

Cardiac troponin I Ser44 is a downstream target for protein kinase C. The current studies show heightened phosphorylation develops during contractile dysfunction in failing human myocardium and in rat models of pressure overload and aging with contractile dysfunction.
Abstract
Increases in protein kinase C (PKC) are associated with diminished cardiac function, but the contribution of downstream myofilament phosphorylation is debated in human and animal models of heart failure. The current experiments evaluated PKC isoform expression, downstream cardiac troponin I (cTnI) S44 phosphorylation (p-S44), and contractile function in failing (F) human myocardium, and in rat models of cardiac dysfunction caused by pressure overload and aging. In F human myocardium, elevated PKCα expression and cTnI p-S44 developed before ventricular assist device implantation. Circulatory support partially reduced PKCα expression and cTnI p-S44 levels and improved cellular contractile function. Gene transfer of dominant negative PKCα (PKCαDN) into F human myocytes also improved contractile function and reduced cTnI p-S44. Heightened cTnI phosphorylation of the analogous residue accompanied reduced myocyte contractile function in a rat model of pressure overload and in aged Fischer 344 × Brown Norway F1 rats (≥26 mo). Together, these results indicate PKC-targeted cTnI p-S44 accompanies cardiac cellular dysfunction in human and animal models. Interfering with PKCα activity reduces downstream cTnI p-S44 levels and partially restores function, suggesting cTnI p-S44 may be a useful target to improve contractile function in the future.
Introduction
More than 26 million people worldwide have heart failure (HF) and the associated increased mortality and morbidity risk (Savarese and Lund, 2017). Clinical treatment of human HF by mechanical circulatory support with ventricular assist devices (VADs) continues to increase (Miller and Rogers, 2018), though long-term improvement in cardiac function and VAD removal remain limited for most patients (Uriel et al., 2017). However, VAD therapy produces some reverse remodeling (Wever-Pinzon et al., 2016) and changes that may help in risk stratification for patients (Morgan et al., 2012; Smith et al., 2015). Further work is needed to identify underlying cellular and molecular targets that improve cardiac function in addition to mechanical unloading.
Elevated expression and activity of PKC are consistently linked to HF in humans (Bowling et al., 1999; Noguchi et al., 2004; Kim et al., 2016). PKC activation modulates cardiac performance via a variety of downstream pathways and targets to influence hypertrophy (Braz et al., 2004), Ca2+ cycling (Piacentino et al., 2003; Braz et al., 2004; Hambleton et al., 2006), and metabolism (Murriel et al., 2004). The promiscuous expression and diverse targets of PKC suggests it functions as a central node for processing and modulating cardiac structure and function. In the sarcomere, which is the functional unit of striated muscle, there is growing evidence that PKC phosphorylation of the molecular switch protein, cardiac troponin I (cTnI), reduces contractile function (Bilchick et al., 2007; Kirk et al., 2009). Other signaling pathways respond to mechanical loading (Castillero et al., 2018), but the relationship between PKC up-regulation, downstream cTnI phosphorylation, contractile function during HF, and the response to VAD therapy remains to be investigated.
Work in myocytes is needed to investigate the relationship between PKC, downstream myofilament phosphorylation, and contractile function. Several investigators demonstrated the feasibility of isolating adult ventricular myocytes from explanted human hearts for contractile function studies (Dipla et al., 1998; Goldberg et al., 2000; Day et al., 2006). Myocytes from nonfailing (NF) human hearts demonstrate a positive force–frequency relationship, while failing (F) human hearts develop a negative staircase response (Chaudhary et al., 2004). Partial reversal of the negative staircase response is reported in myocytes from HF patients after VAD support (Chaudhary et al., 2004). In addition, alterations in Ca2+ cycling proteins are associated with the negative frequency response (Dipla et al., 1998), and VAD improves both Ca2+ handling and the stimulation frequency response in isolated myocytes.
Few studies have examined whether chronic posttranslational modifications within the myofilament contribute to contractile dysfunction. Earlier work indicates increased PKC expression and activity are linked to human HF (Bowling et al., 1999; Noguchi et al., 2004), but the role played by its downstream myofilament targets in dysfunction is not clear. The present study shows PKC-targeted human cTnI p-S44 is associated with contractile dysfunction by analyzing human F myocardium collected before and after VAD treatment and hearts from pressure-overloaded and aging rat myocardium.
Materials and methods
Human cardiac tissue collection
Cardiac tissue collected from F and NF hearts was approved by the Internal Review Board at the University of Michigan, and the consent and protocol for collecting nonmatched NF hearts was approved by Gift of Life-Michigan, as described in detail earlier (Day et al., 2006; Kim et al., 2016). Briefly, the criterion for F hearts was a left ventricular ejection fraction (EF) ≤25% in F hearts, while structurally normal hearts with an EF ≥50% was required for the NF group (Day et al., 2006). Table S1 provides information for the human myocardial tissue. Myocardium not designated for cell isolation was immediately flash frozen in liquid N2 upon removal of the heart and stored at −80°C. Pre- and post-VAD F and NF myocardium were analyzed for protein expression. Contractile function was compared in myocytes from pre- and post-VAD tissue and in post-VAD myocytes after gene transfer of PKCα, dominant negative PKCα (PKCαDN), and in nontreated (NT) cells, as described below.
Animal model studies
Studies performed in rat models were approved by the University of Michigan Institutional Animal Care and Use Committee and followed the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Pressure overload (PO) rat model
The procedures for surgery, postoperative care, cardiac tissue, and isolated Ca2+ tolerant myocytes were performed at 18–20 wk after sham or PO surgery in Sprague–Dawley rats, when the EF begins to decrease (Kim et al., 2016). Myocytes from sham- and PO-treated rats were analyzed for protein expression, immunolabeling of PKCα in cryosections, and cellular contractile function, as described below.
Aging rat model (Fischer 344 × Brown Norway F1 [F344BN] rats)
Female F344BN rats were obtained from the National Institute on Aging colony as described earlier (Wahr et al., 2000). Rats were maintained on a 12-h light/dark cycle and fed normal chow for 2–4 mo before harvesting hearts from 6-, 18-, 26-, and 34-mo-old rats. For studies with these rats, protein expression and myocyte contractile function were analyzed after adenoviral-mediated gene transfer of PKCα, PKCαDN, or GFP and compared with time-matched, NT myocytes. In preliminary studies, there were no differences in contractile function after GFP gene transfer compared with NT myocytes from 6–18- or 34-mo-old rats (Fig. S1).
Myocyte isolation and culture conditions
Human myocytes
The isolation and culture conditions for Ca2+-tolerant myocytes from HF and NF human hearts were described earlier (Day et al., 2006). Briefly, hearts were flushed with ice-cold cardioplegia before explant, and ventricles immediately transported to the laboratory to be minced into 5–10-mm pieces in ice-cold stock buffer (100 mM NaCl, 5.4 mM KCl, 1.5 mM MgSO4, 10 mM NaOAc, 1.5 mM NaH2PO4, 15 mM glucose, 10 mM taurine, and 10 mM HEPES, pH 7.40) supplemented with 2,3-butanedione monoxime (BDM; 15 mM) and nitrilotriacetic acid (5 mM). Then, protease type XXIV (4 U/ml), type II collagenase (400 U/ml), hyaluronidase (200 U/ml), and 1.25 mM CaCl2 were sequentially added at 37°C to digest myocardium. Isolated myocytes were briefly centrifuged and resuspended in stock buffer containing 2% bovine serum albumin plus 10 mM BDM and 5% FBS, and then equal aliquots of CaCl2 were added back every 10 min over 1 h to reach a final concentration of 1.8 mM. Then, 40,000 rod-shaped cells were plated on laminin-coated 22-mm coverslips in MEM (Gibco) containing 5% FBS, 20 mM BDM, 40 mM HEPES, and 50 U/ml penicillin plus 50 µg/ml streptomycin (P/S) for 2 h (MEM+). For gene transfer, media were replaced with MEM+ media (NT), or adenovirus (Ad) containing GFP (AdGFP), PKCα (AdPKCα), or PKCαDN (AdPKCαDN) diluted in MEM+ for 1 h, followed by the addition of MEM containing 5 mM BDM, 40 mM HEPES, 2 mM glutamine, 24 mM NaHCO3, and P/S. Media were changed daily, and then cells were analyzed 24–36 h after gene transfer.
