

SUMMARY
KRAS is one of the driver oncogenes in non-small-cell lung cancer (NSCLC) but remains refractory to current modalities of targeted pathway inhibition, which include inhibiting downstream kinase MEK to circumvent KRAS activation. Here, we show that pulsatile, rather than continuous, treatment with MEK inhibitors (MEKis) maintains T cell activation and enables their proliferation. Two MEKis, selumetinib and trametinib, induce T cell activation with increased CTLA-4 expression and, to a lesser extent, PD-1 expression on T cells in vivo after cyclical pulsatile MEKi treatment. In addition, the pulsatile dosing schedule alone shows superior anti-tumor effects and delays the emergence of drug resistance. Furthermore, pulsatile MEKi treatment combined with CTLA-4 blockade prolongs survival in mice bearing tumors with mutant Kras. Our results set the foundation and show the importance of a combinatorial therapeutic strategy using pulsatile targeted therapy together with immunotherapy to optimally enhance tumor delay and promote long-term anti-tumor immunity.
Graphical Abstract
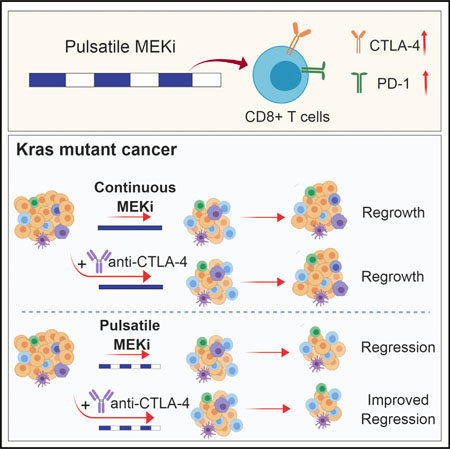
In Brief
KRAS mutant non-small-cell lung cancer (NSCLC) remains refractory to targeted therapeutics. Choi et al. show that pulsatile, rather than continuous, treatment with MEK inhibitors can maintain T cell activity better and prolong survival in mice with Kras mutant cancer. This effect is further enhanced when combined with CTLA-4 blockade.
INTRODUCTION
The RAS-MEK-ERK signaling pathway is hyper-activated in a variety of different cancers, including non-small-cell lung cancer (NSCLC) (Fernández-Medarde and Santos, 2011). Activating mutations of KRAS are common oncogenic drivers, responsible for 20%–30% of lung adenocarcinoma patients (Lovly and Carbone, 2011). However, currently, there are no approved targeted therapies specifically for NSCLC patients with a KRAS mutation. Targeted MEK inhibitors (MEKis), which act downstream of the RAS signaling pathway, are designed to block the hyperactive signaling cascade in KRAS mutant lung cancer patients (Ostrem et al., 2013; Zeng et al., 2017) and block the proliferation and survival program in cancer cells (Riely et al., 2009).
MEKis are in diverse phases of clinical development, including trametinib, which has been approved in combination with the BRAF inhibitor dabrafenib for the treatment of a subset of NSCLC with BRAFV600E mutation (Friday and Adjei, 2008; Greystoke et al., 2017; Planchard et al., 2017; Stinchcombe and Johnson, 2014). However, despite promising co-clinical studies in mouse models and clinical trials (Chen et al., 2012; Greystoke et al., 2017; Planchard et al., 2017), resistance to MEKis is often observed (Soria et al., 2017). This resistance has been attributed to the heterogeneity of the tumor (Jamal-Hanjani et al., 2017; Swanton and Govindan, 2016) and to intrinsic and acquired resistance from both cancer cells and the tumor microenvironment (Ebert et al., 2016; Manchado et al., 2016). Therefore, there is a need to further improve the efficacy of MEKis in KRAS-driven lung cancer. A theoretically promising therapeutic approach would entail simultaneously blocking KRAS signaling and activating tumor-infiltrating T cells, the latter being relevant given the recent demonstration of activity of immune checkpoint blockade of the CTLA-4 and PD-1 pathways in NSCLC and other malignancies (Borghaei et al., 2015; Brahmer et al., 2015; Garon et al., 2015; Reck et al., 2016; Wolchok et al., 2013). Despite the success of immune-based therapies, the need remains for better treatment strategies for the majority of patients with advanced NSCLC, since the response to current single-agent PD-1 pathway blockade is durable only in a subset of patients (Borghaei et al., 2015; Brahmer et al., 2015), and initial results in combination with CTLA-4 blockade showed promising efficacy for the treatment of NSCLC only in a subset of patients (Hellmann et al., 2017).
Based on the limitations of both immune-based therapies and targeted therapies, we sought to rationally combine these two modalities to treat KRAS mutant lung cancers. In addition to the essential role of MEKis in RAS-MEK-ERK signaling suppression during the tumorigenesis of NSCLC, the effect of MEKis on immune cells is complex and context dependent. The RAS-MEKERK signaling cascade is critical in the normal physiologic function of immune cells, especially T cells (Weiss and Littman, 1994). The sequential signaling of RAS-MEK-ERK after T cell receptor (TCR) activation is responsible for the activity of NFAT and the production of interleukin (IL)-2, which are critical for T cell clonal expansion (Kane et al., 2000; Weiss and Littman, 1994). Previous studies have shown that inhibition of MEK signaling by small molecules reduces or regulates paradoxically the proliferation of T cells in vitro (Callahan et al., 2014; Liu et al., 2015a). Nevertheless, it enhances the proliferation of tumor-infiltrating CD8+ T cells in CT26 Kras mutant colorectal cancer, resulting in the expansion of tumor-reactive T cell populations with cytotoxic activity (Ebert et al., 2016; Liu et al., 2015a). However, conventional continuous administration of MEKis achieves an inadequate inhibition of ERK activity, which induces feedback regulation of other proliferation and survival pathways and re-activates MEK-ERK signaling, leading to drug resistance (Samatar and Poulikakos, 2014; Sun et al., 2014). Moreover, prolonged blockade of TCR signaling by MEKis interferes with effector function and proliferation at the tumor site (Dushyanthen et al., 2017). The recent failure of a clinical trial with continuous MEKi (cobimetinib) and anti-PD-L1 (atozolizumab) combination treatment in colorectal cancer (phase III IMblaze370 study, NCT02788279) suggests that the scheduling of these drugs needs optimization.
Unconventional pulsatile treatment schedules using targeted drugs such as the BRAF inhibitor vemurafenib and the EGFR inhibitor gefitinib showed better suppression of tumor growth in melanoma, breast cancer, and leukemia (Das Thakur et al., 2013; Shah et al., 2008; Solit et al., 2005). In support of this pulsatile treatment regimen in the case of MEKis, transient pretreatment or lead-in treatment with MEKis in combination with anti-CTLA-4 or anti-PD-1 showed better survival and lower tumor burden in the CT26 mouse tumor model (Poon et al., 2017). Despite these encouraging observations, the status of T cell activation using these unconventional regimens has not been explored systematically. We hypothesize that through an optimized MEKi dosing schedule, we can maximize the suppression of KRAS-induced proliferation and survival of cancer cells while minimizing the detrimental effects on immune cells.
In this study, we have investigated how a pulsatile dosing schedule of MEKis affects T cell activation in mutant KRAS-driven lung cancer models both ex vivo and in vivo. We observed that pulsatile treatment induced improved proliferation and activation of T cells with higher expression levels of immune checkpoint regulators, including CTLA-4 and PD-1, when compared with the conventional continuous treatment with the same drug. This optimized schedule of pulsatile treatment resulted in delayed tumor growth in KRAS mutant genetically engineered mouse models (GEMMs). Furthermore, the combination of pulsatile MEKis with CTLA-4 blockade resulted in prolonged survival of mice with KRAS mutant lung cancer compared to continuous treatment with MEKis in combination with anti-CTLA-4.
RESULTS
MEK Inhibition Affects Tumor Growth in Kras Mutant Lung Cancer and MAPK Signaling in Both Tumor Cells and T Cells
We have treated various Kras mutant murine lung cancer cell lines (CL13, CL25, IO33, HKP1, and LLC) with selumetinib or trametinib to examine whether they are sensitive to clinically relevant MEKis. The HKP1 cell line was derived from a KrasG12D/+ Trp53−/− mouse (Choi et al., 2015). We characterized the mutations in the IO33, CL13, CL25 and LLC cell lines and identified G12V, Q61R, Q61H, and G12C Kras mutations, respectively (Figure S1A). We observed that MEKis block phosphorylation of ERK in Kras mutant cells effectively (Figure 1A) and that tumor cells show reduced viability after treatment (Figure 1B). Furthermore, selumetinib extends the survival of HKP1 lung tumorbearing mice, suggesting cytotoxic activity on Kras mutant lung cancer in vivo (Figure 1C). When we characterized T cells, we found decreased viability and reduced pERK in CD8+ T cells and CD4+ T cells (Figures 1D and S1B). We also observed increased interferon (IFN)γ expression in lung-infiltrating CD8+ T cells, but not in CD4+ T cells after in vivo selumetinib treatment (Figure S1C), which suggests differential regulation by MEKis in distinct immune cell populations. Taken together, these data confirm that MEKis can dampen signaling in the ERK pathway in both tumor cells and T cells and that combining MEKis with immune modulation should be done carefully with respect to timing.
