

Abstract
Neuronal apoptosis induced by oxidative stress is one of the major pathological processes involved in neurological impairment after hypoxic-ischemic encephalopathy (HIE). Ghrelin, the unique endogenous ligand for the growth hormone secretagogue receptor-1α (GHSR-1α), could take an anti-apoptotic role in the brain. However, whether ghrelin can attenuate neuronal apoptosis by attenuating oxidative stress after hypoxia-ischemia (HI) insult remains unknown. To investigate the beneficial effects of ghrelin on oxidative stress injury and neuronal apoptosis induced by HI, ten-day old unsexed rat pups were subjected to HI injury and exogenous recombinant human ghrelin(rh-Ghrelin) was administered intranasally at 1 h and 24 h after HI induction. [D-Lys3]-GHRP-6, a selective inhibitor of GHSR-1α and Ex527, a selective inhibitor of GHSR-1α were administered intranasally at 1 h before HI induction respectively. Small interfering ribonucleic acid (siRNA) for GHSR-1α were administered by intracerebroventricular (i.c.v) injection at 24 h before HI induction. Neurological tests, immunofluorescence, MitoSox staining, Fluoro-Jade C staining, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, and western blot experiments were performed. Our results indicated that ghrelin significantly improved neurobehavioral outcomes and reduced oxidative stress and neuronal apoptosis. Moreover, ghrelin treatment significantly promoted phosphorylation of AMPK, upregulated the expression of Sirt1, PGC-1α, UCP2 and the ratio of Bcl2/Bax, while it downregulated cleaved caspase-3 levels. The protective effects of ghrelin were reversed by [D-Lys3]-GHRP-6, GHSR-1α siRNA or Ex527. In conclusion, our data demonstrated that ghrelin reduced oxidative stress injury and neuronal apoptosis which was in part via the GHSR-1α/AMPK/Sirt1/PGC-1α/UCP2 signalling pathway after HI. Ghrelin may be a novel therapeutic target for treatment after neonatasl HI injury.
Keywords: Hypoxic-ischemic encephalopathy, Ghrelin, GHSR-1α, Oxidative stress, Neuronal apoptosis
Graphical Abstract
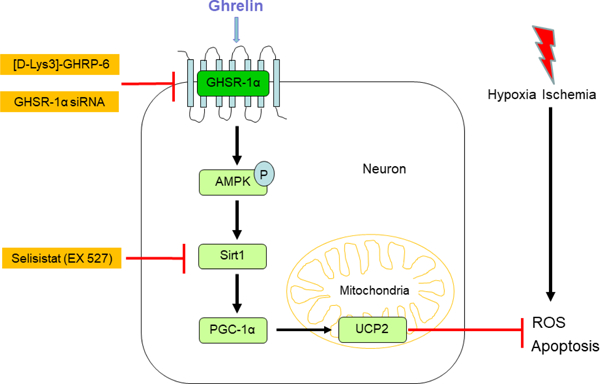
1. Introduction
Hypoxic–ischemic encephalopathy (HIE) is one of the most common causes of brain injury in infants, occurring in 1 to 8 per 1000 live births in developed countries and as high as 26 per 1000 newborns in developing countries (1). Severe HIE has tremendous detrimental effects on the developing brain and leads to long lasting neurological sequelae, including cerebral palsy, mental retardation, seizures, epilepsy and other neurological disabilities (2–5). Despite the effectiveness of current clinical treatments such as therapeutic hypothermia (6), anticonvulsants (4) and fluid and electrolyte management (7), there are still no specific treatments to repair the damage caused by HIE. Therefore, it is imperative to develop effective and safe complementary therapies that will add onto the current therapeutic strategies for infants with HIE.
Ghrelin is a brain-gut hormone with 28 amino acid peptide, which is secreted to regulate the release of growth hormone (GH) and promote adiposity and appetite (8). Ghrelin has been identified as the unique endogenous ligand for the growth hormone secretagogue receptor-1α (GHSR-1α) (9). GHSR-1α is a G-protein-coupled receptor, expressed in peripheral tissues and extensively in the central nervous system, such as pituitary gland, hypothalamus, thalamus, cortex and hippocampus (10–13).
Up until now, myriad studies have shown that neuronal apoptosis, due to oxidative stress, plays a major role in brain injury associated with HIE (14). It has been established that ghrelin could inhibit cell apoptosis in various cells, such as cardiocytes, hepatocytes, epithelial cells and endotheliocytes (15–18). Moreover, ghrelin can also take anti-apoptotic effect on neurons in some experiments in vivo and in vitro. Chung et al reported that ghrelin acts as a survival factor for hypothalamic neuronal cells by inhibiting apoptosis induced by glucose-oxygen deprivation (19). Ghrelin has also been reported to protect cortical neurons and retinal neurons against focal ischemia/reperfusion and glaucoma injured rats respectively (20, 21). However, little is known on ghrelin’s protective and antiapoptotic effects after HIE. We will explore this question in the present study.
Accumulating scientific evidence indicates that most of the biological effects of ghrelin are mediated by the activation of adenosine monophosphate-activated protein kinase (AMPK) (22, 23). AMPK acts as a metabolic stress-sensing protein kinase, switching off biosynthetic pathways to maintain the local and systemic energy balance under energetic stress (24). Recently, activation of AMPK to inhibit neuronal apoptosis has been considered as a therapeutic strategy for neuronal diseases (25, 26). One of the downstream pathways of AMPK activation is Sirt1/PGC-1α. AMPK activates the NAD+-dependent type III deacetylase Sirt1 by increasing the intracellular NAD+/NADH ratio. Once Sirt1 is activated it results in the deacetylation and modulation of the activity of the peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α) (24, 27).
The uncoupling protein 2 (UCP2) is an inner-membrane mitochondrial protein, which has been proposed to be the direct target of PGC-1α transcriptional regulation (28, 29). Recent evidence has shown that UCP2 is involved in reducing reactive oxygen species (ROS) generation and clearance of mitochondrial ROS (30, 31). Concomitantly, it was found that UCP2 can prevent neuronal apoptosis and diminish brain dysfunction after stroke and brain trauma (32).
Based on the aforementioned evidence, we hypothesized that intranasal administration of ghrelin can attenuate neuronal apoptosis via the GHSR-1α/AMPK/Sirt1/PGC-1α/UCP2 pathway in a rat model of neonatal HIE.
2. Material and methods
2.1. Animals
All experimental protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University (IACUC) in accordance with the NIH Guide for the Care and Use of Laboratory Animals. All studies comply with the ARRIVE guidelines.
Sprague Dawley rat mothers, with litters of 12 pups, were purchased from Envigo (Livermore, CA). A total of 196 ten-day old unsexed rat pups (weighing 16–20g) were used for this study. All animals were housed in a controlled humidity and temperature room with a 12 h light and dark cycle and raised with free access to breast milk, water and food. We chose the neonatal rat model in this study for several reasons. Firstly, using the Sprague-Dawley Rat carries several advantages amongst other small species: a) it has been used in hundreds of studies over the past 20 years to study stroke in adults and neonates and can be readily compared to the literature; b) the post brain injury sequelae is similar to humans and HI brain injury induced in P10 rats mimics pre and perinatal brain injuries in humans; c) their small size allows for the use of smaller amounts of drugs for testing; d) it allows us to study mechanisms and signaling pathways and e) we have already established a SD rat HI model with less than 10% mortality that creates significant reproducible neurological deficits. Second, the immunologic and local milieu following HI cannot be mimicked in cell culture or effectively modeled by computers. Furthermore, our interests lie in the pathophysiological (e.g. cell apoptosis) and neurobehavioral outcomes after neonatal HI and these cannot be replicated in-vitro.
2.2. HIE model
The animal model used in this study is the standard HIE Rice-Vannucci model, with some modification, as previously described (26, 33). Briefly, ten-day old neonatal rat pups were placed into a temperature-controlled chamber for induction of general anesthesia. The animals were anesthetized with 3% isoflurane gas in air and maintained at 2.0% isoflurane in air. Throughout the surgical and postoperative period, temperature was controlled with heating blankets and incubators. After induction of anesthesia the neck of the rats was prepared and draped using standard sterile techniques. Skin preparation consisted of swabbing the neck with alcohol. Next a small midline neck incision on the anterior neck was made with a No. 11 blade surgical knife (approximately 3–5 mm in length). Using gentle blunt dissection the right carotid artery was isolated and gently separated from surrounding structures. The carotid artery was ligated with 5-O surgical suture, and cut between the ligatures. All bleeding was controlled with gentle pressure and electrocautery as needed. After ligation of the carotid artery the surgical field was irrigated with several drops of saline, dried, and the skin was closed with sutures. All surgeries were performed aseptically and lasted no longer than 10 minutes. After the surgical procedure was completed, the rats were allowed to wake and recover for 1 hour. Thereafter, they were placed in an airtight jar partially submerged in a 37 °C water bath to maintain a constant thermal environment. A gas mixture of 8% oxygen and 92% nitrogen was delivered into the jars through inlet and outlet portals. The rat pups were exposed to this gas mixture for 150 minutes. Thereafter, the animals were returned to their mothers. Animals were monitored daily post-op for any bleeding, swelling, pain or distress. For the sham animals, CCA was just exposed without ligation or cutting, and pups did not experience the hypoxic insults.
2.3. Experimental design
The experiment was designed as follows.
2.3.1. Experiment I
To evaluate the time course expression of endogenous ghrelin and GHSR-1α after HI, rats were randomly divided into 6 groups: Sham, 6 h HI, 12 h HI, 24 h HI, 72 h HI and the right (ipsilateral) hemisphere was collected for western blot analysis. The Sham group rat pups were euthanized at 24 h post HI.
2.3.2. Experiment II
To assess the short-term outcome of recombinant human ghrelin (rh-Ghrelin) treatment and to establish the optimal dose of rh-Ghrelin treatment for HI insults, pups were randomly divided into 5 groups: Sham, HI + Vehicle, HI + rh-Ghrelin (0.02μg/kg), HI + rh-Ghrelin (0.04μg/kg), and HI + rh-Ghrelin (0.12μg/kg). rh-Ghrelin or Vehicle (10% Glycerol, 20mM Tris HCl, pH 8.0) were administered intranasally at 1 h after HI followed by one more injection at 24 h after HI. Infarct volume, short term neurobehavioral tests, negative geotaxis, and body weight were evaluated at 48 h after HI.
2.3.3. Experiment III
To verify that GHSR-1α is expressed in neurons, the colocalization of GHSR-1α with neuron marker, NeuN, was checked at 48 h post HI. Rats were divided into 3 groups: Sham, HI + Vehicle, HI + rh-Ghrelin (optimal dose).
2.3.4. Experiment IV
To assess the long-term outcomes of exogenous rh-Ghrelin treatment, rats were divided into 3 groups: Sham, HI + Vehicle, HI + rh-Ghrelin (optimal dose). Neurobehavioral test: Foot-fault, Rota-rod and Morris water maze were conducted at 4 weeks post HI, then rats were sacrificed for Nissl staining.
