repeats (100–150) correlate to the most severe infantile form of the disease, while shorter repeats (36–43) correspond to a milder form of the disease with adult onset. The typical disease involves 50–55 repeats, and symptoms begin in adolescence or adulthood.13,46 The protein ataxin-7, a transcription factor, is a component of the STAGA complex involved in chromatin remodelling via histone acetylation and deubiquitination.34,46,95 Polyglutamine-expanded ataxin-7 may exert its deleterious effects through a combination of gene dysregulation in retinal cells and protein aggregation within neurons.
SCA17 is caused by an expanded repeat of CAG and CAA in the gene, TATA-box binding protein (TBP), encoding the ubiquitously expressed transcription factor TBP (normal 25–44, pathological 47–63, incomplete penetrance 45–46).21,96 TBP performs well-known transcriptional functions, although the effect of the polyglutamine expansion on Purkinje cells is still an area of active research.
Ataxias caused by non-coding repeat expansions
The mixed ataxias SCA8, 10, 12 and 36 as well as the pure ataxia SCA31 are caused by non-coding repeat expansions, that is, expanded repeat tracts identified outside of the recognised protein coding regions, such as in introns or 5′ UTRs. SCA8, which presents as either a pure or spastic ataxia, is believed to exert its deleterious effects through a non-coding CTG triplet repeat expansion within the ATXN8 gene (normal 16–37, pathological 107–127),14 with several examples of incomplete penetrance reported.48 Bidirectional transcription of the trinucleotide repeat expansion occurs, resulting in an untranslated CTG expansion in the ATXN8 opposite strand (ATXN8OS) RNA transcript and a translated CAG repeat (C-terminal polyglutamine) expansion in the ATXN8 strand.97 The pathogenesis of the 5′ UTR CTG repeat in ATXN8OS has been most well studied, and leads to RNA toxicity through toxic gain of function, seen as RNA foci co-localising with MBNL1.98
Recent studies in SCA8 have uncovered a novel molecular mechanism of gene expression, known as repeat-associated non-ATG (RAN) translation, which may also contribute significantly to disease. In RAN translation, expanded repeat sequences within mRNA, triplet and otherwise, are the site of initiation of protein translation downstream from the 5′ capped mRNA and initial ATG start codon, and in the case of triplet expansions leading to production of homopolymeric proteins in all three reading frames.99,100 This was initially demonstrated for SCA8, but has now also been shown for other neurological disorders, including myotonic dystrophy type 1, fragile X tremor ataxia syndrome and C9ORF72 ALS with frontotemporal dementia.100 For repeat expansion diseases including SCA8, this discovery raises the question of whether the deleterious effects of the repeat expansion are exerted through RNA, protein, or both. In vitro SCA8 polyserine and polyalanine homopolymeric proteins have been identified, and antibody to these polypeptides detected polyserine and polyalanine deposits in postmortem SCA8 brain specimens. The role of these RAN translation products in SCA8 disease pathogenesis is currently under study.
SCA10 is caused by an untranslated pentanucleotide repeat (ATTCT) expansion, 800–4500 repeats, in the ATXN10 gene (normal 10–29),15 although there is some evidence for incomplete penetrance.101 It has recently been demonstrated that the pathologically expanded RNA sequesters heterogeneous nuclear ribonucleoprotein K (hnRNP K) within mouse neurons, triggering release of protein kinase (PK) C5 and activating apoptosis, suggestive of a gain of toxic RNA function.102
SCA12 is caused by a non-coding triplet CAG expansion in the 5′ UTR of the brain-specific regulatory subunit of the serine/threonine protein phosphatase PP2A, PPP2R2B, (normal 9–28, pathological 55–78.)17 Within a Drosophila SCA12 model it has been demonstrated that the CAG repeat expanded homologue gene results in mitochondrial dysfunction and increased oxidative stress, shortening the organism’s lifespan.103
SCA31 has been attributed to an intronic pentanucleotide (TGGAA) expanded repeat insertion in brain-expressed, associated with NEDD4 (BEAN), (pathological insertion 2.5–3.8 kb).28 Most normal controls in the Japanese population demonstrated no such insertions; the incidence of non-pathogenic uninterrupted pure expansions in control individuals may be somewhat higher in other populations.29 The repeat-expanded RNA was demonstrated to localise to centromeres in vivo, suggesting a role in heterochromatin or chromosomal structure,28 with unknown function in Purkinje cells.