Figure 1. MEK Inhibition Affects Murine Kras Mutant Tumor Growth and Murine T Cell Signaling.

(A) pERK expression in various Kras mutant lung cancer cell lines after trametinib treatment by western blot.
(B) Viability of lung tumor cell lines after selumetinib treatment. Samples were biological replicates.
(C) Survival of HKP1 lung-cancer-bearing mice after 3 weeks selumetinib treatment.
(D) pERK expression in CD4+ and CD8+ T cells from HKP1 tumor-bearing lungs after selumetinib or trametinib treatment by flow cytometry.
Samples were biological replicates. *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001, Welch’s t test. NS, not significant. Error bars represent SD. The experiments were performed 2–3 times, and representative results are shown here.
Short (Pulsatile), but Not Long (Continuous), Treatment with MEKis Activates T Cells Ex Vivo
To investigate how short treatment with MEKis affect T cell activation, we treated splenocytes from HKP1 lung tumor-bearing mice in vitro with selumetinib or trametinib in a long or short schedule while activating T cells using anti-CD3 and anti-CD28 (Figure 2A; Figure S2). Treatment with selumetinib or trametinib reduced expression of Ki-67, 4–1BB, CTLA-4, and PD-1 from T cells in a dose-dependent manner (Figure 2B). However, short schedule treatment maintained them (Ki-67, 4–1BB, CTLA-4, and PD-1) better in CD8+ T cells and CD4+Foxp3– effector T cells, compared to long drug exposure (Figures 2B and S2). This suggests that short treatment with MEKis maintain T cells in a more activated state than continuous treatment.
Figure 2. Short Schedule of MEKi Treatment Alters T Cell Activation Status Ex Vivo.

(A) Schema of ex vivo short versus long treatment experiment.
(B) CTLA-4, PD1, Ki-67, and 4–1BB expression in CD8+ T cells and CD4+Foxp3 cells by flow cytometry after selumetinib (left) or trametinib (right) treatment for 96 hr. *p < 0.05; **p < 0.01, Welch’s test.
Error bars represent SD. Samples were biological replicates. The experiment was performed twice, and representative results are shown here.
Short Treatment with MEKis Increases Effector T Cell Priming
Since it has been shown previously that a MEKi (cobimetinib) reduces priming of T cells in lymph nodes (Ebert et al., 2016), we tested how short treatment with MEKis affects priming of antigen-specific (Pmel-1) CD8+ T cells. Splenocytes from Pmel-1 TCR transgenic mice were pulsed with gp100 (Pmel) peptide and treated with selumetinib or trametinib (Figure 3A). We found that MEKi treatment reduced differentiation of Pmel-1 CD8+ T cells by decreasing T-bet expression in a dose-dependent manner (Figure S3A), as was previously reported (Ebert et al., 2016). However, we have not observed differences in T-bet expression between the two treatment groups (Figure S3A). Selumetinib and trametinib treatment resulted in increased CD44+CD62L effector memory CD8 T cells, as well as reduced CD44+CD62L+ central memory CD8 T cells, compared to untreated samples (Figures 3B and 3C). However, short treatment with MEKis showed more CD44+ cells, specifically CD44+CD62L CD8 T cells, compared to long treatment (Figures 3B–3D). Interestingly, CD44+CD62L+ CD8 T cells and naive cells showed decreased proliferation after selumetinib treatment in both groups, while CD44+CD62L CD8 T cells showed a more proliferative phenotype compared to other subsets, regardless of treatment conditions (Figure S3B). Further-more, analysis of the supernatant after priming showed that the short-treatment group had a higher production of IFNγ, suggesting better cytolytic capability (Figure 3E).
Figure 3. Short Treatment of MEKis Alters T Cell Priming Ex Vivo.

(A) Schema of short treatment on Pmel-1 CD8+ T cells with human gp100 peptide pulse.
(B) Flow cytometry plots of CD44 and CD62L markers on CD8+ T cells after 5 days of priming.
(C) Frequency of CD44 CD62L subsets from CD8+ T cells by flow cytometry analysis. Average percentage of each subset is presented.
(D) Frequency of CD44+ CD62L cell population by flow cytometry.
(E) IFNγ production from supernatant at day 5 by cytokine profiling.
*p < 0.05; **p < 0.01; ***p < 0.001, Welch’s test. Error bars represent SD. Samples were biological replicates. The experiment was performed twice, and representative results are shown here.
Pulsatile Treatment with MEKis Increases CTLA-4 and PD-1 Expression in Tumor-Infiltrating T Cells compared to Continuous Treatment
Next, we investigated whether cyclical pulsatile treatment with MEKis shows differential activation of different subtypes of T cells in vivo. We treated HKP1 lung tumor-bearing mice with selumetinib (Figure 4A). After 2 weeks of treatment, lung tumors were analyzed for the activation phenotype of tumor-infiltrating T cells. Tumors were highly infiltrated by CD8+ T cells in the pulsatile treatment groups (Figures 4B and S4C). More importantly, tumor cells and CD8+ T cells showed differential proliferation (Ki-67+) with the pulsatile treatment. While proliferation of tumor cells was reduced, CD8+ T cells showed increased or stable proliferation (Figure 4C). These observations suggest that individual T cell populations respond to MEKi treatment differently. Furthermore, pulsatile treatment with selumetinib results in the maintenance of a higher level of pERK in both CD8+ and CD4+ T cells, compared to continuous treatment (Figure S4), suggesting reduced suppression of MEK signaling pathways by pulsatile treatment in T cells. Consistent with ex vivo observations, CTLA-4 and PD-1 expression on CD8+ T cells were significantly higher with pulsatile treatment (Figure 4D). Additionally, PD-1 showed a significant increase in CD4+ T cells (CD4+Foxp3 and CD4+Foxp3+) and only a slight increase in CD8+ T cells (Figure 4D). We have observed similar results with pulsatile MEKi treatment at higher doses and a longer lag time (Figures S4B–S4D). T-bet expression is higher with pulsatile treatment in CD4+ T cells and CD8+ T cells from lung tumors (Figure S4C). Many co-stimulatory markers were increased in the regulatory T cell (Treg cell) population, but not in CD4+ T cells or CD8+ T cells, indicating differential regulation in subsets of T cells after pulsatile treatment (Figure S4D). Collectively, these observations indicate that pulsatile treatment with MEKi may establish a tumor microenvironment that suppresses tumor growth while maintaining optimal CD8+ activation and infiltration in the tumor microenvironment.
Figure 4. Pulsatile Treatment of Selumetinib Induces CTLA-4 and PD-1 Expression In Vivo.

HKP1 transplantable lung-tumor-bearing mice were treated with selumetinib (25 mg/kg, BID) as presented in (A). After 2 weeks of treatment, lungs were collected and analyzed by flow cytometry.
(A) Schema of selumetinib treatment in HKP1 lung-tumor-bearing mice in vivo.
(B) Frequency of CD3+ T cell subsets in lung tumors by flow cytometry.
(C) Ki-67 of diverse cell populations in lung tumors by flow cytometry.
(D) Scatterplots of PD-1 and CTLA-4 marker (left) and co-inhibitory marker expression from CD3+ T cell subsets of lung tumors by flow cytometry (right). Gating controls are samples without either PD-1 or CTLA-4 antibodies.
*p < 0.05; **p < 0.01; ***p < 0.001, Mann-Whitney test. Samples were biological replicates. The experiment was performed 3 times, and representative results are shown here.
Pulsatile MEKi Treatment Delays Tumor Growth In Vivo
We next tested whether pulsatile treatment confers better anti-tumor activity compared with continuous dosing. We used the KrasG12D mutated HKP1 transplantable tumor model. Though there was a transient delay of tumor growth (Figure S4E), we did not observe differences in tumor progression and survival between the pulsatile and the continuous treatment groups in these settings (Figure S4F). The absence of differential effects may be due to the highly aggressive nature of these transplantable tumors.
In order to further investigate the effects of MEKi treatment regimens, we utilized the KRASG12C GEMM, an autochthonous model that has a slower tumor progression rate and allows for a better therapeutic window. Intranasal instillation of adenovirus with CRE recombinase expression similarly induces lung tumor formation, as the endogenous KrasG12D lung cancer GEMM model previously reported (Chen et al., 2012). Given the similarity of the data between selumetinib and trametinib, we focused our experiments on one MEKi, selumetinib, in the GEMM experiments. After the mice developed lung tumors, a group of mice was treated continuously with selumetinib, with similar baseline tumor volumes (Figure 5A; Figures S5A and S5B). In the first week of the treatment, these mice responded to MEKi, and tu-mor sizes started to decrease as quantified by MRI imaging (Figure 5B). In the second week, about 50% of the mice developed resistance to treatment, and tumors regrew (Figure 5B; Figure S5B). This is consistent with prior work (Li et al., 2018) . In contrast, when we treated mice with selumetinib following a cyclical pulsatile schedule of 1 week on and 1 week off (Figure 5A), the tumors continued to respond to MEKi and had reduced tumor volume, while untreated control tumors continued to grow (Figure 5C; Figure S5B). Both the continuous and pulsatile treatment groups showed decreased levels of pERK in tumor cells compared to the control group, but the continuous group had the lowest pERK expression (Figure 5D). Supporting this pERK suppression, the MEK signaling activation gene signature (Brant et al., 2017) showed consistent suppression of the MEK signaling pathway in the majority of samples in the pulsatile and continuous groups (Figure S5C). Overall, mice treated with pulsatile selumetinib had significantly prolonged progression-free survival compared with those treated with either continuous selumetinib or vehicle control. (Figure 5E).
