

Abstract
Erdheim–Chester disease is a rare, non-Langerhans cell histiocytosis histologically characterized by multisystemic proliferation of mature histiocytes in a background of inflammatory stroma. The disease can involve virtually any organ system; most commonly the bones, skin, retroperitoneum, heart, orbit, lung, and brain are affected. Although a histiocytic proliferation is the histological hallmark of the disease, a wide range of morphological appearances have been described as part of case studies or small series. A comprehensive review of histopathological features in clinically and molecularly defined Erdheim–Chester disease has yet to be characterized. To address this issue and help guide clinical practice, we comprehensively analyzed the pathological spectrum of Erdheim–Chester disease in a clinically and molecularly defined cohort. We reviewed 73 biopsies from 42 patients showing involvement by histiocytosis from a variety of organ systems, including bone (16), retroperitoneum (11), skin (19), orbit (6), brain (5), lung (6), cardiac structures (2), epidural soft tissue (3), oral cavity (2), subcutaneous soft tissue (2), and testis (2). In eight patients, one or more bone marrow biopsies were performed due to clinical indication and an accompanying myeloid neoplasm was detected in six of them. Thirty-eight cases were investigated for genetic abnormalities. Somatic mutations involving BRAF (25/38), MAP2K1 (6/38), ARAF (2/38), MAP2K2 (1/38), KRAS (1/38), and NRAS (1/38) genes were detected. One of the cases with a MAP2K1 mutation also harbored a PIK3CA mutation. We have observed marked heterogeneity in histology and immunophenotype, identified site-specific features, overlap with other histiocytic and myeloid disorders and potential diagnostic pitfalls. We hope that broadening the spectrum of recognized pathologic manifestations of Erdheim–Chester disease will help practicing clinicians and pathologists to diagnose Erdheim–Chester disease early in the disease course and manage these patients effectively.
Erdheim–Chester disease is a rare, non-Langerhans cell histiocytosis histologically characterized by multi-systemic proliferation of mature histiocytes in a background of inflammatory stroma. The infiltrate can occur in virtually any organ system although most commonly involves the bones, skin, retroperitoneum, heart, orbits, lung, and brain.1–3 Clinical manifestations in Erdheim–Chester disease is mediated by local effects of the histiocytic infiltrate, as well as by chronic systemic inflammation and cytokine perturbations.4 The etiology and pathogenesis of Erdheim–Chester disease were largely unclear until recently; however, the discovery of (1) recurrent BRAFV600E mutations in lesional tissue,5and then (2) an array of other activating mitogen-activated protein kinase (MAPK) pathway mutations6 have established the neoplastic nature of the disease and led to designation of Erdheim–Chester disease as a histiocytic neoplasm. Altogether, Erdheim–Chester disease is now considered a clonal hematopoietic disorder marked by recurrent MAPK pathway genetic alterations, with clinical manifestations caused by neoplastic histiocytic infiltrates and uncontrolled inflammation.
Consensus diagnostic criteria for Erdheim–Chester disease require (1) xanthogranulomatous lesions characterized by foamy CD68(+)/CD1a(- ) histiocytes, often with admixed inflammation and fibrosis, and (2) skeletal findings of bilateral and symmetric abnormalities in the diaphyseal and metaphyseal regions of the long bones of the legs.7 All patients were evaluated at least once in consultation at our center. The bone involvement may appear as osteo-sclerotic lesions by plain film or computed tomography (CT), or it can be visualized by 99Technetium bone scan. Owing to the frequent absence of bone pain, the wide anatomic distribution of ErdheimChester disease lesions, and the protean clinical manifestations of disease, extraosseous lesions are frequently those identified and sampled initially. Furthermore, it has been observed in recent studies that Erdheim–Chester disease lesions can be heterogeneous in their cellular composition and do not invariably demonstrate xanthomatous histiocytes.7 In fact, some lesions in well-established ErdheimChester disease may have a scant presence of lesional histiocytes and variable stromal components of acute and chronic inflammation. For this reason, establishing or more importantly suspecting a diagnosis of Erdheim–Chester disease can be hindered by reliance upon iconic or ‘classic’ morphology in biopsy material.
To our knowledge, a comprehensive and systematic description of histopathological features of Erdheim–Chester disease lesions beyond the classic xanthomatous infiltrate has not been performed. In this report, we present the wide spectrum of histopathologic and immunohistochemical phenotypes in a well-characterized, molecularly defined cohort of Erdheim–Chester disease patients from a single institution. Histopathologic features of lesions from bone, brain, retroperitoneum, skin, orbit, lung, subcutaneous and epidural soft tissues, heart, oral cavity, and testis are demonstrated. When available, the genomic alterations present in lesional tissue are presented. We hope and anticipate that broadening the spectrum of recognized pathologic manifestations of Erdheim–Chester disease will aid in its diagnosis for practicing clinicians and pathologists.
Materials and methods
Patient Selection and Histopathologic Evaluation
This study was conducted according to the Declaration of Helsinki, and human tissues were obtained and analyzed with procedures approved by the Institutional Review Board of Memorial Sloan Kettering Cancer Center. For all patients, ErdheimChester disease was diagnosed in light of published guidelines for the diagnosis and clinical management of Erdheim–Chester disease and in light of institutional experience with this disease.7 In our cohort, all patients but one had radiologic abnormalities in the femora or tibia demonstrable by one or more of the following: CT, MRI, 99Tc nucleotide bone scan, or (18F)-Fluorodeoxyglucose (FDG) PET scan. Biopsy material was retrieved from the pathology archives, or from the evaluation of active clinical cases, at Memorial Sloan Kettering Cancer Center. Available hematoxylin-eosin slides and immunohistochemical slides were examined, and additional immunohistochemical studies were performed, if unstained material was available. Immunohistochemical analysis was performed on paraffin sections for CD163 (Ventana, cat no: #760.4437, clone MRQ-26), CD68 (Ventana, cat no: #790–2931, clone KP1), BRAF V600E (cat no: Spring, #E19294, clone VE1), S100 (DAKO, cat no: #Z0311, rabbit polyclonal), CD1a (Cell Marque, cat no: #760–4525, clone EP3622), Factor XIIIa (Cell Marque, cat no: #760–4384, clone AC-1A1), and Langerin (Leica, cat no: #NCL-L-Langerin, clone 2D6). Immunohisto- chemical staining was performed with BondIII (Leica-Microsystems, Buffalo Groove, IL).
Molecular Analysis of Erdheim–Chester Disease Patient Tissue Samples
Genomic DNA was extracted from formalin-fixed, paraffin-embedded samples after histologic review and enrichment by macrodissection to ≥10% histiocytes, and all materials were from pre-treatment samples. Detection of the BRAFV600E mutation was performed by pyro-sequencing.8 Screening for mutations in other genes was performed with sequenom mass spectrometric-based genotyping assays as previously described (NRAS, KRAS, PIK3CA, and AKT1 hotspot mutations), and whole-exome sequencing analysis.9 For patients undergoing whole-exome sequencing, disease DNA was extracted from frozen biopsies while paired germline DNA was extracted from peripheral blood mononuclear cells.
Urine cfDNA was isolated as previously described.10 Urine cfDNA was quantified by a droplet-digital polymerase chain reaction (ddPCR; QX-100, BioRad) assay to a 44-bp amplicon of RNase P, a single-copy gene, as previously described.10 Quantified DNA up to 60 ng was used for mutation detection of BRAFV600E by ddPCR.
Two of our patients have been previously described in the form of individual case reports,11,12 and 11 patients were also studied by whole-exome sequencing or targeted sequencing previously.9,13
Results
The study population (Table 1) consisted of 42 patients with a total of 73 available biopsy specimens involved by histiocytosis. Sixteen patients had more than one biopsy site.
Table 1.
