

Abstract
Nonalcoholic steatohepatitis (NASH) is emerging as a major public health issue and is associated with significant liver‐related morbidity and mortality. At present, there are no approved drug therapies for NASH. The transcriptional coactivator with PDZ‐binding motif (TAZ; encoded by WW domain‐containing transcription regulator 1 [WWTR1]) is up‐regulated in hepatocytes in NASH liver from humans and has been shown to causally promote inflammation and fibrosis in mouse models of NASH. As a preclinical test of targeting hepatocyte TAZ to treat NASH, we injected stabilized TAZ small interfering RNA (siRNA) bearing the hepatocyte‐specific ligand N‐acetylgalactosamine (GalNAc‐siTAZ) into mice with dietary‐induced NASH. As a preventative regimen, GalNAc‐siTAZ inhibited inflammation, hepatocellular injury, and the expression of profibrogenic mediators, accompanied by decreased progression from steatosis to NASH. When administered to mice with established NASH, GalNAc‐siTAZ partially reversed hepatic inflammation, injury, and fibrosis. Conclusion: Hepatocyte‐targeted siTAZ is potentially a novel and clinically feasible treatment for NASH.
Abbreviations
- +
positive
- α‐SMA+
smooth muscle α‐actin+
- AAV8
adeno‐associated virus 8
- ALT
alanine aminotransferase
- ANOVA
analysis of variance
- BUN
blood urea nitrogen
- Col1/3a1
collagen type I/3, alpha 1
- DMEM
Dulbecco's modified Eagle medium
- FBS
fetal bovine serum
- FDA
U.S. Food and Drug Administration
- GalNAc
N‐acetylgalactosamine
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- H&E
hematoxylin and eosin
- HCC
hepatocellular carcinoma
- HSC
hepatic stellate cell
- IC50
median inhibitory concentration
- IFN
interferon
- Ihh
Indian hedgehog
- IP‐10
interferon‐γ‐induced protein 10
- IVC
inferior vena cava
- MCP‐1
monocyte chemoattractant protein 1
- MMP
matrix metalloproteinase
- mRNA
messenger RNA
- NASH
nonalcoholic steatohepatitis
- NPC
nonparenchymal cell
- PBMC
peripheral blood mononuclear cell
- PBS
phosphate‐buffered saline
- RNAi
RNA interference
- siRNA
small interfering RNA
- sq
subcutaneous
- TAZ
transcriptional coactivator with PDZ‐binding motif
- TGF
transforming growth factor
- Timp1
tissue inhibitor of metalloproteinase 1
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling
- vol
volume
- WWTR1
WW domain‐containing transcription regulator 1
- YAP
yes‐associated protein
Nonalcoholic steatohepatitis (NASH) has emerged as the leading cause of chronic liver disease worldwide due to the ongoing epidemic of obesity.1, 2, 3 This trend is expected to accelerate, particularly with regard to advanced NASH and its devastating consequence, liver fibrosis.4, 5 Because there are no U.S. Food and Drug Administration (FDA)‐approved drugs to treat NASH, there is a critical need for novel therapies that can halt or reverse the progression to hepatic fibrosis, cirrhosis, and hepatocellular carcinoma (HCC).6, 7, 8 We have reported that hepatocytes in human and mouse NASH liver have elevated levels of transcriptional coactivator with PDZ‐binding motif (TAZ; encoded by WW domain‐containing transcription regulator 1 [WWTR1]), a paralogue of yes‐associated protein (YAP) and a transcriptional coactivator of the Hippo pathway that plays a key role in the regulation of organ growth and cell fate.9, 10 We demonstrated that silencing hepatocyte TAZ suppresses steatosis to NASH and NASH‐induced fibrosis progression.11 The mechanism of TAZ‐induced fibrosis in NASH is as follows: TAZ in hepatocytes induces the synthesis and secretion of Indian hedgehog (Ihh), and Ihh activates hepatic stellate cells (HSCs) to promote fibrosis.11
RNA interference (RNAi) technology is a powerful tool for sequence‐specific silencing of disease‐related gene expression and has important clinical applications in diseases for which existing therapies remain insufficient.12, 13 Small interfering RNA (siRNA) can direct sequence‐specific degradation of messenger RNA (mRNA), leading to suppression of synthesis of the corresponding proteins. Nonviral carriers of siRNAs, including polymeric nanospheres and lipid nanoparticles, can deliver nucleic acid molecules and silence genes in vivo. 13, 14 However, the clinical development of nucleic acids as cell type‐specific targeted therapeutics has been stalled because most systemic delivery methods lack organ specificity. Recent studies in mice have shown that siRNA conjugated to N‐acetylgalactosamine (GalNAc), a ligand for the hepatocyte‐specific asialoglycoprotein receptor, is taken up specifically by hepatocytes and causes robust RNAi‐mediated gene silencing in liver after subcutaneous administration.15 Chronic weekly dosing has been shown to be tolerated well in mice. These studies have now moved into humans. In view of the relative instability of siRNA, a key advance has been to chemically modify the RNA structure to enhance stability.16, 17 For example, in subjects with hypercholesterolemia, a single injection with stabilized GalNAc‐siRNA targeting the hepatocyte proprotein convertase subtilisin/kexin type 9 (PCSK9) was effective at lowering low‐density lipoprotein for at least 6 months.17 More recently, the first hepatocyte‐targeted siRNA, patisiran, has been approved for clinical use by the FDA for the treatment of hereditary transthyretin amyloidosis.18, 19
We now show that a stabilized GalNAc‐siRNA targeting hepatocyte TAZ suppresses the progression of steatosis to NASH and partially reverses fibrosis and inflammation in a dietary mouse model of NASH.
Materials and Methods
Cell Culture Studies
Mouse hepatoma Hepa1‐6 cells were purchased from ATCC (CRL‐1830). The cells were grown in Dulbecco's modified Eagle medium (DMEM) (31966047; Gibco) supplemented with 10% fetal bovine serum (FBS), 100 µg/mL streptomycin, and 100 U/mL penicillin and were incubated at 37°C, 5% CO2, and 95% relative humidity. A total of 48 siRNA sequences against mouse Wwtr1 transcript NM_001168281.1 were screened using Hepa1‐6 cells seeded at 1.5 × 105 cells/well in 96‐well plates. Transfections were carried out in quadruplicate at a final siRNA concentration of 10 nmol/L using Lipofectamine 2000 (LP2000) per the manufacturer's instructions (Invitrogen/Life Technologies, Karlsruhe, Germany). The data were normalized to glyceraldehyde 3‐phosphate dehydrogenase (Gapdh) and expressed as percentage of Wwtr1 expression relative to the mean value of three control siRNAs. For dose‐response curves, Hepa1‐6 cells were plated in 96‐well plates by adding 1.5 × 105 cells/well and cultured overnight and then transfected with siRNAs, as described for the initial screening. The final concentrations of each siRNA were 6‐fold dilutions starting at 100 nmol/L for a 10‐point curve. After 24 hours of incubation, the media were removed and the cells were lysed in 150 µL of medium‐lysis mixture (1 volume [vol] lysis mixture, 2 vol cell culture medium) and then incubated at 53°C for 30 minutes. A branched DNA assay was used to assay Wwtr1 mRNA, according to the manufacturer’s instructions. Luminescence was read using a 1420 Luminescence Counter (WALLAC VICTOR Light; Perkin Elmer, Rodgau‐Jügesheim, Germany) following a 30‐minute incubation at room temperature in the dark.