Figure 5. Pulsatile Schedule of MEKi Treatment Delays Tumor Growth In Vivo.

(A) Schema of selumetinib treatment in KRASG12C mutant genetically engineered mouse model (GEMM) of lung cancer. Treatment schedule for continuous treatment (upper panel, 25 mg/kg, BID) and pulsatile treatment (lower panel, 25 mg/kg, BID).
(B) Waterfall plot showing tumor volume change at indicated time points after the continuous treatment of selumetinib.
(C) Waterfall plot showing tumor volume change at indicated time points after the treatment of pulsatile dosing of either vehicle (left panel) or selumetinib (right panel).
(D) Representative images of immunohistochemistry (IHC) staining of pERK (left panels) and multiplicative quick scores for quantification of pERK½ staining with vehicle control, pulsatile selumetinib, or continuous selumetinib for tumor tissue samples at the end of the treatment (right panel). Scale bars, 100 mm. *p < 0.05; ****p < 0.0001.
(E) Progression-free survival of KRASG12C mice treated with vehicle control, pulsatile selumetinib, or continuous selumetinib. **p < 0.01; ***p < 0.001. Samples were biological replicates. This treatment study was performed three times, and results from all mice have been combined as presented.
Pulsatile Treatment Induces Anti-tumor Immunity through T Cells
To understand the potential contribution of T cells in the observed anti-tumor effect, we analyzed the lungs from the KRASG12C GEMMs at the end of each treatment with selumetinib (continuous or pulsatile schedule; Figure 5A). The percentage of tumor-infiltrating T lymphocytes, including CD4+ T cells and CD8+ T cells, did not show significant differences within the total infiltrating CD45+ leukocytes (Figure 6A). The percentage of Treg cells was increased with either continuous or pulsatile selumetinib treatment. However, this was not correlated with tumor volume changes in the pulsatile or continuous dosing schedule (Figures 5B, 5C, and 6A), suggesting that Tregs may not play a major role in this setting, since these two treatment schedules gave different responses despite the increase of Tregs in both conditions. At the end of pulsatile selumetinib treatment, we observed increased levels of CTLA-4 on both CD4+ and CD8+ T cells (Figure 6D; Figure S6A). Although PD-1 levels are also slightly increased in the pulsatile treatment group, the difference between control and treated groups is not statistically significant (Figure 6D; Figure S6A). On the other hand, at the end of continuous treatment with selumetinib, PD-1 expression was decreased in both CD4+ and CD8+ T cells, while CTLA-4 was only found to be downregulated in CD8+ T cells, as determined by both percentage and median fluorescence intensity (MFI) (Figures 6B, 6C, and S6A). The expression levels of both PD-1 and CTLA-4 are high in Treg cells. After pulsatile treatment with selumetinib, the expression levels of PD-1 and CTLA-4 remained unchanged (Figure S6B). In contrast, continuous selumetinib treatment reduced CTLA-4, but not PD-1, levels on Tregs (Figure S6B). The co-inhibitory molecules LAG3 and TIM3 did not show major expression level changes in CD4+ or CD8+ T cells with either treatment schedule (Figures 6C and 6D). Additionally, pulsatile treatment with selumetinib showed a trend toward increased CD69 and Ki-67 expression, indicating that T cells are modestly more activated after pulsatile treatment (Figure S6C). Continuous selumetinib treatment was found to decrease PD-L1 expression only in CD11b+ myeloid cells and increase its expression in CD8+ T cells. Pulsatile selumetinib treatment significantly downregulated PD-L1 levels in EpCAM+, CD11b+, and CD4+ T cells (Figure S6D). Neither pulsatile nor continuous treatment affected the infiltration of CD11b+ myeloid populations significantly (Figure S6E).
Figure 6. Co-inhibitory Signaling Was Altered Differentially by Continuous versus Pulsatile Treatment of MEKis.

(A) Flow cytometry analysis of KRASG12C mutant GEMM lung-tumor-infiltrating T cell subpopulations: CD4+, CD8+, and Tregs (CD4+Foxp3+) after continuous (left) or pulsatile (right) treatment with selumetinib as presented in Figure 5A. Lung tumors were collected at the end of treatment. *p < 0.05. NS, not significant.
(B) Representative flow cytometry analysis of PD-1 levels in both CD4+ and CD8+ tumor-infiltrating T cells after continuous treatment of selumetinib.
(C) Quantification of inhibitory immune checkpoint molecule expression on CD4+ (upper) and CD8+ (lower) T cells after 3 weeks of continuous selumetinib treatment. *p < 0.05; **p < 0.01.
(D) Quantification of inhibitory molecules within CD4+ (left) and CD8+ (right) T lymphocyte subpopulations after 3 cycles of pulsatile selumetinib treatment. **p < 0.01.
Samples were biological replicates. All mice were recruited at the same time for the treatment, and results from all mice are shown here.
Taken together, our data show that pulsatile MEKi treatment results in improved activation of tumor-infiltrating T cells. Inter-estingly, when we used higher doses (>10-fold) of selumetinib with a pulsatile schedule in vivo (Figure 7A), there was no evident detrimental effect on the immune system in terms of T cell frequency and PD-L1 expression (Figure 7B). T cells were activated 3 days after the treatment was stopped, as determined by Ki-67 and CTLA-4 expression on CD8+ T cells (Figures 7B and 7C). In addition, high-dose pulsatile treatment with selumetinib does not seem to restrain the proliferation of T cells (Figure 7B).
Figure 7. Pulsatile Treatment of Selumetinib with High Dosage Impacts Immune Microenvironment Differently and Enhances Survival in Combination with Anti-CTLA-4 Treatment.

(A) Schema of dosing and sample collection after either high-dose (Hi; 600 mg/kg/day; left panel) or low-dose (Lo; 50 mg/kg/day; right panel) selumetinib treatment. KrasG12DTrp53fl/fl murine transplantable tumors were treated with different dosages of selumetinib. Mouse lung tumors were collected at indicated time points. Samples were biological replicates.
(B) Flow cytometry analysis of different tumor-infiltrating T cell subpopulations within total infiltrating CD45+ leukocytes at indicated time points (left). PD-L1 expression levels on tumor cells (EpCAM+), myeloid cells (CD11b+), and T cells (CD4+ and CD8+) (middle); and Ki-67 expression (right).
(C) Quantification of inhibitory immune checkpoint molecules expressed on CD4+ (left) and CD8+ (right) T cells.
(D) Schema of selumetinib and anti-CTLA-4 treatment on LLC transplantable tumor model.
(E) Survival curve from the selumetinib and anti-CTLA-4 treatment combination in immune-competent mice (C57BL/6J).
(F) Survival curve from the selumetinib and anti-CTLA-4 treatment combination in immune-deficient mice (Rag1 / ). The color code is as same as in (F).
(G) Survival of the pulsatile selumetinib and anti-CTLA-4 treatment group.
Survival analysis was done by Log-rank (Mantel-Cox) test. * < 0.05; ** < 0.01. Samples were biological replicates. The experiment was performed 2–3 times, and representative results are shown here.
Combination Therapy with Pulsatile MEKis and CTLA-4 Blockade Enhances Survival in Mice with Kras Mutated Lung Tumors
Based on the aforementioned observations, we combined CTLA-4 blockade with pulsatile selumetinib treatment to investigate whether the enhanced T cell activation associated with the pulsatile schedule can produce a better outcome in combination with the checkpoint blockade in tumor-bearing mice. We chose a high concentration of selumetinib for pulsatile treatment since proliferation and CTLA-4 expression were elevated, per the previous experiments (Figures 7B and 7C), and tested in a transplantable LLC model (same Kras mutation as the GEMM). We found that the combination of pulsatile high doses of selumetinib with a 3-day gap and CTLA-4 blockade provided the longest survival in mice as compared to other monotherapies or combination therapies (Figures 7D and 7E). The survival advantage was lost in Rag / mice, which are deficient in T and B cells (Figures 7F and 7G), suggesting that adaptive immunity contributes to improved survival in this treatment setting. Interestingly, MEKis combined with PD-1 targeting did not improve survival further (Figure S7A). In addition to the contribution of adaptive immune cells in MEKi treatment, we also observed a potential contribution of NK cells to survival in both MEKi treatment groups (Figures S7B and S7C). Although the survival difference in the anti-CTLA-4 combination is modest in our experimental settings, these results bring forth the possibility that the combination of CTLA-4 blockade with pulsatile MEKi might be considered for clinical trials as a treatment strategy for KRAS mutated lung cancers.