Adult rat myocytes
The isolation and culture of Ca2+-tolerant myocytes from each rat heart followed the protocol described earlier (Lang et al., 2015, 2017). Briefly, Ca2+-tolerant adult Sprague–Dawley rat or F344BN rat myocytes were isolated from collagenase-treated hearts (Lang et al., 2015, 2017). Rod-shaped myocytes were plated on laminin-coated coverslips in Dulbecco’s modified Eagle’s medium supplemented with 5% FBS plus P/S at 37°C for 2 h (Lang et al., 2015, 2017) and then replaced by serum-free Dulbecco’s modified Eagle’s medium plus P/S media containing recombinant AdGFP, AdPKCα, or AdPKCαDN or media alone (NT). After an additional hour, M199 media (Sigma-Aldrich) supplemented with 10 mM HEPES, 0.2 mg/ml BSA, and 10 mM glutathione plus P/S (M199+) was added to each coverslip. For contractile function studies, rat myocytes were paced starting the day after isolation, with media exchanged every 12 h. Media were also changed daily for myocytes used in protein expression work.
Protein expression and localization
Myocardial tissue and isolated myocytes
Human myocardium was pulverized to a fine powder in liquid N2, homogenized in ice-cold sample buffer supplemented with proteolysis and phosphatase inhibitors (Roche), and assayed for protein concentration (Zhang et al., 2012; Kim et al., 2016). For Western analysis after gene transfer experiments using human or rat myocytes, cells were scraped from coverslips into the same ice-cold sample buffer and stored at −80°C before protein separation (Lang et al., 2015, 2017). Homogenate samples or isolated myocyte proteins were separated by SDS-PAGE (12%), transferred to polyvinylidene difluoride membrane, and probed with primary Abs followed by an appropriate horseradish peroxidase–conjugated goat anti-mouse or goat anti-rabbit secondary Ab, as described previously (Kim et al., 2016). Previously published Western protocols using primary Ab for PKCα (sc-208; Santa Cruz Biotechnology), phosphorylated PKCα T638 (p-T638; 9375; Cell Signaling Technology), δ (9616; Cell Signaling Technology), ε (2683; Cell Signaling Technology), p-S44 (p2010-43; PhosphoSolutions), phosphorylated S23/24 (p-S23/24; 4004; Cell Signaling Technology), and total cTnI (1:2,000; MAB1691; EMD Millipore) were used to detect proteins in human homogenates and isolated human and rat myocytes (Hwang et al., 2012; Lang et al., 2015, 2017). Phosphorylation of S45 (p-S45) in rat cTnI is equivalent to human cTnI p-S44, while p-S23/24 is the same in cTnI from both species based on the UniProtKB TNNI3 human (P19429) and rat (P23693) sequences. In addition, phospholamban (PLB; EMD Millipore A1), plus phosphorylated PLB (p-PLB; Ser16; 07–052; EMD Millipore) were probed in human myocardium and rat myocytes, as well as phosphorylated cTnI T144 (p-T144; ab58546; Abcam) in rat myocytes. Unless otherwise noted, a titer of 1:1,000 was used for each primary Ab. Protein expression was then detected by enhanced chemiluminescence using a Bio-Rad ChemiDoc MP imager (Bio-Rad) or HyBlot CL film. A portion of each SDS-PAGE gel also was silver-stained (Ag-stained), and expression on Western blots and the silver-stained gel quantified with Quantity One software (Bio-Rad).
In most cases, protein expression detected by Western blot is expressed relative to a silver-stained band on the gel to account for protein loading. These values are then normalized to the expression of the respective protein in NF (human), sham (rat) or NT (human or rat) samples on the same blot to determine fold changes in expression. For PKC isoform detection in human myocardium, these normalized values also were used to determine the ratio of pre- versus post-VAD expression. For phosphorylation studies, the ratio of phosphorylated to total protein is determined and compared for the quantitative analysis unless otherwise specified.
Localization in adult myocytes
Images for GFP and brightfield microscopy were collected from live human myocytes 1–2 d after gene transfer. To determine the PKCα cellular distribution in rat myocytes, paraformaldehyde-fixed myocytes isolated 18–20 wk after sham and PO surgery were immunostained with PKCα (Santa Cruz Biotechnology) and Texas Red–conjugated goat anti-mouse Abs, as described earlier (Hwang et al., 2012; Kim et al., 2016). Images were collected with a Nikon Ti-U inverted fluorescence microscope and DS-Fi1 5 megapixel digital imaging using identical exposure times for each sample.
Contractile function studies
Basal sarcomere shortening measurements
Sarcomere shortening and relengthening were analyzed in signal-averaged traces collected from intact myocytes 24–36 h after isolation and gene transfer using a high-intensity video-based detection system (Ionoptix; Lang et al., 2015, 2017). Cells were washed twice with M199+ and equilibrated for 20 min before measuring contractile function in human myocytes. Resting sarcomere length, peak shortening amplitude, shortening rate, relengthening rate, and time to 50% relengthening (TTR50%) were measured in myocytes perfused with M199+ at 37°C and field-stimulated at 0.2 Hz.
Frequency-dependent changes in steady-state contractile function
Field-stimulated human and rat myocytes paced at 0.2 (basal), 0.5, 1, and/or 2 Hz were monitored for contractile shortening. Contractile function was measured after reaching steady-state contraction at each frequency, and signal-averaged traces were collected as described above for basal shortening measurements. Responses at 0.5–2 Hz were analyzed relative to the results obtained for basal stimulation (0.2 Hz), and then expressed as a percent change from this baseline measurement (% change = [{x Hz – 0.2 Hz}/0.2 Hz] × 100; see Day et al., 2006). Relative shortening in human myocytes is determined by normalizing contractile function to the functional range obtained at 2 Hz in NF myocytes.
Statistical analysis
Samples were compared using an unpaired Student’s t test, one- or two-way ANOVA, and appropriate post hoc tests with statistical significance set at P < 0.05. The specific statistical tests used for each group of data are described in the figure legends.
Online supplemental material
Results in Fig. S1 show that AdGFP gene transfer into rat myocytes does not alter the age-dependent decrease in peak amplitude, and shortening and relengthening rates. Fig. S2, A and B, shows the increased overall and regional PKCα expression in pre- or post-VAD F versus NF human myocardium, with no changes in expression of PKCε or δ. A silver-stained (Ag-stained) band is provided as a loading control. Fig. S2, B and C, shows there are also no significant changes in the p-PLB/PLB ratio in pre- and post-VAD F myocardium. The immunostaining in Fig. S3 shows the redistribution of PKCα in PO (B) compared with sham (A) rat myocardium. Of note, the striated distribution of PKCα increases in PO myocytes. Composite demographic and diagnosis data from F and NF human samples, as well as frequency-dependent shortening data from pre- and post-VAD myocytes, are tabulated in Tables S1 and S2.
Results
PKC isoform expression and contractile function were initially studied in human samples from NF donors and F tissue collected before or after VAD therapy. PKCα expression levels were elevated in pre-VAD F myocardium compared with NF myocardium, and there were no significant changes in PKC δ or ε levels pre- or post-VAD (Fig. 1, A and B; and Fig. S2 A), which agrees with earlier results (Bowling et al., 1999; Noguchi et al., 2004). In post-VAD myocardium, PKCα expression was reduced by 34% compared with pre-VAD levels (Fig. 1 B), although expression still remained higher than in NF hearts (Fig. S2 A). The slowed TTR50% observed in F myocytes at a low stimulation frequency is significantly improved by device support (Fig. 1, C and D). The reduced frequency-dependent amplitude of contraction, and rates of shortening and relengthening observed in pre-VAD myocytes, are also improved in post-VAD myocytes (Fig. 1 E and Table S2), which is consistent with earlier studies (Dipla et al., 1998).
Figure 1.