Summary of the clinical, molecular, and therapeutic features of the study cohort
| Patients | Sex/ Age |
Sites of disease (biopsied sites are bold) | Mutation | Biopsy site with mutation identified |
ECD treatment(s) |
|---|---|---|---|---|---|
| 1 | M/37 | Bone, testis, omentum, retroperitoneum,tongue | MAP2K1 P105_I107del | Testis, Tongue | Anakinra, Methotrexate |
| 2 | F/52 | Bone,subcutaneous soft tissue, skin (xanthelasma), maxilla | BRAF V600E | Subcutaneous soft tissue | Cytarabine, Vemurafenib |
| 3a | F/66 | Bone, right atrium | KRAS G12S | Bone, Right atrium | Interferon-alpha |
| 4 | M/73 | Brain, bone, retroperitoneum | BRAF V600E | Cell-free DNA (Urine) | Anakinra, Vemurafenib |
| 5 | M/55 | Brain, bone, retroperitoneum | BRAF V600E | Femur | Methotrexate, Vemurafenib, Dabrafenib |
| 6 | F/43 | Bone, skin | BRAF V600E | Skin | Vemurafenib |
| 7 | F/74 | Bone, lung, skin | BRAF V600E | Cell-free DNA (Urine) | Imatinib, Anakinra, Vemurafenib |
| 8 | M/59 | Retroperitoneum, bone | MAP2K1 F68L | Retroperitoneum | Anakinra |
| 9 | F/75 | Bone, skin (xanthelasma), lung | BRAF V600E | Skin (xanthelasma) | Vemurafenib, Dabrafenib, Methotrexate |
| 10 | M/61 | Bone, lung, retroperitoneum, skin (xanthelasma) | ARAF A225V | Skin (xanthelasma) | Anakinra, Interferon, clinical trial |
| 11 | F/47 | Bone, orbit, retroperitoneum | BRAF V600E | Retroperitoneum | Vemurafenib |
| 12 | M/51 | Bone, epidural soft tissues, orbit | BRAF V600E | Orbit | Vemurafenib |
| 13a | M/51 | Bone, pericardium, pleura, omentum, retroperitoneum | MAP2K1 K57N | Retroperitoneum | Interferon-alpha, anakinra, Trametinib |
| l4a | M/75 | Bone, peri-aortic soft tissue, retroperitoneum | BRAF V600E | Bone | Anakinra, Vemurafenib |
| 15 | F/66 | Brain, one, retroperitoneum, skin (xanthelasma) | BRAF V600E | Brain | Methotrexate, Vemurafenib |
| 16 | F/64 | Bone | BRAF V600E | Bone | Interferon-alpha |
| 17 | M/54 | Brain, bone, retroperitoneum | Not performed | NA | Methotrexate |
| 18a | M/66 | Dura, aorta, lymph nodes, peri-aortic soft tissue, retroperitoneum, pre-sacral soft tissue, bone | NRAS Q61R | Bone, Pre-sacral soft tissue | Interferon, trametinib |
| 19 | M/50 | Bone, brain, right atrium, peri-aortic soft tissue, retroperitoneum | BRAF V600E | Brain | 6-mercaptopurine, methotrexate, interferon-alpha, vemurafenib |
| 20 | F/63 | Bone, pituitary, retroperitoneum | BRAF V600E | Retroperitoneum | Interferon-alpha, vemurafenib |
| 21 | M/32 | Epidural soft tissue, bone | MAP2K1 E51_G58del, PIK3CA exon 10 | Bone | Mycophenolate, anakinra |
| 22 | M/58 | Bone, retroperitoneum, orbit | BRAF V600E | Orbit | Vemurafenib |
| 23 | F/65 | Dura, temporalis muscle, bones, brain | BRAF V600E | Dural mass | Vemurafenib |
| 24 | M/55 | Bone, retroperitoneum, spleen, peri-aortic soft tissue, cardiac valve, skin | MAP2K1 Q56P | Cardiac valve, Skin | Anakinra, interferon, clinical trial |
| 25a | M/75 | Retroperitoneum, orbit, bone | BRAF V600E | Orbital mass | Vemurafenib, dabrafenib |
| 26a | F/60 | Bone, orbit, cavernous sinus | ARAF S214A | Bone | Clofarabine, anakinra Sorafenib, Trametinib |
| 27 | F/74 | Lung | Not performed | — | Not available |
| 28 | M/15 | Bone, brain | No mutation identified | Brain, bone | Vinblastine, Anakinra, Trametinib |
| 29 | M/59 | Bone, orbit, peri-aortic soft tissue, retroperitoneum, | BRAF V600E | Orbital mass | Vemurafenib |
| 30 | M/68 | Retroperitoneum, bone, subcutaneous soft tissue | MAP2K2 Y134H | Subcutaneous soft tissue | Methotrexate, clinical trial |
| 31 | M/43 | Bone | Not performed | N/A | Not available |
| 32 | M/35 |
Bone, brain, cavernous sinus, skull base, right atrium, peri-aortic soft tissue, retroperitoneum |
BRAF V600E | Bone | Vemurafenib, Dabrafenib |
| 33 | M/41 | Orbit | BRAF V600E | Orbit | Not available |
| 34a | F/64 | Lung, bone | BRAF V600E | Lung | Dabrafenib+Trametinib |
| 35 | M/69 | Brain, peri-aortic soft tissue, retroperitoneum, bone | MAP2K1 C121S | Retroperitoneum | Anakinra |
| 36 | F/45 | Bone | BRAF V600E | Bone | Interferon, Anakinra Vemurafenib |
| 37 | M/47 | Brain, orbit, right atrium, bones | BRAF V600E | Orbital mass | Dabrafenib+Trametinib |
| 38 | F/58 | Bone, brain, skin | BRAF V600E | Skin | Vemurafenib |
| 39 | M/44 | Epidural soft tissue, peri-aortic soft tissue, right atrium, testis | No mutation identified | Epidural soft tissue, Testis | Cladribine |
| 40 | M/53 | Bone, right atrium, peri-aortic soft tissue, retroperitoneum | BRAF V600E | Retroperitoneum | Vemurafenib |
| 41 | F/28 | Lung | Not performed | — | Not available |
| 42a | M/71 | Bone, lung | BRAF V600E | Lung | Dabrafenib |
Abbreviations: M, male; F, female.
Bone marrow biopsy was performed during the disease course.
Clinical Findings and Treatments
The patients include 16 women and 26 men. Ages ranged from 15 to 75 years. Five patients were under the age of 40 years at the time of presentation, and one of them was under the age of 20 years. All patients but one had sclerotic lesions in the femur or tibia demonstrated by either “Technetium (99T) bone scan, 18-flourodeoxyglucose(18F-FDG)-PET scan, CT scan, or magnetic resonance imaging (MRI), and the one patient without long bone lesions has biopsy-proven Langerhans cell histiocytosis of the soft palate, as well as a xanthomatous CD1a( −) histiocytic infiltrate in a sclerotic scapular lesion, and both lesions were found to harbor the BRAFV600E mutation by pyrosequencing. Twenty-one (50%) had retroperitoneal infiltrates, 13 (31%) had involvement of the meninges or brain, 12 (29%) had cardiovascular (right atrium, pericardium, or peri-aorta) involvement, 8 (19%) had skin lesions, and 7 (17%) had diabetes insipidus. Three (7%) patients had disease of the bones only and no other sites of disease demonstrated by full-body PET scan. Four patients had concurrent or prior Langerhans cell histiocytosis.
Initial and subsequent therapy is known for 40 patients, and 2 were evaluated in consultation and then lost to follow-up. Initial therapy was interferonalpha for eight patients; this was the only treatment for two patients. The other six were transitioned to anakinra and/or targeted therapy. Anakinra was initial treatment for six patients and has been maintained in two patients while changed to methotrexate, interferon, or targeted therapy for the others. Targeted therapy (BRAF or MEK inhibition) was initial treatment for 14 patients. The remaining patients’ initial treatments were methotrexate, cytar- abine, imatinib, 6-mercaptopurine, mycophenolate, clofarabine, vinblastine/prednisone, and cladribine. From the time of consultation at our center, the longest period of follow-up is 48 months. Of the 40 patients followed, 4 (10%) have died and the remainder are alive in active follow-up.
Pathological Observations
Morphologic, immunophenotypic and genetic findings are summarized in Table 1 and Supplementary Table S1. Given that there were organ site-specific morphological features, these are discussed separately below and also in Table 2. The bone marrow findings, immunophenotypic, and molecular results are summarized separately at the end of the results section but also referred to in site-specific description where required.
Table 2.
