

In this study, Welte et al. investigated the dual roles of mammalian TRIM71, a phylogenetically conserved regulator of development, in the control of stem cell fate. They demonstrate that TRIM71 shapes the transcriptome of mESCs predominantly through its RNA-binding activity and identify a set of primary targets consistently regulated in various human and mouse cell lines, including MBNL1/Muscleblind.
Keywords: 3′ UTR, 5′ UTR, LIN41, RNA-binding protein, TRIM71, alternative splicing, mRNA degradation, muscleblind, stem cell, translational repression
Abstract
TRIM71/LIN-41, a phylogenetically conserved regulator of development, controls stem cell fates. Mammalian TRIM71 exhibits both RNA-binding and protein ubiquitylation activities, but the functional contribution of either activity and relevant primary targets remain poorly understood. Here, we demonstrate that TRIM71 shapes the transcriptome of mouse embryonic stem cells (mESCs) predominantly through its RNA-binding activity. We reveal that TRIM71 binds targets through 3′ untranslated region (UTR) hairpin motifs and that it acts predominantly by target degradation. TRIM71 mutations implicated in etiogenesis of human congenital hydrocephalus impair target silencing. We identify a set of primary targets consistently regulated in various human and mouse cell lines, including MBNL1 (Muscleblind-like protein 1). MBNL1 promotes cell differentiation through regulation of alternative splicing, and we demonstrate that TRIM71 promotes embryonic splicing patterns through MBNL1 repression. Hence, repression of MBNL1-dependent alternative splicing may contribute to TRIM71's function in regulating stem cell fates.
Organogenesis relies on a balance between self-renewal and differentiation of stem cells and their progeny. Accordingly, stem cell fates are tightly controlled through transcriptional and posttranscriptional mechanisms. TRIM71/LIN-41 is a phylogenetically conserved regulator of stem and progenitor cell fates and developmental events. In vertebrates, TRIM71 regulates proliferation and differentiation of human induced pluripotent and mouse neuronal stem cells (Chen et al. 2012; Worringer et al. 2014), limb development (Lancman et al. 2005), and neurulation (Maller Schulman et al. 2008) and is implicated in human congenital hydrocephalus (Furey et al. 2018). In Caenorhabditis elegans, where TRIM71 is known as LIN-41, it controls differentiation and self-renewal of epidermal progenitor cells (Slack et al. 2000), oocyte maturation (Spike et al. 2014; Matsuura et al. 2016), oocyte-to-embryo transition (Tocchini et al. 2014), reproductive organ formation (Del Rio-Albrechtsen et al. 2006; Ecsedi et al. 2015; Aeschimann et al. 2019), and sexually dimorphic neuron differentiation (Pereira et al. 2019). Trim71 overexpression is associated with rapid disease progression in myxoid liposarcoma (De Cecco et al. 2014), hepatocellular carcinoma (Chen et al. 2013), and nonsmall cell lung cancer (Ren et al. 2018).
Despite its developmental importance, the molecular functions of TRIM71 are poorly understood. TRIM71 contains an N-terminal RING domain typical of a class of E3 ubiquitin ligases and a C-terminal NHL domain with RNA-binding activity, suggesting posttranscriptional and/or posttranslational functions (Ecsedi and Großhans 2013). The NHL domain exhibits RNA-binding activity in vertebrates and C. elegans (Lödige et al. 2013, 2015; Kumari et al. 2018). In contrast, although the RING domain has ubiquitin ligase activity in mammalian cells and is important for neural progenitor cell proliferation (Rybak et al. 2009; Chen et al. 2012; Nguyen et al. 2017), this domain is absent from the Drosophila melanogaster protein and is dysfunctional in C. elegans LIN-41 (Löer et al. 2008; Tocchini et al. 2014).
Manipulation of TRIM71 levels in mammalian cell lines and reporter gene experiments showed that TRIM71 can destabilize mRNAs by binding to 3′ untranslated regions (UTRs) (Chang et al. 2012; Lödige et al. 2013; Mitschka et al. 2015). Moreover, in C. elegans, TRIM71/LIN-41 can bind 5′ UTRs to elicit translational repression without mRNA degradation (Aeschimann et al. 2017). A structure of a truncated zebrafish TRIM71 protein and C. elegans in vivo experiments suggest that TRIM71 functions through binding hairpin elements on target RNAs (Kumari et al. 2018). However, evidence that these are the relevant binding sites of endogenous TRIM71 in mammals is lacking, and the physiological target mRNAs are largely unknown.
Here, we show that the NHL domain of TRIM71 is critical for RNA interaction and maintenance of physiological gene expression levels in mouse embryonic stem cells (mESCs). We reveal that mutations in the NHL domain observed in patients with congenital hydrocephalus disrupt target repression, providing a disease model for this condition and further underlining the importance of RNA binding for TRIM71's function in development. We define the RNA interactome of mouse TRIM71 and show that hairpin motifs mediate silencing of endogenous mRNAs, with repression efficacy depending on hairpin number. We identified a core set of TRIM71 targets in mice and humans, including the splicing regulator Muscleblind-like protein 1 (MBNL1), and found that TRIM71-dependent repression of MBNL1 is required to maintain alternative splicing patterns in stem cells.
Results
TRIM71 acts primarily by repressing target transcripts through binding to 3′ UTRs
Previous gene expression profiling experiments revealed changes in TRIM71 depletion and knockout cells (Lödige et al. 2013; Worringer et al. 2014; Mitschka et al. 2015). However, it is unclear which transcripts are direct primary versus indirect secondary targets and to what extent TRIM71 RNA binding versus protein ubiquitylation contributes to its function. To address the latter question, we generated three mESC lines: Trim71 knockout, RING mutant (RING_mut), and NHL mutant (NHL_mut) mESCs (Fig. 1A). RING_mut contained two point mutations in the conserved catalytic dyad, C12A and C15A (Supplemental Fig. S1A), that inactivate ubiquitin ligase activity (Rybak et al. 2009; Lee et al. 2014). NHL_mut contained an R738A point mutation, corresponding to an R751A mutation in human TRIM71 (Supplemental Fig. S1B) that compromised reporter gene silencing by TRIM71 (Kumari et al. 2018). Western blot analysis confirmed equal levels of full-length TRIM71 in wild-type (WT), RING_mut, and NHL_mut mESCs and full knockout in Trim71 knockout mESCs (Supplemental Fig. S1C,E).
Figure 1.

TRIM71 acts primarily by repressing target transcripts through binding of 3′ UTRs. (A) Schematic view of TRIM71 domain structure with associated functions. Point mutations generated in this study are indicated; the shorthand name is in bold. (B) PCA plot of RNA sequencing (RNA-seq) data of experiments with WT and mutant mESCs. WT and Trim71 knockout: n = 10 biological replicates. A Trim71 knockout outlier replicate with small library size is shown with reduced opacity. Trim71 NHL_mut and RING_mut: n = 5 biological replicates. A corresponding correlation matrix is shown in Supplemental Figure S1F. (C) Scatter plots and correlation matrix showing Pearson's correlation coefficients between CRAC-seq (cross-linking and analysis of cDNAs [CRAC] combined with sequencing) biological replicates based on transcript read counts. (D) Scatter plot showing differential gene regulation in Trim71 knockout versus WT mESCs (n = 10 biological replicates) versus total transcript CRAC-seq enrichment. n = 4 biological WT TRIM71 CRAC-seq replicates, summed by transcript and divided by RPKMs (reads per kilobase per million mapped reads) from n = 3 RNA-seq experiments in WT mESCs. Transcripts with high (red; quantile ≥ 0.98) and medium (orange; quantile 0.95–<0.98) TRIM71 binding are indicated. For regression analysis, a natural regression spline is fitted, with knots at enrichment values of 1–5 (black line). Data source: Supplemental Table S5. (E) Metaplot of mean CRAC-seq coverage around transcript start and stop codons in WT, RING_mut, and NHL_mut mESCs (all replicates combined). (F) Stacked bar chart showing fractions of CRAC-seq reads by genomic regions in WT, RING_mut, and NHL_mut mESCs (all replicates combined). (G) Scatter plot showing the correlation of ribosome profiling (Ribo-seq) versus transcript (RNA-seq) fold changes between Trim71 knockout and WT mESCs (n = 4 biological replicates); 12,718 genes were quantified (marked in gray). Significantly differentially regulated (“Sign. change RNA”; false discovery rate [FDR] < 0.05) genes on the RNA level are marked in blue. n = 2954. Significantly differentially translated (“Sign. change TE”; FDR < 0.05) genes are marked in red. n = 2. Transcripts regulated by degradation are expected to be up-regulated in both RNA-seq and Ribo-seq (blue arrow). Targets regulated by pure translational inhibition are expected to be up-regulated in Ribo-seq only (red arrow). Data source: Supplemental Table S3.
RNA sequencing (RNA-seq) analysis revealed that relative to WT, Trim71 knockout and NHL_mut cells exhibit similar changes in gene expression, whereas RING_mut cells exhibit a separate gene expression pattern (Fig. 1B; Supplemental Fig. S1F; Supplemental Table S1). This suggests that TRIM71 affects gene expression in mESCs primarily through its NHL domain, presumably by RNA binding.
