

Abstract
Recent studies highlight the importance of the RB1 tumor suppressor as a target for cancer therapy. Canonically, RB1 regulates cell cycle progression and represents the down-stream target for CDK4/6 inhibitors that are in clinical use. However, newly discovered features of the RB1-pathway suggest new therapeutic strategies to counter resistance and improve precision medicine. These therapeutic strategies include deepening cell cycle exit with CDK4/6 inhibitor combinations, selectively targeting tumors that have lost RB1, and expanding the therapeutic index by mitigating therapy-associated side effects. In addition, RB1 impacts immunological features of tumors and the microenvironment that can enhance sensitivity to immunotherapy. Lastly, RB1-specifies epigenetically determined cell lineage states that are disrupted during therapy resistance and could be re-installed through the direct use of epigenetic therapies. Thus, new opportunities are emerging to improve cancer therapy by exploiting the RB1-pathway.
Keywords: RB1, retinoblastoma, E2F, CDK4, cyclin, palbociclib, immunotherapy, epigenetics, EZH2
The canonical RB1-pathway and FDA-approved therapy:
Our current understanding of the RB1 pathway is based on decades of research that together provide a frame-work which also accurately describes numerous clinical observations (Figure 1). Conventionally the RB1-pathway is used to describe the mechanisms through which mitogenic or oncogenic signals drive the progression from G1 to S-phase of the cell division cycle (reviewed in [1–4]). These signals elicit the activation of cyclin dependent kinases CDK4 or CDK6. This is believed to represent the key interface between signal transduction pathways (e.g. receptor tyrosine kinases) and the cell cycle. The activation of CDK4 or CDK6 is driven by multiple factors, including the induction of D-type cyclins that are required for catalytic activity[5, 6]. CDK4/6 initiates the phosphorylation and inactivation of the RB1 tumor suppressor. RB1 has multiple functions that will be discussed in more detail. However, one function is clearly the repression of a transcriptional program that includes multiple genes that are essential for DNA-replication and mitotic progression (see Box 1)[7]. Inactivation of RB1 thus allows for the expression of down-stream genes that are necessary for the cell cycle to progress into S-phase and beyond. Genes that drive RB1 inactivation in this circuit are well-established oncogenes (e.g. CDK4 and Cyclin D1). In contrast genes that antagonize CDK4/6 activity (e.g. the CDKN2A gene encoding the p16INK4a protein) are tumor suppressors[5, 8]. This simple linear pathway has stood the test of substantial scrutiny through the years, but two key findings underpin the overall framework. First, preclinical studies demonstrated that RB1 is required for growth inhibition associated with inhibiting CDK4/6 activity. This has been shown by directly targeting CDK4/6 (ie. using antibodies or RNAi)[9], expressing the CDK4/6 inhibitor p16INK4a[10], and more recently by using pharmaceutical CDK4/6 inhibitors[11]. Second, genetic and epigenetic alterations of different components within the RB1-pathway are mutually exclusive in clinical cancer specimens. This finding was first illustrated by immunohistochemistry and targeted analysis in cell lines[12], but has remained a constant feature of essentially all tumor-types that have been subjected to DNA sequencing[13]. Combined, these findings support a linear pathway, highly conserved across cancers, wherein several mechanisms of pathway alteration have similar down-stream effects on the cell division cycle.
Figure 1. Canonical RB1-pathway:

In the canonical pathway mitogenic signals lead to the activation of CDK4/6 complexes with D-type cyclins. These kinases initiate the phosphorylation and inactivation of the retinoblastoma tumor suppressor, thereby leading to the de-repression of E2F regulated genes. These proliferative signals can be antagonized by multiple anti-proliferative signals which can directly limit the activation of CDK4/6 or induce the expression of endogenous inhibitors exemplified by p16ink4a.
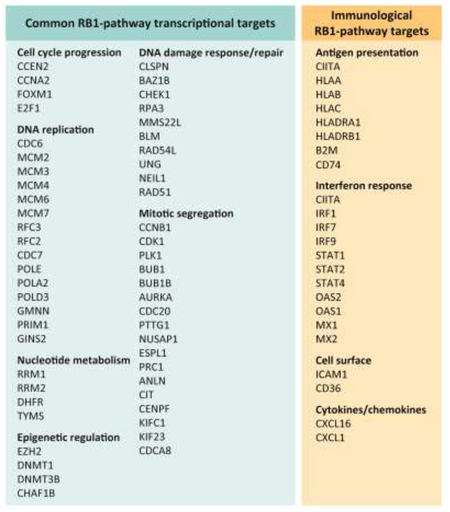
Box 1. RB1 transcriptional targets.
Common repression target genes:
Gene expression analysis from multiple models of RB1 deletion or RB1 activation have identified a highly conserved signature of genes. These genes are involved in multiple processes relevant to the cell cycle, but also play key roles in DNA repair and epigenetic programming. Notably, the genes control multiple different steps in critical features of proliferation control. For example, amongst genes involved in DNA replication are those involved in licensing, initiation, and polymerization. Similarly there are numerous genes that control different sepis in mitosis including entry, exit and cytokinesis. In tumor samples these genes are co-regulated and exhibit a high-degree of correlation indicative of being controlled through a single pathway.
Context dependent activation target genes:
In contrast with genes that are repressed through by RB1, the genes that are upregulated are more variant and a consistent signature has not emerged across the multitude of gene expression studies. However, with CDK4/6 inhibitor mediated RB1 activation there is a signature of antigen presentation and interferon inducible genes that has been identified across several independent studies. While this signature bears some similarities to the senescence-activated secretory phenotype (SASP), key hallmark SASP genes (e.g. IL6, IL8, IL1B) are not significantly induced in the context of CDK4/6 inhibition in several studies.
Multiple targeted therapeutic agents impinge on this canonical RB1 pathway[14, 15]. For example endocrine therapy in breast and prostate cancer is known to elicit clinical activity, at least partially, by inhibiting CDK activity and activating RB1[16, 17]. Similarly, many therapies that impact on mitogen signaling (e.g. MEK or EGFR inhibitors) lead to G1 cell cycle inhibition as part of the mechanism of action. However, EGFR and MEK inhibitors can also elicit tumor cell death by other mechanisms and many pharmaceutical CDK-inhibitors target multiple different CDK complexes and elicit toxicity through mechanisms independent of the RB1 pathway[6]. Only the advent of highly selective CDK4/6 inhibitors allowed an unambiguous case to be made that therapeutic efficacy was a direct feature of targeting the RB1 pathway[6, 18].
Currently, there are three FDA-approved CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib)[18–20]. Interestingly, although CDK4/6 inhibitors were initially evaluated in multiple tumor types, the main clinical activity has emerged in ER+ breast cancer [21, 22]. This likely reflects a unique dependence of luminal breast cancer cells on CDK4/6 activity. As would be expected, the mechanism of action of CDK4/6 inhibitors is largely cytostatic and the drugs limit proliferation of tumor cells typically without inducing substantial cell death in most tumor types[11, 23–25]. This is largely consistent with observations in the clinic. Neoadjuvant studies in ER+ breast cancer show that palbociclib elicits profound suppression of Ki67 as a single agent and in combination with endocrine therapy[26, 27]. Interestingly, the effects on cell proliferation appears to be reversible in clinical cases, illustrating one of the limitations associated with the utilization of CDK4/6 inhibitors[26]. While CDK4/6 inhibitors elicit a profound impact on delaying the progression of ER+ metastatic breast cancer and have transformed the treatment of disease, veritably all patients ultimately progress on treatment [28, 29]. Thus, while targeting the RB1 pathway is clinically useful, there are clearly opportunities to better improve durable disease control and to counter disease progression.