DISCUSSION
In this study, we have demonstrated that, as opposed to conventional continuous MEKi treatment used in the clinic, a pulsatile schedule of MEKi treatment is more effective at controlling tumor progression and enhancing T cell activation with increased levels of CTLA-4 and PD-1 expression. In comparison with continuous treatment, ex vivo pulsatile treatment of T cells with MEKis (selumetinib or trametinib), alongside activation by anti-CD3 and anti-CD28 antibodies, resulted in increased CTLA-4 expression and, to a lesser extent, PD-1 expression. Concurrent changes of Ki-67 and 4–1BB suggest that both CD8+ T cells and CD4+Foxp3 effector cells are more activated by pulsatile treatment with MEKis. While changes in CD4+Foxp3 effector cells and CD4+Foxp3+ T cells are subtle, CD8+ T cells showed high expression levels of immune checkpoint regulators and co-stimulatory markers. This might be due to the fact that CD8+ T cells and CD4+ T cells are regulated through distinct signaling pathways with varying dependence on the mitogen-activated protein (MAP) kinase pathway. In CD8+ T cells, MEK½-ERK½ signaling is critical for cytotoxic activity, proliferation, and survival (Rincón et al., 2001; D’Souza et al., 2008). In CD4+ T cells, MEK4/6/7-p38 and JNK are more important for their activity and development (Rincón et al., 2001). This differential dependency on distinct signaling pathways may regulate responses of CD8+ T cells and CD4+ T cells to MEKis in a different fashion.
In addition to changes in T cells, MEKi increases major histo-compatibility complex class I (MHC I) expression, which is essential for cytotoxic T cell activation, in tumor cells (Brea et al., 2016). In our preliminary data, we observed an increase of H2-Kb MHC I expression in the CD45 cell population after pulsatile MEK inhibition in HKP1 lung tumors. However, after a 2–3 day ‘‘washout’’ period, MHC I expression decreased gradually to the levels of untreated tumors (data not shown). These data further underscore the importance of timing when MEKi treatment and combination approaches are designed.
Our in vivo data with pulsatile MEKis in a transplantable Kras model are consistent with the ex vivo observations. The effects of pulsatile MEKi treatment were more pronounced on CD8+ T cells infiltrating into tumors and their expression of CTLA-4 and PD-1. In vivo, T cells that are being treated have not been pre-selected (as opposed to ex vivo, where cells come from one anatomical location); as a result, we can speculate that cells exposed to MEKis are at different stages of T cell development in vivo. It has been shown previously that the MEK/ERK pathway is fundamental to T cell lineage commitment in the thymus during development. Genetic reduction of ERK induces more CD8+ cells from CD4+ CD8+ double-positive T cells via loss of CD4+ markers in the thymus (Rincón et al., 2001). While pERK reduction is preferable for CD8 expansion in the thymus, pERK positively regulates CD8+ T cell proliferation, survival, and IL-2 cytokine expression from CD8+ cells (Rincón et al., 2001; D’Souza et al., 2008). Furthermore, activation of MEK/ERK signaling downregulates apoptosis in activated primary peripheral T cells (Holmström et al., 2000). Thus, we can speculate that, with pulsatile MEKis, more CD8+ cells will be generated in the thymus during the treatment, then following a period without MEKis treatment may allow proliferation or reduce apoptosis of differentiated CD8+ cells globally, resulting in an expansion of CD8+ populations in the tumor. Considering this intricate role of ERK in CD8+ T cells from development to activation and survival, ERK signaling may be best modulated using a rationally designed schedule in order to maximize the frequency and activation of CD8+ T cells. Our pulsatile schedule of treatment demonstrates better control of this signaling pathway by increasing the frequency of CD8+ T cells and their expression of activation markers and immunotherapy targets in Kras mutated lung tumors.
MEK inhibition in tumor-bearing mice has been previously reported to interfere with the priming of T cells in lymph node yet enhance CD8+ T cell expansion in a CT26 tumor site due to reduced apoptosis (Ebert et al., 2016). To test whether pulsatile MEKi treatment can improve antigen-specific priming of T cells, we utilized antigen-specific, Pmel-1 transgenic CD8+ T cells pulsed with gp100 peptide. We showed that selumetinib and trametinib reduced antigen-specific activation (CD44+) in a dose-dependent manner in both continuous and pulsatile treatments, which suggests a critical role of pERK in CD8+ T cell priming. However, pulsatile MEKi treatment resulted in more CD44+CD62L cells than continuous treatment did. This observation is important, because it suggests that pulsatile treatment leads to maintained activation when compared to conventional MEKi treatment. To clearly understand the effects of MEKis in immune priming, future work to dynamically monitor pERK expression and T cell activation marker changes with diverse types of MEKis pulsation is needed. Additionally, in vivo studies confirming whether the pulsatile schedule of MEKis enhances priming of T cells need to be pursued further.
Given the observations of T cell phenotypic changes ex vivo and in vivo in transplantable models, we further evaluated such changes, using a GEMM harboring the human KRASG12C mutation, one of the most prevalent mutation detected in NSCLC patients. We found that pulsatile treatment with MEKis have a superior anti-tumor effect and delayed drug resistance in comparison with continuous treatment. Consistent with ex vivo and in vivo transplantable model studies, the tumor-infiltrating T cells showed increased CTLA-4 and a modest upregulation of PD-1 levels, with greater proliferation and activation. In contrast, when we treated KRASG12C mutant GEMM mice with MEKis continuously, we observed reduced CTLA-4 and PD-1. Considering that the tumor cells were exposed to equal amounts of MEKis, this confirms that activated T cells resulting from pulsatile MEKi treatment could enhance anti-tumor immunity in KRAS-driven lung cancer.
In all conditions (ex vivo and in vivo in GEMM and transplantable models), we have observed that pulsatile MEKi treatment increases CTLA-4 and PD-1. Although CTLA-4 is a checkpoint molecule (Hardy and Chaudhri, 1997; Thompson and Allison, 1997; Walunas et al., 1994), counterintuitively, it is also known to be induced during early T cell activation (Chambers et al., 1996). In a similar way, while PD-1 is highly expressed on exhausted T cells, it is also a marker of T cell stimulation (Jin et al., 2011; Kansy et al., 2017; Ngiow et al., 2015). Thus, increased CTLA-4 and PD-1 associated with pulsatile MEKi treatment suggests that CD8+ T cells are more activated, and this is supported by data on Ki67 expression. Moreover, considering that exhausted T cells co-express other co-inhibitory markers like TIM-3 and LAG-3 simultaneously with PD-1, the absence of these co-inhibitory markers on T cells after pulsatile MEKi treatment supports that they are activated rather than exhausted cells. Favorable changes in CTLA-4 after pulsatile MEKi treatment warrant considering how CTLA-4 expression is regulated. It is known that CTLA-4 is regulated by ERK (Tsatsanis et al., 2008), but it is not well understood how CTLA-4 expression is regulated temporally in the presence of MEKis. A future study on the mechanism of CTLA-4 expression regulation under MEKis is necessary.
Improved anti-tumor activity and a more favorable phenotype of CD8+ T cells with increased CTLA-4 expression in the pulsatile group inspired us to test the combination of pulsatile MEKi treatment with an anti-CTLA4 antibody in Kras mutant lung cancer. We show that the combination of pulsatile high doses of selumetinib and CTLA-4 blockade prolonged survival to the greatest extent compared to other monotherapies or combination therapies, supporting that the combination of immune checkpoint blockade with pulsatile MEKis may be a more effective approach to treat Kras lung cancer. This prolonged survival was not present in immune-deficient mice, indicating that it was mediated by the adaptive immune system. A similar pulsatile treatment regimen is also currently under evaluation in a clinic for NSCLC patients with intermittent selumetinib and antibodies targeting CTLA-4 and PD-L1 (ClinicalTrial.gov: NCT03581487). It should be noted that, since the trial did not stratify for co-mutations in addition to KRAS, MEKi alone, which targets downstream of KRAS, may not be enough to restrain tumors with a KRAS mutation together with other co-mutations, even when combined with immunotherapy.
In this study, we have tested MEK inhibition with selumetinib and trametinib. However, other MEKis with variations in potency, target specificity, and T½ (elimination half-life) are being used (e.g., cobimetinib and binimetinib) or are in development (Caunt et al., 2015). Based on our findings, these should also be tested using different dosing and scheduling regimens to potentially allow for more effective cancer cell growth control and durable immune microenvironment activation.