PKC expression, contractile function, and PKC-targeted cTnI phosphorylation in human myocardium before and after ventricular assist device (VAD) support. (A) Representative Western blots for PKCα, PKCδ, and PKCε expression in F human heart tissue collected pre- versus post-VAD treatment along with a silver-stained (Ag stain) portion of the SDS-PAGE gel to indicate protein loading. “A” indicates pre-VAD while “B” indicates post-VAD samples for the labels shown below the blot. (B) Quantitative analysis of the fold change (Δ) in PKCα, δ, and ε protein expression in F compared with NF myocardial tissue (left panel) and the fold change in the ratio of post-/pre-VAD isoform expression (right panel). Results are expressed as mean± SEM for quantitative data shown in B and D–F. An unpaired Student’s t test was used to compare F versus NF samples and post- versus pre-VAD levels, with *, P < 0.05, considered statistically significant (n = number of hearts in each group for B and F). (C) Representative shortening traces collected from F myocytes before VAD (upper panel), or after VAD (middle panel) implantation compared with NF (lower panel) myocytes. Individual traces are shown at stimulation frequencies of 0.2 (black) and 2.0 (red) Hz to illustrate the negative frequency response in myocytes from F human hearts before VAD (Goldberg et al., 2000) and partial restoration after VAD therapy. (D) Quantitative analysis of contractile function stimulated at 0.2 Hz in myocytes collected from six pre- versus seven post-VAD hearts (n = number of cells and *, P < 0.05 vs. pre-VAD for D and E). (E) Analysis of the percent change (%Δ) in the shortening amplitude, rates of shortening and relengthening, plus TTR50% measured at 2.0 versus 0.2 Hz. (F) Representative Western blots show cTnI phosphorylation at residue p-S44 relative to total cTnI expression in pre- and post-VAD samples (left panel), and the previously established decrease in p-S23/24 levels in F hearts (middle panel; Messer et al., 2007). Quantitative analysis of cTnI p-S44 shows significantly higher levels before VAD therapy compared with NF tissue using a one-way ANOVA and post hoc Tukey statistical comparison (right panel; *, P < 0.05). After VAD support, cTnI p-S44 levels are not significantly different from NF or pre-VAD F levels.
In pre-VAD tissue, PKC-targeted cTnI p-S44 levels were elevated compared with NF hearts, while post-VAD levels were not significantly different from either NF or pre-VAD levels (Fig. 1 F). This intermediate post-VAD cTnI p-S44 is between NF and pre-VAD expression, and agrees with the partial reduction of PKCα levels in post- versus pre-VAD samples (Fig. S2 A). Together, the post-VAD cTnI p-S44 and reduced p-S23/24 (Fig. 1 F; Milting et al., 2006) help to explain the incomplete restoration of the positive frequency response observed in NF myocytes (Fig. 1 C). In addition, PKC phosphorylates cTnI S42 (p-S42; Noland and Kuo, 1991; Venema and Kuo, 1993), which could also be elevated during HF. However, this site was not examined here due to the lack of specific p-S42 phospho-Abs, and difficulties distinguishing between p-S42 and p-S44 by mass spectrometry (Zhang et al., 2012). Another target of PKCα is protein phosphatase I and the downstream reduction in sarcoplasmic reticulum p-PLB (Braz et al., 2004). As with cTnI p-S44, p-PLB levels were not significantly different in post- versus pre-VAD myocardium, although post-VAD levels tended to improve (Fig. S2, B and C).
In additional studies, contractile function was measured in F human myocytes after gene transfer of PKCα or PKCαDN into F myocytes and compared with time-matched, NT controls (Fig. 2). Initially, AdGFP gene transfer was used to show the feasibility of successful gene transfer in human myocytes (Fig. 2 A). Within 24 h after gene transfer, Western analysis detected increased PKCα or PKCαDN protein expression (Fig. 2 B). Parallel measurements of contractile function showed that PKCαDN improved the frequency-dependent responses compared with NT myocytes (Fig. 2 C). However, gene transfer of PKCα resulted in no shortening at any stimulation frequency, and thus, contractile function was not quantitatively analyzed in these myocytes. In agreement with these functional changes, increased cTnI p-S44 levels were detected by Western analysis after PKCα but not PKCαDN gene transfer (Fig. 2 B).
Figure 2.
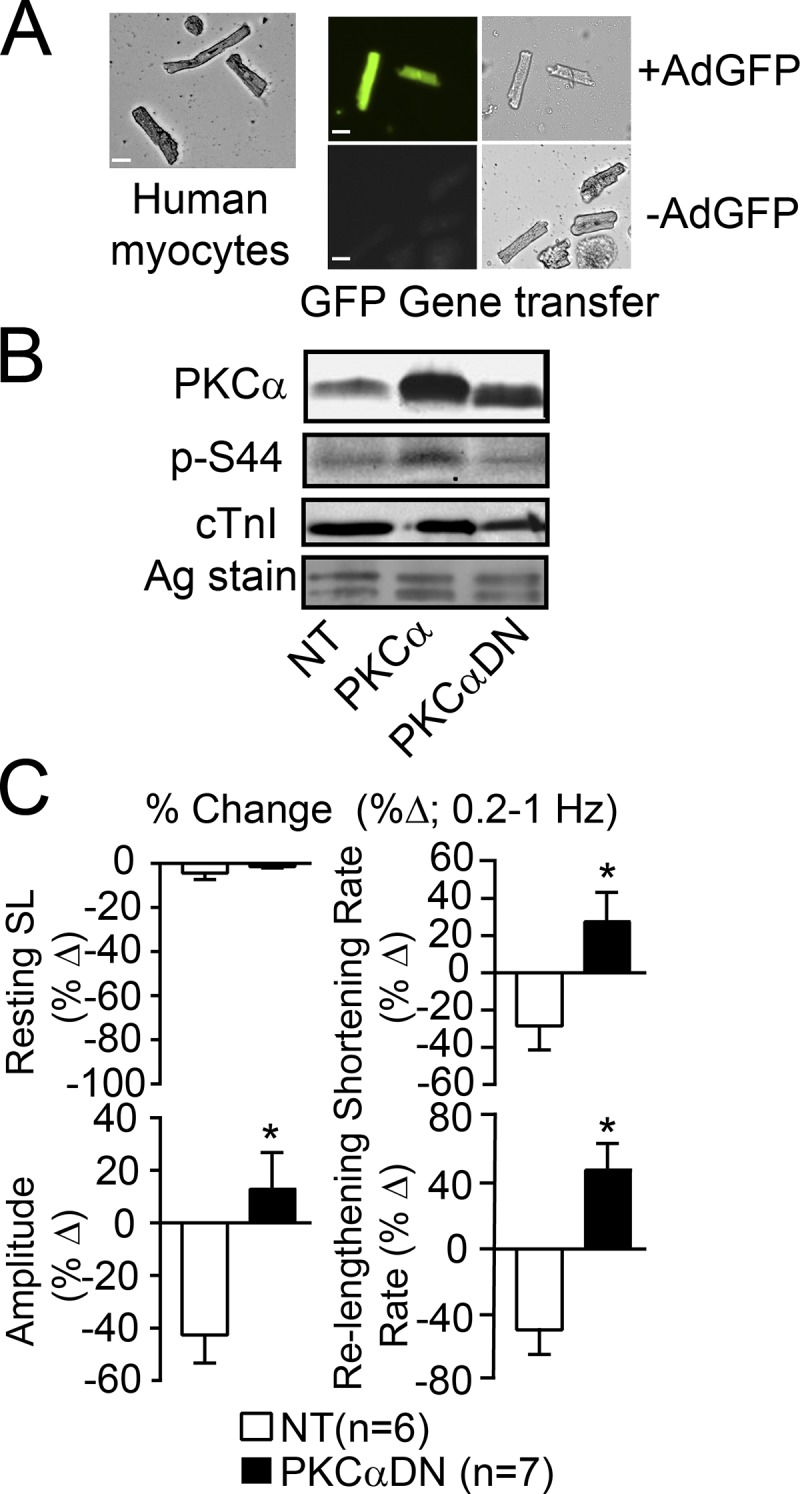
Protein expression and the contractile function frequency response after gene transfer in human myocytes. (A) Representative bright field (left, far right panels) and fluorescence (middle panel) images of F human cardiac myocytes with AdGFP gene transfer (upper panels) compared with NT myocytes (lower panels, -AdGFP). Scale bars, 25 µm. (B) Representative Western blot analysis showing PKCα/PKCαDN, cTnI p-S44, and cTnI expression in isolated F myocytes after AdPKCα or AdPKCαDN gene transfer compared with NT myocytes. The silver-stained (Ag-stain) gel is included to show protein loading in each lane. The limited number of myocytes available for Westerns in each prep did not allow for quantitative analysis of multiple Western blots. (C) Myocyte function after gene transfer of PKCαDN into F myocytes compared with NT myocytes. Results are expressed as the percent change (%Δ) in the resting sarcomere length (SL), amplitude, shortening and relengthening rates collected at 1 versus 0.2 Hz (n = number of cells from three separate hearts in each group). Data are presented as mean ± SEM, with a Student’s unpaired t test used for statistical comparisons (*, P < 0.05 versus NT). Cells no longer contracted after gene transfer of AdPKCα into F myocytes, and therefore, this group was not included in the quantitative analysis.