Cell cycle plasticity and enhancing depth of cell cycle exit as a therapeutic strategy:
While CDK4/6 inhibitors have had clinical success and can clearly suppress the progression of disease, in many disease settings the effect is rather modest or negligible [22]. It had been surmised that RB1-proficient tumors should be largely incapable of bypassing CDK4/6 inhibition, as is supported by the general observation that the vast majority of RB1-proficient tumor cells respond to CDK4/6 inhibitors[24, 30]. However, emerging data indicates that the cell cycle has considerably more plasticity than otherwise appreciated and resistance to CDK4/6 inhibition can clearly occur in the presence of RB1. It now appears that in many tumor contexts, there exist parallel or adaptive pathways that allow for cell cycle progression in spite of pharmacological CDK4/6 inhibition (Figure 2). Multiple studies over the last year have begun to shed-light on this “dark-side” of the cell cycle, where other CDK complexes compensate for the pharmaceutical suppression of CDK4/6 and limit therapeutic efficacy[31–34]. These finding are congruent with prior observations indicating cells can divide in the genetic absence of CDK4 and 6 activity and clearly illustrate the involvement of compensatory pathways [35].
Figure 2. Deepening cell cycle exit with combination therapies:

While the RB1-pathway is generally considered linear, the activity of both CDK4/6 and CDK2 can be induced by oncogenic signaling pathways. In the context of CDK4/6 inhibition, there can be adaptive upregulation of both cyclin D1 and cyclin E. These processes are linked to oncogenic signaling and can be ameliorated by productive combination therapies with agents targeting different signaling pathways. The basis for this cooperation is believed to involve the inhibition of CDK2 activity and maintenance of RB1 activity.
There are several features of the response to CDK4/6 inhibitors that likely represent determinants of transient vs. durable response. A commonly observed feature of response to CDK4/6 inhibition is the upregulation of cyclin D1[31–33, 36]. Limiting this adaptive response enhances cell cycle exit. This can be achieved by combining CDK4/6 inhibitors with endocrine therapy in hormonally dependent cancers, or MEK, EGFR, PI3K, and MTOR inhibitors depending on the tumor context [31, 34, 36, 37] (Figure 2). The presence/accumulation of cyclin E and CDK2 activity represents another common adaptation that contributes to CDK4/6 inhibitor resistance[31, 36]. In fact, in recently published studies cyclin E transcript levels have emerged as a predictor of response to palbociclib and fulvestrant in metastatic ER+ breast cancer[38]. What dictates which of these mechanisms mediate cell cycle plasticity and resistance to CDK4/6 inhibition in RB1-proficient tumors remains unclear. However, it would appear that maintenance of CDK2 activity in the presence of CDK4/6 inhibitors is commonly used by cancer cells to resist therapy[31]. Whether the tissue of origin or the genetic milieu of the tumor dictates the mechanism of resistance is largely unknown. Since this intrinsic resistance can be ameliorated via suppression of specific oncogenic signaling pathways (e.g. EGFR)[39, 40], it suggests oncogenic events prevalent in some tumor types can drive a cell cycle that is only partially dependent on CDK4/6 activity. Conversely, tumor types such as melanoma appear to be more sensitive to CDK4/6 inhibition than other tumor types (e.g. colon cancer and pancreatic cancer) that have analogous oncogenic drivers in the RAS/RAF pathway[22]. Similarly, luminal ER+ breast tumors appear to be particularly sensitive to CDK4/6 inhibition and less prone to adaptation. Thus, the cell of cancer origin likely also influences this cell cycle plasticity.
Interestingly, nearly all drug combinations that cooperate with CDK4/6 inhibition do so by enforcing the full-blockade of RB1 phosphorylation, inhibiting CDK2 activity, and suppressing core RB1 target genes (Figure 2 and Box 1). These combinations include targeting the MEK/RAF, PI3K, MTOR, EGFR, and endocrine pathways and are being tested in multiple clinical settings [34, 36, 37, 39–41]. Importantly, there appears to be a tissue context for cooperation between therapies. For example, PI3K inhibitors appear to be potent in cooperating in breast cancer[37] while MEK inhibitors are particularly potent in RAS-driven tumor[32, 42]. There are approximately 300 genes that have been demonstrated to be repressed by RB/E2F complexes[43]. These genes are involved in a variety of cellular functions including DNA damage repair, chromosome segregation, DNA replication, transcription, and regulation of chromatin structure. Thus, the RB1 pathway function affects multiple, fundamental cancer cell properties. Nonetheless, many of these genes are essential for cellular proliferation and viability as determined by Crispr/Cas9 screens[44]. From a therapeutic perspective, therefore, as long as this feature of the RB1-pathway can be effectively engaged, ostensibly no cell should be able to divide. Deep, RB1-mediated cell cycle arrest has characteristics of senescence, and CDK4/6 inhibition in select combinations has been shown to induce a senescent-like, complete cell cycle arrest [41, 45]. Whether such a response occurs clinically is less clear since ER-positive breast cancers rapidly regain proliferative potential with the cessation of therapy[26]. Presently, a large number of clinical trials are utilizing targeted agents to enhance the durability of clinical response with CDK4/6 inhibitors, potentially to deepen cycle arrest. However, outside of ER-positive breast cancer and endocrine therapy, the results of only a few of these trials have been reported. Therefore, whether the potent synergies that have been observed in pre-clinical studies will make a difference clinically remains largely unknown.
Tumor heterogeneity and targeting RB1-deficient cells:
Loss of RB1 is one of the key mechanisms that yields acquired resistance to endocrine therapies, oncogenic pathway targeted therapies, and CDK4/6 inhibitors [46–48]. Presumably, this reflects evolutionary selection for RB1 loss as a means to circumvent therapies that inhibit the cell cycle as their primary mechanism of action. This acquired resistance may occur through selection of tumor sub-clones with a pre-existing RB1 loss of function mutation (Figure 3). Such sub-clonal selection has been observed in clinical settings[47, 49]. Alternatively, emergence of de novo RB1 loss of function also appears to occur[46, 47, 50]. However, underlying mutagenic processes (either point-mutation or gene deletion) are presumably dependent on cell cycle progression. Therefore, if it is possible to therapeutically yield complete cell cycle exit, tumor evolution would ostensibly cease. Consistent with the importance of ongoing cell cycle progression, experimentally inducing acquired resistance to CDK4/6 inhibition in breast cancer models requires using of low, partially active, concentrations of CDK4/6 inhibitor to enrich for RB1 loss or cyclin E amplification that drive resistance in the clinical setting[46, 51]. Irrespective of how or when RB1 is lost, treating tumors that have lost RB1 is becoming a progressively more significant element of clinical practice.