Silencing efficiency of siRNA6 and siRNA8 on TAZ protein was also analyzed as GalNAc conjugates (named siTAZ‐1 and siTAZ‐2, respectively) in Hepa1‐6 cells. Cells were plated at 6.9 × 105 cells/well in six‐well plates and incubated with siRNAs or controls for 48 hours as above. Cell lysates were generated using radio immunoprecipitation assay (RIPA) buffer (#89900; Thermo Fisher Scientific), and subsequent TAZ protein levels were analyzed by immunoblotting, as described below. The gene‐silencing activity of siTAZ‐1 and siTAZ‐2 was also tested using primary mouse hepatocytes (HepaCur Mouse Hepatocytes [C57BL/6]; Yecuris Corp., OR), which were seeded at 3 × 104 cells/well in 96‐well plates. Using a 7‐point dose‐response curve in 10‐fold dilutions starting at 10 µmol/L, the cells were incubated with siRNA in the absence of a transfection reagent. After 24 hours, Wwtr1 mRNA was assayed as above.
Primary Cell Isolation
Primary hepatocytes and nonparenchymal cells (NPCs) were isolated from 8‐9‐week‐old C57Bl/6J mice. For hepatocytes, the inferior vena cava (IVC) was cannulated, the portal vein was cut, and the IVC was perfused with solution 1 (8 g/L NaCl, 0.4 g/L KCl, 0.12 g/L Na2HPO4, 0.09 g/L NaH2PO4·H2O, 2.38 g/L 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid [Hepes], 0.35 g/L NaHCO3, 0.19 g/L ethylene glycol tetraacetic acid, and 0.9 g/L glucose, pH 7.35) at 5 mL/minute flow rate for 2‐3 minutes. The liver was then perfused with solution 2 (8 g/L NaCl, 0.4 g/L KCl, 0.12 g/L Na2HPO4, 0.09 g/L NaH2PO4·H2O, 2.38 g/L Hepes, 0.35 g/L, NaHCO3, and 0.56 g/L CaCl2·2H2O, pH 7.35) containing 0.3 mg/mL collagenase type 1 (Worthington) for 20 minutes. Hepatocytes were filtered through a 100‐μm filter and harvested after two centrifugations at 90g for 1 minute and an additional purification of viable cells on a Percoll cushion. Cells were seeded on plates coated with collagen type 1 (#354236; Corning) in Williams' E medium supplemented with 10% (vol/vol) FBS, 2 mM glutamine, and 5 μg/mL insulin. Seeding medium was removed after a 5‐hour period and replaced with a Williams' E medium without FBS but with 1 mg/mL bovine serum albumin. NPCs were isolated as described,20 with a few modifications. Briefly, the IVC was perfused sequentially with solutions containing protease (P5147; Sigma Aldrich) and collagenase D (11088866001; Roche). The perfusates were subjected to 17% Nycodenz (#1002424; Accurate Chemical) gradient centrifugation to isolate the NPCs, which were then plated in tissue culture dishes in DMEM containing 10% FBS. Five hours after seeding, cells were washed with the same medium to remove unattached cells. HSCs were isolated from 5‐6‐month‐old BALB/C mice, as described.20 Briefly, the perfusion was carried out as with NPCs, but the perfusates were subjected to 9.7% Nycodenz gradient centrifugation to isolate HSCs, which were then plated in tissue culture dishes and used the next day. For bone marrow‐derived macrophages, mice were euthanized with CO2 and hind legs were removed. Femurs were flushed using a 26‐gauge needle with DMEM containing 4.5 g/L glucose; 20% L‐cell media, which is a source of macrophage‐colony stimulating factor; 10% heat‐inactivated FBS; and 1% penicillin/streptomycin. Cell suspensions were passed over a 40‐μm filter, centrifuged at 500g, resuspended in 50 mL of media, and plated into five 100‐mm dishes. Cells were incubated for 4 days, after which nonadherent cells and debris were aspirated, followed by addition of fresh medium. After 7 to 10 days of differentiation, the macrophages were harvested and replated for experiments.21
IFN‐α and IFN‐γ‐Induced Protein 10 Determination
Interferon‐α (IFN‐α) and IFN‐γ‐induced protein 10 (IP‐10; [also known as C‐X‐C motif chemokine ligand 10 (CXCL10)]) protein concentrations were quantified in the conditioned media of human peripheral blood mononuclear cells (PBMCs) from three different donors. Resuspended PBMCs (1 × 105) in serum‐free Roswell Park Memorial Institute 1640 medium were reversed transfected by incubation for 24 hours at 37°C, 5% CO2, and 95% relative humidity with Lipofectamine 2000 and 100 nmol/L of the indicated targeting siRNA and nontargeting siRNA, negative control siRNA, or positive control RNAs (Mock, LF2000 only; negative control 1, 2′ O‐methyl oligo‐modified siRNA; positive control 1, unmodified siRNA; positive control 2, CpG oligodeoxynucleotides; positive control 3, unmodified single‐stranded RNA). Cell culture medium (25 µL) was used for measurement of IFN‐α or IP‐10 concentrations, using MesoScale Discovery’s electrochemiluminescence technology (Rockville, MD). A human IFN‐α2a isoform‐specific assay (K151VHK) and a human IP‐10‐specific assay (K151UFK) were applied, using MesoScale’s U‐PLEX platform and according to the supplier’s protocol.
Animal Studies
Male wild‐type C57BL/6J mice (#000664, 8‐10 weeks old) were obtained from Jackson Laboratory (Bar Harbor, ME). The mice were housed in Columbia University Medical Center Institute of Comparative Medicine standard cages at 22°C in a 12‐12‐hour light–dark cycle, with typically three to four mice per cage. After adaptation for 1 week, the mice were assigned randomly to experimental groups and fed the NASH diet (Teklad diet, 160785; Teklad) with drinking water containing 23.1 g/L fructose and 18.9 g/L glucose for the times indicated in the figure legends. Nontargeting GalNAc siRNA (siControl), siTAZ‐1, or siTAZ‐2 from Axolabs (Kulmbach, Germany) or phosphate‐buffered saline (PBS) were delivered as a single subcutaneous injection 9 weeks after the initiation of the NASH diet or as once‐weekly injections between 8 and16 weeks or 16 and 28 weeks of NASH‐diet feeding, using the doses indicated in the individual figures. Three days after the last injection, the mice were killed, and blood, plasma, and selected organs were harvested for further analysis. All animal experiments were performed by following institutional guidelines and regulations and approved by the Institutional Animal Care and Use Committee at Columbia University.
