

Abstract
T cell receptor (TCR) ligand discovery is essential for understanding and manipulating immune responses to tumors. We developed a cell-based selection platform for TCR ligand discovery that exploits a membrane transfer phenomenon called trogocytosis. We discovered that T cell membrane proteins are transferred specifically to target cells that present cognate peptide-major histocompatibility complex (MHC) molecules. Co-incubation of T cells expressing an orphan TCR with target cells collectively presenting a library of peptide-MHCs led to specific labeling of cognate target cells, enabling isolation of these target cells and sequencing of the cognate TCR ligand. We validated this method for two clinically employed TCRs and further used the platform to identify the cognate neoepitope for a subject-derived neoantigen-specific TCR. Thus, target cell trogocytosis is a robust tool for TCR ligand discovery that will be useful for studying basic tumor immunology and identifying new targets for immunotherapy.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
T cells mediate adaptive immunity through direct, antigen-specific contact with target cells. The antigenic specificity of each T cell is determined by its unique TCR1, which binds a cognate peptide ligand (epitope) presented on MHC protein molecules on the target cell surface. TCR ligand discovery is fundamental to characterizing adaptive immune responses to pathogens and tumors, as well as inappropriate responses to self- and dietary antigens2,3. This knowledge also enables clinically beneficial immunotherapies (for example, TCR gene transfer and vaccines) that initiate, amplify or attenuate immune responses to target antigens4,5.
Peptide-MHC (pMHC) multimer technologies enable monitoring of T cell-mediated responses to a selected panel of antigens6, but require foreknowledge of those antigenic targets relevant to the response. Unbiased screens with pMHC multimers on the scale of peptide screens used for TCR ligand discovery are precluded because even modern innovations enable the construction of at most a few thousand pMHC reagents in parallel7. In the context of cancer, tumor neoantigens arising from tumor-specific mutations can be discovered through exome sequencing and then used to interrogate T cells using pMHC multimers or neoantigen-transduced antigen-presenting cells8,9. However, this approach limits the characterization of antitumor immunity to private neoantigen-specific clones and cannot be generalized to other immune responses less focused on mutant epitopes (for example, pathogen-specific immunity and autoimmunity). An alternative to interrogating a T cell response with preselected antigens is to identify TCRs mediating that response and use those TCRs to interrogate an antigenic library. TCRs mediating an immune response of interest can be identified by sequencing of T cells that are phenotypically implicated in that response or that are enriched among clonal T cells at the site of that response10–17. However, the antigenic specificities of these orphan TCRs are typically unknown, which limits understanding of antitumor immunity and the potential clinical applications of these TCRs. Thus, there remains a need for technologies that can rapidly and robustly identify ligands reactive to orphan TCRs of interest for both basic and translational research.
Trogocytosis is a biological phenomenon by which cells share membrane and membrane-associated proteins while conjugated18. Although trogocytosis is bidirectional between conjugated FcγR-expressing cells (for example, monocytes) and immunoglobulin-bound cells (for example, anti-CD20-bound B cells)19, it has been described as unidirectional for primary T cells conjugated to unengineered antigen-presenting cells. Specifically, T cells extract membrane and membrane-associated proteins from target antigen-presenting cells with which they are conjugated18,20–23. The reverse process has been reported only for T cells using antigen-presenting cells loaded with cognate peptides24,25. In this study, we show that antigen-presenting target cells genetically engineered to present supraphysiological levels of epitope can extract membrane and membrane-associated proteins from interacting T cells. Moreover, antigen-specific target cell trogocytosis can be tracked by multiple-protein transfer and by loss of proteins from donor cells. We have developed a TCR ligand discovery platform that exploits this phenomenon to selectively mark target cells that present genetically encoded epitopes cognate to orphan TCR-transduced T cells, enabling their isolation from a target cell library.
Results
Trogocytosis occurs from T cell to target cell.
We first establi shed cell lines expressing cognate TCR-antigen pairs, including Jurkat cells expressing either F5-TCR or 1G4-TCR and K562 cells expressing their cognate single-chain trimer (SCT) of HLA-A2/MART126–35(A27L) or A2/NYESO1157–165(C165V) (Fig. 1a and Supplementary Fig. 1a). To test whether T cell membrane is transferred to target cells upon antigen-specific interaction, we labeled T cell surface proteins with N-hydroxysuccinimido (NHS)-biotin and monitored their transfer to target cells during co-incubation. As we expected, low-affinity nerve growth factor receptor (LNGFR)+ Jurkat T cells treated with NHS-biotin stained strongly with streptavidin (Fig. 1b and Supplementary Fig. 1b), whereas ZsGreen+ K562 cells that were not co-incubated with biotinylated Jurkat cells were negative for streptavidin (Fig. 1c and Supplementary Fig. 1b). Co-incubation of biotinylated F5-Jurkat cells with noncognate NYESO1-K562 cells led to a >30-fold shift in streptavidin staining, which is indicative of nonspecific biotin transfer to target cells. However, the co-incubation of biotinylated F5-Jurkat cells with cognate MART1-K562 cells led to a further 3.5-fold shift over this nonspecific shift, demonstrating antigen-specific target cell trogocytosis (Fig. 1c). Notably, both LNGFR and TCR, but not intracellular protein Zsgreen, exhibited a similar two- to threefold shift when MART1-K562 cells were co-incubated with F5-Jurkat cells, without any nonspecific shift for NYESO1-K562 cells (Fig. 1d and Supplementary Fig. 2a). We repeated this experiment without biotinylating the Jurkat T cells, using antibodies alone to monitor the transfer of T cell proteins to target cells upon co-incubation for different periods of time (Fig. 1e and Supplementary Fig. 2b,c). With 30 min of co-incuba tion, NYESO1-K562 and MART1-K562 cells extracted LNGFR and TCR from Jurkat T cells expressing their respective cognate TCRs but not from those expressing the noncognate TCR. Immunofluorescence staining with pMHC dextramers further confirmed the antigenic specificity of target cell trogocytosis (Supplementary Fig. 2d). In addition to LNGFR and TCR, CD3 and CD8 proteins were transferred from Jurkat cells to K562 cells, and HLA-A2 was transferred from K562 cells to Jurkat cells in an antigen-dependent manner (Supplementary Fig. 3a,b). Moreover, we observed an accompanying antigen-dependent reduction of proteins from donor cells, including HLA-A2 from K562 cells and TCR and LNGFR from Jurkat cells (Supplementary Fig. 3c,d). Thus, trogocytosis is bidirectional and mass balance is observed. In addition to F5- and 1G4-TCR, we also observed antigen-specific target cell trogocytosis for each of eight additional TCR-pMHC cognate pairings, including a low-affinity A2-restricted MART1-specific TCR26, four novel A2-restricted NY-ESO-1-specific TCRs of varying affinity and three NY-ESO-1-specific TCRs restricted on various MHC alleles (HLA-B*0702, B*18:01 and C*03:04)27 (Supplementary Fig. 4).