In order to distinguish primary from secondary effects by interaction profiling, we performed CRAC-seq (cross-linking and analysis of cDNAs [CRAC] combined with sequencing) (Granneman et al. 2009; Tuck et al. 2018) on mESCs with endogenously 3xFlag-AVI-tagged Trim71 (Ostapcuk et al. 2018). We also generated cells with tagged RING_mut and NHL_mut Trim71 (Supplemental Fig. S1E). WT and RING_mut TRIM71 bound a specific and similar set of transcripts, whereas binding specificity was severely impaired for NHL_mut (Fig. 1C). The NHL domain is therefore critical for RNA binding in these cells. Comparing CRAC-seq and RNA-seq data revealed a correlation between TRIM71 binding (measured as CRAC-seq enrichment: CRAC-seq counts normalized to RNA-seq) and derepression in Trim71 knockout cells (Fig. 1D; Supplemental Table S2). A regression model confirmed that this relationship was significant (P < 2.2 × 10−16, adjusted R2 = 0.036). Therefore, binding observed by CRAC-seq reveals TRIM71 targets.
We next assessed the relative contributions of TRIM71 binding to different transcript regions; i.e., 5′ UTR, coding DNA sequence (CDS), and 3′ UTR. We calculated TRIM71 CRAC-seq enrichment separately for these regions and compared regression models, including enrichment for 3′ UTR only; 3′ UTR and CDS; or 3′ UTR, CDS, and 5′ UTR. The “3′ UTR only” model fits the data similarly well (residual sum of squares, RSS = 570; total sum of squares = 590) to the whole transcript model (RSS = 569). The “3′ UTR and CDS” (RSS 567) and “3′ UTR, CDS, and 5′ UTR” (RSS 569) models improved the fit only slightly (data source: Supplemental Table S2). Regulation by TRIM71 can therefore be mostly explained by its binding to 3′ UTRs, which is consistent with its preference for binding to 3′ UTRs in our CRAC-seq experiments (Fig. 1E,F) and with reporter gene studies in C. elegans and in mammalian cell lines (Chang et al. 2012; Lödige et al. 2013; Worringer et al. 2014; Mitschka et al. 2015; Aeschimann et al. 2017).
As we recently observed an instance in which C. elegans LIN-41 represses a target exclusively at the level of translation by binding to its 5′ UTR (Aeschimann et al. 2017), we wanted to rule out the possibility of missing targets by focusing only on RNA expression. We therefore performed matched RNA-seq and ribosome profiling (Ribo-seq) experiments in Trim71 knockout and WT mESCs. Changes in endogenous transcript levels and ribosome occupancy (quantified for 12,718 transcripts) (Fig. 1G) were highly correlated (Pearson's correlation coefficient r = 0.73). Although 2954 transcripts were differentially expressed (false discovery rate [FDR] < 0.05; 1890 up-regulated in Trim71 knockout cells), translation efficiency changed for only two (FDR < 0.05), and both of these were down-regulated in Trim71 knockout cells (Supplemental Table S3). We conclude that, in mESCs, TRIM71 functions primarily by reducing target RNA abundance and focused further analyses on RNA-seq experiments.
TRIM71 binds cellular transcripts through mRNA hairpin motifs
Having demonstrated the functional relevance of our binding data for transcript repression, we aimed to identify TRIM71-binding motifs. For this, we calculated TRIM71 CRAC-seq enrichments for 50-nucleotide (nt) windows across transcripts and performed a motif analysis across their 3′ UTRs using a previously described approach (Kumari et al. 2018). We observed a significant enrichment of hairpin motifs with a loop of 3 nt in TRIM71-bound windows (Fig. 2A), in addition to a strong preference for U–A base-pairing in the first position of the stem and a purine at position III in the loop (Fig. 2B,C). These features are strikingly similar to those identified previously by in vitro binding assays with LIN-41/TRIM71 NHL domains (Kumari et al. 2018). This enabled us to use the published in vitro model to scan for hairpin motifs across all transcripts (Supplemental Table S4), assigning each motif a predicted strength based on sequence features and the base-pairing probability of the stem (Supplemental Methods). Total CRAC-seq read coverage was elevated at predicted hairpin sites for WT and RING_mut TRIM71 but not for NHL_mut TRIM71. This confirms that TRIM71 binds to hairpin motifs and that binding depends on the NHL domain (Fig. 2D). Notably, the intensity of TRIM71 binding correlated with predicted hairpin strength (Fig. 2D). In conclusion, structural and sequence elements that determine RNA binding of the LIN-41/TRIM71 NHL domains in vitro also do so remarkably well in an endogenous cellular context with full-length TRIM71.
Figure 2.

TRIM71 binds cellular transcripts through mRNA hairpin motifs. (A) Scatter plot showing enrichment of 11-mer structure motifs in TRIM71-bound 50-nt windows versus their occurrence in TRIM71-unbound 50-nt windows. Data source: Supplemental Table S4. The top 20 enriched motifs are highlighted in red. Corresponding dot bracket strings reveal a stem–loop motif with a loop of 3 nt. Structures containing a loop with 4, 5, or 6 nt (highlighted in cyan, blue, and purple) are not enriched. (B) Heat map showing median-normalized CRAC-seq enrichment values (red to blue color) for groups of predicted 3-nt hairpins; 1536 unique hairpins are vertically ordered by pairing probability in the stem (binned in four groups on log2 scale; grayscale at the left of the heat map) and nucleotide pairing in the loop-proximal stem position (“position −1, +1”; six sequence features; indicated at the right of the heat map). Nucleotide sequence in the loop (“positions I, II, and III”; 64 sequence features) is used for horizontal ordering and is indicated at the bottom of the heat map; purine bases (A/G) at the last position in the loop (position III) are colored in red. (C) Graph displaying structural and sequence features of the TRIM71 response element. (D) Metaplot showing total CRAC-seq read depth around predicted hairpin motifs with varying motif strength according to sequence features identified for C. elegans LIN-41 in Trim71 WT, RING_mut, and NHL_mut mESCs. For each plot, reads were normalized to the depth at position −50.
Repression efficacy correlates with the number of TRIM71-bound hairpins
To test the functional relevance of TRIM71 binding to hairpin motifs, we examined the ability of hairpin binding to predict transcript down-regulation. We calculated CRAC-seq enrichments in windows of 100 nt around predicted hairpins (Supplemental Table S4) and summed these “hairpin enrichments” for each transcript. Similar to our previous analyses with total CRAC-seq signal, high hairpin enrichments correlated with derepression in Trim71 knockout mESCs (Fig. 3A,B). A regression model revealed a similar fit (adjusted R2 = 0.041, P < 2.2 × 10−16) versus the model based on total CRAC-seq signal across transcripts (adjusted R2 = 0.036, P < 2.2 × 10−16), suggesting that TRIM71 binding to hairpin motifs fully explains its effect on RNA abundance.
Figure 3.