Blood and Plasma Assays
Fasting blood glucose was measured using a glucose meter (One Touch Ultra; LifeScan, Inc.) in mice that were fasted for 5 hours with free access to water. The following plasma assay kits were used in this study: cholesterol (#439‐17501) and triglyceride (#465‐09791, #461‐09891) from Wako; and alanine aminotransferase (ALT; #A526‐120) from TECO Diagnostics. Plasma albumin, total bilirubin, blood urea nitrogen (BUN), and creatinine were assayed in the diagnostic laboratory of the Institute of Comparative Medicine at Columbia University Irving Medical Center.
Immunoblotting of Tissue Proteins and Enzyme‐Linked Immunosorbent Assay
Protein from liver and other tissues was extracted using RIPA buffer (#89900; Thermo Fisher Scientific), and the protein concentration was measured by a bicinchoninic acid assay (#23227; Thermo Fisher Scientific). Proteins were separated by electrophoresis on 4%‐20% Tris gels (EC60285; Life Technologies) and transferred to nitrocellulose membranes (#1620115; Bio‐Rad). The membranes were blocked for 30 minutes at room temperature in Tris‐buffered saline and 0.1% Tween 20 containing 5% (weight/vol) nonfat milk and then incubated with primary antibody in the same buffer at 4°C overnight, using 1:1,000 dilution. The following antibodies were used: TAZ (#8418), YAP (#4912), GAPDH (#3683), and β‐actin (#5125) from Cell Signaling and anti‐GAPDH from Sigma (G9295). The protein bands were detected with horseradish peroxidase‐conjugated secondary antibodies (Cell Signaling) and Supersignal West Pico enhanced chemiluminescent solution (#34080; Thermo Fisher Scientific). Cultured cells were lysed in Laemmli sample buffer (#161‐0737; Bio‐Rad) containing 5% β‐mercaptoethanol, heated at 100ºC for 5 minutes, and then electrophoresed and immunoblotted as above. ELISA kits were used to assay liver transforming growth factor (TGF)‐β1 (DY1679‐05; R&D) and monocyte chemoattractant protein 1 (MCP‐1) (88‐7391‐22; Invitrogen) according to the kit instructions. The matrix metalloproteinase (MMP) activity kit was from Abcam (ab112146).
Immunofluorescence Microscopy
Paraffin sections were rehydrated, subjected to antigen retrieval by placing in a pressure cooker for 10 minutes in Target Retrieval Solution (S1699; Dako), and then blocked with serum. Sections were labeled with primary antibodies overnight, using a 1:150 dilution for α‐smooth muscle actin (α‐SMA) (F3777; Sigma) and F4/80 (MCA497G, Bio‐Rad), followed by incubation with a fluorophore‐conjugated secondary antibody (not for α‐SMA) for 1 hour. Stained sections were mounted with 4′,6‐diamidino‐2‐phenylindole‐containing mounting medium (P36935; Life Technologies) and then viewed on an Olympus IX 70 fluorescence microscope.
Histopathological Analysis
Inflammatory cells in hematoxylin and eosin (H&E)‐stained liver section images were quantified as the number of mononuclear cells per field (20× objective). For other parameters involving various stains, computerized image analysis (ImageJ) was used to quantify the stained area as a percentage of the total area of view; the same threshold settings were used for all analyses. For all analyses, we quantified eight randomly chosen fields per section using three sections per mouse, with 1 mm between sections. Liver fibrosis was assessed by picrosirius (sirius) red (#24901; Polysciences) and by fibrous stage scoring of sirius red‐stained sections by a trained pathologist (H.R.) who was blinded to the identity of the samples as follows: stage 1‐2, patchy sinusoidal fibrosis; stage 2, generalized sinusoidal fibrosis; and stage 2‐3, generalized sinusoidal fibrosis with focal bridging fibrosis. Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick‐end labeling (TUNEL) staining was conducted using a kit from Roche (#12156792910). The images were randomly captured, and quantification was conducted without knowledge of mouse cohort assignment.
Quantitative Reverse‐Transcription Polymerase Chain Reaction
Total RNA was extracted from liver tissue using the RNeasy kit (74106; Qiagen). Complementary DNA was synthesized from 1 μg total RNA using oligo (dT) and Superscript II (Invitrogen). Quantitative polymerase chain reaction (qPCR) was performed with a 7500 Real‐Time PCR system (Applied Biosystems), using SYBR Green chemistry (#4367659; Life Technologies); hypoxanthine‐guanine phosphoribosyltransferase (Hprt) served as the internal control. Primer sequences are listed in Supporting Table S1.
Statistical Analysis
Data were tested for normality using the Kolmogorov‐Smirnov test, and statistical significance was determined using GraphPad Prism software. All data presented in the figures were normally distributed except for the fibrosis‐stage data. Unless otherwise indicated in the figure legends, the normally distributed data were analyzed using the Student t test for two groups with one variable tested and equal variances; one‐way analysis of variance (ANOVA) with Tukey’s posttest for multiple groups with only one variable tested; or two‐way ANOVA with Sidak’s posttests for more than two groups with multiple variables tested. Data are shown as mean values ± SEM or ± SD, as indicated in the figure legends. Differences were considered statistically significant at P < 0.05. In graphs with no symbols indicating statistical significance, the P value was > 0.05.
Results
GalNAc‐siTAZ Structure and Efficacy of Silencing in Hepa1‐6 Cells
For identification of siRNAs targeting murine TAZ, which is encoded by the Wwtr1 gene, a screening library comprising 48 sequences was designed against transcript NM_001168281.1. In order to evaluate Taz siRNA silencing activity in vitro, oligonucleotides were synthesized and transfected into Hepa1‐6 hepatoma cells. At a concentration of 10 nmol/L,18 out of 48 siRNAs resulted in a greater than 50% reduction of Wwtr1 mRNA and four showed greater than 80% Wwtr1 knockdown (Fig. 1A). Dose‐response experiments were performed for the top 11 siRNAs to verify the initially observed mRNA reduction and to determine median inhibitory concentration (IC50) values. The two most potent Taz siRNAs with IC50 values of 0.25 nmol/L (siRNA6) and 0.34 nmol/L (siRNA8) (Fig. 1B) were selected, GalNAc‐modified, and scaled up for additional in vitro and in vivo experiments. The GalNAc‐modified conjugates of siRNA6 and siRNA8 were named siTAZ‐1 and siTAZ‐2, respectively (sequences in Table 1).
Figure 1.
