

We outline the reasons why the Roadmap Epigenomics Program, as a group science effort, is a success story.
Abstract
The NIH Roadmap Epigenomics Program was launched to deliver reference epigenomic data from human tissues and cells, develop tools and methods for analyzing the epigenome, discover novel epigenetic marks, develop methods to manipulate the epigenome, and determine epigenetic contributions to diverse human diseases. Here, we comment on the outcomes from this program: the scientific contributions made possible by a consortium approach and the challenges, benefits, and lessons learned from this group science effort.
WHY DEVELOP AN EPIGENOMICS PROGRAM?
The development of the National Institutes of Health (NIH) Roadmap Epigenomics Program was informed by (i) critical scientific gaps and opportunities, (ii) input from the scientific community, and (iii) analysis of funded NIH research in this area. Epigenomics can be operationally defined as the study of structural and functional DNA and histone modifications that alter the reading and writing of the genome, resulting in the regulation of chromatin architecture, gene activity, and expression without changes to the DNA sequence. Epigenetic marks are those DNA and histone modifications that occur at specific loci within the genome. While analyses of epigenetic regulation of gene expression date back to the 1970s and 1980s, methods to analyze epigenetic modifications at a genome-wide scale were not developed until the early 2000s (1, 2). Advances in DNA sequencing technology, the development of methods such as bisulfite sequencing and chromatin immunoprecipitation sequencing, and generation of highly specific antibodies against posttranslationally modified histones created an opportunity to generate cell-specific epigenomic maps.
Furthermore, a portfolio analysis conducted by the NIH in 2006 on epigenetic/epigenomic research revealed increasing numbers of NIH-funded studies and publications associated with altered gene expression profiles and epigenetic processes between 1998 and 2006. This analysis revealed that limited research was being conducted on specific diseases except for cancer, which constituted more than half of disease epigenetics publications. The majority of these publications defined epigenomes as DNA methylation profiles of tumors, and most studies focused on the area of chromatin regulation and DNA methylation in animal models. Genome-wide analysis of primary human tissues was challenging at the time due to low detection sensitivity and the need for large numbers of cells; hence, there was limited effort to analyze human tissues or to establish human reference epigenomes. Overall, the scientific literature suggested that many conditions could involve altered epigenetic mechanisms, but testing those hypotheses—particularly by surveying epigenetic marks across the genome in human samples—remained a challenge.
Further difficulties at that time included (i) the potentially limitless definition of the “human epigenome” because epigenetic marks are dynamic and vary from cell to cell (both between individual cells of the same type and between different cell types) and (ii) the need to describe an epigenomic “ground state” in pluripotent cells and how this state changes during differentiation. In addition, grassroots scientific groups had been encouraging a large-scale effort in the area of epigenomics for some time (3). Several scientific meetings were held to identify the best ways to accelerate epigenomic discovery including one held in 2007 to specifically identify what would ultimately become the themes of this program. These gaps and opportunities, coupled with the analysis of scientific areas already addressed by the NIH grant portfolio and input from the scientific community, ultimately informed plans for a trans-NIH Roadmap Program in Epigenomics. (The NIH Roadmap is now known as the NIH Common Fund, but the Roadmap Epigenomics Program retains its name.)
OVERALL GOALS OF THE ROADMAP EPIGENOMICS PROGRAM
The overarching goal of the program was to establish a set of human reference epigenomes and develop new technologies as fundamental resources for the scientific community to conduct basic and applied research on how epigenetic/epigenomic processes contribute to human development, life span/aging, response to environmental exposures (e.g., physical, chemical, behavioral, and/or social), and disease pathogenesis. The details of the program goals and the funded projects have been previously described (4, 5). The program was designed recognizing that while highly coordinated consortium-based research was required to generate and analyze epigenomic maps across many human cell types and tissues, substantial innovation and discovery through smaller projects, in particular for technology development, were also needed (4). Because of our poor understanding of the epigenomic differences between cell types and between individuals, it was not clear how many samples of a given tissue would need to be analyzed or what epigenomic marks had to be identified to generate a representative epigenomic map. The limited availability of many human cell types and tissues highlighted the need for epigenomic technologies with higher sensitivity. Thus, substantial technology development (including isolating homogeneous cell populations from a given tissue, interrogating extremely limited numbers of cells, and enhancing assay throughput) were necessary to complement the consortium-wide mapping efforts. This combination of integrating large-scale mapping efforts with small-scale discovery-driven approaches and technology development ultimately proved to be critical for the success of this program.