In this study, we show that the pulsatile schedule alters the tumor microenvironment favorably by activating T cells and providing an advantageous anti-tumor effect in Kras-driven lung cancer models. This schedule provides a therapeutic window for immune checkpoint blockade to improve MEKis for the treatment of KRAS-driven NSCLC patients.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Taha Merghoub (merghout@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and transplantable mouse model
CL13, CL25, IO33 cells lines (Jones-Bolin et al., 1998) were obtained from Dr. Phillip A. Dennis (Lastwika et al., 2016). They were cultured in RPMI with 7.5% FBS and Pen/Strep. The HKP1 cell line was obtained from Dr. Vivek Mittal at Weill Cornell Medical College (Choi et al., 2015). LLC (Lewis Lung Carcinoma) was obtained from ATCC (CRL-1642). HKP1 and LLC were cultured in DMEM with 10% fetal calf serum, L-Glutamine, sodium pyruvate, and Pen/Strep. Sex of CL13, CL25, IO33, HKP1, and LLC is female based on our sequencing analysis. KrasG12D p53f/f cell line was derived from male mouse in Dr. Kwok-Kin Wong’s laboratory and cultured in RPMI with 7.5% FBS and Pen/Strep. The cell lines have been kept in culture for a limited number of passage. Cell lines are also routinely mycoplasma tested and each new cell line is mycoplasma tested by the monoclonal core facility at MSK. Cell lines are also tested for Mouse antibody production (MAP) routinely at MSK.
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Memorial Sloan Kettering Cancer Center and Dana-Farber Cancer Institute. All mice for transplantable model were 6 12 week old of age and females and tumor-bearing mice were randomized before selumetinib treatment (5 mice per group for flow cytometry analysis, 10 mice per group for survival analysis).Transplantable lung cancer was generated by intravenous injection of 1 3 105 HKP1 cells into C57BL/6J (Jax 00664) mice and monitored using the IVIS Spectrum in vivo imaging system (Perkin Elmer). 50ul of 30 mg/ml D-luciferine (Perkin Elmer) was injected retro-orbitally under anesthesia using isoflurane and mice were then placed supine in an imaging chamber for imaging. Another transplantable model was generated by subcutaneous injection of 500,000 – 1,000,000 LLC cells in the flank of C57BL/6J (Jax 00664) mice or Rag1 / (Jax 002216) and tumor growth was measured by calipers. Mice were treated with selumetinib (Selleckchem, Houston, TX USA) from day 5 or day 7 to examine effect of MEKis on tumor growth and survival. Selumetinib was prepared in corn oil and administered by oral gavage twice a day as 25mg/kg for the continuous group daily and 25mg/kg or 300mg/kg for the cyclical pulsatile group according to a planned pulsatile schedule. Survival was analyzed based on the approved humane endpoints (distress and tumor size limit). 100 ug of anti-CTLA-4 was administered to a mouse twice a week for 2–3 weeks intraperitoneally with 9H10 clone for first three doses, then 9D9 clone for rest of doses. 250 ug of anti-PD-1 (RMP1–14) was administered to a mouse twice a week for three weeks intraperitoneally. To deplete NK cells, 200 ug of anti-NK1.1 antibody (PK-136) was injected twice a week for three weeks intraperitoneally.
GEMM model studies
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Dana-Farber Cancer Institute and New York University School of Medicine (NYUSoM). The genetically engineered mouse model (GEMM) harbors a conditional activating mutation of the human version of KRAS (KRASLSL-G12C/+) at the collagen I locus (Li et al., 2018) . CRE recombinase was induced through intranasal inhalation of 2.5×106 p.f.u. adeno-Cre (University of Iowa adenoviral core). Lung adenocarcinoma appeared 6 weeks after induction. For drug treatment studies in GEMM models, age matched littermates (15 – 21-week-old) were induced at the same time and tumor burden was monitored by MRI. Once the tumor size reached 500 mm3 (at about 12 weeks after adenoviral inoculation), mice were randomly assigned to experimental groups. We did not observe gender bias response between male and female mice, in terms of tumor growth and response to drug treatment. Mice were evaluated by MRI imaging to quantify lung tumor burden before and after drug treatment. Mice were treated with either vehicle, or 25 mg/kg selumetinib twice daily by oral gavage using either continuous (every day for 3 weeks) or cyclical pulsatile (one week on, one week off) dosing schedule. PFS was analyzed based on the standard criteria in clinical trials. Briefly, PFS was the duration between treatment start and progression, which was defined by increase of tumor size compared to the previous scan of radiological CT and the appearance of new lesions.
METHOD DETAILS
In vitro MEKis treatment
Selumetinib or trametnib (Sellekchem) were added to tumor cells in 96-well plates. Viability was measured using the CellTiter Glo luminescent viability kit (Promega) after 72 hr using a Wallace plate reader (Molecular Probe). To determine pERK after MEKis treatment, Kras mutant lung tumor cell lines and splenocytes collected from HKP1 tumor bearing mice were treated with selumetinib or trametinib for 2 hr at 37C. Selumetinib and trametinib stock solutions were prepared in DMSO and diluted in media. Cells were stimulated with 0.1 ug/ml PMA for 2 min at 37C, then immediately fixed with 4% paraformaldehyde and permeabilized using ice cold 95% methanol. pERK was stained using anti-pERK (#9106, Cell signaling) and goat anti-mouse Ig(H+L)–FITC (SouthernBiotech) by flow cytometry. Washout experiments were done using CD5+ splenocytes from HKP1 tumor bearing mice. CD5+ cells were collected using CD5 microbeads (Miltenyi MACS cell separation system), then seeded in the presence of selumetinib or trametinib into flat bottom 96-well plates pre-coated with anti-CD3 (145–2C11, 1ug/ml) and anti-CD28 (37N, 1ug/ml). After 24hrs, media (RIPA + 7.5% FBS + 0.1% b-mercaptoethanol) was replaced with MEKis (continuous group) or DMSO diluent (wash out group) and the media was changed every day with freshly prepared MEKis. At 72hrs and 96 hr, cells were collected and analyzed by flow cytometry.
Western blot
Trametinib was used in vitro at indicated doses in 1% DMSO. Cells were treated for 24 hours before isolating protein. Protein lysate was isolated from cultured cells using RIPA buffer with protease inhibitor (50mM Tris pH 8, 150 mM NaCl, 0.5% sodium deoxycho-late, 1% NP-40). Protein quantification was performed using DC Lowry assay. Antibodies for pERK (#4370), ERK½ (#9102) were obtained from Cell Signaling Technologies.
Ex vivo T cell priming with MEKis treatment
Pmel-1 TCR transgenic mice (Overwijk et al., 2003) were obtained from Dr. N. Restifo (National Institutes of Health). Splenocytes from a Pmel-1 mouse were stained with 5uM of CFSE (Invitrogen), then seeded at approximately 400,000 cells per well in a U-shape bottom 96 well plate with 1ug/ml of heteroclitic human gp100 peptide (AnaSpec Inc) at day 0 in 0.1% b-mercaptoenthanol supplemented RPMI media with 7.5% FBS. At the same time, splenocytes from another Pmel-1 mouse were set for priming in T175 flask. At day 2 or 3, 20U of mouse IL-2 (eBioscience) was added to the supernatant of the T175 flask that was subsequently used to prepare following treatment groups for the plate. Supernatant with MEKis (selumetinib, trametinib) were added to continuous groups according to the experimental plan. Washout groups were treated with supernatant containing IL-2 and DMSO. At day 4 or 5, 20U/ml of mouse IL-2 supplemented fresh T cell media with or without MEKis was added to support extensive cell growth, and cells were stained and analyzed at day 5 by flow cytometry
MRI quantification
Animals were anesthetized with isoflurane to perform magnetic resonance imaging (MRI) of the lung field using the BioSpec USR70/ 30 horizontal bore system (Bruker) to scan 24 consecutive sections. Overall tumor volumes within the whole lung were quantified using 3D slicer software to reconstruct MRI volumetric measurements as previously described (Chen et al., 2012). Acquisition of the MRI signal was adapted according to cardiac and respiratory cycles to minimize motion effects during imaging.
Tumor-infiltrating immune cells isolation and analysis
Mice were sacrificed, and lungs were perfused using sterile PBS through heart perfusion from the right ventricle. The whole lung was minced into small pieces and digested in collagenase D (Sigma or GIBCO) and DNase I (Sigma) in Hank’s Balanced Salt Solution (HBSS) at 37 C for 30 min. After incubation, the digested tissue was filtered through a 40 um or 70 mm cell strainer (Fisher) to obtain single-cell suspensions. Separated cells were treated with 1X RBC lysis buffer (Biolegend) to lyse red blood cells. Live cells were determined by LIVE/DEAD fixable aqua dead cell stain kit (Molecular Probes) or Fixable viability dye e506 (eBioscience). The cell pellets were re-suspended in PBS with 2% FBS for flow cytometry analysis. Cells were stained with cell surface markers as indicated followed by fixation/permeabilization using foxp3 fixation/permeabilization kit (eBioscience). Lung infiltrating immune cells were stained with different combinations of fluorochrome-coupled antibodies and analyzed by FACS analysis.
Transcriptome analysis
RNA was extracted from 6 – 10 FFPE tissue sections using QUICK RNA FFPE kit (Zymo Research). Expression profiling was performed using Affymetrix Clariom D Pico Assay, mouse and analyzed using Transcriptome Analysis Console as previously described. (Andrzejewski et al., 2017; Nassal et al., 2017). Heatmaps were plotted in R using the Robust Multi-array Average (RMA) procedure (Bolstad et al., 2003; Carvalho and Irizarry, 2010; Irizarry et al., 2003a; Irizarry et al., 2003b). Expression values were normalized using Affymetrix mta10 annotation data and gene names were translated to MGI symbols. Heatmaps of signature gene sets were extracted from a study of Brant et al. (Brant et al., 2017).
