

Summary
Triple-negative breast cancers (TNBCs) display great diversity in cisplatin sensitivity that cannot be explained solely by cancer-associated DNA repair defects. Differential activation of the DNA damage response (DDR) to cisplatin has been proposed to underlie the observed differential sensitivity, but it has not been investigated systematically. Systems-level analysis—using quantitative time-resolved signaling data and phenotypic responses, in combination with mathematical modeling—identifies that the activation status of cell-cycle checkpoints determines cisplatin sensitivity in TNBC cell lines. Specifically, inactivation of the cell-cycle checkpoint regulator MK2 or G3BP2 sensitizes cisplatin-resistant TNBC cell lines to cisplatin. Dynamic signaling data of five cell cycle-related signals predicts cisplatin sensitivity of TNBC cell lines. We provide a time-resolved map of cisplatin-induced signaling that uncovers determinants of chemo-sensitivity, underscores the impact of cell-cycle checkpoints on cisplatin sensitivity, and offers starting points to optimize treatment efficacy.
Keywords: systems biology, checkpoint, cell cycle, DNA damage, modeling, DDR, cisplatin, G3BP2, MK2, mitosis
Graphical Abstract
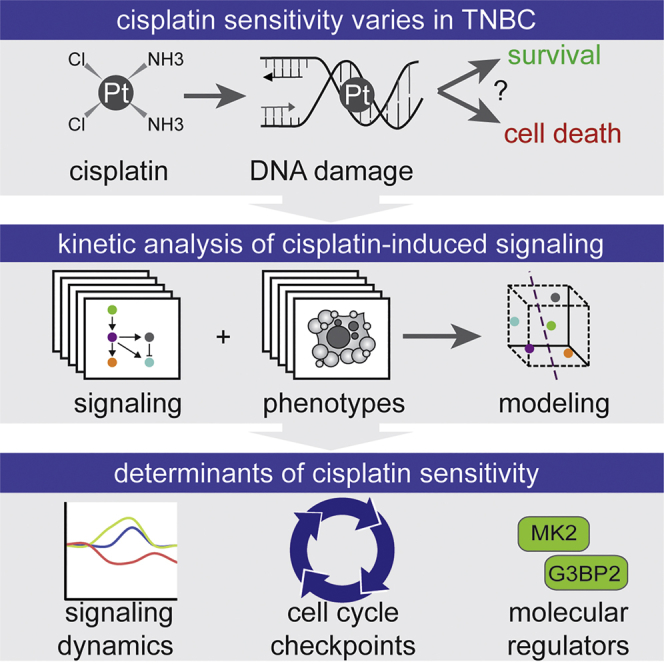
Highlights
-
•
Variation in cisplatin sensitivity in TNBC unrelated to DNA repair deficiencies
-
•
Systems-level analysis of signal transduction in sensitive versus resistant models
-
•
Cell-cycle checkpoint G3BP2 and MK2 activation determines cisplatin sensitivity
-
•
Time-resolved cisplatin-induced signal transduction predicts cisplatin sensitivity
Triple-negative breast cancers show large variation in sensitivity to the chemotherapeutic agent cisplatin that cannot be explained by defects in DNA repair. Heijink et al. conducted a systems-level analysis of cisplatin-induced signal transduction and reveal that signaling dynamics can be used to predict cisplatin sensitivity of TNBC models.
Introduction
In standard care, breast cancers are subtyped based on the expression of the estrogen and progesterone receptors (ER and PR, respectively) and human epidermal growth factor receptor-2 (HER2). These receptors are oncogenic drivers and relevant drug targets. Breast cancers lacking expression of ER, PR, and HER2 are called triple-negative breast cancers (TNBCs); they do not benefit from anti-hormonal or anti-HER2 treatments, and they account for ∼15%–20% of invasive breast cancers (Foulkes et al., 2010). Although patients with TNBC can initially respond to chemotherapy, they have worse overall prognosis compared with other breast cancer subtypes. Unfortunately, TNBCs lack clear targetable driver oncogenes, constituting an unmet need to improve the therapeutic options for these patients.
Apart from chemotherapy, no treatments are proven to be effective for this patient group. Among genotoxic chemotherapeutic agents, platinum-based chemotherapeutics, such as cisplatin, are potential treatment options for TNBC patients and predominantly showed favorable responses in TNBCs with underlying BRCA1/2 mutations (Byrski et al., 2010, Cardoso et al., 2017, Rouzier et al., 2005, Silver et al., 2010). When tested in vitro using panels of TNBC models, platinum-containing agents appeared effective, although the observed sensitivity varied significantly (Lehmann et al., 2011). TNBC is a heterogeneous breast cancer subtype, so identifying molecular features of TNBC that are critical for cisplatin sensitivity will likely be necessary for these drugs to be used effectively. At the molecular level, cisplatin introduces both intra- and inter-strand DNA crosslinks (ICLs), which stall replication forks and are therefore especially toxic in proliferating cells (Siddik, 2002). ICL-induced stalled replication forks activate the DNA damage response (DDR) and initiate DNA repair through multiple DNA repair pathways, including homologous recombination (HR), nucleotide excision repair (NER), and Fanconi anemia (FA) (Kim and D’Andrea, 2012, Shuck et al., 2008). The ability of cells to repair DNA crosslinks is considered a critical determinant for the cytotoxic effect of cisplatin treatment (Bhattacharyya et al., 2000, Kim and D’Andrea, 2012). Consequently, mutations and/or reduced expression of HR and FA genes are robustly linked to sensitivity of platinum-based chemotherapeutics (Taniguchi et al., 2003). Nevertheless, cisplatin sensitivity is not always associated with defective HR, NER, or FA. An important challenge is to unravel which other factors determine the efficacy of cisplatin treatment and to investigate whether such factors could be used as targets to potentiate chemo-sensitivity of TNBC cells.
The complexity of the DDR makes it challenging to predict how cancers will respond to DNA-damaging chemotherapy. For instance, it is becoming clear that the DDR does not function as an isolated linear signaling pathway but rather is a large signaling network that interconnects canonical DDR pathways with additional pro-growth and pro-death signaling pathways (Ciccia and Elledge, 2010, Costelloe et al., 2006, Jackson and Bartek, 2009). In addition, signaling through the DDR occurs non-linearly because of extensive crosstalk and feedback control, including adaptation and rewiring following stimulation (Lee et al., 2012). Differential activation and wiring of the DDR in response to cisplatin has been proposed to underlie the differences in cisplatin sensitivity (Brozovic et al., 2009, Wang et al., 2012). Therefore, it has proven difficult to predict chemo-sensitivity based on the presence or activity of DDR components, which are typically measured at a single static moment after cisplatin treatment. Detailed understanding of how signaling dynamics fluctuate over time and how molecular signals are integrated may be necessary to better understand chemo-sensitivity in TNBCs.
To meet this challenge, we performed a systems-level analysis in cisplatin-sensitive and cisplatin-resistant TNBC cell lines. We collected quantitative time-resolved signaling data on the activation status of several key signaling proteins, together with phenotypic data reporting apoptotic and cell-cycle regulatory responses. These data were integrated using statistical modeling, revealing that cisplatin-induced changes in cell-cycle signaling molecules determine cisplatin-induced initiation of cell death and that these profiles could be useful in predicting cisplatin responses.
Results
Large Variation in Cisplatin Sensitivity in Human TNBC Cell Lines
We assembled a panel of well-described human TNBC cell lines and measured cellular viability after 72 h of continuous cisplatin treatment. To control for potential confounding effects of differences in growth rates, we calculated growth rate inhibition metrics (GR values) (Hafner et al., 2016). Large variations in sensitivity were observed among the nine cell lines, with GR50s ranging from 2.2 μM in HCC1937 to 61 μM in MDA-MB-231 (Figure 1A). The increased cisplatin sensitivity of two TNBC cell lines, SUM149PT and HCC1937, could be rationalized based on defective HR because of BRCA1 mutations. For other cell lines, even within the same molecular TNBC subtype, differences in cisplatin sensitivity could not be explained by underlying BRCA1/2 mutations.
Figure 1.

Heterogeneous Responses to Cisplatin in TNBC Cell Lines
(A) Indicated TNBC cell lines were treated with cisplatin for 72 h. Methyl thiazol tetrazolium (MTT) conversion was measured, and growth rate-adjusted drug responses (GR metrics) were plotted. Error bars indicate SEM of at least three independent experiments with three technical replicates each. MDA-MB-231 and MDA-MB-157 are called MB-231 and MB-157, respectively.
(B) Indicated TNBC cell lines were irradiated (10 Gy) or left untreated and analyzed for RAD51 foci 3 h later. Scale bar represents 10 μM.
(C) Indicated TNBC cell lines were treated with mitomycin C (MMC, 50 ng/mL) for 24 h. FANCD2 ubiquitination was assessed by western blotting.
(D) Characteristics of all 9 tested TNBC cell lines are listed. GR50 values for cisplatin were calculated from averages of three independent experiments. TP53, BRCA1, and BRCA2 mutation status was obtained from the Cosmic database.
See also Figure S1.
To better comprehend the complexity of the cellular response to cisplatin, we aimed to identify factors other than DNA repair-related elements, which determine cisplatin sensitivity. We therefore measured multiple DDR-related signaling nodes in human TNBC cell line models with different levels of sensitivity to cisplatin but similar DNA repair status.