SCA36 has been attributed to an intronic hexanucleotide (GGCCTG) repeat expansion in NOP56 (pathological repeat length 1500–2000).31 NOP56 is predicted to function in an early pre-rRNA processing step104 with unknown effect on Purkinje cell function. With the detection of RAN translation in an increasing number of neurodegenerative diseases caused by non-coding repeat expansions it is likely that there will be additional evidence for this mechanism in other ataxias.
GENETICS AND DISEASE MECHANISMS OF SCAS CAUSED BY CONVENTIONAL MUTATIONS IN CRITICAL GENES
While the identification of coding and non-coding repeat expansions provided initial hope of identifying common molecular mechanisms, the subsequent demonstration that many SCAs are caused by missense mutations in critical proteins promised to reveal common pathogenic themes and key Purkinje cell vulnerabilities. The SCAs attributed to such conventional mutations in critical genes include the mixed cerebellar ataxias SCA13, 14, 19/22, 23, 27, 28 and 35, as well as the pure cerebellar ataxias SCA5, 11, 15/16 and 26.
Although a common downstream outcome for missense mutations in critical proteins may simply be protein misfolding and aggregation, there are some recurring themes of protein function and dysfunction that point to key areas of Purkinje cell vulnerability. The most common theme relates to disturbances of ion channel function either by direct mutation of an ion channel protein or genetic disruption of a pathway that plays a role in modulating ion channel function (SCA13, 15/16 and 19/22) and neuronal excitability (SCA5, 14 and 27).
SCAs due to mutations in ion channel genes
SCA13 is directly attributed to ion channel dysfunction, via a mutation in the widely expressed voltage-gated potassium channel, KCNC3.18,105,106 Three point mutations have been associated with the disease to date, F448L, R420H and R423H. The first (F448L) shifted the activation curve of the channel and slowed channel closing.18 The latter two are located in the voltage-sensing domain and resulted in a dominant negative loss of channel function,18 leading to Purkinje cell degeneration by an unknown mechanism.
SCA15/16 has been associated with deletions or missense mutations in the ITPR1 gene which encodes the type 1 inositol triphosphate receptor.20 SCA15/16 stands alone in implicating a haploinsufficiency mechanism, as some ITPR1 deletions have been demonstrated to result in lower levels of ITPR1 protein.20 Homozygous loss-of-function ITPR1 mutations in mice cause severe ataxia and heterozygous mutations cause motor incoordination.107
SCA19/22 has recently been associated with several point mutations in KCND3, encoding the voltage-gated potassium channel KV4.3.22 These point mutations lead to a misfolded potassium channel subunit that is retained in the ER.22 It is unclear whether the protein misfolding or loss of channel function is responsible for Purkinje cell degeneration.
SCAs affecting neuronal excitability or excitotoxicity
Several SCAs have implicated disruption in neuronal excitability and signalling. SCA5, a pure cerebellar ataxia, has been attributed to a series of mutations in SPTBN2, encoding (β-III spectrin.11 The defective (β-III spectrin fails to stabilise the glutamate transporter, EEAT4, at the membrane, possibly producing degeneration through glutamate toxicity.11 Loss of (β-III spectrin has been demonstrated to stunt development of normal Purkinje cell morphology, specifically of dendritic spines.11
SCA14 has been attributed to a variety of point mutations in the widely expressed PKCγ protein,19,54 Suggested gain-of-function mechanisms from PKCγ mutations include mutant protein aggregation,108 altered calcium homeostasis109 and impaired signalling.110,111
SCA27 is caused by a point mutation, F145S, in the widely expressed fibroblast growth factor 14 (FGF14) protein.26,112 This leads to a loss of FGF14 function in its role of regulating Purkinje cell excitability and plasticity, possibly impairing neuronal signalling.113,114
Other mutations
SCA23 has been attributed to several distinct mutations in the neuron-specific prodynorphin (PDYN), the precursor for several opioid neuropeptides.23 Three mutations are located in DynA, a peptide with opioid and non-opioid activities, resulting in increased DynA production and excessive toxicity in cultured cells.23 A fourth mutation is located in the PDYN domain and affects protein expression patterns in the opioid and glutamate system, potentially pointing to a downstream glutamate toxicity effect.23