The Roadmap Epigenomics Program ended in 2018. We reflect here on the program achievements to date, applying objective measures while acknowledging that the long-term impact of the program is yet to be measured. Given the vast quantities of data generated in the postgenomic era and the ever-increasing need for integrating multidisciplinary approaches to solve today’s biomedical problems, we also evaluate the lessons learned from this large consortium-based approach as well as the many challenges and benefits offered by “group science.”
ROADMAP EPIGENOMICS PROGRAM DELIVERABLES
The Roadmap Epigenomics Program was designed to deliver community resources that would catalyze and expand investigator-initiated epigenomic research across the NIH (4). This effort was distinct from previous efforts in three broad ways: (i) The focus was on human tissues and cells as opposed to animal cells and models; (ii) epigenomic features were studied genome wide rather than at a single locus or a handful of loci; and (iii) an emphasis was placed on discovering novel epigenomic marks. While at this time the ENCODE (Encyclopedia of DNA Elements) Project was cataloging regulatory elements in animal models or human cells grown in culture, the aim of the Roadmap Epigenomics Project was to build on this foundation by analyzing samples taken directly from human tissues and cells (6). The deliverables of the Roadmap Epigenomics Program included (i) reference epigenomic maps, (ii) international epigenomic coordination, (iii) a deeper understanding of the epigenomic basis of disease, (iv) identification of previously unknown epigenetic marks, and (v) improved technologies and methods for monitoring and manipulating epigenomic modifications.
Reference epigenomic maps
One major focus of the Roadmap Epigenomics Program was to provide reference epigenomic maps for normal human cells and tissues (4). This focus required a goal-driven highly coordinated approach that could not be easily accomplished by individual laboratories (7). Fundamental to achieving this effort was the definition of a “reference epigenome” and a plan for the coordinated production of these datasets by investigator groups using different methods. The consortium approach paved the way to establishing global maps of multiple epigenomic modifications across the genome in different types of human cells and tissues. Simultaneously, showing that disease- and trait-associated genetic variants are enriched in tissue-specific epigenetic signatures was critical in revealing important cell types implicated in specific genetic traits as well as in providing a molecular basis for interpreting human disease types as a community resource (8). Similar to the human genome sequences that set the stage for far-reaching studies of genetic variation association with disease phenotypes (9), a set of 111 human reference epigenomes profiling comprehensive histone modification signatures, DNA accessibility and methylation patterns, as well as RNA expression was described in an integrative analysis paper (10). Furthermore, the data from these experiments have been incorporated into the ENCODE Portal and can now be searched with ENCODE and other related data to reveal human regulatory elements and the cognate transcription factors associated with these elements.
The Human Reference Epigenome Maps generated by the Roadmap Epigenomics Program have provided a deeper and more comprehensive view of our regulatory genome in terms of defining regulatory elements, such as promoters and enhancers, for a given tissue or cell type and have complemented the information obtained from programs like ENCODE and GTEx (Genotype-Tissue Expression). These maps have been used to predict tissue-specific patterns of disease and have informed the functional analysis of numerous genome-wide association study (GWAS) hits relevant to many complex human diseases. The prioritization of GWAS findings to identify causal loci for disease pathways has been a challenge in the human genomics field for some time. The maps generated now allow researchers to prioritize disease-associated variants that overlap GWAS-enriched epigenomic annotations. Additional tools have been developed and defined in recent years that allow researchers to continue to explore genomic regions with tissue- or cell-specific epigenomic features, using Roadmap Epigenomics Program data with greater sophistication (11). Many studies have also used the integration of histone marks, chromatin states, and transcription factor binding site data from Roadmap and ENCODE to inform epigenomic signatures or develop epigenomic biomarkers related to complex human diseases (12).
