

Summary
Colorectal cancer (CRC) is associated with metabolic and redox perturbation. The mitochondrial transporter uncoupling protein 2 (UCP2) controls cell proliferation in vitro through the modulation of cellular metabolism, but the underlying mechanism in tumors in vivo remains unexplored. Using murine intestinal cancer models and CRC patient samples, we find higher UCP2 protein levels in tumors compared to their non-tumoral counterparts. We reveal the tumor-suppressive role of UCP2 as its deletion enhances colon and small intestinal tumorigenesis in AOM/DSS-treated and ApcMin/+ mice, respectively, and correlates with poor survival in the latter model. Mechanistically, UCP2 loss increases levels of oxidized glutathione and proteins in tumors. UCP2 deficiency alters glycolytic pathways while promoting phospholipid synthesis, thereby limiting the availability of NADPH for buffering oxidative stress. We show that UCP2 loss renders colon cells more prone to malignant transformation through metabolic reprogramming and perturbation of redox homeostasis and could favor worse outcomes in CRC.
Keywords: uncoupling protein 2, mitochondrial carrier, tumor metabolic reprogramming, oxidative stress, colorectal cancer, lipid synthesis, mitochondria, tumor metabolism
Graphical Abstract
Highlights
-
•
UCP2 protein expression, but not mRNA, is increased in CRC in both mice and humans
-
•
UCP2 loss promotes AOM/DSS-induced CAC and ApcMin-dependent intestinal cancer
-
•
UCP2 loss-induced oxidative stress contributes to increased colon tumorigenesis
-
•
UCP2 deficiency drives an imbalance between lipid metabolism and NADPH homeostasis
Aguilar et al. show that the inhibition of mitochondrial transporter UCP2 increases susceptibility to colon and intestinal tumorigenesis and correlates with poor prognosis. The underlying mechanisms involve deregulation of redox homeostasis through metabolic rewiring. Thus, UCP2 plays a key role in CRC, and its loss is potentially associated with worse outcomes.
Introduction
Colorectal cancer (CRC) is a major public health problem and lethal disease. Globally, CRC is the third most commonly diagnosed cancer in males and the second in females, being the fourth cause of cancer death. The identification of proteins and cellular processes involved in the pathogenesis of CRC is therefore crucial to develop effective targeted treatments that may benefit patient survival rates. Cancer development involves major alterations of cellular metabolism, including increased demands for energy (ATP), reducing equivalents (NADPH) and cellular building blocks to sustain uncontrolled cell proliferation (Vander Heiden and DeBerardinis, 2017, Ward and Thompson, 2012). Thus, cellular metabolism has emerged as a promising Achilles’ heel of different cancer types, including CRC (Kawada et al., 2017), and numerous strategies have been developed to investigate and target tumor metabolism (Marín de Mas et al., 2014). Although several studies have shown that CRC is a cancer type that can rely heavily on glycolysis (Lehuédé et al., 2016), colon cancer cells can adapt and survive under low glucose conditions (Miyo et al., 2016), and a metabolic shift from glycolysis to oxidative phosphorylation (OXPHOS) can lead to acquired therapeutic resistance (Vellinga et al., 2015). Mitochondria produces the majority of the cellular reactive oxygen species (ROS) through OXPHOS, which can significantly affect tumorigenesis through increased nuclear genome instability or by acting as signaling molecules that affect proliferation or differentiation of cells involved in polyp formation (Sabharwal and Schumacker, 2014, Sullivan and Chandel, 2014). A significant increase in oxidative stress markers (e.g., 8-oxodG) has been observed in CRC patients, suggesting a potential role for ROS in colon tumorigenesis (Perše, 2013). These observations point to mitochondria as a central metabolic organelle required for tumor initiation and progression (Vyas et al., 2016).
Uncoupling protein 2 (UCP2) belongs to a family of mitochondrial transporters related to the well-known UCP1 (Enerbäck et al., 1997). Although UCP2 mRNA has a broad tissue distribution, UCP2 protein is only found in specific tissues due to a tight translational regulation (Donadelli et al., 2014, Pecqueur et al., 2001). We previously showed that UCP2 protein expression is restricted to the spleen, lungs, stomach, intestine, white adipose tissue, and immune cells (Arsenijevic et al., 2000, Pecqueur et al., 2001). Besides its controversial uncoupling activity, we recently demonstrated the ability of UCP2 to reprogram cellular metabolism in vitro using different cancer cell types (Esteves et al., 2014). The capacity of UCP2 to modulate glucose and fatty acid oxidation metabolism is directly associated with cell proliferation in various models (Esteves et al., 2014, Pecqueur et al., 2008). In addition, functional characterization through biochemical assays has shown that UCP2 is involved in the transport of four-carbon mitochondrial substrates (e.g., malate, oxaloacetate, aspartate) outside the mitochondria (Vozza et al., 2014), which allows the regulation of mitochondrial substrate oxidation and ROS levels independently of mitochondrial uncoupling (Bouillaud et al., 2016, Pecqueur et al., 2009). Based on these data, UCP2 appears as an excellent candidate for linking metabolism and redox balance, which are intimately linked to the development of CRC.
Some studies previously suggested a link between UCP2 and tumorigenesis since ROS detoxification by UCP2 may confer chemoresistance to cancer cells (Dando et al., 2013, Derdak et al., 2008, Mailloux et al., 2010, Pons et al., 2015). However, these studies were mostly based on RNA-interfering or pharmacological (e.g., genipin) strategies in vitro and did not consider the role of UCP2 within the in vivo tumor complexity. Only a few attempts have been made to show the correlation between UCP2 expression and human colon adenocarcinoma (Horimoto et al., 2004), but the use of non-specific UCP2 antibodies and the lack of proper negative controls limited the interpretation of these results.
We have previously shown that UCP2 overexpression decreases cell proliferation in cancer cell lines and this correlates with reduced tumor growth in a mouse xenograft model (Esteves et al., 2014). In the present study, we have examined the contribution of UCP2 deletion to colon tumorigenesis. We show that UCP2 loss has a tumor-promoting effect both in carcinogen-induced CRC and a mouse model with a genetic predisposition to intestinal tumorigenesis. We decipher the metabolic and redox mechanisms triggered by UCP2 deficiency in colon tumors, which involve the promotion of oxidative stress through the enhancement of fatty acid biosynthesis at the expense of the availability of NADPH. Our study demonstrates that loss of the mitochondrial transporter UCP2 renders cells more prone to malignant transformation and may confer a worse overall outcome in CRC.
Results
UCP2 Protein Expression Is Increased in Established Colon Tumors
As a first step, we wished to address whether our homemade UCP2-605 antibody, which was made in-house and validated for detection of the mitochondrial transporter UCP2 by western blot (Pecqueur et al., 2001), was suitable for immunohistochemistry (IHC) and immunofluorescence (IF) staining. We compared it with other commercial UCP2 antibodies (sc6525 [C-20], Santa Cruz Biotechnology; sc6526 [N-19], Santa Cruz Biotechnology; and ab203244, Abcam) that have been previously described to detect this mitochondrial transporter in tissues (Diao et al., 2008, Horimoto et al., 2004, Zhang et al., 2017). By performing both IHC and IF assays, we observed non-specific binding with all of the antibodies tested in Ucp2−/− colon tissues (Figures S1A and S1B). For western blot analysis, the commercial UCP2 antibodies C-20 and ab203244 failed to specifically detect this mitochondrial transporter (Figure S1C). In fact, the UCP2-specific 33 kDa band was detected only by UCP2-605 and N-19 antibodies, although the UCP2-605 antibody showed a better signal-to-noise ratio in colon mitochondria samples (Figure S1C). After confirming the specificity of our antibody, we next evaluated UCP2 expression levels in colon and small intestinal tumors and paired non-tumoral tissue from two different murine cancer models. In the first model, which closely mimics the location and pathologic appearance of human colitis-associated cancer (CAC) (De Robertis et al., 2011), colon tumorigenesis was chemically induced with a single injection of azoxymethane (AOM) followed by three 5-day cycles of dextran sodium sulfate (DSS) to create a chronic inflammatory state (Figure 1A). The ApcMin/+ model is a genetic murine model that develops spontaneous intestinal cancer upon sporadic loss of heterozygosity (LOH) of the wild-type allele of Apc (Luongo et al., 1994).
Figure 1.

UCP2 Protein Is Overexpressed in Established Colon and Intestinal Tumors
(A) Protocol for AOM/DSS-induced colorectal tumorigenesis in C57BL6/J mice.
(B) UCP2 mRNA expression measured by qPCR in colonic non-tumoral (NT) tissue and colon tumors (T) from AOM/DSS-treated Ucp2+/+ mice. Values are expressed relative to NT (n = 6).
(C) Immunoblot of UCP2 in whole mitochondrial extracts from duodenum (Duo), jejunum (Jej), ileum (Ile), cecum (Cec), and colon. Lung is shown as a positive control for UCP2 expression. Ucp2−/− samples were used as negative controls (n = 3).
(D) Western blot analysis of UCP2 expression in colons of non-treated Ucp2+/+ mice and in colonic NT tissue of AOM/DSS-treated Ucp2+/+ mice. Data represent means ± SEMs (n = 3).
(E) Immunoblot of UCP2 in mitochondrial extracts from colon NT tissue and T from AOM/DSS-treated Ucp2+/+ mice (n = 8).
(F) Immunoblot of UCP2 expression in mitochondrial extracts from jejunum NT tissue and T from ApcMin/+ mice (n = 4).