SCA11 has been attributed to frameshift mutations in the widely expressed t tubulin kinase 2 (TTBK2).16 These mutations have been demonstrated to promote TTBK2 expression while inhibiting its kinase activity and increasing nuclear localisation,115 although the link to Purkinje cell death has not yet been found. Homozygosity for the mutation in mice is notably embryonic lethal.115
SCA26 was recently attributed to a proline to histidine change at residue 596 in the eukaryotic elongation factor 2 protein found in a single kindred.25 In a yeast model of SCA26, P596H eEF2 has been demonstrated to result in an increased rate of frameshifting during protein translation, disrupting proteostasis and rendering yeast more susceptible to unfolded protein response-inducing stressors.25
SCA28 has been attributed to a series of mutations in the widely expressed ATPase family gene 3-like 2 (AFG3L2), encoding the catalytic subunit of the m-AAA protease.27 The AFG3L2 protein is an ATP-dependent protease located in the inner mitochondrial membrane known to degrade misfolded proteins and assist in ribosome assembly.116 The disruption of AFG3L2 function has been linked to both dominant-negative and loss of function mechanisms for disrupting mitochondrial function.116
SCA35 was recently attributed to point mutations in TGM6, the non-neuron specific transglutaminase 6, specifically L517W and D327G,30 but little else is known at this time. Likewise little is presently known about SCA20, which has been mapped to chromosome 11p13-q11. A putative copy number variant has been implied between markers rs4963307 and rs10897193, although this has not yet been demonstrated to be causative.61
CONCLUSION
While a unifying theme in SCA pathology seems elusive, a few trends have emerged—polyglutamine expansions leading to transcriptional dysregulation, RNA toxicity and the novel RAN translation products from expanded RNA repeats, as well as channel dysfunction and signalling disruption. While simple correlations between genetic defect and cell types affected or clinical phenotype seem elusive, the common end point is that these heterogeneous genetic defects predominantly result in a gain-of-toxic function to which Purkinje cells are especially susceptible, either exclusively or in advance of other neuronal cell types.
Several features that distinguish Purkinje cells might contribute to their vulnerability. The Purkinje cell is one of the largest neuronal cell types with a high metabolic activity and a massive dendritic arbour receiving extensive excitatory inputs. It has been demonstrated that impaired proteostasis affects Purkinje cells before other neuronal types in mice119–122 and in human disease.123 Proteostasis, the balance of protein synthesis and degradation, is crucial for synaptic signalling and neuronal function, but is best studied in spherical cells in which all machinery is confined to a limited cytosolic space. The unique Purkinje cell vulnerability to proteostatic insult may be due to an imbalance of synthesis and degradation machinery within its extensive dendritic arbour outside the soma. For example at synapses, protein synthesis regularly occurs, but lysosomes, which are instrumental in autophagic degradation of protein aggregates, are restricted to the soma.124 It seems plausible that some of the common cellular pathways for combating protein misfolding, as well as altered transcription, DNA damage or disrupted ionic gradients, are less well adapted to the size and morphological complexity of the Purkinje cell.
The mechanistic themes that have emerged (transcriptional dysregulation by polyglutamine expanded tracts, RNA toxicity and channel/signalling dysfunction) may represent areas of crucial Purkinje cell function, which when disturbed are more likely to activate the common defence mechanisms. Identification of which intracellular repair or protective pathways might be overwhelmed, such as calcium buffers, DNA repair, the ubiquitin-proteasome system or autophagy may suggest common pathogenic mechanisms or therapeutic strategies. Currently therapy is limited to symptomatic management with as yet no neuroprotective strategies to alleviate the progressive nature of the SCAs. Looking to the future of therapeutics for SCA, emphasis on early diagnosis through genetic testing will be key, as well as further honing in on the aspects of Purkinje cells that renders them so uniquely susceptible to cellular perturbations and how to combat the diverse genetic insults that cause the SCAs.
Acknowledgments
Funding KEH was supported by the Graduate Training in Growth, Development and Disabilities program at the University of Chicago, T32 HD009007.