Figure 3. Distinct forms of genetic heterogeneity that drive RB1 loss with therapeutic selection:

In the presence of different cytostatic therapies, there is a selective pressure for tumors that have lost RB1. This selection can allow for enrichment of pre-existing subclonal mutations or provide the basis for selection of de novo RB1 mutations. Tumors that are deficient in RB1 have been shown to be selectively sensitive to agents that target DNA-replication (CHK1 inhibitors) and mitotic segregation (PLK1 and AURK inhibitors).
One therapeutic approach for treating RB1-deficient tumors is the use of conventional cytotoxic chemotherapy that typically targets rapidly dividing cells (e.g. taxanes). RB1-deficient tumors appear to remain sensitive to standard of care chemotherapy. Indeed, breast cancers that are RB1-deficient are more prone to have a complete response to neoadjuvant chemotherapy[52, 53]. Similarly, small cell lung cancer, which is largely driven by RB1 loss, is highly responsive to chemotherapy (combination chemotherapy overall response rates greater than 50% with multiple regimens). Presumably, this sensitivity reflects the lack of critical cell cycle checkpoints that would protect slower proliferating tumors. While effective initially, it is also clear that RB1-deficient tumors evolve to chemotherapy resistance as is particularly apparent in the context of small cell lung cancer where systemic therapy is rarely curable[54].
Over the last year substantial effort has been directed at defining synthetic lethal vulnerabilities in cancer cells lacking the RB1 tumor suppressor (ie. mutated, deleted, or silenced). From this work a number of clinically testable therapeutic approaches have emerged. Interestingly, the drugs that have emerged from this work fall into categories that largely target elements of the cell cycle checkpoint machinery (ie. DNA replication checkpoint or spindle assembly checkpoint). Preclinical research focused on triple negative breast cancer (which exhibits RB1 loss in approximately 30% of cases) identified sensitivity to checkpoint kinase (CHK), CDC25 phosphatase, polo-like kinase (PLK) or aurora kinase (AURK) inhibitors as being modified by the status of RB1[55, 56]. Independent drug screening efforts identified RB-loss selective sensitivity to AURK inhibitors across large panels of cell lines[57, 58]. Although the mechanisms of sensitivity appear to be largely dependent on changes in cell cycle dynamics manifest through RB1 loss, different studies have focused on distinct targets for RB1-deficient specific activity. For example, AURKB and AURKA were each defined as targets of vulnerability, and in both cases the deregulation of genes controlling the spindle assembly checkpoint (e.g. BUB1B or PTTG1) was associated with increased sensitivity [57, 58]. While provocative and providing important approaches for potentially treating RB1-deficient tumors, there could be a degree of context specificity that will need to be determined for optimal clinical implementation.
Routine clinical evaluation of RB1 status to inform treatment assignment is not currently standard of care. In the context of CDK4/6 inhibitors, it was initially thought that screening for RB1 status would be important to identify patients most likely to respond favorably. Although it is clear from recently published studies that RB1-deficient tumors rapidly progress on CDK4/6 inhibitor therapy[46], evaluation of RB1 status is not part of the FDA-approved indication in ER+/HER2- tumors. At present there is only one clinical trial open that specifically screens for RB1 loss for inclusion, and this is a trial of the CHK1 inhibitor prexasertib for advanced solid tumors (i). This trial is complemented by a clinical trial in small cell lung cancer, where >90% of tumors are expected to be RB1-deficient (ii). While biomarker-directed clinical trials with PLK1 and AURK inhibitors have heretofore not included the synthetic vulnerability of RB1-deficient tumors, there are likely developing trials based on the compelling preclinical data. In general, advancing a precision approach for exploiting the RB1-pathway will require more common assessment RB1-status in clinical practice (especially in recurrent and/or metastatic disease). Only through well-controlled clinical trials can promising pre-clinical findings inform a rational approach to treating tumors that have acquired RB-deficiency in response to targeted therapies.
Role of CDK4/6 inhibitors in reducing treatment related morbidity and expanding therapeutic index against RB1-deficient tumors:
Standard of care cancer treatments based on genotoxic chemotherapy and radiation carry substantial risk of morbidity and mortality [59]. In the case of breast cancer, it has been estimated that one to three deaths from overtreatment occur for every one cancer death avoided [60]. CDK4/6 inhibition has been advanced as a means of suppressing chemotherapy related cytotoxicity for RB1-pathway null cancers[61–63]. Simultaneous administration of the CDK4/6 inhibitor G1T28 with cytotoxic chemotherapy (5-fluorouracil) was shown to suppress hematopoietic exhaustion during repeated treatments and enhance survival in mice where the protective effect is shown to overlap with the period of CDK4/6 inhibitor induced cell cycle arrest[61]. This approach has been extended to clinical trials for metastatic triple negative breast cancer and small cell lung cancer, where the ability of G1T28 to mitigate myelosuppression is being assessed in conjunction with gemcitabine, carboplatin, topotecan, and etoposide in various studies (iii, iv, v, vi). Topline data from these studies have shown significant myeloprotection. Surprisingly, RB1-pathway null status was not an inclusion criterion for these studies and a complete block of cell division by CDK4/6 inhibition in RB1-pathway competent tumor cells might be expected to suppress the efficacy of chemotherapies targeting events during the cell cycle[62, 64]. Nonetheless, approximately 1/3 of metastatic triple negative breast cancers and nearly all small cell lung cancer tumors are expected to have lost RB1 function and re-purposing CDK4/6 inhibitors for myeloprotection may be useful in this subset of tumors.
Most chemotherapeutic agents are administered by infusion and have relatively short half lives in vivo (e.g. gemcitabine, ~1 hr; carboplatin, ~6 hrs; etoposide, ~7.5 hrs; topotecan, ~3 hrs). G1T28 infused with these drugs results in G1 arrest lasting ~32 hrs which is sufficient to allow significant clearance of the cytotoxic drugs and underpins the myeloprotection afforded by G1T28[61]. However, cell cycle position may impact susceptibility to chemotherapeutic treatments, and it is known that cells that have recently entered G0/G1 are protected from the effects of cytotoxic drugs[65, 66]. The incomplete antitumor activity of chemotherapeutics administered intermittently by infusion may, at least in part, reflect the presence of cells that are retractile to drug treatment simply because they are in a phase of the cell cycle in which they are protected from the effects of the drug. Slowly dividing tumors and tumor stem cells are expected to be particularly resistant due to extended lengths of G0/G1 phases[67]. Although CDK4/6 inhibitors are not without side-effects, patients can be maintained on treatment for extended periods with apparent lack of cumulative toxicity. To the extent that prolonged CDK4/6 inhibition affords protection of normal cells in which the RB-pathway is intact, it may be possible to use these drugs to extend the duration over which chemotherapeutic treatments are tolerated by patients and target a larger proportion of the RB-pathway null tumor cells that would otherwise survive.