2.3.5. Experiment V
To explore whether GHSR-1α was involved in the underlying mechanisms of rh-Ghrelin mediated neuroprotective effects, GHSR-1α inhibitor, [D-Lys3]-GHRP-6 was used to inhibit GHSR-1α. Rats were randomly divided into 5 groups: Sham, HI + Vehicle 1 (Vehicle of rh-Ghrelin), HI + rh-Ghrelin, HI + rh-Ghrelin + Vehicle 2 (Vehicle of [D-Lys3]-GHRP-6, 0.9% NaCl), HI + rh-Ghrelin + [D-Lys3]-GHRP-6. Rh-Ghrelin or Vehicle 1 were injected intranasally 1 h and 24 h after HI. [D-Lys3]-GHRP-6 or Vehicle 2 were administered intranasally at 1 h before HI induction. Western blot, mitochondrial oxidative stress, neuronal degeneration and neuronal apoptosis were examined at 48 h after HI.
2.3.6. Experiment VI
To further explore whether GHSR-1α was involved in the underlying mechanisms of rh-Ghrelin mediated neuroprotective effects, GHSR-1α siRNA (Si-GHSR-1α) was used to inhibit GHSR-1α. Rats were randomly divided into 2 groups: HI + rh-Ghrelin + scramble siRNA, HI + rh-Ghrelin + si-GHSR-1α. Rh-Ghrelin was injected intranasally 1 h and 24h after HI. Si-GHSR-1α or scramble siRNA were administered intracerebroventricularly at 24 h before HI. Western blot, mitochondrial oxidative stress, neuronal degeneration and neuronal apoptosis were examined at 48 h after HI.
In addition, to detect the effectiveness of siRNA access to brain, the colocalization of a non-target siRNA with red fluorescence tag on neurons/astrocytes/microglias was evaluated at 24 h post siRNA injection in naïve 10-day-old rats.
2.3.7. Experiment VII
To explore whether Sirt1 was involved in the underlying mechanisms of rh-Ghrelin mediated neuroprotective effects, Sirt1 inhibitor, Ex527 was used to inhibit GHSR-1α. Rats were randomly divided into 2 groups: HI + rh-Ghrelin + Vehicle of Ex527(DMSO), HI + rh-Ghrelin + Ex527. Rh-Ghrelin was injected intranasally 1 h and 24h after HI. Ex527 or Vehicle were administered intranasally at 1 h before HI induction. Western blot, mitochondrial oxidative stress, neuronal degeneration and neuronal apoptosis were examined at 48 h after HI.
2.4. Drug administration
Rh-Ghrelin (0.02, 0.04, 0.12 μg/kg, Abcam, USA) or vehicle of rh-Ghrelin were administered intranasally at 1 and 24 h after HI. [D-Lys3]-GHRP-6 (Abcam, USA), Ex527(Abcam, USA) or vehicle of them were administered intranasally at 1 h before HI. 1.25 μl of rh-Ghrelin, [D-Lys3]-GHRP-6, Ex527 or vehicle per drop was given every 2 min in alternating nares.
2.5. In vivo RNAi
After anesthetized with isoflurane, rat pups were fixed in a stereotactic frame in a prone position. A 10-μl micro syringe (Hamilton Co, USA) was inserted at 1.5 mm posterior, 1.5 mm lateral to the bregma and 1.7 mm deep from the skull surface into the right lateral ventricle. At 24 h before HI induction, 2 μl GHSR-1α siRNA (300 pmol/ μl, Dharmacon, USA) or scramble siRNA (300 pmol/ μl, Dharmacon, USA) was injected slowly over 5 min. To prevent liquid reflux, the syringe was allowed to stand in place for 10 min and then withdrawn slowly for 5 min.
2.6. Infract area measurements
As previously described, 2,3,5-triphenyltetrazolium chloride monohydrate (TTC) staining was used to evaluate the infarct area (34). Rat pups were anesthetized with isoflurane and perfused transcardially with 20 ml 4 ℃ phosphate-buffered saline (PBS) at 48 h post HI. The brains were dissected out and sectioned into coronal brain slices (2 mm) in a rat brain matrix. The slices were stained with 2 % TTC (Sigma Aldrich Inc., USA) solution for 5 min and then washed in PBS, followed by digitally photographed. Un-infarcted area of ipsilateral hemispheres and total area of contralateral hemispheres were traced and analyzed using Image J software (NIH, USA). The percent of infarcted area for each slice was calculated as following formula: [(total area of contralateral hemisphere) - (area of un-infarcted area of ipsilateral hemisphere)] / (total area of contralateral hemisphere × 2). The average value of each slice in one brain was taken to represent the percentage of infarcted area for that animal.
2.7. Neurobehavioral tests
Negative geotaxis tests were performed at 48 h post HI for evaluating short-term neurological function. Foot-fault, Rotarod and Morris water maze were performed at 4 weeks post HI for evaluating long-term neurological function.
2.7.1. Negative geotaxis
The pups were put head downward on a 40-degree sloping board, the time taken for the pups to turn their body around and face upward was recorded. The maximum testing time was 60 s and the time taken more than 60 s was recorded as 60 s.
2.7.2. Rotarod test
Rat pups were placed on a rotating horizontal rod (Columbus Instruments Rotamex, USA) and the time it took for them to fall was recorded. The rotation speed started from 5 or 10 rpm separately with an acceleration of 2 rpm per 5 s. The maximum testing time was 60 s and the time taken more than 60 s was recorded as 60 s.
2.7.3. Foot-fault test
Rat pups were placed on an elevated (1 m above the floor) horizontal grid piece (square size 20 × 40 cm with a mesh size of 4cm2, wire diameter 0.4 cm) for 1 min. Foot-fault was defined as when the pup could not place a for- or hind limb accurately and it fell down between the grid bars. The number of foot faults of each animal were recorded by video device and analyzed by an investigator blinded to the experimental groups.
2.7.4. Morris water maze test
Morris water maze testing was used to evaluate the animal ability of spatial locations learning and memory (35). Rats performed a 6-day test and 5 trials were applied to each rat per day. Between successive trials, there was a 10 min interval. All trials lasted no more than 60 s. On day 1, rats were trained using a visible platform with 10 cm diameter (cued test, block 1). If the rats had not discovered the platform in 60s, they were manually guided to the platform. On days 2 – 5, the time taken to find a platform submerged 1 cm below the water was measured (memory test, blocks 2–5). On day 6, the platform was removed, and the time rats took in the platform quadrant were tested (probe trial, block 6). All of the animals’ swimming paths, the quantification of distance, latency and swimming speed was recorded by the Video Tracking system SMART-2000 (San Diego Instruments Inc, USA).
2.8. Western blotting analysis
Western blot was performed as previously described (36). After TTC staining and images recording at 48 h after HI, brain slices were separated immediately into the contralateral and ipsilateral cerebrums and stored instantly at −80 °C freezer until lysis. The right/ipsilateral hemisphere tissue was homogenized by RIPA lysis buffer (Santa Cruz Biotechnology, USA) with protease inhibitor cocktail and then centrifuged at 14,000 g at 4 °C for 20 min, followed by the supernatant was collected. After protein concentration assay (DC™ Protein Assay, Bio-Rad, USA), equal amounts of protein were loaded into a 7.5 % - 12 % sodium dodecyl sulfate–polyacrylamide (SDS-PAGE) gel for electrophoresis and then transferred to a nitrocellulose membrane, which was blocked with 5% non-fat blocking grade milk (Bio-Rad, USA) for 1h at room temperature. The membranes were incubated with the primary antibody overnight at 4 °C. The following primary antibodies were used: anti-GHSR-1α (1:1000, Abcam, USA), anti-ghrelin (reacts with human and rats, 1:2000, Thermo fisher, USA), anti-ghrelin (reacts specifically with human, 1:2000, Abcam, USA), anti-AMPK (1:1000, Cell Signaling Technology, USA), anti-p-AMPK (1:500, Cell Signaling Technology, USA), anti-Sirt1 (1:1000, Abcam, USA), anti-PGC1α (1:1000, Cell Signaling Technology, USA), anti-UCP2 (1:1000, Cell Signaling Technology, USA), anti-Bcl2 (1:1000, Abcam, USA), anti-Bax (1:500, Abcam, USA), and anti-cleaved caspase-3(1:500, Cell Signaling Technology, USA). Goat anti-β-actin (1:3000, Santa Cruz Biotechnology, USA) was used on the same membrane as loading control. The membranes were then incubated with appropriate secondary antibodies (1:3000, Santa Cruz Biotechnology, USA) for 1 h at room temperature. Immunoreactive bands were visualized with an ECL Plus kit (American Bioscience, UK) followed by exposure to X-ray films, then analyzed using Image J software (NIH, USA).
2.9. Tissue processing
Rats were deeply anesthetized and transcardially perfused with 4 °C PBS and 10% formalin. The brains were removed and fixed with 10% formalin for 24 h, and then immersed in 30% sucrose until they sank. After being embedded into OCT compound (Scigen Scientific, USA) and frozen, serial 10 μm thick coronal sections through the infarct region were cut sequentially at −20 °C with a cryostat (LM3050S; Leica Microsystems, Germany) for double immunofluorescence, Fluoro-Jade C, and TUNEL staining.
2.10. Double immunofluorescence staining
The prepared sections were permeabilized with 0.3% Triton X-100 for 15 min at room temperature and then blocked with 5% donkey serum at 37 °C for 30 min. Subsequently, each coronal section was incubated at 4 °C overnight with primary antibodies: rabbit anti-Ghrelin (1:100, Abcam, USA), and mouse anti-NeuN (1:400, Abcam, USA). Sections were washed in PBS and appropriate secondary antibodies fluorescence-conjugated were applied at the dilution of 1:200 for 1 h at 37 °C and then washed in PBS. To facilitate proper photographic orientation, sections were mounted with Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories Inc., USA). Images were then captured by a fluorescence microscope (Leica DMi8, Leica Microsystems, Germany).
2.11. MitoSOX staining
To assess the oxidative stress level in the mitochondria, freshly prepared frozen brain sections were incubated with 5 μmol/L MitoSOX (Thermo Fisher Scientific, USA) at 37 °C for 10 min in a dark humidified chamber. Sections were mounted with Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories Inc., USA). Digital images were captured and the number of MitoSOX-positive cells per field of view were counted manually in the ipsilateral cortex. Six sections with ischemic regions were picked in each brain to be analyzed. Data was expressed as ratio of MitoSOX-positive cells (%).
2.12. Fluoro-Jade C staining
Fluoro-Jade C (FJC) staining was performed according to the manufacturer’s protocol (Fluoro-Jade C Ready-to-Dilute Staining Kit, Biosensis, USA). The number of FJC-positive cells per field of view were counted manually in the ipsilateral cortex. Six sections with ischemic regions were picked in each brain to be analyzed. The data were presented as the average number of FJC-positive cells/mm2 in the microscopic field of 20 ×.