Significant morphological features of Erdheim-Chester disease according to the most commonly involved regions
| Location | Typical features of histiocytic infiltrate |
Typical features of stroma and reactive infiltrate |
Differential diagnosis |
|---|---|---|---|
| Bone | - In mild fibrotic background; loose clusters of classical foamy or granular histiocytes with well-defined cell borders - In prominent fibrotic background; scant infiltrate including amorphous lipid-laden or granular histiocytes |
- Process replaces normal bone marrow - Sclerotic changes in trabecular bone with scant inflammatory cells -Inflammatory infiltrate can be prominent in focal areas which may be problematic related to the sampling - A certain degree of fibrosis present, relatively more extensive than other regions -Storiform fibrosis can be seen |
- Benign fibrous histiocytoma of bone - Osteomyelitis - Xanthogranulomatous inflammations - RDD - LCH |
| CNS | - Mononuclear infiltration as single cells or small clusters especially denser in perivascular areas - The typical histiocyte has indistinct cellular margins and non-lipidized, eosinophilic cytoplasm or only faintly foamy |
- Reactive astrocytic proliferation, sometimes associated with prominent Rosenthal fiber formation - Absent to mild reactive inflammatory infiltrate - Focal demyelination - No fibroplasia |
- Organizing infarct - Demyelinating diseases - Alexander’s disease - LCH - JXG - Granular cell astrocytoma - Regressed primary CNS lymphoma |
| Lung | - Characteristic subpleural, septal, perivascular interstitium and peribronchiolar localization of infiltrate - Lesional histiocytes with eosinophilic cytoplasm and distorted shape due to fibrosis |
- Absent to mild reactive inflammatory infiltrate - Marked fibrosis in pleura and interlobular septa |
- Drug side effect - Interstitial lung diseases in particular, usual interstitial pneumonitis - LCH - RDD |
| Skin | - Diffuse or interstitial/perivascular infiltration pattern can be seen - In both, typical xanthomatous features are seen; however, lesional histiocytes can be subtle in interstitial pattern - High possibility of accompanying Touton-type giant cells in diffuse infiltration pattern |
- Occasionally, small focus of neutrophilic abscesses - Occasional eosinophils as isolated cells - Perivascular dense lymphoid aggregates can be seen - No change in epidermis |
- Benign xanthoma - Dermatofibroma - Hypersensitivity reactions - Granulomatous mycosis fungoides - LCH - RDD - JXG |
| Orbit | - Nodular to diffuse sheets of lipid-laden or eosinophilic histiocytes with classical xanthomatous features - High possibility of accompanying Touton-type giant cells |
- Nodular lymphoplasmacytic aggregates without germinal center formation - Moderate to marked fibrosis |
- Extranodal marginal zone lymphoma - IgG4-related disease - Adult-onset asthma and periocular xanthogranuloma |
| Retroperitoneum | - Mostly, histiocytic infiltrate with xanthomatous quality - Sometimes, scant infiltrate including amorphous lipid-laden or granular histiocytes in prominent fibrotic background |
- Reactive infiltrate with prominent plasma cells - IgG4 expression may reach the cut off value of IgG4-related disease (>40%) - Occasionally, prominent fibrosis |
- IgG4-related disease - Lymphoma - Idiopathic retroperitoneal fibrosis |
| Cardiac structures | - Abundant histiocytic infiltrate with xanthomatous quality - Diffuse myocardial infiltration with extension to the depth of myocardium |
- Prominent chronic fibro-inflammatory exudation with moderate amount of fibrosis | - Myocarditis and other inflammatory processes |
Abbreviations: RDD, Rosai-Dorfman disease; CNS, central nervous system; LCH, Langerhans cell histiocytosis; JXG, juvenile xanthogranuloma- tosis; Ig, immunoglobulin; ECD, Erdheim–Chester disease.
Bone.
Fifteen bone biopsies were performed on 14 patients. The lesions were predominantly sampled from the tibia and femur, but one patient had a sacral lesion, one had a scapular lesion and another had a calvarial lesion. Histologically, all lesions involved the medullary cavities of the affected bones with marrow replacement. All lesions had various degrees of histiocytic content; however, classical foamy histiocytes were not necessarily seen in all cases. Cases with mild fibrotic background, loose clusters of classical foamy or granular histiocytes with well- defined cell borders were present (Figure 1a). In some cases, however, prominent fibrotic background obscured the morphology, and scant infiltrates comprised of amorphous lipid-laden or granular histiocytes were seen instead of classical morphology (Figure 1b). In addition, the calvarial lesion (showed moderate S100 expression) had focally dense aggregates of lymphocytes and plasma cells, mimicking Rosai–Dorfman disease (Figure 1c). A certain degree of fibrosis was seen more extensively in all bone biopsies when compared to other tissue sites. Some cases had prominent fibrosis and osteosclerosis (Figure 1d). Touton-type giant cells were seen in one case, and reactive bone formation was common. No emperipolesis or necrobiosis was seen and eosinophils were rare.
Figure 1.
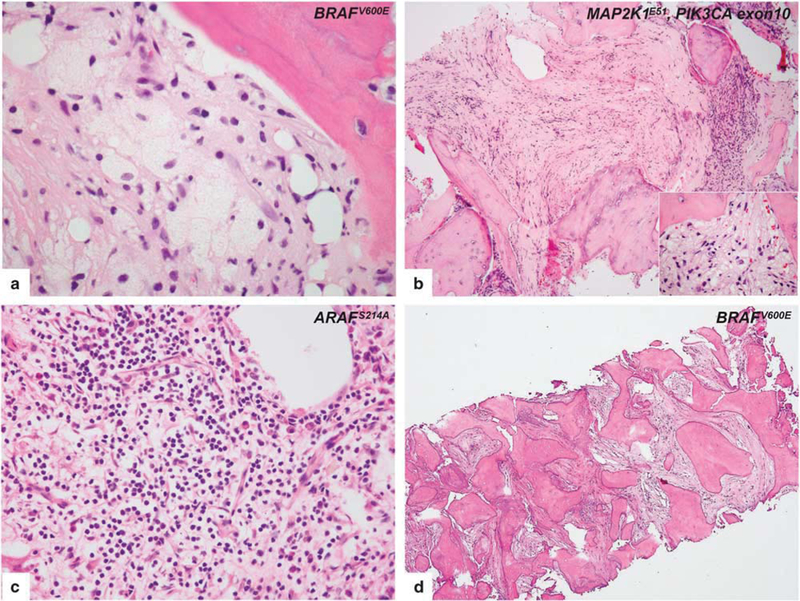
Histopathologic features of intraosseous Erdheim-Chester disease. (a) A case, harboring BRAFV600E mutation, with mild fibrotic background reveals the classical morphology of Erdheim-Chester disease involvement. Lipid-laden histiocytes tended to form loose clusters with scant inflammatory cells. (b) In a case, a prominent fibrotic background obscured the morphology, and scant infiltrate including amorphous lipid-laden and granular histiocytes (inset) were seen instead of the classical morphology. MAP2K1 and PIK3CA mutations are co-detected in this specimen. (c) In a calvarium biopsy of an Erdheim-Chester disease patient, areas of dense lymphoplasmacytic infiltrate whose presence may prompt the diagnostic consideration of osteomyelitis or Rosai-Dorfman disease. Whole exome sequencing of tumor tissue demonstrates ARAFS214A mutation in the tumor. (d) Femur biopsy of an Erdheim-Chester disease patient shows prominent fibrosis and osteosclerosis. Despite the decalcification procedure, BRAFV600E mutation is detected in that specimen.
Retroperitoneum.
Eleven retroperitoneum biopsies were performed on 11 patients. Microscopically, the typical xanthomatous quality of the Erdheim–Chester disease infiltrate was present in all the retroperitoneal biopsies examined (Figure 2a), although the variability in lesional composition and microscopic appearance sometimes made the interpretation difficult. In this region, the disease was occasionally accompanied by numerous plasma cells (Figure 2b). Other inflammatory cells included small lymphocytes, and in rare cases, very few eosinophils. In five cases, immunohistochemical staining for IgG4 was performed due to the presence of a prominent lymphoplasmacytic infiltrate and was either completely negative or highlighted only rare plasma cells. The plasma cells were often located around small vessels. The amount of fibrosis varied from mild to moderate. No extensive ‘storiform’ fibrosis was seen. Touton-type giant cells were seen in one case. No emperipolesis or necrobiosis was seen.
Figure 2.
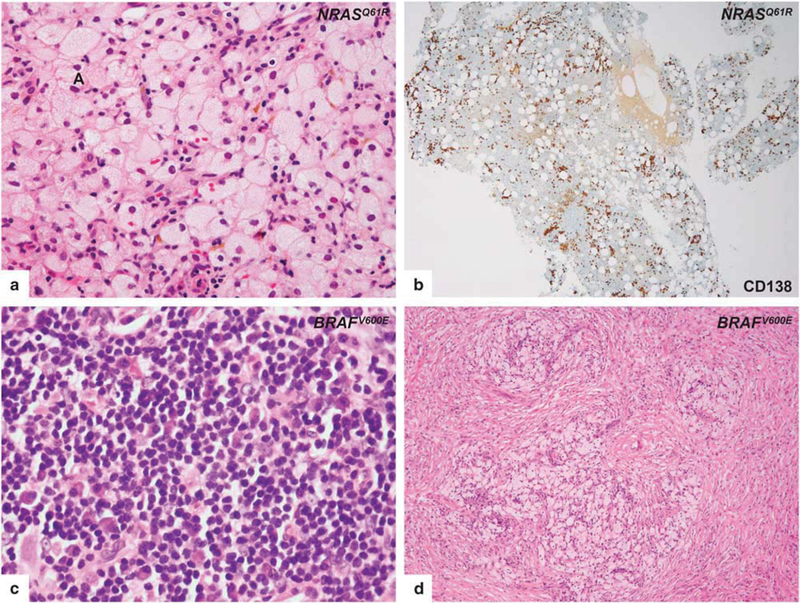
Histopathologic features of retroperitoneal (upper row) and orbital (lower row) lesions of Erdheim-Chester disease. Biopsy from retroperitoneal region, harboring NRASQ61R mutation, with typical foamy histiocytic infiltrate (a) reveals abundant plasma cell infiltration by CD138 immunostain (b). Biopsy from the orbital region, harboring the BRAFV600E mutation, without characteristic lesional histiocytes in areas containing a dense lymphoplasmacytic infiltrate (c). Representative biopsy from orbit shows nodular aggregates of foamy histiocytes (d).