International epigenomics coordination
The Roadmap Epigenomics Program was also a founding member of the International Human Epigenome Consortium (IHEC) created in 2010. The primary goal of IHEC was to pursue an international effort in coordinating the production of reference maps of human epigenomes for key cellular states relevant to human health and diseases. A critical component of IHEC goals was also to coordinate the development of common bioinformatics standards, data models, and analytical tools to organize, integrate, and display the epigenomic data generated by the broad international community. An important step toward this goal was the first installment of 41 coordinated papers showcasing the achievements and scientific progress made by IHEC in core areas of current epigenomic investigation (13). Both Roadmap and IHEC were designed to improve compatibility and interoperability of diverse datasets while ensuring that data generation efforts would not be duplicative. Accordingly, the Roadmap Epigenomics Mapping Consortium developed a common set of metadata standards and data standards that were, in part, based on standards developed by the ENCODE program. These standards have been adopted by IHEC and further refined in collaboration with other stakeholder groups. Roadmap Epigenomics Program participation in IHEC also helped spur significant additional international investment in epigenomic mapping and disease research. As of September 2018, IHEC members, including Roadmap and ENCODE, have generated 8870 epigenomic datasets (http://epigenomesportal.ca/ihec/).
Epigenomic basis of diseases and novel epigenomic marks
It is important to point out that in addition to coordinated mapping efforts, the Roadmap Epigenomics Program included discovery-driven investigations into the role of epigenomic changes in a wide array of diseases. These projects generated foundational knowledge necessary for translation of epigenomic information into improved preventive, diagnostic, and therapeutic strategies. Here, we provide some notable examples of program activities driven by individual efforts. For instance, De Jager et al. (14) took advantage of two prospective studies of aging. They identified 11 regions of the genome in which brain frontal cortex methylation status was associated with Alzheimer’s disease pathology in both a discovery cohort and a smaller replication cohort. This study also identified eight nearby genes that were differentially expressed in Alzheimer’s disease brains relative to unaffected controls. In a second study, Reynolds et al. (15) identified age-associated methylation changes in monocytes and T cells, which correlated with both local gene expression changes and clinical measures of vascular aging. These epigenomic changes were enriched in regions predicted to have roles in regulating gene expression programs, such as enhancers. A third example was provided by Yang et al. (16) who compared DNA methylation profiles of peripheral blood mononuclear cells from inner city children with asthma to a healthy control population and identified 11 genes with methylation and gene expression changes specifically associated with asthma, all of which were validated in an independent cohort. Many of these genes are related to immune function, specifically T cell maturation and TH2 (T helper 2) immunity, raising the possibility that in the future, epigenetic therapies could be effective for reversing immune dysfunction in children with asthma. In 2010, the Ren laboratory published a highly cited paper comparing the epigenomes of pluripotent and lineage-committed human embryonic stem cells (17), one of the many advances made by the Roadmap Consortium in stem cell epigenomics.
In addition, a search for novel epigenomic marks was fundamental to the design of the overall program to help more fully understand the diversity of epigenomic modifications that might need to be considered as part of a reference epigenome and provide a critical balance between large and small projects contributing to a collective goal. Studies funded by this program identified at least 70 additional novel histone modifications, including lysine crotonylation (Kcr), which was shown to be evolutionarily conserved (18). In addition, lysine β-hydroxybutyrylation (Kbhb) was discovered, which was markedly induced in response to elevated β-hydroxybutyrate levels in cultured cells and in livers from mice subjected to prolonged fasting or streptozotocin-induced diabetic ketoacidosis (19). The extent to which the novel marks identified are associated with human maladies is a current area of further exploration.
Epigenomic technology development
The technology development projects supported by this program were focused on enabling novel or markedly improved epigenomic monitoring or manipulation. Among the innovative epigenomic monitoring technologies developed were the MethylC-seq assay, chromatin affinity purification with mass spectrometry, and the development of ligands for in vivo imaging of epigenetic enzymes in humans (20–22). Methods for locus- or mark-specific manipulation of the epigenome include a light-inducible CRISPR-Cas9 system for control of endogenous gene activation (23), a CRISPR-Cas9 acetyltransferase approach to perform epigenome editing (24), and epigenome editing to silence distal regulatory elements (25). These studies are important contributions to the epigenome editing tool box, which will aid in modern genome- and epigenome-based therapies and which, in part, helped spawn a new Common Fund program on Somatic Cell Genome Editing (https://commonfund.nih.gov/editing).