2.13. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
Double staining of TUNEL and neuron marker NeuN was performed for quantification of neuronal cell death (37). TUNEL staining was performed according to the manufacturer’s instructions at 48 h after HI (in situ Apoptosis Detection Kit, Roche, USA). NeuN staining was performed as the immunofluorescence procedure above mentioned. The number of TUNEL-positive neurons and NeuN-stained positive cells were counted manually in the ipsilateral cortex. Six sections with ischemic regions were picked in each brain to be averaged. Data was expressed as ratio of TUNEL-positive neurons (%).
2.14. Tissue loss measurement
Nissl staining was performed to evaluate the brain tissue loss. The process of coronal brain sections (20mm thick) were prepared as that in the tissue processing of immunofluorescence, Fluoro-Jade C, and TUNEL staining. The sections were dehydrated in 95% and 70% ethanol for 1 min successively, and then stained with 0.2 % cresyl violet (Sigma-Aldrich, USA) for 2 min. Following dehydrated in 100% ethanol and xylene for 1.5 min, respectively, sections were mounted with DPX (Sigma-Aldrich, USA). A total of 4 slices were used to average percent tissue loss in each brain. The percentage of brain tissue loss = [(contralateral hemisphere − ipsilateral hemisphere) / 2 × contralateral hemisphere] × 100%.
2.15. Statistical analysis
Statistical analyses were performed with SPSS v. 21.0 software (IBM, USA). The data were expressed as means ± SD. Differences between individual groups were first compared using analysis of variance (one-way ANOVA) and then post hoc testing were analyzed with Tukey or Student-Newman-Keuls multiple comparisons. Differences between two groups were compared using Student’s t test. All reported P values were two-sided, and a value of P < 0.05 was considered statistically significant.
3. Results
3.1. Expression levels of endogenous ghrelin and GHSR-1α decreased in time-dependent manner post HI
The endogenous expression levels of ghrelin and GHSR-1α in the brain were measured at 6 h, 12 h, 24 h, 48 h and 72 h after HI. Ghrelin expression level decreased significantly from 6 h to 72 h post HI when compared with sham group (P < 0.05), reaching the lowest point at 24h post HI (Fig. 1A and B). GHSR-1 α expression level decreased significantly from 6 h to 72 h post HI when compared with sham group (P < 0.05), reaching the lowest point at 48 h post HI (Fig. 1A and C).
Fig. 1.
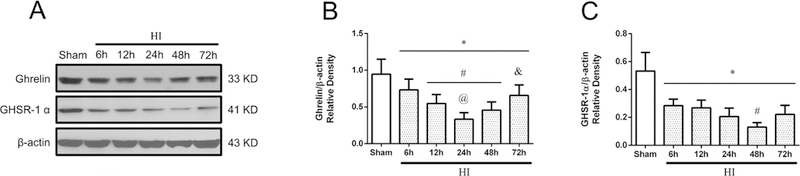
Temporal expression of endogenous Ghrelin and GHSR-1α in the brain after HI. (A) Representative pictures of western blot data. (B) Western blot data analysis showed that endogenous ghrelin expression levels significantly decreased in a time-dependent manner from 6 h to 72 h reaching the lowest point at 24 h post HI. (C) Western blot data analysis showed that endogenous GHSR-1α expression levels significantly decreased in a time-dependent manner from 6 h to 72 h reaching the lowest point at 48 h post HI. *P < 0.05 vs. Sham; #P < 0.05 vs. 6 h HI; @P< 0.05 vs. 12 h HI; &P < 0.05 vs. 24 h HI. Data are represented as mean ± SD, n= 6 for each group.
3.2. Intranasal administration of exogenous rh-Ghrelin reduced infarct area, improved short-term neurological function, and reduced body weight loss at 48 h post HI
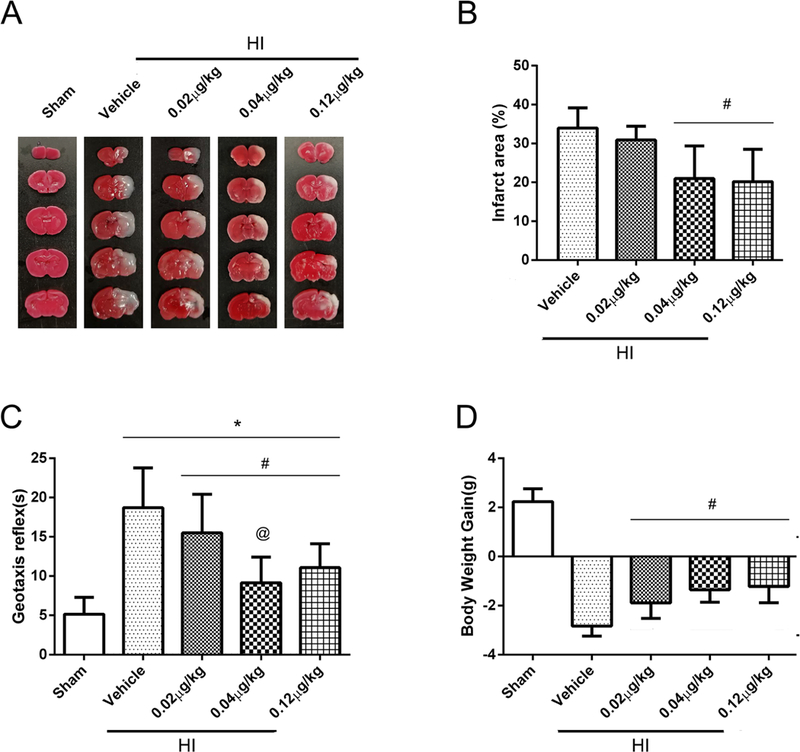
To determine the best dosage of rh-Ghrelin treatment, three doses were used: low (0.02 μg/kg), medium (0.04 μg/kg) and high (0.12 μg/kg). TTC staining results (Fig. 2A and B) showed that the medium (0.04μg/kg) and high (0.12 μg/kg) doses of rh-Ghrelin treatment significantly reduced percent infarct area when compared with vehicle group (P < 0.05). No significant differences in infarcted area were found in the low dose (0.02 μg/kg) group when compared with vehicle group (P > 0.05).
Fig. 2.
Effect of intranasal administration of rh-Ghrelin on brain infarct area (A-B), short-term neurological function (C) and body weight (D) at 48 h post HI. (A-B) TTC staining showed that medium (0.04μg/kg) and high (0.12μg/kg) doses of rh-Ghrelin treatment significantly reduced infarct area when compared with vehicle. (C) Geotaxis test showed reflex time increased significantly after HI when compared with sham group. All three doses of rh-Ghrelin decreased the reflex time significantly compared with the Vehicle group, and the medium dose group (0.12 μg/kg) showed the shortest time in the three doses groups. (D) Animals in vehicle group showed to lose significant weight compared with sham and all three treatment groups after HI. *P < 0.05 vs. sham; #P < 0.05 vs. vehicle; @P < 0.05 vs. HI+ rh-Ghrelin (0.02 μg/kg). Data are represented as mean ± SD, n= 6 for each group.
Geotaxis test results (Fig. 2C) showed that pups reflex time increased significantly after HI when compared with sham group (P < 0.05). All three doses of rh-Ghrelin decreased the reflex time significantly compared with the vehicle group (P < 0.05), while the medium dose group (0.12 μg/kg) showed the best performance compared to the other two dose groups.
Body weight measurement results (Fig. 2D) showed that vehicle group had significant body weight loss at 48 h post HI when compared with sham and rh-Ghrelin treated groups (P < 0.05, Fig. 2D). No difference in body weight loss was observed between treatment groups (P > 0.05).
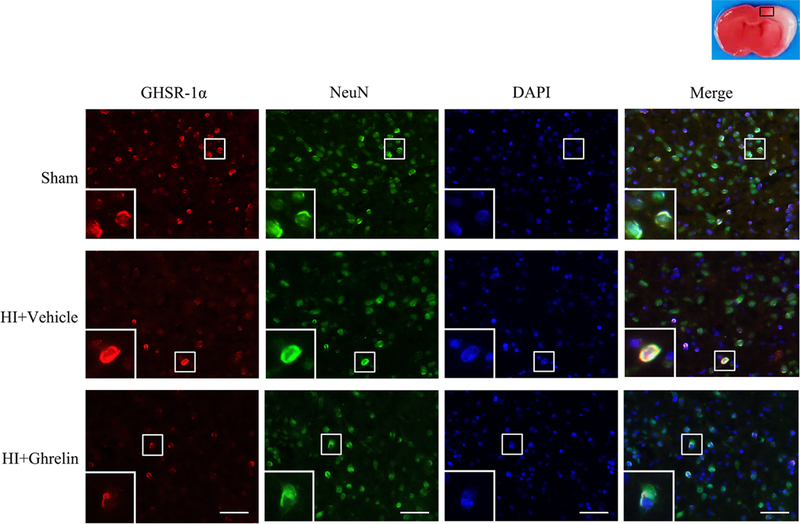
3.3. Immunofluorescence staining showed the colocaliztion of GHSR-1α with neurons at 48h post HI
Immunofluorescence staining showed that GHSR-1α colocalized with the neuronal marker, NeuN, at 48h post HI in the following three groups: sham, vehicle, and rh-Ghrelin treatment (0.04 μg/kg). Expressions of GHSR-1α on neurons was lower in vehicle and rh-Ghrelin treatment group compared to the sham group (Fig 3).
Fig. 3.
Representative immunofluorescence staining of GHSR-1α and the neuron marker NeuN in the brain at 48 h post HI. GHSR-1α was colocalized with NeuN in the sham, vehicle, and rh-Ghrelin treatment (0.04 μg/kg) group respectively. Compared with the sham group, there were lower expressions of GHSR-1α on neurons in vehicle and rh-Ghrelin treatment group. Red was for GHSR-1α, green was for NeuN, and blue was for DAPI. Top panel indicates the location of staining (small black box). 1 rat for each group. Scale bar = 50 μm.
3.4. Rh-Ghrelin reduced mitochondria oxidative stress injury and neuronal apoptosis at 48 h post HI
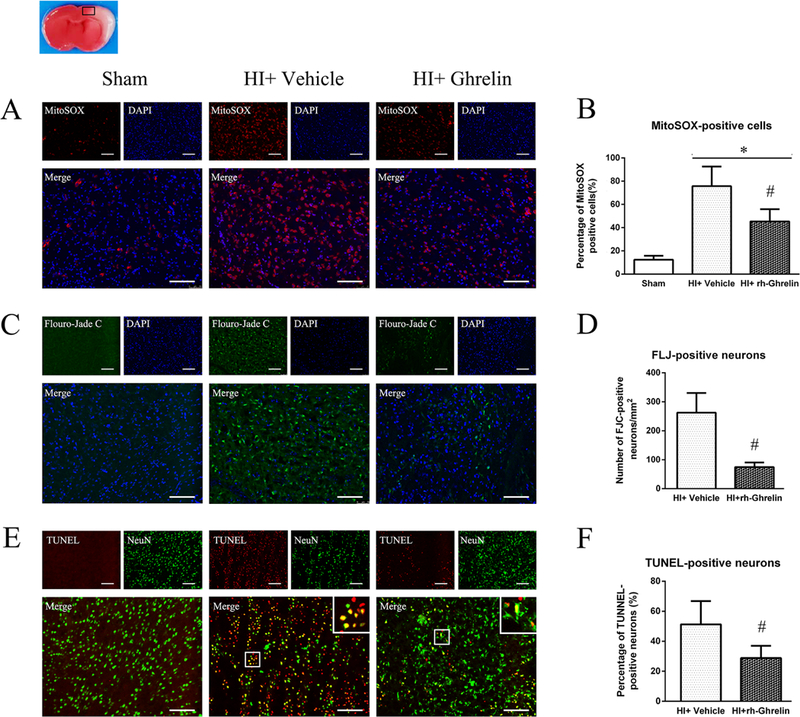
Mitochondria oxidative stress level in the ipsilateral hemisphere after HI was measured by MitoSOX staining (Fig. 4 A, B). MitoSOX-positive cells in vehicle group were more evident compared with sham group at 48 h after HI. Intranasal administration of rh-Ghrelin (0.04 μg/kg) significantly reduced MitoSOX-positive cells when compared to the vehicle group.