RB1-pathway status: implications to immune-evasion and immunotherapy:
RB1 is an important mediator of specific functions in both tumor and normal cells that can be leveraged to improve response to immunotherapy (Figure 4). RB1 plays a pivotal, albeit complex, role in modulating the expression of genes associated directly with immune system function (Box 1). Notably, several studies have indicated that multiple interferon-response genes cannot be efficiently activated in the absence of RB1[68–71]. This property of “non-inducibility” is believed to represent one important consequence of RB1 disruption by viral oncoproteins (e.g. HPV-E7) that allow escape from immune surveillance[72]. Multiple mechanisms have been ascribed to how RB1 regulates these immunological related genes. RB1 contributes to CIITA and NF-KappaB activity that are key determinants of such responses[45, 71].
Figure 4. Impact of RB-pathway on tumor immunology and the tumor microenvironment:

CDK4/6 inhibition and the activation of RB1 can lead to cell cycle inhibition in the tumor and tumor cell intrinsic effects relative to antigen presentation (e.g. MHC expression), interferon response (e.g. cytokine release). These events can enhance the mobilization of immune cells and NK cells into the tumor microenvironment. In parallel with these effects, the CDK4/6 inhibition has critical effects on T-regulatory cells, Myeloid-derived suppressors, and T-effector cells that have distinct effects on tumor biology.
These findings have gained more attention recently with the advent of means to activate RB1 with CDK4/6 inhibitors in the clinic. Multiple recent studies have shown that treatment with CDK4/6 inhibitors and activation of RB1 induces a number of immunologically relevant genes[45, 73]. These induced genes include those involved in canonical interferon response as well as multiple genes associated with antigen presentation (Box 1). This gene expression signature is generally considered immunogenic, and exhibits some similarities to the senescence activated secretory phenotype (SASP) activated as part of the senescence program[45], although this remains controversial[73, 74]. It has been shown that the gene-expression signature induced by CDK4/6 inhibitors alone, and in select combinations, is dependent on RB1, further illustrating the importance of this tumor-suppressive node beyond cell cycle regulation[45, 73]. While the immunological gene expression signatures have been observed by multiple groups, the exact mechanism of activation and its consequence remains a matter of debate. One hypothesis is that RB1 activation elicits the transcriptional repression of DNMT1, which in turn is critical for the suppression of endogenous retroviruses that are known to elicit an interferon response[73]. Interestingly EZH2, which is also a target for RB1-mediated transcriptional repression, has also been implicated in pro-immunogenic responses that are similarly linked to aberrant expression of endogenous retroviral elements[75]. Paradoxically, RB1 loss has been associated with de-silencing of specific repeat elements that can also lead to the engagement of an interferon response[76]. Thus, it is possible that RB-deficient cells become desensitized to such signals over the course of extended culture. In spite of this complexity, multiple factors involved in epigenetic regulation controlled through RB1-pathway could drive an immunological response via de-repression of aberrant/pro-inflammatory transcripts. Alternatively, it has been proposed that cytoplasmic nucleases are down-regulated during senescence, and the accumulation of cytoplasmic DNA drives the response[77]. Irrespective of the specific mechanism, nucleic-acid sensing mechanisms in the cytoplasm (ie STING or MAVS) are believed to transduce this signal to the activation of NF-Kappa B and interferon regulatory factors (IRFs) to effect the down-stream activation of the overall interferon response and antigen presentation. Importantly, these responses are tumor cell intrinsic and clearly link the activation state of RB1 to features (ie. MHC Class1 expression) that are positive determinants of the response to immunotherapy. Correspondingly, multiple studies have illustrated that CDK4/6 inhibitors enhance the response to contemporary immune checkpoint inhibitors[73, 74, 78, 79].
Importantly, the effects of RB1-pathway transcend the tumor and have wide-ranging impact within the tumor microenvironment and systemically. Within the tumor microenvironment, the use of CDK4/6 inhibitors has been associated with reduction in T-regulatory and myeloid-derived suppressor cells that are immune-suppressive. This effect of CDK4/6 in suppressing T-regulatory cells is clearly linked to the suppression of their proliferation. The impact of CDK4/6 inhibition on myeloid-derived suppressor cells remains less clear. Conflicting studies suggest CDK4/6 inhibitor stroma can function to facilitate chemotaxis into the tumor microenvironment; while systemic treatments with CDK4/6 inhibitors can limit myeloid-derived suppressor cells. Potent RB1-dependent cell cycle inhibition has been associated with an ICAM1 mediated engagement of NK cells that would drive a more potent immunological anti-tumor response[45, 73]. Additionally, it has been proposed that treatment with CDK4/6 inhibitors increases influx of cytotoxic T cells and limits intrinsic immunological exclusion in tumors [79]. Interestingly, it would appear that these features are not completely consistent across disease models and likely are modified by tumor context. Whether this heterogeneity of response is related to tumor genetics or the tissue of origin remains unclear, but will obviously have significance in considering clinically targeting such features of response to CDK4/6 inhibition alone and in combination.
Interestingly, these effects in the tumor environment would seem at odds with the critical role of CDK4/6 and cell cycle progression in the activation and maturation of T-cell response. However, effects of CDK4/6 inhibition on NFAT and differential dependence of various T cells subsets on CDK6 levels appear to enhance rather than limit anti-tumor T cell responses[78]. Thus, over the last year a number of studies have been published illustrating a clear cooperative effect between CDK4/6 inhibition and immune checkpoint inhibitors. While it is appealing to consider that direct effects of CDK4/6 inhibitor on the immune system are responsible for these effects, it is also possible that slower tumor cell cycling by CDK4/6 inhibition could also be contributing to effective cooperation with immunotherapies. In spite of the complexities related to systemic action CDK4/6 inhibitors, there are now multiple clinical trials that have been advanced based on published preclinical studies that have been published (vii, viii, ix).
These trials are all in the earliest phases monitoring safety and preliminary efficacy.
RB1 and cancer lineage plasticity:
Molecularly targeted cancer therapies often rely on cell lineage specific dependencies for their superior cancer selective effects. Breast and prostate tissue, for example, requires hormone receptor signaling for normal growth and development;cancers deriving from these tissues often retain this cell lineage specific dependence. Therapeutic hormone signaling blockade, therefore, can be very effective in treating these cancers, but is rarely curative.
Acquired resistance to such molecularly targeted therapies is often associated with genetic alteration of the molecular target itself, blocking or circumventing drug action. For example, lung adenocarcinomas driven by EGFR mutation are effectively treated with EGFR selective tyrosine kinase inhibitors like erlotinib, but disease relapse can occur upon acquisition of second site EGFR mutations that reduce drug binding (e.g. T790M)[80]. With development and application of newer generation therapies countering these genetic resistance mechanisms (e.g. osimertinib selective for T790M EGFR), it is becoming increasingly apparent that a significant fraction of cancers can acquire therapeutic resistance via cancer lineage plasticity[81, 82], the transcriptional reprogramming of cancer cells to a state no longer dependent on the lineage specific therapeutic target. In some cases these resistant cell states express markers of alternative cell lineages like the neuroendocrine variants observed in lung and prostate adenocarcinoma patients relapsing from anti-EGFR or anti-androgen therapies, respectively[83, 84].