Fig. 1 |. Target cell trogocytosis is antigen specific.
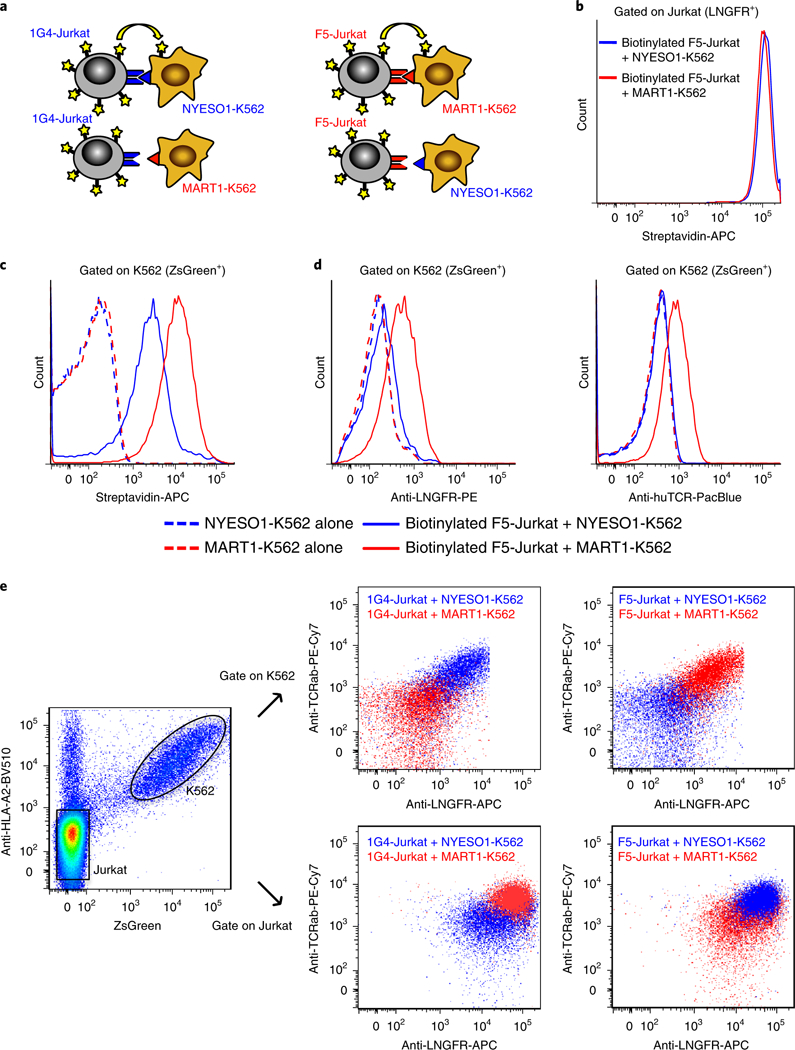
a, Schematic of cognate TCR-antigen pairs. Red/blue coloring indicates cognate TCR-antigen pairs. F5-TCR (red) is paired with the SCT expressing melanoma antigen MARTI peptide, and 1G4-TCR (blue) is paired with the SCT expressing cancer-testis antigen NYESO1 peptide. Yellow stars represent membrane proteins. b-d, FACS plots of biotinylated F5-Jurkat T cells (b) after co-incubation with cognate MART1-K562 target cells (ZsGreen+) expressing SCT of HLA-A2/MART126–35(A27L) or noncognate NYEOS1-K562 target cells expressing SCT of HLA-A2/NYESO1157–165(C165V). c, Transfer of biotinylated membrane proteins from Jurkat T cells (LNGFR+) to K562 target cells (ZsGreen+). d, Antigen-specific transfer of LNGFR and TCR from F5-Jurkat T cells to cognate MART1-K562 target cells. e, FACS plot overlay of either NYESO1-K562 or MART1-K562 cells after co-incubation with 1G4-Jurkat or F5-Jurkat T cells. SCT-K562 cells (ZsGreen+) were assessed for acquisition of TCR using an anti-human TCRα-β (TCRαβ) antibody. Data are representative of three independent experiments.
Trogocytosis scales with pMHC density and is enhanced by CD8.
To determine whether trogocytosis intrinsically depends on the identities of donor and acceptor cells, we generated K562 cell lines and Jurkat cell lines expressing either TCR or SCT and performed co-incubation experiments using all pairing combinations. We observed antigen-specific TCR transfer for all cell pairings (Supplementary Fig. 5), indicating that target cell trogocytosis does not depend on the identities of donor and acceptor cells.
Target cell trogocytosis has been reported in experiments using supraphysiological levels of pulsed peptide24,25. Our observation of target cell trogocytosis may therefore be explained by the sup-raphysiological expression of SCTs on the target cell surface. To test whether trogocytosis scales with increasing pMHC density, we pulsed K562 cells expressing HLA-A2 (A2-K562) with either MART1 heteroclitic peptide (ELAGIGILTV) or NYESO1 heteroclitic peptide (SLLMWITQV) at various concentrations and performed coculture experiments. Target cells pulsed with MART1 peptide exhibited a peptide-concentration-dependent increase in TCR transfer from cognate F5-Jurkat cells (but not from noncognate 1G4-Jurkat cells) (Fig. 2a, top, and Supplementary Fig. 6a). Similarly, target cells pulsed with NYESO1 peptide exhibited increased trogocytosis with increasing peptide concentration only when co-incubated with cognate 1G4-Jurkat cells (Fig. 2a, bottom, and Supplementary Fig. 6b). Thus, target cell trogocytosis is antigen specific and its magnitude depends on the density of pMHC.
Fig. 2 |. Trogocytosis is titratable and augmented by the presence of CD8.