Fig. 4.
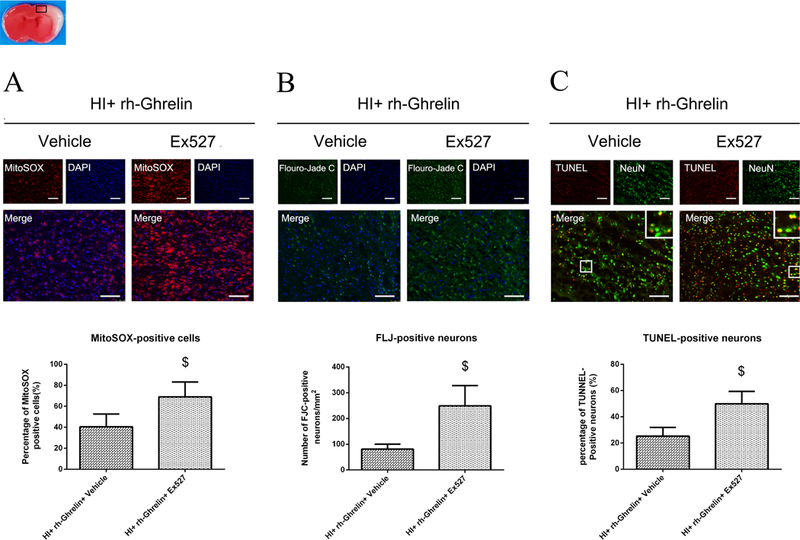
Effects of rh-Ghrelin on mitochondria oxidative stress, neuronal degeneration and apoptosis after HI. (A) Representative microphotographs of MitoSOX staining in the ipsilateral cortex of rat brain. (B) Quantitative analysis of MitoSOX-positive cells in the ipsilateral cortex 48 h after HI. (C) Representative microphotographs of FJC-positive cells in the ipsilateral cortex 48 h after HI. (D) Quantitative analysis of FJC-positive cells in the ipsilateral cortex 48 h after HI. (E) Representative microphotographs of TUNEL-positive neurons in the ipsilateral cortex 48 h after HI. (F) Quantitative analysis of TUNEL-positive neurons in the ipsilateral cortex 48 h after HI. Top panel indicates the location of staining (small black box). *P < 0.05 vs. sham; #P < 0.05 vs. HI+ vehicle. Data are represented as mean ± SD, n= 3 for each group. scale bar = 100μm.
Neuronal degeneration in the ipsilateral hemisphere, after HI, was measured by Flouro-Jade C (FLJ) staining (Fig. 4 C, D). No FJC-positive cell was detected in sham group, while degenerated neurons were detected easily in the vehicle group at 48 h after HI. Rh-Ghrelin (0.04 μg/kg) treatment significantly reduced FJC-positive neurons in comparison to the vehicle group.
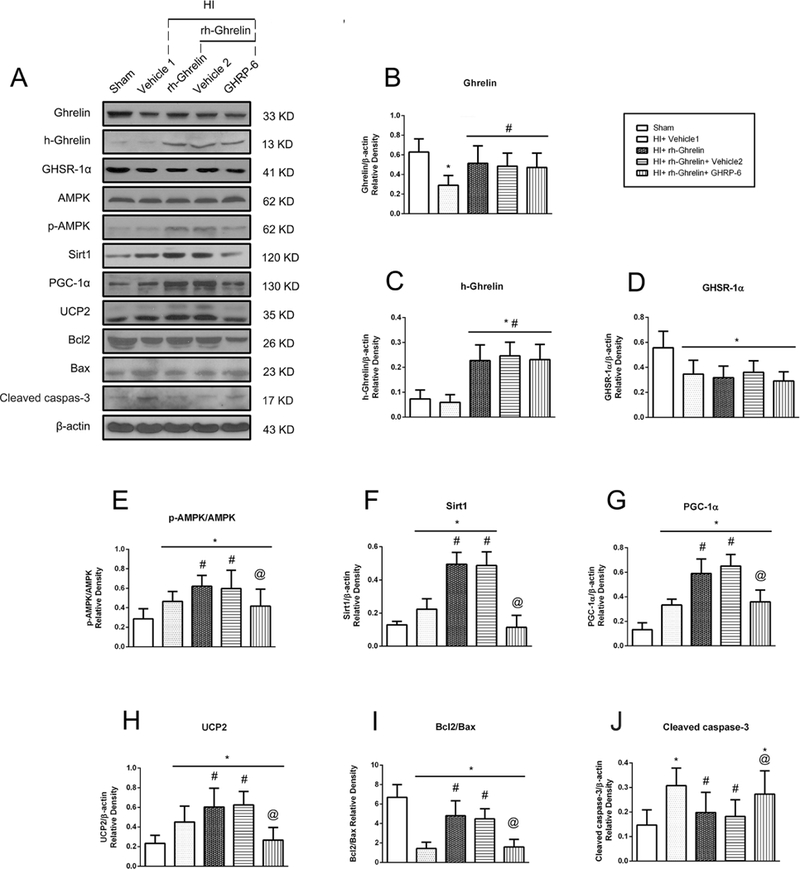
Neuronal apoptotic cells in the ipsilateral hemisphere after HI were measured by TUNEL staining (Fig. 4 E, F). There was a higher level of TUNEL-positive cells in vehicle group compared to sham at 48 h after HI. Rh-Ghrelin (0.04 μg/kg) treatment significantly reduced TUNEL-positive cells in comparison with vehicle group. In addition, western blot data showed that cleaved caspase-3 and Bax were significantly increased and Bcl-2 was significantly decreased after HI, and rh-Ghrelin (0.04 μg/kg) treatment could reduce the expression of Bax while upregulating the expression of cleaved caspase-3 and Bcl-2 (Fig. 10).
Fig. 10.
Representative picture of western blot bands (A) and quantification of data(B-I) showing the effects of [D-Lys3]-GHRP-6 intervention on proteins in proposed signaling pathway and proteins related to apoptosis in rh-Ghrelin treatment animals at 48h post HI. Vehicle 1 is the vehicle of rh-Ghrelin, Vehicle 2 is the vehicle of [D-Lys3]-GHRP-6. *P < 0.05 vs. sham, #P < 0.05 vs. HI+ Vehicle 1, @P < 0.05 vs. HI+ rh-Ghrelin+ Vehicle Data are represented as mean ± SD, n= 6 for each group.
3.5. Rh-Ghrelin reduced brain atrophy and improved long-term neurological functions at 4 weeks post HI
To test the effects of rh-Ghrelin treatment on brain atrophy at 4 weeks after HI, tissue loss was assessed by Nissl’s staining. There was no tissue loss in the sham group. Intranasal administration of rh-Ghrelin (0.04 μg/kg) significantly reduced brain tissue loss in the ipsilateral hemisphere in comparison to vehicle group (P < 0.05, Fig. 5A, B).
Fig. 5.
Effects of rh-Ghrelin treatment on brain atrophy and neurological function at 4 weeks after HI. (A) Representative pictures of Nissl’s stained brain slices. (B) Quantitative analysis of tissue loss showed by Nissl’s staining. (C) The falling latency at 5 rpm and 10 rpm acceleration in rotarod test. (D) Percentage of foot-faults in each limb on each side. (E) Swimming distance of Morris Water Maze. (F) Escape latency of Morris Water Maze. (G) Representative heatmaps of the probe trial. The probe platform was located at southwest quadrant. (H) Quantification of the probe quadrant duration in the probe trial. *P < 0.05 vs. sham; #P < 0.05 vs. HI+ vehicle. Data are represented as mean ± SD, n= 8 for each group.
In the rotarod test, animals in vehicle group had significantly lower falling latency when compared with sham group; rh-Ghrelin treatment (0.04 μg/kg) markedly improved rotarod latency at both 5 rpm and 10 rpm acceleration when compared with vehicle group (P < 0.05, Fig. 5C).
In the foot-fault test, vehicle group displayed more foot-faults on contralateral side(left) compared to the sham group (P < 0.05); the performance was significantly improved in rh-Ghrelin (0.04 μg/kg) group compared to the vehicle group (P < 0.05, Fig. 5D).
In water maze test, vehicle group showed substantial spatial memory loss in terms of swimming longer distance (Fig. 5E), longer time taken to find the platform (Fig. 5F) and less time spent in the platform quadrant (Fig. 5G, H) when compared with sham group (P < 0.05). Rh-Ghrelin treated group (0.04 μg/kg) showed significant memory function recovery compared with vehicle group as seen from the decreased swimming distance(Fig. 5E), time to find platform(Fig. 5F), and more time spent in platform quadrant (Fig. 5G, H).
3.6. Rh-Ghrelin treatment upregulated p-AMPK, Sirt1, PGC-1α, UCP2 levels at 48h post HI
To investigate whether AMPK, Sirt1, PGC-1α or UCP2 are involved in the mechanism of ghrelin treatment after HI, these protein expression levels were tested by western blot. Western blot data showed that p-AMPK, Sirt1, PGC-1α and UCP2 expression significantly increased in vehicle group when compared with sham group (P < 0.05, Fig. 10). Rh-Ghrelin treatment further upregulated p-AMPK, Sirt1, PGC-1α and UCP2 expression when compared with vehicle group (P < 0.05, Fig. 10).