We selected two cisplatin-sensitive TNBC cell lines (HCC38 and BT549) and two cisplatin-resistant TNBC cell lines (MDA-MB-231 and MDA-MB-157) of the same Claudin-low breast cancer subtype. To test whether the differential cisplatin sensitivity was caused by defective HR, NER, or FA pathways, we analyzed RAD51 foci formation after irradiation as a measure of HR proficiency (Figure 1B), assessed FANCD2 ubiquitination after mitomycin C (MMC) treatment as measure of FA proficiency (Figure 1C), and screened for mutations in NER and mismatch repair (MMR) genes. In all four selected cell lines, RAD51 foci were clearly induced after irradiation, confirming HR proficiency (Figures 1B and 1D). In addition, all four cell lines showed mono-ubiquitinated FANCD2 upon MMC treatment (Figure 1C), illustrating FA pathway functionality (Figures 1C and 1D). We did not find pathogenic mutations in NER or MMR pathway components or genomic scars associated with MMR deficiency (Figure S1), which are described as contributing to repair of cisplatin-induced DNA lesions. Thus, the selected Claudin-low TNBC cell lines show differences in cisplatin sensitivity that do not appear to be caused by deficiencies in DNA repair.
Creation of a Signal-Response Dataset for Cisplatin Sensitivity
To identify which DDR-related signals determine cisplatin sensitivity, we aimed to determine the relationship between changes in DDR-related signaling proteins and cellular responses to cisplatin. We measured the levels or activation states of 22 signaling proteins that comprise the DDR, cell-cycle machinery, and/or apoptotic cell death pathways and six phenotypic responses (Figure 2A). Each signal was quantified at 11 time points following exposure to 2 or 20 μM cisplatin, resulting in 44 measurements of each signal protein (green) with corresponding loading controls (red) (Figure 2B, upper panel). Fold changes (FLDs) across all cell lines and cisplatin concentrations were quantified (Figures 2B, bottom panel, and 2C). To identify the potential relationship between signaling dynamics and differential sensitivity to cisplatin, we concomitantly measured phenotypes related to cisplatin treatment, including induction of DNA damage, changes in cell-cycle progression, and cell death (Figure 2D). These responses were quantified at 12 time points between 0 and 120 h after cisplatin exposure using flow cytometry (Figures 2D and 2E). All signaling and phenotypic response measurements were performed in biological and experimental duplicates in HCC38, BT549, MDA-MB-157, and MDA-MB-231 cells, yielding a dataset of 3,872 molecular signals measurements and 1,044 cellular response measurements (Figures 2C and 2E; Table S1).
Figure 2.

A Systems-Level Signal-Response Dataset following Cisplatin
(A) An expanded DNA damage signaling-response network, including canonical components of the DDR, growth, and stress response pathways. Signals integrated in the model are green, and responses are blue.
(B) Protein abundance and activation levels were analyzed by western blotting using two-color infrared detection (top). Signal intensity was quantified, normalized to actin, and plotted as FLD compared with the lowest measurement across all cell lines and treatments. The signaling time course plot is presented from the western blot shown on top. Mean values ± SD of two experiments are shown.
(C) The complete signaling dataset for four TNBC cell lines following 2 or 20 μM cisplatin treatment. Each box represents an 11-point time course of biological duplicate experiments. Grayscale reflects signal strength. Background color indicates signaling profile: sustained increase in green, late increase in red, transient increase in yellow, and sustained decrease in blue, as explained in the STAR Methods section. Numbers below each plot report the maximum FLD on the y axis.
(D) Measurements of response data. DNA content, percentages of mitotic cells, and level of DNA damage were measured by flow cytometry. Left panel: example fluorescence-activated cell sorting (FACS) plot showing cell-cycle profiles based on DNA content. Percentage of cells in G1, S, and G2 phases and cell death measured by sub-G1 were quantified. Middle panel: percentage of mitotic cells as measured by phospho-histone H3 positivity. Right panel: level of DNA damage in G1 cells was quantified as phospho-H2AX mean fluorescence intensity in 2n cells.
(E) The complete response dataset colored as in (C).
The addition of cisplatin caused clear dose-dependent increases in the percentage of sub-G1 cells and in the magnitude of many molecular signals (Figures 2C and 2E). For example, after exposure to 20 μM cisplatin, phosphorylation of H2AX rose to a maximum fold increase of 55, while with 2 μM cisplatin, this level increased 22-fold. Baseline protein levels or activation states of most individual signals were poorly correlated with sub-G1 levels after 120 h of cisplatin treatment (Figure S2), indicating that sensitivity to cisplatin is poorly predicted by overall DDR activity states before drug exposure. In addition, the activation patterns of the molecular signals did not show clear dose-dependent changes. The activation patterns of signals differed strongly between cell lines, without clear distinctions between cisplatin-sensitive and cisplatin-resistant cell lines. Thus, clear dose-dependent changes could be observed in the magnitude of most signals, but the duration and pattern of activation differed strongly between cell lines.
Statistical Modeling Using Partial Least-Squares Regression (PLSR)
To more rigorously analyze the DDR signaling data after cisplatin treatment, we used PLSR (Geladi and Kowalski, 1986). PLSR functions by identifying a reduced set of metavariables (or principal components [PCs]) that maximize co-variation between molecular signaling input variables and cellular response output responses (Figure S3; Janes and Yaffe, 2006). The first PC captures the greatest amount of information within the data. Additional PCs are identified iteratively to maximally capture residual variance until additional PCs cease to capture meaningful data (relative to the technical error of measurements). This approach can be used to simplify complex data and to uncover hidden associations between signals and phenotypic outcomes that may be missed visually.
Prior quantitative analysis of signaling has revealed that many networks respond to changes in levels of protein activation, rather than to absolute activation levels (Gaudet et al., 2005, Janes et al., 2008). These dynamic features can be obscured by large differences between cell lines in the overall magnitude of signal activation. To highlight signaling dynamics in our models, we derived six metavariables-metrics from our time-staggered signaling dataset. These signaling dynamic metrics were (1) FLD, (2) slope between adjacent time points (SLP), (3) maximum slope (SMX), (4) dynamic range (DYN), (5) total activity (area under the curve, AUC), and (6) average level of activity (AVE). In addition, time-dependent measurements were divided into three time frames—early, ranging from 0 to 2 h; middle, spanning 2 to 12 h; and late, ranging from 12 to 24 h—to capture specific time regimes in which fluctuation in signal dynamics best correlated with cellular response. In total, 132 metrics were composed, based on 22 molecular signals and 6 metavariables. Using this approach, signaling dynamics were included in the input variables (signals), while output variables (responses) were encoded using uncoupled time points. Each response variable was represented by the average value calculated for each time frame.
We initially explored these data by building a model composed of the data from all four cell lines. The resulting model reduced the dataset into four PCs (Figure S3B). Altogether, these four PCs explained 60% of the overall variance (R2), and predicted 29% of the variation, using a cross-validation scheme (Q2) (Figure S3B). These low model-fitness parameters reflected that the underlying data were not well captured in a single model. Based on this observation, we speculated that the signaling proteins were used in a fundamentally different manner in cisplatin-sensitive and cisplatin-resistant cell lines. Thus, we next separated the data to build two separate models: one for cisplatin-sensitive cell lines and one for cisplatin-resistant cell lines. Our partial least-squares (PLS) model of cisplatin-sensitive cell lines HCC38 and BT549 captured 81.6% of the co-variance between signals and responses with the first two PCs (Figure S3C). Likewise, 81.4% of the co-variance in the data of the cisplatin-resistant cell lines MDA-MB-231 and MDA-MB-157 was explained by the first two PCs of the cisplatin-resistant model (Figure S3D). In both cases, we observed significant improvements in model prediction accuracy, with Q2 parameters increasing to more than 80% for both models. In both cisplatin-sensitive and cisplatin-resistance models, PC1 largely captured the variation associated with the different time regimens (Figure 3A), whereas PC2 captured cell line-specific variance (Figure 3A). To determine whether the improvements in model fitness in the cisplatin-sensitive and cisplatin-resistance models reflect similarities in the biological responses within these cells, we created models from all random pairs of cell lines. These other models produced substantially reduced fitting parameters, with R2 and Q2 values of ∼30% and 10%, respectively (Figure S3E), suggesting that model-fitness improvements emerged because of similar biological responses within cisplatin-sensitive and cisplatin-resistant cells.
Figure 3.

PLSR Correctly Predicts Sub-G1 from Molecular Signals Activated by Cisplatin
(A) PLSR analysis of covariation between molecular signals and cellular responses. Score plots represent the signaling response of each TNBC cell line at a specified time, as indicated by the colors and symbols in the legend. Scores are plotted for the sensitive and resistant PLS models.
(B) Correlation between measured sub-G1 (flow cytometry, y axis) and model-predicted sub-G1 (x axis).
(C) PLS loadings plotted for signals and responses and colored by signaling class.
(D) PC1 loading scores of the dynamic signaling metrics (FLD, fold change; DYN, dynamic range; SMX, maximum slope; SLP, slope) are plotted. Loading scores of the four dynamic metrics of pMK2 and their average are shown in the upper panel. Loading scores of the dynamic metrics of all cell cycle-related signals (PLK1, Aurora-A, CyclinB1, CDC25C, and CDC25A) and their averages are shown in the bottom panel.
(E) Cisplatin sensitivity of BT549 and MDA-MB-231 cell lines, transduced with indicated shRNAs measured by MTT conversion. Inset bar graphs depict MTT conversion upon treatment with 7.5 or 15 μM cisplatin of BT549 and MDA-MB-231, respectively.
Error bars indicate SEM of three independent experiments. The p values were calculated using two-tailed Student’s t test. ∗∗∗∗p < 0.0001. See also Figures S3 and S4.