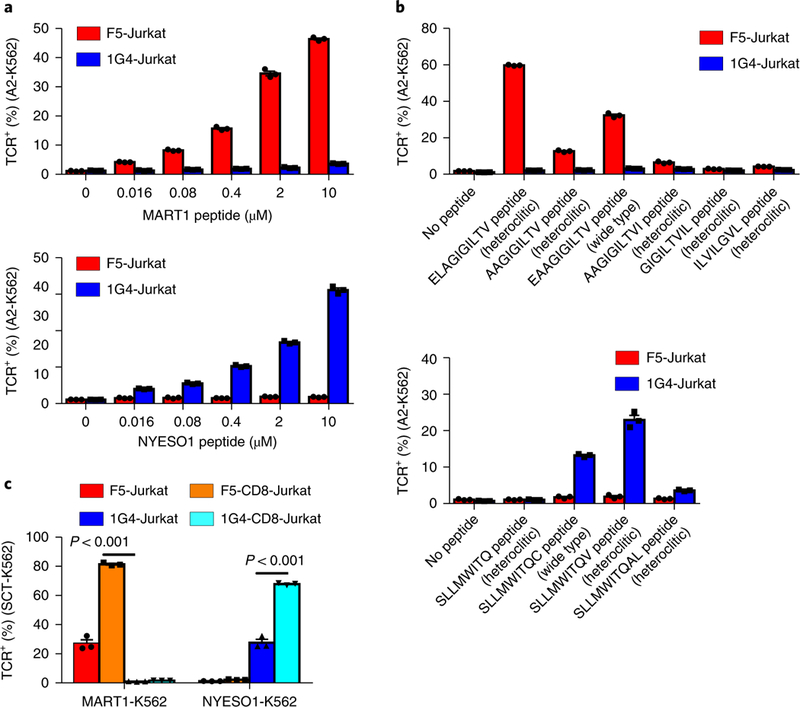
a-c, Comparison of trogocytosis capability of (a) F5-Jurkat or 1G4-Jurkat cells with HLA-A2 expressing K562 cells (A2-K562) loaded with different doses of MART1 (ELAGIGILTV; top) or NYESO1 (SLLMWITQV; bottom) heteroclitic peptide; (b) F5-Jurkat or 1G4-Jurkat cells with A2-K562 cells loaded with different MART1 (top) or NYESO1 (bottom) peptide variants; and (c) CD8+ or CD8- F5-Jurkat or 1G4-Jurkat cells with MART1-K562 or NYESO1-K562 cells (n = 3). Statistical analysis of quantification was performed using unpaired two-tailed Student’s t-test. Data are presented as mean± s.e.m. and are representative of two independent experiments.
F5-TCR-expressing T cells respond to different MART1 peptide variants to different extents28. For example, F5-TCR is more responsive to the heteroclitic MART1 peptide (ELAGIGILTV) than to the native MART1 peptide (EAAGIGILTV). We examined the extent to which different MART1 peptide variants elicit trogocytosis by incubating F5-Jurkat or 1G4-Jurkat cells with A2-K562 cells that were pulsed with different MART1 peptide variants at the same dose. We found that there were more trogocytosis+ target cells in the mixture of F5-Jurkat and ELAGIGILTV peptide-pulsed A2-K562 cells than in F5-Jurkat and A2-K562 cells pulsed with native EAAGIGILTV peptide or other peptide variants (Fig. 2b, top, and Supplementary Fig. 6c). Similarly, there was more TCR transfer to A2-K562 cells pulsed with heteroclitic NYESO1 peptide than to those pulsed with native peptide or other peptide variants (Fig. 2b, bottom, and Supplementary Fig. 6d).
Coexpression of CD8 increases the avidity of the TCR-pMHC interaction by binding to MHC I directly, enabling lower-affinity TCRs to engage29. We found that Jurkat cells coexpressing CD8 mediated significantly enhanced trogocytosis relative to those expressing only TCR for each of three TCRs tested, including the low-affinity M1W-TCR (Fig. 2c and Supplementary Fig. 7). Thus, our results suggest that target cell trogocytosis depends on the density of the presented epitope and that it is enhanced when donor cells coexpress CD8.
Trogocytosis enables isolation of cognate target cells.
In addition to providing the supraphysiological levels of pMHC presentation necessary for target cell trogocytosis, SCTs genetically encode the epitope presented. We devised an epitope discovery strategy in which donor cells expressing an orphan TCR label target cells presenting their cognate epitope via trogocytosis, facilitating isolation of these target cells from a library by fluorescence-activated cell sorting (FACS) and epitope identification by next-generation sequencing (NGS). To determine whether target cell trogocytosis enables separation of cognate epitope-presenting target cells from noncognate epitope-presenting cells, we co-incubated either 1G4-Jurkat or F5-Jurkat T cells with a 1:1 mixture of NYESO1-K562 and MART1-K562 target cells and then labeled the cell mixture with soluble 1G4- or F5-TCR multimers. This allowed us to determine the antigenic composition of target cells undergoing trogocytosis (Supplementary Fig. 8a). When a mixture of NYESO1-K562 and MART1-K562 target cells was co-incubated with 1G4-Jurkat cells, trogocytosis+ target cells were highly enriched (95.5%) for cognate NYESO1-K562 cells (Supplementary Fig. 8b,c). Similarly, co-incubation of the target cell mixture with F5-Jurkat cells enriched MART1-K562 (94.6%) among trogocytosis+ target cells. Thus, target cell trogocytosis is itself antigen specific and enables resolution of antigen-expressing target cells from non-antigen-expressing cells.
To determine whether detection of target cell trogocytosis is sufficiently sensitive for the identification of a cognate TCR ligand from a library of noncognate ligands, we conducted a proof-of-concept library simulation. We diluted ZsGreen+ NYESO1-K562 cells 1:10,000 into CD80+HLA-A2+ K562 cells, co-incubated this mixture with either 1G4-Jurkat or F5-Jurkat cells, and compared trogocytosis among CD80ZsGreen+ (NYESO1-K562) and CD80+ZsGreen− (control) target cells (Fig. 3a). We found that there was a marked difference in the percentage of trogocytosis+ NYESO1-K562 target cells between NYESO1-K562 cells coincubated with cognate 1G4-Jurkat cells (70.8%) and noncognate F5-Jurkat cells (0.3%). By contrast, there was no significant trogocytosis observed for control CD80+HLA-A2+ K562 cells after co-incubation with either Jurkat cell type. The experiment was repeated using one MART1-K562 cell per 10,000 control cells, yielding a similar result (Fig. 3b). Thus, target cell trogocytosis can be used to detect cognate antigen-expressing target cells at a ratio at least as low as 1:10,000.
Fig. 3 |. Target cell trogocytosis is sufficiently sensitive to identify one cognate-antigen-expressing target cell from 10,000 non-antigen-expressing cells.