MEASURING SUCCESS
It can be extremely difficult to measure the ultimate success of a scientific program because it is difficult to disentangle the effects of the program versus other nonprogrammatic effects. In addition, scientific advances often take many years to achieve their ultimate impact on human health and disease. In our evaluation of the Roadmap Epigenomics Program, we focused on the following quantitative measures of success: (i) use of data, publications, and resources; (ii) new discoveries of epigenomic mechanisms in human disease; and (iii) new high-value technologies. On the basis of a bibliometric analysis, publications from the Roadmap Epigenomics Program were found to be highly influential. A total of 857 articles published between 2008 and 30 November 2017 were identified using an internal NIH database linking grants to publications. All 857 articles were analyzed for their influence using iCite (26), which provides a Relative Citation Ratio (RCR) for each publication. Of these publications, 699 were categorized as research articles and received 44,245 citations. The mean RCR value of the Epigenomics Program’s research publications is 3.60 and the median RCR is 1.44 compared to an NIH-wide benchmark median RCR value of 1.0. This indicates that publications from the Epigenomics Program tended to be more influential (Fig. 1). Both the number of “Epigenomics” publications in PubMed as well as the number of NIH-funded grants increased significantly since the program started in 2007 (Fig. 2, A and B). However, it is unclear whether this is a direct consequence of the start of the program or whether this increase would have happened independently. Likewise, a substantial increase in epigenomics research in specific disease areas, in addition to cancer, was also evident since 2007 (Fig. 2C), which is an indication of how this program catalyzed new discoveries related to epigenomic mechanisms of human diseases. Last, we analyzed patent applications as an indicator of commercialization potential. As of December 2017, there were 27 granted patents out of 55 patent applications from the Epigenomics Program.
Fig. 1. Mean and median RCR (Relative Citation Ratio) of Roadmap Epigenomics Program research articles for each year.
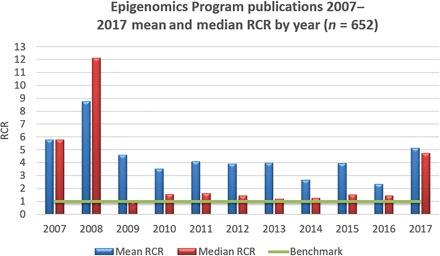
The RCR benchmark of 1.0 is based on the median RCR for all NIH-funded publications, including reviews. This excludes 2017 articles that did not have an assigned RCR value at the time of analysis.
Fig. 2. Influence of the Roadmap Epigenomics Program on the field of epigenomics research.
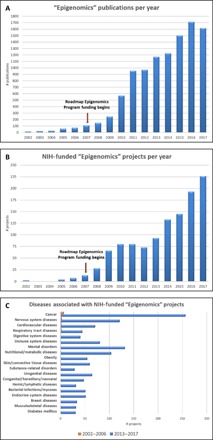
(A) Epigenomics publications per year identified by searching PubMed for “epigenom*.” There were 10,439 epigenomics publications identified. (B) NIH-funded Epigenomics projects per year identified by searching an NIH grant database for new awards with epigenom* in the title or specific aims. Excludes subprojects and intramural projects. (C) Number of projects per year for top 10 conditions of NIH-funded Epigenomics projects. Projects were identified by searching an NIH grant database for new awards with epigenom* in the title or specific aims. Excludes subprojects and NIH intramural research projects.