Flow cytometry analysis
Immune cells obtained via ex vivo treatment or in vivo were collected and processed as single-cell suspensions and stained with antibodies against mouse CD3 (145–2C11), CD62L (MEL-14), CD44 (IM7), T-bet (eBio4B10), CD45 (30-F11), PD-1 (J43), 4–1BB (17B5), CTLA4 (UC10–4B9), Ki67 (SolA15), Foxp3 (FJK-16 s), CD4 (GK1.5), CD8 (3B5), and fixable viability dye 506 (eBioscience) for ex vivo studies; and antibodies against mouse CD3 (17A2), CD4 (GK1.5), CD8 (53–6.7, 5H19), Foxp3 (FJK-16 s), CD11b (M1/70), CD11c (N418), Epcam (G8.8), CD279 (PD-1, 29F.1A12, J43), CD152 (CTLA-4, UC10–4B9), TIM-3 (RMT3–23), CD223 (Lag-3, C9B7W), ICOS (c398.4A), 4–1BB (17B5), GITR (DTA-1), OX-40 (OX-86), PD-L1 (10F.9G2) for in vivo studies. Staining signals were acquired on BD LSRFortessa or BD LSR II (BD Biosciences) and analyzed using FlowJo software (FlowJo, LLC).
Cytokine profiling analysis
The supernatant of primed Pmel-1 with or without selumetinib was collected at day 5 after human gp-100 peptide addition and subjected to Luminex cytokine analysis using Luminex MAGPIX system and Milliplex multiplex assays mouse panel (Millipore) and analyzed using xPOTENT software (Millipore Sigma). Mouse lung bronchoalveolar lavage (BAL) from GEMM KRASG1C mice after the treatment was performed by intratracheal injection of 2ml of sterile PBS and collected by aspiration. Cytokines were measured using Mouse Cytokine 23-plex Assay (Bio-Rad) and measured on Bio-Plex 200 system (Bio-Rad). Concentrations [pg/ml] of each protein were derived from 5-parameter curve fitting models. Fold changes relative to the control were calculated and plotted as log2FC. Lower and upper limits of quantitation (LLOQ/ULOQ) were derived from standard curves for cytokines above or below detection.
Immunohistochemistry (IHC) staining and analyses
Paraffin-embedded sections were deparaffinized, followed by immunohistochemical staining with antibodies against pERK (Cell Signaling) and examined using a Leica upright microscope (Liu et al., 2015b). H&E sections were examined by a pathologist at Dana-Faber Cancer Institute. Results were independently scored by two pathologists using multiplicative quick systems (Liu et al., 2015b). Briefly, the expression score of each marker was calculated by multiplying a score indicating percentage of positively stained cells within tumor cells counted (1 = 0%–4%; 2 = 5%–19%; 3 = 20%–39%; 4 = 40%–59%; 5 = 60%–79%; 6 = 80%–100%) by the intensity grade of staining (0 = negative; 1 = weak; 2 = moderate; 3 = strong).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Data are presented as mean with SEM unless otherwise specified. Statistical comparisons were performed using unpaired Student’s t tests for two tailed p value unless otherwise specified. *p < 0.05, **p < 0.01, ***p < 0.001. Survival was analyzed by Log-rank analysis by Graphad Prism 7.
DATA AND SOFTWARE AVAILABILITY
Data Resources
The accession number for the Affymetrix transcriptome analysis reported in this paper is GEO: GSE126202.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| phospho-p44/42 MAPK (Erk1/2) (20G11) | Cell Signaling Technology | Cat# 4376; RRID:AB_331772 |
| phospho-p44/42 MAPK (Erk1/2) (E10) | Cell Signaling Technology | Cat# 9106; RRID:AB_331768 |
| phospho-p44/42 MAPK (Erk1/2) (D13.14.4E) | Cell Signaling Technology | Cat# 4370; RRID:AB_2315112 |
| P44–42 MAPK (Erk1/2) | Cell Signaling Technology | Cat#9102; RRID:AB_330744 |
| CD11b (M1/70) | BioLegend | Cat# 101224; RRID:AB_755986 |
| CD11b (M1/70.15) | Invitrogen | Cat# RM2817; RRID:AB_1464525 |
| CD11c (N418) | BioLegend | Cat# 117324; RRID:AB_830649 |
| PD-1 (J43) | eBioscience | Cat# 11–9985-85: RRID:AB_465473 |
| PD-1 (29F.1A12) | Biolegend | Cat# 135215; RRID:AB_10696422 |
| CTLA-4 (UC10–4B9) | ThermoFisher | Cat# 12–1522-81; RRID:AB_465878 |
| CTLA-4 (UC10–4B9) | eBioscience | Cat# 17–1522-82; RRID:AB_2016700 |
| LAG-3 (C9B7W) | BioLegend | Cat# 125209; RRID:AB_10639935 |
| LAG-3 (eBioC9B7W) | eBioscience | Cat# 48–2231-82; RRID:AB_11149866 |
| PD-L1 (10F.9G2) | BioLegend | Cat# 124311; RRID:AB_10612935 |
| Armenian Hamster IgG (eBio299Arm) | ThermoFisher | Cat# 12–4888-81; RRID:AB_470073 |
| CD3 (17A2, for flow cytometry) | BioLegend | Cat# 100214; RRID:AB_493645 |
| CD3 (17A2, for flowcytometry) | BioLegends | Cat# 100221; RRID:AB_2057374 |
| CD3 (145–2C11) | BD PharMingen | Cat# 561108/551163; RRID:AB_10562558/ RRID:AB_394082 |
| EpCAM (G8.8) | BioLegend | Cat# 118215; RRID:AB_1236477 |
| CD4 (GK1.5) | BioLegend | Cat# 100411; RRID:AB_312696 |
| CD4 (GK1.5) | BioLegend | Cat# 100406; RRID:AB_312691 |
| CD45 (30-F11) | BioLegend | Cat# 103108; RRID:AB_312973 |
| CD8 (53–6.7) | BioLegends | Cat# 100734; RRID:AB_2075238 |
| CD8 (5H19) | Invitrogen | Cat # MCD0817; RRID:AB_10374589 |
| FoxP3 (FJK-16 s) | eBioscience | Cat# 17–5773-80; RRID:AB_469456 |
| FoxP3 (FJK-16 s) | eBioscience | Cat# 25–5773-82; RROD:AB_891552 |
| IFN-γ (XMG1.2) | BioLegend | Cat# 505825; RRID:AB_1595591 |
| Ki-67 (16A8) | BioLegend | Cat# 652411; RRID:AB_2562663 |
| Ki-67 (SolA15) | Invitrogen | Cat# 48–5698-82; RRID:AB_11149124 |
| IgG1 (RTK2071) | BioLegend | Cat# 400415; RRID:AB_326521 |
| IgG1 (RTK2071) | BioLegend | Cat# 400411; RRID:AB_326517 |
| IgG2a (eBR2a) | eBioscience | Cat# 12–4321-81; RRID:AB_470051 |
| IgG2a (eBR2a) | eBioscience | Cat# 17–4321-81; RRID:AB_470181 |
| IgG2a (RTK2758) | BioLegend | Cat# 400521; RRID:AB_326542 |
| IgG2a (RTK2758) | BioLegend | Cat# 400535; RRID:AB_10933427 |
| TIM3 (RMT3–23) | eBioscience | Cat# 12–5870-81; RRID:AB_465973 |
| Tbet (eBio4B10) | eBioscience | Cat# 12–5825-82; RRID:AB_925761 |
| ICOS (c398.4A) | eBioscience | Cat# 11–9949-82; RRID:AB_465458 |
| 4–1BB (17B5) | eBioscience | Cat# 12–1371-83; RRID:AB_465865 |
| GITR (DTA-1) | eBioscience | Cat# 48–5874-82; RRID:AB_1944394 |
| OX40 (OX-86) | eBioscience | Cat# 17–1341-82; RRID:AB_10717260 |
| CD62L (MEL-14) | eBioscience | Cat# 56–0621-82; RRID:AB_494003 |
| CD44 (IM7) | BD PharMingen | Cat# 559250; RRID:AB_398661 |
| Anti Fcgamma, purified | MSK Antibody and Bioresource Core | Clone: 2.4G2 |
| CD3 (for in vitro T cell activation) | MSK Antibody and Bioresource Core | Clone: 145–2C11 |
| CD28 (for in vitro T cell activation) | MSK Antibody and Bioresource Core | Clone: 37N |
| mouse anti-CTLA-4 (9H10, in vivo antibody) | Bioxcell | Cat# BE0131; RRID:AB_10950184 |
| mouse anti-CTLA-4 (9D9, in vivo antibody) | Bioxcell | Cat# BE0164; RRID:AB_10949609 |