To examine the quality of our models, we used jack knife-based cross-validation to compare each measured cellular response in isolation with the responses predicted by our models (Gong, 1986). Both models were particularly accurate in predicting the sub-G1 apoptotic response, cell-cycle state, and extent of γH2AX phosphorylation following cisplatin treatment. The correlations between measured responses and those predicted by our model were above 0.97 (Figures 3B, S3F, and S3G). Thus, the combination of signaling metrics and responses was adequate to build two well-fit models that could predict cellular responses, including at sub-G1 levels, in response to cisplatin. Because model fitness required sensitive and resistant cells to be modeled separately, the underlying differences between cisplatin-sensitive and cisplatin-resistant cells were not likely to be different levels of activation within similarly functioning networks but instead were likely to be caused by signaling through fundamentally different networks.
PLS Model Identifies Determinants of Cisplatin Sensitivity
To better understand how specific signal transduction proteins influence the responses to cisplatin, we projected the loading vectors for each model feature into the PC vector space (Janes and Yaffe, 2006). Vector loadings report the contribution of each signal to the variation captured by a specific PC. This information can be used to highlight critical features that differentiate between cisplatin responses in sensitive and resistant cells. In both models, we observed a strong anti-correlation between sub-G1 and G1, which was captured by PC1 in both instances (Figure 3C). Thus, signals that contribute strongly to PC1 are likely to be important for cisplatin sensitivity in these cells. The vector loading plot revealed many signals and signaling features that are strongly co-variant with sub-G1 cells, suggesting that multiple signaling features, rather than a single signal, are critical for predicting cisplatin sensitivity in TNBC cells.
Because both models could accurately predict cell death, we next wished to determine whether specific signal proteins contributed to this differential accumulation of sub-G1 in response to cisplatin in our models. Our strategy was to identify the signals that were the most differentially weighted in sensitive versus resistant cells, because these signals might underlie the difference in cisplatin sensitivity in TNBC cell lines. A particularly interesting example was MK2, an inflammation-related and cell-cycle checkpoint kinase, whose role in the DDR remains unclear. In the model of cisplatin-sensitive cells, pMK2 showed positive co-variance with subsequent emergence of sub-G1 cells, suggesting that this protein contributes to cisplatin-induced cell death (Figure 3D). In contrast, in the cisplatin-resistant model, dynamic MK2-related metrics were negatively correlated with sub-G1, suggesting that activation of MK2 promotes cell death in sensitive cells but paradoxically inhibits cell death in resistant cells.
To test these model-generated predictions, cisplatin-sensitive (BT549) and cisplatin-insensitive (MDA-MB-231) cell lines were transduced with short hairpin RNAs (shRNAs) targeting MK2 (Figure S4A). Consistent with our model-based predictions, knockdown of MK2 reduced cisplatin sensitivity in cisplatin-sensitive BT549 cells (Figure 3E, left panel). The cisplatin-resistant cell line MDA-MB-231 showed contrasting results. Consistent with the model’s paradoxical prediction that MK2 activation prevents cell death in cisplatin-resistant cell lines, knockdown of MK2 resulted in enhanced cisplatin sensitivity in MDA-MB-231 cells (Figure 3E, right panel).
Among the signals that showed the largest differences in PC1 scores between the sensitive and the resistant PLS models, many were linked to cell-cycle regulation (Figures 3D and S4B). Whereas other signal classifications showed a similar distribution of PC1 scores in the sensitive and the resistant models, PC1 scores of cell cycle-related signals showed a differential distribution (Figures 3D and S4C). These data underscore that cisplatin sensitivity is linked to the ability of cancer cells to activate cell-cycle checkpoint signaling, which is in line with a role for cell-cycle checkpoints in preventing transmission of DNA lesions to daughter cells to protect genome integrity.
Cisplatin-Induced Changes in Cell-Cycle Progression and Cell Death in TNBC Cell Lines
To test whether altered cell-cycle checkpoint activation could differentiate between the selected cisplatin-sensitive and the selected cisplatin-resistant TNBC cell lines, we monitored cell-cycle dynamics at several time points after treatment with cisplatin (Figure 4A). Both insensitive cell lines showed transient S/G2 cell-cycle arrest, after which proliferation was resumed (Figures 4A and 4B). In contrast, cisplatin-sensitive cell lines ceased cell-cycle progression at the G2 stage and remained with 4n DNA for the remainder of the experiment (Figures 4A and 4B). Similar results were obtained when synchronized cell cultures were treated with cisplatin (Figures S5A and S5B).
Figure 4.

Cisplatin-Induced Changes in Cell-Cycle Progression and Cell Death in TNBC Cell Lines
(A–C) Quantitative cell-cycle analysis. Cells were treated with 2 or 20 μM cisplatin, and cell-cycle profiles were analyzed at indicated time points. (A) Representative cell-cycle profiles of MDA-MB-157 (red) and BT549 (blue) cells after treatment with 2 or 20 μM cisplatin. (B) Quantification of G1 cells from two independent experiments. Error bars indicate SEM. (C) Quantification of sub-G1-cells from two independent experiments. Error bars indicate SEM.
(D and E) TNBC cell lines stably expressing GFP-MDC1 were treated with cisplatin (2 μM) for 24 h before time-lapse imaging, and cell fate was assessed. (D) Representative cells are shown, with time point M−1 showing the last frame before mitosis, M1 indicating the onset of mitosis, M2 denoting mitotic exit, and M+1 presenting the first time frame after cytokinesis. Scale bar represents 17 μM. (E) Quantification of MDC1 foci before mitosis (open circles) and after mitosis (filled circles). At least 10 cells have been analyzed per condition. Error bars indicate SEM.
(F) Gene Ontology (GO) pathway analysis of differentially expressed genes (DEGs). MDA-MB-231, MDA-MB-157, HCC38, and BT549 cells were left untreated or were treated with 2 μM cisplatin for 72 h. For each cell line, DEGs were classified based on GO enrichment analysis. GO terms that appeared in both cisplatin-sensitive and cisplatin-insensitive cell lines are indicated. Upregulated GO terms are yellow, and downregulated GO terms are blue. Color intensity is based on the p value.
(G) Overlap between DEGs of cisplatin-sensitive and cisplatin-resistant TNBC cell lines. Genes with a FLD ≥ 1.75 in sensitive cell lines, as well as in resistant cell lines, are red.
When TNBC cell lines were treated with high-dose cisplatin (20 μM), both sensitive and resistant cell lines entered prolonged cell-cycle arrest (Figures 4A and 4B). In line with their high sensitivity to cisplatin, BT549 and HCC38 displayed clear induction of apoptosis, as judged by the proportion of cells with sub-G1 DNA content, in contrast to MDA-MB-231 and MDA-MB-157 cells (Figure 4C). Thus, in line with our modeling data, cisplatin-sensitive and cisplatin-resistant TNBC cell lines show different cell-cycle distributions in response to cisplatin.
To explore whether the cell-cycle arrest kinetics were related to dynamics of DNA damage resolution, TNBC cell lines were transduced with GFP-tagged MDC1, which binds γH2AX and therefore serves as a marker for DNA breaks (Stucki et al., 2005). Live cell imaging revealed that cisplatin-resistant cell lines accumulated DNA damage in response to cisplatin treatment, as evidenced by GFP-MDC1 foci, but only entered mitosis when DNA damage foci were resolved (Figures 4D and 4E). In contrast, cisplatin-sensitive cell lines often entered mitosis in the presence of GFP-MDC1 foci. This was particularly pronounced in HCC38 cells, which entered mitosis with very high levels of DNA damage that remained visible even after cells exited mitosis (Figure 4D and 4E). These findings suggest that cisplatin-sensitive TNBC cells are unable to properly repair DNA breaks before mitotic entry, possibly caused by slippage through prolonged DNA damage-induced G2/M cell-cycle arrest.
Our prior data suggested that differences in DDR and cell-cycle checkpoint signaling may account for the observed differences in cisplatin sensitivity. We next explored cisplatin-induced gene expression changes to reiterate this notion and potentially highlight signals that may contribute to the observed differences in drug sensitivity. To investigate this, we analyzed changes in gene expression 72 h after low-dose cisplatin (2 μM) in both sensitive and resistant TNBC cell lines (Figure S5C). Gene Ontology (GO) pathway analysis of differentially expressed genes (DEGs) revealed a strong enrichment for genes involved in cell-cycle regulation, DNA repair, mRNA processing, and apoptosis (Figure 4F), although the DEGs showed limited overlap between cell lines (Figure 4G; Table S2). In line with our cell-cycle progression data, gene expression analysis showed decreased expression of G2/M cell-cycle pathway components and lowered levels of DNA repair genes in cisplatin-resistant cell lines after 72 h of treatment. In contrast, cisplatin-sensitive cell lines consistently showed upregulated expression of G2/M cell-cycle pathways (Figure 4F). These data suggest that cell-cycle progression or the ability to install damage-induced cell-cycle checkpoint arrest determines the cellular response to cisplatin. However, the limited numbers of DEGs and the lack of significant overlap of altered genes between cell lines suggested that cisplatin sensitivity is not predominantly transcriptionally controlled but rather is driven by post-translational modifications.
G3BP2 Depletion Promotes Cell-Cycle Arrest in Cisplatin-Resistant Cell Lines
Among the genes that revealed contrasting regulation between cisplatin-sensitive versus cisplatin-resistant TNBC cell lines, three genes were identified, G3BP2, HMMR, and NEK2, that were previously remotely linked to DNA damage but were not associated to cisplatin response (Figure 4G; Fletcher et al., 2004, Isabelle et al., 2012, Sohr and Engeland, 2008). We measured their levels after cisplatin treatment in our selected TNBC cell lines (Figure S6A) and added these data to our previously collected dataset. PLSR modeling using this expanded dataset resulted in improved predictive models, with Q2 parameters of 91% and 92% for the sensitive and resistant models, respectively (Figure S6B).