KEY CHALLENGES AND LESSONS LEARNED FROM THE EPIGENOMICS ROADMAP CONSORTIUM
When the NIH Roadmap Epigenomics Program was being developed and implemented, there were several issues that needed to be addressed: (i) the overall structure of the program (e.g., the balance between smaller discovery and technology development projects versus larger goal-directed mapping efforts), (ii) how best to coordinate the activities of the consortium as a whole and address the challenges of working in a group, and (iii) how to establish and best achieve the goals of the consortium. A “consortium approach” can bring to mind large projects with NIH-mandated goals that offer little or no opportunity for investigator-initiated innovation. Concerns about this type of approach were voiced at the outset of the Epigenomics Program (27). However, the design of the Epigenomics Program included both large data-generating projects as well as smaller innovation- and discovery-focused projects. Common Fund programs are intended to change paradigms, develop innovative tools and technologies, and/or provide fundamental foundations for research that can be used by the global biomedical research community. These programs often involve a combination of highly coordinated research to develop large datasets or tools, innovative technology development to further enhance the capabilities of the consortium, and demonstration projects to road test the value of the data/tools. As stated earlier, the combination of large-scale data-generating projects and smaller, nimble technology-driven methods for which the primary goals are innovation and discovery allows the work of the consortium as a whole to evolve while maintaining an overall focus on delivering large amounts of high-quality data to end users. However, the value of coordinated efforts to deliver community resources that could not be provided by individual groups versus a need for more focused, hypothesis-testing investigator-initiated approaches was also debated (27). At the root of the debate was whether the field was ready for a large mapping effort; mapping epigenetic modifications was more daunting than sequencing the human genome because there was no clear definition of a single reference epigenome. Nevertheless, the program was launched in 2008 with the expectation that coordinated mapping efforts would ultimately lead to the creation of reference epigenomes and that these would be “durable goods” for the benefit of the community in the long run. International data standards, benchmarking across laboratories as methods were developed, development of computational tools, and data management that addressed the needs of different types of users were worthwhile goals that would not have been achieved without a coordinated effort. Last, community outreach and engagement via presentations at meetings and workshops was strongly emphasized by external program consultants who represented the interests of the broad scientific community, because community resources are only valuable if the community knows about them.
A second major issue was how best to coordinate the activities of the consortium as a whole and address the challenges of working in a group. As rewarding as consortium science can be, the formation of a consortium also presents several challenges. Investigators are asked to adopt a community-centric focus rather than concentrating on their own individual research interests. This represents a shift in thinking as well as decision-making for themselves and their laboratories. Consistent with this perspective, instead of maximizing their own benefit, the Roadmap Epigenomics Consortium adopted a data- and resource-sharing policy that emphasized early sharing of tools and methods. For instance, MethylC-seq for whole-genome bisulfite sequencing of mammalian cells (20) was readily adopted by the consortium after the steering committee and NIH staff decided that all mapping centers should use it for their reference epigenomes. While this initially caused some feasibility concerns, rapid sharing of data and tools facilitated this shift without diminishing the recognition of the data/tool developers.
A third important issue was how best to establish and achieve the goals of the consortium. Newly funded members of large consortia may be uncomfortable with the goals established by the NIH. Moving from the high-level objectives established via funding opportunities to the articulation of specific consortium goals, development of plans for benchmarking between laboratories, and establishment of integrated analysis plans requires iterative discussion so that consensus can be established. The Epigenomics Consortium was governed by a steering committee, consisting of consortium members and NIH staff. Substantial input was also received from the external program consultants regarding consortium goals and expectations. Although it took extensive time and effort, the investigators ultimately collectively coordinated among themselves by focusing on high-achieving goals leading to prominent publications that everyone recognized could only be accomplished through a considerable consortium effort that was greater than the sum of which all the laboratories could complete individually.
MOVING FORWARD
In addition to the obvious synergistic relation between Roadmap Epigenomics, IHEC, and ENCODE, several other Common Fund programs have benefited from the lessons learned by the Epigenomics Program. In particular, having established a successful blueprint for mapping efforts, programs like 4D Nucleome (4DN) also ventured into this space in attempting to create, among other things, three-dimensional (3D) maps of genomic interactions as a long-term community tool. Although creating 3D maps of genomic interactions is a far more complex task, direct comparison of Epigenomics and ENCODE datasets with 3D maps generated by the 4DN program and ENCODE would ultimately provide the scientific community with a powerful framework to integrate structural and functional genomic, epigenomic, and transcription factor binding datasets to better understand human health and disease. Likewise, the newly minted Common Fund Human Biomolecular Atlas Program, focusing on the ambitious goal of providing the map of the human body at cellular resolution, may also benefit from the Epigenomics Program data. Our understanding of the human genome, sequenced in the early 1990s, has been further untangled by epigenomic and nucleomic research and may culminate in an unprecedented view of the human body at subcellular resolution. Scientific progress will continue to require the judicious use of large goal-driven programs that produce technologies and tools that can accelerate discovery from individual laboratory science. The Common Fund is committed to continued evaluation and refining of best practices for design and management of consortia to maximize their scientific and health impact.