3.7. Inhibition of GHSR-1α attenuated the neuroprotective, anti-oxidative and anti-apoptotic effects induced by rh-Ghrelin treatment at 48 h post HI
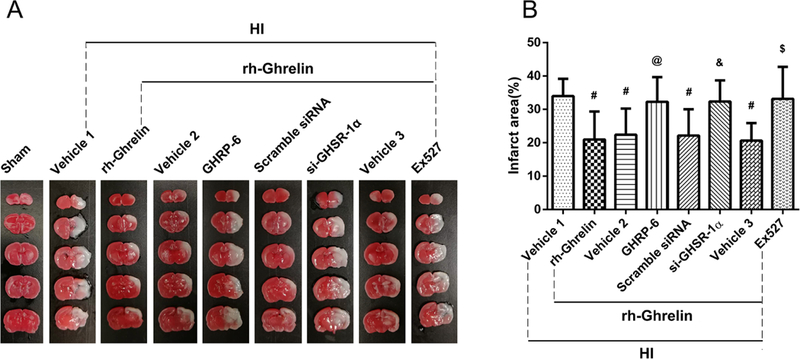
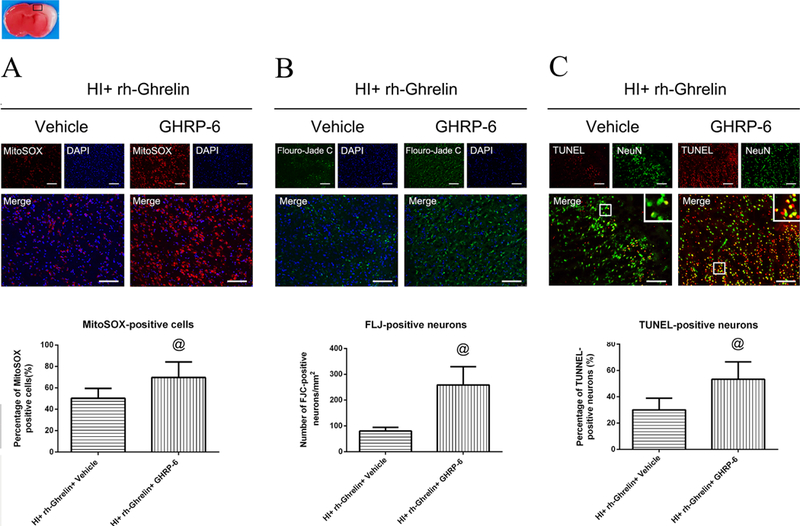
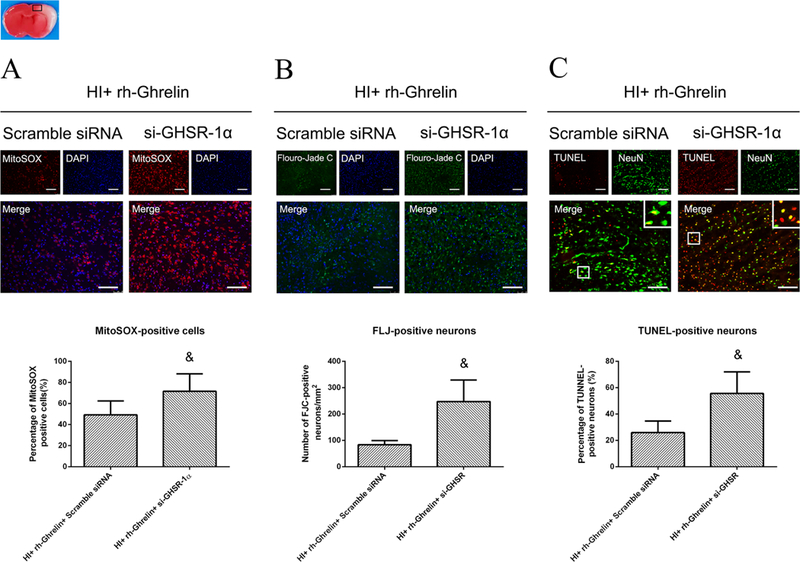
To determine GHSR-1α’s role in rh-Ghrelin’s neuroprotective effects we used a specific inhibitor and a siRNA to inhibit GHSR-1α: the specific GHSR-1α inhibitor, [D-Lys3]-GHRP-6, was administered intranasally (i.n.) and the GHSR-1α siRNA was administered via an intracerebroventricular (i.c.v.) injection. Both [D-Lys3]-GHRP-6 and si-GHSR-1α reversed rh-Ghrelin’s neuroprotective effects as seen from the increase in infarction area (Fig. 6), MitoSOX-positive cells, FJC-positive neurons and the TUNEL-positive cells (Fig. 7, 8). Furthermore, we also observed that [D-Lys3]-GHRP-6 and si-GHSR-1α made a decrease in the Bcl2/Bax ratio (Fig. 10, 11) and an increase in cleaved caspase-3 expression when compared with their respective control group in the rh-Ghrelin treated HI rats (P < 0.05).
Fig. 6.
Effect of GHSR-1α inhibitor, si-GHSR-1α and Sirt1 inhibitor on brain infarction area in the rh-Ghrelin treated HI rats respectively. (A) Representative pictures of TTC stained brain slices. (B) Quantitative analysis of infarction area showed in TTC stained brain slices. Vehicle 1 is the vehicle of rh-Ghrelin, Vehicle 2 is the vehicle of [D-Lys3]-GHRP-6, Vehicle 3 is the vehicle of Ex527. #P < 0.05 vs. Vehicle 1; @P < 0.05 vs. Vehicle 2; &P < 0.05 vs. Scramble siRNA; $P < 0.05 vs. Vehicle 3. Data are represented as mean ± SD, n= 6 for each group.
Fig. 7.
Representative microphotographs and quantification data showing effects of [D-Lys3]-GHRP-6 intervention on oxidative stress, neuronal degeneration and apoptosis in rh-Ghrelin treated animals at 48h post HI. The MitoSOX-positive cells (A), FJC-positive neurons (B) and TUNEL-positive cells (C) were significantly increased in [D-Lys3]-GHRP-6 intervention group when compared with [D-Lys3]-GHRP-6 vehicle control group. Top panel indicates the location of staining (small black box). @P < 0.05 vs. HI+ rh-Ghrelin+ Vehicle. Data are represented as mean ± SD, n= 3 for each group. Scale bar = 100μm.
Fig. 8.
Representative microphotographs and quantification data showing effects of locking down GHSR-1α on oxidative stress, neuronal degeneration and apoptosis in rh-Ghrelin treated animals at 48h post HI. The MitoSOX-positive cells (A), FJC-positive neurons (B) and TUNEL-positive cells (C) were significantly increased in si-GHSR-1α group when compared with scramble siRNA control group. Top panel indicates the location of staining (small black box). &P < 0.05 vs. HI+ rh-Ghrelin+ Vehicle. Data are represented as mean ± SD, n= 3 for each group. Scale bar = 100μm.
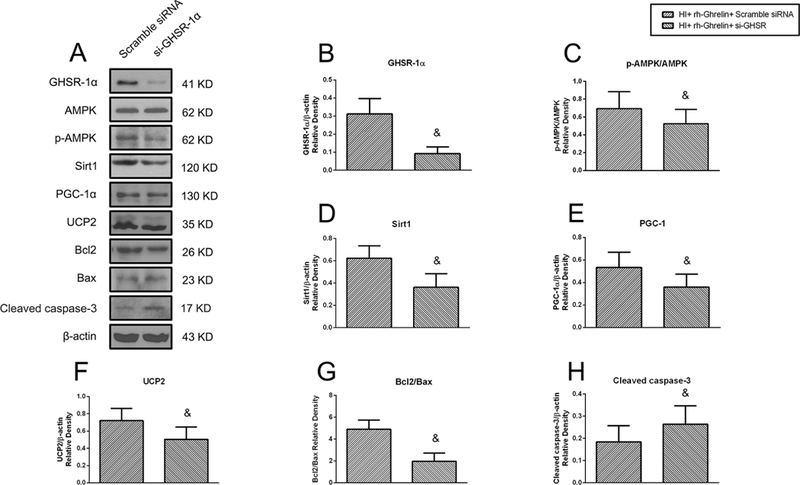
Fig. 11.
Representative picture of western blot bands (A) and quantification of data(B-H) showing the effects of locking down GHSR-1α on proteins in proposed signaling pathway and proteins related to apoptosis in rh-Ghrelin treatment animals at 48h post HI. &P < 0.05 vs. HI+ rh-Ghrelin+ scramble siRNA. Data are represented as mean ± SD, n= 6 for each group.
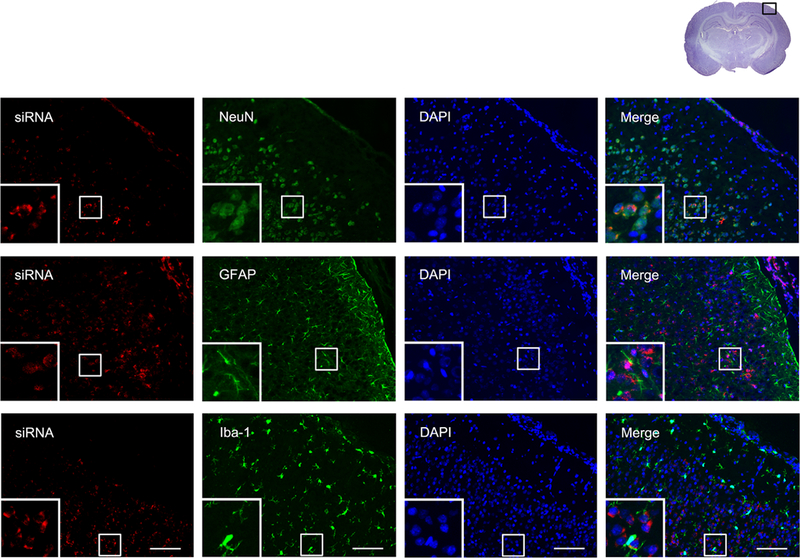
To detect the effectiveness of siRNA’s access to the brain, we injected scramble siRNA with red fluorescence tag intracerebroventricularly to the naïve rat brain. Results showed that the red fluorescence colocalized with the neuronal marker (NeuN), astrocyte marker (GFAP), and microglial marker (Iba-1), respectively (Fig. 9).
Fig. 9.

Representative microphotographs of siRNA staining in neurons, astrocytes and microglia in naïve rats. The upper panel showed the colocalization of siRNA red fluorescence tag with neurons marker NeuN; the middle panel showed the colocalization of siRNA red fluorescence tag with astrocytes marker GFAP; the lower panel showed the colocalization of siRNA red fluorescence tag with neurons marker Iba-1. Top panel indicates the location of staining (small black box). Scale bar = 100μm.
3.8. Inhibition of GHSR-1α reversed the upregulation of p-AMPK, Sirt1, PGC-1α and UCP2 induced by rh-Ghrelin treatment at 48 h post HI
To investigate the role of GHSR-1α and its downstream signaling molecules in rh-Ghrelin’s protective effects we administered a selective inhibitor or siRNA and measured their expression levels. Both [D-Lys3]-GHRP-6 and si-GHSR-1α significantly decreased expression of p-AMPK, Sirt1, PGC-1α, UCP2 and the ratio of Bcl2/Bax and increased expression of cleaved caspase-3 when compared to the control groups (P < 0.05, Fig. 10, 11).