(G) UCP2 mRNA levels in human T from stages II (n = 4) and III (n = 6) and matched normal mucosa (N) from colorectal cancer (CRC) patients. Values are expressed relative to N.
(H) Immunoblot of UCP2 in six pairs of T samples and matched N from CRC patients (n = 6).
In all of the panels, data are the means ± SEMs. See also Figure S1 and Table S1.
We found similar relative UCP2 mRNA levels in non-tumoral (NT) colonic tissue and colon tumors from AOM/DSS-treated mice (Figure 1B). Under non-pathological conditions, the UCP2 protein can be detected along the entire small and large intestines (Figure 1C). Whereas no significant difference was detected between colons from untreated mice and NT colon tissue from AOM/DSS-treated mice (Figure 1D), UCP2 protein expression was three times higher in colon tumors compared to their NT counterparts (Figure 1E). Similar results were obtained in intestinal tumors and NT samples from ApcMin/+ mice (Figure 1F).
In accordance with our observations in mouse models, analysis of human CRC samples showed no difference in UCP2 mRNA expression between stage II and stage III colon tumors and their normal mucosa counterparts (Figure 1G). Analysis using the public Oncomine clinical cohort database (https://www.oncomine.com) also revealed no clear correlation in UCP2 mRNA expression between normal and tumor colorectal tissues (Table S1). However, UCP2 protein expression was significantly increased in colon tumors from human CRC patients compared to their matched normal mucosa (Figure 1H). These results show that UCP2 protein, not RNA, is significantly upregulated within the local intestinal tumor microenvironment, suggesting a potential role of the mitochondrial transporter UCP2 in the pathogenesis of CRC.
Targeting UCP2 Expression Increases Initiation of Colorectal Tumorigenesis In Vivo
UCP2 expression inversely correlates with cell proliferation (Esteves et al., 2014, Pecqueur et al., 2008). As UCP2 overexpression in mouse and human cancer cell lines decreases cell proliferation in vitro (Esteves et al., 2014), we used UCP2 knockout mouse models to investigate the contribution of UCP2 deletion to malignant transformation in vivo of the small intestine and colon. We observed that the loss of this mitochondrial transporter did not change the abundance of intestinal stem cells that may be associated with CRC initiation (Figure S2A) and the proliferative capacity of intestinal epithelial cells before and after malignant transformation (Figures S2B–S2D). We then investigated tumor development using the two different murine models described above. AOM/DSS-treated mice were sacrificed 15 weeks after starting the treatment, and colon tumors were harvested. UCP2 deletion significantly increased the total number of colon tumors (Figure 2A), indicating a higher susceptibility of Ucp2−/− mice to develop tumors following AOM/DSS treatment. Histologically, we found no difference in the grade of dysplasia between both genotypes (Figure 2B). Comparison of the expression levels of cleaved caspase-3 between Ucp2+/+ and Ucp2−/− colon tumors showed no differences at the level of cell death (Figure S2E). Ucp2−/− mice displayed a higher number of smaller colon tumors (<4 mm) than Ucp2+/+ mice (Figure 2C). To further characterize the impact of UCP2 invalidation on intestinal tumorigenesis, we generated ApcMin/+ Ucp2−/− mice. Concomitant invalidation of UCP2 and Apc LOH did not change significantly the total number of tumors in the small intestine and colon (Figure 2D), but it did significantly increase the number of jejunum tumors (Figure 2E), which also presented reduced size (<2 mm) compared to those tumors from Ucp2+/+ mice (Figure 2F). UCP2 invalidation in ApcMin/+ mice significantly reduced the survival rate (Figure 2G). Our results indicate that UCP2 loss plays a major role in promoting colon tumorigenesis independently of the presence of a pro-inflammatory stimulus, and it could represent a potential indicator of worse outcomes when accompanied by a predisposition to develop intestinal cancer.
Figure 2.

Targeting UCP2 Expression Increases Colorectal Tumorigenesis in AOM/DSS-Treated Mice
(A) Total number of colon tumors in AOM/DSS-treated mice quantified by blue methylene staining. Data indicate means ± SEMs (Ucp2+/+ n = 8, Ucp2−/− n = 13).
(B) Representative H&E-stained sections from colon tumors from AOM/DSS-treated Ucp2+/+ and Ucp2−/− mice. Scale bar, 200 μm.
(C) Size of individual colon tumors in AOM/DSS-treated mice, presented as means ± SEMs (Ucp2+/+ n = 8, Ucp2−/− n = 13).
(D) Total number of tumors along the small intestine and colon in Ucp2+/+ApcMin/+ and Ucp2−/−ApcMin/+ mice. Data represent means ± SEMs (n = 7).
(E) Number of tumors in the duodenum, jejunum, ileum, and colon in Ucp2+/+ApcMin/+ and Ucp2−/−ApcMin/+ mice. Data represent means ± SEMs (n = 7).
(F) Size of individual jejunum tumors from ApcMin/+ mice, presented as means ± SEMs (n = 7).
(G) Kaplan-Meier survival curve. Each survival curve consists of data from 11 mice.
See also Figure S2.
Differential Expression of Metabolic Genes Triggered by UCP2 Loss in Colon Tumors
Our data suggest that the molecular mechanism beyond tumor initiation upon the loss of UCP2 does not involve dysregulation of cell proliferation and cell death in the colon (Figure S2). Since AOM/DSS is a powerful and reproducible model of CAC, we performed a transcriptomic analysis on Ucp2+/+ and Ucp2−/− colon tumors from AOM/DSS-treated mice to identify cellular pathways altered in established colon tumors lacking UCP2 expression. Among 312 genes differentially expressed (fold change >1.2 and ANOVA P < 0.05), 182 and 130 genes were up- and downregulated, respectively, in Ucp2−/− tumors compared to Ucp2+/+ tumors (Table S2). As shown in Figure 3A, unsupervised hierarchical clustering revealed a distinct gene expression pattern between the two tumor types. Gene Ontology analysis using the bioinformatic PANTHER tool revealed a significant fold enrichment of genes involved in mitochondrial function in Ucp2−/− colon tumors (Figure 3B). The metabolic process was identified as one of the main biological processes altered upon UCP2 deficiency, with 19.4% of the total genes differentially expressed belonging to this category (Figure 3C). Within this metabolic process, the genes involved in primary cell metabolism, including carbohydrate, lipid, nucleobase-containing compound, and protein metabolism, were predominant (Figure 3D). The core analysis with Ingenuity Pathway Analysis (IPA) predicted downstream effects on several molecular and cellular functions, including key metabolic pathways such as lipid, amino acid, and carbohydrate metabolism, as well as reactions involved in redox balance such as free radical scavenging (Figure 3E).
Figure 3.

UCP2 Loss in AOM/DSS-Treated Mice Leads to a Differential Metabolic Gene Expression in Colon Tumors
(A) Heatmap of the log2 relative expression values for each gene in each tumor (T) type plotted in red–blue color scale, with red indicating high gene expression and blue indicating low gene expression (n = 5).
(B) Bar plot ranking of the top 10 Gene Ontology (GO) cellular components based on fold enrichment.
(C) Pie chart of the biological processes associated with the upregulated genes after UCP2 loss in colon tumors. Biological categories were obtained using GO annotations from the PANTHER classification system.
(D) GO analysis of the total number of differentially expressed genes involved in metabolic processes.
(E) Gene function analysis using the software Ingenuity Pathway Analysis (IPA). Graph shows top molecular and cellular functions altered in Ucp2−/− tumors compared to Ucp2+/+ tumors. Bars above the line are statistically significant (p < 0.05) (n = 5).
See also Table S2.
UCP2 Loss in Colon Tumors Decreases Antioxidant Defenses and Increases Oxidative Stress Levels
Results obtained from the microarray analysis prompted us to further explore how UCP2 invalidation affects tumor redox state and cellular metabolism in vivo. UCP2 levels are inversely correlated with oxidative stress in several in vitro models (Arsenijevic et al., 2000, Pecqueur et al., 2009). However, it remains unknown whether there is a correlation between UCP2 expression and redox imbalance driving in vivo tumorigenesis. Although we did not detect different levels of the end product of lipid peroxidation 4-hydroxy-2-nonenal (4-HNE) (Figure 4A), analysis of the protein oxidative damage revealed increased protein carbonylation levels in Ucp2−/− colon tumors (Figure 4B). Of note, there were no significant differences in NT tissues between Ucp2+/+ (n = 6) and Ucp2−/− (n = 9) protein carbonylation levels (11.82 ± 1.96 versus 14.77 ± 1.01, p = 0.1122), supporting that the induction of oxidative stress by UCP2 deficiency was only appreciated at the tumor level. The balance between reduced glutathione (GSH) and oxidized GSH (GSSG) has long been recognized as an indicator of oxidative stress. Although the total GSH content was not affected, the proportion of oxidized GSSG significantly increased, confirming a pro-oxidant status of Ucp2−/− colonic tumors (Figures 4C–4E). Similar to the protein oxidation results, loss of UCP2 did not change the GSH:GSSG ratio in NT colon tissues between Ucp2+/+ (n = 4) and Ucp2−/− (n = 5) (6.65 ± 0.69 versus 5.11 ± 0.54, p = 0.1165). In addition, NADPH is an essential cofactor for maintaining the cellular redox balance. UCP2 loss led to a dramatic decrease in intracellular NADPH content, without altering NADP+ levels (Figures 4F and 4G), resulting in a significant reduction in the NADPH:NADP+ ratio (Figure 4H). These findings are in accordance with the GSH results, as NADPH is an essential cofactor in keeping GSH in the reduced state. Consequently, UCP2 loss in vivo leads to specific enhanced tumor oxidative damage that may contribute to the increased tumor initiation observed in Ucp2−/− AOM/DSS-treated mice. To investigate this, mice were fed with a diet containing 0.7% (w/w) antioxidant butylated hydroxyanisole (BHA) and subsequently subjected to AOM/DSS-induced colorectal tumorigenesis. While BHA-fed Ucp2−/− mice displayed a similar total number of tumors as BHA-fed Ucp2+/+ mice in the colon (Figure 4I), they exhibited a significantly reduced number of small colon tumors (<4 mm) compared to BHA-fed Ucp2+/+ mice (Figure 4J). These data indicate that antioxidant treatment prevents the increased colon tumor development triggered by UCP2 loss.