Highlights
-
1)
Critical analysis of current funding reveals gaps where Common Fund support can have the greatest impact;
-
2)
Scientific gaps often occur around issues that cannot be addressed by a single researcher and that require a multidisciplinary and coordinated effort to achieve;
-
3)
Development of community resources requires consistent input from scientists who represent the user community;
-
4)
Community resources need to be developed in the context of the international community—this requires dedicated time and effort;
-
5)
Principal investigators funded to generate a community resource need to commit to consortium goals and community outreach;
-
6)
Early sharing of large datasets and tools does not impede the work of data/tool generators—it enhances the impact of the data/tools
-
7)
The combination of consortium-driven community resource development in addition to discovery and technology development projects can result in rapid advances for the field as a whole.
Acknowledgments
We thank M. Pazin (NHGRI/NIH) and E. Marcotte (CIHR) for critically reading the manuscript. We also acknowledge the efforts of many researchers, including the consortium members, who generated the foundational knowledge that made the epigenomics program possible. Author contributions: A.L.R., J.S.S., and E.L.W. wrote the article with input from J.M.A., L.H.C., F.L.T., K.M., L.B., and N.D.V. J.B. did the analysis that led to the figures. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the materials cited herein. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Bird A., Perceptions of epigenetics. Nature 447, 396–398 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T., Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Beck S., Olek A., Walter J., From genomics to epigenomics: A loftier view of life. Nat. Biotechnol. 17, 1144 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Bernstein B. E., Stamatoyannopoulos J. A., Costello J. F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M. A., Beaudet A. L., Ecker J. R., Farnham P. J., Hirst M., Lander E. S., Mikkelsen T. S., Thomson J. A., The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 28, 1045–1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satterlee J. S., Beckel-Mitchener A., McAllister K., Procaccini D. C., Rutter J. L., Tyson F. L., Chadwick L. H., Community resources and technologies developed through the NIH Roadmap Epigenomics Program. Methods Mol. Biol. 1238, 27–49 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Romanoski C. E., Glass C. K., Stunnenberg H. G., Wilson L., Almouzni G., Roadmap for regulation. Nature 518, 314–316 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Satterlee J. S., Schübeler D., Ng H. H., Tackling the epigenome: Challenges and opportunities for collaboration. Nat. Biotechnol. 28, 1039–1044 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Maurano M. T., Humbert R., Rynes E., Thurman R. E., Haugen E., Wang H., Reynolds A. P., Sandstrom R., Qu H., Brody J., Shafer A., Neri F., Lee K., Kutyavin T., Stehling-Sun S., Johnson A. K., Canfield T. K., Giste E., Diegel M., Bates D., Hansen R. S., Neph S., Sabo P. J., Heimfeld S., Raubitschek A., Ziegler S., Cotsapas C., Sotoodehnia N., Glass I., Sunyaev S. R., Kaul R., Stamatoyannopoulos J. A., Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lander E. S., Linton L. M., Birren B., Nusbaum C., Zody M. C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., Funke R., Gage D., Harris K., Heaford A., Howland J., Kann L., Lehoczky J., LeVine R., McEwan P., McKernan K., Meldrim J., Mesirov J. P., Miranda C., Morris W., Naylor J., Raymond C., Rosetti M., Santos R., Sheridan A., Sougnez C., Stange-Thomann Y., Stojanovic N., Subramanian A., Wyman D., Rogers J., Sulston J., Ainscough R., Beck S., Bentley D., Burton J., Clee C., Carter