Footnotes
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCES
- 1.Muzaimi MB, Thomas J, Palmer-Smith S, et al. Population based study of late onset spinocerebellar ataxia in south east Wales. J Neurol Neurosurg Psychiatry 2004;75:1129–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Erichsen AK, Koht J, Stray-Pedersen A, et al. Prevalence of hereditary ataxia and spastic paraplegia in southeast Norway: a population-based study. Brain 2009;132:1577–88. [DOI] [PubMed] [Google Scholar]
- 3.Coutinho P, Ruano L, Loureiro JL, et al. Hereditary ataxia and spastic paraplegia in Portugal: a population-based prevalence study. JAMA Neurol 2013;70:746–55. [DOI] [PubMed] [Google Scholar]
- 4.Craig K, Keers SM, Archibald K, et al. Molecular epidemiology of spinocerebellar ataxia type 6. Ann Neurol 2004;55:752–5. [DOI] [PubMed] [Google Scholar]
- 5.Hersheson J, Haworth A, Houlden H. The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum Mutat 2012;33:1324–32. [DOI] [PubMed] [Google Scholar]
- 6.Zoghbi HY, Orr HT. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J Biol Chem 2009;284:7425–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orr HT, Chung MY, Banfi S, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 1993;4:221–6. [DOI] [PubMed] [Google Scholar]
- 8.Pulst SM, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally biallelic tricnucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996;14:269–76. [DOI] [PubMed] [Google Scholar]
- 9.Sanpei K, Takano H, Igarashi S, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct idenfication of repeat expansion and cloning technique, DIRECT. Nat Genet 1996;14:277–84. [DOI] [PubMed] [Google Scholar]
- 10.Kawaguchi Y, Okamoto T, Taniwaki M, et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 1994;8:221–8. [DOI] [PubMed] [Google Scholar]
- 11.Ikeda Y, Dick KA, Weatherspoon MR, et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet 2006;38:184–90. [DOI] [PubMed] [Google Scholar]
- 12.Zhuchenko O, Bailey J, Bonnen P, et al. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet 1997;15:62–9. [DOI] [PubMed] [Google Scholar]
- 13.David G, Abbas N, Stevanin G, et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 1997;17:65–70. [DOI] [PubMed] [Google Scholar]
- 14.Koob MD, Moseley ML, Schut LJ, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet 1999;21:379–84. [DOI] [PubMed] [Google Scholar]
- 15.Matsuura T, Yamagata T, Burgess DL, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet 2000;26:191–4. [DOI] [PubMed] [Google Scholar]
- 16.Houlden H, Johnson J, Gardner-Thorpe C, et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet 2007;39:1434–6. [DOI] [PubMed] [Google Scholar]
- 17.Holmes SE, O’Hearn EE, McInnis MG, et al. Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat Genet 1999;23:391–2. [DOI] [PubMed] [Google Scholar]
- 18.Waters MF, Minassian NA, Stevanin G, et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet 2006;38:447–51. [DOI] [PubMed] [Google Scholar]
- 19.Yabe I, Sasaki H, Chen DH, et al. Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma. Arch Neurol 2003;60:1749–51. [DOI] [PubMed] [Google Scholar]
- 20.van de Leemput J, Chandran J, Knight MA, et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet 2007;3:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koide R, Kobayashi S, Shimohata T, et al. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet 1999;8:2047–53. [DOI] [PubMed] [Google Scholar]
- 22.Duarri A, Jezierska JJ, Fokkens M, et al. Mutations in potassium channel KCND3 cause spinocerebellar ataxia type 19. Ann Neurol 2012;72:870–80. [DOI] [PubMed] [Google Scholar]
- 23.Bakalkin G, Watanabe H, Jezierska J, et al. Prodynorphin mutations cause the neurodegenerative disorder spinocerebellar ataxia type 23. Am J Hum Genet 2010;87:593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevanin G, Broussolle E, Streichenberger N, et al. Spinocerebellar ataxia with sensory neuropathy (SCA25). Cerebellum 2005;4:58–61. [DOI] [PubMed] [Google Scholar]