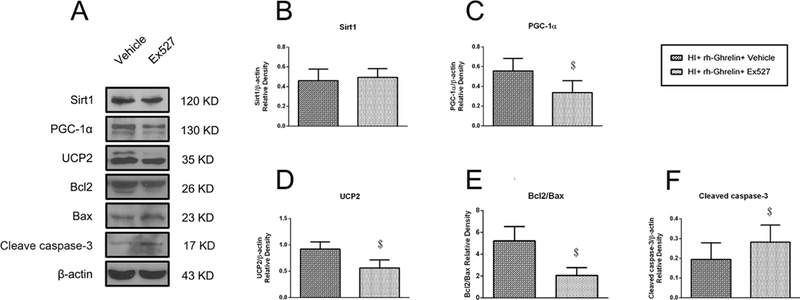
3.9. Inhibition of Sirt1 reversed rh-Ghrelin’s protective effects at 48 h post HI
To determine the role of Sirt1 in rh-Ghrelin’s signaling mechanism, we administered Ex527 to inhibit Sirt1. Inhibition of Sirt1 significantly increased infarction area (Fig. 6), MitoSOX-positive cells, FJC-positive neurons, TUNEL-positive cells (Fig. 12) and the expression of cleaved caspase-3 (Fig. 13), while the ratio of Bcl2/Bax decreased (Fig. 13) in the rh-Ghrelin treated HI rats when compared with the vehicle group (P < 0.05).
Fig. 12.
Representative microphotographs and quantification data showing effects of Ex527 intervention on oxidative stress, neuronal degeneration and apoptosis in rh-Ghrelin treated animals at 48h post HI. The MitoSOX-positive cells (A), FJC-positive neurons (B) and TUNEL-positive cells (C) were significantly increased in Ex527 intervention group when compared with Ex527 vehicle control group. Top panel indicates the location of staining (small black box). $P < 0.05 vs. HI+ rh-Ghrelin+ vehicle. Data are represented as mean ± SD, n= 3 for each group. Scale bar = 100μm.
Fig. 13.
Representative picture of western blot bands (A) and quantification of data(B-F) showing the effects of Ex527 intervention on proteins in proposed signaling pathway and proteins related to apoptosis in rh-Ghrelin treatment animals at 48h post HI. Vehicle is the vehicle of Ex527. $P < 0.05 vs. HI+ rh-Ghrelin+ Vehicle. Data are represented as mean ± SD, n= 6 for each group.
3.10. Inhibition of Sirt1 reversed the upregulation of PGC-1α and UCP2 induced by rh-Ghrelin treatment at 48 h post HI
To investigate whether Sirt1 is involved in the mechanism between ghrelin treatment with the downstream proteins PGC-1α and UCP2, Ex527 was administered to inhibit Sirt1 in the rh-Ghrelin treated rats. Inhibition of Sirt1 resulted in a significant decrease in PGC-1α and UCP2 expression levels (P < 0.05, Fig. 13).
4. Discussion
In this study, we demonstrated that intranasal administration of exogenous rh-Ghrelin was able to significantly prevent HI-induced brain injury by reducing oxidative stress and neuronal apoptosis which was at least in part mediated via the GHSR1α/AMPK/Sirt1/PGC-1α/UCP2 signaling pathway. Summary of our findings showed that: Firstly, the level of endogenous ghrelin and GHSR-1α decreased in a time-dependent manner post HI and that they were mainly expressed on neurons. Secondly, administration of 0.04ug/kg rh-Ghrelin remarkably reduced oxidative stress and neuronal apoptosis after HI, which was accompanied by a reduction in infarcted area and improvement in short-term and long-term neurobehavioral outcomes. Western blot assays showed that rh-Ghrelin upregulated the expression of p-AMPK, Sirt1, PGC-1α, and UCP2. Finally, Inhibition of GHSR-1α using GHSR-1α siRNA or inhibitor [D-Lys3]-GHRP-6 abolished the anti-oxidative and anti-apoptotic effects of rh-Ghrelin, and reversed the upregulation of p-AMPK, Sirt1, PGC-1α, and UCP2. Inhibition of Sirt1 by the specific inhibitor, Ex527, in the presence of rh-Ghrelin also offset the rh-Ghrelin beneficial effects and reversed the upregulation of PGC-1α and UCP2 induced by rh-Ghrelin treatment.
Ghrelin influences many physiological and pathological processes including food intake, adiposity, memory, neuronal apoptosis, neuroinflammation and so on (8, 38, 39). Given the multifunction of ghrelin by binding and activating its endogenous receptor GHSR-1α in the central nervous system of humans and animals, GHSR-1α is extensively distributed within the brain organs (thalamus, hypothalamus, cortex, hippocampus, etc.) as well as in peripheral organs (heart, stomach, adrenals, etc.) (20, 40, 41). Our present data showed that GHSR-1α immunoreactivity was colocalized with neurons in the per-infarcted area, which was consistent with previous study (20). GHSR-1α upregulation was reported previously in a myocardial injury induced by isoproterenol rat models (42); however, the other two studies showed slightly different findings with downregulation in GHSR-1 α following ischemia/reperfusion injury in spinal cord and brain respectively (20, 43). In our study, we found that GHSR-1α expression in the brain was decreased and reached to the lowest level at 48 h after HIE. According to previous studies, the brain injury was much worse at 48 h in experimental HIE models, of which the damaging factors may include oxidative stress, mitochondria dysfunction, neuronal apoptosis, and inflammation (44). The lowest expression of GHSR-1α at 48h after HIE corresponds to the severity of brain injury in HIE. It is likely that the declining appetite caused by the brain injury after HIE played a feedback effect on the induction of GHSR-1α expression. Taken together, current results indicate that the GHSR-1α was suppressed by deleterious stress at 48 h after HIE.
Ghrelin is principally released into bloodstream by endocrine gastric cells of the stomach and demonstrates a very limited ability to cross the blood brain barrier (BBB) from the circulation into the brain (40, 45, 46). Although some insist that ghrelin is likewise produced in the brain, the validity of its central synthesis is a topic of contention (40). Since its predominant peripheral production and poor ability to cross the BBB, one of the issues needed to be considered in this study is the effectiveness of the exogenous ghrelin administration delivery route to CNS. Intranasal delivery is an innovative approach since it is non-invasive and clinically relevant (47, 48). Drugs can be rapidly absorbed through the large surface area of the nasal mucosa, resulting in a rapid onset of action and avoiding degradation in the gastrointestinal tract and first-pass metabolism in the liver. Furthermore, the nose also serves as a direct route to the brain. It has been shown that drugs administered with the intranasal method can be transported from the nose to brain along the olfactory and trigeminal nerve pathways (49). The intranasal method of delivery circumvents the BBB and minimizes systemic exposure, since it is transported directly to the brain. Results indicate that the brain uptake of intranasal delivered therapeutics is 5 times greater than that after intraperitoneal delivery (50). Based on the above advantages of intranasal administration, we have selected it as the optimal delivery rout of ghrelin treatment for the neonatal HIE rat model in our study.
The results (Fig. 10) showed that intranasal administration of rh-Ghrelin increased ghrelin expression in the brain after intranasal administration, which indicates the effectiveness of ghrelin delivery to the brain in the HIE rat model. To determine the best dose of intranasal ghrelin delivery, three doses of ghrelin (0.02 μg/kg, 0.04 μg/kg, 0.12 μg/kg) treatment effects were investigated by measuring percent infarcted area and short-term neurobehavioral tests. TTC staining results showed that 0.04 μg/kg and 0.12 μg/kg ghrelin treatment significantly reduced the percent infarcted area when compared to the vehicle group. Geotaxis reflex results showed that all three doses of ghrelin treatment significantly decreased the reflex time compared with vehicle group animals, with 0.04 μg/kg showing to be the most effective. At the same time, the three doses of ghrelin treatment can significantly reduce body weight loss compared with the vehicle animals. Based on the above results we select the medium dose of 0.04 μg/kg ghrelin as the most effective treatment for the remainder of our studies.
Growing evidence suggests that neuronal apoptosis is one of the major pathological processes involved in neurological impairment after HIE (51). Oxidative stress induced by hypoxia/ischemic could lead to neuronal apoptosis via various signaling pathway, such as PI3K/Akt/Caspase, MAPK/p38/cPLA2 (52, 53). Over production of reactive oxygen species (ROS) in mitochondria leads to an overwhelmed antioxidant system and oxidative stress (54). Previous studies explored many important mechanistic details involved in mitochondrial ROS metabolism and factors that regulate ROS generation and removal (55). The components related with mitochondrial ROS production consists of cytochrome b5 reductase, monoamine oxidases, dihydroorotate dehydrogenase, dehydrogenase of α-glycerophosphate, succinate dehydrogenase and NADH-ubiquinone oxidoreductase, etc. (55) The antioxidant systems for ROS clearance system include enzymes and non-enzymatic antioxidants, such as phospholipid hydroperoxide glutathione peroxidase, MnSOD, cytochrome c, catalase, and glutathione, etc. (55) Recently, uncoupling the mitochondrial respiratory chain mediated by UCP2 has been proposed as an important mechanism to reduce mitochondrial ROS levels and attenuate cell death (56, 57). It was suggested that compounds inducing UCP2 expression or activity would be neuroprotective and prevent neuronal apoptosis via inhibiting oxidative stress after stroke, epilepsy or neurodegenerative disease (58–60). UCP2 was demonstrated to be involved in ghrelin treatment effects in various experiments in vivo and in vitro. For example, Ghrelin suppressed Ang II-induced renal damages through its UCP2 dependent anti-oxidative stress effect and mitochondria maintenance (61). The inhibitory effects of ghrelin on the inflammatory response relied on its ability to induce the accumulation of cellular UCP2 levels in human umbilical vein endothelial cells (HUVECs) (62). In this study, exogenous rh-Ghrelin treatment could promote the upregulation of UCP2 expression and reduce the damage caused by oxidative stress and apoptosis, which is consistent with previous studies. In addition, inhibition of the unique receptor of ghrelin, GHSR-1, with either GHRP-6 inhibitor or siRNA silencer, significantly reduced UCP2 levels and the beneficial effects of ghrelin on the marker of oxidative stress and apoptosis deteriorated correspondingly. These mentioned results indicated that ghrelin can enhance mitochondrial uncoupling by upregulating UCP2 expression and reduce oxidative stress injury and neuronal apoptosis in a GHSR-1 α dependent manner after HI injury. However, the exact mechanism between ghrelin/GHSR-1 α and UCP2 has not yet been elucidated.
Given the high metabolic energy demands of the brain and the relative intolerance to energy depletion induced by ischemia and hypoxic insults, mounting evidence suggests that AMPK, which serves as an energy sensor and master regulator of metabolism, plays a pivotal role in modulating cell survival in vivo and in vitro (63). Until now, the role of AMPK activation on neuronal apoptosis are controversial. Some studies results showed that AMPK activation is associated with the de-phosphorylation of mTOR, Akt, FOXO3a and upregulation of Bim and the induction of neuronal apoptosis (64, 65); while in other studies, pharmacological activation or genetic upregulation of AMPK has been shown to be neuroprotective against glucose deprivation, ischemia, and hypoxia insults (25, 26, 66). AMPK activation may have dual roles in the regulation of neuronal apoptosis. The transient versus persistent activation kinetics may critically determine the functions of AMPK and cell fate (65). A transient activation of AMPK protected the neurons from glucose deprivation, metabolic, excitotoxic and oxidative insults (25),whereas prolonged activation would trigger Bim-dependent apoptosis in neurons (64, 65). Studies have shown that ghrelin could transiently promote AMPK phosphorylation and inhibit neuronal apoptosis in various cell lines and tissues (21, 67). Consistent with these findings, our study showed that intranasal administration of rh-Ghrelin increased AMPK phosphorylation. Collectively, these observations suggest that the ghrelin/AMPK regulatory signaling axis functions to modulate neuronal survival under ischemic conditions, protecting against cerebral injury after HI.