N., Coulson A., Deadman R., Deloukas P., Dunham A., Dunham I., Durbin R., French L., Grafham D., Gregory S., Hubbard T., Humphray S., Hunt A., Jones M., Lloyd C., McMurray A., Matthews L., Mercer S., Milne S., Mullikin J. C., Mungall A., Plumb R., Ross M., Shownkeen R., Sims S., Waterston R. H., Wilson R. K., Hillier L. W., McPherson J. D., Marra M. A., Mardis E. R., Fulton L. A., Chinwalla A. T., Pepin K. H., Gish W. R., Chissoe S. L., Wendl M. C., Delehaunty K. D., Miner T. L., Delehaunty A., Kramer J. B., Cook L. L., Fulton R. S., Johnson D. L., Minx P. J., Clifton S. W., Hawkins T., Branscomb E., Predki P., Richardson P., Wenning S., Slezak T., Doggett N., Cheng J. F., Olsen A., Lucas S., Elkin C., Uberbacher E., Frazier M., Gibbs R. A., Muzny D. M., Scherer S. E., Bouck J. B., Sodergren E. J., Worley K. C., Rives C. M., Gorrell J. H., Metzker M. L., Naylor S. L., Kucherlapati R. S., Nelson D. L., Weinstock G. M., Sakaki Y., Fujiyama A., Hattori M., Yada T., Toyoda A., Itoh T., Kawagoe C., Watanabe H., Totoki Y., Taylor T., Weissenbach J., Heilig R., Saurin W., Artiguenave F., Brottier P., Bruls T., Pelletier E., Robert C., Wincker P., Smith D. R., Doucette-Stamm L., Rubenfield M., Weinstock K., Lee H. M., Dubois J., Rosenthal A., Platzer M., Nyakatura G., Taudien S., Rump A., Yang H., Yu J., Wang J., Huang G., Gu J., Hood L., Rowen L., Madan A., Qin S., Davis R. W., Federspiel N. A., Abola A. P., Proctor M. J., Myers R. M., Schmutz J., Dickson M., Grimwood J., Cox D. R., Olson M. V., Kaul R., Raymond C., Shimizu N., Kawasaki K., Minoshima S., Evans G. A., Athanasiou M., Schultz R., Roe B. A., Chen F., Pan H., Ramser J., Lehrach H., Reinhardt R., McCombie W. R., de la Bastide M., Dedhia N., Blöcker H., Hornischer K., Nordsiek G., Agarwala R., Aravind L., Bailey J. A., Bateman A., Batzoglou S., Birney E., Bork P., Brown D. G., Burge C. B., Cerutti L., Chen H. C., Church D., Clamp M., Copley R. R., Doerks T., Eddy S. R., Eichler E. E., Furey T. S., Galagan J., Gilbert J. G., Harmon C., Hayashizaki Y., Haussler D., Hermjakob H., Hokamp K., Jang W., Johnson L. S., Jones T. A., Kasif S., Kaspryzk A., Kennedy S., Kent W. J., Kitts P., Koonin E. V., Korf I., Kulp D., Lancet D., Lowe T. M., McLysaght A., Mikkelsen T., Moran J. V., Mulder N., Pollara V. J., Ponting C. P., Schuler G., Schultz J., Slater G., Smit A. F., Stupka E., Szustakowki J., Thierry-Mieg D., Thierry-Mieg J., Wagner L., Wallis J., Wheeler R., Williams A., Wolf Y. I., Wolfe K. H., Yang S. P., Yeh R. F., Collins F., Guyer M. S., Peterson J., Felsenfeld A., Wetterstrand K. A., Patrinos A., Morgan M. J., de Jong P., Catanese J. J., Osoegawa K., Shizuya H., Choi S., Chen Y. J., Szustakowki J.; International Human Genome Sequencing Consortium , Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y. C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K. H., Feizi S., Karlic R., Kim A. R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L. H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He Y., Wang T., EpiCompare: An online tool to define and explore genomic regions with tissue or cell type-specific epigenomic features. Bioinformatics 33, 3268–3275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pérez R. F., Tejedor J. R., Bayón G. F., Fernández A. F., Fraga M. F., Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell 17, e12744 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stunnenberg H. G.; International Human Epigenome Consortium, Hirst M., The International Human Epigenome Consortium: A blueprint for scientific collaboration and discovery. Cell 167, 1145–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 14.De Jager P. L., Srivastava G., Lunnon K., Burgess J., Schalkwyk L. C., Yu L., Eaton M. L., Keenan B. T., Ernst J., McCabe C., Tang A., Raj T., Replogle J., Brodeur W., Gabriel S., Chai H. S., Younkin