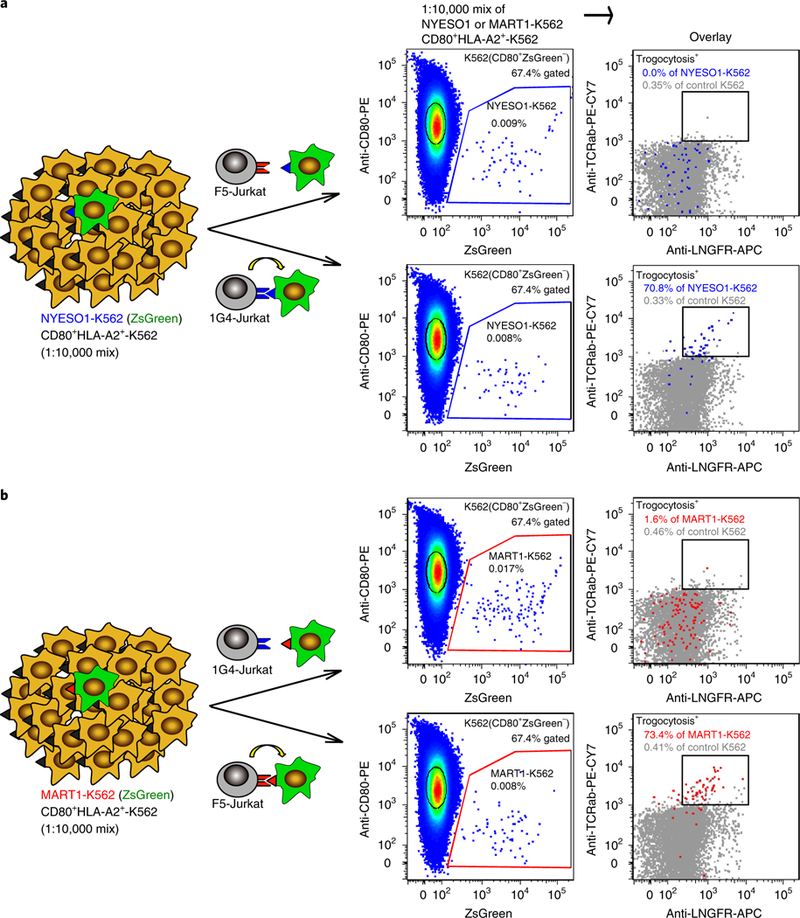
a,b, Schematic and representative FACS plots for the mixture of cognate NYESO1-K562 target cells and noncognate A2-K562 cells (a) and the mixture of cognate MART1-K562 target cells to noncognate A2-K562 cells (b) co-incubated with either F5-Jurkat (top) or 1G4-Jurkat (bottom) cells. An overlay of the noncognate A2-K562 cells (gray) provides a clear comparison of trogocytosis in cognate versus noncognate K562 cells. Data are representative of three independent experiments.
Having determined the sensitivity with which target cell trogocytosis resolves cognate target cells from those presenting a non-cognate ligand, we next determined the specificity with which trogocytosis separates cognate target cells from cells presenting a more extensive library of ligands. We generated an A2-restricted SCT cDNA library containing 12,055 public T cell epitopes from the Immune Epitope Database ranging from 8–12 amino acids in length, including the epitopes specific to the F5- and 1G4-TCRs. We then transduced K562 cells with the A2-restricted SCT library, co-incubated the transduced target cell library with F5-Jurkat or 1G4-Jurkat cells, and separated trogocytosis+ target K562 cells by FACS (Fig. 4a and Supplementary Fig. 9a,b). After two rounds of trogocytosis selection, the specificity of sorted K562 cells was validated by trogocytosis experiments (Fig. 4b). Furthermore, target K562 cells sorted after co-incubation with F5-Jurkat cells were 95% positive for F5-TCR dextramer staining (increased from 37% after one round of sorting) (Fig. 4c, top) and target K562 cells sorted after co-incubation with 1G4-Jurkat cells were 86% positive for 1G4-TCR dextramer staining (increased from 25% after one round of sorting) (Fig. 4c, bottom). In contrast, sorted cells were negative for dextramer staining in noncognate pairings for both rounds of sorting (Fig. 4c). Target cells from the second round of each trogocytosis selection were subjected to NGS to identify the enriched epitopes. Eight of the top nine enriched ligands from the F5-Jurkat selection were the cognate native and heteroclitic ligands (EAAGIGILTV and ELAGIGILTV) for F5-TCR or closely related ligands. The top two ligands from the 1G4-Jurkat selection were likewise the cognate native and heteroclitic ligands (SLLMWITQC and SLLMWITQV) for 1G4-TCR (Fig. 4d). These data demonstrate that target cell trogocytosis provides a platform for identifying cognate ligands and cross-reactive ligands of similar sequence for orphan TCRs.
Fig. 4 |. Target cell trogocytosis validates ligands for public F5- and 1G4-TCR.
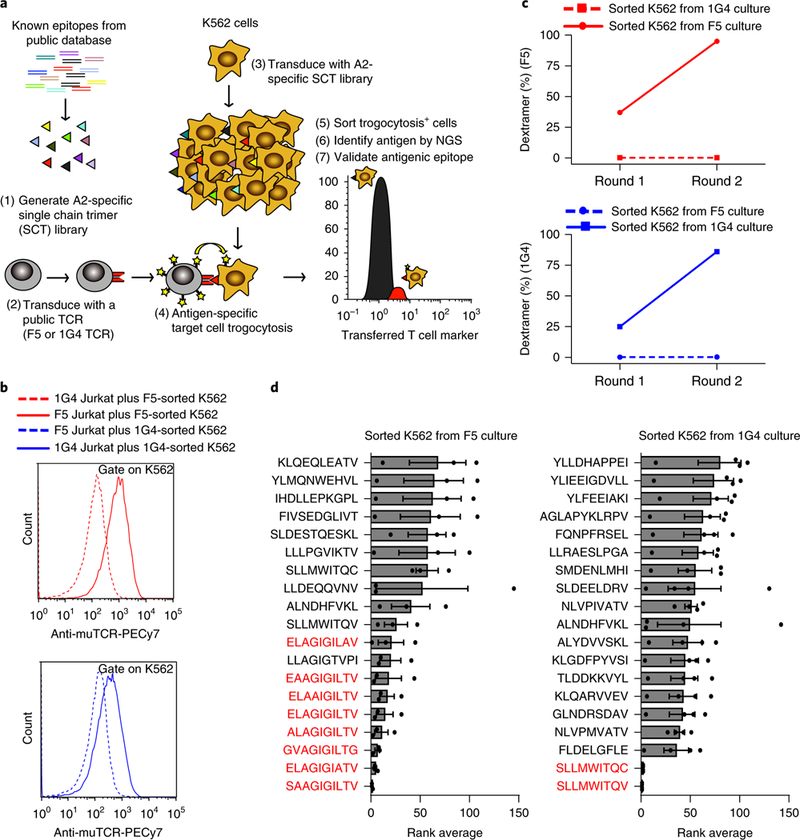
a, Schematic outline of trogocytosis used in the context of an A2-retricted library containing 12,055 epitopes. b, Histograms representing the mean fluorescence of second-round-sorted trogocytosis+ SCT-K562 cells after a validating co-incubation with F5-Jurkat or 1G4-Jurkat. SCT-K562 cells (eGFP+) were assessed for acquisition of TCR with an anti-mouse TCR (muTCR) antibody. Data are representative of two independent experiments. c, Representative FACS plots of 1G4-TCR or F5-TCR dextramer binding by sorted trogocytosis+ SCT-K562 cells (n = 1). d, Identification of enriched peptides after two rounds of trogocytosis selection by NGS. Rank average represents the ranking of enriched peptides, calculated based on the abundance of each peptide among all peptides, from experimental triplicates or quadruplicates
Trogocytosis identifies the ligand of a subject-derived neoTCR.