One of the mechanisms employed by AMPK to regulate metabolism is protein acetylation (68). AMPK activation induces expression of nicotinamide phosphoribosyl transferase (NAMPT), the rate-limiting enzyme in the NAD+ salvage pathway that converts nicotinamide to nicotinamide mononucleotide to enable NAD+ biosynthesis. AMPK activation thus increases NAD+ level, elevating NAD+-dependent type III deacetylase, Sirt1 activity (27, 68, 69). Sirt1-mediated protein deacetylation subsequently activates downstream targets, including peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α) and forkhead box protein O1 (FOXO1) (27, 68). In addition to being an important functional regulator of PGC-1α, Sirt1 could also regulate PGC-1α target genes under conditions of nutrient restriction in myotubes (70). Other researchers found that the change of Sirt1 expression is related to the change of PGC-1α expression as well, in myocardial (71), human pancreatic cancer cells (72), male germ cells (73), and kidneys (74), etc. Our results demonstrated that rh-Ghrelin treatment resulted in upregulation of Sirt1 and PGC-1α expression, which suggested that Sirt1/PGC-1α signaling axis is involved in the beneficial effects of ghrelin treatment after neonatal HI injury.
To date, limited information is available on the regulation of UCP-2 expression. PGC-1α and cAMP response element binding protein (CREB) were proposed as the transcriptional regulators in previous studies (58, 75). In this study, we only focused on PGC-1α since AMPK could be activated by ghrelin in various cells and Sirt1/PGC-1α is one of the downstream pathways of AMPK. In the present study, ghrelin treatment could upregulate Sirt1, PGC-1α and UCP2 expression. In addition, the Sirt1 inhibitor, EX527, offset the upregulation of UCP2 induced by ghrelin treatment. These results indicated that Sirt1/PGC-1α was involved in the mechanism between ghrelin and UCP2 expression. To this end, the current results shed new light on the role of ghrelin in reducing oxidative stress and neuronal apoptosis through the GHSR-1α/AMPK/Sirt1/PGC-1α/UCP2 pathway.
Anti-inflammatory properties of ghrelin are well documented and in vivo data suggests that ghrelin has activity in a number of disease models that are partly driven by the inflammatory response (76–78). The excess of ROS production induced by hypoxia-ischemia insults will modify or degenerate cellular macromolecules, such as membranes, proteins, lipids, and DNA, and lead to a cascading inflammatory response(14). Therefore, neuroinflammation is also a substantial component of HI brain injury as neuronal apoptosis. We speculate it is possible that some part of the observed protective effect of ghrelin may be attributed to the regulation of cytokine production and attenuation of neuroinflammation, which we will focus on in future studies.
In conclusion, intranasal administration of rh-Ghrelin reduced the percent infarcted area, improved short term neurobehavioral deficits, as well as long-term neurological function after HI. The neuroprotective effects of ghrelin could be mediated via the GHSR-1α/AMPK/Sirt1/PGC-1α/UCP2 signaling pathway. Due to limited current therapies and failed trails, this study provides a basis for ghrelin as a promising therapeutic candidate for patients with HIE.
Highlights.
The level of endogenous ghrelin and GHSR-1α decreased in a time-dependent manner post HI.
Rh-Ghrelin administration remarkably reduced oxidative stress and neuronal apoptosis after HI.
Rh-Ghrelin taking its anti-oxidative stress and anti-apoptotic effects in neurons after HI at least in part is mediated by GHSR1α/AMPK/Sirt1/PGC-1α/UCP2 signaling pathway.
Acknowledgments
This article is supported by a grant from the National Institutes of Health (NS104083) to Dr. Zhang and a grant from National Natural Science Foundation of China (NO. 81601051) to Dr. Juan Huang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
The authors declare that they have no conflict of interest.
References
- 1.Douglas-Escobar M, Weiss MD. Hypoxic-ischemic encephalopathy: a review for the clinician. JAMA pediatrics 2015;169(4):397–403. [DOI] [PubMed] [Google Scholar]
- 2.Battin MR, Dezoete JA, Gunn TR, Gluckman PD, Gunn AJ. Neurodevelopmental outcome of infants treated with head cooling and mild hypothermia after perinatal asphyxia. Pediatrics 2001;107(3):480–4. [DOI] [PubMed] [Google Scholar]
- 3.Mwaniki MK, Atieno M, Lawn JE, Newton CR. Long-term neurodevelopmental outcomes after intrauterine and neonatal insults: a systematic review. Lancet (London, England) 2012;379(9814):445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shetty J Neonatal seizures in hypoxic-ischaemic encephalopathy--risks and benefits of anticonvulsant therapy. Developmental medicine and child neurology 2015;57 Suppl 3:40–3. [DOI] [PubMed] [Google Scholar]
- 5.Lugli L, Balestri E, Berardi A, Guidotti I, Cavalleri F, Todeschini A, et al. Brain cooling reduces the risk of post-neonatal epilepsy in newborns affected by moderate to severe hypoxic-ischemic encephalopathy. Minerva pediatrica 2018. [DOI] [PubMed]
- 6.Silveira RC, Procianoy RS. Hypothermia therapy for newborns with hypoxic ischemic encephalopathy. Jornal de pediatria 2015;91(6 Suppl 1):S78–83. [DOI] [PubMed] [Google Scholar]
- 7.Prempunpong C, Efanov I, Sant’anna G. The effect of the implementation of therapeutic hypothermia on fluid balance and incidence of hyponatremia in neonates with moderate or severe hypoxic-ischaemic encephalopathy. Acta paediatrica (Oslo, Norway : 1992) 2013;102(11):e507–13. [DOI] [PubMed] [Google Scholar]
- 8.Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 2000;141(11):4255–61. [DOI] [PubMed] [Google Scholar]
- 9.Kojima M, Kangawa K. Ghrelin: structure and function. Physiological reviews 2005;85(2):495–522. [DOI] [PubMed] [Google Scholar]
- 10.Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003;37(4):649–61. [DOI] [PubMed] [Google Scholar]
- 11.Date Y, Murakami N, Kojima M, Kuroiwa T, Matsukura S, Kangawa K, et al. Central effects of a novel acylated peptide, ghrelin, on growth hormone release in rats. Biochemical and biophysical research communications 2000;275(2):477–80. [DOI] [PubMed] [Google Scholar]
- 12.Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, et al. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain research Molecular brain research 1997;48(1):23–9. [DOI] [PubMed] [Google Scholar]
- 13.Hou Z, Miao Y, Gao L, Pan H, Zhu S. Ghrelin-containing neuron in cerebral cortex and hypothalamus linked with the DVC of brainstem in rat. Regulatory peptides 2006;134(2–3):126–31. [DOI] [PubMed] [Google Scholar]
- 14.Zhao M, Zhu P, Fujino M, Zhuang J, Guo H, Sheikh I, et al. Oxidative Stress in Hypoxic-Ischemic Encephalopathy: Molecular Mechanisms and Therapeutic Strategies. International journal of molecular sciences 2016;17(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eid RA, Zaki MSA, Al-Shraim M, Eleawa SM, El-Kott AF, Al-Hashem FH, et al. Subacute ghrelin administration inhibits apoptosis and improves ultrastructural abnormalities in remote myocardium post-myocardial infarction. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2018;101:920–8. [DOI] [PubMed] [Google Scholar]
- 16.Ezquerro S, Mocha F, Fruhbeck G, Guzman-Ruiz R, Valenti V, Mugueta C, et al. Ghrelin Reduces TNF-alpha-Induced Human Hepatocyte Apoptosis, Autophagy, and Pyroptosis: Role in Obesity-Associated NAFLD. The Journal of clinical endocrinology and metabolism 2019;104(1):21–37. [DOI] [PubMed] [Google Scholar]
- 17.Zeng M, Huang C, Zheng H, Chen Q, He W, Deng Y. Effects of Ghrelin on iNOS-Derived NO Promoted LPS-Induced Pulmonary Alveolar Epithelial A549 Cells Apoptosis. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 2018;49(5):1840–55. [DOI] [PubMed] [Google Scholar]
- 18.Liao P, Yang D, Liu D, Zheng Y. GLP-1 and Ghrelin Attenuate High Glucose/High Lipid-Induced Apoptosis and Senescence of Human Microvascular Endothelial Cells. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 2017;44(5):1842–55. [DOI] [PubMed] [Google Scholar]
- 19.Chung H, Kim E, Lee DH, Seo S, Ju S, Lee D, et al. Ghrelin inhibits apoptosis in hypothalamic neuronal cells during oxygen-glucose deprivation. Endocrinology 2007;148(1):148–59. [DOI] [PubMed] [Google Scholar]
- 20.Miao Y, Xia Q, Hou Z, Zheng Y, Pan H, Zhu S. Ghrelin protects cortical neuron against focal ischemia/reperfusion in rats. Biochemical and biophysical research communications 2007;359(3):795–800. [DOI] [PubMed] [Google Scholar]
- 21.Zhu K, Zhang ML, Liu ST, Li XY, Zhong SM, Li F, et al. Ghrelin Attenuates Retinal Neuronal Autophagy and Apoptosis in an Experimental Rat Glaucoma Model. Investigative ophthalmology & visual science 2017;58(14):6113–22. [DOI] [PubMed] [Google Scholar]
- 22.Yuan MJ, Wang T, Kong B, Wang X, Huang CX, Wang D. GHSR-1a is a novel pro-angiogenic and anti-remodeling target in rats after myocardial infarction. European journal of pharmacology 2016;788:218–25. [DOI] [PubMed] [Google Scholar]
- 23.Rivas PMS, Vechiato FMV, Borges BC, Rorato R, Antunes-Rodrigues J, Elias LLK. Increase in hypothalamic AMPK phosphorylation induced by prolonged exposure to LPS involves ghrelin and CB1R signaling. Hormones and behavior 2017;93:166–74. [DOI] [PubMed] [Google Scholar]
- 24.Corton JM, Gillespie JG, Hardie DG. Role of the AMP-activated protein kinase in the cellular stress response. Current biology : CB 1994;4(4):315–24. [DOI] [PubMed] [Google Scholar]
- 25.Weisova P, Davila D, Tuffy LP, Ward MW, Concannon CG, Prehn JH. Role of 5’-adenosine monophosphate-activated protein kinase in cell survival and death responses in neurons. Antioxidants & redox signaling 2011;14(10):1863–76. [DOI] [PubMed] [Google Scholar]
- 26.Xu N, Zhang Y, Doycheva DM, Ding Y, Zhang Y, Tang J, et al. Adiponectin attenuates neuronal apoptosis induced by hypoxia-ischemia via the activation of AdipoR1/APPL1/LKB1/AMPK pathway in neonatal rats. Neuropharmacology 2018;133:415–28. [DOI] [PubMed] [Google Scholar]
- 27.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009;458(7241):1056–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006;127(2):397–408. [DOI] [PubMed] [Google Scholar]