Cancer lineage plasticity can have profound effects on disease progression because transcriptional reprogramming is an adaptive, albeit potentially reversible, response to therapy[85–87]. Analogous to plasticity-dependent evolution of species[88], it allows more rapid and directed sampling of phenotypic space compared to evolution driven by stochastic genetic mutation. This is particularly useful in highly selective, rapidly changing environments like those faced by cancer cells. This hypothesis suggests there may be a vulnerable window in which cancers adapting to therapy require lineage plasticity for survival. Indeed, small numbers of drug tolerant cells have long been recognized in cultured cancer cell lines that are otherwise sensitive to therapy as a population. In the absence of therapeutic selection, these tolerant cells revert to drug sensitivity. Analogous observations have been observed in the clinic upon drug re-challenge [89]. Targeting the lineage plastic state[90] would thus be expected to suppress acquired therapeutic resistance. Effectively targeting of lineage plasticity will require a better understanding of the underlying molecular mechanisms.
As noted above, RB1 has traditionally been thought to suppress tumorigenesis by virtue of its ability to restrain the cell division cycle. It does so by binding E2F transcription factors and recruiting chromatin regulatory factors to E2F regulated genes. In general, these factors modify a gene’s chromatin state to one that is non-permissive for transcription. Thus RB1/E2F complexes repress gene expression required for continuous cell cycling. While this paradigm is well established, there has been increasing focus on RB1-mediated chromatin regulation as fundamental to its function, with potential effects beyond the cell cycle. Emerging evidence suggests one of these effects is restricting lineage plasticity.
RB1 can suppress lineage plasticity in at least three ways. One, it represses the cell division cycle. Since the cell’s epigenome has to be re-established subsequent to each round of DNA replication, reducing cell division cycles indirectly stabilizes the epigenome. Two, RB1 is required for the organization and maintenance of constitutive and facultative heterochromatin.[91–94] Heterochromatin serves not only to stabilize gene expression patterns defining differentiated cell states, it also provides chromosome structure necessary for centromere and telomere function. Thus RB1 loss can compromise cell differentiation[95], chromosome segregation[96], and telomere maintenance[97]. Three, RB1/E2F complexes directly repress pluripotency genes including SOX2[98] and EZH2[99, 100]. In normal differentiated fibroblasts, RB1 loss promotes reprogramming into induced pluripotent stem cells, essentially replacing the requirement for the Yamanaka factor SOX2[98]. In prostate cancer cells, RB1 loss also induces SOX2 and EZH2 activity that drives reprogramming of prostate adenocarcinoma to neuroendocrine lineage variants[101, 102]. Consistent with the clinical observation that RB1 pathway dysregulation is ubiquitous in neuroendocrine cancers arising in several tissues[103], suppression of RB1 pathway activity can facilitate direct reprograming of normal human lung and prostate epithelial cells to neuroendocrine cancers[104].
Given this emerging role for RB1 in maintaining epigenetic stability (Figure 5), can the RB1 pathway be targeted to suppress cancer progression and therapeutic resistance associated with cancer lineage plasticity? In theory, activating the RB1 pathway could in inhibit both the cell cycle and pluripotency gene networks to stabilize the cancer epigenome, slowing cancer progression. Experimental evidence characterizing effects of CDK4/6 inhibitors on pluripotency gene networks or chromatin structure in cancers retaining RB1 is lacking. Nonetheless, CDK4/6 inhibitors are being evaluated in several different cancers by combining them with therapies targeting cell lineage specific factors (e.g. x, xi, xii). While suppressing cancer lineage plasticity is not the rationale cited for these trials, how the addition of CDK4/6 inhibitors affects the durability of therapeutic responses and the nature of therapeutic resistance that develops may provide evidence relevant to this therapeutic concept.
Figure 5. Impact of the RB1 pathway on cancer lineage plasticity and acquired therapeutic resistance.

The figure depicts the increasingly appreciated effects of RB1 on epigenetic instability mediated by the cell cycle and pluripotency regulatory networks aberrantly re-activated in cancer. This epigenetic instability can facilitate adaptation of cancer cells to therapies targeting lineage specific dependencies (e.g. androgen receptor, EGFR, etc.). Points of possible therapeutic intervention to counter cancer lineage plasticity include activation of the RB1 when it is present or suppressing downstream effects of RB1 loss using epigenetic modulating drugs.
Cancers lacking RB1 are predicted to have elevated lineage plasticity. Inhibiting the downstream epigenetic regulators mediating these effects would thus be predicted to suppress phenotypic reprogramming and cancer progression. Again, clinical evidence directly relevant to this hypothesis is not yet available, but preclinical experiments support potential feasibility. In experimental models of prostate cancer lineage plasticity associated with RB1 and TP53 loss, genetic or pharmacological inhibition of SOX2 or EZH2 can reverse reprogramming to neuroendocrine lineage variants and restore sensitivity to anti-androgen therapies[101, 102]. A clinical trial is underway to test the utility of combining EZH2 inhibitors with anti-androgen therapy for the treatment of prostate cancers that have progressed on anti-androgen therapy alone (xiii). Another potential therapeutic approach for exploiting lineage plasticity is to encourage cancer cell reprogramming to a more benign or treatable phenotype by manipulating cues that normally influence epithelial differentiation. For example, bipolar androgen therapy, rapid cycling between high and low testosterone concentrations, has demonstrated some efficacy in prostate cancer patients progressing on the anti-androgen enzalutamide (xiv)[105]. While the molecular mechanisms underlying observed clinical responses are unclear, brief exposure to high serum androgen concentrations clearly re-sensitizes the prostate cancer to enzalutamide. This implies androgen signaling may drive prostate cancer differentiation to a more luminal epithelial phenotype, consistent with its role in normal prostate development. Targeting the RB1 pathway to more effectively counter or exploit cancer lineage plasticity will require a more detailed understanding of how RB1 loss affects chromatin in different cancer contexts as well as the identification of the epigenetic regulators mediating these effects.
Concluding Remarks:
While the text-book description of the RB1-pathway remains largely valid today in describing processes of cell cycle control, there are ever increasing complexities that provide insights into therapeutic failure and new opportunities for therapeutic intervention. Leveraging these advances to develop new precision therapies based on targeting the RB1-pathway represents an opportunity to improve and extend clinical responses across multiple tumor sites. However, important questions remain that will need to be addressed in the clinic to further guide our implementation of targeting this crucial pathway for cancer therapy.
Since deregulated cellular proliferation is the central hallmark of tumor biology, it is appealing to specifically prevent cell division as a therapeutic strategy. However, in the face of tumor heterogeneity and tumor evolution it seems that such a strategy would represent a “ticking time-bomb”. Presumably a permanent and complete tumor cell cycle exit could block tumor evolution; however, whether such a state can be achieved clinically remains unknown and feasibility will be dependent on outcomes from multiple clinical studies. Complementary, ongoing efforts are focused on finding means to selectively target dormant tumor cells and perhaps the use of senolytic agents or immunotherapy which are not dependent on tumor proliferation for efficacy may represent a key approach to leverage the delay in tumor progression for long-term therapeutic responses.