Recent advances in genomics and proteomics, along with supportive bioinformatics and in silico prediction tools, have enabled the rapid identification of mutated neoepitopes from subject-derived tumors30,31. We next determined whether this platform can identify the cognate neoepitope for a tumor-infiltrating lymphocyte-derived orphan TCR from a custom library of privately mutated, subject- specific neoepitopes. We identified private mutations by exome and RNA sequencing of tumor material from a subject with metastatic melanoma, predicted which of these mutations generated neoepitope ligands presented by HLA-A*02:01 and used a pMHC multimer panel to isolate a neoepitope-reactive TCR (S. Peng, J.M. Zaretsky, A.H.C. Ng, W. Chour, M.T. Bethune, A. Hsu, E. Holman, X. Ding, K. Guo, J. Kim, A.M. Xu, J.E. Heath, W. Noh, J. Zhou, Y. Su, Y. Lu, J. Mclaughlin, D. Cheng, O.N. Witte, D. Baltimore, A. Ribas and J.R. Heath, unpublished data). We then generated a neoepitope SCT cDNA library comprising 3,251 unique neoepitopes ranging from 8 to 12 amino acids in length, co-incubated K562 cells transduced with this library with neo-TCR-transduced Jurkat cells (neo-TCR-Jurkat), and performed two rounds of sorting for trogocytosis+ target cells (Supplementary Fig. 9c). The top five ligands enriched after two rounds of selection were overlapping mutated peptides derived from ubiquitin-specific peptidase 7 (USP7) (Fig. 5a). To verify that the neo-TCR recognizes the enriched neoepitopes, K562 cells were transduced with an SCT encoding the core nonamer mutant peptide YLYHRVDVI (mutUSP7- K562) contained in all enriched hits. As we expected, we observed significantly more trogocytosis+ target cells after coincubation of mutUSP7-K562 with neo-TCR-Jurkat cells than with noncognate F5-Jurkat cells (Fig. 5b), confirming that this USP7 mutant epitope is recognized by the neo-TCR. This was further validated by measuring the ability of the neo-TCR to induce cytotoxicity upon recognition of mutUSP7-K562 target cells (Fig. 5c). Notably, mutUSP7 is a mutation that affects MHC binding rather than TCR recognition31. We found that both wild-type USP7-K562 and mutUSP7-K562 target cells exhibited target cell trogocytosis when co-incubated with neo-TCR T cells (Supplementary Fig. 10). This is probably because the employed disulfide-trapped SCT architecture enables presentation of the weak-binding wild-type peptide32, leading to TCR recognition despite the poor natural affinity of this peptide for the HLA-A2 binding groove.
Fig. 5 |. Target cell trogocytosis identifies the cognate ligand for a neoantigen-specific, subject-derived TCR.
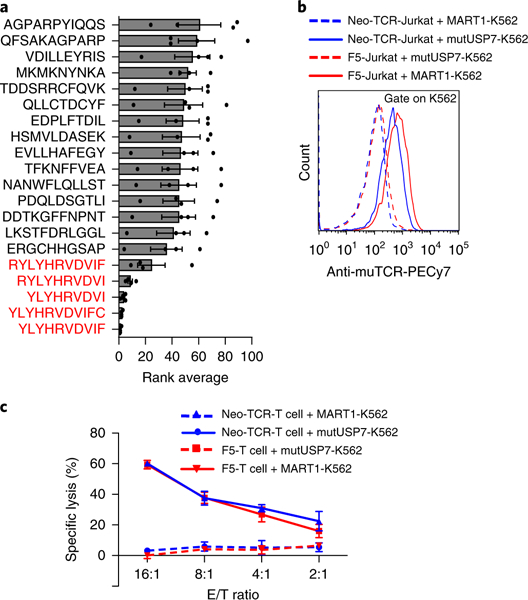
a, Identification of enriched peptides after two rounds of trogocytosis selection by NGS. Rank average represents the ranking of enriched peptides, calculated based on the abundance of each peptide among all peptides, from experimental quadruplicates. b, Validation of TCR-neoantigen pairing via trogocytosis. Data are representative of two independent experiments. c, Cytotoxicity of neo-TCR T cells against mutUSP7-K562 or MART1-K562 cells in various effector-to-target (E/T) ratios (presented as percentage specific lysis). Data are presented as mean± s.e.m. and are representative of two independent experiments.
Discussion
Our key discovery is that target cells presenting supraphysiological levels of a peptide epitope extract membrane-associated proteins from interacting T cells in an antigen-specific and pMHC-density-dependent manner. We have exploited this phenomenon to mark ligand-presenting target cells with extracted TCRs (and other membrane proteins) from cognate T cells, enabling ligand discovery, and we have validated this method using both public and neoepitope-specific TCRs. In a companion study33, we described the use of signaling and antigen-presenting bifunctional receptors in a second cell-based platform for TCR ligand discovery. These platforms exploit distinct biological mechanisms for the purpose of TCR ligand discovery, and the results obtained in our studies cross-validate each other.
The trogocytosis-based platform has several key advantages over existing technologies for de novo TCR ligand discovery. In contrast to pMHC yeast display34, our method does not require producing orphan TCR protein reagents, but rather uses orphan TCRs expressed in their natural context. Similarly, whereas multimer-based approaches rely on purified pMHC protein reagents that are expensive and time-consuming to generate, we genetically encode the peptide ligand and MHC, enabling rapid, modular assembly of pMHC libraries presented on mammalian cells. We recently described an unbiased TCR-ligand discovery method using fluo-rescently labeled reagents derived from orphan TCR ectodomains to screen a yeast cell-surface-displayed pMHC library34. However, this method requires the production of soluble TCR ectodomains, which is laborious, nonrobust and low throughput. Furthermore, neither yeast display nor multimer libraries can be applied to additional MHC alleles without extensive development of allele-specific reagents (that is, optimization of each human MHC allele library on yeast cells or design and synthesis of exchange ligands unique to each MHC allele for multimer library production). Because membrane receptor recognition is interrogated in the cell-to-cell context, our trogocytosis-based platform can be readily extended to additional TCRs or MHC alleles in high throughput and without further optimization. This is key for clinical applications, as the MHC I locus is the most polymorphic in the human genome, and MHC haplotypes vary widely across individuals and between ethnic groups35.