- 29.Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovascular research 2005;66(3):562–73. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz-Ramirez A, Lopez-Acosta O, Barrios-Maya MA, El-Hafidi M. Cell Death and Heart Failure in Obesity: Role of Uncoupling Proteins. Oxidative medicine and cellular longevity 2016;2016:9340654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubattu S, Stanzione R, Volpe M. Mitochondrial Dysfunction Contributes to Hypertensive Target Organ Damage: Lessons from an Animal Model of Human Disease. Oxidative medicine and cellular longevity 2016;2016:1067801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehta SL, Li PA. Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2009;29(6):1069–78. [DOI] [PubMed] [Google Scholar]
- 33.Rice JE 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Annals of neurology 1981;9(2):131–41. [DOI] [PubMed] [Google Scholar]
- 34.Liu NW, Ke CC, Zhao Y, Chen YA, Chan KC, Tan DT, et al. Evolutional Characterization of Photochemically Induced Stroke in Rats: a Multimodality Imaging and Molecular Biological Study. Translational stroke research 2017;8(3):244–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang B, Li L, Chen Q, Tao Y, Yang L, Zhang B, et al. Role of Glibenclamide in Brain Injury After Intracerebral Hemorrhage. Translational stroke research 2017;8(2):183–93. [DOI] [PubMed] [Google Scholar]
- 36.Guo D, Wilkinson DA, Thompson BG, Pandey AS, Keep RF, Xi G, et al. MRI Characterization in the Acute Phase of Experimental Subarachnoid Hemorrhage. Translational stroke research 2017;8(3):234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Zhou F, Dou Y, Tian X, Liu C, Li H, et al. Melatonin Alleviates Intracerebral Hemorrhage-Induced Secondary Brain Injury in Rats via Suppressing Apoptosis, Inflammation, Oxidative Stress, DNA Damage, and Mitochondria Injury. Translational stroke research 2018;9(1):74–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiao Q, Du X, Li Y, Gong B, Shi L, Tang T, et al. The neurological effects of ghrelin in brain diseases: Beyond metabolic functions. Neuroscience and biobehavioral reviews 2017;73:98–111. [DOI] [PubMed] [Google Scholar]
- 39.Beynon AL, Brown MR, Wright R, Rees MI, Sheldon IM, Davies JS. Ghrelin inhibits LPS-induced release of IL-6 from mouse dopaminergic neurones. Journal of neuroinflammation 2013;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edwards A, Abizaid A. Clarifying the Ghrelin System’s Ability to Regulate Feeding Behaviours Despite Enigmatic Spatial Separation of the GHSR and Its Endogenous Ligand. International journal of molecular sciences 2017;18(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Chen Q, Li G, Ke D. Ghrelin ameliorates impaired angiogenesis of ischemic myocardium through GHSR1a-mediated AMPK/eNOS signal pathway in diabetic rats. Peptides 2015;73:77–87. [DOI] [PubMed] [Google Scholar]
- 42.Li L, Zhang LK, Pang YZ, Pan CS, Qi YF, Chen L, et al. Cardioprotective effects of ghrelin and des-octanoyl ghrelin on myocardial injury induced by isoproterenol in rats. Acta pharmacologica Sinica 2006;27(5):527–35. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Q, Huang C, Meng B, Tang T, Shi Q, Yang H. Acute effect of Ghrelin on ischemia/reperfusion injury in the rat spinal cord. International journal of molecular sciences 2012;13(8):9864–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dixon BJ, Reis C, Ho WM, Tang J, Zhang JH. Neuroprotective Strategies after Neonatal Hypoxic Ischemic Encephalopathy. International journal of molecular sciences 2015;16(9):22368–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banks WA, Tschop M, Robinson SM, Heiman ML. Extent and direction of ghrelin transport across the blood-brain barrier is determined by its unique primary structure. The Journal of pharmacology and experimental therapeutics 2002;302(2):822–7. [DOI] [PubMed] [Google Scholar]
- 46.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999;402(6762):656–60. [DOI] [PubMed] [Google Scholar]
- 47.Wolfe TR, Braude DA. Intranasal medication delivery for children: a brief review and update. Pediatrics 2010;126(3):532–7. [DOI] [PubMed] [Google Scholar]
- 48.Doyle KP, Yang T, Lessov NS, Ciesielski TM, Stevens SL, Simon RP, et al. Nasal administration of osteopontin peptide mimetics confers neuroprotection in stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2008;28(6):1235–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pardeshi CV, Belgamwar VS. Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood-brain barrier: an excellent platform for brain targeting. Expert opinion on drug delivery 2013;10(7):957–72. [DOI] [PubMed] [Google Scholar]
- 50.Chauhan MB, Chauhan NB. Brain Uptake of Neurotherapeutics after Intranasal versus Intraperitoneal Delivery in Mice. Journal of neurology and neurosurgery 2015;2(1). [PMC free article] [PubMed] [Google Scholar]
- 51.Banasiak KJ, Xia Y, Haddad GG. Mechanisms underlying hypoxia-induced neuronal apoptosis. Progress in neurobiology 2000;62(3):215–49. [DOI] [PubMed] [Google Scholar]
- 52.Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, et al. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxidants & redox signaling 2011;14(8):1505–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarmah D, Kaur H, Saraf J, Vats K, Pravalika K, Wanve M, et al. Mitochondrial Dysfunction in Stroke: Implications of Stem Cell Therapy 2018. [DOI] [PubMed]
- 54.Munro D, Treberg JR. A radical shift in perspective: mitochondria as regulators of reactive oxygen species 2017;220(Pt 7):1170–80. [DOI] [PubMed] [Google Scholar]
- 55.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry Biokhimiia 2005;70(2):200–14. [DOI] [PubMed] [Google Scholar]
- 56.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005;120(4):483–95. [DOI] [PubMed] [Google Scholar]
- 57.Li N, Karaca M, Maechler P. Upregulation of UCP2 in beta-cells confers partial protection against both oxidative stress and glucotoxicity. Redox biology 2017;13:541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mattiasson G, Sullivan PG. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxidants & redox signaling 2006;8(1–2):1–38. [DOI] [PubMed] [Google Scholar]
- 59.Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 2008;454(7206):846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bechmann I, Diano S, Warden CH, Bartfai T, Nitsch R, Horvath TL. Brain mitochondrial uncoupling protein 2 (UCP2): a protective stress signal in neuronal injury. Biochemical pharmacology 2002;64(3):363–7. [DOI] [PubMed] [Google Scholar]
- 61.Fujimura K, Wakino S, Minakuchi H, Hasegawa K, Hosoya K, Komatsu M, et al. Ghrelin protects against renal damages induced by angiotensin-II via an antioxidative stress mechanism in mice. PloS one 2014;9(4):e94373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang R Ghrelin suppresses inflammation in HUVECs by inhibiting ubiquitin-mediated uncoupling protein 2 degradation. International journal of molecular medicine 2017;39(6):1421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. The Journal of physiology 2006;574(Pt 1):85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li D, Luo L, Xu M, Wu J, Chen L, Li J, et al. AMPK activates FOXO3a and promotes neuronal apoptosis in the developing rat brain during the early phase after hypoxia-ischemia. Brain research bulletin 2017;132:1–9. [DOI] [PubMed] [Google Scholar]
- 65.An JY, Zhou LL, Sun P, Pang HG, Li DD, Li Y, et al. Role of the AMPK signaling pathway in early brain injury after subarachnoid hemorrhage in rats. Acta neurochirurgica 2015;157(5):781–92. [DOI] [PubMed] [Google Scholar]
- 66.Yu J, Li X, Matei N, McBride D, Tang J, Yan M, et al. Ezetimibe, a NPC1L1 inhibitor, attenuates neuronal apoptosis through AMPK dependent autophagy activation after MCAO in rats. Experimental neurology 2018;307:12–23. [DOI] [PubMed] [Google Scholar]
- 67.Bayliss JA, Lemus MB, Stark R, Santos VV, Thompson A, Rees DJ, et al. Ghrelin-AMPK Signaling Mediates the Neuroprotective Effects of Calorie Restriction in Parkinson’s Disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2016;36(10):3049–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vancura A, Nagar S, Kaur P, Bu P, Bhagwat M, Vancurova I. Reciprocal Regulation of AMPK/SNF1 and Protein Acetylation. International journal of molecular sciences 2018;19(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Developmental cell 2008;14(5):661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. The EMBO journal 2007;26(7):1913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tian L, Cao W, Yue R, Yuan Y, Guo X, Qin D, et al. Pretreatment with Tilianin improves mitochondrial energy metabolism and oxidative stress in rats with myocardial ischemia/reperfusion injury via AMPK/SIRT1/PGC-1 alpha signaling pathway. Journal of pharmacological sciences 2019;139(4):352–60. [DOI] [PubMed] [Google Scholar]
- 72.Huang B, Cheng X, Wang D, Peng M, Xue Z, Da Y, et al. Adiponectin promotes pancreatic cancer progression by inhibiting apoptosis via the activation of AMPK/Sirt1/PGC-1alpha signaling. Oncotarget 2014;5(13):4732–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang X, Chen J, Zhang C, Zhang Z, Tan Y, Feng W, et al. The protective effect of FGF21 on diabetes-induced male germ cell apoptosis is associated with up-regulated testicular AKT and AMPK/Sirt1/PGC-1alpha signaling. Endocrinology 2015;156(3):1156–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hou S, Zhang T, Li Y, Guo F, Jin X. Glycyrrhizic Acid Prevents Diabetic Nephropathy by Activating AMPK/SIRT1/PGC-1alpha Signaling in db/db Mice. Journal of diabetes research 2017;2017:2865912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Turnham RE, Scott JD. Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene 2016;577(2):101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao Y, Shen Z, Zhang D, Luo H, Chen J, Sun Y, et al. Ghrelin ameliorates nerve growth factor Dysmetabolism and inflammation in STZ-induced diabetic rats. Metabolic brain disease 2017;32(3):903–12. [DOI] [PubMed] [Google Scholar]
- 77.Kawaguchi M, Kanemaru A, Fukushima T, Yamamoto K, Tanaka H, Haruyama Y, et al. Ghrelin administration suppresses inflammation-associated colorectal carcinogenesis in mice. Cancer science 2015;106(9):1130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Santos VV, Stark R, Rial D, Silva HB, Bayliss JA, Lemus MB, et al. Acyl ghrelin improves cognition, synaptic plasticity deficits and neuroinflammation following amyloid beta (Abeta1–40) administration in mice 2017;29(5). [DOI] [PubMed] [Google Scholar]