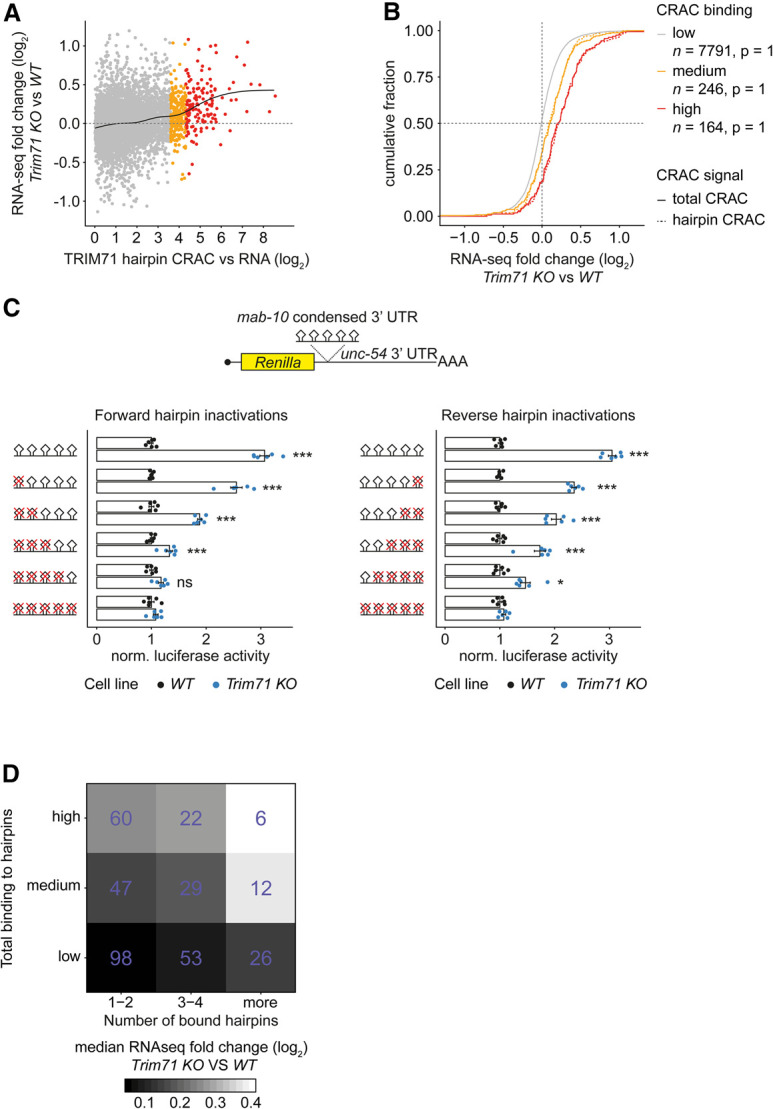
Repression efficacy correlates with the number of TRIM71-bound hairpins. (A) Scatter plot showing differential gene regulation in Trim71 knockout versus WT mESCs (n = 10 biological replicates) versus hairpin CRAC-seq enrichment (from n = 4 biological replicates in 100-nt windows around hairpins, summed by transcript and divided by RPKMs from n = 3 RNA-seq experiments in WT mESCs). Transcripts with high (red) and medium (orange) TRIM71 binding are indicated. This assignment and regression analyses were done as described in Figure 1D. (B) Cumulative density functions showing similar transcript repression efficacy in respective binding categories for total CRAC-seq enrichment and hairpin CRAC-seq enrichment. Significance was tested with a two-sided Kolmogorov-Smirnov test between total CRAC and hairpin CRAC for each binding category as defined in A. (C) Luciferase reporter gene assay with Renilla::mab-10 condensed 3′ UTR and hairpin inactivation mutants in WT and Trim71 knockout mESCs. Data were normalized to WT and represent mean ± SEM. n = 6 biological replicates. Significance was tested using a two-tailed Student's t-test. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (ns) not significant (P ≥ 0.05). (D) Heat map showing median differential gene regulation in Trim71 knockout versus WT mESCs (n = 10 biological replicates) for transcripts grouped by total TRIM71 binding to all hairpins and the number of bound hairpins. Hairpin binding categories are as in A. Bound hairpins per transcript are defined as hairpins with at least half as many CRAC-seq read counts as the hairpin with the highest CRAC-seq read count. Color gradient represents the median RNA-seq (log2) fold change of Trim71 knockout versus WT mESCs in each group. Numbers of transcripts in each group are indicated in purple. Data source for A, B, and D: Supplemental Table S5.
As the extent of mRNA silencing varied considerably between transcripts with similar levels of TRIM71 binding (Fig. 3A), we suspected that additional factors determine TRIM71 activity on its targets. Specifically, we wondered whether the number of TRIM71-binding sites was relevant. To test this experimentally, we used a Renilla luciferase reporter gene assay with a previously described variant of the C. elegans mab-10 3′ UTR containing five annotated hairpins (“mab-10 condensed 3′ UTR”) (Kumari et al. 2018). This Renilla::mab-10 reporter was repressed by endogenous TRIM71 in mESCs in luciferase reporter gene assays (Supplemental Fig. S2A). We generated a series of mutant constructs in which we successively disrupted more hairpins by inserting point mutations starting from either the most upstream or the most downstream hairpin. Testing these constructs in WT and Trim71 knockout mESCs revealed a “dose dependence”; i.e., repression increased with increasing hairpin number (Fig. 3C). We conclude that the extent to which TRIM71 binds transcripts through hairpins determines the magnitude of their silencing.
To uncouple the effects of overall TRIM71 binding on a transcript from effects resulting from the number of bound sites, we assigned transcripts bound by TRIM71 to nine categories based on the total level of TRIM71 binding (low, medium, or high summed hairpin enrichments) and the number of hairpins bound (one to two, three to four, and more). Comparing transcripts with similar overall binding, the extent of silencing depended on the number of bound hairpins. For instance, in the “medium” binding group (Fig. 3D, middle row), average silencing is low for targets with one to two bound hairpins, higher for those containing three to four bound hairpins, and highest for those with more than four bound hairpins. Hence, target repression is favored when a given binding activity is distributed over several rather than few binding sites.
Identification of a core set of TRIM71 target candidates in mouse and human cell lines
Having defined candidate TRIM71 targets in mESCs, we next sought to determine those likely to explain shared functions in stem and tumor cells by identifying transcripts reproducibly regulated by TRIM71 across different cell lines. Hence, we knocked out Trim71 in mouse neuronal stem cells (Ne4C) and human hepatocellular carcinoma cells (Huh-7) (Supplemental Fig. S1C,D) and performed RNA-seq to measure changes in endogenous transcript levels (Supplemental Table S1). Transcripts bound by TRIM71 in mESCs (defined as the 5% of transcripts with the highest total CRAC-seq enrichment) were globally derepressed in all cell lines, indicating reasonable target conservation (Fig. 4A,B). Applying an additional log2 fold change cutoff of ≥0.5 and an FDR cutoff of <0.05 in all cell lines identified 12 genes as consistent direct target candidates (Fig. 4C). As expected, we predict “strong” and “medium” hairpins in both mouse and human 3′ UTR homologs for most of these genes, indicating conservation of binding motifs (Supplemental Table S5).
Figure 4.

Identification of a core set of TRIM71 target candidates in mouse and human cell lines. (A,B) Scatter plot of differential gene expression in Trim71 knockout versus WT in mESCs and Ne4C cells (A) and Trim71 knockout versus WT in mESCs and Huh-7 cells (B). TRIM71-bound genes (top 5% total CRAC-seq enrichment) with significant expression change (FDR < 0.05) are shown in red (or black if not bound). Red numbers indicate the number of significantly differentially expressed TRIM71-bound transcripts in each quadrant as a percentage of all TRIM71-bound transcripts, including those not significantly expressed. (C) “Core” TRIM71 target candidates. Significantly up-regulated (FDR < 0.05; log2 fold change ≥0.5 in Trim71 knockout vs. WT mESCs, Ne4C cells, and Huh-7 cells) TRIM71-bound genes (as defined in A) were ranked descending according to CRAC enrichment. (D) Luciferase reporter gene assay with Renilla::Mbnl1, Plxnb2, and Mllt1 3′ UTR constructs in the indicated mESC lines. Data were normalized to WT and represent mean ± SEM. n = 3 biological replicates. (E, top left panel) Schematic view of expected gene expression levels in WT (straight lines) and Trim71 knockout (dashed lines) cells for Trim71 (black) and TRIM71 (red) targets over the course of differentiation. (All other panels) mRNA profiles during neuronal differentiation of WT and Trim71 knockout mESCs measured by RT-qPCR. Cell culture medium conditions for the five differentiation steps (I–V) are indicated. Trim71, Mbnl1, Plxnb2, Mllt1, and Pou5f1 levels were tested. Values were normalized to Eif5 and are shown relative to expression levels in WT mESCs. Data represent mean ± SEM. n = 2 biological replicates. (F) Luciferase reporter gene assay with Renilla::Mbnl1, Plxnb2, and Mllt1 3′ UTR constructs in Trim71 knockout mESCs overexpressing HsTRIM71 (positive control), mCherry (negative control), HsTRIM71 R751A, R608H, or R796H. Data were normalized to HsTRIM71-overexpressing cells and represent mean ± SEM. n = 3 biological replicates. Data source for A–C: Supplemental Table S5. Significance in D and F was tested as in Figure 3C.
We focused on Mbnl1 (a splicing regulator), Mllt1 (a histone acetylation reader), and Plxnb2 (an Angiogenin cell surface receptor), as these are developmental regulators (Chen et al. 2013; Han et al. 2013; Maethner et al. 2013). CRAC-seq peaks overlapped with predicted hairpins in their 3′ UTRs (Supplemental Fig. S2B), indicating the validity of our hairpin model for individual targets. Consistent with being direct TRIM71 targets, luciferase reporter gene assays with 3′ UTRs of Mbnl1, Plxnb2, and Mllt1 caused an approximately twofold TRIM71-dependent repression of luciferase activity in mESCs. Silencing depended on a functional NHL but not the RING domain (Fig. 4D). We used the 3′ UTR of Pou5f1 as a negative control, as this did not bind TRIM71 or change in abundance in Trim71 knockout cells, in agreement with a previous study (Mitschka et al. 2015).
We hypothesized that Mbnl1, Mllt1, and Plxnb2 are regulated by TRIM71 during development. To test this, we conducted neuronal differentiation assays with WT and Trim71 knockout mESCs and analyzed gene expression by RT-qPCR. As reported previously (Chen et al. 2012; Mitschka et al. 2015), Trim71 mRNA levels decreased throughout differentiation. In WT cells, mRNA levels of Mbnl1, Plxnb2, and Mllt1 increased as cells differentiated, and Trim71 levels decreased. Conversely, in Trim71 knockout cells, Mbnl1, Plxnb2, and Mllt1 mRNA levels were already high in undifferentiated cells and showed at most moderate increases during differentiation. The expression of Pou5f1 mRNA was unaltered by loss of TRIM71 (Fig. 4E). In combination with the 3′ UTR reporter assays, this demonstrates that MBNL1, PLXNB2, and MLLT1 are bona fide TRIM71 targets in mESCs.