A similar level of trogocytosis was observed with A2-restricted TCRs of varying affinity for the same epitope, which suggests that individual TCR-pMHC affinity is not a critical parameter. The extent of trogocytosis could be augmented through coexpression of CD8 on the donor cells, presumably by increasing the avidity between T cells and cognate target cells. Thus, the trogo-cytosis-based cell platform is a robust tool for discovering ligands for orphan TCRs of various, unknown affinities, such as CD8-dependent TCRs (which would be poor candidates for antigen discovery via yeast display). We found that target cells presenting cognate pMHC could be identified by the transfer of any of multiple proteins that were involved in the T cell synapse (TCR, CD3 and CD8) or that were not (LNGFRΔ). The ability to identify cognate TCR-pMHC interactions using multiple independent, commercially available reagents (antibodies) is yet another key advantage of trogocytosis over pMHC multimers or display technologies, for which TCR-pMHC binding is the sole selective axis. Unlike yeast display, in which bioinformatics is required to infer true biological ligands from the sequences of top synthetic ligand hits34, the top epitopes identified in our experiments are reliably the biological target ligands for the TCR being tested.
Although we have validated our method only using MHC I-restricted TCRs, it is amenable to application to MHC II-restricted responses. Such responses are central to autoimmune disease, and are emerging as important components of cancer immunotherapy36,37. A similar platform could be conceived for other immuno-receptors (for example, chimeric antigen receptors and antibodies) using target cells expressing a library of suitable surface target proteins. Finally, this method can be used to determine whether a therapeutic candidate TCR of known specificity cross-reacts with self epitopes, thereby reducing the risk of serious adverse events38.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41592–018-0305–7.
Methods
A step-by-step protocol for trogocytosis-based TCR ligand identification is available at Protocol Exchange39 and in the Supplementary Protocol.
Cell lines and primary cells.
HEK-293T, Jurkat E6–1 and K562 cells were obtained from the American Type Culture Collection. Primary human peripheral blood mononuclear cells were purchased from the CFAR Virology Core Lab at the UCLA AIDS Institute. HEK-293T cells were cultured in DMEM (Mediatech) supplemented with 10% (v/v) FBS and 100 U ml−1 penicillin-streptomycin (Mediatech). Jurkat and K562 cells were cultured in RPMI 1640 medium (Mediatech) supplemented with 10% (v/v) FBS, 10 mM HEPES (Thermo Fisher), 50 μM β-mercaptoethanol (Sigma-Aldrich), 1× MEM non-essential amino acid (Thermo Fisher), and 1 mM sodium pyruvate (Mediatech). All cells were cultured at 37 °C with 5% atmospheric CO2.
DNA constructs.
A murine-stem-cell-virus-based retroviral vector encoding F5-TCR, 1G4-TCR or neo-TCR genes carrying either human or murine TCR constant regions had the format TCRα-F2A-TCRβ-P2A-LNGFRΔ. LNGFRΔ is a transduction marker comprising low-affinity nerve growth factor receptor with the intracellular domain truncated. A lentiviral vector encoding ZsGreen/eGFP and peptide-MHC SCT composed of antigenic peptide (NY-ESO-1, SLLMWITQV; MART1, ELAGIGILTV; wild-type USP7, DLYHRVDVI; mutant USP7, YLYHRVDVI), β2-microglobulin, and HLA-A2 domains via flexible glycine-serine linkers was prepared with a disulfide trap modification as described40. E2-Crimson and a CD3 fusion gene encoding CD3δ, CD3ε, CD3γ and CD3ξ were subcloned into a lentiviral vector. CD8 was subcloned into a retroviral vector.
Cell line construction.
Retroviruses encoding F5-TCR, 1G4-TCR, neo-TCR or CD8 were produced in HEK-293T cells by transient transfection of retroviral-based plasmids and their packaging vectors (pRD114 and pHIT60) using TransIT-293 (Mirus Bio) according to the manufacturer’s protocol. Lentiviruses encoding E2-Crimson, SCT or CD3 were produced in HEK-293T cells by transient transfection of lentiviral-based vectors and their packaging vectors (psPAX2 and pMD2.G). At 48 h after transfection, the virus was collected, filtered through a 0.45-μm syringe filter, and used for infection. The Jurkat or K562 cells were spin-infected with viral supernatant supplemented with 10 μg ml−1 Polybrene at 2,500 r.p.m. at 30 °C for 90 min. On day 3 post-infection, TCRhi, TCRhi E2-Crimson+, TCRhiCD8hi, A2hieGFPhi, or A2hiZsgreenhi K562 (CD3− or CD3+) or Jurkat cells were sorted by flow cytometry to establish derivative cell lines as indicated. For the SCT-K562 library cells, eGFP+ E2-Crimson− K562 cells were sorted.
T cell activation and transduction.
T cells were stimulated, transduced and cultured as described40. To transduce primary human T cells, peripheral blood mononuclear cells (2 × 106 cells ml−1) were activated in 24-well plates coated with 1μg ml−1 antibody to CD3 (clone OKT3, eBioscience) in the presence of 1μg ml−1 soluble antibody to CD28 (clone CD28.2, eBioscience) and 300 U ml−1 interleukin-2 (IL-2; Fisher Scientific) in T cell medium (AIM-V medium supplemented with 5% (v/v) human AB serum and antibiotics (penicillin-streptomycin)). After 48 h of activation, the cells were spin-infected with viral supernatant supplemented with 10 μg ml−1 Polybrene for 90 min at 2,500 r.p.m. at 30 °C. After spin-infection, the retroviral supernatant was replaced with fresh T cell medium containing 300 U ml−1 IL-2 and 1 μg ml−1 anti-CD28. The transduced primary T cells were cultured for 48 h and then used for cytotoxicity assays.
Surface biotin labeling.
F5-Jurkat cells were resuspended at 10 million cells per ml in freshly prepared biotin solution (1 mg ml−1 in sterile PBS) and incubated for 10 min at room temperature. The cells were quenched with one equivalent of FBS and incubated for 10 min at 4 °C, then washed three times with 10 ml RPMI 1640 medium supplemented with FBS. The labeled cells were then used for co-incubation experiments.
Peptide loading of A2-K562 cells.
Lyophilized peptides (Thermo Fisher) were reconstituted in dimethylsulfoxide to 10 mM and then further diluted in water to a 2 mM working stock. Five-fold serial dilutions of the peptides were performed using a 50 μM starting solution (5 μl of 2 mM working stock in 195 μl of serum-free medium) until 80 nM was reached. Target A2-K562 cells (50,000 total, 0.5 ×106 cells ml−1) were pulsed with 25 μl of 5× serial peptide dilution in a 96-well U-bottom plate and incubated for 2 h at 37 °C. After incubation, 100 μl of medium was added to each well and then centrifuged for 5 min at 1,500 r.p.m. The cell pellets were washed once with 200 μl of medium and then resuspended in 100 μl of medium for co-incubation experiments.
Co-incubation assay and cell staining and sorting.
For experiments using F5-Jurkat, 1G4-Jurkat, MART1-K562 and NYESO-K562 cells, co-incubations were set up in 96-well U-bottom plates at a ratio of 5:1 Jurkat to K562 (300,000 cells per well) and co-incubated for 45 min at 37 °C. Ratios for co-incubation experiments involving SCT-Jurkat cells and TCR-K562 cells are indicated in the text and figure legends. The co-incubation was then centrifuged for 5 min at 1,500 r.p.m. and the medium was aspirated by vacuum. The cell pellets were resuspended with cold PBS solution containing 2 mM EDTA and then centrifuged. Cells were stained with different antibodies at 4 °C for 20 min, washed twice, and then analyzed by flow cytometry using MACSQuant Analyzer 10 (Miltenyi Biotec). The following antibodies were used to detect expression of surface markers: anti-CD3-PE, anti-CD8-BV510, anti-TCRαβ-PacificBlue, anti-TCRαβ-PE/Cy7, anti-LNGFR-PE, anti-LNGFR-APC, anti-HLA-A2-PacificBlue, anti-HLA-A2-BV510, and anti-muTCRa β-PE/Cy7 (all from Biolegend). Flow cytometry gating strategies are described in the Supplementary Note.
Co-incubation experiments with A2-restricted SCT and neoantigen SCT libraries.
For the co-incubations of F5-Jurkat or 1G4-Jurkat cells (E2-Crimson+) with A2-restricted SCT library cells (eGFP+), 4.8 million F5-Jurkat or 1G4-Jurkat cells were co-incubated with 2.4 million K562 cells expressing the A2-restricted SCT library for 45 min at 37 °C. The cells were stained as previously described and then sorted by FACS. For A2-restricted neoantigen SCT library co-incubations, 2million donor cells were co-incubated with 1 million acceptor cells for 45 min at 37 °C. The cells were prepared for sorting by FACS analogously to the A2-restricted SCT library co-incubations.
Dextramer binding on SCT-K562 cells.
We prepared F5-TCR or 1G4-TCR dextramers by using fluorescently labeled streptavidin (Life Technologies) as described26. SCT-transduced K562 cells were stained with TCR dextramer and 7AAD (eBioscience) for 20 min at room temperature. Stained cells were analyzed by flow cytometry using MACSQuant Analyzer 10 (Miltenyi Biotec).
Immunofluorescence staining.
Co-incubated cells were stained with MART1 pMHC dextramer (ELAGIGILTV) conjugated to PE and then treated with 4% formaldehyde, followed by incubation at room temperature for 10 min. Cells were washed three times with PBST (PBS plus 0.1% Triton X-100) for 5 min and then placed on the slide with mounting medium. Images were acquired on Zeiss LSM880.
Cytotoxicity assays with primary human T cells using calcein AM.
To measure the cytotoxic response of effector T cells to target cells, we carried out a calcein AM release-based cytotoxicity assay as described41. Briefly, 1 × 106 SCT-K562 target cells were resuspended in 1 ml of calcein AM (Thermo Fisher) in cell medium (5 μlml−1), incubated for 1 h at 37 °C while being shaken occasionally, and then washed twice with cell medium to remove residual dye. Effector T cells transduced with F5-TCR or neo-TCR and target SCT cells were plated on a 96-well U-bottom plate in various effector-to-target ratios (16:1, 8:1, 4:1, 2:1, 1:1, 0.5:1 and 0:1) and co-incubated for 4 h at 37 °C. After co-incubation, cell-free supernatant was carefully removed and analyzed using a fluorescence plate reader (excitation filter, 485 nm; emission filter, 530 nm). The percentage of specific lysis was calculated using the formula [(test release - spontaneous release)/(maximum release - spontaneous release)] × 100 based on the optical density measured. Target cells in the spontaneous release well were incubated with medium alone (considered as 0% lysis), and target cells in the maximum release well were incubated with 2% Triton X-100 (considered as 100% lysis).
Library generation.
The A2-restricted SCT cDNA library contains ~12,055 public T cell epitopes from the Immune Epitope Database (https://www.iedb.org/). The neoantigen SCT cDNA library contains ~3,000 unique neoantigens that had been identified independently by exome and RNA sequencing of tumor material from a melanoma subject and predicting neoantigen presentation by HLA-A*02:01 (data not shown). The oligonucleotide pool encoding A2-restricted public and neoantigen neoepitopes was synthesized (Twist Bioscience) and used as the template for PCR amplification using primers 5´-CAGGAGGGCTCGGCA-3´ and 5´-GATGAGCGGCCGCCGGACCCTCCGCATCC-3´ with the following program: initial denaturation step at 95 °C for 2 min followed by six cycles of 95 °C for 20 s, 61 °C for 10 s, and 70 °C for 15 s, then a final extension at 70 °C for 1 min, followed by a 4 °C hold. After PCR amplification of the oligonucleotide library, the PCR amplicons were digested with NotI and then inserted into a BsmBI-digested lentiviral vector with internal-ribosome-entry-site-mediated coexpression of a GFP gene by In-Fusion cloning. The lists of epitopes in the A2-restricted SCT cDNA library and the neoantigen SCT cDNA library are in Supplementary Tables 1 and 2.
PCR amplification and deep sequencing.
Genomic DNA of sorted SCT-K562 cells was extracted using the PureLink Genomic DNA Mini Kit (Invitrogen) and used as template in barcoded PCR amplification in a Mastercycler Pro S PCR System (Eppendorf) with the following program: initial denaturation step at 95 °C for 2 min followed by 35 cycles of 95 °C for 20 s, 66 °C for 10 s, and 70 °C for 15 s, then a final extension at 70 °C for 2 min, followed by a 4 °C hold. The primers used in PCR amplification were the following: TruSeq-Univ-SCTfixed-Forward, 5´- AATGATACGGCGACCACC GAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGGCCTGCTTTGTTTGCC-3´; TruSeq-Read2- SCTfixed-Reverse, 5´-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT CCTCCACCACCGCTACCTC-3´; and Truseq-Adaptor-Index Reverse primer. The amplified PCR was purified using the DNA Clean & Concentrator-5 Kit (Zymo Research) according to the manufacturer’s protocol. The amplified barcoded DNA was quantified by Bioanalyzer (Agilent Genomics) and sequenced by the Illumina Genome Analyzer IIx System (Illumina).