Due to a limited number of myocytes available from each human heart preparation, the relationship between PKCα, cTnI p-S45 (e.g., analogous to human cTnI S44), and function was further explored in rat models with cardiac dysfunction. Increased PKC expression or enhanced PKC-mediated cTnI p-S45 developed in earlier PO animal models (Bayer et al., 2003; Dong et al., 2012), but PKCα, cTnI p-S45, and cellular function have not been analyzed in one model. Here, analysis of myocardium from an established rat model of PO (Kim et al., 2016) showed increased PKCα and PKCα p-T638 levels in PO versus sham rat myocytes (Fig. 3 A). Increased PKCα signaling was previously linked to elevated p-T638 levels in earlier studies on human HF (Lange et al., 2016). Immunolabeling showed PKCα primarily localized to the intercalated disks in sham myocardium, but developed a more striated distribution in PO myocytes (Fig. 3 B and Fig. S3). A slight increase in PKCδ and decrease in PKCε expression observed in this rat PO model (Fig. 3 C) also are consistent with the changes reported in earlier PO models (Bayer et al., 2003; Dong et al., 2012). In functional studies, PO caused a significant reduction in basal shortening amplitude at 0.2 Hz (Fig. 3 D) along with diminished frequency-dependent amplitude and rates of shortening and relengthening (Fig. 3 E) compared with myocytes from sham rats. Heightened cTnI p-S45 (Fig. 3 F) levels accompanied the PO-induced increase in PKCα levels and diminished function, while there was no significant change in cTnI p-T144 (Fig. 3 G) or basal cTnI p-S23/24 levels (Fig. 3 H), which are also targets for PKC (Noland and Kuo, 1991; Venema and Kuo, 1993).
Figure 3.

Comparison of PKC expression and localization, contractile function, and cTnI phosphorylation in hearts from PO- versus sham-treated rats. (A) Representative Western blots for PKCα (upper left panel) and phosphorylated PKCα T638 (PKCα p-T638; lower left panel) in PO compared with sham rat hearts. A silver-stained (Ag-stain) portion of the gel also is shown as a loading control. The quantitative analyses of PKCα and PKCα p-T638 are shown in the upper and lower right panels, respectively. There were increases in the ratio of PKCα p-S657/total PKCα in two samples from PO (1.42, 2.64) when normalized to the sham control ratio (1.00, 1.00). Quantitative data presented in A and D–H are expressed as mean ± SEM. The n values in A and F–H equal the number of rat samples. Statistical analysis in A, D, F, and G used an unpaired Student’s t test (*, P < 0.05). (B) Fluorescence images collected with comparable exposure times after immunostaining for PKCα show the redistribution of PKCα in two sham (panels 1, 2) compared with two PO (panels 3, 4) rat hearts. Scale bars, 10 µm. (C) Representative Westerns show PKCδ and PKCε expression in PO compared with sham-treated rat hearts. Actin immunostaining is shown as a loading control for each lane. (D) Composite shortening traces collected from sham (black) and PO (red) myocytes at 0.2 Hz (upper panel) and the quantitative analysis of traces showed PO produced a significant reduction in shortening amplitude versus sham myocytes (lower panel; *, P < 0.05; n = number of myocytes in D and E). No significant changes were detected in resting sarcomere length (SL) or the rates of shortening and relengthening (data not shown). (E) The percent change (%Δ) in resting SL, peak shortening amplitude, and the rates of shortening and relengthening at 0.5, 1, and 2 Hz were compared with the response at 0.2 Hz in sham and PO myocytes. Results are analyzed with a two-way ANOVA and Tukey’s post hoc tests, with significance set to P < 0.05 (*, versus 0.2 Hz response; ♦, versus sham at the same frequency). PO and stimulation frequency each caused significant effects in the peak amplitude, shortening, and relengthening rates, and there were interaction effects for peak amplitude and relengthening rate. Overall, shortening amplitude and relengthening rate are significantly more impaired at higher frequencies in PO compared with sham myocytes. (F) Representative cTnI p-S45 and cTnI Western blots (left panels) and the quantitative analysis of expression (middle, right panels) show that PO increases cTnI p-S45 without a change in total cTnI compared with sham rat myocytes. The cTnI expression is normalized to a silver-stained band in each gel. *, P < 0.05 versus sham.(G) Representative Western blot for cTnI p-T144 and an silver-stained portion of the gel, which is used as a loading control (left panels) and quantitative analysis of cTnI p-T144/silver (Ag) ratio (right panel) show there are no significant differences in cTnI p-T144 levels for PO compared with sham rat myocytes. (H) Representative Western blots for cTnI p-S23/24 and a silver-stained portion of the gel (left panels) and quantitative analysis of cTnI p-S23/24 levels (right panel) in sham and PO rat myocytes under basal conditions and in response to 1, 10, and 100 nM of the β-adrenergic receptor agonist ISO, and/or the β-antagonist, propranolol (PRO; 10 µM). Results are analyzed with a two-way ANOVA and post hoc Tukey’s test. The quantitative analysis shows that 10 nM ISO produced a significant increase in p-S23/24 above basal levels in sham myocytes (*, P < 0.05 versus basal sham), but not PO myocytes (P > 0.05 versus basal PO), and this ISO response is reduced in PO compared with sham myocytes (♦, P < 0.05 versus sham+ISO).
In addition to PKC, β-adrenergic receptor (β-AR) activation of PKA phosphorylates cTnI S23/24. More severe PO-induced cardiac dysfunction increases cTnI p-S23/24 (Belin et al., 2006), while β-AR down-regulation also develops with hypertension (Böhm et al., 1995). Thus, p-S23/24 responses to increasing doses of isoproterenol (ISO) were evaluated, and the β-AR–mediated cTnI p-S23/24 levels are attenuated in PO compared with sham myocytes (Fig. 3 H). Overall, these data are consistent with increased p-S45 contributing to basal and frequency-dependent systolic and diastolic dysfunction during PO (Fig. 3, D–F), and the attenuated ISO-induced p-S23/24 working with p-S45 to further slow relaxation in the PO myocytes (Fig. 3 H).
The F344BN rat has a significantly longer life span than many other rat strains but also develops cardiac myocyte dysfunction with age (Turturro et al., 1999; Wahr et al., 2000; Chung and Diffee, 2011). Isolated myocytes from F344BN rats were studied at 6, 18, 26, and 34 mo of age to determine whether alterations in PKC and/or downstream p-S45 phosphorylation are linked with age-dependent contractile dysfunction. In support of this idea, peak shortening as well as shortening and relengthening rates were significantly reduced in older 26–34-mo-old compared with 6-mo-old rat myocytes (Fig. 4 B). Although there were no significant changes in basal PKC α, δ, or ε expression detected by quantitative Western analysis in older rats (Fig. 4 A), enhanced downstream cTnI p-S45 accompanied cellular contractile dysfunction in these myocytes (Fig. 4 C). Representative Western blots showing basal PKC isoform expression and phosphorylated cTnI in 6- and 26-mo-old NT myocytes are provided in Fig. 5, A, B, E, and F.
Figure 4.

PKC isoform expression, basal contractile function, and cTnI phosphorylation in young compared with older F344BN rat hearts. (A) Quantitative analysis showed no significant change in basal expression of PKCα, δ, and ε protein levels in myocytes from young adult (6 mo) versus older (26 +34 mo data = ≥26 mo) rats. PKC isoform expression is normalized to a portion of the silver-stained (Ag stain) gel. Quantitative results in each panel are expressed as mean ± SEM (n = number of rat samples in A and C). Statistical comparisons in A and C used an unpaired Student’s t test (*, P < 0.05). (B) Analysis of composite myocyte contractile function in 6-, 18-, 26-, and 34-mo-old rat myocytes paced at 0.2 Hz (n = number of myocytes from four or more different rats). Resting sarcomere length, peak shortening amplitude, the rates of shortening and relengthening, and the TTR50% were analyzed with a one-way ANOVA and post hoc Dunnett’s tests (*, P < 0.05 versus 6 mo). (C) Quantitative analysis of cTnI phosphorylation at cTnI p-S23/24, p-S45, and total cTnI expression in 6- versus ≥26-mo-old rat hearts. Total cTnI levels are normalized to a portion of the silver-stained gel. The p-S45 levels are significantly elevated in rat hearts from ≥26-mo-olds compared with 6-mo-olds (middle panel), while there were no differences in cTnI p-S23/24 (left panel) or total cTnI (right panel) expression. Note that representative Western blots for A are shown in Fig. 5, A, B, and E, and the representative Western results for basal cTnI p-S23/24, p-S45, and cTnI are shown in Fig. 5 F.