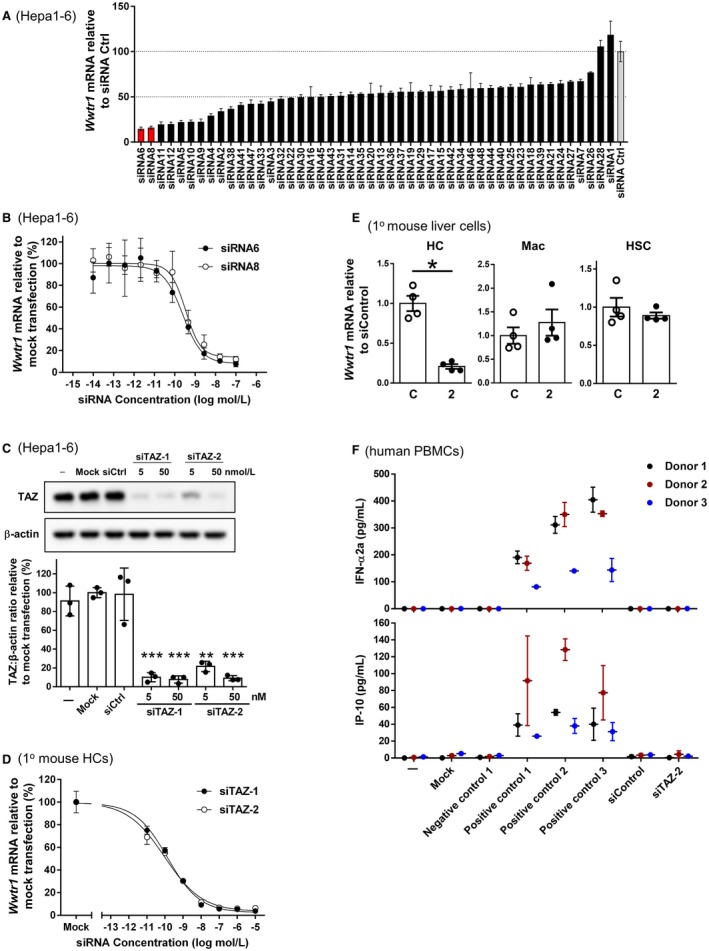
Taz siRNA screen in mouse liver cells. (A) Forty‐eight siRNAs designed against mouse Taz (Wwtr1) were transfected into mouse Hepa1‐6 cells using an siRNA concentration of 10 nM; Wwtr1 mRNA was assayed 24 hours later. Data were normalized to Gapdh, and the values in the graph are shown as percentage of Wwtr1 expression relative to the mean value of three nonspecific control siRNAs (= 100%). The average of four biological replicas ± SD is shown. (B) Dose‐response curves of siRNA6 and siRNA8; the average of four biological replicas ± SD is shown. (C) Hepa1‐6 cells were left untreated (−) or incubated under mock conditions with siCtrl or with 5 or 50 nM siTAZ‐1 or siTAZ‐2, which are the GalNAc conjugates of siRNA6 and siRNA8, respectively. Taz protein was analyzed by immunoblot and quantified by densitometry. (D) Primary mouse hepatocytes were incubated with the indicated concentrations of siTAZ‐1 and siTAZ‐2 and then assayed for Wwtr1 mRNA. The average of four biological replicas ± SD is shown. (E) Primary mouse hepatocytes, bone marrow‐derived macrophages, and HSCs were incubated with 100 nM siControl or siTAZ‐2 (shown as 2) and then assayed for Wwtr1 mRNA. The average of four biological replicas ± SEM is shown. (F) Human PBMCs were left untreated (−); incubated under mock conditions; with a negative control or three positive controls, as defined in Materials and Methods; with siCtrl; or with 100 nM siTAZ‐2. After 24 hours, IFN‐α2a and IP‐10 were assayed. Data are shown for PBMCs from three healthy donors as mean average of two to six biological replicas ± SD; *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: C, siControl; Ctrl, control; HC, primary hepatocyte; HSC, hepatic stellate cell; Mac, bone marrow‐derived macrophage.
Table 1.
Sequences of siTAZ‐1 and siTAZ‐2
| siRNA | Strand Direction | Sense Strand* | Antisense Strand* |
|---|---|---|---|
| siTAZ‐1 | 5′ → 3′ | cscsaGgGcUuGuAgUuUcUuUaAs(GalNAc3) | UsUsaAaGaAaCuAcAaGcCcsdTsdT |
| siTAZ‐2 | 5′ → 3′ | cscsaGuUaAuUuAaGuUuCgAcAs(GalNAc3) | UsGsuCgAaAcUuAaAuUaAcsdTsdT |
Small letters (a, c, g, u), 2′ O‐methyl oligo modification; capital letters, 2′‐F modification; s, phosphorothioate.
The silencing efficiencies of siTAZ‐1 and siTAZ‐2 were tested in Hepa1‐6 cells with Lipofectamine 2000 as the transfection reagent and showed a reduction in TAZ protein of 80%‐90% (Fig. 1C). In primary mouse hepatocytes, siTAZ‐1 and siTAZ‐2 in the absence of any transfection reagent dose dependently lowered Wwtr1 mRNA with IC50 values of 0.14 nmol/L and 0.095 nmol/L, respectively (Fig. 1D). siTAZ‐2 was selected for further in vitro and in vivo characterization. In vitro, we showed that GalNAc‐siTAZ‐2 did not lower Wwtr1 mRNA in macrophages and HSCs (Fig. 1E). To test for an immune response, PBMCs from three different donors were incubated with siTAZ‐2 or nontargeting siRNA, followed by assay of the media for secreted IFN‐α and IP‐10. In this assay, three different positive controls showed a response, but there were no signs of immune stimulation with either siTAZ‐2 or the nontargeting siRNA (Fig. 1F).
GalNAc‐siTAZ Decreases TAZ in the Livers of NASH Mice
In a previous study, we characterized and validated a model of diet‐induced obesity, insulin resistance, and NASH in C57BL/6J mice.11 The diet, which is rich in fructose, palmitic acid, and cholesterol, causes steatosis after 8 weeks and increased TAZ, inflammation, cell death, and early stage fibrosis between 8 and 16 weeks.11 Mice were fed the NASH diet for 9 weeks to induce TAZ expression in liver and then injected subcutaneously with GalNAc‐siTAZ‐1 or ‐siTAZ‐2 (5 and 15 mg/kg), nontargeting GalNAc‐siRNA (siControl; 15 mg/kg), or PBS. Organs were harvested 3 days after injection (Fig. 2A). We found that TAZ protein was ~90% lower in the livers of all four groups of mice injected with GalNAc‐siTAZ, while the related protein YAP was not affected by GalNAc‐siTAZ (Fig. 2B). GalNAc‐siTAZ did not alter TAZ expression in the heart, lung, muscle, spleen, or kidney (Supporting Fig. S1A‐E).
Figure 2.
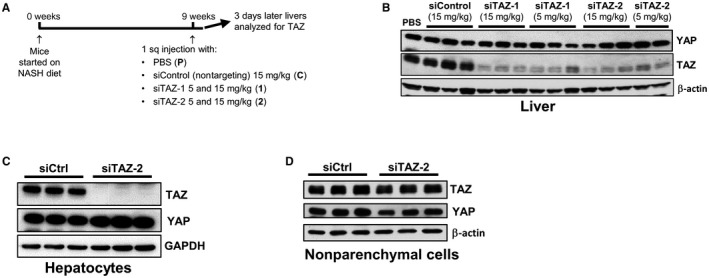
TAZ silencing using GalNAc‐siTAZ in NASH mice. (A) Experimental scheme. Male C57BL/6J mice were fed the NASH diet for 9 weeks and injected with PBS, GalNAc‐siControl, or GalNAc‐siTAZ‐1 or GalNAc‐siTAZ‐2 at the indicated doses. (B) Immunoblots of YAP and TAZ in the livers of the treated mice. (C,D) Immunoblots of TAZ and YAP in primary hepatocytes and NPCs isolated from mice treated with GalNAc‐siControl or GalNAc‐siTAZ‐2. Abbreviations: Ctrl, control; HC, primary hepatocyte.