To identify the minimal subset of signaling features that are required to accurately predict cisplatin sensitivity, we iteratively removed signals, beginning with those contributing the least to model fitness (lowest variable importance in projection [VIP] score) (Gaudet et al., 2005). For PLS models of either sensitive or resistant cells, we found that the full predictive capacity of our models required only the 4 or 6 most informative metrics (Figures 5A and S6C). In parallel, we performed this analysis in the inverse order, iteratively removing signals starting with the highest VIP score. These models were also resilient to this type of perturbation, because the full predictive capacity of the model was unchanged even when the top 60 most informative metrics were eliminated (Figure 5B). Thus, accurate predictions could be generated using models that contained either the most or the least informative signals, albeit with a substantially larger number of signals required when the least informative signals are used. Altogether, these data highlight that predictive information is not rare in signaling data but rather is redundantly encoded throughout the signaling network.
Figure 5.

Robustness of PLSR Models, and Validation of G3BP2 as a Determinant of Cisplatin Sensitivity
(A and B) The minimal number of signaling metrics required for predicting sub-G1 was calculated by iteratively removing metrics. The fraction of G3BP2-related metrics among metrics with the highest VIP scores is indicated in pie charts. (A) Metrics were eliminated sequentially from the models of cisplatin-sensitive cell lines (left panel) or cisplatin-resistant cell lines (right panel) based on the relative magnitude of their coefficients in the model, from highest to lowest VIP score. (B) Metrics were sequentially eliminated from the model of cisplatin-sensitive cell lines from lowest to highest VIP score.
(C) PC1 loading scores of the dynamic signaling metrics of G3BP2 and their average are plotted for the sensitive and resistant models individually.
(D) MDA-MB-231 and BT549 cells were transduced with indicated shRNAs and MTT conversion after cisplatin treatment was measured. Inset bar graphs depict MTT conversion upon treatment with 7.5 μM cisplatin. sh#1 and sh#2 refer to shG3BP2#1 and shG3BP2#2, respectively. Error bars indicate SEM of three independent experiments. The p values were calculated using two-tailed Student’s t test. ∗∗∗p < 0.001.
(E and F) MDA-MB-231 cells with doxycycline-inducible shRNAs targeting luciferase or G3BP2 were treated with 2 μM cisplatin. At indicated time points, cell-cycle profiles were determined by flow cytometry (E). Means and SDs of percentages of G2 cells from three independent experiments are plotted (F).
(G) γH2AX levels after 2 μM cisplatin treatment for 72 h. MDA-MB-231 cells expressing inducible shRNAs against luciferase or G3BP2 were fixed and stained with anti-γH2AX antibody and propidium iodide. γH2AX levels and DNA content were determined by flow cytometry of two independent experiments.
See also Figure S6.
The metrics with the highest predictive accuracy differed between the sensitive and the resistant models. Signaling metrics of G3BP2 were critical for model predictive accuracy in the sensitive model, while these were absent within the top VIP scores of the resistant model (Figure 5A). In addition, the dynamic metrics of G3BP2 had the opposite PC1 score in sensitive versus resistant models (Figure 5C). These data indicate that G3BP2 promotes cell death in sensitive cells but paradoxically inhibits cell death in resistant cells. To test this prediction, cisplatin-sensitive (BT549) and cisplatin-insensitive (MDA-MB-231) cell lines were transduced with shRNAs targeting G3BP2 (Figure S6D). Consistent with our modeling-based predictions in cisplatin-insensitive TNBC cells, depletion of G3BP2 sensitized MDA-MB-231 cells to cisplatin (Figure 5D). However, knockdown of G3BP2 did not significantly alter cisplatin sensitivity in BT549 cells (Figure 5D).
To examine whether G3BP2 knockdown changed the behavior of MDA-MB-231 to resemble other aspects of the behavior observed for cisplatin-sensitive cell lines, we analyzed cell-cycle distribution after cisplatin treatment. To this end, MDA-MB-231 cells were transduced with doxycycline-inducible shRNAs targeting G3BP2. Although control cell lines were only transiently arrested in G2, G3BP2-depleted cells maintained G2 arrest (Figures 5E and 5F). In line with this observation, G3BP2 knockdown cells accumulated more cisplatin-induced DNA damage when compared with control cells (Figure 5G). Although MDA-MB-231 cells are described as displaying mesenchymal features (Lombaerts et al., 2006), their morphology changed upon G3BP2 knockdown into a mobile phenotype with extensive protrusions (Figure S6E). To test whether knockdown of G3BP2 had influence on the epithelial-mesenchymal transition (EMT) in MDA-MB-231 cells, we analyzed the abundance of different EMT-related factors. Although control MDA-MB-231 cells showed expression of the mesenchymal markers Fibronectin and ZEB1, knockdown of G3BP2 resulted in a decrease in their expression (Figure S6F). Conversely, the expression of the epithelial marker E-cadherin increased after knockdown of G3BP2 (Figure S6F).
PLS Models Trained on Cisplatin-Sensitive and Cisplatin-Resistant Cells Accurately Predict Cisplatin Sensitivity in a Panel of TNBC Cells
To validate whether cisplatin-induced signaling dynamics of pMK2, RPA, and G3BP2 can predict cisplatin sensitivity beyond the model training set of four TNBC cell lines, we measured the abundance of these three signals, together with the levels of BCL-xL and pKAP1—the two highest scoring signals in our original models—following cisplatin treatment in three untested TNBC cell lines (MDA-MB-468, HCC1806, and HCC1143). Although these cell lines were all relatively sensitive to cisplatin, they displayed a significant range in GR50 (MDA-MB-468, 0.7 μM; HCC1806, 3.8 μM; and HCC1143, 14.7 μM) (Figure 6A). Based on our PLS modeling, we anticipated that signaling metrics collected for these five signals alone would be sufficient to predict cisplatin sensitivity. To test this notion, we generated a new PLS model with data from the original dataset (i.e., four TNBC cell lines treated with 20 μM) in combination with data from the additional three cell lines. The score plot for this combined dataset showed that PC1 separated all cell lines based on their cisplatin sensitivity, including the three additional cell lines (Figure 6B). The least-sensitive cell line of the validation panel, HCC1143, was located between the resistant and the sensitive cell lines of the original model, while the projection of HCC1806 and MDA-MB-468 was similar to that in the cisplatin-sensitive cell lines (Figure 6B). We used this minimal model to predict the sub-G1 percentage for the validation panel in response to cisplatin. Sub-G1 was accurately predicted for the newly included cell lines (R2 = 0.849) (Figure 6C). Thus, using only the five most important signals for distinguishing cisplatin-sensitive from cisplatin-insensitive cell lines, PLS modeling successfully captured levels of cisplatin sensitivity. Altogether, our data highlight a small compendium of signals—including RPA, pMK2, and G3BP2—that are used differently in the context of cisplatin-sensitive and cisplatin-resistant TNBC cells to promote the observed differences in drug sensitivity.
Figure 6.

Validation of PLS Model-Generated Predictions in Additional TNBC Cell Lines
(A) Cisplatin sensitivity of the validation cell lines (colored) compared with the original four cell lines (gray). After cells were treated with cisplatin for 72 h, MTT conversion was measured and growth rate-adjusted drug responses (GR metrics) were plotted. Averages and error bars of at least three replicates are shown.
(B) Score plot of the general PLS model comprehended with signaling (pMK2, RPA, G3BP2, pKAP1, and BCL-xL) and response data of additional TNBC cell lines (MDA-MB-468, HCC1806, and HCC1143).
(C) Correlation plot between measured sub-G1 by flow cytometry (y axis), and cross-validated predictions of sub-G1 (x axis) by the PLS model.
Discussion
In this study, we describe a systematic time-resolved approach to identify molecular signals that can distinguish cisplatin-sensitive from cisplatin-resistant TNBC cell lines. We found that cell-cycle checkpoint factors appeared to determine cisplatin sensitivity in TNBC cell line models that do not harbor obvious DNA repair defects. These findings are in line with earlier observations that expression levels of the WEE1 and CHK1 kinases are related to cisplatin sensitivity (Pouliot et al., 2012) and that targeting of cell-cycle checkpoints, including ATR, CHK1, and WEE1, can be used to sensitize cancer cells to cisplatin (Gadhikar et al., 2013, Hirai et al., 2009, Perez et al., 2006, Reaper et al., 2011, Sangster-Guity et al., 2011).
At the cellular level, we observed that both sensitive and resistant cell lines engage cisplatin-induced S/G2 cell-cycle arrest. However, whereas resistant cell lines recommence cell-cycle progression, cisplatin-sensitive models did not. Our data indicate that cisplatin-induced changes in signaling, rather than static states of signaling molecules before treatment, are important in determining cell fate.
Our finding that differences in treatment-induced signaling dynamics determine phenotypic outcomes is in apparent contrast to the finding that kinase-effector signaling is stable across models from the same lineage (Miller-Jensen et al., 2007). This discrepancy could be caused by the different origins of the cell line models. TNBCs are highly genomically instable (Curtis et al., 2012), and individual TNBCs may have evolved contrarily in their ability to deal with DNA lesions, possibly influenced by the various baseline levels of endogenous DNA lesions. In addition, previous quantitative modeling studies of the DNA damage signaling in single models demonstrated context dependence, especially involving mitogen-activated protein kinase (MAPK) signaling (Lee et al., 2012, Tentner et al., 2012). The stability of signal processing across models may be different for inflammatory stress versus genotoxic agents (Miller-Jensen et al., 2007).
Changes in G3BP2 expression after cisplatin treatment were identified as one of the signals that correlated strongly with cell death following cisplatin exposure. Previously, G3BP2 was described as playing a role in stress granule formation (Gupta et al., 2017) and was found to be involved in Twist-induced EMT (Wei et al., 2015). In line with these reports, G3BP2 depletion in the cisplatin-resistant cell line MDA-MB-231 reduced mesenchymal cell morphology, resulted in prolonged cisplatin-induced G2 cell-cycle arrest, and led to increased sensitivity to cisplatin (Wei et al., 2015). These results underscore a role for mesenchymal transition in reduced chemo-sensitivity. Our data suggest that such a mesenchymal transition is versatile and that targeting G3BP2 in mesenchymal-like cisplatin-resistant TNBC cells may increase chemo-sensitivity.