Figure 5.

PKC expression, contractile function, and phosphorylation in young and aging F344BN rats after gene transfer. (A) Representative Western blot detecting PKCα in NT myocytes and after AdGFP gene transfer along with PKCαDN after AdPKCαDN gene transfer in myocytes from 6- and 26-mo-old rats. A portion of silver-stained gel also is included as a loading control. (B) Representative Western blots showing gene transfer of AdPKCα or AdPKCαDN increases PKCα or PKCαDN expression, respectively, compared with NT myocytes. These representative blots also illustrate that gene transfer produces similar increases in PKCαDN expression in 6-mo-old (left panel) versus 26-mo-old (right panel) rat myocytes. As shown here and in E, the levels of PKCα expression after gene transfer of AdPKCα also were similar in 6- and >26-mo-old myocytes. These Western blots also were reprobed for PKCδ and ε expression in 6- and ≥26-mo-old myocytes. (C) Quantitative analysis of PKCαDN/PKCα protein expression after gene transfer. In the left panel, PKCαDN protein expression after gene transfer is normalized to a silver-stained gel band from young (6 mo) and older (≥26 mo) samples. Results are expressed as mean + SEM in C–E, and n = number of rat hearts in C and E. PKCαDN expression is similar in myocytes from the two age groups when compared using an unpaired Student’s t test (P > 0.05). Gene transfer also produced similar increases in PKCα expression in myocytes from 6- and ≥26-mo-old rats (data not shown). Thus, expression in the two age groups was pooled to quantitatively compare the level of PKCα versus PKCαDN expression levels after gene transfer and relative to NT myocytes using one-way ANOVA and post hoc Dunnett’s tests (*, P < 0.05 versus NT; right panel). This comparison shows gene transfer results in comparable increases in PKCα and PKCαDN expression in adult myocytes. (D) Sarcomere length, peak shortening amplitude, and the rates of shortening and relengthening in NT myocytes and after gene transfer of AdPKCα or AdPKCαDN for 6–18- versus ≥26-mo-old rats (n = number of myocytes). A two-way ANOVA (PKC, age) and post hoc Tukey’s tests were used for the statistical comparison (*, P < 0.05 versus NT for PKC effect; ♦, P < 0.05 versus 6–18-mo-old myocytes for age effect). (E) Representative Western analysis of PKCα, p-PLB, and PLB expression plus a silver-stained portion of the gel in 6- and 26-mo-old rat myocytes after AdPKCα or AdPKCαDN gene transfer compared with NT myocytes (left, middle panel). Quantitative analysis of p-PLB/PLB ratio in 6–18- versus ≥26-mo-old rat myocytes (right panel) after PKCαDN gene transfer is compared using a Student’s unpaired t test (P > 0.05; right panel). (F) Representative Western blots show cTnI p-S23/24, p-S45, and total cTnI expression in 6-, 26-, and 34-mo-old rat myocytes after AdGFP or AdPKCαDN gene transfer compared with time-matched NT myocytes.
PKCα and PKCαDN in 6–18- and ≥26-mo-old F344BN rat myocytes also was analyzed after gene transfer of AdGFP, AdPKCα, and AdPKCαDN and compared to time-matched NT myocytes to gain further insight into the relationship between PKC and aging-related contractile dysfunction. PKC isoform expression in either young or old myocytes did not change after AdGFP gene transfer (Fig. 5 A), and similar increases in PKCα/PKCαDN expression levels developed in both young and old myocytes after AdPKCα/AdPKCαDN gene transfer (Fig. 5, A–C). Age did not change the level of increased PKCαDN (Fig. 5 C, left panel) or PKCα expression after gene transfer. In addition, the increases in PKCα and PKCαDN were similar (Fig. 5 C, right panel). Thus, any functional differences are not caused by age-related differences in PKCαDN expression after gene transfer or a difference in PKCαDN versus PKCα expression after gene transfer. In functional studies, the shortening and relengthening rates in myocytes from young rats (6–18 mo) were consistent with earlier results after gene transfer of PKCα and PKCαDN (Braz et al., 2004; Fig. 5 D). However, there were interesting age-related differences in the functional response of older myocytes (≥26 mo) after gene transfer. Specifically, the increased peak shortening amplitude observed after AdPKCαDN gene transfer into younger myocytes was absent in myocytes from ≥26-mo-old rats, and in fact further decreased compared with younger myocytes (Fig. 5 D). The enhanced p-PLB/PLB produced by PKCαDN gene transfer is comparable for both age groups (Fig. 5 E), but there is a sustained elevation in cTnI p-S45 levels in myocytes from older rats regardless of gene transfer group (Fig. 5 F). This age-related elevation in cTnI p-S45 levels is consistent with the inability of PKCαDN to improve peak shortening in the older rat myocytes.
Discussion
The current results show that elevated cTnI p-S44 levels accompany chronic contractile dysfunction in myocytes from F human hearts and the equivalent p-S45 in a PO rat model, when there is up-regulation of PKC (Figs. 1 A and 3 A). Previously, the contribution of PKC-targeted cTnI p-S42/44 (rat p-S43/45) to progressive contractile dysfunction has received little attention even though rodent cTnI S43/45 is a known target for PKC (Noland and Kuo, 1991; Venema and Kuo, 1993), and phospho-mimetic cTnI S43 and/or S45 substitutions significantly reduce myocyte contractile function (Lang et al., 2015, 2017). The improved cellular function and reduced cTnI p-S45 after gene transfer of PKCαDN into F human myocytes (Fig. 2) further support the idea that this cTnI phosphorylation site contributes significantly to chronic cardiac dysfunction associated with elevated PKC levels. Chronic PKC up-regulation and impaired cardiac function have been more often linked to changes in other cellular functions not directly responsible for contractile function (Braz et al., 2004; Hambleton et al., 2006; Li et al., 2014; Newton et al., 2016; Singh et al., 2017). The current studies suggest chronic cTnI p-S44 in human myocardium contributes to progressive dysfunction and thus, therapeutically targeting this site may improve cardiac performance in HF.
Relationship between PKC expression and contractile dysfunction
The increased PKCα levels observed in F human hearts and in a rat model of PO (Figs. 1 A and 3 A) are consistent with earlier work showing that chronic PKC activity is associated with up-regulation and produces contractile dysfunction (Bowling et al., 1999; Noguchi et al., 2004; Belin et al., 2007; Dong et al., 2007). Improved contractile function in post-VAD hearts with reduced PKCα expression (Fig. 1, B–E) and after PKCαDN gene transfer in F human myocytes (Fig. 2 C) also agrees with the cardiac phenotype in genetically targeted PKCα knockout mice (Braz et al., 2004). However, in aging rat myocytes, contractile dysfunction and increased cTnI p-S45 developed even though PKC isoform expression did not change (Fig. 4, A–C). The PO-related redistribution of PKCα in rat myocytes (Fig. 3 B) suggests spatiotemporal localization of cardiac PKC could produce diverse responses by targeting different patterns of downstream proteins (Mukherjee et al., 2016). This possibility could also help to explain age-associated increases in cTnI p-S45 and contractile dysfunction in F344BN rats (Fig. 4, B and C), especially given that there is no significant age-related change in basal p-S23/24 observed here and in previous work (Fig. 4 C; Sakai et al., 1989). The potential for changes in downstream targeting patterns also indicates that in addition to monitoring PKC, there is a need to identify downstream proteins in the PKC pathway and their contribution to chronic cardiac dysfunction.