- 25.Hekman KE, Yu GY, Brown CD, et al. A conserved eEF2 coding variant in SCA26 leads to loss of translational fidelity and increased susceptibility to proteostatic insult. Hum Mol Genet 2012;21:5472–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Swieten JC, Brusse E, de Graaf BM, et al. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am J Hum Genet 2003;72:191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Bella D, Lazzaro F, Brusco A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet 2010;42:313–21. [DOI] [PubMed] [Google Scholar]
- 28.Sato N, Amino T, Kobayashi K, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet 2009;85:544–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishikawa K, Durr A, Klopstock T, et al. Pentanucleotide repeats at the spinocerebellar ataxia type 31 (SCA31) locus in Caucasians. Neurology 2011;77:1853–5. [DOI] [PubMed] [Google Scholar]
- 30.Wang JL, Yang X, Xia K, et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 2010;133(Pt 12):3510–18. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi H, Abe K, Matsuura T, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 2011;89:121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harding AE. Clinical features and classification of inherited ataxias. Adv Neurol 1993;61:1–14. [PubMed] [Google Scholar]
- 33.Perlman SL. Spinocerebellar degenerations. Handb Clin Neurol 2011;100:113–40. [DOI] [PubMed] [Google Scholar]
- 34.Matilla-Duenas A, Ashizawa T, Brice A, et al. Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum 2013;13:269–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manto MU. The wide spectrum of spinocerebellar ataxias (SCAs). Cerebellum 2005;4:2–6. [DOI] [PubMed] [Google Scholar]
- 36.Yu GY, Howell MJ, Roller MJ, et al. Spinocerebellar ataxia type 26 maps to chromosome 19p13.3 adjacent to SCA6. Ann Neurol 2005;57:349–54. [DOI] [PubMed] [Google Scholar]
- 37.Serrano-Munuera C, Corral-Juan M, Stevanin G, et al. New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32. JAMA Neurol 2013;70:764–71. [DOI] [PubMed] [Google Scholar]
- 38.di Donato SD, Mariotti C, Taroni F. Spinocerebellar ataxia type 1. Handb Clin Neurol 2012;103:399–421. [DOI] [PubMed] [Google Scholar]
- 39.Auburger GWJ. Spinocerebellar ataxia type 2. Handb Clin Neurol 2012;103:423–36. [DOI] [PubMed] [Google Scholar]
- 40.Geschwind DH, Perlman S, Figueroa CP, et al. The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet 1997;60:842–50. [PMC free article] [PubMed] [Google Scholar]
- 41.Paulson H Spinocerebellar ataxia type 3. Handb Clin Neurol 2012;103:437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cloud LJ, Wilmot G. Other spinocerebellar ataxias. Handb Clin Neurol 2012;103:581–6. [DOI] [PubMed] [Google Scholar]
- 43.Flanigan K, Gardner K, Alderson K, et al. Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy (SCA4): clinical description and genetic localization to chromosome 16q22.1. Am J Hum Genet 1996;59:392–9. [PMC free article] [PubMed] [Google Scholar]
- 44.Gardner K, Alderson K, Galster B, et al. Autosomal dominant spinocerebellar ataxia: clinical description of a distinct hereditary ataxia and genetic localization to chromosome 16 (SCA4) in a Utah kindred [abstract]. Neurology 1994;44:A361. [Google Scholar]
- 45.Hellenbroich Y, Bubel S, Pawlack H, et al. Refinement of the spinocerebellar ataxia type 4 locus in a large German family and exclusion of CAG repeat expansions in this region. J Neurol 2003;250:668–71. [DOI] [PubMed] [Google Scholar]
- 46.Martin JJ. Spinocerebellar ataxia type 7. Handb Clin Neurol 2012;103:475–91. [DOI] [PubMed] [Google Scholar]
- 47.Ikeda Y, Ranum LPW, Day JW. Clinical and genetic features of spinocerebellar ataxia type 8. Handb Clin Neurol 2012;103:493–505. [DOI] [PubMed] [Google Scholar]
- 48.Day JW, Schut LJ, Moseley ML, et al. Spinocerebellar ataxia type 8: clinical features in a large family. Neurology 2000;55:649–57. [DOI] [PubMed] [Google Scholar]
- 49.Grewal RP, Achari M, Matsuura T, et al. Clinical features and ATTCT repeat expansion in spinocerebellar ataxia type 10. Arch Neurol 2002;59:1285–90. [DOI] [PubMed] [Google Scholar]
- 50.Ashizawa T Spinocerebellar ataxia type 10. Handb Clin Neurol 2012;103:507–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Hearn E, Holmes SE, Calvert PC, et al. SCA-12: Tremor with cerebellar and cortical atrophy is associated with a CAG repeat expansion. Neurology 2001;56:299–303. [DOI] [PubMed] [Google Scholar]
- 52.O’Hearn E, Holmes SE, Margolis RL. Spinocerebellar ataxia type 12. Handb Clin Neurol 2012;103:535–47. [DOI] [PubMed] [Google Scholar]
- 53.Stevanin G, Durr A. Spinocerebellar ataxia 13 and 25. Handb Clin Neurol 2012;103:549–53. [DOI] [PubMed] [Google Scholar]
- 54.Chen DH, Raskin WH, Bird TD. Spinocerebellar ataxia 14. Handb Clin Neurol 2012;103:555–9. [DOI] [PubMed] [Google Scholar]
- 55.Nakamura K, Jeong S, Uchihara T, et al. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 2001;10:1441–8. [DOI] [PubMed] [Google Scholar]
- 56.Brkanac Z, Fernandez M, Matsushita M, et al. Autosomal dominant sensory/motor neuropathy with Ataxia (SMNA): linkage to chromosome 7q22-q32. Am J Med Genet 2002;114:450–7. [DOI] [PubMed] [Google Scholar]
- 57.Schelhaas HJ, Ippel PF, Hageman G, et al. Clinical and genetic analysis of a four-generation family with a distinct autosomal dominant cerebellar ataxia. J Neurol 2001;248:113–20. [DOI] [PubMed] [Google Scholar]
- 58.Chung MY, Lu YC, Cheng NC, et al. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain 2003;126:1293–9. [DOI] [PubMed] [Google Scholar]
- 59.Lee YC, Durr A, Majczenko K, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol 2012;72:859–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Storey E, McKinlay Gardner RJ. Spinocerebellar ataxia 20. Hanb Clin Neurol 2012;103:567–73. [DOI] [PubMed] [Google Scholar]
- 61.Knight MA, Hernandez D, Diede SJ, et al. A duplication at chromosome 11q12.2–11q12.3 is associated with spinocerebellar ataxia type 20. Hum Mol Genet 2008;17:3847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Devos D, Schraen-Maschke S, Vuillaume I, et al. Clinical features and genetic analysis of a new form of spinocerebellar ataxia. Neurology 2001;56:234–8. [DOI] [PubMed] [Google Scholar]
- 63.Verbeek DS, van de Warrenburg P, Wesseling P, et al. Mapping of the SCA23 locus involved autosomal dominant cerebellar ataxia to chromosome region 20p13–12.3. Brain 2004;127:2551–7. [DOI] [PubMed] [Google Scholar]
- 64.Stevanin G, Bouslam N, Thobois S, et al. Spinocerebellar ataxia with sensory neuropathy (SCA25) maps to chromosome 2p. Ann Neurol 2004;55:97–104. [DOI] [PubMed] [Google Scholar]
- 65.Mariotti C, Di Bella D, Di Donato S, et al. Spinocerebellar ataxia type 28. Hanb Clin Neurol 2012;103:575–9. [DOI] [PubMed] [Google Scholar]
- 66.Dudding TE, Friend K, Schofield PW, et al. Autosomal dominant congenital non-progressive ataxia overlaps with the SCA15 locus. Neurology 2004;63:2288–92. [DOI] [PubMed] [Google Scholar]
- 67.Huang L, Chardon JW, Carter MT, et al. Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia. Orphanet J RareDis 2012;7:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang H, Zhu H, Gomez CM. SCA32: an autosomal dominant cerebellar ataxia with azoospermia maps to chromosome 7q32-q33 [abstract]. Mov Disord 2010;25:S192 only. [Google Scholar]
- 69.Dick KA, Ikeda Y, Day JW, et al. Spinocerebellar ataxia type 5. Handb Clin Neurol 2012;103:451–9. [DOI] [PubMed] [Google Scholar]
- 70.Ranum LP, Schut LJ, Lundgren JK, et al. Spinocerebellar ataxia type 5 in a family descended from the grandparents of President Lincoln maps to chromosome 11. Nat Genet 1994;8:280–4. [DOI] [PubMed] [Google Scholar]
- 71.Solodkin A, Gomez CM. Spinocerebellar ataxia type 6. Handb Clin Neurol 2012;103:461–73. [DOI] [PubMed] [Google Scholar]
- 72.Giunti P, Houlden H, Gardner-Thorpe C, et al. Spinocerebellar ataxia type 11. Handb Clin Neurol 2012;103:521–34. [DOI] [PubMed] [Google Scholar]
- 73.Storey E, Bahlo M, Fahey M, et al. A new dominantly inherited pure cerebellar ataxia, SCA 30. J Neurol Neurosurg Psychiatry 2009;80:408–11. [DOI] [PubMed] [Google Scholar]
- 74.Ouyang Y, Sakoe K, Shimazaki H, et al. 16q-linked autosomal dominant cerebellar ataxia: a clinical and genetic study. J Neurol Sci 2006;247:180–6. [DOI] [PubMed] [Google Scholar]
- 75.Giroux JM, Barbeau A. Erythrokeratodermia with ataxia. Arch Dermatol 1972;106:183–8. [PubMed] [Google Scholar]
- 76.Fujioka S, Sundal C, Zbigniew KW. Autosomal dominant cerebellar ataxia type III: a review of the phenotypic and genotypic characteristics. Orphanet J Rare Dis 2013;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kordasiewicz HB, Gomez CM. Molecular pathogenesis of spinocerebellar ataxia type 6. Neurotherapeutics 2007;4:285–94. [DOI] [PubMed] [Google Scholar]
- 78.Zoghbi HY, Orr HT. Polyglutamine diseases: protein cleavage and aggregation. Curr Opin Neurobiol 1999;9:566–70. [DOI] [PubMed] [Google Scholar]
- 79.Tong X, Gui H, Jin F, et al. Ataxin-1 and Brother of ataxin-1 are components of the Notch signalling pathway. EMBO Rep 2011;12:428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Orr HT. SCA1-Phosphorylation, a regulator of Ataxin-1 function and pathogenesis. Prog Neurobiol 2012;99:179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lam YC, Bowman AB, Jafar-Nejad P, et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 2006;127:1335–47. [DOI] [PubMed] [Google Scholar]
- 82.Lim J, Crespo-Barreto J, Jafar-Nejad P, et al. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008;452:713–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vig PJ, Wei J, Shao Q, et al. Suppression of calbindin-D28k expression exacerbates SCA1 phenotype in a Disease Mouse Model. Cerebellum 2012;11:718–32. [DOI] [PubMed] [Google Scholar]
- 84.Cummings CJ, Mancini MA, Antalffy B, et al. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 1998;19:148–54. [DOI] [PubMed] [Google Scholar]
- 85.Charles P, Camuzat A, Benammar N, et al. Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology 2007;69:1970–5. [DOI] [PubMed] [Google Scholar]
- 86.Paciorkowski AR, Shafrir Y, Hrivnak J, et al. Massive expansion of SCA2 with autonomic dysfunction, retinitis pigmentosa, and infantile spasms. Neurology 2011;77:1055–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bezprozvanny I Calcium signaling in neurodegenerative diseases. Trends Mol Med 2009;15:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ouyang H, Ali YO, Ravichandran M, et al. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem 2012;287:2317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kuhlbrodt K, Janiesch PC, Kevei E, et al. The Machado-Joseph disease deubiquitylase ATX-3 couples longevity and proteostasis. Nat Cell Biol 2011;13:273–81. [DOI] [PubMed] [Google Scholar]
- 91.Riess O, Rub U, Pastore A, et al. SCA3: neurological features, pathogenesis and animal models. Cerebellum 2008;7:125–37. [DOI] [PubMed] [Google Scholar]
- 92.Chou AH, Yeh TH, Kuo YL, et al. Polyglutamine-expanded ataxin-3 activates mitochondrial apoptotic pathway by upregulating Bax and downregulating Bcl-xL. Neurobiol Dis 2006;21:333–45. [DOI] [PubMed] [Google Scholar]
- 93.Tsai HF, Tsai HJ, Hsieh M. Full-length expanded ataxin-3 enhances mitochondrial-mediated cell death and decreases Bcl-2 expression in human neuroblastoma cells. Biochem Biophys Res Commun 2004;324:1274–82. [DOI] [PubMed] [Google Scholar]
- 94.Du X, Wang J, Zhu H, et al. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell 2013;154:118–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Helmlinger D, Hardy S, Abou-Sleymane G, et al. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol 2006;4:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gao R, Matsuura T, Coolbaugh M, et al. Instability of expanded CAG/CAA repeats in spinocerebellar ataxia type 17. Eur J Hum Genet 2008;16:215–22. [DOI] [PubMed] [Google Scholar]
- 97.Ikeda Y, Daughters RS, Ranum LP. Bidirectional expression of the SCA8 expansion mutation: one mutation, two genes. Cerebellum 2008;7:150–8. [DOI] [PubMed] [Google Scholar]
- 98.Daughters RS, Tuttle DL, Gao W, et al. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet 2009;5:e1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zu T, Gibbens B, Doty NS, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci 2011;108:260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cleary JD, Ranum LPW. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum Mol Genet 2013;22:R41–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raskin S, Ashizawa T, Teive HA, et al. Reduced penetrance in a Brazilian family with spinocerebellar ataxia type 10. Arch Neurol 2007;64:591–4. [DOI] [PubMed] [Google Scholar]