Figure 4.

The Absence of UCP2 in Colon Tumors Promotes Glutathione Oxidation and Protein Carbonylation and Depletes NADPH Levels
(A) Western blot analysis for 4-hydroxy-2-nonenal (4-HNE)-modified proteins in colon tumor (T) samples. Data indicate means ± SEMs (n = 5).
(B) Representative immunoblot of protein oxidation levels (total carbonyl groups with dinitrophenyl [DNP]) in colon T samples. Data indicate means ± SEMs (n = 6).
(C–E) Total glutathione (C), oxidized glutathione (GSSG) (D), and ratio of reduced glutathione (GSH) versus GSSG (E). Total GSH and GSSG were normalized by protein content. Data indicate means ± SEMs (Ucp2+/+ T n = 7, Ucp2−/− T n = 9).
(F–H) NADPH (F) and NADP+ (G) levels normalized to protein content and ratio of reduced NADPH versus oxidized NADP+ (H). Data indicate means ± SEMs (n = 6).
(I) Total number of colon tumors in mice fed with BHA and AOM/DSS treated. Data indicate means ± SEMs (Ucp2+/+ n = 6, Ucp2−/− n = 7).
(J) Size of individual colon tumors in mice fed with BHA and AOM/DSS treated, presented as means ± SEMs (Ucp2+/+ n = 6, Ucp2−/− n = 7).
UCP2 Loss in Colon Tumors Affects Glycolysis with No Effect on Oxidative Metabolism and Mitochondrial Respiration
Based on the transcriptomic analysis and the mitochondrial subcellular localization of UCP2, we determined whether the increased oxidative stress levels were associated with an enhanced mitochondrial oxidative metabolism. We previously reported that despite UCP2’s sharing homology with the uncoupling protein UCP1, it does not show uncoupling activity (Couplan et al., 2002, Esteves et al., 2014). Accordingly, we observed that UCP2 invalidation in colon tumors affected neither the proton leak rate nor the mitochondrial oxygen consumption rate in the presence of malate, pyruvate, and ADP (Figure 5A). No difference in citrate synthase (CS) activity (i.e., mitochondrial content) was detected between Ucp2+/+ and Ucp2−/− tumors (Figure 5B). We further characterized the mitochondrial respiratory capacity of these colonic tumors. Protein levels of the OXPHOS complexes and the activities of complex I and II were not significantly affected (Figures S3A and S3B). Correspondingly with the PANTHER analysis (Figure 3B), complex IV activity was significantly reduced in Ucp2−/− tumors (Figure S3B) but without reaching a critical threshold to effectively affect mitochondrial respiration. Next, we measured the rates of [U-14C]-glucose and [2-14C]-pyruvate oxidation into 14CO2. 14CO2 production from [U-14C]-glucose depends on the rates of both glycolysis and pyruvate oxidation, whereas with [2-14C]-pyruvate, 14CO2 is generated only during the second round of the tricarboxylic acid (TCA) cycle (Figure 5C). Our results showed that UCP2 invalidation did not modify the global glucose and pyruvate oxidation fluxes in tumors (Figure 5D). This was in accordance with no differences at the level of pyruvate dehydrogenase (PDH) activity measured as 14CO2 release from [1-14C]-pyruvate (Figure S3C).
Figure 5.

UCP2 Invalidation Increases HK2 Protein Expression and Decreases LDHA and G6PDH Activities in Colon Tumors
(A) Mitochondrial respiration rate was determined in the presence of 10 mM pyruvate, 5 mM malate, and 1.4 mM ADP. The leak was measured after adding 1 μg/mL oligomycin (Ucp2+/+ T n = 10, Ucp2−/− T n = 9).
(B) CS enzyme activity normalized to protein content. Data represent means ± SEMs (Ucp2+/+ T n = 11, Ucp2−/− T n = 7).
(C) Scheme showing the 14CO2-producing reactions from [U-14C]-glucose (red) and [2-14C]-pyruvate (blue).
(D) [U-14C]-glucose (Ucp2+/+ n = 11, Ucp2−/− n = 12) and [2-14C]-pyruvate (Ucp2+/+ n = 7, Ucp2−/− T n = 9) oxidation into 14CO2 of Ucp2+/+and Ucp2−/− colon tumors (T). Data indicate means ± SEMs.
(E) Immunoblots and quantifications of HK2 and LDHA expression using whole-cell extracts from colon tumors. Data indicate means ± SEMs (n = 8–12).
(F) LDH enzyme activity. Data represent means ± SEMs (Ucp2+/+ n = 5, Ucp2−/− n = 6).
(G) G6PDH enzyme activity relative to Ucp2+/+ T (Ucp2+/+ n = 7, Ucp2−/− n = 8).
See also Figures S3 and S4.
We also examined whether the absence of UCP2 in established colon tumors affected glucose catabolism. We observed an increased protein expression of hexokinase 2 (HK2) in colon Ucp2−/− tumors (Figure 5E), whereas no significant changes were detected in other rate-limiting glycolytic enzymes (Figure S4). Since we excluded changes in complete glucose oxidation by the TCA cycle, once HK2 converts glucose to glucose-6-phosphate, it can be either fully catabolized to pyruvate and yield lactate or enter the oxidative branch of the pentose phosphate pathway (PPP), the latter being one of the major sources of NADPH through the reaction catalyzed by glucose-6-phosphate dehydrogenase (G6PDH). Both the expression and the activity of lactate dehydrogenase A (LDHA), as well as the activity of G6PDH, were decreased after UCP2 invalidation (Figures 5E–5G). The decrease in G6PDH activity was in accordance with the lower levels of NADPH observed in Ucp2−/− colon tumors. Here, we show that the rate-limiting first step of glycolysis was significantly increased in Ucp2−/− colon tumors in contrast to either pyruvate to lactate conversion (i.e., LDHA reaction), complete mitochondrial oxidation, or PPP fluxes. These results suggest that UCP2 loss may trigger a metabolic shift in glucose utilization, one potential avenue being lipid synthesis through citrate produced in the TCA cycle.
UCP2 Loss Promotes Fatty Acid Synthesis in Colon Tumors
In addition to bioenergetics, mitochondrial metabolism displays an important biosynthetic role that can sustain tumorigenesis. Fatty acid synthesis is coordinated with other metabolic pathways and redox reactions to sustain the production of both acetyl-coenzyme A (CoA) and reducing power (i.e., NADPH). Since glucose yields pyruvate that represents one source of acetyl-CoA in cells, we analyzed whether UCP2 deletion in AOM/DSS-derived colon tumors directed pyruvate into fatty acid synthesis by measuring the incorporation of the radiolabeled carbon from [2-14C]-pyruvate into phospholipids (Figure 6A). We found that the phospholipid synthesis rate was significantly increased after UCP2 invalidation in colon tumors (Figure 6B). We also observed that the expression of the lipogenic enzymes acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) was increased in Ucp2−/− tumors compared to Ucp2+/+ tumors (Figure 6C), supporting an increased flux of glycolytic carbons into phospholipids.
Figure 6.

UCP2 Loss Promotes Phospholipid Synthesis and Increases Expression of Lipogenic Enzymes in Colon Tumors
(A) Scheme showing the incorporation of radioactivity from [2-14C]-pyruvate into phospholipids.
(B) [2-14C]-pyruvate esterification into phospholipids. Data indicate means ± SEMs (n = 7).
(C) Representative immunoblot analysis of ATP citrate lyase (ACLY), ACC, and FAS using whole-cell extracts. Data indicate means ± SEMs (n = 7–10).
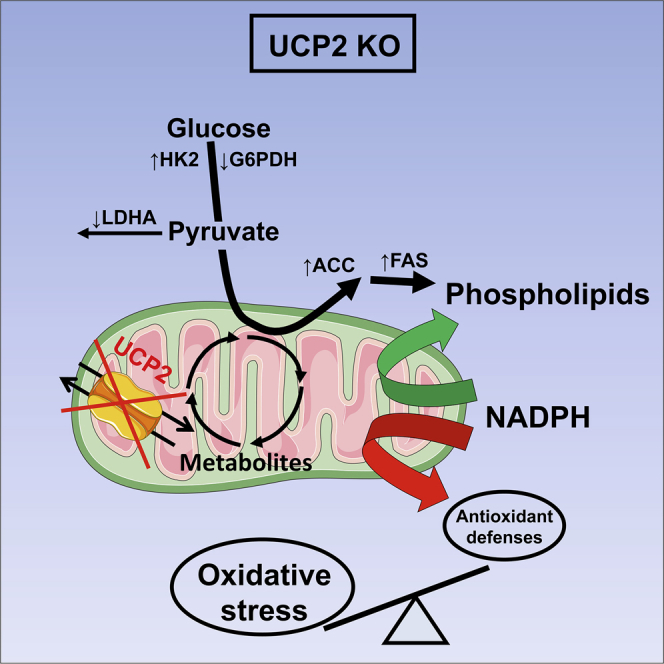
(D) Altered metabolic and redox landscape accompanying UCP2 loss favoring colon tumor initiation. Green arrows indicate upregulated reactions, and red arrows indicate downregulated reactions. UCP2 loss in colon tumors redirects glucose metabolism toward phospholipid synthesis. This is achieved by increasing the protein expression of HK2, ACC, and FAS and decreasing the enzyme activity of LDHA and G6PDH. The high demand of NADPH for fatty acid synthesis depletes the levels of this cofactor within Ucp2−/− colon tumors, becoming limiting for the antioxidant defense system. Consequently, the levels of oxidative stress increase in Ucp2−/− colon tumors, impairing the cellular redox balance.