PKC and downstream cTnI phosphorylation
Sarcomere proteins play a pivotal role in cardiac pressure development and relaxation (Li et al., 2002; Stehle and Iorga, 2010). Thus, myofilament signaling targets such as cTnI are especially important to investigate in the context of understanding pump dysfunction. Our results show chronic cardiac contractile dysfunction is consistently associated with increased PKC-targeted rat cTnI p-S45 levels (Figs. 3 and 4). PKC preferentially phosphorylates rat cTnI S23, S24, S43, S45, and T144 (Venema and Kuo, 1993). The best understood site on cTnI is S23/24, which is phosphorylated by PKA, PKC, and other kinases (Westfall, 2014) to accelerate in vivo cardiac relaxation by increasing Ca2+ dissociation from cTnC and reducing myofilament Ca2+ sensitivity (Takimoto et al., 2004; Dong et al., 2007; Yasuda et al., 2007). In contrast, cTnI p-S43/45 reduces maximum actomyosin adenosine triphosphatase (ATPase), force, and shortening in addition to decreasing myofilament calcium sensitivity (Noland and Kuo, 1991; Burkart et al., 2003; Takimoto et al., 2004; Liu et al., 2014). Thus, this cluster is expected to diminish in vivo function.
Endogenous cTnI has been replaced with cTnI containing phospho-mimetic S43/45 combined with other PKC-targeted residues (e.g., S23/24, T144) in genetic mouse models to determine the impact of PKC phosphorylation on cTnI and myofilament function. However, phenotypes in these mice range from minimal to severe cardiac dysfunction (Sakthivel et al., 2005; Bilchick et al., 2007; Kirk et al., 2009). These findings are not easily explained by the other modified residue and/or cTnI replacement levels, which has led to ambiguity in the role cTnI S43/45 plays in modulating in vivo contractile performance. There is no published genetic animal model expressing cardiac-specific phospho-mimetic cTnI S43/45 alone, but work in isolated myocytes provides some insight (Westfall, 2014). Specifically, gene transfer and replacement of endogenous cTnI with phospho-mimetic cTnI S43D, S45D, or S43/45D reduce myocyte contraction and relaxation (Lang et al., 2015), which is consistent with the enhanced cTnI p-S44/p-S45 and contractile dysfunction observed in human hearts and in PO and aging rat myocytes (Figs. 1, 3, and 4). The enhanced cTnI p-S45 linked to dysfunction in older F344BN myocytes and the inability of PKCαDN gene transfer to improve shortening amplitude and reduce p-S45 levels in older myocytes (Figs. 4 and 5) further support a significant role for cTnI p-S45 in chronic cardiac dysfunction. Cellular studies also showed that secondary phosphorylation develops at other myofilament protein sites over time and coincides with a partial attenuation of myocyte dysfunction (Lang et al., 2017). This adaptive mechanism may contribute to the partial improvement in post-VAD myocyte contractile function, when there were no significant improvements in pre- versus post-VAD cTnI p-S44 and p-PLB (Fig. 1 F and Fig. S2 C). Most importantly, the current set of studies shows that elevated cTnI p-S44 is consistently observed during contractile dysfunction (Figs. 1, 3, and 4). Future work is needed to determine if there is a similar relationship between human cTnI p-S42 and chronic in vivo cardiac dysfunction, and to better understand the intricate modulatory nature of the myofilament.
Our results also show that elevated cTnI p-S44 is present in addition to the well-documented reduction in cTnI p-S23/24 during cardiac dysfunction and end-stage human HF (van der Velden et al., 2004, 2006; Messer et al., 2007; Fig. 1). Heightened rat cTnI p-S45 develops under circumstances when there is chronic contractile dysfunction without decreases in basal p-S23/24 levels (Fig. 3, F and H, and Fig. 4 C). As a result, data from both human and rat models support the idea that cTnI p-S45 significantly contributes to chronic cardiac dysfunction.
Therapeutically targeting downstream PKC sites in the myofilament
Recent clinical trials tested whether molecular inotropes targeted to myofilament myosin can improve cardiac performance (Cleland et al., 2011). The elevated PKC-targeted cTnI p-S44/p-S45 that accompanies myocyte dysfunction (Figs. 1, 3, and 4) indicates reduced human cTnI p-S44 may be functionally beneficial. Thus, this site should be considered as an additional myofilament target for therapeutically treating cardiac dysfunction. Therapeutic strategies designed to broadly modify multiple posttranslational sites and thus target the rheostat-like function of cTn may optimize improvements in both systolic and diastolic function, and potentially increase the relatively small improvement observed in cardiac performance during VAD support (Ogletree et al., 2010; Kirklin et al., 2015).
Limitations
The current myocyte studies do not include measurement of Ca2+ transients, but the peak amplitude and rate of Ca2+ decay do improve and contribute to the functional improvement in F human myocytes after VAD therapy (Dipla et al., 1998). However, there is also earlier work showing contractile dysfunction can develop independent of changes in Ca2+. For example, no significant changes in diastolic Ca2+ or the peak Ca2+ transient were detected at a point when peak shortening was impaired in a rat hypertensive model with a comparable duration of suprarenal aortic banding as the myocytes in Fig. 3 (McCall et al., 1998). Significant reductions in peak shortening and the rates of shortening and relengthening also were accompanied by higher peak Ca2+ transients in rat myocytes after aortic banding (Kagaya et al., 1996). There also were no significant changes in the Ca2+ transient in the same aging rodent model used here (Wahr et al., 2000), and a similar outcome was reported in Wistar rats (Orchard and Lakatta, 1985). Although these findings collectively suggest elevated human cTnI p-S44 (or rat p-S45) contributes to chronic reductions in contractile function, future work is needed to test the impact of cTnI p-S43/45 alone on in vivo contractile performance.
Supplementary Material
Acknowledgments
Henk L. Granzier served as editor.
We gratefully acknowledge the technical contributions of Michelle Tracy.
This work was supported by National Institutes of Health grants R01-HL-067254 (M.V. Westfall) and NIGMS T32 GM007315 (V. Ravichandran).
The authors declare no competing financial interests.
Author contributions: V.S. Ravichandran, H.J. Patel, and M.V. Westfall collected and analyzed data. H.J. Patel and F.D. Pagani provided human tissue. V.S. Ravichandran wrote the original draft and edited the manuscript with M.V. Westfall.
References
- Bayer A.L., Heidkamp M.C., Patel N., Porter M., Engman S., and Samarel A.M.. 2003. Alterations in protein kinase C isoenzyme expression and autophosphorylation during the progression of pressure overload-induced left ventricular hypertrophy. Mol. Cell. Biochem. 242:145–152. 10.1023/A:1021106232511 [DOI] [PubMed] [Google Scholar]
- Belin R.J., Sumandea M.P., Kobayashi T., Walker L.A., Rundell V.L., Urboniene D., Yuzhakova M., Ruch S.H., Geenen D.L., Solaro R.J., and de Tombe P.P.. 2006. Left ventricular myofilament dysfunction in rat experimental hypertrophy and congestive heart failure. Am. J. Physiol. Heart Circ. Physiol. 291:H2344–H2353. 10.1152/ajpheart.00541.2006 [DOI] [PubMed] [Google Scholar]
- Belin R.J., Sumandea M.P., Allen E.J., Schoenfelt K., Wang H., Solaro R.J., and de Tombe P.P.. 2007. Augmented protein kinase C-alpha-induced myofilament protein phosphorylation contributes to myofilament dysfunction in experimental congestive heart failure. Circ. Res. 101:195–204. 10.1161/CIRCRESAHA.107.148288 [DOI] [PubMed] [Google Scholar]