Further, we isolated hepatocytes and NPCs from C57BL/6J mice from mice treated with GalNAc‐siControl or GalNAc‐siTAZ‐2 and assayed TAZ and YAP. GalNAc‐siTAZ‐2 inhibited TAZ expression in hepatocytes but not in NPCs, and YAP was not decreased in either cell type (Fig. 2C,D). Thus GalNAc‐siTAZ efficiently and specifically lowers liver hepatocyte TAZ in NASH diet‐fed mice. We also tested TAZ silencing efficiency after one injection of GalNAc‐siTAZ in mice fed a high‐fat/cholesterol/fructose diet enriched in transfatty acids (amylin liver NASH [AMLN] diet) for 61 weeks, which induces substantial fibrotic NASH after long‐term feeding.22 As with the NASH model above, GalNAc‐siTAZ was very effective at silencing liver TAZ protein and Wwtr1 mRNA (Supporting Fig. S2).
GalNAc‐siTAZ Decreases the Progression From Steatosis to NASH in Mice
To test the efficacy of TAZ siRNAs in NASH development, we fed male C57BL/6J mice with the NASH diet for 8 weeks, which we showed previously causes steatosis but not NASH.11 The mice were then injected once weekly with 10 mg/kg GalNAc‐siTAZ‐1, GalNAc‐siTAZ‐2, or siControl or an equal volume of PBS subcutaneously for an additional 8 weeks while still on the NASH diet (Fig. 3A). Liver TAZ protein was significantly decreased by GalNAc‐siTAZ‐1 and ‐siTAZ‐2 but not by siControl (Fig. 3B,C). Serum ALT, a marker of liver injury, was unexpectedly decreased by nontargeting siControl, but it was decreased to a greater extent by GalNAc‐siTAZ (Fig. 3D). Body weight, liver:body weight ratio, fasting blood glucose, plasma triglycerides, plasma cholesterol, and liver triglyceride were similar among the four groups of mice (Supporting Fig. S3A‐F).
Figure 3.
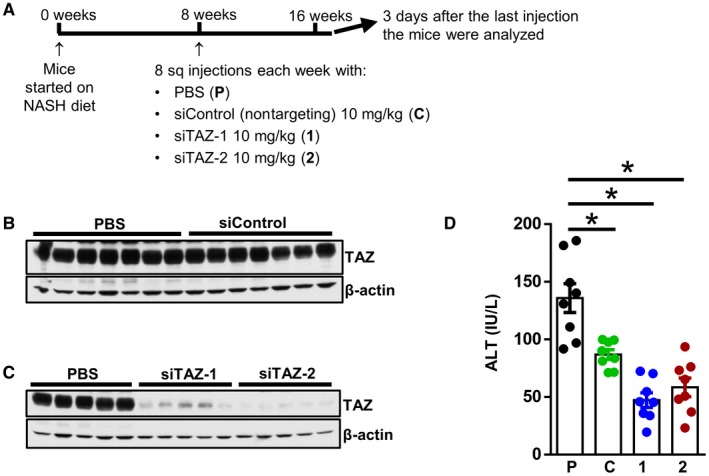
Mice treated with GalNAc‐siTAZ during steatosis to NASH progression have markedly lower liver TAZ without affecting metabolic endpoints, liver triglyceride, or plasma lipids. (A) Experimental scheme. Male C57BL/6J mice were fed the NASH diet for 8 weeks and then injected once weekly for 8 additional weeks with PBS, GalNAc‐siControl, GalNAc‐siTAZ‐1, or GalNAc‐siTAZ‐2 at 10 mg/kg. (B,C) Immunoblots of liver TAZ in mice from the various experimental groups. (D) Assay of plasma ALT. For panel D, n = 8 mice/group, and values shown are mean ± SEM; *P < 0.05 using one‐way ANOVA. Abbreviations: 1, siTAZ‐1; 2, siTAZ‐2; C, siControl; P, phosphate‐buffered saline.
We next evaluated the livers from these mice. As expected,11 livers from PBS‐treated mice that were fed the NASH diet for 16 weeks accumulated inflammatory cells and demonstrated early fibrosis, and both of these parameters were decreased by GalNAc‐siTAZ (Fig. 4A; Supporting Fig. S4A,B). Consistent with the plasma ALT data (above), there was slightly lower inflammation and fibrosis in livers from siControl‐treated mice than in livers from PBS‐treated mice. At the mRNA level, GalNAc‐siTAZ caused a marked decrease in the expression of mRNAs related to both hepatic inflammation (tumor necrosis factor α [Tnfα] and Mcp1) and fibrosis (tissue inhibitor of metalloproteinase 1 [Timp1], collagen, type I, alpha 1 [Col1a1], Col3a1, and Tgfb1) (Fig. 4B). These changes were accompanied by decreases in both F4/80‐positive (+) macrophages and smooth muscle α‐actin+ (α‐SMA+) cells (Fig. 4C; Supporting Fig. S4C,D). In addition, MCP‐1 and TGF‐β1 protein levels were decreased in GalNAc‐siTAZ‐treated livers (Fig. 4D). There was also a decrease in TUNEL+ cells in the livers of GalNAc‐siTAZ‐treated livers (Fig. 4E; Supporting Fig. S4E), indicating that TAZ silencing reduced cell death. Thus, treatment of mice having established steatosis with GalNAc‐siTAZ decreases the progression to NASH.
Figure 4.
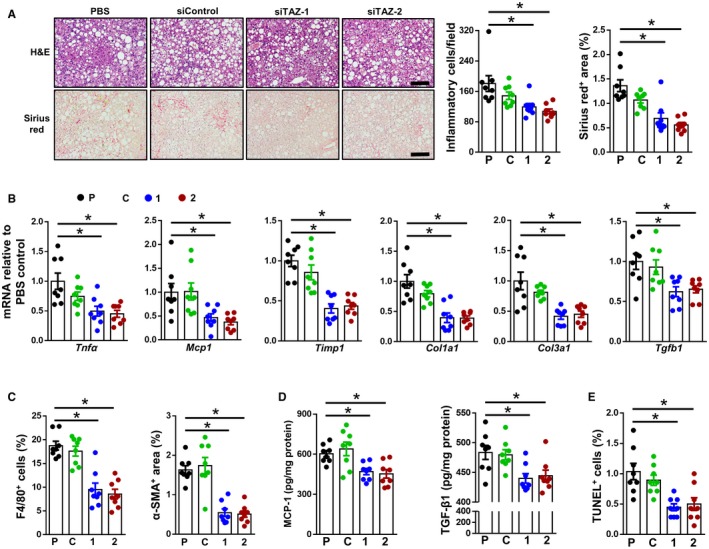
Mice treated with GalNAc‐siTAZ during steatosis to NASH progression show improvements in liver inflammation and fibrosis and less liver cell death. (A) Livers of the mice described in Fig. 3 were stained with H&E (upper row of images) and sirius red (lower row of images) and then quantified for inflammatory cells per field and percentage sirius red area. Scale bar, 200 μm; * indicates difference from P. (B‐E) The following endpoints were assayed in the livers of these mice: (B) indicated mRNAs; (C) percent of F4/80+ cells and α‐SMA+ area; (D) MCP‐1 and TGF‐β1 concentration; and (E) percentage of liver cells that stained for TUNEL. For all panels, n = 8 mice per group; values shown are means ± SEM; *P < 0.05 using one‐way ANOVA. Abbreviations: 1, siTAZ‐1; 2, siTAZ‐2; C, siControl; P, phosphate‐buffered saline; Tnfa, tumor necrosis factor a mRNA.