C., Younkin S. G., Zou F., Szyf M., Epstein C. B., Schneider J. A., Bernstein B. E., Meissner A., Ertekin-Taner N., Chibnik L. B., Kellis M., Mill J., Bennett D. A., Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 17, 1156–1163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reynolds L. M., Taylor J. R., Ding J., Lohman K., Johnson C., Siscovick D., Burke G., Post W., Shea S., Jacobs D. R. Jr., Stunnenberg H., Kritchevsky S. B., Hoeschele I., McCall C. E., Herrington D., Tracy R. P., Liu Y., Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun. 5, 5366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang I. V., Pedersen B. S., Liu A., O'Connor G. T., Teach S. J., Kattan M., Misiak R. T., Gruchalla R., Steinbach S. F., Szefler S. J., Gill M. A., Calatroni A., David G., Hennessy C. E., Davidson E. J., Zhang W., Gergen P., Togias A., Busse W. W., Schwartz D. A., DNA methylation and childhood asthma in the inner city. J. Allergy Clin. Immunol. 136, 69–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hawkins R. D., Hon G. C., Lee L. K., Ngo Q., Lister R., Pelizzola M., Edsall L. E., Kuan S., Luu Y., Klugman S., Antosiewicz-Bourget J., Ye Z., Espinoza C., Agarwahl S., Shen L., Ruotti V., Wang W., Stewart R., Thomson J. A., Esker J. R., Ren B., Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6, 479–491 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan M., Luo H., Lee S., Jin F., Yang J. S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N., Lu Z., Ye Z., Zhu Q., Wysocka J., Ye Y., Khochbin S., Ren B., Zhao Y., Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Z., Zhang D., Chung D., Tang Z., Huang H., Dai L., Qi S., Li J., Colak G., Chen Y., Xia C., Peng C., Ruan H., Kirkey M., Wang D., Jensen L. M., Kwon O. K., Lee S., Pletcher S. D., Tan M., Lombard D. B., White K. P., Zhao H., Li J., Roeder R. G., Yang X., Zhao Y., Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell 62, 194–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lister R., Pelizzola M., Dowen R. H., Hawkins R. D., Hon G., Tonti-Filippini J., Nery J. R., Lee L., Ye Z., Ngo Q. M., Edsall L., Antosiewicz-Bourget J., Stewart R., Ruotti V., Millar A. H., Thomson J. A., Ren B., Ecker J. R., Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrum S. D., Raman A., Taverna S. D., Tackett A. J., ChAP-MS: A method for identification of proteins and histone posttranslational modifications at a single genomic locus. Cell Rep. 2, 198–205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wey H. Y., Gilbert T. M., Zürcher N. R., She A., Bhanot A., Taillon B. D., Schroeder F. A., Wang C., Haggarty S. J., Hooker J. M., Insights into neuroepigenetics through human histone deacetylase PET imaging. Sci. Transl. Med. 8, 351ra106 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yumerefendi H., Lerner A. M., Zimmerman S. P., Hahn K., Bear J. E., Strahl B. D., Kuhlman B., Light-induced nuclear export reveals rapid dynamics of epigenetic modifications. Nat. Chem. Biol. 12, 399–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klann T. S., Black J. B., Chellappan M., Safi A., Song L., Hilton I. B., Crawford G. E., Reddy T. E., Gersbach C. A., CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 35, 561–568 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thakore P. I., D’Ippolito A. M., Song L., Safi A., Shivakumar N. K., Kabadi A. M., Reddy T. E., Crawford G. E., Gersbach C. A., Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutchins B. I., Yuan X., Anderson J. M., Santangelo G. M., Relative Citation Ratio (RCR): A new metric that uses citation rates to measure influence at the article level. PLOS Biol. 14, e1002541 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pennisi E., Research funding. Are epigeneticists ready for big science? Science 319, 1177 (2008). [DOI] [PubMed] [Google Scholar]