These data show that in vivo UCP2 loss promotes the use of pyruvate to sustain lipogenesis, which is required for cell growth. Consequently, the metabolic adaptation following UCP2 invalidation leads to decreased NADPH levels, which may limit the use of these reducing equivalents by the antioxidant defense system (Figure 6D).
Discussion
Tumorigenesis depends on cellular metabolic reprogramming for cell proliferation, growth, survival, and increased malignant phenotype (Hanahan and Weinberg, 2011). In addition, mitochondria, the function of which involves free radical generation and the consequent increase in oxidative stress, have recently emerged as central bioenergetic and biosynthetic organelles required for tumor transformation (Corbet and Feron, 2017, Iommarini et al., 2017, Vyas et al., 2016). Colon tumorigenesis is one of the best examples of cancer tightly associated with oxidative stress and chronic inflammation, which can be present from the earliest stages of tumor onset (Lasry et al., 2016). In this context, the mitochondrial transporter UCP2 appears as an excellent candidate to understand the crosstalk between mitochondrial metabolic perturbation, oxidative stress, and colon tumorigenesis.
Controversial studies about both the protecting and promoting roles of UCP2 in cancer have their bases in the ability of UCP2 to shift the balance between aerobic glycolysis (Warburg effect) and OXPHOS in vitro, which can subsequently affect cell proliferation. Our present study using UCP2 knockout mice models represents a fundamental step forward in understanding the in vivo effect of UCP2 expression on the CRC landscape. Derdák et al. (2006) previously addressed the impact of UCP2 invalidation on CRC development and proposed the enhanced risk of malignant transformation due to increased oxidative stress levels and nuclear factor κB (NF-κB) activation in colon tissue. However, in this study, only a minor fraction of the AOM-induced aberrant crypt foci progressed into CRC, and the UCP2-specific antioxidant and metabolic functions in colon tumors were not characterized. Another recent study reported that downregulation of UCP2 expression through the overexpression of the microRNA (miRNA) miR-15a-3p correlates with colorectal adenoma-to-carcinoma progression (de Groen et al., 2015). In our work, we demonstrate that UCP2 invalidation in vivo increases the initiation of colon tumorigenesis by using a mouse model that accurately recapitulates the pathogenesis observed in human CRC (De Robertis et al., 2011). This pro-tumoral role accompanying UCP2 loss involves the rewiring of glucose metabolism toward fatty acid synthesis in colon tumors, which conversely depletes NADPH availability, an essential cofactor in fighting against oxidative stress (Figure 6D). It is worth noting that in the context of UCP2 deficiency, the increased colon tumor initiation may be independent of changes in cell proliferation and cell death and relies more exclusively on metabolic reprogramming and unbalanced redox homeostasis.
While we observed that UCP2 deficiency promotes intestinal tumorigenesis, its expression is higher in murine and human colon tumors, as demonstrated by western blot analyses using our specific homemade UCP2-605 antibody. However, the fact that UCP2 expression is enhanced upon intestinal tumorigenesis does not preclude from a tumor-suppressive role for UCP2, as exemplified for other antioxidant proteins (Konzack et al., 2015, Ritchie et al., 2009). Ritchie et al. (2009) point out that invalidation of the GSH transferase Pi (GSTP), which is commonly overexpressed in human tumors, increases intestinal tumor initiation and decreases survival in ApcMin mice. These studies support that increased ROS production is crucial for the initiation of tumorigenesis and renders cells more prone to malignant transformation. In addition, we have previously demonstrated that UCP2 expression is increased by inflammation and ROS in epithelial cells (Alves-Guerra et al., 2003). Tumor metabolic reprogramming leading to increased glutamine availability could also promote the translation of UCP2 protein in tumors versus adjacent non-tumoral tissue. Therefore, we believe that UCP2 elevated expression may be a consequence of increased inflammation and/or higher immune cell infiltration rather than causal for tumor progression. The precise tumor microenvironmental factors upregulating UCP2 expression in established small intestinal and colon tumors are not clear and still require further investigation.
Based on UCP2 mitochondrial substrate exchange activity (Bouillaud et al., 2016, Vozza et al., 2014), its loss may modify levels of cellular metabolites. We observed by mass spectrometry (MALDI-TOF) that the loss of UCP2 leads to a decrease in α-ketoglutarate (α-KG) levels in colon tumors harvested from AOM/DSS-treated mice (Ucp2+/+ 3.773 ± 0.8868 a.u. (n = 6) versus Ucp2−/− 1.752 ± 0.4569 a.u. (n = 7); p < 0.05), which in turn can modulate a wide variety of molecular pathways, including epigenetic modifications, and therefore affect gene expression (Wong et al., 2017). Recent studies emphasize the importance of tumor metabolites in the activity of most chromatin-modifying enzymes (Miranda-Gonçalves et al., 2018). Tumor cells modulate the expression of several metabolic enzymes altering α-KG levels, enabling an epigenetic profile that is favorable to less differentiation and higher proliferative capacity (Schvartzman et al., 2018). Thus, UCP2 invalidation results in metabolic fluxes rewiring to limit the accumulation of TCA cycle intermediates and/or cofactors that could result in cellular collapse. Our data indicate that UCP2 loss promotes the redirection of citrate toward phospholipid synthesis, which may benefit the generation of membranes to sustain rapid cell proliferation, and tumors become less dependent on circulating dietary fatty acids (Notarnicola et al., 2012). Of note, FAS overexpression and higher FAS activity, among other lipogenic enzymes, contributes to the pathogenesis of CRC (Notarnicola et al., 2006, Notarnicola et al., 2012). Nowinski et al. (2015) demonstrated that UCP3 overexpression in basal epidermis, a close UCP2 homolog, enhanced lipid catabolism and impeded tumor promotion, suggesting that the increased expression of UCPs could limit the intermediates available for macromolecule biosynthesis. Therefore, the loss of UCP2 contributes to this metabolic hallmark that underlies the initiation of CRC.
Enhancement of fatty acid synthesis and a parallel decrease in G6PDH activity, one of the major reactions that supply NADPH, can dramatically disturb redox homeostasis. The promotion of oxidative stress after UCP2 invalidation was reported previously in two in vivo studies. Derdák et al. (2006) showed increased malondialdehyde levels, a by-product of lipid peroxidation, in distal and proximal colon tissue of AOM-treated Ucp2−/− mice. Similarly, Pi et al. (2009) observed increased levels of oxidized GSH after UCP2 invalidation in lung, spleen, and pancreatic islets, which are tissues that express high UCP2 levels. However, how UCP2 modulates oxidative stress levels in vivo was not documented. As UCP2 clearly does not exhibit uncoupling activity in our model, we propose that the pro-oxidative activity due to UCP2 deficiency involves a metabolic rewiring that decreases the NADPH pool, an essential component of the cellular antioxidant system. Feeding mice the antioxidant BHA before and during the AOM/DSS protocol reduces the increased number of colon tumors associated with the Ucp2−/− phenotype.
We further highlight that UCP2 deficiency accelerates intestinal tumorigenesis in mice carrying germline Apc mutations. Dysregulation of Wnt/β-catenin signaling in intestinal epithelial cells is a well-characterized event initiating intestinal polyposis (White et al., 2012). The additive pro-tumoral effect observed may be due to the induction of oxidative stress driven by UCP2 deficiency, as suggested previously in other models in which the antioxidative system was also compromised (Cheung et al., 2014, Liu et al., 2017). Our study demonstrates the in vivo role of UCP2 invalidation in promoting colorectal and intestinal tumorigenesis and decreasing the survival rate, which could be exploited for predicting the outcomes of patients with an identified colon cancer predisposition.