Although a complete block of cell division by CDK4/6 inhibition in RB-pathway competent tumor cells is expected to suppress the efficacy of genotoxic chemotherapies, cells which are re-entering the cell cycle following a CDK4/6 block, or cycling slowly due to partial CDK4/6 inhibition may show increased susceptibility to such agents. For example, cells reentering the cell cycle from G0 show a unique requirement for cyclin E in DNA replication origin licensing[106] and a heighted sensitivity to reduced levels of replication licensing factors (minichromosome maintenance proteins, MCM) resulting in genome instability[107]. In fact, multiple studies are evaluating “metronomic” approaches of using CDK4/6 inhibitors with genotoxic chemotherapies (e.g. gemcitabine (xv, xvi), carboplatin (xvii), and taxanes (xviii). While preliminary indications of efficacy are emerging, to date, no randomized studies have reported findings.
In many instances RB1 loss occurs during the course of disease progression and selection for therapy resistance. In this context one must expect that a tumor contains both RB1-proficient and deficient tumor cells. Therapeutically, with progression on targeted therapies (e.g. hormonal therapy, CDK4/6 inhibitors, or EGFR inhibitors) ostensibly an RB1-deficient population can drive resistance. However, in this same tumor there would be expected to be a dormant RB1-proficient tumor population. How to coordinately target both features of the tumor is complicated, but potentially measuring intra-tumoral heterogeneity with treatment could represent a means to tailor therapies in such a way to ultimately control the heterogeneous tumor.
Given the multi-faceted effect of CDK4/6 inhibition on immunological features of the tumor cell and the microevironment, there is substantial interes in combinations with immunotherapy. Based on the preclinical findings, it would seem that pre-treatment with an active regimen to activate RB1 within the tumor would heighten the subsequent efficacy of immune checkpoint inhibitors. However, since engagement of an immunological anti-tumor response requires time to mature, there is a strong argument to delay tumor progression with the CDK4/6 inhibitor based therapy while the immunotherapy is being delivered. Since immune checkpoint therapy can elicit complete or durable response, it would be predicted that at the point immunotherapy is discontinued there would be no need to maintain treatment with CDK4/6 inhibitors. Conversely whether CDK4/6 inhibition to activate RB1 can make a non-responsive tumor responsive is currently unknown, although recent studies suggest that it could be used as a general strategy to increase the immunological “hotness” of the tumor which would significantly expand efficacy of immune checkpoint inhibitors.
The RB1 pathway has long been recognized to regulate key aspects of chromatin structure that influence gene expression patterns[108, 109]. RB1 has also been linked to the suppression of pluripotency regulatory networks[98, 99]. Cancer lineage plasticity is emerging as an important mechanism of acquired resistance to therapies targeting lineage specific dependencies[101, 102, 110]. In principle, therefore, CDK4/6 inhibitors could be used to suppress acquired therapeutic resistance in cancers retaining RB1. In cancers lacking RB1, epigenetic modulating drugs, either approved or in development for clinical use, could be applied to blunt effects of RB1 loss on pluripotency regulatory networks. However, the context dependent effects of RB1 loss or hyperactivation on cancer lineage plasticity are not well characterized at the molecular level, nor are the effects of most epigenetic modulating drugs. These gaps will likely need to be filled before the RB1 pathway can be effectively targeted to counter acquired therapeutic resistance mediated by cancer lineage plasticity.
Multiple new approaches for considering RB1 as a key therapeutic node in the treatment of cancer have emerged. These findings are challenging the simple canonical role of RB1 and suggest varied and complex mechanisms to exploit the status of this key tumor suppressor in cancer treatment (see Outstanding Questions)
Highlights:
Sensitivity to CDK4/6 inhibitors can be expanded by ameliorating cell cycle plasticity and deepening cell cycle exit with combination therapies.
Targeting the vulnerabilities of tumors lacking RB1 provides a new precision means to attack tumors that escape from cytostatic interventions.
RB1-pathway activation in normal tissue can limit the side-effects of specific therapeutic interventions and provide a means to expand the therapeutic index against RB-deficient tumors.
Exploiting the impact of RB1 on immunological features of the tumor compartment and microenvironment can expand sensitivity to immunotherapy and provides hope for yielding highly durable combinatorial therapies.
Understanding disparate roles of RB1 in coordinating epigenetic states that drive histologic transformation and resistance to targeted therapies will provide new opportunities to prevent resistance and intercede in advanced disease.
Acknowledgements:
The authors would like to thank all of their colleagues in the field for the thought-provoking discussions that support the concepts of this article. Any omission is regretted and we attempted to incorporate as many studies as possible in the discussion of the RB1-pathway. The authors are supported by grants: R01CA130995 to SCP, R01CA188650 to ESK, R01CA211878 to AKW, and CA207757 to DWG. Additionally, the authorship team is supported by funding from the Herd of Hope Campaign of the Roswell Park Alliance Foundation and would like to thank all of the donors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: DWG, PAH, and SCP have nothing to report. AKW and ESK have received past research funding from Eli Lilly, Novartis, and Pfizer in the analysis of CDK4/6 inhibitors.