| Syrian Hamster IgG | Bioxcell | Cat# BE0087; RRID:AB_1107782 |
| InVivoMAb mouse IgG2b isotype control (MCP-11) | Bioxcell | Cat# BE0086; RRID:AB_1107791 |
| mouse anti-PD-1 (RMP1–14, for in vivo experiment) | Bioxcell | Cat# BE0146: RRID:AB_10949053 |
| InVivoMAb rat IgG2a isotype control (2A3) | Bioxcell | Cat# BE0089; RRID:AB_1107769 |
| NK1.1 (PK136, in vivo antibody) | Bioxcell | Cat#BE0036; RRID: AB_1107737 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| Ad5CMVCre | UI Viral Vector Core Web | VVC-U of Iowa-5 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Selumetinib | Selleckchem | Cat# S1008 |
| Trametinib | Selleckchem | Cat# S2673 |
| gp100 peptide, human | Anaspec Inc | Cat# AS-62589 |
| Recombinant mouse IL-2 | eBioscience | Cat# 14–8021-64 |
| Fixable viability dye (for flow cytometry) | eBioscience | Cat# 65–0866-14 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | ThermoFisher | Cat# L34966 |
| CD5 (Ly-1) MicroBeads, mouse | Miltenyi | Cat# 130–049-301 |
| Luminex cytokine analysis (mouse)-MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel - Immunology Multiplex Assay | Millipore | Cat# MCYTMAG-70K-PX32 |
| Mouse Cytokine 23-plex Assay | Bio-Rad | #m60009rdpd |
| Cell titer Glo luminescent cell viability assay | Promega | G7571 |
| QUICK RNA FFPE Kit | Zymo Research | R1008 |
|
| ||
| Deposited Data | ||
|
| ||
| Affymetrix transcriptome data | NCBI Gene Expression Omnibus | GEO: GSE126202 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Mouse; CL13 | Dr. Phillip A. Dennis | PMID: 26637667 |
| Mouse; CL25 | Dr. Phillip A. Dennis | PMID: 26637667 |
| Mouse; IO33 | Dr. Phillip A. Dennis | PMID: 26637667 |
| Mouse; HKP1 | Dr. Vivek Mittal laboratory (WCMC) | PMID: 25704820 |
| Mouse; LLC | ATCC | ATCC® CRL-1642 |
| Mouse; KrasG12DTrp53fl/fl cell line from GEMM | Dr. Kwok-Kin Wong laboratory (NYU) | N/A |
| Mouse: primary T lymphocytes | This study | N/A |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse; KRASLSL-G12C GEMM | Dr. Kwok-Kin Wong laboratory (NYU) | PMID: 29945997 |
| Mouse; Pmel TCR transgenic mouse | Dr. Nicholas Restifo laboratory (NIH) | PMID: 12925674 |
| Mouse; Rag1–/–; B6.129S7-Rag1tm1Mom/J | Jax lab | Jax 002216 |
| Mouse; C57BL/6J; C57BL/6J | Jax lab | Jax 000664 |
|
| ||
| Software and Algorithms | ||
|
| ||
| 3D slicer | Online download | https://www.slicer.org/ |
| GraphGad Prism 7 | Online download | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo v.10 | Online download | https://www.flowjo.com/solutions/flowjo/downloads |
| IVIS Living image software v. 4.4 | Perkin Elmer | http://www.perkinelmer.com/category/in-vivo-imaging-software?gclid=EAIaIQobChMIoK2N_LKW4AIVk4TICh0LeAU9EAAYASACEgLwr_D_BwE |
| Transcriptome Analysis Console (TAC) Software v.4.0 | ThermoFisher Scientific | https://www.thermofisher.com/us/en/home/life-science/microarray-analysis/microarray-analysis-instruments-software-services/microarray-analysis-software/affymetrix-transcriptome-analysis-console-software.html |
| Robust Multi-array Average (RMA) procedure | Irizarry et al., 2003a | https://www.rdocumentation.org/packages/affy/versions/1.50.0/topics/rma |
|
| ||
| Other | ||
|
| ||
| MRI imaging | Dana-Farber Cancer Institute’s Lurie Family Imaging Center | http://www.lfic.dfci.harvard.edu/about |
| Affymetrix microarray Assay | ThermoFisher Scientific | Mouse Clariom D Pico assay |
Highlights.
Pulsatile treatment with MEK inhibitors maintains T cell activation
Pulsatile dosing of MEK inhibitor delays growth rebound of KRAS mutant lung tumors
Combining CTLA-4 blockade with pulsatile MEK inhibition extends survival
The enhanced survival was conferred by adaptive immunity
ACKNOWLEDGMENTS
We thank Nouraiz Falik for the work on validation animal experiments. This research was funded in part through a Stand Up To Cancer-American Cancer Society Lung Cancer Dream Team Translational Research Grant (SU2C-AACR-DT17–15); NIH-NCI Cancer Center Support grant P30 CA008748; the Ludwig Collaborative and Swim Across America Laboratory; Memorial Sloan Kettering Cancer Center, United States; the Parker Institute for Cancer Immunotherapy, Memorial Sloan Kettering Cancer Center; the Department of Medicine, Memorial Sloan Kettering Cancer Center; and Weill Cornell Medicine, United States.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.066.
DECLARATION OF INTERESTS
P.D.S. is an employee and shareholder of AstraZeneca.
T.M. is a cofounder and holds an equity in IMVAQ Therapeutics. He is a consultant of Immunos Therapeutics and Pfizer. He has research support from Bristol-Myers Squibb; Surface Oncology; Kyn Therapeutics; Infinity Pharmaceuticals, Inc.; Peregrine Pharmeceuticals, Inc.; Adaptive Biotechnologies; Leap Therapeutics, Inc.; and Aprea. He has patents on applications related to work on oncolytic viral therapy, alpha virus-based vaccine, neo antigen modeling, CD40, GITR, OX40, PD-1, and CTLA-4.
K.K.W. is a founder and equity holder of G1 Therapeutics. He has sponsored research agreements with MedImmune, Takeda, TargImmune, and BMS. He also has consulting and sponsored research agreements with AstraZeneca, Janssen, Pfizer, Novartis, Merck, Ono, and Array.
J.D.W. is a consultant for Adaptive Biotech; Advaxis; Amgen; Apricity; Array BioPharma; Ascentage Pharma; Astellas; Bayer; Beigene; Bristol Myers Squibb; Celgene; Chugai; Elucida; Eli Lilly; F Star; Genentech; Imvaq; Janssen; Kleo Pharma; Linneaus; MedImmune; Merck; Neon Therapuetics; Ono; Polaris Pharma; Polynoma; Psioxus; Puretech; Recepta; Trieza; Sellas Life Sciences; Serametrix; Surface Oncology; and Syndax. He has research support from Bristol Myers Squibb, Medimmune, and Genentech. He holds equity in Potenza Therapeutics; Tizona Pharmaceuticals; Adaptive Biotechnologies; Elu-cida; Imvaq; Beigene; Trieza; and Linneaus and has an honorarium from Esanex. He has patents of xenogeneic DNA vaccines (royalties); alphavirus replicon particles expressing TRP2; myeloid-derived suppressor cell (MDSC) assay (royalties); Newcastle disease viruses for cancer therapy; a genomic signature to identify responders to ipilimumab in melanoma; engineered vaccinia viruses for cancer immunotherapy; an anti-CD40 agonist monoclonal antibody (mAb) fused to monophosphoryl lipid A (MPL) for cancer therapy; CAR T cells targeting differentiation antigens as means to treat cancer; an anti-PD1 antibody; anti-CTLA4 antibodies; and anti-GITR antibodies and methods of use thereof.
The other authors declare no competing interests.