The characterization of UCP2 invalidation in colon and intestinal tumorigenesis emphasizes the tumor-suppressing role of UCP2 in CRC. Our findings provide evidence for an antioxidant mechanism of UCP2 involving the control of NADPH levels through modulating anabolic pathways in colon tumors. Our results further support the potential relevance of UCP2 deficiency as an indicator of worse outcomes in patients with a predisposition to develop intestinal cancer.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ACC | Cell Signaling | Cat# 3662; RRID: AB_2219400 |
| Rabbit polyclonal anti-ACLY | Cell Signaling | Cat# 4332S; RRID: AB_2223744 |
| Mouse monoclonal anti-b-actin | Sigma-Aldrich | Cat# A5316; RRID: AB_476743 |
| Rabbit polyclonal anti-GAPDH | Santa Cruz | Cat# sc-25778; RRID: AB_10167668 |
| Rabbit monoclonal antibody anti-GLUT1 | Cell Signaling | Cat# 12939; RRID: AB_2687899 |
| Rabbit monoclonal anti-FAS | Cell Signaling | Cat# 3180; RRID: AB_2100796 |
| Rabbit monoclonal anti-HK2 | Cell Signaling | Cat# 2867S; RRID: AB_2232946 |
| Rabbit polyclonal Anti-LDHA | Cell Signaling | Cat# 2012S; RRID: AB_2137173 |
| Donkey polyclonal anti-mouse IgG/HRP | Jackson ImmunoResearch | Cat# 715-035-150; RRID: AB_2340770 |
| Mouse monoclonal anti-PFK2 | Santa Cruz | Cat# sc-377416; RRID: N/A |
| Rabbit polyclonal anti-phospho-PFK2 | Santa Cruz | Cat# sc-32966; RRID: AB_2162845 |
| Rabbit monoclonal anti-PKM2 | Cell Signaling | Cat# 4053S; RRID: AB_1904096 |
| Rabbit polyclonal anti-phospho-PKM2 | Cell Signaling | Cat# 3827S; RRID: AB_1950369 |
| Mouse monoclonal anti-porin | Abcam | Cat# ab14734; RRID: AB_443084 |
| Donkey polyclonal anti-rabbit IgG/HRP | Jackson ImmunoResearch | Cat# 711-035-152; RRID: AB_10015282 |
| Rodent anti-total-OXPHOS | Abcam | Cat# ab110413; RRID: AB_2629281 |
| Rabbit polyclonal anti-UCP2 | Pecqueur et al., 2001 | N/A |
| Rabbit polyclonal anti-UCP2 | Abcam | Cat# ab203244; RRID: N/A |
| Goat polyclonal anti-UCP2 | Santa Cruz | Cat# sc-6525 (C-20); RRID: AB_2213585 |
| Goat polyclonal anti-UCP2 | Santa Cruz | Cat# sc-6526 (N-19); RRID: AB_2213582 |
| Rabbit monoclonal anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Anti-4-HNE | Ahmed et al., 2010 | N/A |
| Rabbit polyclonal anti-Caspase-3 | Cell Signaling | Cat# 9662; RRID: AB_331439 |
| Rabbit polyclonal anti-cleaved-Caspase-3 (Asp175) | Cell Signaling | Cat# 9661; RRID: AB_2341188 |
| Alexa Fluor 488 goat anti-rabbit | Invitrogen | Cat# A11034; RRID: AB_2576217 |
| Alexa Fluor 488 donkey anti-goat | Invitrogen | Cat# A11055; RRID: AB_2534102 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dextran sulfate sodium salt (DSS) | MP Biomedicals | 160110 |
| Azoxymethane (AOM) | Sigma-Aldrich | A5486 |
| Open standard Diet with 0.7% BHA | Research Diet | D11011401i |
| Light Cycler 480 SYBR Green 1 Master | Roche | 17109320 |
| SuperScript II Reverse transcriptase | Invitrogen | 18064-014 |
| D-glucose, [U-14C] | PerkinElmer | NEC042V250UC |
| Pyruvic Acid, Sodium Salt, [1-14C] | PerkinElmer | NEC255050UC |
| Pyruvic Acid, Sodium Salt, [2-14C] | PerkinElmer | NEC256050UC |
| Critical Commercial Assays | ||
| Oxyblot Protein Oxidation Detection Kit | Millipore | S7150 |
| NADP/NADPH Glo Assay | Promega | G9081 |
| RNeasy kit | QIAGEN | 74104 |
| Roti-Histofix® 4% solution (pH 7.0) | Roth | P087.5 |
| Pierce BCA Protein Assay kit | Thermo scientific | 23227 |
| Deposited Data | ||
| Gene expression data | This paper | GEO: GSE104403 |
| Experimental Models: Organisms/Strains | ||
| Mouse C57BL/6J | Charles River Laboratories | N/A |
| Mouse C57BL/6J Ucp2+/+/ Ucp2−/− | Blanc et al., 2003 | N/A |
| Mouse C57BL/6J ApcMin/+ | The Jackson Laboratory | 002020 |
| Mouse C57BL/6J ApcMin/+Ucp2+/+/ Ucp2−/− | This paper | N/A |
| Oligonucleotides | ||
| Primers for mouse UCP2, see Table S3 | This paper | N/A |
| Primers for human UCP2, see Table S3 | This paper | N/A |
| Primers for mouse Cyclophilin, see Table S3 | This paper | N/A |
| Primers for human Cyclophilin, see Table S3 | This paper | N/A |
| Primers for mouse Ki-67, see Table S3 | This paper | N/A |
| Primers for mouse Lgr5, see Table S3 | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 5 and 7 software | GraphPad Software | N/A |
| ImageJ software | https://imagej.nih.gov/ij/ | N/A |
| FUSION Capt Advance software | Vilber Lourmat | N/A |
| Other | ||
| Oxygraph O2 k | Oroboros Instruments | N/A |
| Criterion Cell and Criterion Blotter (Western Blot) | BIORAD | N/A |
| NIKON Eclipse E600 microscope | NIKON | N/A |
| AxioObserver Z1 inverted fluorescence microscope coupled with the Zeiss Axiocam MRm | Zeiss | N/A |
| Tissue Lyser | Retsch | MM301 |
| Tissue Shredder | Oroboros Instruments | N/A |
| LightCycler 480 System | Roche | N/A |
| FUSION FX Western Blot & Chemi imaging | Vilber Lourmat | N/A |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marie-Clotilde Alves-Guerra (clotilde.alves-guerra@inserm.fr). Mouse lines generated in this study are available for distribution through a Material Transfer Agreement.
Experimental Model and Subject Details
Experimental mouse models
All animal procedures were carried out according to French legal regulations and were approved by an ethics committee (n°CEEA34.MCAG.101.12). UCP2 knockout (Ucp2−/−) mice on the C57BL6/J background have been previously described (Blanc et al., 2003). All experiments were carried out in males, and treatments were started when mice were 10-14 weeks old. To analyze UCP2 basal expression along the gut of Ucp2+/+ mice, animals were sacrificed and different segments of the intestine were collected (duodenum, jejunum, ileum, cecum and colon).
Chemically-induced colorectal cancer model was achieved by a single intraperitoneal injection of azoxymethane (AOM, 10 mg kg-1 body weight) (Sigma-Aldrich, St. Louis, MO, USA) followed by addition of dextran sodium sulfate (DSS) (MP Biomedicals, Solon, OH) to the drinking water (neutral pH) at a concentration of 2% (w/v) for 5 days (first DSS cycle). This was followed by 2 weeks of regular neutral pH water for recovery, and this DSS cycle was repeated twice. The mice were sacrificed by cervical dislocation at week 15 and the colons were removed.
C57BL/6 ApcMin/+ mice, originally obtained from the Jackson Laboratory, were a kind gift from Dr. Sandra Guilmeau (Institut Cochin). Both ApcMin/+ Ucp2+/+ and ApcMin/+ Ucp2−/− mice were generated in our laboratory. Animals were sacrificed for the collection of intestinal samples when mice were between 20 and 30 weeks of age. Only males were used for this study.
To investigate the effect of the antioxidant compound butylated hydroxyanisole (BHA), Ucp2+/+ and Ucp2−/− mice were fed with a diet containing 0.7% (w/w) antioxidant butylated hydroxyanisole (BHA) for 3 weeks prior and all along the AOM/DSS protocol described above. Previously, it has been shown that BHA treatment can enhance glutathione (GSH) levels (Sciuto and Moran, 1999).
For the collection of mouse samples, colons were cut open longitudinally along the main axis, washed with cold PBS (pH 7.4) and then macroscopically inspected. Colon tumors were removed and kept in different conditions depending on the subsequent analysis. Briefly, for western blotting, determination of enzyme activities, analysis of glutathione and NADPH, colon tumors were snap-frozen in liquid nitrogen and after stored at −80°C. For RNA isolation, samples were kept in RNAlater solution (Sigma-Aldrich) at 4°C. For mitochondria isolation, 14C fluxes analysis and mitochondrial respiration, samples were kept in TES buffer (5 mM Tris, pH 7.4, 1 mM EGTA, 300 mM sucrose) and processed immediately. Finally, some pieces of colon tumors were fixed in Roti-Histofix® 4% solution (pH 7.0) (Roth, Karlsruhe, Germany) overnight at 4°C for further immunohistochemical or immunofluorescence studies.
Human tissues samples
Colorectal samples of tumor and adjacent mucosa were collected in the surgery unit at Avicenne Hospital (Bobigny, France). Tissue samples were obtained from 10 male patients ranged from 54 to 90 years (mean 69.6 years) who underwent surgical resection because of colon adenocarcinomas. Patients did not receive any neoadjuvant or COXIBs therapy before colonic tissues collection. Immediately after excision, tissue samples were snap frozen and stored at −80°C for further analysis. The 10 samples of colorectal mucosa were from the surgical resection margin. According to TNM classification (Gunderson et al., 2010), within all cancer samples analyzed, 4 were stage II and 6 were stage III. From all patients, 10 paired tumor and normal mucosa samples were available for RNA extraction and qPCR analysis, whereas 6 paired tumor and normal mucosa samples were available for protein extraction and western blot analysis. The protocol of the present study was reviewed and approved by the local ethical committee and a written informed consent was obtained from all subjects (Schumacher et al., 2016).
Method Details
Methylene blue staining
Colons were removed from AOM/DSS-treated mice, cut longitudinally and washed with cold PBS (pH 7.4). Samples were fixed in 4% paraformaldehyde (w/v) for 45 min, after specimens were dipped in 0.2% (w/v) methylene blue for 3 min and thoroughly washed with 70% (v/v) methanol. Colons were placed on fresh 70% (v/v) methanol overnight before examination. Quantification of colonic tumors was performed by two independent observers.
RNA isolation and quantitative RT-PCR
RNA isolation was performed using RNeasy Mini Kit (QIAGEN, Hilden, Germany). RNA (1 μg) was reverse-transcribed by SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) and cDNA samples were analyzed by qPCR using LightCycler 480 SYBR Green I Master mix through the LightCycler 480 System (Roche). The primers that were used in this study are listed in Table S3.
Transcriptomic analysis
RNA from colon tumors was isolated as described above. After validation of the RNA quality with the Bioanalyzer 2100 (Agilent RNA6000 nano chip kit), 240 ng of total RNA were reverse transcribed using the GeneChip® WT Plus Reagent Kit (Affymetrix, Santa Clara, CA, USA). Briefly, the resulting double strand cDNA was used for in vitro transcription with T7 RNA polymerase (all these steps are included in the WT cDNA synthesis and amplification kit of Affymetrix). After purification according to Affymetrix protocol, 5.5 μg of Sense Target DNA were fragmented and labeled with biotin using the Encore Biotin Module kit (NuGEN Technologies, Inc. San Carlos, CA). Fragmentation was verified using the Bioanalyzer 2100, and cDNA was hybridized to a GeneChip® Mouse Gene 2.0 ST array (Affymetrix) at 45°C for 17 h. Following overnight hybridization, chips were washed on the fluidic station FS450 following specific protocols (Affymetrix) and scanned using the GCS 3000 7G scanner. The images were then analyzed with the Expression Console software (Affymetrix) to obtain raw data (CEL files) and metrics for Quality Controls. The observations of some of these metrics and the study of the distribution of raw data showed no outlier sample. CEL files were then normalized and processed to signal intensities using the RMA algorithm from the Bioconductor library and the cdf file V19 from BrainArray. All data for subsequent analysis were then log transformed (base 2) in Partek Genomics Suite. Unsupervised analysis and Anova were used to detect eventual outlier samples and to identify differentially expressed genes.
GO PANTHER classification system (http://pantherdb.org/) was used to categorize differentially expressed genes by biological processes. Gene networks and canonical pathways representing key genes were also identified using the curated Ingenuity Pathways Analysis® (IPA) database. Fisher’s exact test was performed to calculate a P value determining the probability that each biological function and/or disease assigned to the dataset was due to chance alone.
The data discussed in this work have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE. Microarray analysis supporting these studies can be found at GEO: GSE104403.
Western blotting
For whole cell extracts, colon tumors were disrupted in lysis buffer (50 mM HEPES pH 7.4, 1.5 mM EDTA, 150 mM NaCl, 10% (v/v) glycerol and 1% (v/v) NP40) supplemented with protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific Inc., Middletown, VA, USA) using a Tissue Lyser (model MM301; Retsch, Haan, Germany). Mitochondrial fractions were isolated as described previously (Pecqueur et al., 2001). Total cellular lysates and mitochondrial fractions were loaded into a precast gradient SDS-PAGE gel (4%–20%; BioRad, Hercules, CA, USA), transferred to a nitrocellulose membrane. Western blot analysis was done using colon samples from independent mice. Primary antibodies to ACC, ACLY, Caspase 3, cleaved Caspase 3, GLUT1, FAS, HK2, LDHA, PKM2 and p-PKM2 were obtained from Cell Signaling (Danvers, MA, USA). Anti-β-actin was obtained from Sigma-Aldrich (Saint Louis, MA, USA). Anti-GAPDH, anti-PFK2, anti-p-PFK2, anti-UCP2 (C-20) and anti-UCP2 (N-19) antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-porin, anti-total-OXPHOS and anti-UCP2 (ab203244) were purchased from Abcam (Cambridge, MA, USA). Homemade anti-UCP2-605 antibody (Pecqueur et al., 2001) and homemade anti-HNE antibody (Ahmed et al., 2010) were also used. Horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA).
Immunohistochemical staining
Colon sections from Ucp2+/+ and Ucp2−/− AOM/DSS-treated mice were fixed in Roti-Histofix® 4% solution (pH 7.0) (Roth) overnight at 4°C and then transferred to 70% (v/v) ethanol. The tissues were embedded in paraffin, sectioned at 4 μm thickness and placed on slides. Slides were deparaffinized through three changes of xylene each for 3 min, then rehydrated through descending grades of ethanol to water. Endogenous peroxidases were blocked with 3% (v/v) hydrogen peroxide for 10 min. Antigen retrieval was performed by heating the sections in sodium citrate buffer (10 mM sodium citrate, 0.05% (v/v) Tween-20, pH 6.0) in a rice steamer at 95°C for 10 min. After cooling at room temperature, the slides were incubated with blocking solution (TBS, 5% (w/v) BSA, 0.1% (v/v) Triton) for 2 h at room temperature and then incubated with the primary antibodies diluted in blocking solution overnight at 4°C. The primary antibodies used were our homemade anti-UCP2-605 as well as the anti-UCP2 (ab203244) and anti-Ki67 (ab15580) from Abcam. After rinsing in TBS, 0.1% (v/v) Tween-20, slides were incubated with biotinylated secondary antibodies (Vector Laboratories, Burlingame, CA, USA) for 90 min at room temperature, followed by incubation with Vectastain ABC system (Vector Laboratories) for 30 min. The detection was made using the DAB substrate kit and slides were counterstained with hematoxylin (Vector Laboratories), dehydrated and coverslipped using VectaMount (Vector Laboratories).
Immunofluorescence
Colon tissues were fixed in Roti-Histofix® 4% solution (pH 7.0) (Roth, Karlsruhe, Germany) as detailed above. The same procedure as indicated for immunohistochemical staining was followed (except for the inactivation of endogenous peroxidases using hydrogen peroxide). The following primary antibodies were used: homemade anti-UCP2-605 and anti-UCP2 from Santa Cruz (N-19, sc-6526). The fluorescent secondary antibodies were Alexa Fluor 488 goat anti-rabbit and Alexa Fluor 488 donkey anti-goat (1:400; Invitrogen). Fluorescent image acquisition was performed using the Zeiss AxioObserver Z1 inverted fluorescence microscope coupled with the Zeiss Axiocam MRm (Zeiss, Marly-le-Roi, France).
Protein oxidation assay
Samples were stored frozen at −80°C until processing. Protein oxidation of whole colon tumors extracts were determined by quantifying the formation of carbonyl groups on protein side chains using the Oxyblot Protein Oxidation Detection Kit (Millipore, Billerica, MA, USA) according to manufacturer recommendations. Protein extracts were obtained as described above, and samples were protected from further oxidation by adding 50 mM DTT. Briefly, 20 μg of tissue lysate were treated with 2,4-dinitrophenylhydrazine (DNPH) to derivatize the carbonyl groups on proteins to 2,4-dinitrophenylhydrazone-tagged products. Samples were loaded and separated into a precast gradient SDS-PAGE gel (4%–20%; BioRad) and then transferred to nitrocellulose membranes. After blockade with TBS, 1% (w/v) BSA, 0.1% (v/v) Tween-20 solution, membranes were incubated with antibodies against dinitrophenylhydrazone-modified carbonyl groups, followed by HRP-conjugated secondary antibodies.
Enzyme activities
Frozen colonic tumors were disrupted in mannitol buffer (225 mM mannitol, 75 mM saccharose, 10 mM Tris-HCl, 0.1 mM EDTA, pH 7.2) using a Tissue Lyser (model MM301; Retsch). After centrifugation at 1,000 g for 20 min at 4°C, specific enzyme activities were determined on supernatants by spectrophotometric monitoring of product production or substrate disappearance at different UV/visible light wavelengths. LDH specific activity was measured by adding diluted sample to a cuvette containing 0.25 mM NADH in 100 mM KH2PO4/K2HPO4, pH 7.4, at 25°C. Reaction was initiated by the addition of pyruvate at a final concentration of 0.9 mM. LDH activity was monitored by measuring absorbance at 340 nm. G6PDH specific activity was measured by adding samples to a cuvette containing 0.65 mM NADP+ in 50 mM Tris-HCl, ph 7.6 at 37°C. Reaction was initiated by the addition of glucose-6-phosphate at a final concentration of 2.5 mM. G6PDH activity was monitored by measuring absorbance at 340 nm. Respiratory chain complex activities were measured as previously described (Medja et al., 2009). All enzyme activities were normalized by protein content in the samples, determined by the BCA assay.
Glucose and pyruvate metabolism
Freshly excised colon tumors were incubated for 1h in TES buffer (5 mM Tris, pH 7.4, 1 mM EGTA, 300 mM sucrose) containing either 5 mM [U-14C]-glucose (0.133 mCi/mmol) or 0.1 mM [2-14C]-pyruvate (1.667 mCi/mmol). Rates of oxidation (14CO2 production) and esterification into phospholipids (14C phospholipids) were determined. For measurement of 14C phospholipids, cellular lipids were extracted in chloroform/methanol (2:1, v/v) with vigorous shaking for 10 min. After centrifugation for 25 min at 1200 g, the lower organic phase was collected, dried, and solubilized in chloroform/methanol. Lipid classes were separated by TLC on silica-gel plates using petroleum ether/diethyl ether/acetic acid (85:15:0.5, v/v/v) as the mobile phase. Lipids were visualized with iodine vapor. Bands of phospholipids were cut and 14C radioactivity was determined using radioactive counter. PDH activity was measured by the release of 14CO2 from 0.1 mM [1-14C]-pyruvate.
Oxygen consumption
Mitochondrial respiration was assessed using freshly excised colon tumors that were homogenized using a tissue shredder (Oroboros Instruments, Innsbruck, Austria). Samples were introduced in the respiratory chamber of an oxygraph O2 k (Oroboros Instruments). Mitochondrial respiration rate was determined in the presence of 10 mM pyruvate, 5 mM malate and 1.4 mM ADP and calculated after subtracting the oxygen consumption value measured in the presence of 1 mM potassium cyanide. The leak was calculated in the presence of 1 μg mL-1 oligomycin, an inhibitor of ATPase synthase.
Intracellular glutathione content
Total glutathione content was determined by the glutathione reductase enzymatic method (Rahman et al., 2006). Frozen colon tumors were lysed with 0.1% (v/v) Triton X-100, 0.6% (w/v) 5-sulfosalicylic acid in assay buffer (100 mM KH2PO4/K2HPO4 buffer with 5 mM EDTA, pH 7.5), and disrupted using a Tissue Lyser (model MM301; Retsch). Cell extracts were centrifuged at 8,000 g at 4°C for 10 min and resulting supernatants were used for quantification of total glutathione and oxidized glutathione (GSSG) levels alone (pre-incubation with 2-vinyl-pyridine for 1h at room temperature). A working solution of 3.3 U/mL of glutathione reductase and 1.68 mM of 5,5′-dithiobis(2-nitrobenzoic acid) was prepared in assay buffer. Glutathione standards were prepared from a 3.28 mM GSSG stock solution. Reaction was initiated by mixing 120 μL of working solution with 20 μL of cell extract (1:10 dilution for total glutathione; no dilution for GSSG) or 20 μL of GSSG standard (final concentrations from 0 to 13.2 μM). Next, 60 μL of 0.8 mM NADPH solution were added and the increase in absorbance was recorded at 405 nm wavelength. Glutathione concentration was normalized by protein content, as determined by BCA assay.
Intracellular NADPH content
NADP+ and NADPH analyses from frozen colon tumor samples were performed directly in 96-well plate culture plates according to the manufacturers’ instructions (NADP/NADPH Glo Assay, Promega, Madison, WI, USA). NADP+ and NADPH values were normalized by protein content.
Quantification and Statistical Analysis
Differences between experimental groups were analyzed by non-parametric Mann-Whitney U-test or ANOVA. Differences among survival in the groups were investigated by means of the Gehan-Breslow-Wilcoxon test. Results are expressed as the mean ± SEM. All n-values per group are reported in the figure legends. Statistical significance is represented in figures as follows: ∗p < 0.05 and ∗∗p < 0.01. Statistical analyses were performed using GraphPad Prism 5 software.
Data and Code Availability
The accession number for the microarray data reported in this paper is GEO: GSE104403.
Acknowledgments
We gratefully thank all of the members of the Frédéric Bouillaud and Anne Lombès team (Institut Cochin) for helpful discussion and support of this project, and Catherine Postic for critical evaluation of the work. The authors gratefully acknowledge members from the Institut Cochin Animal Facility, especially Matthieu Bénard, for excellent mouse care. We thank Bertrand Duvillié from Cochin Institute (Scharfmann team) for the acquisition and analysis of IF data and Ophélie Renoult for α-KG levels measurement (CRCINA - INSERM U1232, Université de Nantes). We thank Franck Letourneur and Jacques Sebastien from the Institut Cochin Genomic Platform for microarray experiments and analysis, and Maryline Favier from the Institut Cochin Histology Facility. E.A. was supported by a postdoctoral fellowship from the Cancéropôle Île-de-France (2014-2-APD-03-INSERM-5-1) and by the European Research Council (ERC-2013-StG-336629). This work was financially supported by grants from La Ligue Contre le Cancer (LCC RS17/75-63), the Fondation ARC (PJA 20151203347), the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé et de la Recherche Médicale (INSERM) and the Université Paris Descartes.
Author Contributions
E.A. and M.-C.A.-G. conceived the project, designed and performed the experiments, and interpreted the results. P.E., C.P.-B., and S.G. also aided with the experimental design and interpretation of the results. P.E., T.S., and V.L. performed the experiments. T.A., F.B., R.D., and D.R. provided administrative, technical, and material support. The manuscript was written by E.A. and edited by M.-C.A.-G., C.P., S.G., R.D., and C.P.-B. The study was supervised by M.-C.A.-G.
Declaration of Interests
The authors declare no competing interests.
Published: August 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.097.
Supplemental Information
References
- Ahmed E.K., Rogowska-Wrzesinska A., Roepstorff P., Bulteau A.L., Friguet B. Protein modification and replicative senescence of WI-38 human embryonic fibroblasts. Aging Cell. 2010;9:252–272. doi: 10.1111/j.1474-9726.2010.00555.x. [DOI] [PubMed] [Google Scholar]
- Alves-Guerra M.C., Rousset S., Pecqueur C., Mallat Z., Blanc J., Tedgui A., Bouillaud F., Cassard-Doulcier A.M., Ricquier D., Miroux B. Bone marrow transplantation reveals the in vivo expression of the mitochondrial uncoupling protein 2 in immune and nonimmune cells during inflammation. J. Biol. Chem. 2003;278:42307–42312. doi: 10.1074/jbc.M306951200. [DOI] [PubMed] [Google Scholar]
- Arsenijevic D., Onuma H., Pecqueur C., Raimbault S., Manning B.S., Miroux B., Couplan E., Alves-Guerra M.C., Goubern M., Surwit R. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- Blanc J., Alves-Guerra M.C., Esposito B., Rousset S., Gourdy P., Ricquier D., Tedgui A., Miroux B., Mallat Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation. 2003;107:388–390. doi: 10.1161/01.cir.0000051722.66074.60. [DOI] [PubMed] [Google Scholar]
- Bouillaud F., Alves-Guerra M.C., Ricquier D. UCPs, at the interface between bioenergetics and metabolism. Biochim. Biophys. Acta. 2016;1863:2443–2456. doi: 10.1016/j.bbamcr.2016.04.013. [DOI] [PubMed] [Google Scholar]
- Cheung K.L., Lee J.H., Khor T.O., Wu T.Y., Li G.X., Chan J., Yang C.S., Kong A.N. Nrf2 knockout enhances intestinal tumorigenesis in Apc(min/+) mice due to attenuation of anti-oxidative stress pathway while potentiates inflammation. Mol. Carcinog. 2014;53:77–84. doi: 10.1002/mc.21950. [DOI] [PubMed] [Google Scholar]
- Corbet C., Feron O. Cancer cell metabolism and mitochondria: nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta Rev. Cancer. 2017;1868:7–15. doi: 10.1016/j.bbcan.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Couplan E., del Mar Gonzalez-Barroso M., Alves-Guerra M.C., Ricquier D., Goubern M., Bouillaud F. No evidence for a basal, retinoic, or superoxide-induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J. Biol. Chem. 2002;277:26268–26275. doi: 10.1074/jbc.M202535200. [DOI] [PubMed] [Google Scholar]
- Dando I., Fiorini C., Pozza E.D., Padroni C., Costanzo C., Palmieri M., Donadelli M. UCP2 inhibition triggers ROS-dependent nuclear translocation of GAPDH and autophagic cell death in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta. 2013;1833:672–679. doi: 10.1016/j.bbamcr.2012.10.028. [DOI] [PubMed] [Google Scholar]
- de Groen F.L., Timmer L.M., Menezes R.X., Diosdado B., Hooijberg E., Meijer G.A., Steenbergen R.D., Carvalho B. Oncogenic Role of miR-15a-3p in 13q Amplicon-Driven Colorectal Adenoma-to-Carcinoma Progression. PLoS One. 2015;10:e0132495. doi: 10.1371/journal.pone.0132495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Robertis M., Massi E., Poeta M.L., Carotti S., Morini S., Cecchetelli L., Signori E., Fazio V.M. The AOM/DSS murine model for the study of colon carcinogenesis: from pathways to diagnosis and therapy studies. J. Carcinog. 2011;10:9. doi: 10.4103/1477-3163.78279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdák Z., Fülöp P., Sabo E., Tavares R., Berthiaume E.P., Resnick M.B., Paragh G., Wands J.R., Baffy G. Enhanced colon tumor induction in uncoupling protein-2 deficient mice is associated with NF-kappaB activation and oxidative stress. Carcinogenesis. 2006;27:956–961. doi: 10.1093/carcin/bgi335. [DOI] [PubMed] [Google Scholar]
- Derdak Z., Mark N.M., Beldi G., Robson S.C., Wands J.R., Baffy G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 2008;68:2813–2819. doi: 10.1158/0008-5472.CAN-08-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J., Allister E.M., Koshkin V., Lee S.C., Bhattacharjee A., Tang C., Giacca A., Chan C.B., Wheeler M.B. UCP2 is highly expressed in pancreatic alpha-cells and influences secretion and survival. Proc. Natl. Acad. Sci. USA. 2008;105:12057–12062. doi: 10.1073/pnas.0710434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadelli M., Dando I., Fiorini C., Palmieri M. UCP2, a mitochondrial protein regulated at multiple levels. Cell. Mol. Life Sci. 2014;71:1171–1190. doi: 10.1007/s00018-013-1407-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enerbäck S., Jacobsson A., Simpson E.M., Guerra C., Yamashita H., Harper M.E., Kozak L.P. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 1997;387:90–94. doi: 10.1038/387090a0. [DOI] [PubMed] [Google Scholar]