In the context of defective p53, cancer cells were previously shown to increasingly depend on p38MAPK/MK2 for proper cell-cycle checkpoint control and survival after DNA damage (Manke et al., 2005). Specifically, inactivation of MK2 was reported to abrogate cell-cycle checkpoint responses and to sensitize tumor cells to cisplatin in vitro and in vivo (Dreaden et al., 2018, Morandell et al., 2013, Reinhardt et al., 2010). Other studies demonstrated MK2 to be involved in DNA damage-induced replication fork stalling (Köpper et al., 2013). In the latter case, MK2 knockdown rescued gemcitabine-induced replication stalling and increased cell survival. Our data also show opposite roles for MK2 in dictating cell survival after DNA damage. Knockdown of MK2 resulted in increased cisplatin sensitivity in the cisplatin-resistant cell line MDA-MB-231 but reduced cisplatin-sensitive of BT549 cells. The impact of MK2 modulation on cisplatin sensitivity was surprising, because we observed only limited changes in pMK2 levels upon cisplatin treatment. These data underscore the utility of our modeling approach in identifying key regulators of signaling outcome and suggest a context-dependent requirement for MK2 in checkpoint responses. Although modulation of MK2 activity resulted in the predicted opposite effects on cisplatin sensitivity, the effect sizes were relatively modest. This may again reflect the redundant wiring of DNA damage-induced cell-cycle checkpoints, as well as pro-apoptotic signaling, in which the effects of MK2 inactivation are partially buffered by parallel signaling axes. Further research is warranted to uncover which tumors may benefit from combined treatment with platinum-containing chemotherapeutics and MK2 inhibitors.
In finding predictive biomarkers for chemo-response, the status of a signaling molecule is typically assessed in treatment-naive tumors. We measured signaling flux at various time points in response to cisplatin treatment and found signaling dynamics to be of key importance in predicting cellular response to cisplatin. These results underscore that steady-state levels of signaling molecules in untreated tumor cells or clinical tumor material may have low predictive value, and in an ideal scenario, samples before and shortly after start of treatment should be analyzed. Moreover, the notion that predictive information is redundantly encoded in our models implies that a limited set of signaling features may be sufficient to probe signaling dynamics in response to cisplatin treatment.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-RAD51 | GeneTex | Cat# GTX70230; RRID:AB_372856 |
| Anti-FANCD2 | Santa Cruz | Cat# sc-20022; RRID:AB_2278211 |
| Anti-G3BP2 | Bethyl | Cat# A302-040A, RRID:AB_1576545 |
| Anti-MAPKAPK-2, phospho (Thr334) | Cell Signaling | Cat# 3041, RRID:AB_330726 |
| Anti-RPA32/RPA2 | Abcam | Cat# ab2175, RRID:AB_302873 |
| Anti-B-actin | MP Biomedicals | Cat# 08691001, RRID:AB_2335127 |
| Anti-Histone H2A.X, phospho (Ser139) | Cell Signaling | Cat# 9718, RRID:AB_2118009 |
| Anti-Histone H3, phospho (Ser10) | Cell Signaling | Cat# 9706, RRID:AB_331748 |
| Anti-CDC25C | Cell Signaling | Cat# 4688, RRID:AB_560956 |
| Anti-Phospho-Chk1 (Ser345) (133D3) | Cell Signaling | Cat# 2348, RRID:AB_331212 |
| Anti-Phospho-Chk2 (Thr68) (C13C1) | Cell Signaling | Cat# 2197, RRID:AB_2080501 |
| Anti-ATR, phospho (Ser428) | Cell Signaling | Cat# 2853, RRID:AB_2290281 |
| Anti-Phospho-Akt (Ser473) (736E11) | Cell Signaling | Cat# 3787, RRID:AB_331170 |
| Anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (20G11) | Cell Signaling | Cat# 4376, RRID:AB_331772 |
| Anti-Phospho-p38 MAPK (Thr180/Tyr182) (D3F9) | Cell Signaling | Cat# 4511, RRID:AB_2139682 |
| Anti-Phospho-SAPK/JNK (Thr183/Tyr185) | Cell Signaling | Cat# 9251, RRID:AB_331659 |
| Anti-NF-KappaB p65, phospho (Ser536) | Cell Signaling | Cat# 3033, RRID:AB_331284 |
| Anti-RIPK1 | Cell Signaling | Cat# 3493, RRID:AB_2305314 |
| Anti-Aurora A | Cell Signaling | Cat# 3092, RRID:AB_2061342 |
| Anti-Bcl-xL | Cell Signaling | Cat# 2762, RRID:AB_10694844 |
| Anti-MCL-1 | Cell Signaling | Cat# 4572, RRID:AB_2281980 |
| Anti-Cyclin B1 | Santa Cruz | Cat# sc-752, RRID:AB_2072134 |
| Anti-CDC25A | Santa Cruz | Cat# sc-7389, RRID:AB_627226 |
| Anti-CDK1, phospho (Y15) | Abcam | CAT# ab133463 |
| Anti-Phospho KAP-1 (S824) | Bethyl | Cat# A300-767A, RRID:AB_669740 |
| Anti-NEK2 | BD Biosciences | Cat# 610593, RRID:AB_397933 |
| Anti-HMMR | Origene | Cat# TA307117, RRID:AB_10620394 |
| Anti-PLK1 | Millipore | Cat# 06-813, RRID:AB_310254 |
| Anti-E-cadherin | Cell Signaling | Cat# 3195, RRID:AB_2291471 |
| Anti-ZEB1 | Santa Cruz | Cat# sc-10572, RRID:AB_2273177 |
| Anti-Fibronectin | BD Biosciences | Cat# 610077, RRID:AB_2105706 |
| IRDye 680RD Goat anti-Mouse | LI-COR | Cat# 925-68070, RRID:AB_2651128 |
| IRDye 800CW Goat anti-Rabbit | LI-COR | Cat# 925-32211, RRID:AB_2651127 |
| HRP-conjugated swine anti-rabbit | DAKO/Agilent | Cat# P0217, RRID:AB_2728719 |
| HRP-conjugated rabbit anti-mouse | DAKO/Agilent | Cat# P0260, RRID:AB_2636929 |
| Alexa Fluor 647 goat anti-mouse | Thermo Fisher Scientific | Cat# A-21235, RRID:AB_2535804 |
| Alexa Fluor 488 goat anti-rabbit | Thermo Fisher Scientific | Cat# A-11008, RRID:AB_143165 |
| Alexa Fluor 488 goat anti-mouse | Thermo Fisher Scientific | Cat# A-11001; RRID:AB_2534069 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cisplatin | Accord Healthcare Ltd | Dutch drug database ZI# 15683354 |
| Doxycycline | Sigma Aldrich | Cat. D9891 |
| Thiazolyl Blue Tetrazolium Bromide (MTT) | Sigma Aldrich | Cat. M2128 |
| Halt Protease Inhibitor Cocktail | Thermo Fisher Sci. | Cat. 78425 |
| Halt Phosphatase Inhibitor Cocktail | Thermo Fisher Sci. | Cat. 78426 |
| Mitomycin C | Sigma Aldrich | Cat. M4287 |
| Propidium Iodide | Sigma Aldrich | Cat. P4170 |
| RNaseH | Thermo Fisher Sci. | Cat. EN0201 |
| Odyssey Blocking Buffer | LI-COR | Cat. 927-40000 |
| Critical Commercial Assays | ||
| RNAeasy Kit | QIAGEN | Cat. 74104 |
| HumanHT-12 v4 Expression BeadChip Kit | Illumina | N/A |
| Bradford Protein assay | Thermo Fisher Sci. | Cat. 23200 |
| Deposited Data | ||
| Raw mRNA expression data | This paper | GEO: GSE103115 |
| Growth rate inhibition (GR) metrics of breast cancer cell lines | (Hafner et al., 2016) | LINCS dataset #20268 |
| Experimental Models: Cell Lines | ||
| MDA-MB-231 | ATCC | Cat# CRL-12532, RRID:CVCL_0062 |
| MDA-MB-157 | ATCC | Cat# HTB-24, RRID:CVCL_0618 |
| MDA-MB-468 | ATCC | Cat# HTB-132, RRID:CVCL_0419 |
| BT549 | ATCC | Cat# HTB-122, RRID:CVCL_1092 |
| HCC38 | ATCC | Cat# CRL-2314, RRID:CVCL_1267 |
| HCC70 | ATCC | Cat# CRL-2315, RRID:CVCL_1270 |
| HCC1806 | ATCC | Cat# CRL-2335, RRID:CVCL_125 |
| HCC1937 | ATCC | Cat# CRL-2336, RRID:CVCL_0290 |
| SUM149PT | BIOIVT | RRID:CVCL_3422 |
| CAL120 | DSMZ | Cat# ACC-459, RRID:CVCL_1104 |
| Hs578T | ATCC | Cat# CRL-7849, RRID:CVCL_0332 |
| HEK293T | ATCC | Cat. CRL-3216; RRID:CVCL_0063 |
| Recombinant DNA | ||
| pLenti CMV/TO GFP-MDC1 (779-2) | Addgene | CAT# 26285, RRID:Addgene_26285 |
| Tet-pLKO-puro | Addgene | CAT# 21915, RRID:Addgene_21915 |
| pLKO.1 puro | Addgene | CAT# 8453 RRID:Addgene_8453 |
| pCMV-VSV-G | Addgene | CAT# 8454 RRID:Addgene_8454 |
| pCMV-dR8.2 dvpr | Addgene | CAT# 8455 RRID:Addgene_8455 |
| pLKO.1-MK2#1 | This paper | N/A |
| pLKO.1-MK2#2 | This paper | N/A |
| pLKO.1-G3BP2#1 | This paper | N/A |
| pLKO.1-G3BP2#2 | This paper | N/A |
| pLKO.1-SCR | Heijink et al., 2015 | N/A |
| pLKO.1-LUC | Heijink et al., 2019 | N/A |
| Software and Algorithms | ||
| GeneSpring GX software | Agilent Technologies | https://www.agilent.com/ |
| FlowJo software (version 10) | FlowJo | https://www.flowjo.com/ |
| MATLAB | MathWorks | https://www.mathworks.com/ |
| Odyssey | LI-COR | https://www.licor.com/ |
| GraphPad Prism 6 | GraphPad Software | https://www.graphpad.com/ |
| SIMCA-P | Umetrics | https://umetrics.com/ |
| SoftWorX | Applied Precision/GE Healthcare | N/A |
| growth rate inhibition (GR) calculator | (Hafner et al., 2016) | http://www.grcalculator.org/grtutorial/Home.html |
| Other | ||
| E-PAGE 8% Protein Gels, 48-well | Invitrogen | Cat. EP04808 |
| iBlot Transfer Stack, nitrocellulose | Invitrogen | Cat. IB301001 |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marcel A.T.M. van Vugt (m.vugt@umcg.nl). Plasmids generated in this study will be provided upon request.