Patient-related mutations in the human TRIM71 NHL domain impair RNA silencing
The identification of TRIM71 targets enabled us to examine mutations in the TRIM71 NHL domain found in congenital hydrocephalus patients (Furey et al. 2018). These patients exhibit brain developmental defects resembling those in Trim71 knockout mice (Mitschka et al. 2015). Together with our finding that the TRIM71 NHL domain is critical for posttranscriptional gene regulation, this suggests that the RNA-binding activity of TRIM71 may be of particular importance for its developmental function. To test this further, we explored whether point mutations in the Homo sapiens (Hs)TRIM71 NHL domain, present heterozygously in three patients with congenital hydrocephalus (Furey et al. 2018), affect TRIM71 target mRNA repression. Supporting this, the mutated amino acids R608H and R796H are highly conserved (Supplemental Fig. S2C,D), located in the RNA-binding groove, and in contact with a bound RNA hairpin [based on the Danio rerio (Dr)TRIM71 NHL domain structure] (Supplemental Fig. S2E; Kumari et al. 2018). We performed luciferase reporter gene assays for the 3′ UTRs of Mbnl1, Plxnb2, and Mllt1 using Trim71 knockout mESCs complemented with transgenes expressing WT HsTRIM71, HsTRIM71_R608H, or HsTRIM71_ R796H. As controls, we expressed mCherry or the R751A NHL point mutant HsTRIM71, described above. Although all TRIM71 constructs were expressed to similar levels (Supplemental Fig. S2F), the three NHL point mutant proteins failed to provide effective reporter gene repression (Fig. 4F).
Collectively, our results demonstrate the importance of the TRIM71 NHL domain for target repression and physiological gene expression. Combined with the Trim71 knockout mouse phenotype, this suggests that derepression of TRIM71 targets might contribute to the disease phenotype of the hydrocephalus patients.
Repression of Mbnl1 by TRIM71 is mediated by a 3′ UTR hairpin
As our results indicated that Mbnl1, a known regulator of stem cell fates, was among the most strongly regulated TRIM71 targets, we analyzed it in greater detail. First, we performed RT-qPCR analysis of Mbnl1 in mESCs, Ne4C cells, and Huh-7 cells, confirming the RNA-seq results (Supplemental Fig. S3A). Second, we tested the Mbnl1 3′ UTR luciferase reporter construct in the three cellular backgrounds and observed derepression in Trim71 knockout cells but not for the Pou5f1 3′ UTR control (Fig. 5A). Third, we showed that re-expression of HsTRIM71 in Trim71 knockout mESCs rescued Mbnl1 repression in luciferase reporter assays (Fig. 5B) and at the level of endogenous mRNA and protein (Supplemental Fig. S3B,C).
Figure 5.

Repression of Mbnl1 by TRIM71 is mediated by a 3′ UTR hairpin. (A) Luciferase reporter gene assay with Renilla::Pou5f1 3′ UTR (negative control) and Renilla::Mbnl1 in WT and Trim71 knockout mESCs, Ne4C cells, and Huh-7 cells. Data were normalized to WT and represent mean ± SEM. n = 3 biological replicates. (B) Luciferase reporter gene assay with Renilla::Pou5f1 3′ UTR (negative control) and Renilla::Mbnl1 in WT mESCs expressing mCherry (WT + mCherry), Trim71 knockout cells expressing mCherry (KO + mCherry), and Trim71 knockout cells overexpressing HsTRIM71 (KO + HsTRIM71). Data were normalized to “WT + mCherry” levels and represent mean ± SEM. n = 3 biological replicates. (C) Integrative Genomics Viewer snapshot showing merged reads from four TRIM71 WT CRAC-seq experiments aligned to the Mbnl1 3′ UTR. Stretches 1 and 2 (boxed in gray) were chosen for deletion mutants. Predicted strong, medium, and weak/minimal hairpin motifs are shown as vertical lines and colored according to strength. (D) Luciferase reporter gene assay with Mbnl1 3′ UTR deletion mutants. Data were normalized to WT and represent mean ± SEM. n = 3 biological replicates. (E) Schematic view of stretch 1 and the four stretch 1 parts. Part 2 contains a predicted hairpin motif. All stretches were cloned into the Pou5f1 3′ UTR luciferase reporter gene construct. (F) Luciferase reporter gene assay with Mbnl1 3′ UTR insertion constructs. Data were normalized to WT and represent mean ± SEM. n = 3 biological replicates. Data for “Pou5f1 3′ UTR” are identical in D and F, as experiments were performed together. Significance in A, B, D, and F was tested as in Figure 3C.
To identify TRIM71-responsive elements in the Mbnl1 3′ UTR, we analyzed CRAC-seq reads. We identified two regions (stretch 1 [178 nt] and stretch 2 [351 nt]) with strong TRIM71 binding (Fig. 5C). Deletion of stretch 1 severely impaired reporter repression in luciferase assays, whereas deletion of stretch 2 did not alter repression (Fig. 5D), suggesting that a primary functional TRIM71-binding site is in stretch 1. To dissect this further, we cloned the entire stretch 1 or one of four overlapping subsections (“parts”) into the TRIM71-independent Pou5f1 3′ UTR (Fig. 5E) and compared repression efficacy. The complete stretch 1—or part 2 alone—showed repressive activity, while parts 1, 3, and 4 did not (Fig. 5F). Part 2 contains a consensus hairpin motif with sequence conservation from mice to humans (Supplemental Fig. S3D). Deleting this motif from part 2 led to complete abrogation of repression in the luciferase assay (Fig. 5F). We conclude that TRIM71 binding to this hairpin motif in the Mbnl1 3′ UTR results in repression.
To validate the luciferase reporter results in an endogenous context, we deleted stretch 1 [Mbnl13′UTR(mut)] in 3xFlag-AVI-Trim71 mESCs using CRISPR–Cas9. RNA immunoprecipitations (RIPs) revealed reduced binding of TRIM71 to Mbnl1 but not Plxnb2 and Mllt1 mRNAs in Mbnl13′UTR(mut) cells (Fig. 6A; Supplemental Fig. S3E). Furthermore, the same UTR deletion in Trim71 WT cells resulted in partial derepression of Mbnl1 mRNA and protein (Fig. 6B; Supplemental Fig. S4A,B). This confirms that MBNL1 is a direct target of TRIM71 via the hairpin motif in its 3′ UTR (Fig. 6C). Notably, elimination of the binding motif does not fully abrogate binding or repression. Hence, additional (less strongly bound) TRIM71-binding sites on Mbnl1 are likely to be identified in future experiments, consistent with our observation that TRIM71 preferentially silences targets with multiple hairpins (Fig. 3C,D).
Figure 6.

MBNL1 is a primary TRIM71 target. (A, top panel) Quantification of RNA immunoprecipitating with 3xFlag-AVI-TRIM71 by RT-qPCR. Data were normalized to WT mESC input levels and represent mean ± SEM. n = 3 technical replicates. (Bottom panel) Western blot of the RNA coimmunoprecipitation experiment. 3xFlag-AVI-tagged TRIM71 migrates more slowly than WT TRIM71, and its recognition by the TRIM71 antibody is impaired (asterisk). Second biological replicate of RNA coimmunoprecipitation (Supplemental Fig. S3E). (B) RT-qPCR for Mbnl1 in WT, Trim71 knockout, and Mbnl13′UTR(mut) mESCs. Data were normalized to WT and represent mean ± SEM. n = 4 biological replicates. Significance was tested as in Figure 3C. Data are an excerpt of Supplemental Figure S4B. (C) Schematic view of TRIM71-dependent Mbnl1 repression. Mbnl1 WT 3′ UTR is bound by TRIM71, leading to repression. In Trim71 knockout cells, Mbnl1 is not repressed. Mbnl1 lacking the hairpin region in the 3′ UTR is impaired for binding and repression by TRIM71.
TRIM71 modulates alternative splicing by repressing MBNL1
As frequently observed for RNA-binding proteins (Takada et al. 2009; Cho et al. 2012; Yu et al. 2014), changes in mRNA levels caused by TRIM71 on its targets were modest. Nonetheless, targets may be very dose-sensitive, with small changes in abundance having large effects. To test this for MBNL1, we focused on its function as a regulator of differentiation-specific alternative splicing that can influence reprogramming of induced pluripotent stem cells (iPSCs) (Han et al. 2013). We used paired-end RNA-seq to analyze splicing events in WT, Trim71 knockout, Mbnl3′UTR(mut), and Mbnl1 overexpression (Mbnl1O/Ex) cell lines, calculating percentage spliced in (PSI) values for all splicing events. We observed correlation of global splicing changes in Mbnl13′UTR(mut), Trim71 knockout, and Mbnl1O/Ex cell lines, consistent with them all having elevated MBNL1 levels (Fig. 7A,B; Supplemental Table S6). This indicates that splicing changes detected in Trim71 knockout cells can be explained by MBNL1 deregulation.