RESOURCES:
https://clinicaltrials.gov/ct2/show/NCT02873975: A Study of LY2606368 (Prexasertib) in Patients With Solid Tumors With Replicative Stress or Homologous Repair Deficiency
https://clinicaltrials.gov/ct2/show/NCT02735980: A Study of Prexasertib (LY2606368) in Participants With Extensive Stage Disease Small Cell Lung Cancer
https://clinicaltrials.gov/ct2/show/NCT02978716: Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Gemcitabine and Carboplatin in Metastatic Triple Negative Breast Cancer (mTNBC)
https://clinicaltrials.gov/ct2/show/NCT02514447: Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Patients With Previously Treated Extensive Stage SCLC Receiving Topotecan Chemotherapy
https://clinicaltrials.gov/ct2/show/NCT02499770: Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Combination With Etoposide and Carboplatin in Extensive Stage Small Cell Lung Cancer (SCLC)
https://clinicaltrials.gov/ct2/show/NCT03041311: Carboplatin, Etoposide, and Atezolizumab With or Without Trilaciclib (G1T28), a CDK 4/6 Inhibitor, in Extensive Stage Small Cell Lung Cancer (SCLC)
https://clinicaltrials.gov/ct2/show/NCT02791334: A Study of Anti-PD-L1 Checkpoint Antibody (LY3300054) Alone and in Combination in Participants With Advanced Refractory Solid Tumors (PACT)
https://clinicaltrials.gov/ct2/show/NCT03294694: Ribociclib + PDR001 in Breast Cancer and Ovarian Cancer
https://clinicaltrials.gov/ct2/show/NCT03498378: Avelumab, Cetuximab, and Palbociclib in Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma
https://clinicaltrials.gov/ct2/show/NCT02555189: Enzalutamide With and Without Ribociclib for Metastatic, Castrate-Resistant, Chemotherapy-Naive Prostate Cancer That Retains RB Expression
https://clinicaltrials.gov/ct2/show/NCT03455829: G1T38, a CDK 4/6 Inhibitor, in Combination With Osimertinib in EGFR-Mutant Non-Small Cell Lung Cancer
https://clinicaltrials.gov/ct2/show/NCT02747004: A study of Abemaciclib plus tamoxifen or abemaciclib alone in patients with metastatic breast cancer
https://clinicaltrials.gov/ct2/show/NCT03480646: ProSTAR: a study evaluating CPI-1205 in patients with metastatic castration resistant prostate cancer
https://clinicaltrials.gov/ct2/show/NCT02090114: re-sensitizing with supraphysiologic testosterone to overcome resistance (the RESTORE study)
https://clinicaltrials.gov/ct2/show/NCT03434262: SJDAWN: St. Jude Children’s Research Hospital Phase 1 Study Evaluating Molecularly-Driven Doublet Therapies for Children and Young Adults With Recurrent Brain Tumors
https://clinicaltrials.gov/ct2/show/NCT03237390: Ribociclib and gemcitabine hydrochloride in treating patients with advanced or metastatic solid tumors
https://clinicaltrials.gov/ct2/show/NCT03056833: Ribociclib (Ribociclib (LEE-011)) With Platinum-based Chemotherapy in Recurrent Platinum Sensitive Ovarian Cancer
XVIII.: https://clinicaltrials.gov/ct2/show/NCT01320592: PD0332991/Paclitaxel in Advanced Breast Cancer
REFERENCES:
- 1.Goodrich DW (2006) The retinoblastoma tumor-suppressor gene, the exception that proves the rule. Oncogene 25 (38), 5233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burkhart DL and Sage J (2008) Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer 8 (9), 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sherr CJ (2000) Cell cycle control and cancer. Harvey Lect 96, 73–92. [PubMed] [Google Scholar]
- 4.Nevins JR (2001) The Rb/E2F pathway and cancer. Hum Mol Genet 10 (7), 699–703. [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ (1995) D-type cyclins. Trends Biochem Sci 20 (5), 187–90. [DOI] [PubMed] [Google Scholar]
- 6.Asghar U et al. (2015) The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 14 (2), 130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cam H and Dynlacht BD (2003) Emerging roles for E2F: beyond the G1/S transition and DNA replication. Cancer Cell 3 (4), 311–316. [DOI] [PubMed] [Google Scholar]
- 8.Sherr CJ (1996) Cancer cell cycles. Science 274 (5293), 1672–7. [DOI] [PubMed] [Google Scholar]
- 9.Lukas J et al. (1994) DNA tumor virus oncoproteins and retinoblastoma gene mutations share the ability to relieve the cell’s requirement for cyclin D1 function in G1. J Cell Biol 125 (3), 625–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lukas J et al. (1995) Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature 375 (6531), 503–6. [DOI] [PubMed] [Google Scholar]
- 11.Fry DW et al. (2004) Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3 (11), 1427–38. [PubMed] [Google Scholar]
- 12.Aagaard L et al. (1995) Aberrations of p16Ink4 and retinoblastoma tumour-suppressor genes occur in distinct sub-sets of human cancer cell lines. Int J Cancer 61 (1), 115–20. [DOI] [PubMed] [Google Scholar]
- 13.Witkiewicz AK et al. (2011) The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 10 (15), 2497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knudsen ES and Wang JY Targeting the RB-pathway in cancer therapy. Clin Cancer Res 16 (4), 1094–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knudsen ES and Knudsen KE (2008) Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 8 (9), 714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosco EE et al. (2007) The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest 117 (1), 218–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sutherland RL and Musgrove EA (2004) Cyclins and breast cancer. J Mammary Gland Biol Neoplasia 9 (1), 95–104. [DOI] [PubMed] [Google Scholar]
- 18.Sherr CJ et al. (2016) Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 6 (4), 353–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khatib ZA et al. (1993) Coamplification of the CDK4 gene with MDM2 and GLI in human sarcomas. Cancer Res 53 (22), 5535–41. [PubMed] [Google Scholar]
- 20.Knudsen ES and Witkiewicz AK (2017) The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 3 (1), 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeMichele A et al. (2015) CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res 21 (5), 995–1001. [DOI] [PubMed] [Google Scholar]
- 22.Patnaik A et al. (2016) Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov 6 (7), 740–53. [DOI] [PubMed] [Google Scholar]
- 23.Gelbert LM et al. (2014) Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finn RS et al. (2009) PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 11 (5), R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dean JL et al. (2010) Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 29 (28), 4018–32. [DOI] [PubMed] [Google Scholar]
- 26.Ma CX et al. (2017) NeoPalAna: Neoadjuvant palbociclib, a cyclin-dependent kinase 4/6 inhibitor, and anastrozole for clinical stage 2 or 3 estrogen receptor positive breast cancer. Clin Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnston S et al. (2018) Randomized Phase II Study Evaluating Palbociclib in Addition to Letrozole as Neoadjuvant Therapy in Estrogen Receptor-Positive Early Breast Cancer: PALLET Trial. J Clin Oncol, JCO1801624. [DOI] [PubMed] [Google Scholar]
- 28.Turner NC et al. (2015) Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med. [DOI] [PubMed] [Google Scholar]
- 29.Turner NC et al. (2018) Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N Engl J Med 379 (20), 1926–1936. [DOI] [PubMed] [Google Scholar]
- 30.Konecny GE et al. (2011) Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res 17 (6), 1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herrera-Abreu MT et al. (2016) Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franco J et al. (2016) Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haines E et al. (2018) Palbociclib resistance confers dependence on an FGFR-MAP kinase-mTOR-driven pathway in KRAS-mutant non-small cell lung cancer. Oncotarget 9 (60), 31572–31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen SH et al. (2018) RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 37 (6), 821–832. [DOI] [PubMed] [Google Scholar]
- 35.Malumbres M et al. (2004) Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118 (4), 493–504. [DOI] [PubMed] [Google Scholar]
- 36.Franco J et al. (2014) CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 5 (15), 6512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vora SR et al. (2014) CDK 4/6 Inhibitors Sensitize PIK3CA Mutant Breast Cancer to PI3K Inhibitors. Cancer Cell 26 (1), 136–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner NC et al. (2019) Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol, JCO1800925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou J et al. (2017) CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nat Commun 8, 13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu M et al. (2016) PD 0332991, a selective cyclin D kinase 4/6 inhibitor, sensitizes lung cancer cells to treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Oncotarget 7 (51), 84951–84964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshida A et al. (2016) Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res 76 (10), 2990–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi S et al. (2006) Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res 66 (23), 11389–98. [DOI] [PubMed] [Google Scholar]
- 43.Fischer M et al. (2016) Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res 44 (13), 6070–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang T et al. (2015) Identification and characterization of essential genes in the human genome. Science 350 (6264), 1096–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruscetti M et al. (2018) NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science 362 (6421), 1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z et al. (2018) Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 34 (6), 893–905 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Leary B et al. (2018) The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov 8 (11), 1390–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dick FA et al. (2018) Non-canonical functions of the RB protein in cancer. Nat Rev Cancer 18 (7), 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Leary B et al. (2018) Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun 9 (1), 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Condorelli R et al. (2018) Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 29 (3), 640–645. [DOI] [PubMed] [Google Scholar]