Statistical analysis.
All statistical analysis was done in GraphPad Prism software using an unpaired Student’s t-test. Data are reported as mean ± s.e.m. *P <0.05; **P < 0.01; ***P < 0.001; NS, not significant.
Supplementary Material
Acknowledgements
We thank I. Antoshechkin (Millard and Muriel Jacobs Genetics and Genomics Laboratory, Caltech) for deep DNA sequencing, and D. Perez, J. Tijerina and R.A. Diamond (Flow Cytometry Facility, Caltech) for cell sorting. This work was supported by the Prostate Cancer Foundation Challenge Award 15CHAL02 to D.B., O.N.W, L.Y. and M.T.B., and the National Cancer Institute (grant 1U54 CA199090–01 to J.R.H.). M.T.B. is the recipient of a Jane Coffin Childs Postdoctoral Fellowship. A.R. was supported by National Institutes of Health (NIH) grant R35 CA197633. G.L. was supported by the Parker Institute for Cancer Immunotherapy. J.T.K. was supported by NIH/National Center for Advancing Translational Science UCLA CTSI grant KL2TR001882.
Footnotes
Competing interests
M.T.B., G.L., J.T.K., S.W. and D.B. are co-inventors on a patent application concerning the described technology, which is licensed to PACT Pharma, Inc. J.R.H. and A.R. are directors and consultants of PACT; D.B. is a consultant of PACT and head of their scientific advisory board; M.T.B. and S.P. are employees of PACT; J.M.Z. is a consultant of PACT; and each of the foregoing individuals has equity interests in PACT.
Data availability
The original NGS DNA-seq data have been deposited in the Sequence Read Archive under accession numbers SRR8217181, SRR8217182 and SRR8217183. The data that support the findings of this study are available from the corresponding author upon request. Source data for Figs. 2, 4 and 5 are available online.
Supplementary information is available for this paper at https://doi.org/10.1038/s41592–018-0305–7.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Dembic Z et al. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature 320, 232–238 (1986). [DOI] [PubMed] [Google Scholar]
- 2.Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Sollid LM et al. Small bowel, celiac disease and adaptive immunity. Dig. Dis. 33, 115–121 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SA & Restifo NP Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kontos S, Grimm AJ & Hubbell JA Engineering antigen-specific immunological tolerance. Curr. Opin. Immunol. 35, 80–88 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Dolton G et al. More tricks with tetramers: a practical guide to staining T cells with peptide-MHC multimers. Immunology 146, 11–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bentzen AK et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 34, 1037–1045 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Gros A et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 22, 433–438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strønen E et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Briggs AW et al. Tumor-infiltrating immune repertoires captured by single-cell barcoding in emulsion. bioRxiv Preprint at https://www.biorxiv.org/content/early/2017/05/05/134841 (2017). [Google Scholar]
- 11.Duhen T et al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 9, 2724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gros A et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 124, 2246–2259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han A, Glanville J, Hansmann L & Davis MM Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol. 32, 684–692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linnemann C, Mezzadra R & Schumacher TN TCR repertoires of intratumoral T-cell subsets. Immunol. Rev. 257, 72–82 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Pasetto A et al. Tumor- and neoantigen-reactive T-cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol. Res. 4, 734–743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prickett TD et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol. Res. 4, 669–678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simoni Y et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Joly E & Hudrisier D What is trogocytosis and what is its purpose? Nat. Immunol. 4, 815 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Daubeuf S, Lindorfer MA, Taylor RP, Joly E & Hudrisier D The direction of plasma membrane exchange between lymphocytes and accessory cells by trogocytosis is influenced by the nature of the accessory cell. J. Immunol. 184, 1897–1908 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Cone RE, Sprent J & Marchalonis JJ Antigen-binding specificity of isolated cell-surface immunoglobulin from thymus cells activated to histocompatibility antigens. Proc. Natl Acad. Sci. USA 69, 2556–2560 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang JF et al. TCR-mediated internalization of peptide-MHC complexes acquired by T cells. Science 286, 952–954 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Hwang I et al. T cells can use either T cell receptor or CD28 receptors to absorb and internalize cell surface molecules derived from antigen-presenting cells. J. Exp. Med. 191, 1137–1148 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uzana R et al. Human T cell crosstalk is induced by tumor membrane transfer. PLoS ONE 10, e0118244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He T et al. Bidirectional membrane molecule transfer between dendritic and T cells. Biochem. Biophys. Res. Commun. 359, 202–208 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Kim HR et al. T cell microvilli constitute immunological synaptosomes that carry messages to antigen-presenting cells. Nat. Commun. 9, 3630 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bethune MT, Comin-Anduix B, Hwang Fu YH, Ribas A & Baltimore D Preparation of peptide-MHC and T-cell receptor dextramers by biotinylated dextran doping. Biotechniques 62, 123–130 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Bethune MT et al. Isolation and characterization of NY-ESO-1-specificT cell receptors restricted on various MHC molecules. Proc. Natl Acad. Sci. USA 115, E10702–E10711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skipper JC et al. Mass-spectrometric evaluation of HLA-A*0201-associated peptides identifies dominant naturally processed forms of CTL epitopes from MART-1 and gp100. Int. J. Cancer 82, 669–677 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Wooldridge L et al. Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor-antigen complexes at the cell surface. J. Biol. Chem. 280, 27491–27501 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bassani-Sternberg M & Coukos G Mass spectrometry-based antigen discovery for cancer immunotherapy. Curr. Opin. Immunol. 41, 9–17 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Bethune MT & Joglekar AV Personalized T cell-mediated cancer immunotherapy: progress and challenges. Curr. Opin. Biotechnol. 48, 142–152 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Truscott SM et al. Disulfide bond engineering to trap peptides in the MHC class I binding groove. J. Immunol. 178, 6280–6289 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Joglekar AV et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. Methods 10.1038/s41592-018-0304-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gee MH et al. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell 172, 549–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.González-Galarza FF et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 43, D784–D788 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran E et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu YC & Robbins PF Cancer immunotherapy targeting neoantigens. Semin. Immunol. 28, 22–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan RA et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 36, 133–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li G, Wong S, Bethune TM & Baltimore D Trogocytosis-based cell platform for TCR ligand discovery. Protocol Exchange 10.1038/protex.2018.127 (2019). [DOI] [Google Scholar]
- 40.Bethune M et al. Domain-swapped T cell receptors improve the safety of TCR gene therapy. eLife 5, e19095 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neri S, Mariani E, Meneghetti A, Cattini L & Facchini A Calcein-acetyoxymethyl cytotoxicity assay: standardization of a method allowing additional analyses on recovered effector cells and supernatants. Clin. Diagn. Lab. Immunol. 8, 1131–1135 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.