Figure 7.

TRIM71 modulates alternative splicing by repressing MBNL1. (A) Heat map of differences in PSI values in Trim71 knockout, Mbnl13′UTR(mut), and Mbnl1O/Ex mutants relative to WT cells. (B, top panel) Correlation of differences in PSIs of Trim71 knockout versus WT mESCs and differences in PSI of Mbnl13′UTR(mut) versus WT mESCs. Spearman's correlation coefficient ρ = 0.71. P < 2.2 × 10−16, one-sided binomial test between quadrants. A median squared error (between biological replicates for all conditions) of <40 was applied to filter out highly noisy events within the same condition, resulting in 3264 splice events. ΔPSIs of ≥10 or ≤−10 in Trim71 knockout versus WT and/or Mbnl13′UTR(mut) versus WT cells are colored in red and were used as input data for subsequent plots (965 splice events). (Bottom left panel) Correlation of differences in PSIs of Mbnl1O/Ex versus WT mESCs and differences in PSIs of Trim71 knockout versus WT mESCs. Spearman's correlation coefficient ρ = 0.67. P < 2.2 × 10−16, one-sided binomial test between quadrants. (Bottom right panel) Correlation of differences in PSIs of Mbnl1O/Ex versus WT mESCs and differences in PSIs of Mbnl13′UTR(mut) versus WT mESCs. Spearman's correlation coefficient ρ = 0.65. P < 2.2 × 10−16, one-sided binomial test between quadrants. Data source: Supplemental Table S6. (C) Validation of individual splice events by RT-qPCR. Data represent mean ± SEM. n = 3–4 biological replicates. Specificity of amplicons for individual splice isoforms was validated by sequencing. Significance of PSI change relative to WT mESCs was tested as in Figure 3C. Displayed Foxp1 data are an excerpt from Supplemental Fig. S4C.
We further confirmed the MBNL1-dependent PSI changes for a set of differential splicing events by RT-qPCR (Fig. 7C). We also investigated splicing of Foxp1. This was not identified in our splicing analysis due to insufficient sequence coverage, but a previous study showed that MBNL1 promotes accumulation of the adult isoform of FOXP1, leading to differentiation of stem cells (Gabut et al. 2011; Han et al. 2013). As for the other splicing events, PSI values for the Foxp1 embryonic isoform inversely correlated with MBNL1 levels in WT, Trim71 knockout, NHL_mut, RING_mut, Mbnl13′UTR(mut), and Mbnl1O/Ex mESCs (Fig. 7C; Supplemental S4A–C). Also, overexpression of HsTRIM71 in Trim71 knockout mESCs led to a reversal of Foxp1 from an adult to an embryonal splicing pattern (Supplemental Fig. S4D). Finally, while the FOXP1 embryonic isoform was not expressed in Huh-7 cells and so could not be quantified, TRIM71-dependent alternative splicing at Foxp1 was confirmed in Ne4C cells (Supplemental Fig. S4E). We thus conclude that TRIM71 globally shapes alternative splicing by repressing MBNL1.
Discussion
Although the functions of TRIM71 in controlling stem and progenitor cell fates are well documented, its mechanism of action has been poorly understood. This is because (1) it has been unclear to what extent the two TRIM71 molecular activities—RNA binding and protein ubiquitylation—contribute to its functions (Rybak et al. 2009; Chang et al. 2012; Lödige et al. 2013), (2) the magnitudes of TRIM71-dependent changes in target RNA abundance are often low (Lödige et al. 2013; Worringer et al. 2014; Mitschka et al. 2015), and (3) few direct targets have been extensively validated.
Our data show that in mESCs, RNA binding explains most effects on the transcriptome. We note that the transcriptome of RING_mut cells does not resemble that of WT or Trim71 knockout cells. Speculatively, this could suggest that lack of RING domain activity causes some neomorphic or dominant-negative TRIM71 activity. Although the RING domain does not explain the transcriptome changes in Trim71 knockout cells, we cannot exclude important cellular functions of TRIM71-mediated protein ubiquitylation, and indirect but compelling evidence suggests that TRIM71 promotes mouse neural progenitor maintenance through protein ubiquitylation (Chen et al. 2012). Nevertheless, several lines of evidence support the importance of the RNA silencing activity of TRIM71/LIN-41. First, in C. elegans LIN-41, the NHL domain is a hot spot for phenotypically detectable mutations (Slack et al. 2000), and various developmental phenotypes are explained through its target transcripts (Aeschimann et al. 2019). Second, mutations in the NHL domain of human TRIM71 are detected in patients with congenital hydrocephalus (Furey et al. 2018), the most prevalent developmental human brain defect (Tully and Dobyns 2014), which resembles the neurulation phenotype in Trim71 knockout mice (Mitschka et al. 2015). Third, we show that these patient-derived mutations impair mRNA silencing by TRIM71. Notably, the hydrocephalus phenotype is associated with heterozygous mutations in the NHL domain. As Trim71 full knockout leads to embryonic lethality in mice (Mitschka et al. 2015) and analysis of genetic variation in 60,076 humans has shown TRIM71 to be highly intolerant to loss of function or missense variation (Lek et al. 2016), homozygous mutations in the NHL domain likely lead to embryonic lethality. It will be of great interest to study the phenotypes of analogous NHL, but also RING domain, mutant mice.
By combining functional and interaction profiling, we found that TRIM71 predominantly silences transcripts in mESCs by inducing degradation, mostly via binding to 3′ UTRs. Interestingly, we did not observe evidence of translational repression (Lödige et al. 2013; Aeschimann et al. 2017), although we cannot rule out isolated instances. Transcript binding occurred through a trinucleotide loop hairpin within endogenous targets that gratifyingly matches the motif found to confer in vitro RNA-binding by the C. elegans, D. rerio, and H. sapiens LIN-41/TRIM71 NHL domains (Lödige et al. 2015; Kumari et al. 2018). Together with the extensive loss of specific CRAC-seq signal in Trim71 NHL_mut cells, we conclude that the RNA-binding activity and target specificity of full-length TRIM71 in cells are indeed predominantly conferred by the NHL domain.
The extent of target repression depends on the number of TRIM71 target sites. Sites appear to have mostly additive effects, which would seem to argue against cooperative binding or function. Instead, a greater number of binding sites might increase the probability of at least one (or two) TRIM71 being bound to a given transcript. Nonetheless, the fact that at a given level of experimentally determined binding, distributed binding across more sites is beneficial for silencing suggests that some level of cooperativity exists. Given the low to inexistent levels of silencing achieved with only a single site, it seems possible that efficient silencing depends on occupation of at least two binding sites, which may be more readily achieved by increasing the number rather than the quality of binding sites. Systematic variations of binding site quality, quantity, and distance and in particular in vitro assays might help to elucidate this in future experiments.
Despite the presence of a clear binding motif, many predicted hairpins did not show CRAC-seq binding or derepression in RNA-seq experiments. Hence, only some potential targets elicit TRIM71-dependent regulation. This may be due to their adopting alternative RNA structures in the cell compared with those predicted in silico and/or competing binding by other RNA-binding proteins. Although our ability to computationally predict TRIM71 targets is thus limited, our insights into transcript binding help to overcome the small effects observed upon experimental manipulation of TRIM71 levels. Many other RNA-binding proteins are similarly challenging to study for the same reason, and our approach thus highlights how combining functional and interaction studies can be of huge benefit. Importantly, the TRIM71-binding sites that we validated in mESCs also explain a large extent of TRIM71 target repression in other mouse and human cell lines, with approximately two thirds of the binding sites functionally conserved (Fig. 4A,B). Hence, we predict that our data will be useful for TRIM71 target identification in other biological contexts.
Here, we focused on identifying a set of “core” targets broadly regulated across cell types to reveal some fundamental rather than cell type-specific function of TRIM71. Two previously reported candidate TRIM71 targets—EGR1 (Worringer et al. 2014) and CDKN1A (Chang et al. 2012)—did not fulfill our criteria for “core” targets, as they were not up-regulated in all tested cell lines, suggesting either cell type-specific regulation or secondary effects. In contrast, PLXNB2 did pass our criteria and has been proposed previously as a TRIM71 target (Mitschka et al. 2015). PLXNB2 and two other targets that we validated—MLLT1/ENL and MBNL1—have important functions in development and disease: PLXNB2 is the receptor of the secreted ribonuclease Angiogenin, which mediates intracellular RNA processing (Yu et al. 2017), and MLLT1/ENL is a histone acetylation reader that regulates oncogenic transcriptional programs in acute myeloid leukemia (Wan et al. 2017). The three MBNL paralogs (MBNL1–3) are regulators of alternative splicing, mRNA stability and localization (Wang et al. 2012), and alternative polyadenylation (Batra et al. 2014), with MBNL1 and MBNL2 expressed in the adult muscle and brain, respectively, and MBNL3 expressed in embryonal tissues (Holt et al. 2009; Charizanis et al. 2012). MBNL1 and MBNL2 have been shown to promote diverse developmental transitions, such as ESC differentiation (Han et al. 2013), muscle and heart development (Lin et al. 2006; Kalsotra et al. 2008), erythroid and monocyte–macrophage differentiation (Cheng et al. 2014; Liu et al. 2018), and brain development (Charizanis et al. 2012). Sequestration of MBNL proteins on RNA expanded trinucleotide repeats leads to the neuromuscular disease myotonic dystrophy type 1 (DM1) by causing a splicing shift in MBNL targets (Lin et al. 2006).