Experimental Model and Subject Details
Human TNBC cell lines MDA-MB-468, MDA-MB-157, MDA-MB-231, CAL120 and Hs578T, and HEK293T human embryonic kidney cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM). Hs578T were further supplemented with 10 μg/ml insulin. HCC38, BT549, HCC70, HCC1806 and HCC1937 were grown in RPMI 1640 media. SUM-149PT cells were grown in Ham’s F12 media supplemented with 5 μg/ml insulin and 1 μg/ml hydrocortisone. All culture media were supplemented with 10% fetal calf serum (FCS), 100 units/ml penicillin, and 100 μg/ml streptomycin. The cell lines were cultured at 37°C in a humidified incubator supplied with 5% CO2. When indicated, cells were treated with cisplatin (Accord). If indicated, cells were treated with 1 μg/ml doxycycline.
Method Details
Viral Infection
To obtain stable MDC-GFP-expressing HCC38, BT549, MDA-MB-231 and MDA-MB-157 cell lines, cells were infected with pLenti CMV/TO GFP-MDC1 (779-2), which was a gift from Eric Campeau (Addgene plasmid # 26285). GFP-positive cells were subsequently sorted into polyclonal cell lines using a Moflo cell sorter.
shRNAs against MK2 and G3BP2 or a scrambled sequence (SCR) were cloned into pLKO.1 vectors using the Age1 and EcoR1 restriction sites. The hairpin targeting sequences that were used are: MK2#1 5′-CCAGCACTCGATTGTTGTAAA-3′, MK2#2 5′-AGAAAGAGAAGCATCCGAAAT-3′, G3BP2#1 5′-GACTCTGACAACCGTAGAATA-3′, G3BP2#2 5′-GTGATGATCGCAGGGATATTA-3′, SCR 5′-CAACAAGATGAAGAGCACCAA-3′ and luciferase (‘shLUC’), 5′-AAGAGCTGTTTCTGAGGAGCC-3′) (Heijink et al., 2019). Lentiviral particles were produced as described previously (Heijink et al., 2015). In brief, HEK293T packaging cells were transfected with 4 μg plasmid DNA in combination with the packaging plasmids VSV-G and ΔYPR. Virus-containing supernatant was harvested at 48 and 72 hours after transfection and filtered through a 0.45 μm syringe filter, and used to infect target cells in three consecutive 12 hour periods.
MTT Assays
Cells were seeded in 96-well plates at 3,000 to 4,000 cells per well and treated with indicated concentrations of cisplatin. After 72 hours of treatment, methyl thiazol tetrazolium (MTT) was added to a final concentration of 0.5 mg/ml and cells were incubated for four additional hours. Cells were then dissolved in DMSO and the produced formazan was measured at 520 nm with a Bio-Rad iMark spectrometer. Growth rate inhibition (GR) metrics were calculated using a custom MATLAB script (Data S1), based on a previously described R script (Hafner et al., 2016). Average growth rates for each cell line were calculated based on LINCS dataset #20268 (Hafner et al., 2016).
Immunofluorescence Microscopy
Cells were left untreated or were irradiated with 10Gy using a Cesium137 source (CIS international/IBL 637 irradiator, dose rate: 0.01083 Gy/s). Three hours later, cells were fixed in 2% paraformaldehyde with 0.1% Triton X-100 in PBS for 30 minutes at room temperature. Cells were permeabilized in 0.5% Triton X-100 in PBS for 10 minutes. To block nonspecific binding, cells were incubated with PBS containing 0.05% Tween-20 and 4% BSA (Fraction V) (PBS-Tween-BSA) for 1 hour. Cells were incubated overnight at 4°C with mouse anti-RAD51 (GeneTex, GTX70230, 1:400) in PBS-Tween-BSA. Cells were extensively washed and incubated for 45 minutes with Alexa488-conjugated secondary antibodies. Images were acquired on a Leica DM-6000RXA fluorescence microscope, equipped with Leica Application Suite software.
Low-Throughput Western Blotting
Knockdown efficiencies and the ubiquitination of FANCD2 were analyzed by western blotting. Cells were lysed in Mammalian Protein Extraction Reagent (MPER, Thermo Scientific), supplemented with protease and phosphatase inhibitor mixtures (Thermo Scientific). Forty micrograms of protein extract was used for separation by SDS/PAGE. Separated proteins were transferred to Polyvinylidene fluoride (PVDF) membranes and blocked in 5% (wt/vol) BSA in Tris-buffered saline (TBS), with 0.05% Tween20. Immunodetection was done with antibodies directed against FANCD2 (sc-20022, Santa Cruz), G3BP2 (A302-040A, Bethyl), phospho-MK2 (#3041, Cell Signaling), E-cadherin (#3195, Cell Signaling), ZEB1 (sc-10572, Santa Cruz), Fibronectin (610077, BD Biosciences) and β-actin (0869100, MP Biomedicals). Appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (DAKO) were used for enhanced chemiluminescence (Lumi-Light, Roche Diagnostics) on a Bio-Rad bioluminescence device, equipped with Quantity One/ChemiDoc XRS software (Bio-Rad). Knockdown efficiency was quantified using Adobe Photoshop. To this end, Biorad SCN files were exported into TIF files after which gray scale values were inverted. Mean intensity of a fixed-size rectangular marquee surrounding bands was assessed. Mean intensity was corrected for mean local background, and related to action intensity which was measured similarly. For G3BP2, shRNAs were designed to target all transcript variants. We assessed the bands at the indicated size, which represent the long isoform of G3BP2, encoded by mRNA variants 1 and 2, which only differ in their 5′ UTR. The G3BP2 antibody that we used (Bethyl-A302-040A) was raised against a 51 amino acid peptide of the long isoform, and detects a band with a predicted molecular weight of ∼54 kDa of the long G3BP2 isoform.
RNA Expression Analysis by Microarray Analysis
Total RNA was extracted from MDA-MB-231, MDA-MB-157, HCC38 and BT549 cells after treatment with 2 μM cisplatin for 0, 24 and 72 hours using the RNAeasy Kit (QIAGEN). The Illumina whole-genome expression array HumanHT-12 v4.0 (Illumina) was used and processed at the UMCG Medical Genetics department on a fee-for-service basis. Microarray data were obtained from two independent biological replicates per time point. Data were normalized using percentile shift normalization using GeneSpring GX software (Agilent Technologies). The cut-off for differential gene expression (DEG) was greater than 1.75-fold change and a p value less than 0.05. Expression data can be found in the GEO repository under the accession number GSE103115.
Live Cell Microscopy
GFP-MDC1-expressing TNBC cells were seeded in 8-chambered cover glass plates (Lab-Tek-II, Nunc). 16 hours after plating, cells were treated with 2 μM cisplatin for 24 hours. After media replacement, GFP and DIC images were obtained every 5 minutes over a period of 16 hours on a DeltaVision Elite microscope, equipped with a CoolSNAP HQ2 camera and a 20x immersion objective (U-APO 340, numerical aperture: 1.35) as described previously (Heijink et al., 2015). In the Z-plane, 6 images were acquired at 0.5-micron interval. Image analysis was done using SoftWorX software (Applied Precision/GE Healthcare). The number of MDC1-foci before, during and just after mitosis was scored.
Flow Cytometric Analysis
Cells were plated 24 hours prior to treatment with 2 or 20 μM cisplatin. Following treatment, cells were washed in PBS, trypsinized, and fixed in ice-cold 70% ethanol. Cells were stained with anti-phospho-Ser139-histone-H2A.X (#9718, Cell Signaling) or anti-phospho-histone-H3 (#9706, Cell Signaling) and subsequently stained with Alexa488-conjugated secondary antibodies, in combination with propidium iodide/RNase treatment. Samples were measured on a FACSCalibur (Becton Dickinson) and data were analyzed using the FlowJo software. The level of cell death was measured as fractionated DNA (sub-G1).
Cells were synchronized at the G1/S transition using a 24-hour incubation with 2.5 mM thymidine. After extensive washing, cells were released and harvested at indicated time points and subsequently fixed in ice-cold 70% ethanol.
High-Throughput Western Blotting
Cells were washed twice in PBS and lysed directly on the plate in Mammalian Protein Extraction Reagent (MPER, Thermo Scientific), supplemented with protease inhibitor and phosphatases inhibitor mixture (Thermo Scientific). Cell lysates were normalized for protein content using the BCA protein assay (Pierce). Lysates were run on 48-well 8% pre-cast poly-acrylamide gels (E-PAGE, Invitrogen) and transferred using the iBlot gel transfer device onto nitrocellulose membranes (Invitrogen). Blots were blocked in Odyssey Blocking Buffer (LiCOR Biosciences), incubated overnight with primary antibody at 4 degrees, stained with secondary antibodies conjugated to an infrared dye for 1 hour at RT, and visualized using an Odyssey scanner (LiCOR Biosciences).
Most antibodies used in this study were purchased from Cell Signaling, including those targeting γH2AX (#9718), CDC25C (#4688), p-CHK1-Ser345 (#2348), p-CHK2-Thr68 (#2197), p-MK2-Thr334 (#3041), p-ATR-Ser428 (#2853), p-AKT-Ser473 (#3787), p-ERK-Thr202/Tyr204 (#4376), p-P38-Thr180/Tyr182 (#4511), p-JNK-Thr183/Tyr185 (#9251), p-NFκB p65-Ser536 (#3033), RIPK1 (#3493), Aurora A (#3092), Bcl-xL (#2762) and MCL1 (#4572). Antibodies against CyclinB1 (sc-752), FANCD2 (sc-20022) and CDC25A (sc-7389) were purchased from Santa Cruz, antibodies against p-CDK1-Y15 (ab133463) and RPA32 (ab2175) from Abcam, antibodies against p-KAP1-ser824 (A300-767A) and G3BP2 (A302-040A) from Bethyl, antibodies against NEK2 (610593) from BD Bioscience, antibodies against HMMR (TA307117) from Origene, antibodies against β-actin (0869100) from MP Biomedicals and antibodies against PLK1 (06-813) were from Millipore.
For computational modeling, raw signals for each protein were quantified and background was subtracted using Li-COR Odyssey software. For each gel, loading differences were corrected through normalization to β-actin signals. For gel-to-gel normalization, samples were normalized using a reference sample, which was loaded on each gel. The reference sample for all signals was derived from MCF-7 cells, lysed at 3 hours after irradiation (10Gy). For time-course plots, signal averages were calculated from biological duplicate experiments. The data were then plotted as a fold change to the lowest value that was measured of that particular signal across all cell lines and treatments. Signaling plots for each individual metric were shaded in gray scale, with darkness reflecting signal strength (Figures 2C, 2E and S6A). The background of each individual box is colored according to signaling profile: ‘sustained increase’ in green, ‘late increase’ in red, ‘transient increase’ in yellow and ‘sustained decrease’ in blue. A signaling profile was assigned if the maximum signal of a time-course plot reached ≥ 33% of the maximum signal measured across all cell lines and treatments. Plots are colored as ‘sustained increase’ or ‘sustained decrease’ when signal strength increased or decreases for at least six subsequent time points, respectively. In case the fold change was higher than two-fold increase within the last three time points, plots are colored as ‘late increase’. Plots are colored as ‘transient increase’ when the difference between the minimum and maximum value was ≥ 2-fold and the maximum value is neighbored by ascending or descending values respectively.
Computational Data-Driven Modeling
Data-driven modeling and the application of partial-least-squares to biological data have been described in detail previously (Janes and Yaffe, 2006). In PLS modeling, the goal is to use X (signals) to predict Y (responses) and to describe their covariance. The data were divided into two matrices: E (a matrix containing the X variables) and F (a matrix containing the Y variables). In our study, the dimensions of E are 176 × 25 signals (2 treatments x 4 cell lines x biological duplicate measurements x 11 time points) and the dimensions of F are 176 × 6 (six cellular responses). Signal dynamics can be lost in PLS modeling due to uncoupling of temporal data. Therefore, we quantified several aspects of each signaling trajectory of which the approach was similar to that used previously by Janes et al. (2008). In the current study, all data were mean centered and unit variance scaled to non-dimensionalize the different measurements. PLSR analyses were performed using the program SIMCA-P (Umetrics), using the combined dataset as provided in Table S1. The PLS model was constructed using the following iterative formulas:
E represents the residual of the principal component, with score vector t, weight vector w, loading vector p, and T represents transpose. F represents the residuals of the dependent principal component, with score vector t, loading vector q, and b represents the inner relation between the independent and dependent principal components. Model predictions were made via cross-validation by leaving out a random sample of 1/6th of the observations and predicting that 1/6th from the remaining 5/6th of the data. The process was reiterated until each of the data were omitted and predicted. Model fitness was calculated using R2 and Q2, which were calculated as described previously (Gaudet et al., 2005). VIP scores were calculated as previously described (Janes et al., 2008). Metavariables that captured signal dynamics were calculated as 1) average, 2) area-under-the curve, 3) slope (between first time point and last time point of time frame), 4) maximum slope (highest slope observed in time frame), 5) fold change (maximum value divided by minimum value in time frame) and 6) dynamical range (maximum value minus lowest value in time frame). For Figure 6, the original training model was extended with signaling and response data of MDA-MB-468, HCC1806 and HCC1143 cells. Signaling data were included for five signals (pMK2, RPA, G3BP2, pKAP1 and BCL-xL), which were measured at 11 time points in response to treatment with 20 μM cisplatin (5 signals x 3 cell line models x 2 biological replicate measurements x 11 time points x1 treatment = 330 additional signal dimensions of ‘E’). In total, 4 responses (sub-G1, G1, S, G2/M) were measured (1 treatment x 3 cell lines x biological duplicate measurements x 11 time points x 4 responses = 264 additional phenotype dimensions of ‘F’).
Quantification and Statistical Analysis
Information about statistical details can be found in the figure legends. Differences with p value < 0.05 were considered significant, with ∗ representing p < 0.05, ∗∗ representing p < 0.01, ∗∗∗ representing p < 0.001 and ∗∗∗∗ representing p < 0.0001. Throughout the manuscript, graphs represent means, with error bars representing standard error of the means (SEM) or standard deviation (SD) as indicated. P values were calculated using two-tailed Student’s t test. For RNA expression analysis, the cut-off for differential gene expression (DEG) was a fold change greater than 1.75-fold, and a p value less than 0.05 as assessed using GeneSpring GX software.
Data and Code Availability
The mRNA expression data generated during this study are available at the GEO repository under the accession number GSE103115.
Acknowledgments
This work was supported by the Netherlands Organization for Scientific Research (NWO-VIDI 916-76062), the European Research Council (ERC CoG 682421), and the Dutch Cancer Society (RUG 2011-5093) (to M.A.T.M.v.V.); the René Vogels Foundation and the De Boer-Merema Foundation (to A.M.H.); the American Cancer Society (RSG-17-011-01) and the NIGMS/NIH (R01GM127559) (to M.J.L.); and the Translational Cancer Biology Training Grant (T32-CA130807) (to R.R.). We thank Floris Foijer, Arne Lindqvist, and Michael B. Yaffe for comments and members of the van Vugt and Lee laboratories for feedback.
Author Contributions
A.M.H. and M.A.T.M.v.V. conceived the study. A.M.H., M.E., M.E.H., and R.R. performed experiments. A.M.H., M.E., M.E.H., R.R., Y.P.K., M.J.L., and M.A.T.M.v.V. analyzed data. M.J.L. supervised modeling studies. A.M.H., M.J.L., and M.A.T.M.v.V. wrote the manuscript. E.G.E.d.V. advised on the project. All authors edited the manuscript and approved the final version before submission.
Declaration of Interests
M.A.T.M.v.V. has acted on the Scientific Advisory Board of RepareTx.
Published: August 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.070.
Contributor Information
Michael J. Lee, Email: michael.lee@umassmed.edu.
Marcel A.T.M. van Vugt, Email: m.vugt@umcg.nl.
Supplemental Information
All signaling and response measurements were performed in duplicates (A and B), measured at two cisplatin concentrations (2 or 20 μM) and measured at different time points after treatment in 7 TNBC cell lines. Orange columns indicate the different conditions, green columns list all measured signals, and yellow columns list response measurements.
Genes with contrasting or similar expression changes after cisplatin treatment (2 μM, 72 hours), in sensitive (‘sens’) versus resistant (‘res’) cell lines are indicated. Genes highlighted in yellow were selected for follow-up analysis.
Custom MATLAB script in text form to calculate growth rate inhibition (GR) metrics.