GalNAc‐siTAZ Decreases Inflammation and Fibrosis in Mice With Established NASH
NASH treatment in humans would have to be effective in halting the progression of NASH or reversing NASH in subjects that present with early NASH. To test the efficacy of GalNAc‐siTAZ in this scenario, we placed mice on the NASH diet for 16 weeks to allow development of early stage NASH and then injected them with PBS or 10 mg/kg GalNAc‐siTAZ‐2 subcutaneously each week for an additional 12 weeks while still on the NASH diet (Fig. 5A). GalNAc‐siTAZ‐2 efficiently lowered TAZ protein and Wwtr1 mRNA but not YAP protein in the livers of these mice (Fig. 5B). Body weight, fasting blood glucose, plasma cholesterol, and plasma triglyceride were similar among the two groups of mice (Supporting Fig. S5A‐D). As expected for 28 weeks of NASH diet feeding, the control PBS‐treated mice had substantial hepatic inflammation and fibrosis as assessed by inflammatory cell content, sirius red area, and fibrosis grading by a liver pathologist, and both of these key endpoints were markedly improved by GalNAc‐siTAZ‐2 (Fig. 5C,D; Supporting Fig. S6A,B). These changes were accompanied by decreases in hepatic mRNAs encoding proteins that mediate inflammation and fibrosis in NASH (Fig. 5E). In addition, GalNAc‐siTAZ caused decreases in both F4/80+ macrophages and α‐SMA+ area (Fig. 5F; Supporting Fig. S6C,D). MCP‐1 and TGF‐β1 protein levels were also decreased in GalNAc‐siTAZ‐2‐treated livers (Fig. 5G). GalNAc‐siTAZ also decreased TUNEL+ cells in the liver and plasma ALT (Fig. 5H,I; Supporting Fig. S6E), indicating less liver damage.
Figure 5.
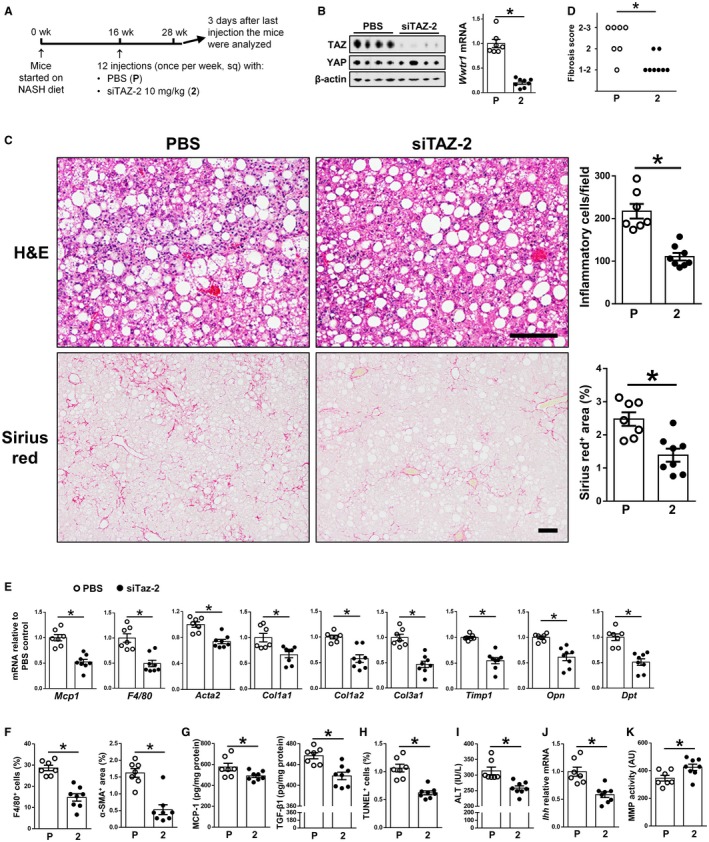
GalNAc‐siTAZ treatment after the development of NASH reduces liver inflammation and fibrosis. (A) Experimental scheme. Male C57BL/6J mice were fed the NASH diet for 16 weeks and then injected once weekly for 12 additional weeks with PBS or GalNAc‐siTAZ‐2 at 10 mg/kg. (B) Livers were immunoblotted for TAZ and β‐actin and assayed for Wwtr1 mRNA. (C) Livers were stained with H&E (upper row of images) and sirius red (lower row of images) and then quantified for inflammatory cells per field and percentage sirius red area. Scale bars, 200 μm. (D) Livers were scored for fibrosis stage as described in Materials and Methods. P value was calculated from the average score for each group using the Wilcoxon rank‐sum test. (E‐K) The following endpoints were measured in the livers or plasma from these mice: (E) indicated mRNAs; (F) percentage of F4/80+ cells and the α‐SMA+ area; (G) MCP‐1 and TGF‐β1 concentrations; (H) percentage of liver cells that stained for TUNEL; (I) plasma ALT; (J) Ihh mRNA; and (K) MMP activity. For all graphs, n = 7 (PBS) and n = 8 (GalNAc‐siTAZ‐2) mice. Values shown for all graphs except panel D are means ± SEM; *P < 0.05 using the two‐tailed Student t test. Abbreviations: 2, siTAZ‐2; Acta2, smooth muscle aortic α‐actin; AU, arbitrary unit; Dpt, dermatopontin; Opn, osteopontin; P, phosphate‐buffered saline.