The identification of a TRIM71-binding site enabled us to disrupt Mbnl1 regulation by TRIM71 without affecting other TRIM71 targets and thus specifically test the contribution of the TRIM71–Mbnl1 interaction to the overall effect of TRIM71. This revealed that not only does TRIM71 shape alternative splicing in ESCs but that this is largely attributable to its regulation of MBNL1. This analysis also emphasizes the importance of precise control of MBNL1 levels, as small expression changes lead to dose-dependent splicing defects. This dose dependence has been demonstrated in previous studies and was proposed to explain the phenotypic variability in DM1 (Nakamori et al. 2013; Wagner et al. 2016).
Given that both depletion of MBNL1 proteins and overexpression of TRIM71 enhance reprogramming of mouse embryonic fibroblasts to iPSCs (Han et al. 2013; Worringer et al. 2014) and that MBNL1 promotes differentiation-specific alternative splicing (Han et al. 2013), we propose that these events may be linked and that TRIM71 may at least in part promote stem cell fates through repression of MBNL1 and maintenance of embryonic alternative splicing patterns. As we observed substantial TRIM71 binding and repression for MBNL1 but not for its paralogs, which differ in function and expression patterns (Squillace et al. 2002; Holt et al. 2009; Charizanis et al. 2012), we hypothesize that posttranscriptional regulation of MBNL1 expression by TRIM71 might enable precise spatial and temporal control of alternative splicing to help regulate and shape development.
Materials and methods
Cell culture
mESCs (129xC57BL/6 Rosa26Cre-ERT2/BirA-V5 [cMB063]) (Ostapcuk et al. 2018) were cultured as described previously (Flemr and Bühler 2015).
Ne4C cells were cultured on poly-L-lysine-coated dishes in Ne4C medium (DMEM [Gibco, 11885084], 10% fetal bovine serum [FBS] [Sigma, F7524], 2 mM L-glutamine [Gibco, 25030024], 50 µg/mL penicillin, 80 µg/mL streptomycin, 1× MycoZap prophylactic [Lonza, VZA-2032]) at 37°C in 5% CO2.
Huh-7 cells (JCRB0403) (Nakabayashi et al. 1982) were cultured in Huh-7 medium (DMEM [Gibco, 31966021], 10% FBS, antibiotic supplements as in Ne4C cells).
Neuronal differentiation assay
mESCs were differentiated to embryoid bodies (EBs) and treated with retinoic acid (RA) as described previously (Bibel et al. 2007). The culture of neuroprogenitors as neurospheres from RA-treated EBs was performed as described previously (Iwamaru et al. 2013). For generation of neurons, neurospheres were dissociated and plated on laminin/poly-DL-ornithine-coated adherent plates as described previously (Bibel et al. 2007).
Genome editing
Genome editing of mESCs and Ne4C cells was performed as described previously (Flemr and Bühler 2015). Sequences of TALENs, single-stranded oligodeoxynucleotides (ssODNs; ordered as ultramers from Integrated DNA Technologies), and sgRNAs are in Supplemental Table S7A. Genome-edited clones were characterized by genotyping, sequencing, and Western blotting. Sequences of genotyping primers are in Supplemental Table S7B. A list of generated cell lines is in Supplemental Table S8.
As we failed to achieve clonal selection with Huh-7 cells, we integrated EGFP with a previously described variant of the C. elegans mab-10 3′ UTR containing five annotated hairpins (“mab-10 condensed 3′ UTR”) (Kumari et al. 2018) into the AAVS locus. Based on the resulting cell line (Huh-7 AAVS1EGFP::unc-54::mab-10 condensed), we generated TRIM71−/− cells by CRISPR genome editing and enriched those by FACS sorting for increased EGFP expression in comparison with the parental cell line (Supplemental Fig. S5A–C).
Descriptions for the generation of individual cell lines are in the Supplemental Material.
Plasmid generation
For SpCas9-2A-puromycin (named pC2P in our study), mCherry from SpCas9-2A-mCherry (Knuckles et al. 2017) was deleted and replaced with puromycin. sgRNAs were cloned into the BsaI restriction site of pC2P. TALENs and loxP recombination reporters were assembled as described previously (Flemr and Bühler 2015). pCAGGS overexpression constructs were generated by inserting HsTRIM71, Mbnl1, and mCherry into the BglII site of pCAGGS expression vectors containing a C-terminal T2A puromycin tag. HsTRIM71 point mutants were generated by cloning mutation-bearing gBlocks into BaeI/EcoRI (R753A and R796H) or Bpu10I/EcoRI (R608H) sites in HsTRIM71. Luciferase constructs were generated by cloning respective 3′ UTRs into the NotI site of psiCHECK-2 (Promega, C8021).
For generating pAAVS1 reporter constructs, sequences of unc-54 3′ UTR (pCM5.37; Addgene, 17253; gift from Geraldine Seydoux) and unc-54::mab-10 condensed 3′ UTR (pFA113) (Kumari et al. 2018) were cloned 3′ of EGFP in pcDNA3.1. EGFP 3′ UTR reporter constructs were then cloned into the EcoRI site of pAAVS1-Nst-CAG-DEST (Addgene, 80489) (Oceguera-Yanez et al. 2016).
Generated plasmids with insert sources and primer and gBlock sequences are in Supplemental Tables S9 and S10.
Luciferase reporter gene assays
Luciferase assays were performed as described (Mitschka et al. 2015), except 100 ng of plasmid DNA per well was transfected. For luciferase assays with Trim71 knockout cells overexpressing HsTRIM71 constructs, cells were transfected with overexpression plasmid followed by puromycin selection at 1 µg/mL for 24 h for 5 d. Expression levels of transfected cells were analyzed by Western blotting. Cells were then split and transfected with luciferase plasmids (see above).
RNA extraction, RT-qPCR, and RNA-seq
Total RNA was extracted using the Norgen single-cell RNA purification kit (Norgen, 51800) with on-column DNase digestion (Norgen, 25710). Four-hundred nanograms of total RNA was transcribed to cDNA using the reverse transcription system (Promega, A3500) and random primers. qPCRs were performed using the SYBR Green PCR master mix (Applied Biosystems, 4309155) with standard cycling conditions. Unless indicated otherwise, data were normalized to Gapdh expression levels.
For splice isoform RT-qPCRs, the specificity of PCR products was analyzed by sequencing of PCR products. PSI values were calculated as described previously (Han et al. 2013; Camacho Londoño and Philipp 2016) by the following formula:
qPCR primer sequences are in Supplemental Table S11.
Sequencing library preparation was performed using either ScriptSeq 2 (Epicentre, SSV21106) or TruSeq 2 (Illumina, RS-122-2001) kits.
Western blotting
Cells were lysed with lysis buffer (25 mM Tris-HCL at pH 7.5, 100 mM KCl, 0.5% [v/v] NP-40, 1 mM EDTA, EDTA-free cOmplete miniprotease inhibitor [Roche]). Forty micrograms of protein was loaded onto NuPAGE-Novex Bis-Tris 4%–12% gradient gels (Invitrogen), transferred semidry to PVDF membranes, blocked for 1 h with 5% milk in TBS plus 0.1% Tween 20 (TBST), and stained with primary antibodies. Antibodies, dilutions, and incubation conditions are in Supplemental Table S12.
RIPs
Cells were grown in 10-cm dishes, washed in PBS, and lysed in 500 µL of lysis buffer (see “Western Blotting”) with 0.25 U/mL RNaseOUT (Invitrogen, 10777019). Equal amounts of protein were incubated with 20 µL of Flag M2 magnetic beads (Sigma, M8823) for 3 h at 4°C. Beads were washed three times with 200 µL of washing buffer (50 mM Tris-HCl at pH 7.5, 150 mM KCl, 5 mM MgCl2, 0.05% NP-40, EDTA-free cOmplete miniprotease inhibitor [Roche, 11836153001], 0.25 U/mL RNaseOUT [Invitrogen, 10777019]) followed by RNA extraction and qPCR.
Ribo-seq
Ribo-seq was performed as published previously (McGlincy and Ingolia 2017), except RNase digestion was carried out for 30 min at 23°C using 4.8 U/µL RNase T1 (Thermo Fisher, EN0541) and 8 ng/µL RNase A (Thermo Fisher, EN0531). Monosome isolation, extraction of ribosome RNA footprints, and library preparation were performed as described in Aeschimann et al. (2015), except an rRNA contaminant was depleted by annealing the library with 10 pM of an oligonucleotide harboring a 3′ C3 spacer group (sequence GGCCTCGATCAGAAGGACTTGGGCCCCCC spacer C3; ordered from Integrated DNA Technologies) before the reverse transcription step.