- 102.White M, Xia G, Gao R, et al. Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure: a toxic RNA gain-of-function model. J Neurosci Res 2012;90:706–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang YC, Lee CM, Lee LC, et al. Mitochondrial dysfunction and oxidative stress contribute to the pathogenesis of spinocerebellar ataxia type 12 (SCA12). J Biol Chem 2011;286:21742–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hayano T, Yanagida M, Yamauchi Y, et al. Proteomic analysis of human Nop56p-associated preribosomal ribonucleoprotein complexes. Possible link between Nop56p and the nucleolar protein treacle responsible for Treacher Collins syndrome. J Biol Chem 2003;278:34309–19. [DOI] [PubMed] [Google Scholar]
- 105.Stevanin G, Durr A, Benammar N, et al. Spinocerebellar ataxia with mental retardation (SCA13). Cerebellum 2005;4:43–6. [DOI] [PubMed] [Google Scholar]
- 106.Figueroa KP, Minassian NA, Stevanin G, et al. KCNC3: phenotype, mutations, channel biophysics-a study of 260 familial ataxia patients. Hum Mutat 2010;31:191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iwaki A, Kawano Y, Miura S, et al. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet 2008;45:32–5. [DOI] [PubMed] [Google Scholar]
- 108.Seki T, Takahashi H, Adachi N, et al. Aggregate formation of mutant protein kinase C gamma found in spinocerebellar ataxia type 14 impairs ubiquitin-proteasome system and induces endoplasmic reticulum stress. Eur J Neurosci 2007;26:3126–40. [DOI] [PubMed] [Google Scholar]
- 109.Adachi N, Kobayashi T, Takahashi H, et al. Enzymological analysis of mutant protein kinase C gamma causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J Biol Chem 2008;283:19854–63. [DOI] [PubMed] [Google Scholar]
- 110.Sakai N, Saito N, Seki T. Molecular pathophysiology of neurodegenerative disease caused by gamma PKC mutations. World J Biol Psychiatry 2011;12(Suppl 1):95–8. [DOI] [PubMed] [Google Scholar]
- 111.Shuvaev AN, Horiuchi H, Seki T, et al. Mutant PKC gamma in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J Neurosci 2011;31:14324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brusse E, de Koning I, Maat-Kievit A, et al. Spinocerebellar ataxia associated with a mutation in the fibroblast growth factor 14 gene (SCA27): a new phenotype. Mov Disord 2006;21:396–401. [DOI] [PubMed] [Google Scholar]
- 113.Laezza F, Gerber BR, Lou JY, et al. The FGF14 (F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci 2007;27:12033–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xiao M, Xu L, Laezza F, et al. Impaired hippocampal synaptic transmission and plasticity in mice lacking fibroblast growth factor 14. Mol Cell Neurosci 2007;34:366–77. [DOI] [PubMed] [Google Scholar]
- 115.Bouskila M, Esoof N, Gay L, et al. TTBK2 kinase substrate specificity and the impact of spinocerebellar-ataxia-causing mutations on expression, activity, localization and development. Biochem J 2011;437:157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Koppen M, Langer T. Protein degradation within mitochondria: versatile activities of AAA proteases and other peptidases. Crit Rev Biochem Mol Biol 2007;42:221–42. [DOI] [PubMed] [Google Scholar]
- 117.Schols L, Bauer P, Schmidt T, et al. Autosomal dominant cerebellar ataxias: clinical features, genetics and pathogenesis. Lancet Neurol 2004;3:291–304. [DOI] [PubMed] [Google Scholar]
- 118.Verbeek DS. Spinocerebellar ataxia type 23: a genetic update. Cerebellum 2009;8:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhao L, Longo-Guess C, Harris BS, et al. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat Genet 2005;37:974–9. [DOI] [PubMed] [Google Scholar]
- 120.Lee JW, Beebe K, Nangle LA, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 2006;443:50–5. [DOI] [PubMed] [Google Scholar]
- 121.Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006;441:885–9. [DOI] [PubMed] [Google Scholar]
- 122.Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441:880–4. [DOI] [PubMed] [Google Scholar]
- 123.Howes J, Shimizu Y, Feige MJ, et al. C-terminal mutations destabilize SIL1/BAP and can cause marinesco-sjogren syndrome. J Biol Chem 2012;287:8552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 2010;11:247–58. [DOI] [PMC free article] [PubMed] [Google Scholar]