- Esteves P., Pecqueur C., Ransy C., Esnous C., Lenoir V., Bouillaud F., Bulteau A.L., Lombès A., Prip-Buus C., Ricquier D., Alves-Guerra M.C. Mitochondrial retrograde signaling mediated by UCP2 inhibits cancer cell proliferation and tumorigenesis. Cancer Res. 2014;74:3971–3982. doi: 10.1158/0008-5472.CAN-13-3383. [DOI] [PubMed] [Google Scholar]
- Gunderson L.L., Jessup J.M., Sargent D.J., Greene F.L., Stewart A.K. Revised TN categorization for colon cancer based on national survival outcomes data. J. Clin. Oncol. 2010;28:264–271. doi: 10.1200/JCO.2009.24.0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Horimoto M., Resnick M.B., Konkin T.A., Routhier J., Wands J.R., Baffy G. Expression of uncoupling protein-2 in human colon cancer. Clin. Cancer Res. 2004;10:6203–6207. doi: 10.1158/1078-0432.CCR-04-0419. [DOI] [PubMed] [Google Scholar]
- Iommarini L., Ghelli A., Gasparre G., Porcelli A.M. Mitochondrial metabolism and energy sensing in tumor progression. Biochim. Biophys. Acta Bioenerg. 2017;1858:582–590. doi: 10.1016/j.bbabio.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Kawada K., Toda K., Sakai Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int. J. Clin. Oncol. 2017;22:651–659. doi: 10.1007/s10147-017-1156-4. [DOI] [PubMed] [Google Scholar]
- Konzack A., Jakupovic M., Kubaichuk K., Görlach A., Dombrowski F., Miinalainen I., Sormunen R., Kietzmann T. Mitochondrial Dysfunction Due to Lack of Manganese Superoxide Dismutase Promotes Hepatocarcinogenesis. Antioxid. Redox Signal. 2015;23:1059–1075. doi: 10.1089/ars.2015.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasry A., Zinger A., Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016;17:230–240. doi: 10.1038/ni.3384. [DOI] [PubMed] [Google Scholar]
- Lehuédé C., Dupuy F., Rabinovitch R., Jones R.G., Siegel P.M. Metabolic Plasticity as a Determinant of Tumor Growth and Metastasis. Cancer Res. 2016;76:5201–5208. doi: 10.1158/0008-5472.CAN-16-0266. [DOI] [PubMed] [Google Scholar]
- Liu H., Liu X., Zhang C., Zhu H., Xu Q., Bu Y., Lei Y. Redox Imbalance in the Development of Colorectal Cancer. J. Cancer. 2017;8:1586–1597. doi: 10.7150/jca.18735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo C., Moser A.R., Gledhill S., Dove W.F. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54:5947–5952. [PubMed] [Google Scholar]
- Mailloux R.J., Adjeitey C.N., Harper M.E. Genipin-induced inhibition of uncoupling protein-2 sensitizes drug-resistant cancer cells to cytotoxic agents. PLoS One. 2010;5:e13289. doi: 10.1371/journal.pone.0013289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín de Mas I., Aguilar E., Jayaraman A., Polat I.H., Martín-Bernabé A., Bharat R., Foguet C., Milà E., Papp B., Centelles J.J., Cascante M. Cancer cell metabolism as new targets for novel designed therapies. Future Med. Chem. 2014;6:1791–1810. doi: 10.4155/fmc.14.119. [DOI] [PubMed] [Google Scholar]
- Medja F., Allouche S., Frachon P., Jardel C., Malgat M., Mousson de Camaret B., Slama A., Lunardi J., Mazat J.P., Lombès A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion. 2009;9:331–339. doi: 10.1016/j.mito.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Miranda-Gonçalves V., Lameirinhas A., Henrique R., Jerónimo C. Metabolism and Epigenetic Interplay in Cancer: Regulation and Putative Therapeutic Targets. Front. Genet. 2018;9:427. doi: 10.3389/fgene.2018.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyo M., Konno M., Nishida N., Sueda T., Noguchi K., Matsui H., Colvin H., Kawamoto K., Koseki J., Haraguchi N. Metabolic Adaptation to Nutritional Stress in Human Colorectal Cancer. Sci. Rep. 2016;6:38415. doi: 10.1038/srep38415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notarnicola M., Altomare D.F., Calvani M., Orlando A., Bifulco M., D’Attoma B., Caruso M.G. Fatty acid synthase hyperactivation in human colorectal cancer: relationship with tumor side and sex. Oncology. 2006;71:327–332. doi: 10.1159/000107106. [DOI] [PubMed] [Google Scholar]
- Notarnicola M., Messa C., Caruso M.G. A significant role of lipogenic enzymes in colorectal cancer. Anticancer Res. 2012;32:2585–2590. [PubMed] [Google Scholar]
- Nowinski S.M., Solmonson A., Rundhaug J.E., Rho O., Cho J., Lago C.U., Riley C.L., Lee S., Kohno S., Dao C.K. Mitochondrial uncoupling links lipid catabolism to Akt inhibition and resistance to tumorigenesis. Nat. Commun. 2015;6:8137. doi: 10.1038/ncomms9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecqueur C., Alves-Guerra M.C., Gelly C., Levi-Meyrueis C., Couplan E., Collins S., Ricquier D., Bouillaud F., Miroux B. Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J. Biol. Chem. 2001;276:8705–8712. doi: 10.1074/jbc.M006938200. [DOI] [PubMed] [Google Scholar]
- Pecqueur C., Bui T., Gelly C., Hauchard J., Barbot C., Bouillaud F., Ricquier D., Miroux B., Thompson C.B. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2008;22:9–18. doi: 10.1096/fj.07-8945com. [DOI] [PubMed] [Google Scholar]
- Pecqueur C., Alves-Guerra C., Ricquier D., Bouillaud F. UCP2, a metabolic sensor coupling glucose oxidation to mitochondrial metabolism? IUBMB Life. 2009;61:762–767. doi: 10.1002/iub.188. [DOI] [PubMed] [Google Scholar]
- Perše M. Oxidative stress in the pathogenesis of colorectal cancer: cause or consequence? BioMed Res. Int. 2013;2013:725710. doi: 10.1155/2013/725710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi J., Bai Y., Daniel K.W., Liu D., Lyght O., Edelstein D., Brownlee M., Corkey B.E., Collins S. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic beta-cell function. Endocrinology. 2009;150:3040–3048. doi: 10.1210/en.2008-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons D.G., Nadal-Serrano M., Torrens-Mas M., Valle A., Oliver J., Roca P. UCP2 inhibition sensitizes breast cancer cells to therapeutic agents by increasing oxidative stress. Free Radic. Biol. Med. 2015;86:67–77. doi: 10.1016/j.freeradbiomed.2015.04.032. [DOI] [PubMed] [Google Scholar]
- Rahman I., Kode A., Biswas S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- Ritchie K.J., Walsh S., Sansom O.J., Henderson C.J., Wolf C.R. Markedly enhanced colon tumorigenesis in Apc(Min) mice lacking glutathione S-transferase Pi. Proc. Natl. Acad. Sci. USA. 2009;106:20859–20864. doi: 10.1073/pnas.0911351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharwal S.S., Schumacker P.T. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher Y., Aparicio T., Ourabah S., Baraille F., Martin A., Wind P., Dentin R., Postic C., Guilmeau S. Dysregulated CRTC1 activity is a novel component of PGE2 signaling that contributes to colon cancer growth. Oncogene. 2016;35:2602–2614. doi: 10.1038/onc.2015.283. [DOI] [PubMed] [Google Scholar]
- Schvartzman J.M., Thompson C.B., Finley L.W.S. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 2018;217:2247–2259. doi: 10.1083/jcb.201803061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciuto A.M., Moran T.S. BHA diet enhances the survival of mice exposed to phosgene: the effect of BHA on glutathione levels in the lung. Inhal. Toxicol. 1999;11:855–871. doi: 10.1080/089583799196772. [DOI] [PubMed] [Google Scholar]
- Sullivan L.B., Chandel N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., DeBerardinis R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellinga T.T., Borovski T., de Boer V.C., Fatrai S., van Schelven S., Trumpi K., Verheem A., Snoeren N., Emmink B.L., Koster J. SIRT1/PGC1α-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015;21:2870–2879. doi: 10.1158/1078-0432.CCR-14-2290. [DOI] [PubMed] [Google Scholar]
- Vozza A., Parisi G., De Leonardis F., Lasorsa F.M., Castegna A., Amorese D., Marmo R., Calcagnile V.M., Palmieri L., Ricquier D. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc. Natl. Acad. Sci. USA. 2014;111:960–965. doi: 10.1073/pnas.1317400111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S., Zaganjor E., Haigis M.C. Mitochondria and Cancer. Cell. 2016;166:555–566. doi: 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P.S., Thompson C.B. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White B.D., Chien A.J., Dawson D.W. Dysregulation of Wnt/β-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012;142:219–232. doi: 10.1053/j.gastro.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C.C., Qian Y., Yu J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene. 2017;36:3359–3374. doi: 10.1038/onc.2016.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Zhang Y., Li W., Wang P., Gu R., Feng Y., Wei S., Peng K., Zhang Y., Su L. Uncoupling Protein 2 Inhibits Myointimal Hyperplasia in Preclinical Animal Models of Vascular Injury. J. Am. Heart Assoc. 2017;6:e002641. doi: 10.1161/JAHA.117.006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the microarray data reported in this paper is GEO: GSE104403.