- Bilchick K.C., Duncan J.G., Ravi R., Takimoto E., Champion H.C., Gao W.D., Stull L.B., Kass D.A., and Murphy A.M.. 2007. Heart failure-associated alterations in troponin I phosphorylation impair ventricular relaxation-afterload and force-frequency responses and systolic function. Am. J. Physiol. Heart Circ. Physiol. 292:H318–H325. 10.1152/ajpheart.00283.2006 [DOI] [PubMed] [Google Scholar]
- Böhm M., Castellano M., Agabiti-Rosei E., Flesch M., Paul M., and Erdmann E.. 1995. Dose-dependent dissociation of ACE-inhibitor effects on blood pressure, cardiac hypertrophy, and beta-adrenergic signal transduction. Circulation. 92:3006–3013. 10.1161/01.CIR.92.10.3006 [DOI] [PubMed] [Google Scholar]
- Bowling N., Walsh R.A., Song G., Estridge T., Sandusky G.E., Fouts R.L., Mintze K., Pickard T., Roden R., Bristow M.R., et al. 1999. Increased protein kinase C activity and expression of Ca2+-sensitive isoforms in the failing human heart. Circulation. 99:384–391. 10.1161/01.CIR.99.3.384 [DOI] [PubMed] [Google Scholar]
- Braz J.C., Gregory K., Pathak A., Zhao W., Sahin B., Klevitsky R., Kimball T.F., Lorenz J.N., Nairn A.C., Liggett S.B., et al. 2004. PKC-α regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10:248–254. 10.1038/nm1000 [DOI] [PubMed] [Google Scholar]
- Burkart E.M., Sumandea M.P., Kobayashi T., Nili M., Martin A.F., Homsher E., and Solaro R.J.. 2003. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J. Biol. Chem. 278:11265–11272. 10.1074/jbc.M210712200 [DOI] [PubMed] [Google Scholar]
- Castillero E., Ali Z.A., Akashi H., Giangreco N., Wang C., Stöhr E.J., Ji R., Zhang X., Kheysin N., Park J.S., et al. 2018. Structural and functional cardiac profile after prolonged duration of mechanical unloading: potential implications for myocardial recovery. Am. J. Physiol. Heart Circ. Physiol. 315:H1463–H1476. 10.1152/ajpheart.00187.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary K.W., Rossman E.I., Piacentino V. III, Kenessey A., Weber C., Gaughan J.P., Ojamaa K., Klein I., Bers D.M., Houser S.R., and Margulies K.B.. 2004. Altered myocardial Ca2+ cycling after left ventricular assist device support in the failing human heart. J. Am. Coll. Cardiol. 44:837–845. 10.1016/j.jacc.2004.05.049 [DOI] [PubMed] [Google Scholar]
- Chung E., and Diffee G.M.. 2011. Effect of aging on power output properties in rat skinned cardiac myocytes. J. Gerontol. A Biol. Sci. Med. Sci. 66:1267–1273. 10.1093/gerona/glr150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland J.G., Teerlink J.R., Senior R., Nifontov E.M., Mc Murray J.J., Lang C.C., Tsyrlin V.A., Greenberg B.H., Mayet J., Francis D.P., et al. 2011. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet. 378:676–683. 10.1016/S0140-6736(11)61126-4 [DOI] [PubMed] [Google Scholar]
- Day S.M., Westfall M.V., Fomicheva E.V., Hoyer K., Yasuda S., La Cross N.C., D’Alecy L.G., Ingwall J.S., and Metzger J.M.. 2006. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nat. Med. 12:181–189. 10.1038/nm1346 [DOI] [PubMed] [Google Scholar]
- Dipla K., Mattiello J.A., Jeevanandam V., Houser S.R., and Margulies K.B.. 1998. Myocyte recovery after mechanical circulatory support in humans with end-stage heart failure. Circulation. 97:2316–2322. 10.1161/01.CIR.97.23.2316 [DOI] [PubMed] [Google Scholar]
- Dong W.J., Jayasundar J.J., An J., Xing J., and Cheung H.C.. 2007. Effects of PKA phosphorylation of cardiac troponin I and strong crossbridge on conformational transitions of the N-domain of cardiac troponin C in regulated thin filaments. Biochemistry. 46:9752–9761. 10.1021/bi700574n [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X., Sumandea C.A., Chen Y.C., Garcia-Cazarin M.L., Zhang J., Balke C.W., Sumandea M.P., and Ge Y.. 2012. Augmented phosphorylation of cardiac troponin I in hypertensive heart failure. J. Biol. Chem. 287:848–857. 10.1074/jbc.M111.293258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg A.T., Bond B.R., Mukherjee R., New R.B., Zellner J.L., Crawford F.A. Jr., and Spinale F.G.. 2000. Endothelin receptor pathway in human left ventricular myocytes: relation to contractility. Ann. Thorac. Surg. 69:711–715, discussion:716. 10.1016/S0003-4975(99)01515-5 [DOI] [PubMed] [Google Scholar]
- Hambleton M., Hahn H., Pleger S.T., Kuhn M.C., Klevitsky R., Carr A.N., Kimball T.F., Hewett T.E., Dorn G.W. II, Koch W.J., and Molkentin J.D.. 2006. Pharmacological- and gene therapy-based inhibition of protein kinase Calpha/beta enhances cardiac contractility and attenuates heart failure. Circulation. 114:574–582. 10.1161/CIRCULATIONAHA.105.592550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H., Robinson D.A., Stevenson T.K., Wu H.C., Kampert S.E., Pagani F.D., Dyke D.B., Martin J.L., Sadayappan S., Day S.M., and Westfall M.V.. 2012. PKCβII modulation of myocyte contractile performance. J. Mol. Cell. Cardiol. 53:176–186. 10.1016/j.yjmcc.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagaya Y., Hajjar R.J., Gwathmey J.K., Barry W.H., and Lorell B.H.. 1996. Long-term angiotensin-converting enzyme inhibition with fosinopril improves depressed responsiveness to Ca2+ in myocytes from aortic-banded rats. Circulation. 94:2915–2922. 10.1161/01.CIR.94.11.2915 [DOI] [PubMed] [Google Scholar]
- Kim E.H., Galchev V.I., Kim J.Y., Misek S.A., Stevenson T.K., Campbell M.D., Pagani F.D., Day S.M., Johnson T.C., Washburn J.G., et al. 2016. Differential protein expression and basal lamina remodeling in human heart failure. Proteomics Clin. Appl. 10:585–596. 10.1002/prca.201500099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk J.A., MacGowan G.A., Evans C., Smith S.H., Warren C.M., Mamidi R., Chandra M., Stewart A.F., Solaro R.J., and Shroff S.G.. 2009. Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ. Res. 105:1232–1239. 10.1161/CIRCRESAHA.109.205427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirklin J.K., Naftel D.C., Pagani F.D., Kormos R.L., Stevenson L.W., Blume E.D., Myers S.L., Miller M.A., Baldwin J.T., and Young J.B.. 2015. Seventh INTERMACS annual report: 15,000 patients and counting. J. Heart Lung Transplant. 34:1495–1504. 10.1016/j.healun.2015.10.003 [DOI] [PubMed] [Google Scholar]
- Lang S.E., Schwank J., Stevenson T.K., Jensen M.A., and Westfall M.V.. 2015. Independent modulation of contractile performance by cardiac troponin I Ser43 and Ser45 in the dynamic sarcomere. J. Mol. Cell. Cardiol. 79:264–274. 10.1016/j.yjmcc.2014.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang S.E., Stevenson T.K., Schatz T.M., Biesiadecki B.J., and Westfall M.V.. 2017. Functional communication between PKC-targeted cardiac troponin I phosphorylation sites. Arch. Biochem. Biophys. 627:1–9. 10.1016/j.abb.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S., Gehmlich K., Lun A.S., Blondelle J., Hooper C., Dalton N.D., Alvarez E.A., Zhang X., Bang M.-L., Abassi Y.A., et al. 2016. MLP and CARP are linked to chronic PKCα signalling in dilated cardiomyopathy. Nat. Commun. 7:12120–12130. 10.1038/ncomms12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Fultz M.E., and Wright G.L.. 2002. PKC-α shows variable patterns of translocation in response to different stimulatory agents. Acta Physiol. Scand. 174:237–246. 10.1046/j.1365-201x.2002.00945.x [DOI] [PubMed] [Google Scholar]
- Li Z., Abdullah C.S., and Jin Z.-Q.. 2014. Inhibition of PKC-θ preserves cardiac function and reduces fibrosis in streptozotocin-induced diabetic cardiomyopathy. Br. J. Pharmacol. 171:2913–2924. 10.1111/bph.12621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Lopez J.J., Biesiadecki B.J., and Davis J.P.. 2014. Protein kinase C phosphomimetics alter thin filament Ca2+ binding properties. PLoS One. 9:e86279 10.1371/journal.pone.0086279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall E., Ginsburg K.S., Bassani R.A., Shannon T.R., Qi M., Samarel A.M., and Bers D.M.. 1998. Ca flux, contractility, and excitation-contraction coupling in hypertrophic rat ventricular myocytes. Am. J. Physiol. 274:H1348–H1360. [DOI] [PubMed] [Google Scholar]