CRAC-seq
CRAC-seq was performed as described in Tuck et al. (2018), except (1) 50 mM Tris-HCl (pH 7.8), 150 mM NaCl, and 0.5% (v/v) Nonidet P-40 substitute were used for cell lysis and binding to Flag beads, and (2) SDS-PAGE and nitrocellulose transfer steps were omitted. Instead, streptavidin beads were incubated directly with proteinase K to release RNA.
Bioinformatic analysis
A detailed description of bioinformatic analysis is in the Supplemental Material.
Data availability
Sequencing data have been deposited in the Gene Expression Omnibus under accession code GSE134125. A BED file containing genome-wide hairpin predictions is in the Supplemental Material.
Computer codes are available on request.
Acknowledgments
We thank Anca Neagu and Kathrin Kunzer for technical support. We are grateful to Jörg Betschinger and Sebastien Smallwood for input on optimizing experiments. We thank Kirsten Jacobeit, Sirisha Aluri, Eliza Pandini Figueiredo Moreno, and Stéphane Thiry for help with library preparation and high-throughput sequencing, and Hubertus Kohler for FACS sorting. We are thankful to Dimos Gaidatzis for sharing computer codes and discussing TRIM71 motif analysis. We thank Witold Filipowicz and Florian Aeschimann for feedback on the manuscript. This project has received funding from the Swiss National Science Foundation through the National Center of Competence in Research RNA and Disease (to M.B. and H.G.), the Wellcome Trust (WT103977 to A.C.T.), and the Novartis Research Foundation through Friedrich Miescher Institute for Biomedical Research core funding.
Author contributions: T.W. conceived the project; designed, performed, and computationally analyzed Ribo-seq, RNA-seq, CRAC-seq, and RNA splicing experiments; performed luciferase reporter gene assays, RT-qPCR, Western blot experiments, RIPs, and differentiation assays; created genome-edited cell lines; and wrote the manuscript. A.C.T. designed and performed CRAC-seq, computationally analyzed CRAC-seq and RNA-seq experiments, and wrote the manuscript. P.P. computationally analyzed RNA-seq and CRAC-seq experiments and RNA splicing. S.H.C. computationally analyzed Ribo-seq, RNA-seq, and CRAC-seq experiments. M.F. created genome-edited cell lines and plasmids. P.K. performed differentiation assays and generated plasmids. A.R. performed CRAC-seq experiments. M.B. conceived the project and critically edited the manuscript. H.G. conceived the project, designed and analyzed experiments, and wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.328492.119.
References
- Aeschimann F, Xiong J, Arnold A, Dieterich C, Großhans H. 2015. Transcriptome-wide measurement of ribosomal occupancy by ribosome profiling. Methods 85: 75–89. 10.1016/j.ymeth.2015.06.013 [DOI] [PubMed] [Google Scholar]
- Aeschimann F, Kumari P, Bartake H, Gaidatzis D, Xu L, Ciosk R, Großhans H. 2017. LIN41 post-transcriptionally silences mRNAs by two distinct and position-dependent mechanisms. Mol Cell 65: 476–489.e4. 10.1016/j.molcel.2016.12.010 [DOI] [PubMed] [Google Scholar]
- Aeschimann F, Neagu A, Rausch M, Großhans H. 2019. let-7 coordinates the transition to adulthood through a single primary and four secondary targets. Life Sci Alliance 2: e201900335. 10.26508/lsa.201900335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, et al. 2014. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell 56: 311–322. 10.1016/j.molcel.2014.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Richter J, Lacroix E, Barde YA. 2007. Generation of a defined and uniform population of CNS progenitors and neurons from mouse embryonic stem cells. Nat Protoc 2: 1034–1043. 10.1038/nprot.2007.147 [DOI] [PubMed] [Google Scholar]
- Camacho Londoño J, Philipp SE. 2016. A reliable method for quantification of splice variants using RT-qPCR. BMC Mol Biol 17: 8. 10.1186/s12867-016-0060-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HM, Martinez NJ, Thornton JE, Hagan JP, Nguyen KD, Gregory RI. 2012. Trim71 cooperates with microRNAs to repress Cdkn1a expression and promote embryonic stem cell proliferation. Nat Commun 3: 923. 10.1038/ncomms1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. 2012. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75: 437–450. 10.1016/j.neuron.2012.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lai F, Niswander L. 2012. The ubiquitin ligase mLin41 temporally promotes neural progenitor cell maintenance through FGF signaling. Genes Dev 26: 803–815. 10.1101/gad.187641.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Yuan RH, Yang WC, Hsu HC, Jeng YM. 2013. The stem cell E3-ligase Lin-41 promotes liver cancer progression through inhibition of microRNA-mediated gene silencing. J Pathol 229: 486–496. 10.1002/path.4130 [DOI] [PubMed] [Google Scholar]
- Cheng AW, Shi J, Wong P, Luo KL, Trepman P, Wang ET, Choi H, Burge CB, Lodish HF. 2014. Muscleblind-like 1 (Mbnl1) regulates pre-mRNA alternative splicing during terminal erythropoiesis. Blood 124: 598–610. 10.1182/blood-2013-12-542209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Chang H, Kwon SC, Kim B, Kim Y, Choe J, Ha M, Kim YK, Kim VN. 2012. LIN28A is a suppressor of ER-associated translation in embryonic stem cells. Cell 151: 765–777. 10.1016/j.cell.2012.10.019 [DOI] [PubMed] [Google Scholar]
- De Cecco L, Negri T, Brich S, Mauro V, Bozzi F, Dagrada G, Disciglio V, Sanfilippo R, Gronchi A, D'Incalci M, et al. 2014. Identification of a gene expression driven progression pathway in myxoid liposarcoma. Oncotarget 5: 5965–5977. 10.18632/oncotarget.2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio-Albrechtsen T, Kiontke K, Chiou SY, Fitch DH. 2006. Novel gain-of-function alleles demonstrate a role for the heterochronic gene lin-41 in C. elegans male tail tip morphogenesis. Dev Biol 297: 74–86. 10.1016/j.ydbio.2006.04.472 [DOI] [PubMed] [Google Scholar]
- Ecsedi M, Großhans H. 2013. LIN-41/TRIM71: emancipation of a miRNA target. Genes Dev 27: 581–589. 10.1101/gad.207266.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi M, Rausch M, Großhans H. 2015. The let-7 microRNA directs vulval development through a single target. Dev Cell 32: 335–344. 10.1016/j.devcel.2014.12.018 [DOI] [PubMed] [Google Scholar]
- Flemr M, Bühler M. 2015. Single-step generation of conditional knockout mouse embryonic stem cells. Cell Rep 12: 709–716. 10.1016/j.celrep.2015.06.051 [DOI] [PubMed] [Google Scholar]
- Furey CG, Choi J, Jin SC, Zeng X, Timberlake AT, Nelson-Williams C, Mansuri MS, Lu Q, Duran D, Panchagnula S, et al. 2018. De novo mutation in genes regulating neural stem cell fate in human congenital hydrocephalus. Neuron 99: 302–314.e4. 10.1016/j.neuron.2018.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabut M, Samavarchi-Tehrani P, Wang X, Slobodeniuc V, O'Hanlon D, Sung HK, Alvarez M, Talukder S, Pan Q, Mazzoni EO, et al. 2011. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 147: 132–146. 10.1016/j.cell.2011.08.023 [DOI] [PubMed] [Google Scholar]
- Granneman S, Kudla G, Petfalski E, Tollervey D. 2009. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc Natl Acad Sci 106: 9613–9618. 10.1073/pnas.0901997106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Irimia M, Ross PJ, Sung HK, Alipanahi B, David L, Golipour A, Gabut M, Michael IP, Nachman EN, et al. 2013. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature 498: 241–245. 10.1038/nature12270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt I, Jacquemin V, Fardaei M, Sewry CA, Butler-Browne GS, Furling D, Brook JD, Morris GE. 2009. Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle. Am J Pathol 174: 216–227. 10.2353/ajpath.2009.080520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamaru Y, Takenouchi T, Imamura M, Shimizu Y, Miyazawa K, Mohri S, Yokoyama T, Kitani H. 2013. Prion replication elicits cytopathic changes in differentiated neurosphere cultures. J Virol 87: 8745–8755. 10.1128/JVI.00572-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. 2008. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci 105: 20333–20338. 10.1073/pnas.0809045105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckles P, Carl SH, Musheev M, Niehrs C, Wenger A, Bühler M. 2017. RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nat Struct Mol Biol 24: 561–569. 10.1038/nsmb.3419 [DOI] [PubMed] [Google Scholar]