References
- Bhattacharyya A., Ear U.S., Koller B.H., Weichselbaum R.R., Bishop D.K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 2000;275:23899–23903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- Brozovic A., Damrot J., Tsaryk R., Helbig L., Nikolova T., Hartig C., Osmak M., Roos W.P., Kaina B., Fritz G. Cisplatin sensitivity is related to late DNA damage processing and checkpoint control rather than to the early DNA damage response. Mutat. Res. 2009;670:32–41. doi: 10.1016/j.mrfmmm.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Byrski T., Gronwald J., Huzarski T., Grzybowska E., Budryk M., Stawicka M., Mierzwa T., Szwiec M., Wisniowski R., Siolek M. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J. Clin. Oncol. 2010;28:375–379. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- Cardoso F., Costa A., Senkus E., Aapro M., André F., Barrios C.H., Bergh J., Bhattacharyya G., Biganzoli L., Cardoso M.J. 3rd ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 3) Ann. Oncol. 2017;28:3111. doi: 10.1093/annonc/mdx036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A., Elledge S.J. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costelloe T., Fitzgerald J., Murphy N.J., Flaus A., Lowndes N.F. Chromatin modulation and the DNA damage response. Exp. Cell Res. 2006;312:2677–2686. doi: 10.1016/j.yexcr.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Curtis C., Shah S.P., Chin S.-F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y., METABRIC Group The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreaden E.C., Kong Y.W., Quadir M.A., Correa S., Suárez-López L., Barberio A.E., Hwang M.K., Shi A.C., Oberlton B., Gallagher P.N. RNA-Peptide nanoplexes drug DNA damage pathways in high-grade serous ovarian tumors. Bioeng. Transl. Med. 2018;3:26–36. doi: 10.1002/btm2.10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher L., Cerniglia G.J., Nigg E.A., Yend T.J., Muschel R.J. Inhibition of centrosome separation after DNA damage: a role for Nek2. Radiat. Res. 2004;162:128–135. doi: 10.1667/rr3211. [DOI] [PubMed] [Google Scholar]
- Foulkes W.D., Smith I.E., Reis-Filho J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- Gadhikar M.A., Sciuto M.R., Alves M.V.O., Pickering C.R., Osman A.A., Neskey D.M., Zhao M., Fitzgerald A.L., Myers J.N., Frederick M.J. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol. Cancer Ther. 2013;12:1860–1873. doi: 10.1158/1535-7163.MCT-13-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet S., Janes K.A., Albeck J.G., Pace E.A., Lauffenburger D.A., Sorger P.K. A compendium of signals and responses triggered by prodeath and prosurvival cytokines. Mol. Cell. Proteomics. 2005;4:1569–1590. doi: 10.1074/mcp.M500158-MCP200. [DOI] [PubMed] [Google Scholar]
- Geladi P., Kowalski B.R. Partial least-squares regression: a tutorial. Anal. Chim. Acta. 1986;185:1–17. [Google Scholar]
- Gong G. Cross-Validation, the Jackknife, and the Bootstrap: Excess Error Estimation in Forward Logistic Regression. J. Am. Stat. Assoc. 1986;81:108–113. [Google Scholar]
- Gupta N., Badeaux M., Liu Y., Naxerova K., Sgroi D., Munn L.L., Jain R.K., Garkavtsev I. Stress granule-associated protein G3BP2 regulates breast tumor initiation. Proc. Natl. Acad. Sci. USA. 2017;114:1033–1038. doi: 10.1073/pnas.1525387114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M., Niepel M., Chung M., Sorger P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods. 2016;13:521–527. doi: 10.1038/nmeth.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijink A.M., Blomen V.A., Bisteau X., Degener F., Matsushita F.Y., Kaldis P., Foijer F., van Vugt M.A. A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proc. Natl. Acad. Sci. USA. 2015;112:15160–15165. doi: 10.1073/pnas.1505283112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijink A.M., Talens F., Jae L.T., van Gijn S.E., Fehrmann R.S.N., Brummelkamp T.R., van Vugt M.A.T.M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019;10:100. doi: 10.1038/s41467-018-07927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H., Iwasawa Y., Okada M., Arai T., Nishibata T., Kobayashi M., Kimura T., Kaneko N., Ohtani J., Yamanaka K. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- Isabelle M., Gagné J.-P., Gallouzi I.-E., Poirier G.G. Quantitative proteomics and dynamic imaging reveal that G3BP-mediated stress granule assembly is poly(ADP-ribose)-dependent following exposure to MNNG-induced DNA alkylation. J. Cell Sci. 2012;125:4555–4566. doi: 10.1242/jcs.106963. [DOI] [PubMed] [Google Scholar]
- Jackson S.P., Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes K.A., Yaffe M.B. Data-driven modelling of signal-transduction networks. Nat. Rev. Mol. Cell Biol. 2006;7:820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- Janes K.A., Reinhardt H.C., Yaffe M.B. Cytokine-induced signaling networks prioritize dynamic range over signal strength. Cell. 2008;135:343–354. doi: 10.1016/j.cell.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., D’Andrea A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012;26:1393–1408. doi: 10.1101/gad.195248.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köpper F., Bierwirth C., Schön M., Kunze M., Elvers I., Kranz D., Saini P., Menon M.B., Walter D., Sørensen C.S. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proc. Natl. Acad. Sci. USA. 2013;110:16856–16861. doi: 10.1073/pnas.1304355110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.J., Ye A.S., Gardino A.K., Heijink A.M., Sorger P.K., MacBeath G., Yaffe M.B. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012;149:780–794. doi: 10.1016/j.cell.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann B.D., Bauer J.A., Chen X., Sanders M.E., Chakravarthy A.B., Shyr Y., Pietenpol J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombaerts M., van Wezel T., Philippo K., Dierssen J.W.F., Zimmerman R.M.E., Oosting J., van Eijk R., Eilers P.H., van de Water B., Cornelisse C.J., Cleton-Jansen A.M. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br. J. Cancer. 2006;94:661–671. doi: 10.1038/sj.bjc.6602996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manke I.A., Nguyen A., Lim D., Stewart M.Q., Elia A.E.H., Yaffe M.B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell. 2005;17:37–48. doi: 10.1016/j.molcel.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Miller-Jensen K., Janes K.A., Brugge J.S., Lauffenburger D.A. Common effector processing mediates cell-specific responses to stimuli. Nature. 2007;448:604–608. doi: 10.1038/nature06001. [DOI] [PubMed] [Google Scholar]
- Morandell S., Reinhardt H.C., Cannell I.G., Kim J.S., Ruf D.M., Mitra T., Couvillon A.D., Jacks T., Yaffe M.B. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013;5:868–877. doi: 10.1016/j.celrep.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez R.P., Lewis L.D., Beelen A.P., Olszanski A.J., Johnston N., Rhodes C.H., Beaulieu B., Ernstoff M.S., Eastman A. Modulation of cell cycle progression in human tumors: a pharmacokinetic and tumor molecular pharmacodynamic study of cisplatin plus the Chk1 inhibitor UCN-01 (NSC 638850) Clin. Cancer Res. 2006;12:7079–7085. doi: 10.1158/1078-0432.CCR-06-0197. [DOI] [PubMed] [Google Scholar]
- Pouliot L.M., Chen Y.-C., Bai J., Guha R., Martin S.E., Gottesman M.M., Hall M.D. Cisplatin sensitivity mediated by WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer Res. 2012;72:5945–5955. doi: 10.1158/0008-5472.CAN-12-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaper P.M., Griffiths M.R., Long J.M., Charrier J.-D., Maccormick S., Charlton P.A., Golec J.M.C., Pollard J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011;7:428–430. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- Reinhardt H.C., Hasskamp P., Schmedding I., Morandell S., van Vugt M.A.T.M., Wang X., Linding R., Ong S.-E., Weaver D., Carr S.A., Yaffe M.B. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell. 2010;40:34–49. doi: 10.1016/j.molcel.2010.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzier R., Perou C.M., Symmans W.F., Ibrahim N., Cristofanilli M., Anderson K., Hess K.R., Stec J., Ayers M., Wagner P. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin. Cancer Res. 2005;11:5678–5685. doi: 10.1158/1078-0432.CCR-04-2421. [DOI] [PubMed] [Google Scholar]
- Sangster-Guity N., Conrad B.H., Papadopoulos N., Bunz F. ATR mediates cisplatin resistance in a p53 genotype-specific manner. Oncogene. 2011;30:2526–2533. doi: 10.1038/onc.2010.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuck S.C., Short E.A., Turchi J.J. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res. 2008;18:64–72. doi: 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddik Z.H. Biochemical and molecular mechanisms of cisplatin resistance. Cancer Treat. Res. 2002;112:263–284. doi: 10.1007/978-1-4615-1173-1_13. [DOI] [PubMed] [Google Scholar]
- Silver D.P., Richardson A.L., Eklund A.C., Wang Z.C., Szallasi Z., Li Q., Juul N., Leong C.-O., Calogrias D., Buraimoh A. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010;28:1145–1153. doi: 10.1200/JCO.2009.22.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohr S., Engeland K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle. 2008;7:3448–3460. doi: 10.4161/cc.7.21.7014. [DOI] [PubMed] [Google Scholar]
- Stucki M., Clapperton J.A., Mohammad D., Yaffe M.B., Smerdon S.J., Jackson S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Taniguchi T., Tischkowitz M., Ameziane N., Hodgson S.V., Mathew C.G., Joenje H., Mok S.C., D’Andrea A.D. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003;9:568–574. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- Tentner A.R., Lee M.J., Ostheimer G.J., Samson L.D., Lauffenburger D.A., Yaffe M.B. Combined experimental and computational analysis of DNA damage signaling reveals context-dependent roles for Erk in apoptosis and G1/S arrest after genotoxic stress. Mol. Syst. Biol. 2012;8:568. doi: 10.1038/msb.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Mosel A.J., Oakley G.G., Peng A. Deficient DNA damage signaling leads to chemoresistance to cisplatin in oral cancer. Mol. Cancer Ther. 2012;11:2401–2409. doi: 10.1158/1535-7163.MCT-12-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S.C., Fattet L., Tsai J.H., Guo Y., Pai V.H., Majeski H.E., Chen A.C., Sah R.L., Taylor S.S., Engler A.J., Yang J. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015;17:678–688. doi: 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All signaling and response measurements were performed in duplicates (A and B), measured at two cisplatin concentrations (2 or 20 μM) and measured at different time points after treatment in 7 TNBC cell lines. Orange columns indicate the different conditions, green columns list all measured signals, and yellow columns list response measurements.
Genes with contrasting or similar expression changes after cisplatin treatment (2 μM, 72 hours), in sensitive (‘sens’) versus resistant (‘res’) cell lines are indicated. Genes highlighted in yellow were selected for follow-up analysis.
Custom MATLAB script in text form to calculate growth rate inhibition (GR) metrics.
Data Availability Statement
The mRNA expression data generated during this study are available at the GEO repository under the accession number GSE103115.