Orbit.
Six orbital biopsies were performed on six patients. Nodular to diffuse sheets of foamy histiocytes infiltrating the subcutaneous adipose tissue, accompanied by variable numbers of dispersed and/ or aggregates of lymphocytes and plasma cells were seen. Nodular lymphoid aggregates were commonly scattered throughout, without reactive germinal center formation sometimes focally mimicking extranodal marginal zone lymphoma (Figure 2c). Thin sclerotic bands separated the histiocytic infiltrate into pseudonodules (Figure 2d). Variable degrees of fibrosis were often present. Three of the six cases included Touton-type giant cells. No emperipolesis, necrobiosis, or eosinophils were seen. In four cases, IgG4 immunohistochemistry was performed due to the presence of prominent lymphoplasmacytic infiltrates, and were negative.
Central nervous system.
We had five patients with biopsy-proven brain involvement. Histologically, brain lesions were less characteristic in their histology compared to Erdheim–Chester disease lesions of other localizations from the same patients. Four cases showed mild to florid cellular infiltrates composed of mononuclear cells arranged in loosely aggregated, formless clusters or present as single cells without associated fibroplasia. The infiltrates were denser in the perivascular areas and consisted of an intermixture of small lymphocytes (mostly T-cells by immunohistochemistry) and histiocytes. Other types of inflammatory cells, notably eosinophils, were found rarely as isolated cells. The typical histiocyte in these lesions had indistinct cellular margins and non-lipidized cytoplasm, which was mostly pale or more densely eosinophilic (Figure 3a). CD68 and/or CD163 outlined the histiocytic infiltrate (Figure 3b) without expression of CDla and Lan- gerin. Apart from perivascular localization, the distribution of histiocytes within the background of mononuclear cells was generally haphazard as individual cells or small foci. Many nuclei were round or oval. Grooved or lobulated nuclei were not prominent. Reactive astrocytic proliferation was a constant feature and sometimes was prominent. In the biopsy from two patients, astrogliosis with focally prominent Rosenthal fiber formation was evident and one of them was initially thought to possibly represent Alexander’s disease (Figure 3c). In another patient, areas of myelin deficits with relative preservation of axons were seen (Figures 3e and f). This finding in the context of perivascular chronic inflammation with surrounding histiocytic infiltrate and gliosis raised question of multiple sclerosis, though the total and sharply circumscribed type of myelin loss that characterizes classic demyelinating disease was not apparent. The classical foamy and xanthomatous appearance and Touton-type giant cells were seen in only one patient Figure 3d). Emperipolesis, necrosis, vascular proliferation, well-formed granulomas, and mitotic figures were consistently absent. Fibrosis, in contrast to Erdheim–Chester disease lesions from other regions, was only found in sections of cerebellum from one patient in the context of a typical Erdheim–Chester disease infiltrate (Figure 3e). Of note, expression of S100 could not be judged clearly, given the extensive staining of endogenous central nervous system structures.
Figure 3.
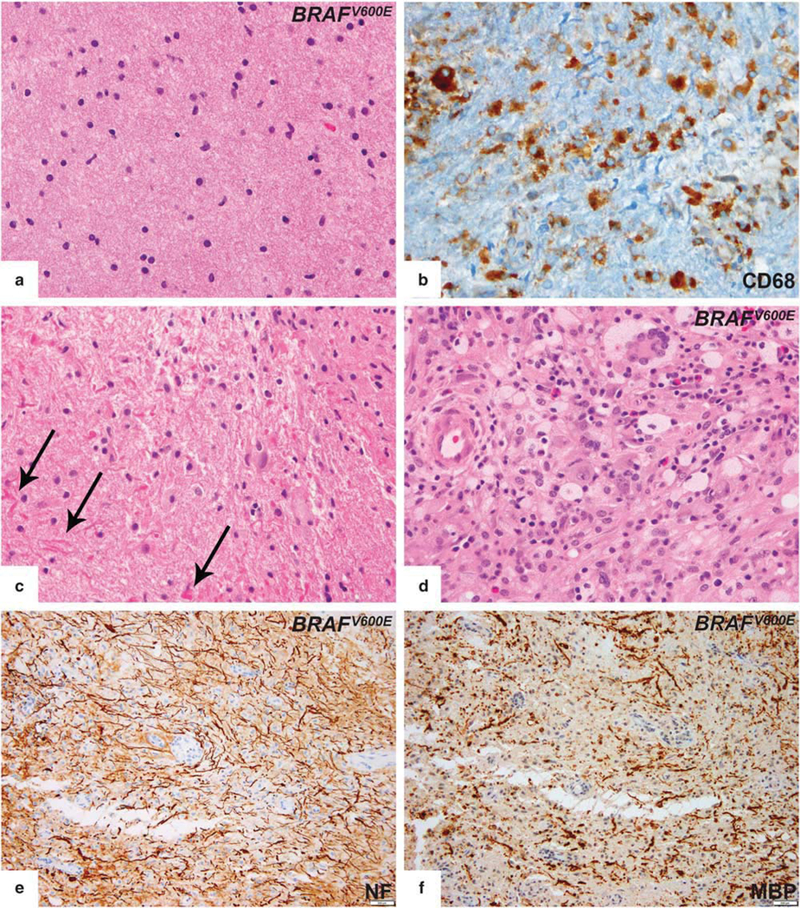
Histopathologic features of central nervous system lesions in Erdheim-Chester disease. (a) Typical brain lesion showing subtle histiocytic infiltrate without aggregate formation. Lesional histiocytes had indistinct cellular borders and non-lipidized cytoplasm. (b) Histiocytic markers (CD68 or CD163) were required to highlight the infiltrate. (c) One patient’s biopsy showed astrogliosis with focally prominent Rosenthal fiber formation (arrows). (d) Classical foamy and xanthomatous appearance in fibrotic background and Touton-type giant cells were seen in only one patient. (e,f) Neurofilament (NF) and myelin basic protein (MBP) immunostain showed areas of myelin loss with relative axonal preservation in the same region. However, this was not the total and sharply circumscribed type of myelin loss that characterizes classic demyelinating disease.
Lung.
Biopsy-proven lung involvement was found in six patients. Five of these patients were never- smokers, whereas 1 patient reported to smoke 1 pack per day for 20 years and had quit 9 years before the Erdheim–Chester disease diagnosis.
The morphologic appearances in all six patients were similar to one another and showed marked thickening of the pleura, interlobular septa, and perivascular interstitium with lesser involvement of the peribronchiolar areas (Figures 4a and b). The infiltrates were characterized by varying amounts of histiocytic infiltrates and fibrosis (Figure 4c). The most distinctive feature was an infiltrate of mononuclear histiocytic cells with associated fibrosis in an exquisitely lymphangitic distribution. Well-formed granulomas were not seen. However, in the biopsy from one patient, a loose granuloma-like structure with multinucleated giant cells as well as accompanying intense plasma cell infiltration raised the differential diagnosis with sarcoidosis. But subtle interstitial foamy histiocytic infiltration in adjacent parenchyma was present. The histiocytic cells constantly lacked nuclear grooves typical of Langer-hans’ cells. No emperipolesis or necrobiosis was seen and eosinophils were rare.
Figure 4.
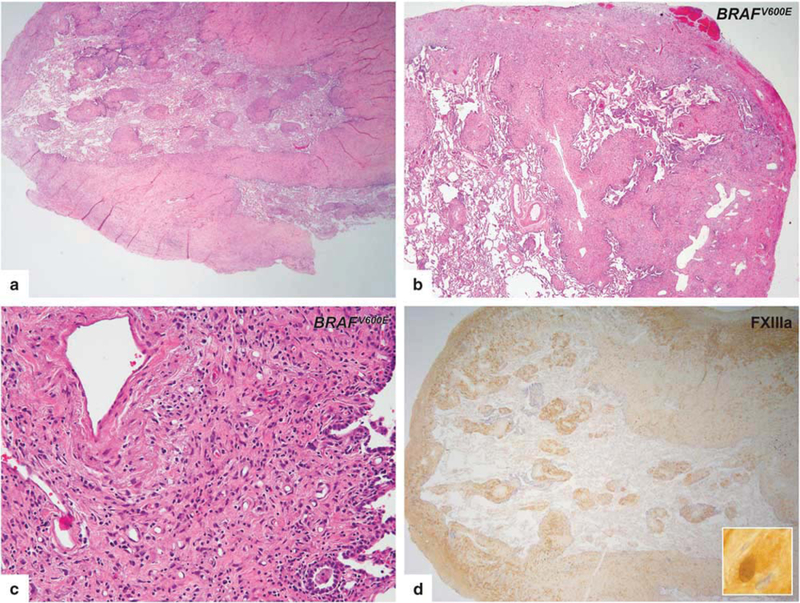
Histopathologic features of lung involvement in Erdheim-Chester disease. (a,b) Lung lesions had a characteristic septal and subpleural pattern. Perivascular interstitium with involvement of the peribronchiolar areas were also seen. (c) The infiltrates were characterized by histiocytic infiltrates in the background of fibrosis. (d) FXIIIa immunostain showing strong cytoplasmic and nuclear expression in lesional histiocytes.
Skin.
All of the patients in this group had multiple skin lesions, manifesting as papules or papulonodular lesions. No lesion was ulcerated and no large nodular masses were seen. The anatomic distribution varied, but the face (peri-orbital region) was most commonly affected, followed by the lower extremity. In the absence of therapy, no lesion resolved spontaneously. All but one patient had at least one extracutaneous biopsy documenting Erdheim-Chester disease involvement. This patient had long standing skin lesions in both lower extremities and was diagnosed as multicentric reticulohistiocytosis. Although her clinical status was initially stable, after 10 years, the patient developed dysarthria, ataxia, and bone pain. Previous skin biopsies, systemic clinical findings, and imaging studies were suspicious for Erdheim–Chester disease. Mutation analysis of a skin lesion confirmed the presence of BRAF V600E.
The microscopic findings of 19 biopsies from 7 patients were reviewed. Ten lesions showed classical features of Erdheim–Chester disease characterized by small aggregates of histiocytes in the dermis with xanthomatous features and Touton-type giant cells (Figures 5a and b). Two lesions contained a neutrophilic infiltrate with microabscesses (Figure 5c). In four lesions the histiocytes were displayed in an interstitial/perivascular pattern (Figure 5d). Five lesions from two patients displayed unusual features, including dermatofibroma-like morphology (Figure 5e). In some lesions, dense perivascular lymphoid aggregates were seen (Figure 5f). None of the dermal infiltrates was associated with epidermal changes. The extent of stromal fibrosis was variable. None of the lesions showed emperipolesis or necrobiosis.
Figure 5.
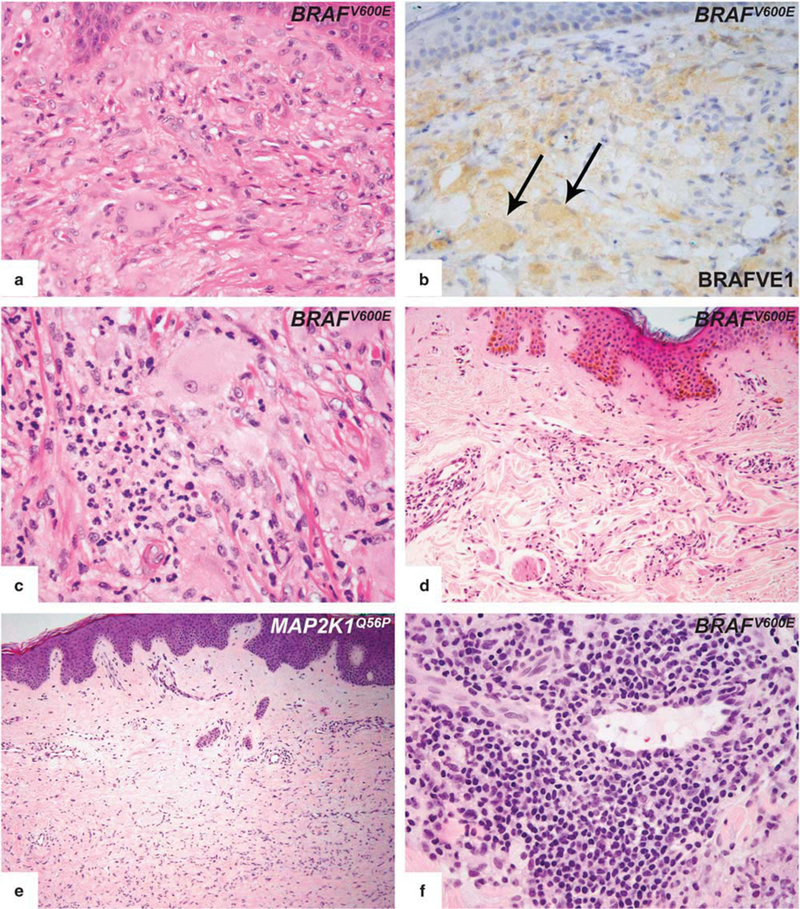
Histopathologic features of cutaneous involvement in Erdheim–Chester disease. (a) Representative cutaneous infiltrate in Erdheim–Chester disease demonstrated xanthogranulomatous quality and was dominated by large histiocytes with Touton-type giant cells. (b) BRAF VE1 immunostain disclosed cytoplasmic staining of both histiocytes and Touton cells (arrows), which confirmed that Touton cells are also part of the neoplastic process rather than reactive. (c) In some lesions, focal clusters of neutrophils were present. (d) Some lesions showed a subtle perivascular/interstitial histiocytic infiltration pattern. (e) One of the patients developed multiple dermatofibroma-like lesions during the disease course. Detection of a MAP2K1 mutation in such a skin lesion, which is morphologically unusual for an Erdheim–Chester disease involvement, confirmed the diagnosis. (f) Dense lymphoplasmacytic infiltrate constantly in perivascular location was seen in some biopsies.
Cardiac structures.
Biopsies of one atrial mass and one cardiac valve (pulmonary) were performed in two patients. Both of the lesions showed exuberant histiocytic and chronic fibro-inflammatory exudation, which was composed of numerous histiocytes with large vesicular nuclei and moderate amounts of lightly eosinophilic cytoplasm, all with areas of fibrosis seen. Typical foamy histiocytes were seen only in the cardiac valve biopsy (Figure 6a). In the atrial mass lesion, there was diffuse myocardial involvement, with extension to the depth of the myocardium (Figure 6b). The reactive lymphoid infiltrate was seen but not prominent. Other types of inflammatory cells were not seen. The extent of stromal fibrosis was variable.
Figure 6.
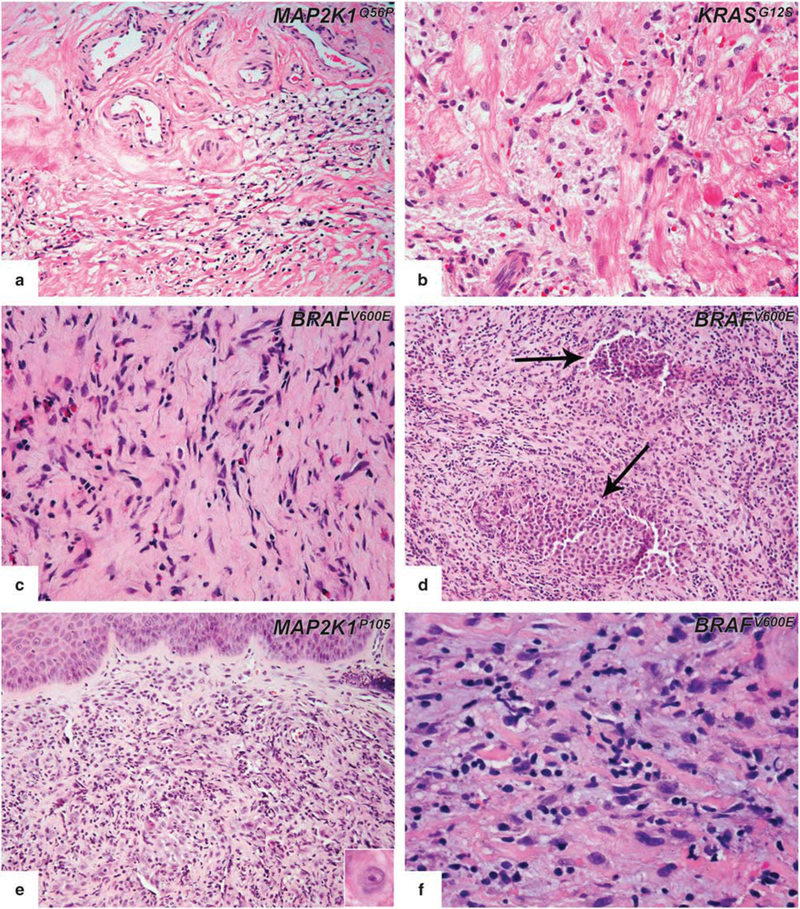
Rare biopsy sites in Erdheim–Chester disease. Pulmonary valve (a) and atrial mass (b) biopsies showed exuberant histiocytic infiltrate. In atrial mass lesion (b), the cells are infiltrating between muscle fibers and replacing the myocardial connective tissue. (c) Biopsy from an epidural mass lesion showed amorphous histiocytic infiltrate within the fibrous tissue. (d) Langerhans cell histiocytosis (arrows) and Erdheim–Chester disease lesions were present but distinguishable in the same biopsy from a patient with mixed histiocytosis. (e) Tongue involvement of Erdheim–Chester disease harbored prominent neutrophils and was mimicking reactive granulation tissue. Infiltrating histiocytes were cytologically atypical (e, inset). (f) Teeth extraction material; histiocytes with xanthomatous quality spreading into extracellular basophilic matrix.
Exceptional Sites of Involvement
Epidural soft tissues.
Three biopsies were performed on three patients. Microscopically, the lesions had ill- defined margins and revealed a fibrous tumor with rarefied sheets of intermediate-sized, eosinophilic and amorphous histiocytes (Figure 6c). Interspersed lymphocytes and plasma cells without any aggregate or germinal center formation are seen. In focal areas, foamy histiocytes were present in all three cases. No giant cells, emperipolesis, or necrobiosis were seen; and eosinophils were rare. One of the patients also had multifocal dense deposits of Langerhans cells with indented nuclei and eosinophilic cytoplasm in the same biopsy (Figure 6d). Immunohistochemical studies proved the co-existence of Langerhans cell histiocytosis aggregates with CD68+/S100+/CD1a+ expression within the Erdheim–Chester disease lesion.
Oral cavity.
One tongue biopsy and one tooth extraction were performed on two patients who developed symptomatic lesions. The intraoral lesions were observed after the pathologic diagnosis from omental lesion and chest wall mass, respectively. The first patient presented with a nodule on the anterior ventral tip of the tongue. Excisional biopsy showed a benign squamous mucosa and underlying diffuse infiltration of atypical histiocytes intermixed with moderate amounts of neutrophils resembling granulation tissue (Figure 6e). Histiocytes were medium to large sized with abundant eosinophilic cytoplasm, round to oval nuclei, and prominent nucleoli. Typical foamy histiocytes were not seen. In the second patient, upon teeth extraction, a round soft tissue lesion with a cystic lumen was noted attached to the mesial wall of the extraction site. Biopsy showed a dispersed histiocytic infiltrate with xanthomatous features among hyalinized stroma, extracellular basophilic matrix, and accompanying giant cells (Figure 6f).
Subcutaneous soft tissues.
One patient initially presented with a chest wall mass, and another patient developed a soft tissue mass on the upper back during the course of the disease. Following core biopsy, both of the lesions were composed of solid sheets of histiocytic cells with no discernible architecture. All showed similar cytomorphology and classical appearance, being composed of medium-sized, foamy histiocytes with well-defined cell borders in the background of fibrosis. The soft tissue mass from the back was also infiltrated by numerous plasma cells. IgG4 staining was positive in < 40% of the IgG-positive infiltrating plasma cells. The plasmacytic infiltrate was diffuse throughout the biopsy specimen.
Testis.
Orchiectomy was performed on two patients who developed testicular lesions seen on PET/CT during the course of disease. Both of the patients had at least one extra-testicular biopsy revealing Erdheim–Chester disease involvement. Both of the testicular biopsies showed lesions with histological features indistinguishable from those of classical Rosai-Dorfman disease (Figures 7a and b). These include a mixed lympho-plasmacytic infiltrate with interspersed histiocytes with voluminous cytoplasm, emperipolesis, and extensive S100 expression (Figure 7c). The infiltrates were interstitial and well circumscribed. The surrounding testicular parenchyma was largely unremarkable. One biopsy (Table 1; Patient 1) was analyzed for genetic mutations by whole-exome sequencing and showed the same MAP2K1mutation, which was found in the Erdheim–Chester disease lesion in the tongue biopsy.
Figure 7.
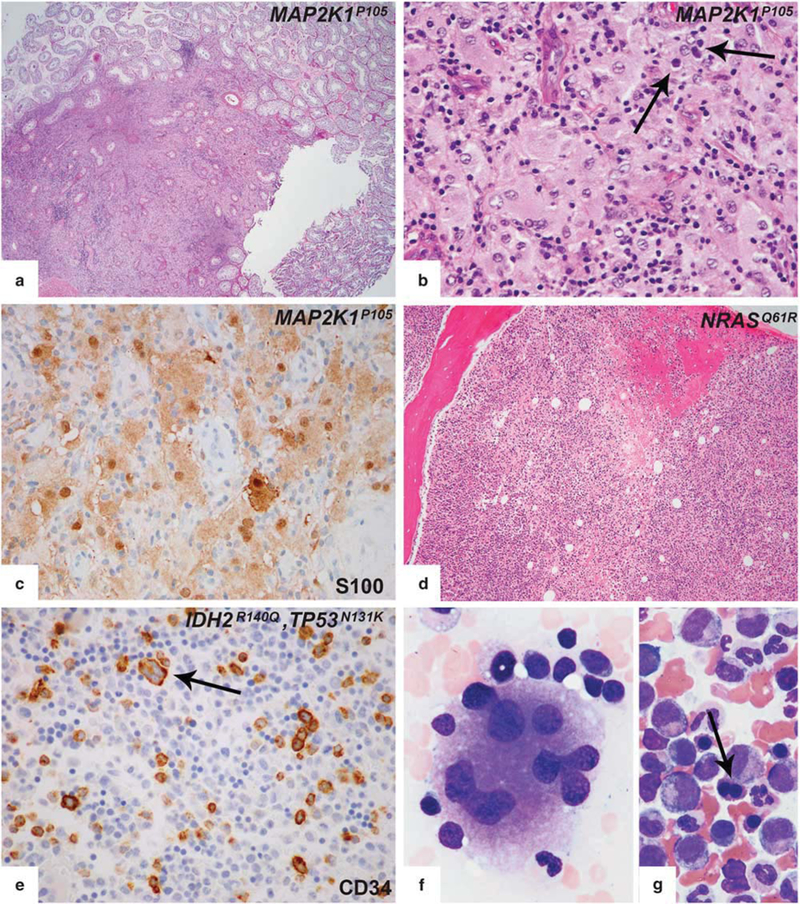
Arising testicular and bone marrow lesions in the course of Erdheim–Chester disease. (a) Testis biopsies showed replacement of the testis by Rosai-Dorfman disease with a well-defined border separating Rosai-Dorfman disease and adjacent testis. (b) Higher magnification revealed emperipolesis (arrow). (c) Significant S100 expression was seen in testis lesions. (d) Bone marrow biopsy from a patient with NRASQ61R-mutant Erdheim–Chester disease revealed accompanying chronic myelomonocytic leukemia. (e-g) Bone marrow biopsy from a patient with BRAFV600E mutant mixed histiocytosis (Erdheim–Chester disease/Langerhans cell histiocytosis) showed myelodysplastic syndrome. Immunohistochemical studies showed increased CD34-positive blasts (e). Aberrant CD34 expression was also noted in dysplastic megakaryocytes (e, arrows). BM aspirate smear of the same patient demonstrated dysplastic megakaryocytes with widely separated nuclei (f) and nuclear abnormalities in erythroid precursors (binucleation, nuclear budding) (arrow) (g).
Bone Marrow Findings
Bone marrow biopsy was concurrently or subsequently performed on eight Erdheim–Chester disease patients. Four patients had chronic myelomonocytic leukemia-1 (Figure 7d), one patient had JAK2+ unclassified myeloproliferative neoplasm, one patient had myelodysplastic syndrome with excess blasts-1 (Figures 7e–g), and two of them had only some reactive changes. Two of the patients who were diagnosed as chronic myelomonocytic leukemia-1 had asymptomatic monocytosis before they were diagnosed as Erdheim–Chester disease. In the follow-up bone marrow biopsy, the patient diagnosed with myelodysplastic syndrome progressed to acute myeloid leukemia with myelodysplasia-related changes within 6 months. Clinical and molecular features of these cases have been reported.13
Summary of Immunohistochemical Features
All specimens were positive for one or more of the histiocytic markers including CD68, CD163, or FXIIIa, while all of the specimens were completely negative for CD1a (except the case with concomitant Langerhans cell histiocytosis infiltration). S100 was positive in 12/40 (30%) cases of Erdheim–Chester disease samples (brain and testis biopsies were excluded). IgG4 immunostaining was performed in 10 cases, and none of them reached the 40% cutoff value often required for the diagnosis IgG4-related disease. BRAF V600E immunostaining was performed in 28 samples of 23 patients, 16 with BRAFV600E mutation detected by molecular studies. BRAF V600E immunostain was positive in 14 samples; all BRAFV600E-mutated cases. BRAF V600E immunostain was negative in 14 samples including 2 samples from two different BRAFV600E- mutated cases (one was detected from the lesional tissue, while the other one was detected from the urinary cell-free DNA but not on the lesional tissue by PCR) (Supplementary Table S1).
Two cases exhibited features of Erdheim–Chester disease and Langerhans cell histiocytosis. In the first case, the morphologic appearance and immunophe- notypic findings were typical for Erdheim–Chester disease in lung and bone and for Langerhans cell histiocytosis in the oropharynx. A second case had aggregates of Langerhans cell histiocytosis in a background of Erdheim–Chester disease infiltration in the same epidural mass lesion.
Summary of Molecular Features
Mutational analysis was performed in 38 cases. Somatic mutations involving BRAF (25/38), MAP2K1 (6/38), ARAF (2/38), MAP2K2 (1/38), KRAS (1/38), and NRAS (1/38) genes were detected (Figure 8). One of the cases with MAP2K1 mutation also harbored a concurrent PIK3CA mutation. In two cases, no mutation was identified. Molecular analysis detected the same MAP2K1 mutation in the tongue and testis of a patient (Table 1; Patient 1) who has an Erdheim–Chester disease-like appearance in the omental and tongue lesions and Rosai-Dorfman disease-like features in the testis biopsy.
Figure 8.
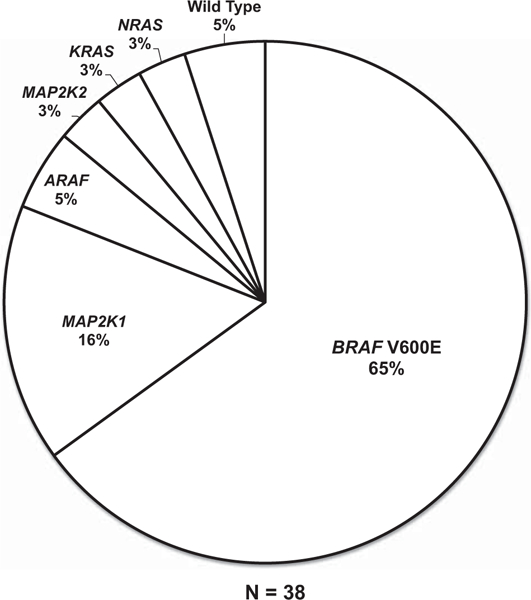
Graphical summary of diverse kinase alterations in the Erdheim–Chester disease study cohort. Pie chart illustrating the activating kinase alterations uncovered in the Erdheim–Chester disease study cases by whole exome sequencing and targeted mutational profiling.
Discussion
In this study, we provide a detailed description of the histopathology of Erdheim–Chester disease in clinical biopsy specimens to help establish pathological diagnosis of Erdheim–Chester disease early in the disease course. Our study shows variability of histopathology from patient to patient but also establishes site-specific patterns, and we also identify important pitfalls in the evaluation of potential Erdheim–Chester disease specimens.
We describe the morphological, immunohisto-chemical, and molecular spectrum of 42 Erdheim–Chester disease patients. On the basis of our series, similar with previous reports, Erdheim–Chester disease is predominantly a disease of adulthood, with an age range of 15–75 years (mean 58) with male predominance. In our cohort, two patients had a childhood history of Langerhans cell histiocytosis and developed Erdheim–Chester disease at relatively early ages (28 and 43) (Table 1; Patients 6 and 41). This observation suggests that the Erdheim–Chester disease may possibly represent an end point for some of the Langerhans cell histiocytosis patients.
We also show that myeloid neoplasms can be seen in the course of Erdheim–Chester disease in approximately 15% (6/42) of our case cohort, consistent with what has been recently demonstrated in a multi-center series.13 Since bone marrow biopsies are not a routine procedure in the work up of Erdheim–Chester disease patients, the exact ratio of accompanying myeloid neoplasms is not known. The presence of concomitant myeloid neoplasms can cause discordant and adverse responses to targeted therapies and also can be associated with a worse prognosis in Erdheim–Chester disease.13 Therefore, we recommend that a bone marrow biopsy should be routinely performed for the work up of these patients. The pathogenesis of myeloid neoplasms in Erdheim–Chester disease remains unclear but it is likely that at least a subset of the cases may share a common clonal precursor.13
The results of our study establish a broad range of morphology and clinical behavior in Erdheim–Chester disease harboring activating mutations of the MAPK pathway. While confirming the well- known features of this disease, this article documents presentation of Erdheim–Chester disease in previously unreported or unusual sites and unusual infiltration patterns which creates challenges for early and accurate diagnosis.
The histological appearances of Erdheim–Chester disease considerably vary depending on anatomical location of lesions, even in the same patient. If there are any suspicions of a possible Erdheim–Chester disease diagnosis in a particular lesion, the pathologist should request an additional biopsy on another lesion in order to confirm the diagnosis. Brain and skin lesions are especially problematic for an initial Erdheim–Chester disease diagnosis without a corroborating genomic finding.
Since the most common clinical presentation of Erdheim–Chester disease is bone pain and radiographic findings of bone lesions are pathognomonic, bone biopsies are most commonly encountered in clinical practice. Typically, marrow spaces were infiltrated by a cellular population of histiocytic cells showing xanthomatous features in the background of prominent fibrosis, which mostly lacked inflammatory cells. In some lesions, however, mixed inflammatory cells including lymphocytes and plasma cells were focally present, which caused diagnostic challenges in differentiation from Rosai-Dorfman disease and osteomyelitis. Histologically, the bone lesions must be distinguished from benign fibrous histiocytoma of bone (fibrous xanthoma or xanthofibroma, nonossifying fibroma), Langerhans cell histiocytosis, Rosai-Dorfman disease, xanthogranulomatous inflammation, and fibrous dysplasia with xanthomatous reaction secondary to hemorrhage.
Cutaneous involvement occurs in approximately 20% of cases. Skin lesions are neither characteristic nor specific. Most of the cases show a nodulardiffuse infiltration with foamy histiocytes and— more commonly than other locations—with accompanying Touton-type giant cells. We also noticed that some cases show subtle interstitial-perivascular histiocytic infiltration with variable cytologic features. In one case (Table 1; Patient 24) we initially considered an incidental dermatofibroma, since foamy histiocytes and Touton-type giant cells were absent. However, the molecular analysis identified the same MAP2K1 mutation in the dermatofibroma-like skin lesion as previously detected in a cardiac valve biopsy with classical morphology, thereby revealing it as an Erdheim–Chester disease lesion. In skin biopsies most important differential diagnoses include other histiocytic disorders and lymphomas. For patients who already have an Erdheim–Chester disease diagnosis, biopsy of skin lesions, even if not classic, may be a minimally invasive means to establish the mutational status of the patient for targeted therapies.
In contrast to Erdheim–Chester disease involvement in other tissue sites, the histology of lung involvement by Erdheim–Chester disease is distinct. The low-power appearance of Erdheim–Chester disease in pulmonary wedge biopsy specimens is characterized by a strikingly lymphangitic distribution. The infiltrate is detected in the visceral pleura, in the interlobular septa, and around the broncho-vascular bundles. Owing to this lymphangitic pattern, the transbronchial biopsy may not be diagnostic and also not appropriate for molecular testing due to lack of lesional histiocytes. Fibrosis typically involves the pleura and interlobular septa but is much less pronounced in the interstitial, interalveolar septa. The histiocytes can be scarce within the fibrosis, which can make the diagnosis of Erdheim–Chester disease very difficult, especially if small biopsies are performed. Most of the lesional histiocytes show nuclear, as well as strong cytoplasmic expression for FXIIIa immunostain in Erdheim–Chester disease, whereas only nuclear expression is seen in intraalveolar macrophages (Figure 4d). This finding is especially helpful in highlighting lesional histiocytes in Erdheim–Chester disease and differentiating Erdheim–Chester disease from the lesions of drug side effects and interstitial usual pneumonitis, which also harbor exuberant reactive histiocytic infiltrates and fibrosis. Of note, while pulmonary Langerhans cell histiocytosis is found almost exclusively in cigarette smokers, only one of the six patients with pulmonary Erdheim–Chester disease in the present study was a heavy smoker.
It merits particular emphasis that brain biopsy material from patients with Erdheim–Chester disease often fails to display the overtly xanthomatous histiocytic infiltration characteristic of this disorder in bony and other locations. Also, in contrast to meningeal and other tissue site involvement by Erdheim–Chester disease, fibroplasia typically does not occur within the brain parenchyma. Immuno-histochemical assessment for expression of CD163 or other histiocytic ‘markers’ may be required to demonstrate both the presence and the enumeration of histiocytes, which may display ambiguous morphologic features leading to their misidentification as glial. The presence of perivascular lymphocytic cuffing may prompt misdiagnosis as a non-specific inflammatory process and florid astrogliosis with Rosenthal fiber formation may further obscure the picture. All relevant immunohistochemical methods must be employed to address a broad range of differential diagnostic considerations such as organizing infarct, demyelinating disease, Langerhans cell histiocytosis, and granular cell astrocytoma. The distinction of Erdheim–Chester disease from regressed primary central nervous system lymphoma is practically impossible in the absence of corroborating molecular diagnostic findings, radiologic and histologic confirmation of the expected skeletal involvement, or other patterns of extraneural disease that would support Erdheim–Chester disease. While Erdheim–Chester disease may involve the dura and cerebrum, a particularly high level of suspicion should be attached to histiocyte-rich lesions presenting in the pons and cerebellum.
Although Erdheim–Chester disease lesions generally lack a significant plasma cell infiltrate, some of the cases from retroperitoneal, orbital, and subcutaneous sites have abundant plasma cell infiltrate. This may be related to the special microenvironment of these sites. This finding often mimics IgG4-related disease, which also can frequently involve retroperitoneal and orbital tissues. The similarities between the Erdheim–Chester disease and IgG4-related disease include abundant fibrosis, the pseudonodular inflammatory infiltrate rich in lympho- plasmacytic cells, and the absence of cellular atypia. The presence of CD68(+)/CDla( − ) foamy histiocytes and the absence IgG4 expressing plasma cells favor Erdheim–Chester disease. Fibrosis can be seen in both disorders but is often more prominent and dense in IgG4-related disease. Although none of our cases had an IgG4+ plasma cell rich infiltrate greater than the 40% cutoff value, some recent studies reported that IgG4+ plasma cells could exceed 40% in Erdheim–Chester disease lesions.14 Therefore, it is important to take Erdheim–Chester disease into the differential for those lesions presenting in the retroperitoneum and orbit, even with large numbers of infiltrating IgG4+ plasma cells, with the suggestion for clinical and radiographical correlation.
In our cohort, there were only two testicular biopsies from known Erdheim–Chester disease patients, and both demonstrated Rosai-Dorfman disease-like morphology. There was no other biopsy sample from another site showing entirely Rosai- Dorfman disease-like morphological features in the remaining cases. In one of those cases (see Tables 1 and 2, Patient 1) the same MAP2K1 mutation was detected in the testis (Rosai-Dorfman disease-like) and tongue (Erdheim–Chester disease-like) lesions of the patient. In the absence of a larger cohort of testicular biopsies, we can speculate whether this finding is truly an accompanying Rosai-Dorfman disease with a common progenitor sharing the same MAP2K1 mutation, as analogous to the Langerhans cell histiocytosis-Erdheim–Chester disease association with a common BRAFV600E mutation or a morphological manifestation of Erdheim–Chester disease in the testis microenvironment.
Immunophenotypic features of Erdheim–Chester disease are non-specific. Therefore immunohisto-chemical studies are supportive rather than diagnostic. Although S100 expression has been described in several Erdheim–Chester disease case reports, the exact frequency is not known, and it seems that nearly one third of Erdheim–Chester disease patients show some overexpression for this marker. Nevertheless, we did not note any Erdheim–Chester disease lesion with extensive expression of S100. Altogether our experience confirms the notion that S100 expression does not exclude Erdheim–Chester disease by any means, although robust expression was not found in any of our cases.
Immunohistochemistry using the VE1 antibody for detecting BRAF V600E mutant protein has been previously shown to be highly reliable in evaluation of melanoma, thyroid carcinoma, and hairy cell leukemia specimens. However, in BRAF V600E- mutated Erdheim–Chester disease, VE1-staining is generally very weak, scattered, and sometimes limited to only a few histiocytes. Furthermore, VE1 was negative in two of our biopsies harboring the BRAF V600E mutation by molecular analysis. Therefore, VE1-negative Erdheim–Chester disease cases have to be confirmed by BRAF V600E mutational analysis when adequate material is available.
Genetic data were available for 38 cases. Various molecular abnormalities were found, but strikingly, all of the mutations were within the MAPK pathway. Molecular studies are invaluable for supporting a diagnosis of Erdheim–Chester disease occurring in unusual sites or showing unusual morphological features, although we could not find any correlation between molecular findings, morphological or demographic features, or the sites of involvement.
Detection of somatic MAPK pathway mutations can also assist in the differential diagnosis of Erdheim–Chester disease and related histiocytic neoplasms. Both Erdheim–Chester disease and Langerhans cell histiocytosis share BRAF V600E mutation but unique patterns have emerged for other disorders. BRAF V600E mutations are not seen in juvenile xanthogranuloma and Rosai-Dorfman disease, whereas 18% of juvenile xanthogranuloma cases have been found to have an ARAF mutation9 often co-occurring with activating NRAS mutations. Similarly, NRAS and KRAS mutations are present in 12.5 and 25% of Rosai-Dorfman disease patients; respectively.9 Given these findings, the presence of a BRAF V600E mutation would support the diagnosis of Erdheim–Chester disease rather than juvenile xanthogranuloma, which are otherwise indistinguishable from each other by conventional histopathology.
A total of 42 cases have been included in our overview, and this is the largest number of Erdheim–Chester disease patients analyzed from a pathological perspective to date. The rarity of this disease and diversity of clinical and pathological features is a challenge for clinicians and pathologists alike. We hope that recognition of the broad spectrum of pathologic manifestations of Erdheim–Chester disease described in this study will help practicing clinicians and pathologists to diagnose Erdheim–Chester disease early in the disease course and manage these patients effectively.
Supplementary Material
Acknowledgments
This study was supported in part by the Sloan Kettering Institute for Cancer Research Cancer Center Support Grant (P30CA008748). BHD is supported by the American Society of Hematology Senior Research Training Award for Fellows and the New York State Council on Graduate Medical Education Empire Clinical Research Investigator Program Fellowship. ELD and OA-W are supported by grants from the Erdheim–Chester Disease Global Alliance and the Histiocytosis Association. OA-W is supported by grants from the Edward P. Evans Foundation, the Dept. of Defense Bone Marrow Failure Research Program (BM150092 and W81XWH-12–1-0041), NIH/NHLBI (R01 HL128239), an NIH K08 Clinical Investigator Award (1K08CA160647–01), the Josie Robertson Investigator Program, a Damon Runyon Clinical Investigator Award, an award from the Starr Foundation (I8-A8– 075), the Leukemia and Lymphoma Society, and the Pershing Square Sohn Cancer Research Alliance.
Presented in part on 14 March 2016 at the 105th Annual Meeting of the United States and Canadian Academy of Pathology (USCAP), Seattle, WA, USA.
Footnotes
Disclosure/conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on Modern Pathology website (http://www.nature.com/modpathol)
References
- 1.Estrada-Veras JI, O’Brien KJ, Boyd LC, et al. The clinical spectrum of Erdheim–Chester disease: an observational cohort study. Blood Adv 2017;1:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mazor RD, Manevich-Mazor M, Shoenfeld Y. Erdheim–Chester disease: a comprehensive review of the literature. Orphanet J Rare Dis 2013;8:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavalli G, Guglielmi B, Berti A, et al. The multifaceted clinical presentations and manifestations of Erdheim–Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis 2013;72:1691–1695. [DOI] [PubMed] [Google Scholar]
- 4.Arnaud L, Gorochov G, Charlotte F, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim–Chester disease: a single-center series of 37 patients. Blood 2011;117:2783–2790. [DOI] [PubMed] [Google Scholar]
- 5.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim–Chester disease but not in other non-Langerhans cell histiocytoses. Blood 2012;120:2700–2703. [DOI] [PubMed] [Google Scholar]
- 6.Ozkaya N, Dogan A, Abdel-Wahab O. Identification and targeting of kinase alterations in histiocytic neoplasms. Hematol/Oncol Clin 2017;31:705–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim–Chester disease. Blood 2014;124:483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colomba E, Hélias-Rodzewicz Z, Von Deimling A, et al. Detection of BRAF p.V600E mutations in melanomas: comparison of four methods argues for sequential use of immunohistochemistry and pyrosequencing. J Mol Diagn 2013;15:94–100. [DOI] [PubMed] [Google Scholar]
- 9.Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov 2016;6:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hyman DM, Diamond EL, Vibat CRT, et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 2015;5:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diamond EL, Abdel-Wahab O, Pentsova E, et al. Detection of an NRAS mutation in Erdheim–Chester disease. Blood 2013;122:1089–1091. [DOI] [PubMed] [Google Scholar]
- 12.Diamond EL, Abdel-Wahab O, Durham BH, et al. Anakinra as efficacious therapy for two cases of intracranial Erdheim–Chester disease. Blood 2016;128: 1896–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papo M, Diamond EL, Cohen-Aubart F, et al. High prevalence of myeloid neoplasms in adults with non-langerhans cell histiocytosis. Blood 2017;130: 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gianfreda D, Musetti C, Nicastro M, et al. Erdheim–Chester disease as a mimic of IgG4-related disease: a case report and a review of a single-center cohort. Medicine (Baltimore) 2016;95:e3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.