- 51.Guarducci C et al. (2018) Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witkiewicz AK et al. (2014) Systematically defining single-gene determinants of response to neoadjuvant chemotherapy reveals specific biomarkers. Clin Cancer Res 20 (18), 4837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Witkiewicz AK et al. (2012) RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res 18 (18), 5110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gazdar AF et al. (2017) Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer 17 (12), 725–737. [DOI] [PubMed] [Google Scholar]
- 55.Witkiewicz AK et al. (2018) Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep 22 (5), 1185–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu JC et al. (2018) Identification of CDC25 as a Common Therapeutic Target for Triple-Negative Breast Cancer. Cell Rep 23 (1), 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oser MG et al. (2018) Cells Lacking the RB1 Tumor Suppressor Gene are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong X et al. (2018) Aurora-A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. [DOI] [PubMed] [Google Scholar]
- 59.Darby SC et al. (2005) Long-term mortality from heart disease and lung cancer after radiotherapy for early breast cancer: prospective cohort study of about 300,000 women in US SEER cancer registries. Lancet Oncol 6 (8), 557–65. [DOI] [PubMed] [Google Scholar]
- 60.Baum M (2013) Harms from breast cancer screening outweigh benefits if death caused by treatment is included. BMJ 346, f385. [DOI] [PubMed] [Google Scholar]
- 61.He S et al. (2017) Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med 9 (387). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roberts PJ et al. (2012) Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst 104 (6), 476–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnson SM et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest 120 (7), 2528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McClendon AK et al. (2012) CDK4/6 inhibition antagonizes the cytotoxic response to anthracycline therapy. Cell Cycle 11 (14), 2747–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ryl T et al. (2017) Cell-Cycle Position of Single MYC-Driven Cancer Cells Dictates Their Susceptibility to a Chemotherapeutic Drug. Cell Syst 5 (3), 237–250 e8. [DOI] [PubMed] [Google Scholar]
- 66.Arora M and Spencer SL (2017) A Cell-Cycle “Safe Space” for Surviving Chemotherapy. Cell Syst 5 (3), 161–162. [DOI] [PubMed] [Google Scholar]
- 67.Kreso A and Dick JE (2014) Evolution of the cancer stem cell model. Cell Stem Cell 14 (3), 275–91. [DOI] [PubMed] [Google Scholar]
- 68.Hutcheson J et al. (2014) Retinoblastoma protein potentiates the innate immune response in hepatocytes: Significance for hepatocellular carcinoma. Hepatology 60 (4), 1231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Markey MP et al. (2007) Loss of the retinoblastoma tumor suppressor: differential action on transcriptional programs related to cell cycle control and immune function. Oncogene 26 (43), 6307–18. [DOI] [PubMed] [Google Scholar]
- 70.Zhu X et al. (1999) pRB is required for interferon-gamma-induction of the MHC class II abeta gene. Oncogene 18 (35), 4940–7. [DOI] [PubMed] [Google Scholar]
- 71.Lu Y et al. (1994) Evidence for retinoblastoma protein (RB) dependent and independent IFN-gamma responses: RB coordinately rescues IFN-gamma induction of MHC class II gene transcription in noninducible breast carcinoma cells. Oncogene 9 (4), 1015–9. [PubMed] [Google Scholar]
- 72.Park JS et al. (2000) Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem 275 (10), 6764–9. [DOI] [PubMed] [Google Scholar]
- 73.Goel S et al. (2017) CDK4/6 inhibition triggers anti-tumour immunity. Nature 548 (7668), 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schaer DA et al. (2018) The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep 22 (11), 2978–2994. [DOI] [PubMed] [Google Scholar]
- 75.Canadas I et al. (2018) Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med 24 (8), 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ishak CA et al. (2016) An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol Cell 64 (6), 1074–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takahashi A et al. (2018) Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9 (1), 1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deng J et al. (2018) CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 8 (2), 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jerby-Arnon L et al. (2018) A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175 (4), 984–997 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tan CS et al. (2015) Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol 16 (9), e447–59. [DOI] [PubMed] [Google Scholar]
- 81.Flavahan WA et al. (2017) Epigenetic plasticity and the hallmarks of cancer. Science 357 (6348). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brock A et al. (2009) Non-genetic heterogeneity--a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet 10 (5), 336–42. [DOI] [PubMed] [Google Scholar]
- 83.Sequist LV et al. (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3 (75), 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beltran H et al. (2011) Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 1 (6), 487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pisco AO et al. (2013) Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun 4, 2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sharma SV et al. (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141 (1), 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shaffer SM et al. (2017) Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546 (7658), 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pennisi E (2018) Buying time. Science 362 (6418), 988–991. [DOI] [PubMed] [Google Scholar]
- 89.Kuczynski EA et al. (2013) Drug rechallenge and treatment beyond progression--implications for drug resistance. Nat Rev Clin Oncol 10 (10), 571–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Viswanathan VS et al. (2017) Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547 (7664), 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Siddiqui H et al. (2007) Loss of RB compromises specific heterochromatin modifications and modulates HP1alpha dynamics. J Cell Physiol 211 (1), 131–7. [DOI] [PubMed] [Google Scholar]
- 92.Isaac CE et al. (2006) The retinoblastoma protein regulates pericentric heterochromatin. Mol. Cell. Biol 26 (9), 3659–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Narita M et al. (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113 (6), 703–716. [DOI] [PubMed] [Google Scholar]
- 94.Gonzalo S et al. (2005) Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nature Cell Biology 7 (4), 420–428. [DOI] [PubMed] [Google Scholar]
- 95.Korenjak M et al. (2005) E2F-Rb complexes regulating transcription of genes important for differentiation and development. Curr Opin Genet Dev 15 (5), 520–7. [DOI] [PubMed] [Google Scholar]
- 96.Manning AL et al. (2010) Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 24 (13), 1364–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Garcia-Cao M et al. (2002) A role for the Rb family of proteins in controlling telomere length. Nat. Genet 32, 415–419. [DOI] [PubMed] [Google Scholar]
- 98.Kareta MS et al. (2015) Inhibition of pluripotency networks by the rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16 (1), 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ishak CA et al. (2016) An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bracken AP et al. (2003) EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22 (20), 5323–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mu P et al. (2017) SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355 (6320), 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ku SY et al. (2017) Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355 (6320), 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rickman DS et al. (2017) Biology and evolution of poorly differentiated neuroendocrine tumors. Nat Med 23 (6), 1–10. [DOI] [PubMed] [Google Scholar]
- 104.Park JW et al. (2018) Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 362 (6410), 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Teply BA et al. (2018) Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol 19 (1), 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Geng Y et al. (2003) Cyclin E ablation in the mouse. Cell 114 (4), 431–43. [DOI] [PubMed] [Google Scholar]
- 107.Kunnev D et al. (2010) DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 29 (25), 3630–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Talluri S and Dick FA (2012) Regulation of transcription and chromatin structure by pRB: Here, there and everywhere. Cell Cycle 11 (17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Longworth MS and Dyson NJ (2009) pRb, a local chromatin organizer with global possibilities. Chromosoma 119 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Niederst MJ et al. (2015) RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 6, 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]