Data Resources
REFERENCES
- Andrzejewski S, Klimcakova E, Johnson RM, Tabariés S, Annis MG, McGuirk S, Northey JJ, Chénard V, Sriram U, Papadopoli DJ, et al. (2017). PGC-1a promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab 26, 778–787.e5. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, and Speed TP (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193. [DOI] [PubMed] [Google Scholar]
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. (2015). Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med 373, 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. (2015). Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med 373, 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brant R, Sharpe A, Liptrot T, Dry JR, Harrington EA, Barrett JC, Whalley N, Womack C, Smith P, and Hodgson DR (2017). Clinically viable gene expression assays with potential for predicting benefit from MEK inhibitors. Clin. Cancer Res 23, 1471–1480. [DOI] [PubMed] [Google Scholar]
- Brea EJ, Oh CY, Manchado E, Budhu S, Gejman RS, Mo G, Mondello P, Han JE, Jarvis CA, Ulmert D, et al. (2016). Kinase regulation of human MHC class I molecule expression on cancer cells. Cancer Immunol. Res 4, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, Russell V, Gordon RA, Vyas S, Yuan J, et al. (2014). Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Im-munol. Res 2, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho BS, and Irizarry RA (2010). A framework for oligonucleotide microarray preprocessing. Bioinformatics 26, 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caunt CJ, Sale MJ, Smith PD, and Cook SJ (2015). MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat. Rev. Cancer 15, 577–592. [DOI] [PubMed] [Google Scholar]
- Chambers CA, Krummel MF, Boitel B, Hurwitz A, Sullivan TJ, Fournier S, Cassell D, Brunner M, and Allison JP (1996). The role of CTLA-4 in the regulation and initiation of T-cell responses. Immunol. Rev 153, 27–46. [DOI] [PubMed] [Google Scholar]
- Chen Z, Cheng K, Walton Z, Wang Y, Ebi H, Shimamura T, Liu Y, Tupper T, Ouyang J, Li J, et al. (2012). A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 483, 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Sheng J, Gao D, Li F, Durrans A, Ryu S, Lee SB, Narula N, Rafii S, Elemento O, et al. (2015). Transcriptome analysis of individual stromal cell populations identifies stroma-tumor crosstalk in mouse lung cancer model. Cell Rep 10, 1187–1201. [DOI] [PubMed] [Google Scholar]
- D’Souza WN, Chang C-F, Fischer AM, Li M, and Hedrick SM (2008). The Erk2 MAPK regulates CD8 T cell proliferation and survival. J. Immunol 181, 7617–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, and Stuart DD (2013). Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dushyanthen S, Teo ZL, Caramia F, Savas P, Mintoff CP, Virassamy B, Henderson MA, Luen SJ, Mansour M, Kershaw MH, et al. (2017). Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat. Commun 8, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA, Kim JM, et al. (2016). MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 44, 609–621. [DOI] [PubMed] [Google Scholar]
- Fernández-Medarde A, and Santos E (2011). Ras in cancer and developmental diseases. Genes Cancer 2, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friday BB, and Adjei AA (2008). Advances in targeting the Ras/Raf/MEK/ Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin. Cancer Res 14, 342–346. [DOI] [PubMed] [Google Scholar]
- Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. ; KEYNOTE-001 Investigators (2015). Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- Greystoke A, Steele N, Arkenau HT, Blackhall F, Md Haris N, Lindsay CR, Califano R, Voskoboynik M, Summers Y, So K, et al. (2017). SELECT-3: a phase I study of selumetinib in combination with platinum-doublet chemotherapy for advanced NSCLC in the first-line setting. Br. J. Cancer 117, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy K, and Chaudhri G (1997). Activation and signal transduction via mitogen-activated protein (MAP) kinases in T lymphocytes. Immunol. Cell Biol 75, 528–545. [DOI] [PubMed] [Google Scholar]
- Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, Ready NE, Gerber DE, Chow LQ, Juergens RA, et al. (2017). Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol 18, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmström TH, Schmitz I, Söderström TS, Poukkula M, Johnson VL, Chow SC, Krammer PH, and Eriksson JE (2000). MAPK/ERK signaling in activated T cells inhibits CD95/Fas-mediated apoptosis downstream of DISC assembly. EMBO J 19, 5418–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, and Speed TP (2003a). Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, and Speed TP (2003b). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. [DOI] [PubMed] [Google Scholar]
- Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, et al. ; TRACERx Consortium (2017). Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med 376, 2109–2121. [DOI] [PubMed] [Google Scholar]
- Jin HT, Ahmed R, and Okazaki T (2011). Role of PD-1 in regulating T-cell immunity. Curr. Top. Microbiol. Immunol 350, 17–37. [DOI] [PubMed] [Google Scholar]
- Jones-Bolin SE, Johansson E, Palmisano WA, Anderson MW, Wiest JS, and Belinsky SA (1998). Effect of promoter and intron 2 polymorphisms on murine lung K-ras gene expression. Carcinogenesis 19, 1503–1508. [DOI] [PubMed] [Google Scholar]
- Kane LP, Lin J, and Weiss A (2000). Signal transduction by the TCR for antigen. Curr. Opin. Immunol 12, 242–249. [DOI] [PubMed] [Google Scholar]
- Kansy BA, Concha-Benavente F, Srivastava RM, Jie H-B, Shayan G, Lei Y, Moskovitz J, Moy J, Li J, Brandau S, et al. (2017). PD-1 status in CD8+ T cells associates with survival and anti-PD-1 therapeutic outcomes in head and neck cancer. Cancer Res 77, 6353–6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, et al. (2016). Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res 76, 227–238. [DOI] [PubMed] [Google Scholar]
- Li S, Liu S, Deng J, Akbay EA, Hai J, Ambrogio C, Zhang L, Zhou F, Jenkins RW, Adeegbe DO, et al. (2018). Assessing therapeutic efficacy of MEK inhibition in a KRAS<SUP>G12C</SUP>-driven mouse model of lung cancer. Clin. cancer res 24, 4854–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, Yang J, Seestaller-Wehr L, Zhang SY, Hopson C, et al. (2015a). The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 21, 1639–1651. [DOI] [PubMed] [Google Scholar]
- Liu X, Yu X, Xie J, Zhan M, Yu Z, Xie L, Zeng H, Zhang F, Chen G, Yi X, and Zheng J (2015b). ANGPTL2/LILRB2 signaling promotes the propagation of lung cancer cells. Oncotarget 6, 21004–21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovly CM, and Carbone DP (2011). Lung cancer in 2010: one size does not fit all. Nat. Rev. Clin. Oncol 8, 68–70. [DOI] [PubMed] [Google Scholar]
- Manchado E, Weissmueller S, Morris JP 4th, Chen CC, Wullenkord R, Lujambio A, de Stanchina E, Poirier JT, Gainor JF, Corcoran RB, et al. (2016). A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 534, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassal DM, Wan X, Liu H, Maleski D, Ramirez-Navarro A, Moravec CS, Ficker E, Laurita KR, and Deschênes I (2017). KChIP2 is a core transcriptional regulator of cardiac excitability. eLife 6, e17304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, and Smyth MJ (2015). A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res 75, 3800–3811. [DOI] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, Dellemijn TA, Antony PA, Spiess PJ, Palmer DC, et al. (2003). Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J. Exp. Med 198, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, Giannone V, D’Amelio AM Jr., Zhang P, Mookerjee B, and Johnson BE (2017). Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 18, 1307–1316. [DOI] [PubMed] [Google Scholar]
- Poon E, Mullins S, Watkins A, Williams GS, Koopmann JO, Di Genova G, Cumberbatch M, Veldman-Jones M, Grosskurth SE, Sah V, et al. (2017). The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J. Immunother. Cancer 5, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. ; KEYNOTE-024 Investigators (2016). Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med 375, 1823–1833. [DOI] [PubMed] [Google Scholar]
- Riely GJ, Marks J, and Pao W (2009). KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc 6, 201–205. [DOI] [PubMed] [Google Scholar]
- Rincón M, Flavell RA, and Davis RJ (2001). Signal transduction by MAP kinases in T lymphocytes. Oncogene 20, 2490–2497. [DOI] [PubMed] [Google Scholar]
- Samatar AA, and Poulikakos PI (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov 13, 928–942. [DOI] [PubMed] [Google Scholar]
- Shah NP, Kasap C, Weier C, Balbas M, Nicoll JM, Bleickardt E, Nicaise C, and Sawyers CL (2008). Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell 14, 485–493. [DOI] [PubMed] [Google Scholar]
- Solit DB, She Y, Lobo J, Kris MG, Scher HI, Rosen N, and Sirotnak FM (2005). Pulsatile administration of the epidermal growth factor receptor inhibitor gefitinib is significantly more effective than continuous dosing for sensitizing tumors to paclitaxel. Clin. Cancer Res 11, 1983–1989. [DOI] [PubMed] [Google Scholar]
- Soria JC, Fülop A, Maciel C, Fischer JR, Girotto G, Lago S, Smit E, Ostoros G, Eberhardt WEE, Lishkovska P, et al. (2017). SELECT-2: a phase II, double-blind, randomised, placebo-controlled study to assess the efficacy of selumetinib plus docetaxel as a second-line treatment for patients with advanced or metastatic non-small cell lung cancer. Ann. Oncol 28, 3028–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe TE, and Johnson GL (2014). MEK inhibition in non-small cell lung cancer. Lung Cancer 86, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, Cottino F, Prahallad A, Grernrum W, Tzani A, et al. (2014). Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep 7, 86–93. [DOI] [PubMed] [Google Scholar]
- Swanton C, and Govindan R (2016). Clinical implications of genomic discoveries in lung cancer. N. Engl. J. Med 374, 1864–1873. [DOI] [PubMed] [Google Scholar]
- Thompson CB, and Allison JP (1997). The emerging role of CTLA-4 as an immune attenuator. Immunity 7, 445–450. [DOI] [PubMed] [Google Scholar]
- Tsatsanis C, Vaporidi K, Zacharioudaki V, Androulidaki A, Sykulev Y, Margioris AN, and Tsichlis PN (2008). Tpl2 and ERK transduce antiproliferative T cell receptor signals and inhibit transformation of chronically stimulated T cells. Proc. Natl. Acad. Sci. USA 105, 2987–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, and Bluestone JA (1994). CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405–413. [DOI] [PubMed] [Google Scholar]
- Weiss A, and Littman DR (1994). Signal transduction by lymphocyte antigen receptors. Cell 76, 263–274. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. (2013). Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med 369, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Lu J, Li L, Feru F, Quan C, Gero TW, Ficarro SB, Xiong Y, Ambrogio C, Paranal RM, et al. (2017). Potent and selective covalent quina-zoline inhibitors of KRAS G12C. Cell Chem. Biol 24, 1005–1016.e3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data Resources
The accession number for the Affymetrix transcriptome analysis reported in this paper is GEO: GSE126202.