A key mechanism through which hepatocyte TAZ promotes fibrosis is by inducing the expression and secretion of Ihh, which then activates HSCs to produce collagen.11 In line with this mechanism, GalNAc‐siTAZ‐2 lowered the expression of hepatic Ihh (Fig. 5J). In addition, MMP activity was increased in GalNAc‐siTAZ‐treated livers (Fig. 5K), which may indicate that siTAZ promotes collagen degradation during NASH regression. GalNAc‐siTAZ‐2 did not increase hepatic expression of IP‐10 with tetratricopeptide repeats 1 (Ifit1), Cxcl10, Ifna, or Ifnb (Supporting Fig. S5E), which, like the PBMC data in Fig. 1F, suggests absence of an immune response to the siRNA. GalNAc‐siTAZ‐2 also did not alter thymus cell antigen 1 (Thy1) mRNA, which encodes the HCC‐specific cancer stem cell marker clusters of differentiation (CD)9023 (Supporting Fig. S5F). Further, mice treated with GalNAc‐siTAZ‐2 had similar plasma concentrations of albumin, total bilirubin, creatinine, and BUN as PBS‐treated mice (Supporting Fig. S5G), and GalNAc‐siTAZ treatment was not associated with overt signs of toxicity, e.g., body weights and activity levels were similar to those of control mice. In summary, GalNAc‐siTAZ treatment improves hepatic inflammation and fibrosis and liver damage even in mice with pre‐established NASH.
Discussion
Understanding the pathophysiology of steatosis to NASH progression at a deep mechanistic level has the potential to identify novel therapeutic targets for this widespread and devastating disease for which no FDA‐approved drugs currently exist. Our previous work has identified hepatocyte TAZ as a potential NASH target based on multiple pieces of evidence, including mechanistic and causation studies using human and mouse primary hepatocytes; causation studies in several mouse models of NASH; and supportive evidence from the analysis of liver specimens from humans with various stages of nonalcoholic fatty liver disease.11 The fact that this target is in hepatocytes provides the potential for cell‐specific targeting as siRNA‐based therapies targeting hepatocyte genes have shown efficacy in humans for a variety of diseases,24, 25, 26 including hemophilia,27 hypercholesterolemia and atherosclerotic heart disease,17 and hard to cure cancers.28 Most importantly, the FDA has recently approved the first hepatocyte‐targeted RNAi drug, patisiran, for the treatment of hereditary transthyretin amyloidosis.18 In this context, we have shown here that GalNAc‐siTAZ inhibited several key parameters that drive NASH, including fibrogenesis, inflammation, and cell death, and that GalNAc‐siTAZ reduced fibrosis, the main determinant of outcomes in human NASH,6, 29 both in a preventative setting as well as a therapeutic setting with more advanced fibrosis, i.e., F2‐F3 stage, after 28 weeks. Our previous study showed that the ability of adeno‐associated virus 8 histone H1 (AAV8‐H1)‐siTaz to inhibit NASH fibrosis was mechanistically linked to suppression of hepatocyte Ihh,11 and we show here that GalNAc‐siTAZ lowers hepatic Ihh. However, cell death and inflammation were also lowered by GalNAc‐siTAZ, and these processes have been shown in other studies to contribute to liver fibrogenesis.30, 31 Although our previous study presented detailed mechanistic work linking TAZ to NASH fibrosis,11 the mechanism linking TAZ to cell death and inflammation remains to be investigated.
For hepatocyte targeting, GalNAc has been proven to be effective owing to its being a highly efficient ligand for the hepatocyte asialoglycoprotein receptor.15, 32, 33 In the case of TAZ, hepatocyte‐specific targeting is critical as it has other functions in other cell types that would likely introduce safety concerns to the use of systemic TAZ‐inhibitory therapy.34, 35, 36, 37, 38 Moreover, previously developed small molecule inhibitors target not only TAZ but also the related transcription regulator YAP or the TAZ‐YAP common cofactors TEA domain transcription factor (TEAD)1‐4,39, 40, 41 thus further limiting specificity. Although there could be safety concerns even about targeting only TAZ specifically in hepatocytes, TAZ expression in healthy liver is very low or undetectable.11 Moreover, we observed no changes in body weight or activity level and no other physical signs of illness in NASH mice using three different methods of hepatocyte TAZ targeting (AAV8‐H1‐short hairpin [sh]Taz and treatment of Wwtr1flox [fl]/fl mice with AAV8‐thyroxine binding globulin promoter‐Cre [TBG‐cre] in our previous study11 and GalNAc‐siTAZ here). Moreover, the mice treated with GalNAc‐siTAZ‐2 in the fibrosis reversal experiment showed no decrease in plasma albumin and no increases in total bilirubin, creatinine, or BUN. Nonetheless, safety issues related to both the target TAZ and the therapeutic agent (hepatocyte‐specific stabilized siRNA) will need to be closely monitored. A limitation of our study is that our NASH model induced mild to moderate fibrosis, which equated F2‐F3 fibrosis at 28 weeks at the end of our intervention study. Most studies on NASH‐induced fibrosis focus on patients with F3‐F4 fibrosis in order to measure changes in clinical outcomes. Hence, studying the effect of GalNAc‐siTAZ in mouse models of NASH with F3‐F4 NASH fibrosis may be desirable to confirm that TAZ inhibition leads to fibrosis regression even in these very advanced stages. In the current study, we were also surprised to see a benefit, albeit modest, with nontargeting GalNAc‐siControl versus PBS in certain endpoints (Figs. 3 and 4). This finding will require further investigation as more advanced formulations of hepatocyte‐targeted siTAZ are investigated.
Systemically delivered therapy can be a challenge in humans, but stabilization of siRNA using enhanced stabilization chemistry enables therapeutic siRNAs to be administered at relatively infrequent intervals, e.g., once every 6 months, depending also on the half‐life of the targeted protein.17 In the current study, we used once weekly injections as these were tool compounds that were not yet optimized for a long half‐life. Moreover, the subcutaneous route of therapeutic siRNAs, i.e., rather than intramuscular or intravenous administration, further favors good compliance. Finally, future developments in this field may lead to the possibility of enteral administration.42
In summary, we have demonstrated in a preclinical model that silencing hepatocyte TAZ using a platform shown so far to be safe and effective in humans can both block steatosis to NASH progression and help reverse features of already established NASH. The importance of further work in this area is underscored by the fact that NASH is emerging worldwide as the leading cause of liver transplantation and HCC43 and that no drugs are currently approved to treat this devastating illness.
Supporting information
Acknowledgment
We thank the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center, Columbia University, for histologic analysis and all involved staff members from the respective chemistry, analytical, in vitro, and in vivo laboratories at Sanofi for excellent technical support. Notably, this includes Christiane Metz‐Weidmann, Elke Reinhardt, Claire Kammermeier, Pamela Bindel, Volker Adam, and Bodo Brunner.
Supported by the National Institutes of Health (grants K99‐DK115778 to B.C. and R01‐DK116620 to R.F.S. and I.T.), American Liver Foundation Liver Scholar Award (to X.W.), National Cancer Institute (grant #P30 CA013696 to the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center, Columbia University), and the Sanofi iAwards program (to I.T.).