- Kumari P, Aeschimann F, Gaidatzis D, Keusch JJ, Ghosh P, Neagu A, Pachulska-Wieczorek K, Bujnicki JM, Gut H, Großhans H, et al. 2018. Evolutionary plasticity of the NHL domain underlies distinct solutions to RNA recognition. Nat Commun 9: 1549. 10.1038/s41467-018-03920-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancman JJ, Caruccio NC, Harfe BD, Pasquinelli AE, Schageman JJ, Pertsemlidis A, Fallon JF. 2005. Analysis of the regulation of lin-41 during chick and mouse limb development. Dev Dyn 234: 948–960. 10.1002/dvdy.20591 [DOI] [PubMed] [Google Scholar]
- Lee SH, Cho S, Kim MS, Choi K, Cho JY, Gwak HS, Kim YJ, Yoo H, Lee SH, Park JB, et al. 2014. The ubiquitin ligase human TRIM71 regulates let-7 microRNA biogenesis via modulation of Lin28B protein. Biochim Biophys Acta 1839: 374–386. 10.1016/j.bbagrm.2014.02.017 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. 2006. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 15: 2087–2097. 10.1093/hmg/ddl132 [DOI] [PubMed] [Google Scholar]
- Liu H, Lorenzini PA, Zhang F, Xu S, Wong MSM, Zheng J, Roca X. 2018. Alternative splicing analysis in human monocytes and macrophages reveals MBNL1 as major regulator. Nucleic Acids Res 46: 6069–6086. 10.1093/nar/gky401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lödige I, Gaidatzis D, Sack R, Meister G, Filipowicz W. 2013. The mammalian TRIM-NHL protein TRIM71/LIN-41 is a repressor of mRNA function. Nucleic Acids Res 41: 518–532. 10.1093/nar/gks1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lödige I, Jakob L, Treiber T, Ray D, Stotz M, Treiber N, Hennig J, Cook KB, Morris Q, Hughes TR, et al. 2015. The crystal structure of the NHL domain in complex with RNA reveals the molecular basis of Drosophila brain-tumor-mediated gene regulation. Cell Rep 13: 1206–1220. 10.1016/j.celrep.2015.09.068 [DOI] [PubMed] [Google Scholar]
- Löer B, Bauer R, Bornheim R, Grell J, Kremmer E, Kolanus W, Hoch M. 2008. The NHL-domain protein Wech is crucial for the integrin-cytoskeleton link. Nat Cell Biol 10: 422–428. 10.1038/ncb1704 [DOI] [PubMed] [Google Scholar]
- Maethner E, Garcia-Cuellar MP, Breitinger C, Takacova S, Divoky V, Hess JL, Slany RK. 2013. MLL-ENL inhibits polycomb repressive complex 1 to achieve efficient transformation of hematopoietic cells. Cell Rep 3: 1553–1566. 10.1016/j.celrep.2013.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maller Schulman BR, Liang X, Stahlhut C, DelConte C, Stefani G, Slack FJ. 2008. The let-7 microRNA target gene, Mlin41/Trim71 is required for mouse embryonic survival and neural tube closure. Cell Cycle 7: 3935–3942. 10.4161/cc.7.24.7397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura R, Ashikawa T, Nozaki Y, Kitagawa D. 2016. LIN-41 inactivation leads to delayed centrosome elimination and abnormal chromosome behavior during female meiosis in Caenorhabditis elegans. Mol Biol Cell 27: 799–811. 10.1091/mbc.E15-10-0713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlincy NJ, Ingolia NT. 2017. Transcriptome-wide measurement of translation by ribosome profiling. Methods 126: 112–129. 10.1016/j.ymeth.2017.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschka S, Ulas T, Goller T, Schneider K, Egert A, Mertens J, Brüstle O, Schorle H, Beyer M, Klee K, et al. 2015. Co-existence of intact stemness and priming of neural differentiation programs in mES cells lacking Trim71. Sci Rep 5: 11126. 10.1038/srep11126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. 1982. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42: 3858–3863. [PubMed] [Google Scholar]
- Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, et al. 2013. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 74: 862–872. 10.1002/ana.23992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DTT, Richter D, Michel G, Mitschka S, Kolanus W, Cuevas E, Wulczyn FG. 2017. The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differentiation pathways during stem cell differentiation. Cell Death Differ 24: 1063–1078. 10.1038/cdd.2017.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oceguera-Yanez F, Kim SI, Matsumoto T, Tan GW, Xiang L, Hatani T, Kondo T, Ikeya M, Yoshida Y, Inoue H, et al. 2016. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods 101: 43–55. 10.1016/j.ymeth.2015.12.012 [DOI] [PubMed] [Google Scholar]
- Ostapcuk V, Mohn F, Carl SH, Basters A, Hess D, Iesmantavicius V, Lampersberger L, Flemr M, Pandey A, Thoma NH, et al. 2018. Activity-dependent neuroprotective protein recruits HP1 and CHD4 to control lineage-specifying genes. Nature 557: 739–743. 10.1038/s41586-018-0153-8 [DOI] [PubMed] [Google Scholar]
- Pereira L, Aeschimann F, Wang C, Lawson H, Serrano-Saiz E, Portman DS, Großhans H, Hobert O. 2019. Timing mechanism of sexually dimorphic nervous system differentiation. Elife 8: e42078. 10.7554/eLife.42078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Xu Y, Wang Q, Jiang J, Wudumuli HL, Zhang Q, Zhang X, Wang E, Sun L, et al. 2018. E3 ubiquitin ligase tripartite motif-containing 71 promotes the proliferation of non-small cell lung cancer through the inhibitor of κB-α/nuclear factor κB pathway. Oncotarget 9: 10880–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak A, Fuchs H, Hadian K, Smirnova L, Wulczyn EA, Michel G, Nitsch R, Krappmann D, Wulczyn FG. 2009. The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat Cell Biol 11: 1411–1420. 10.1038/ncb1987 [DOI] [PubMed] [Google Scholar]
- Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. 2000. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell 5: 659–669. 10.1016/S1097-2765(00)80245-2 [DOI] [PubMed] [Google Scholar]
- Spike CA, Coetzee D, Eichten C, Wang X, Hansen D, Greenstein D. 2014. The TRIM-NHL protein LIN-41 and the OMA RNA-binding proteins antagonistically control the prophase-to-metaphase transition and growth of Caenorhabditis elegans oocytes. Genetics 198: 1535–1558. 10.1534/genetics.114.168831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squillace RM, Chenault DM, Wang EH. 2002. Inhibition of muscle differentiation by the novel Muscleblind-related protein CHCR. Dev Biol 250: 218–230. 10.1006/dbio.2002.0798 [DOI] [PubMed] [Google Scholar]
- Takada H, Kawana T, Ito Y, Kikuno RF, Mamada H, Araki T, Koga H, Asashima M, Taira M. 2009. The RNA-binding protein Mex3b has a fine-tuning system for mRNA regulation in early Xenopus development. Development 136: 2413–2422. 10.1242/dev.029165 [DOI] [PubMed] [Google Scholar]
- Tocchini C, Keusch JJ, Miller SB, Finger S, Gut H, Stadler MB, Ciosk R. 2014. The TRIM-NHL protein LIN-41 controls the onset of developmental plasticity in Caenorhabditis elegans. PLoS Genet 10: e1004533. 10.1371/journal.pgen.1004533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck AC, Natarajan KN, Rice GM, Borawski J, Mohn F, Rankova A, Flemr M, Wenger A, Nutiu R, Teichmann S, et al. 2018. Distinctive features of lincRNA gene expression suggest widespread RNA-independent functions. Life Sci Alliance 1: e201800124. 10.26508/lsa.201800124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully HM, Dobyns WB. 2014. Infantile hydrocephalus: a review of epidemiology, classification and causes. Eur J Med Genet 57: 359–368. 10.1016/j.ejmg.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SD, Struck AJ, Gupta R, Farnsworth DR, Mahady AE, Eichinger K, Thornton CA, Wang ET, Berglund JA. 2016. Dose-dependent regulation of alternative splicing by MBNL proteins reveals biomarkers for myotonic dystrophy. PLoS Genet 12: e1006316. 10.1371/journal.pgen.1006316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YE, Erb MA, et al. 2017. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543: 265–269. 10.1038/nature21687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. 2012. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150: 710–724. 10.1016/j.cell.2012.06.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worringer KA, Rand TA, Hayashi Y, Sami S, Takahashi K, Tanabe K, Narita M, Srivastava D, Yamanaka S. 2014. The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 14: 40–52. 10.1016/j.stem.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Jin L, Yang G, Ji L, Wang F, Lu Z. 2014. Post-transcriptional repression of FOXO1 by QKI results in low levels of FOXO1 expression in breast cancer cells. Oncol Rep 31: 1459–1465. 10.3892/or.2013.2957 [DOI] [PubMed] [Google Scholar]
- Yu W, Goncalves KA, Li S, Kishikawa H, Sun G, Yang H, Vanli N, Wu Y, Jiang Y, Hu MG, et al. 2017. Plexin-B2 mediates physiologic and pathologic functions of angiogenin. Cell 171: 849–864.e25. 10.1016/j.cell.2017.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequencing data have been deposited in the Gene Expression Omnibus under accession code GSE134125. A BED file containing genome-wide hairpin predictions is in the Supplemental Material.
Computer codes are available on request.