- Messer A.E., Jacques A.M., and Marston S.B.. 2007. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J. Mol. Cell. Cardiol. 42:247–259. 10.1016/j.yjmcc.2006.08.017 [DOI] [PubMed] [Google Scholar]
- Miller L.W., and Rogers J.G.. 2018. Evolution of left ventricular assist device therapy for advanced heart failure: A review. JAMA Cardiol. 3:650–658. 10.1001/jamacardio.2018.0522 [DOI] [PubMed] [Google Scholar]
- Milting H., Scholz C., Arusoglu L., Freitag M., Cebulla R., Jaquet K., Körfer R., V Lewinski D., Kassner A., Brodde O.-E., et al. 2006. Selective upregulation of β1-adrenergic receptors and dephosphorylation of troponin I in end-stage heart failure patients supported by ventricular assist devices. J. Mol. Cell. Cardiol. 41:441–450. 10.1016/j.yjmcc.2006.04.010 [DOI] [PubMed] [Google Scholar]
- Morgan J.A., Brewer R.J., Nemeh H.W., Murthy R., Williams C.T., Lanfear D.E., Tita C., and Paone G.. 2012. Left ventricular reverse remodeling with a continuous flow left ventricular assist device measured by left ventricular end-diastolic dimensions and severity of mitral regurgitation. ASAIO J. 58:574–577. 10.1097/MAT.0b013e31826e4267 [DOI] [PubMed] [Google Scholar]
- Mukherjee A., Roy S., Saha B., and Mukherjee D.. 2016. Spatio-temporal regulation of PKC isoforms imparts signaling specificity. Front. Immunol. 7:45 10.3389/fimmu.2016.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murriel C.L., Churchill E., Inagaki K., Szweda L.I., and Mochly-Rosen D.. 2004. Protein kinase Cdelta activation induces apoptosis in response to cardiac ischemia and reperfusion damage: a mechanism involving BAD and the mitochondria. J. Biol. Chem. 279:47985–47991. 10.1074/jbc.M405071200 [DOI] [PubMed] [Google Scholar]
- Newton A.C., Antal C.E., and Steinberg S.F.. 2016. Protein kinase C mechanisms that contribute to cardiac remodelling. Clin. Sci. (Lond.). 130:1499–1510. 10.1042/CS20160036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi T., Hünlich M., Camp P.C., Begin K.J., El-Zaru M., Patten R., Leavitt B.J., Ittleman F.P., Alpert N.R., LeWinter M.M., and VanBuren P.. 2004. Thin-filament-based modulation of contractile performance in human heart failure. Circulation. 110:982–987. 10.1161/01.CIR.0000139334.43109.F9 [DOI] [PubMed] [Google Scholar]
- Noland T.A. Jr., and Kuo J.F.. 1991. Protein kinase C phosphorylation of cardiac troponin I or troponin T inhibits Ca2(+)-stimulated actomyosin MgATPase activity. J. Biol. Chem. 266:4974–4978. [PubMed] [Google Scholar]
- Ogletree M.L., Sweet W.E., Talerico C., Klecka M.E., Young J.B., Smedira N.G., Starling R.C., and Moravec C.S.. 2010. Duration of left ventricular assist device support: Effects on abnormal calcium cycling and functional recovery in the failing human heart. J. Heart Lung Transplant. 29:554–561. 10.1016/j.healun.2009.10.015 [DOI] [PubMed] [Google Scholar]
- Orchard C.H., and Lakatta E.G.. 1985. Intracellular calcium transients and developed tension in rat heart muscle. A mechanism for the negative interval-strength relationship. J. Gen. Physiol. 86:637–651. 10.1085/jgp.86.5.637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacentino V. III, Weber C.R., Chen X., Weisser-Thomas J., Margulies K.B., Bers D.M., and Houser S.R.. 2003. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 92:651–658. 10.1161/01.RES.0000062469.83985.9B [DOI] [PubMed] [Google Scholar]
- Sakai M., Danziger R.S., Staddon J.M., Lakatta E.G., and Hansford R.G.. 1989. Decrease with senescence in the norepinephrine-induced phosphorylation of myofilament proteins in isolated rat cardiac myocytes. J. Mol. Cell. Cardiol. 21:1327–1336. 10.1016/0022-2828(89)90678-0 [DOI] [PubMed] [Google Scholar]
- Sakthivel S., Finley N.L., Rosevear P.R., Lorenz J.N., Gulick J., Kim S., VanBuren P., Martin L.A., and Robbins J.. 2005. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J. Biol. Chem. 280:703–714. 10.1074/jbc.M409513200 [DOI] [PubMed] [Google Scholar]
- Savarese G., and Lund L.H.. 2017. Global public health burden of heart failure. Card. Fail. Rev. 3:7–11. 10.15420/cfr.2016:25:2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.M., Cummings E., Pantos C., and Singh J.. 2017. Protein kinase C and cardiac dysfunction: a review. Heart Fail. Rev. 22:843–859. 10.1007/s10741-017-9634-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L.A., Yarboro L.T., and Kennedy J.L.W.. 2015. Left ventricular assist device implantation strategies and outcomes. J. Thorac. Dis. 7:2088–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle R., and Iorga B.. 2010. Kinetics of cardiac sarcomeric processes and rate-limiting steps in contraction and relaxation. J. Mol. Cell. Cardiol. 48:843–850. 10.1016/j.yjmcc.2009.12.020 [DOI] [PubMed] [Google Scholar]
- Takimoto E., Soergel D.G., Janssen P.M., Stull L.B., Kass D.A., and Murphy A.M.. 2004. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ. Res. 94:496–504. 10.1161/01.RES.0000117307.57798.F5 [DOI] [PubMed] [Google Scholar]
- Turturro A., Witt W.W., Lewis S., Hass B.S., Lipman R.D., and Hart R.W.. 1999. Growth curves and survival characteristics of the animals used in the Biomarkers of Aging Program. J. Gerontol. A Biol. Sci. Med. Sci. 54:B492–B501. 10.1093/gerona/54.11.B492 [DOI] [PubMed] [Google Scholar]
- Uriel N., Kim G., and Burkhoff D.. 2017. Myocardial recovery after LVAD implantation: a vision or simply an illusion? J. Am. Coll. Cardiol. 70:355–357. 10.1016/j.jacc.2017.06.001 [DOI] [PubMed] [Google Scholar]
- van der Velden J., Merkus D., Klarenbeek B.R., James A.T., Boontje N.M., Dekkers D.H., Stienen G.J., Lamers J.M., and Duncker D.J.. 2004. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ. Res. 95:e85–e95. 10.1161/01.RES.0000149531.02904.09 [DOI] [PubMed] [Google Scholar]
- van der Velden J., Narolska N.A., Lamberts R.R., Boontje N.M., Borbély A., Zaremba R., Bronzwaer J.G., Papp Z., Jaquet K., Paulus W.J., and Stienen G.J.. 2006. Functional effects of protein kinase C-mediated myofilament phosphorylation in human myocardium. Cardiovasc. Res. 69:876–887. 10.1016/j.cardiores.2005.11.021 [DOI] [PubMed] [Google Scholar]
- Venema R.C., and Kuo J.F.. 1993. Protein kinase C-mediated phosphorylation of troponin I and C-protein in isolated myocardial cells is associated with inhibition of myofibrillar actomyosin MgATPase. J. Biol. Chem. 268:2705–2711. [PubMed] [Google Scholar]
- Wahr P.A., Michele D.E., and Metzger J.M.. 2000. Effects of aging on single cardiac myocyte function in Fischer 344 x Brown Norway rats. Am. J. Physiol. Heart Circ. Physiol. 279:H559–H565. 10.1152/ajpheart.2000.279.2.H559 [DOI] [PubMed] [Google Scholar]
- Westfall M.V. 2014. Post-translational modifications of troponin. In Troponin: Regulator of muscle contraction. Jin J.-P., editor. Nova Science Publishers, New York: 163–202. [Google Scholar]
- Wever-Pinzon O., Drakos S.G., McKellar S.H., Horne B.D., Caine W.T., Kfoury A.G., Li D.Y., Fang J.C., Stehlik J., and Selzman C.H.. 2016. Cardiac recovery during long-term left ventricular assist device support. J. Am. Coll. Cardiol. 68:1540–1553. 10.1016/j.jacc.2016.07.743 [DOI] [PubMed] [Google Scholar]
- Yasuda S., Coutu P., Sadayappan S., Robbins J., and Metzger J.M.. 2007. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ. Res. 101:377–386. 10.1161/CIRCRESAHA.106.145557 [DOI] [PubMed] [Google Scholar]
- Zhang P., Kirk J.A., Ji W., dos Remedios C.G., Kass D.A., Van Eyk J.E., and Murphy A.M.. 2012. Multiple reaction monitoring to identify site-specific troponin I phosphorylated residues in the failing human heart. Circulation. 126:1828–1837. 10.1161/CIRCULATIONAHA.112.096388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.