Potential conflict of interest: Dr. Jahn‐Hofmann, Dr. Kannt, and Dr. Sommerfeld are employed by Sanofi. Dr. Kannt owns stock in Sanofi. Dr. Tabas received a research grant from Sanofi. Dr. Tabas and Dr. Wang have a pending patent application. The other authors have nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Corey KE, Kaplan LM. Obesity and liver disease: the epidemic of the twenty‐first century. Clin Liver Dis 2014;18:1‐18. [DOI] [PubMed] [Google Scholar]
- 2. Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA 2015;313:2263‐2273. [DOI] [PubMed] [Google Scholar]
- 3. Kabbany MN, Conjeevaram Selvakumar PK, Watt K, Lopez R, Akras Z, Zein N, et al. Prevalence of nonalcoholic steatohepatitis‐associated cirrhosis in the United States: an analysis of National Health and Nutrition Examination Survey data. Am J Gastroenterol 2017;112:581‐587. [DOI] [PubMed] [Google Scholar]
- 4. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018;67:123‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Estes C, Anstee QM, Arias‐Loste MT, Bantel H, Bellentani S, Caballeria J, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016‐2030. J Hepatol 2018;69:896‐904. [DOI] [PubMed] [Google Scholar]
- 6. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lombardi R, Onali S, Thorburn D, Davidson BR, Gurusamy KS, Tsochatzis E. Pharmacological interventions for non‐alcohol related fatty liver disease (NAFLD): an attempted network meta‐analysis. Cochrane Database Syst Rev 2017;3:CD011640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. White DL, Kanwal F, El‐Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol 2012;10:1342‐1359.e1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao B, Li L, Lei Q, Guan KL. The Hippo‐YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 2010;24:862‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma S, Meng Z, Chen R, Guan KL. The Hippo pathway: biology and pathophysiology. Annu Rev Biochem 2019;88:577‐604. [DOI] [PubMed] [Google Scholar]
- 11. Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 2016;24:848‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004;432:173‐178. [DOI] [PubMed] [Google Scholar]
- 13. Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol 2006;2:711‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bobbin ML, Rossi JJ. RNA interference (RNAi)‐based therapeutics: delivering on the promise? Annu Rev Pharmacol Toxicol 2016;56:103‐122. [DOI] [PubMed] [Google Scholar]
- 15. Nair JK, Willoughby JL, Chan A, Charisse K, Alam MR, Wang Q, et al. Multivalent N‐acetylgalactosamine‐conjugated siRNA localizes in hepatocytes and elicits robust RNAi‐mediated gene silencing. J Am Chem Soc 2014;136:16958‐16961. [DOI] [PubMed] [Google Scholar]
- 16. Fitzgerald K, Frank‐Kamenetsky M, Shulga‐Morskaya S, Liebow A, Bettencourt BR, Sutherland JE, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single‐blind, placebo‐controlled, phase 1 trial. Lancet 2014;383:60‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med 2017;376:41‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ledford H. Gene‐silencing technology gets first drug approval after 20‐year wait. Nature 2018;560:291‐292. [DOI] [PubMed] [Google Scholar]
- 19. Adams D, Gonzalez‐Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018;379:11‐21. [DOI] [PubMed] [Google Scholar]
- 20. Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High‐yield and high‐purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc 2015;10:305‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin CS, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci U S A 2016;113:6526‐6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kristiansen MN, Veidal SS, Rigbolt KT, Tolbol KS, Roth JD, Jelsing J, et al. Obese diet‐induced mouse models of nonalcoholic steatohepatitis‐tracking disease by liver biopsy. World J Hepatol 2016;8:673‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayashi H, Higashi T, Yokoyama N, Kaida T, Sakamoto K, Fukushima Y, et al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell‐like behaviors contributing to disease progression. Cancer Res 2015;75:4985‐4997. [DOI] [PubMed] [Google Scholar]
- 24. Adams D, Suhr OB, Dyck PJ, Litchy WJ, Leahy RG, Chen J, et al. Trial design and rationale for APOLLO, a phase 3, placebo‐controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol 2017;17:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, et al. Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med 2017;376:1430‐1440. [DOI] [PubMed] [Google Scholar]
- 26. Liebow A, Li X, Racie T, Hettinger J, Bettencourt BR, Najafian N, et al. An investigational RNAi therapeutic targeting glycolate oxidase reduces oxalate production in models of primary hyperoxaluria. J Am Soc Nephrol 2017;28:494‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med 2018;9:135‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim HJ, Kim A, Miyata K, Kataoka K. Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv Drug Deliv Rev 2016;104:61‐77. [DOI] [PubMed] [Google Scholar]
- 29. Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology 2015;61:1547‐1554. [DOI] [PubMed] [Google Scholar]
- 30. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765‐783.e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsuda S, Keiser K, Nair JK, Charisse K, Manoharan RM, Kretschmer P, et al. siRNA conjugates carrying sequentially assembled trivalent N‐acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem Biol 2015;10:1181‐1187. [DOI] [PubMed] [Google Scholar]
- 33. Prakash TP, Graham MJ, Yu J, Carty R, Low A, Chappell A, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N‐acetyl galactosamine improves potency 10‐fold in mice. Nucleic Acids Res 2014;42:8796‐8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hagenbeek TJ, Webster JD, Kljavin NM, Chang MT, Pham T, Lee HJ, et al. The Hippo pathway effector TAZ induces TEAD‐dependent liver inflammation and tumors. Sci Signal 2018;11pii:eaaj1757. [DOI] [PubMed] [Google Scholar]
- 35. Howell M, Borchers C, Milgram SL. Heterogeneous nuclear ribonuclear protein U associates with YAP and regulates its co‐activation of Bax transcription. J Biol Chem 2004;279:26300‐26306. [DOI] [PubMed] [Google Scholar]
- 36. Kaan H, Chan SW, Tan S, Guo F, Lim CJ, Hong W, et al. Crystal structure of TAZ‐TEAD complex reveals a distinct interaction mode from that of YAP‐TEAD complex. Sci Rep 2017;7:2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gandhirajan RK, Jain M, Walla B, Johnsen M, Bartram MP, Huynh Anh M, et al. Cysteine S‐glutathionylation promotes stability and activation of the Hippo downstream effector transcriptional co‐activator with PDZ‐binding motif (TAZ). J Biol Chem 2016;291:11596‐11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Plouffe SW, Lin KC, Moore JL 3rd, Tan FE, Ma S, Ye Z, et al. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J Biol Chem 2018;293:11230‐11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brodowska K, Al‐Moujahed A, Marmalidou A, Meyer Zu Horste M, Cichy J, Miller JW, et al. The clinically used photosensitizer Verteporfin (VP) inhibits YAP‐TEAD and human retinoblastoma cell growth in vitro without light activation. Exp Eye Res 2014;124:67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feng J, Gou J, Jia J, Yi T, Cui T, Li Z. Verteporfin, a suppressor of YAP‐TEAD complex, presents promising antitumor properties on ovarian cancer. Onco Targets Ther 2016;9:5371‐5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu‐Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD‐YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012;26:1300‐1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murakami M, Nishina K, Watanabe C, Yoshida‐Tanaka K, Piao W, Kuwahara H, et al. Enteral siRNA delivery technique for therapeutic gene silencing in the liver via the lymphatic route. Sci Rep 2015;5:17035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Younossi Z, Stepanova M, Ong JP, Jacobson IM, Bugianesi E, Duseja A, et al. Global Nonalcoholic Steatohepatitis Council. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin Gastroenterol Hepatol 2019;17:748‐755.e753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials