

Abstract
Inductively coupled plasma mass spectrometry (ICP-MS) is an analytical technique that can be used to measure elements at trace levels in biological fluids. Although older techniques such as atomic absorption and atomic emission are still in use by some laboratories, there has been a slow shift toward ICP-MS, particularly in the last decade. As this shift is likely to continue, clinical scientists should be aware of the analytical aspects of ICP-MS, as well as the potential for both spectroscopic and non-spectroscopic interference, and strategies that can be employed to eliminate or mitigate these issues.
Introduction
The measurement of trace elements in biological samples is useful in a large number of clinical settings. Table 1 lists a number of elements of clinical interest, along with an approximate guide to the concentration ranges which may be encountered in biological samples. Elements monitored for nutritional purposes include essential elements such as iodine, manganese, copper, selenium and zinc.1 These elements play important roles in a wide range of biological processes including electron transport, oxygen transport, hormone synthesis and catalysis of biological reactions.1 Disturbances in normal homeostasis of these elements may cause (or be a symptom of) one or more pathophysiological conditions. Other elements such as arsenic, cadmium, mercury and lead are known to exert toxic effects (often through a variety of different mechanisms), and are therefore measured to assess exposure.2 A wide range of analytical techniques have historically been used for trace element analysis. This review will focus on the analytical aspects underlying ICP-MS.
Table 1.
Element concentration ranges which may be encountered in biological samples (blood or urine).
| Element | Clinical application | Approximate concentration range* |
|---|---|---|
| Aluminium | Toxic | 0.1–10 μmol/L |
| Antimony | Toxic | 1–30 nmol/L |
| Arsenic | Toxic | 0.01–80 μmol/L |
| Barium | Toxic | 7–700 nmol/L |
| Beryllium | Toxic | 1–150 nmol/L |
| Bismuth | Toxic, Therapeutic | 1–200 nmol/L |
| Bromide | Toxic, Therapeutic | 0.1–40 mmol/L |
| Cadmium | Toxic | 1–100 nmol/L |
| Chloride(sweat) | Metabolic | 10–200 mmol/L |
| Chromium | Toxic | 1–5000 nmol/L |
| Cobalt | Toxic | 1–5000 nmol/L |
| Copper | Nutritional, Metabolic | 1–50 μmol/L |
| Gold | Therapeutic | 1–10 μmol/L |
| Iodine | Toxic, Nutritional | 0.008–200 μmol/L |
| Lead | Toxic | 0.01–10 μmol/L (~0.2–200 μg/dL) |
| Manganese | Nutritional | 1–400 nmol/L |
| Mercury | Toxic | 1–1000 nmol/L |
| Molybdenum | Toxic | 1–20 nmol/L |
| Nickel | Toxic | 1–200 nmol/L |
| Selenium | Toxic, Nutritional | 0.1–10 μmol/L |
| Silver | Skin Pigmentation | 1–500 nmol/L |
| Thallium | Toxic | 1–50 nmol/L |
| Tin | Toxic | 0.2–500 nmol/L |
| Vanadium | Toxic | 1–1000 nmol/L |
| Zinc | Nutritional | 1–40 μmol/L |
These ranges are meant as a guide only; toxic levels may occasionally exceed the upper limit of the quoted range.
The Analytical Shift Toward ICP-MS
Although ICP-MS was first developed over 30 years ago, a number of older techniques are still in use by some laboratories.3 Table 2 lists advantages and disadvantages of ICP-MS compared to other techniques such as inductively coupled plasma atomic emission spectroscopy (ICP-AES), flame atomic emission (flame photometry), flame atomic absorption, graphite furnace atomic absorption and cold vapour/hydride generation atomic absorption. From a laboratory perspective, perhaps the most significant advantage of ICP-MS is its multi-element capability, which allows multiple elements to be measured simultaneously in a single analysis. This is in contrast to flame and graphite furnace atomic absorption where the lamp is specific for a limited number of elements, therefore only one (or a few) elements can be measured at a time. Coupled with short analysis time and simple sample preparation, ICP-MS offers the opportunity for very high sample throughput in the laboratory.
Table 2.
Advantages and disadvantages of selected methods for analysis of trace elements.
| Technique | Advantages | Disadvantages |
|---|---|---|
| ICP-MS | Multi-element technique Large analytical range Low detection limit High sample throughput Low sample volume Simple sample preparation High-resolution and tandem mass spectrometry (triple-quadrupole) instruments offer a very high level of interference control |
Equipment cost Operating cost (argon) Multiple high purity gases required High level of staff expertise Interferences need to be controlled Laboratory set up costs (air-conditioning, HEPA filters, pipe work, dust reduction measures) |
| ICP-AES (also known as ICP-OES) | Multi-element technique Large analytical range High sample throughput Low sample volume Simple sample preparation |
High detection limit Equipment cost High level of staff expertise Laboratory set up costs |
| Flame Atomic Emission | Equipment cost Low level of staff expertise Simple sample preparation Low laboratory set up cost |
Single element technique Limited analytical range High detection limit Higher sample volume Flammable gases |
| Flame Atomic Absorption | Equipment cost Low level of staff expertise Simple sample preparation Low laboratory set up cost Reasonably high sample throughput Few interferences |
Single element technique Limited analytical range High detection limit Higher sample volume Flammable gases |
| Graphite Furnace Atomic Absorption | Low detection limit Equipment cost Few interferences Low sample volume Low laboratory set up cost Simple sample preparation (in most cases) |
Single element technique Limited analytical range Low sample throughput Some elements require acid digestion prior to analysis |
| Atomic Absorption (cold vapour/ hydride generation) | Low detection limit Equipment cost Few interferences Low laboratory set up cost |
Suitable for limited elements Limited analytical range Low sample throughput High sample volume Complex acid digestions required for biological samples |
ICP-MS: inductively coupled plasma mass spectrometry; ICP-AES: inductively coupled plasma atomic emission spectroscopy; ICP-OES: inductively coupled plasma optical emission spectrometry; HEPA filter: high efficiency particulate air filter.
A wide range of factors are worth considering when evaluating the suitability of a technique for a particular clinical application. ICP-MS instruments are quite expensive, and this cost may be prohibitive to laboratories. For a low-volume laboratory it may be more economical to purchase a graphite furnace instrument, whereas an ICP-MS is more cost-effective for a high-volume laboratory. The required lower limit of detection of the analyte(s) will also dictate the technique of choice. For example, flame atomic absorption instruments do not have the necessary detection limits to measure certain elements of clinical interest.
A Brief Overview of ICP-MS
There are six fundamental compartments of a single quadrupole ICP-MS: the sample introduction system, inductively coupled plasma (ICP), interface, ion optics, mass analyser and detector. Figure 1 shows a simple diagram of the instrument. Liquid samples are first nebulised in the sample introduction system, creating a fine aerosol that is subsequently transferred to the argon plasma. The high-temperature plasma atomises and ionises the sample, generating ions which are then extracted through the interface region and into a set of electrostatic lenses called the ion optics. The ion optics focuses and guides the ion beam into the quadrupole mass analyser. The mass analyser separates ions according to their mass-charge ratio (m/z), and these ions are measured at the detector.
Figure 1.
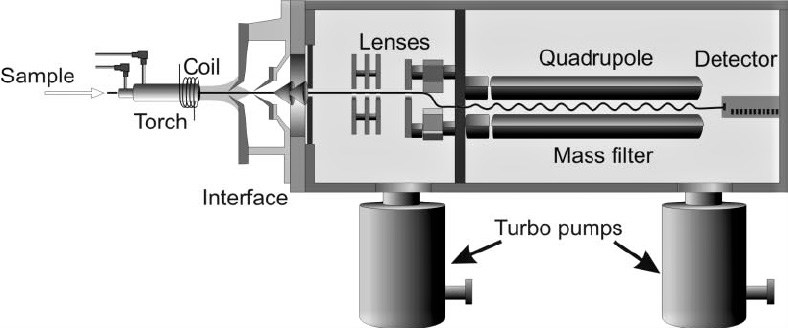
Cross section schematic of an ICP-MS (adapted from ref. 86).
Sample Preparation
Sample preparation for ICP-MS is relatively simple; biological samples are usually diluted or thermally digested before analysis. Common diluents include dilute acids (e.g. nitric acid, hydrochloric acid) or alkali (e.g. ammonium hydroxide, tetramethylammonium hydroxide).4–8 Deionised water has been used as a diluent, however some elements are unstable in pure water, therefore acidic or alkaline diluents are preferred in most cases.9,10 The isoelectric point of many proteins is approximately 5–6, therefore the addition of an acidic diluent to a highly proteinaceous sample such as blood may cause protein precipitation, which may co-precipitate certain analytes or cause an obstruction in the nebuliser.10 Proteins tend to be more tolerant of dilute alkali, however not all elements are soluble at alkaline pH, therefore a chelating agent such as EDTA is often incorporated into alkaline diluents. Surfactants such as Triton-X100 are also commonly added to help solubilise and disperse lipid and membrane proteins in the sample.
A total dissolved solids (TDS) content in the sample of <0.2% (2 g/L) is usually recommended in ICP-MS to reduce sample-specific matrix effects and the potential for nebuliser blockage. Given that inorganic components of serum (such as Na+, Cl−, HCO3−, K+, Ca2+, PO43− and Mg2+) account for approximately 1% of the total mass, a dilution factor between 10 and 50 is usually adequate for biological fluids.11 Solid samples such as tissue (e.g. liver), hair and nails require chemical digestion to dissolve the sample before analysis. The digestion can be carried out using strong acid(s) or alkali, either at room temperature or heated in a hot water bath, dry heating block or high-pressure microwave to assist digestion.12–14
Sample Introduction
ICP-MS instruments are primarily designed to analyse liquids. Solid samples can be analysed directly using electrothermal vaporisation or laser-ablation, however these techniques are not routinely used in clinical biochemistry laboratories and are hence outside the scope of this review.15,16
Liquid Sample Introduction
Before being introduced into the ICP, liquid samples are first aerosolised by a nebuliser. A common configuration uses an autosampler and a peristaltic pump to deliver the sample to the nebuliser, although self-aspirating nebulisers are also available.
Nebuliser
A number of different nebulisers are commercially available, including pneumatic, ultrasonic and desolvating types. Pneumatic nebulisers which use gas flow to generate the aerosol are the most common type for routine clinical applications. There are several different types of pneumatic nebulisers, including concentric, cross-flow, Babington and v-groove (a variant of the Babington). Each has its own advantages and disadvantages; the choice is application dependent. Concentric nebulisers are ideal for low matrix (low TDS) samples, whereas cross-flow and v-groove nebulisers are more rugged, tolerating high matrix samples. In contrast to the pneumatic design, an ultrasonic nebuliser utilises sound energy (from a piezoelectric transducer) to generate the aerosol. These nebulisers improve analytical sensitivity by an order of magnitude compared to pneumatic nebulisers, however they are significantly more expensive.16 Desolvating nebuliser systems use a heated spray chamber to desolvate the sample before it reaches the plasma.17 In addition to increasing sensitivity, desolvating nebulisers also decrease the formation of oxide species in the plasma which can interfere with the measurement of certain analytes (discussed in Interference section).17
Spray Chamber
After being aerosolised by the nebuliser, the sample enters the spray chamber. The spray chamber has a simple design but serves a few important purposes: it selectively filters out the larger aerosol droplets that are generated by the nebuliser and acts to smooth out nebulisation ‘pulses’ produced by the peristaltic pump.18 This is important because the plasma is inefficient at dissociating large droplets (>10 μm diameter). In a double-pass spray chamber, aerosol droplets emerge from the nebuliser and travel down a central tube in the spray chamber. At the end of the tube, larger aerosol droplets exit the spray chamber under the influence of gravity and are drained to waste while smaller droplets, roughly <10 μm diameter, are transferred to the plasma. Cyclonic spray chambers have a different design but operate in a similar manner. In contrast to techniques such as graphite furnace atomic absorption, the sample introduction process in ICP-MS is quite inefficient. Normally only 1–2% of the sample reaches the plasma; the remainder is drained to waste.18 Therefore factors which influence the efficiency of sample introduction even slightly can have marked effects on instrument response. One such factor is the spray chamber temperature, which is normally maintained at around 2 °C using a thermoelectric cooling device or water jacket. Operating the spray chamber at this temperature minimises the formation of oxides, and prevents overloading the plasma with solvent (which can have a deleterious effect on analytical performance).
Hyphenated ICP-MS (HPLC-ICP-MS, GC-ICP-MS)
As is the case with older techniques such as flame and graphite furnace atomic absorption, ICP-MS completely atomises the sample, hence different chemical forms (‘species’) of an element are indistinguishable after the sample reaches the plasma. If information on the different forms of an element are sought, chromatographic systems such as ion-exchange HPLC or gas chromatography can be coupled to the ICP-MS by simply connecting the end of the analytical column to the nebuliser with a capillary tube.19 By optimising chromatographic conditions, different species of an element can be effectively separated before the sample reaches the plasma, allowing each fraction to be measured individually. This process is called speciation. Speciation is useful in certain clinical contexts because different species can have remarkably disparate toxicological profiles. For example, inorganic forms of arsenic such as arsenite (AsIII) or dimethylarsinic acid are considerably more toxic than organic forms such as arsenobetaine which is found in seafood.20
Inductively Coupled Plasma
A plasma is essentially an ionised gas, consisting of positively-charged ions and free (unbound) electrons. The role of the plasma (ICP) in ICP-MS is to ionise the sample. In contrast to so-called ‘soft’ ionisation sources used in other forms of mass spectrometry (such as electrospray) which impart relatively little energy to the analyte, the ICP is considered a ‘hard’ ionisation technique because it completely atomises most molecules in the sample.21 ICP-MS instruments use an argon plasma, although helium plasmas have also been described.22,23 Although there are several advantages to using helium, argon is preferred as the cost of helium is prohibitive.
The plasma is formed in the end of a set of three concentric quartz tubes, collectively referred to as the torch. Argon gas flows through all three tubes. The inner tube is called the injector, and contains the sample aerosol in a stream of argon which delivers the sample to the plasma. Concentric to this tube is a tangential flow of argon called the auxiliary gas, which forms the plasma. The outer tube contains a flow of argon which serves as a cooling layer to prevent the torch from melting. The far end of the torch is surrounded by a copper induction coil (or ‘load coil’), which is connected to a radio frequency (RF) generator. The RF generator supplies power to the load coil, creating a high-frequency alternating current which in turn induces a time-varying electromagnetic field in the torch. With argon gas flowing through the torch, a high-voltage discharge (called a tesla spark) is applied, which ionises a fraction of the argon atoms generating ions and electrons. Ions and electrons in the torch are influenced by the electromagnetic field, and are accelerated and collide with other argon atoms. If these collisions impart sufficient energy, additional atoms are ionised creating electrons and ions which propagate the cascade. The movement of electrons and ions in the torch generate a tremendous amount of heat. The result is called an ICP, which can reach a temperature of up to 10,000 Kelvin (hotter than the surface of the sun).16
Ionisation
After the sample is nebulised, the so-called tertiary aerosol exiting the spray chamber (comprising the smallest droplets of the primary aerosol generated by the nebuliser) is swept in a stream of argon gas along the injector and into the plasma. After reaching the high-temperature plasma, the sample is desolvated, vaporised, atomised and ionised. Most elements form singly charged positive ions, however some elements may also form a small fraction of double charged ions.24 The degree to which an element is ionised depends on the temperature of the plasma and the ionisation potential of the element. This relationship is described by the Saha equation.16 At a given temperature, the degree of ionisation (%) in an argon plasma decreases as the ionisation potential increases, declining to almost zero at values higher than 15.76 eV (the ionisation potential of argon itself). Fortunately, most elements have a first ionisation potential much lower than argon, and are therefore efficiently ionised in the plasma. ICP-MS is therefore able to measure nearly all elements in the periodic table, a significant advantage over techniques such as flame atomic absorption which struggles with refractory elements in the oxide form due to the lower temperatures encountered in the flame.
The analytical sensitivity of an analyte in ICP-MS is linked to its overall ionisation as well as the isotopic abundance of the measured isotope. While negative ion detection in other forms of mass spectrometry (namely LC-MS) has proven particularly useful for analytes which exhibit poor ionisation in positive ion mode, this approach in ICP-MS has been mostly unsuccessful.25
Interface
A pair of coaxial nickel (or platinum) cones separate the plasma from the mass spectrometer vacuum chamber (Figure 2). The first cone (in contact with the plasma) is called the sample cone, and the second is called the skimmer cone. Ions, photons and neutral atoms or molecules are extracted from the plasma into the interface region via a small orifice at the tip of the sample cone (~1 mm diameter). A mechanical roughing pump is used to maintain an interface pressure (between the cones) of ~150–300 Pa. As ions enter this interface region, the dramatic reduction in pressure causes a supersonic expansion of the ions, generating a so-called free jet.26 Ions are subsequently extracted through an even smaller orifice in the skimmer cone (~0.45 mm diameter), and into the main vacuum chamber which is held under high vacuum (~7x10−5–1x10−3 Pa) by a turbomolecular pump. At this pressure, ions can be guided effectively by charged surfaces called electrostatic lenses. A cooling fluid circulates continuously between a chiller unit and the instrument (particularly the RF coil and the interface cones) to prevent these components from overheating.
Figure 2.
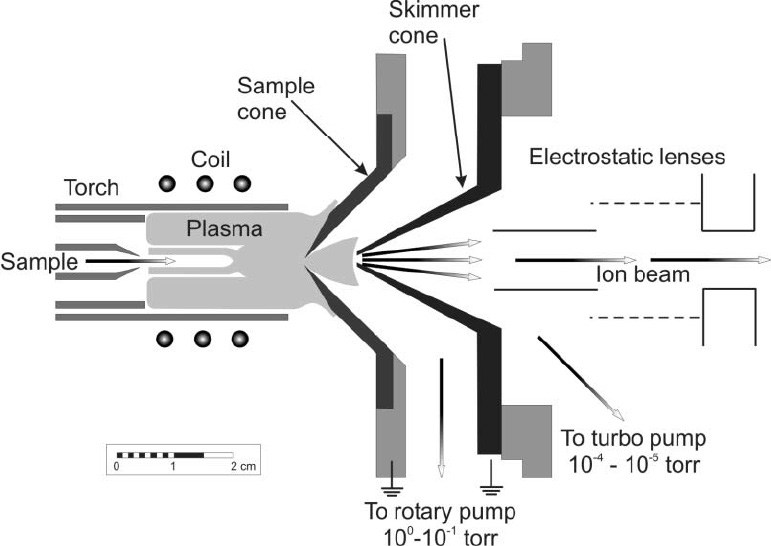
Cross section schematic of the interface region (adapted from ref. 86).
Ion Optics
The set of electrostatic lenses located behind the skimmer cone are collectively referred to as the ion optics. The role of the ion optics system is to guide the ion beam toward the mass analyser, and to prevent photons and other neutral species (such as non-ionised matrix components) from reaching the detector. While photons are the basis of measurement in atomic emission techniques such as flame photometry and ICP-AES, in ICP-MS they are a source of noise and signal instability if they are permitted to reach the detector. The ion optics usually prevent this from occurring by positioning a ‘photon-stop’ (or ‘shadow-stop’) in the ion path, or by guiding the ion beam off-axis.27 For example, Agilent instruments often have an ‘omega lens’ (so-named because it resembles the Greek letter Ω) which shifts the ion beam slightly off-axis, as shown in Figure 1. The design of the interface and ion optics may differ between manufacturers; for example the Perkin Elmer NexION 2000 has a triple cone interface, with a third cone (called a hyperskimmer) positioned behind the skimmer cone, and an ion-beam deflector in lieu of traditional ion optics.
Mass Analyser
After transiting through the ion optics system, ions arrive at the mass analyser. Several different types of mass analyser have been used for ICP-MS; these include quadrupole, magnetic sector and (rarely) time-of-flight (TOF).7,28 By far the most common type used for routine clinical biochemistry applications is the quadrupole mass analyser.
Quadrupole
As is the case with all mass analysers, a quadrupole is essentially a mass filter, separating ions based on their m/z ratio (defined as the mass of an ion divided by its charge). A quadrupole consists of four parallel hyperbolic or cylindrical metallic rods (normally 15–20 cm long) positioned in a square array. Radio frequency alternating current (AC) and direct current (DC) potentials are applied to the rods, creating a time-varying electric field in the centre through which ions pass. For an ion with a particular m/z ratio, only specific combinations of AC and DC potentials result in a stable ion flight trajectory through the quadrupole.29 Ions with unstable trajectories collide with the rods and are not transmitted through the quadrupole. These voltages can be ramped very rapidly, allowing the entire mass range to be scanned within a matter of milliseconds. The high scan speed of modern instruments allows data to be acquired for multiple elements virtually simultaneously. For the clinical laboratory, this confers a considerable advantage over other techniques such as flame and graphite furnace atomic absorption, which are single element techniques.
The length of time the instrument spends acquiring data at a particular m/z value is called the dwell time. Longer dwell times allow for more accurate (and sensitive) measurements at the detector by averaging the signal for a longer period of time. A number of other quadrupole parameters such as the mode of data acquisition (continuous scanning or peak-hopping) and the number of scans or replicates can also be defined by the operator; a detailed discussion of these parameters is outside the scope of this review.
Resolution
The resolution of a mass analyser is a measure of its ability to resolve adjacent masses. There are two ways of defining resolution in ICP-MS. The first is to measure the width of the mass peak at a particular peak height (usually 10% of the maximum). For quadrupole analysers, the resolution here is normally approximately 0.75 amu (atomic mass units). The second measure involves calculating the ratio M/ΔM, where M is the mass of the analyte peak and ΔM is the difference in mass to the nearest peak that can be separated. Using this second measure, quadrupole analysers for ICP-MS usually operate at a resolution of approximately 300 (unit-less measure).30 This resolution is adequate for most clinical applications, however quadrupole instruments are unable to resolve many cases of spectroscopic interferences (discussed in Interference section).
Abundance Sensitivity
Another property of a mass analyser worth mentioning is the abundance sensitivity. The abundance sensitivity is the contribution that a signal for an isotope at a certain mass (M) makes to adjacent masses (M-1, and M+1). In other words, abundance sensitivity is a measure of peak tailing, which can cause interference when the signal at one mass is considerably higher than the signal at an adjacent mass (discussed in Interference section). A typical value for abundance sensitivity of a quadrupole is 10−7 (i.e. if the signal for an isotope at mass M is 107 units, there will be a contribution of 1 unit at M±1). Mass peaks normally have a slight negative skew, hence the abundance sensitivity is worse on the low mass side.
Detector
The most common detector used for ICP-MS is an electron multiplier (EM). Positively-charged analyte ions strike the first dynode of the detector which is held at a high negative voltage. The impact of the ion on the detector causes the emission of several electrons from the surface, which, in turn, strike the next dynode releasing more electrons. This process (called secondary emission) continues, generating an amplification cascade that culminates in a signal large enough to be measured reliably as an ion ‘count’. In this way, an EM can generate a measurable signal pulse from the impact of a single ion on the detector, conferring very high analytical sensitivity. In fact, detection limits in ICP-MS are far superior to flame atomic absorption, and are comparable (or superior) to graphite furnace atomic absorption (Figure 3). Typical limits of detection in ICP-MS are in the nmol/L range for most elements; the exact value being dependent on the element, the type of biological matrix, the dilution factor employed during sample preparation, the design of the sample introduction system, instrument operating conditions (including plasma temperature) and background signals (reagent purity etc.). Most detectors are able to operate in both a pulse (digital) and analogue mode. These so-called dual detectors automatically switch from pulse to analogue mode when the signal intensity exceeds a certain threshold, allowing the linear dynamic range of the detector to be extended to approximately 8–12 orders of magnitude. These two detector modes require cross-calibration to ensure optimum linear response across this range.
Figure 3.
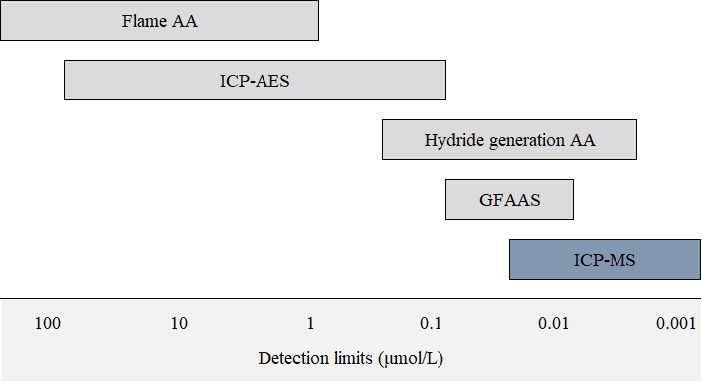
Typical detection limits for common elemental techniques.
AA: atomic absorption; ICP-AES: inductively coupled plasma atomic emission spectroscopy; GFAAS: graphite furnace atomic absorption; ICP-MS: inductively coupled plasma mass spectrometry.
Calibration Strategies
External Calibration
The signal measured by the ICP-MS detector is in units of ‘counts per second’ (CPS). This corresponds to the number of ions striking the detector every second. In order to convert this data to a concentration value, calibration standards containing known concentrations of elements can be used to construct a calibration curve. This technique is called external calibration. Figure 4 shows an example of a calibration curve for arsenic in blood. The relationship between signal (CPS) and concentration is linear over approximately 8–12 orders of magnitude. This is in contrast to atomic absorption techniques where the calibration can become non-linear at high analyte concentration.31
Figure 4.
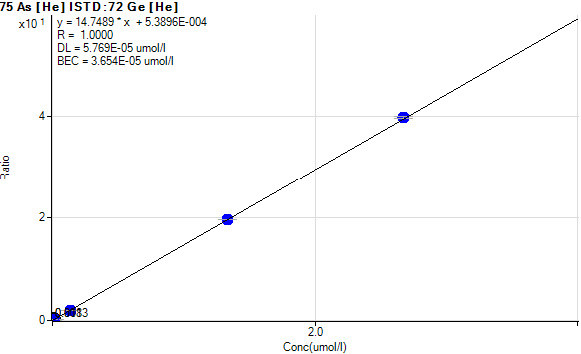
Calibration curve for arsenic in blood.
Compound-Independent Calibration
As stated earlier, biological molecules are nearly completely atomised in the high-temperature plasma. Therefore, the signal for an element will theoretically be independent of the molecular form of the element (i.e. 1 nmol of arsenite should generate the same signal for arsenic as 1 nmol of its methylated derivative dimethylarsinic acid). This presents the unique opportunity to perform compound independent calibration (CIC) for elemental speciation methods. This technique involves calibrating one chemical species of an element using another as the calibrant. This is useful for situations in which an analyte is not available commercially or is prohibitively expensive. CIC also allows unknown peaks in a chromatogram to be quantified and non-targeted screening approaches for speciation methods. For example, non-targeted screening of organofluorine compounds using ICP-MS detection offers a promising alternative to traditional targeted electrospray-LCMS methods.32 Although fluorine is widely considered not measurable by ICP-MS due to insignificant ionisation, post-column addition of a solution containing barium allows fluorine to be measured indirectly as BaF (at m/z 157).32 A significant proportion of the mass-fraction of fluorine in blood is derived from unknown compounds, therefore non-targeted screening using HPLC-ICPMS could prove particularly useful.33
Caution should be exercised, however; in some instances CIC may produce inaccurate results. For example, inorganic and organic tin compounds have been shown to exhibit compound-dependent responses with ICP-MS.34 Elemental responses were correlated with compound volatility. As might be expected, higher volatility compounds (such as tin tetrachloride) produced the highest signals. This effect can reasonably be ascribed to the increased delivery of volatile compounds from the sample introduction system to the plasma. In addition to differences in compound volatility, elemental response may vary as a function of mobile phase composition with gradient elution HPLC-ICPMS methods.35,36 In light of these issues, the suitability of CIC should be evaluated for each method before use.
Internal Standardisation
Role of an Internal Standard
Internal standardisation is usually employed to correct for changes in instrument operating conditions and sample-specific matrix effects which may enhance or suppress the analyte signal. The same quantity of internal standard is added to each sample, standard and blank, and results are calculated using the ratio of the analyte and internal standard signal. An ideal internal standard will have similar physical and chemical properties to the analyte, and will therefore behave in a similar manner to the analyte; hence the analyte-internal standard ratio should be independent of the sample matrix or fluctuations in instrument operating conditions (‘drift’). An internal standard is often incorporated into the diluent used for sample preparation (usually at a concentration of 50–100 μg/L), however it may also be added via a T-piece in the sample introduction system.7 For methods which employ internal standards, the accuracy of the measured result is clearly dependent on the suitability of the internal standard.
Internal Standard Selection
In ICP-MS, the mass and ionisation potential of an element are two of the most significant determinants of matrix effects. Therefore an ideal internal standard will have a similar mass and ionisation potential to the analyte (although the latter appears to be of secondary importance).37,38 For example, copper has an atomic mass of 63 amu and an ionisation potential of 7.73 eV. Germanium is a popular choice as an internal standard for copper because it has both a similar mass (72 amu) and ionisation potential (7.90 eV).
In addition to mass and ionisation potential, a number of other factors should be considered when selecting an internal standard. For example, the internal standard should not suffer from spectroscopic interference from the sample matrix and should not itself cause spectroscopic interference with the analyte. Moreover, the internal standard should not be a source of analyte contamination. For example, rhodium internal standard stock solutions are supplied in dilute hydrochloric acid by some manufacturers (e.g. SPEX CertiPrep). The presence of chloride in this internal standard precludes its use in sweat chloride assays. This point is particularly pertinent because ICP-MS has recently been proposed as a candidate reference method for sweat chloride testing for the diagnosis of cystic fibrosis.39
Common Internal Standards and Other Analytical Issues
Internal standards commonly used in ICP-MS include lithium (6Li), scandium (45Sc), germanium (72Ge), yttrium (89Y), rhodium (103Rh), indium (115In), tellurium (125Te), terbium (159Tb), rhenium (185Re) and iridium (191Ir). In a multi-element method, a combination of internal standards is often used to cover the analytical mass range. Note that these elements are not normally present (at least in appreciable quantities) in biological fluids. Bismuth (209Bi) has also been used as an internal standard for high mass analytes such as lead (208Pb), for which it has a similar ionisation potential.40,41 However, the authors recommend against this as bismuth is still used in some medications such as bismuth subsalicylate (an aspirin derivative sold under the brand names Pepto-Bismol® and Kaopectate®) and ranitidine bismuth citrate (brand name Tritec®). Patients taking these medications may have an elevated level of bismuth in their blood; therefore if bismuth is used as an internal standard for ICP-MS, falsely low analyte concentrations may be reported. A similar situation could arise in workers exposed to indium. A mean blood indium concentration of 4.66 μg/L from 80 workers at an indium-tin oxide production facility has previously been reported.42 These levels would be sufficient to cause a significant error if indium was used as an internal standard for such patients.
Issues such as these could be detected by carefully comparing the internal standard signal intensity in the samples, standards and blanks. Normal variation due to matrix effects is usually <20% (where the recovery of internal standard in one sample is used as a reference). Figure 5 shows a plot of internal standard recovery for different blood samples in an analytical run (each coloured line represents a different internal standard). The inter-sample variation in recovery shown here is a good demonstration of sample-specific matrix effects. A significant outlier (>20% difference in recovery) is an alert to potential analytical error, such as an issue with the sample introduction system (causing inappropriate delivery of the sample to the ICP), a sample preparation error (e.g. incorrect volume of internal standard added to the sample vial), spectroscopic interference on the internal standard, or the situation described above (in which an internal standard is endogenously present in the sample). Theoretically, if the analysis was performed in matrix-free samples and instrument operating conditions were constant, Figure 5 would be a horizontal line.
Figure 5.
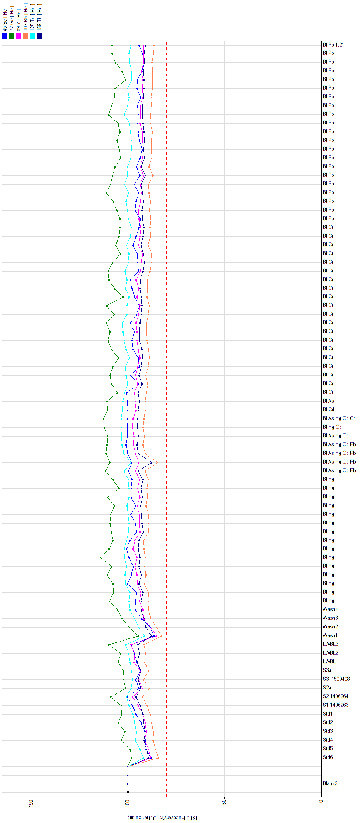
Internal standard recovery in an analytical run (whole blood samples).
Matrix-Matched Calibration
Even with the use of internal standards, analytical bias may occur if samples and calibration standards are not matrix-matched. Calibration standards are therefore often spiked with a ‘matrix-match solution’ in an attempt to mimic the matrix of the sample. A common approach for the analysis of blood is to simply add human or animal blood to the calibration standards (which are otherwise usually aqueous solutions).43,44 The same approach has been successfully used for urine, and serum.9,45 These matrix-matched standards can be prepared in-house (for example by pooling previous patient samples) or may be commercially available, although the latter option can be somewhat expensive.8 Unfortunately the use of biological samples for matrix-matching purposes will create analytical bias if the analyte(s) of interest are present in appreciable quantities in the matrix-match solution. In such a case, calibrations need to be adjusted to account for this. Alternatively, matrix-matching may be achieved using a synthetic matrix with a similar ionic strength to the sample matrix. Good accuracy and precision have been obtained using a solution consisting of NaCl (7.5 g/L) and CaCl2 (0.5 g/L) for blood.46 Theoretically, matrix-matching for urine is more difficult owing to the large inter-individual variation in urine concentration, pH and ionic strength, which are affected by patient hydration status, renal function, diet and pathophysiological conditions. In spite of this, a synthetic matrix for urine consisting of NaCl (0.95% w/v) is reported to be an effective matrix-match solution.6
Method of Standard Additions
In complex sample matrices where external calibration with matrix-matched standards and internal standardisation are found to be inadequate to correct for matrix effects, the method of standard additions may be used. This technique involves adding known and increasing amounts of analyte to several aliquots of the sample. When the analyte signal is plotted against added concentration (as opposed to absolute concentration), the negative x-intercept of the calibration curve corresponds to the analyte concentration in the unspiked sample. Standard addition is normally performed offline, however online addition has also been described.47 Unfortunately, this method of quantification is quite laborious and time consuming, however it has proven to be highly accurate for biological matrices such as serum.48 Although able to correct (at least in part) for matrix effects, the method of standard addition fails to account for errors which may arise due to instrument drift. Standard addition is therefore often used in conjunction with internal standards.
Isotope Dilution
The most effective method to correct for both matrix effects and instrument drift in ICP-MS is isotope dilution. In this technique, a known amount of an isotopically-enriched isotope of the analyte is added to the sample, and the measured change in isotope ratio is used to calculate the original composition of the sample. The enriched isotope acts as both a calibration standard and an internal standard.
Unfortunately, different isotopes may exhibit differential transmission in the instrument (due in part to the space-charge effect), therefore the measured isotope ratio must be corrected for mass bias.49,50 This technique has been successfully applied to the analysis of analytes in urine, blood and plasma.51–54 A caveat here is that isotope dilution requires the analyte to have at least two isotopes which are free of spectroscopic interference. Therefore the method is not applicable to monoisotopic elements such as beryllium, manganese, arsenic and thorium. Although these methods are highly accurate and robust, they are also both laborious and expensive. For these reasons isotope dilution methods are rarely used in routine clinical laboratories.
Interference
Interference in ICP-MS is classified as either spectroscopic or non-spectroscopic. Spectroscopic interference arises when non-analyte ions have the same m/z ratio as the analyte, whereas non-spectroscopic interference refers to effects attributable to the sample matrix or instrument drift. Both types of interference will be discussed, as well as strategies to attenuate them.
Spectroscopic Interferences
There are four types of spectroscopic interference in ICP-MS: isobaric elements, double charged ions, polyatomic ions and tailing interference.
Isobaric Elements
When two isotopes of different elements have the same mass to within the resolution of the mass spectrometer, they are said to be isobaric. For example, iron and nickel both have isotopes at a mass of 58 amu. Therefore iron will interfere with the measurement of nickel at this isotope, and vice versa. The significance of this type of interference is dependent on the relative concentration of the two isobars. For example, 58Fe has a very low isotopic abundance (0.28%); however, in biological samples such as blood, the concentration of iron is very high relative to nickel, therefore the interference can be significant. Other examples of isobaric elemental overlap include 204Hg/204Pb, 115Sn/115In, 114Sn/114Cd, 87Rb/87Sr, 48Ca/48Ti and 40Ca/40Ar.16
Double Charged Interference
Most elements form singly charged ions in the ICP, however elements with a second ionisation potential lower than the first ionisation potential of argon also form a small but significant fraction of double charged ions. Such elements include calcium, barium, strontium, lanthanide elements (including gadolinium and samarium) and the lighter actinides.24 Recall that mass spectrometers separate ions on the basis of m/z ratio, therefore an analyte at a particular mass (M) will be indistinguishable from a double charged ion at twice the mass (2M). From a clinical biochemistry point of view, perhaps the most significant example is the interference of gadolinium with selenium. Four isotopes of selenium (76Se, 77Se, 78Se, and 80Se) suffer interference from double charged gadolinium isotopes (152Gd, 154Gd, 156Gd and 160Gd respectively). Gadolinium isn’t normally present in biological samples, however it is present in some intravenous contrast agents used for magnetic resonance imaging (MRI) including Gadobutrol (GadovistTM) and Gadodiamide (OmniscanTM). The elimination half-life of both compounds is normally <2 h, however this may be prolonged with renal impairment.55 The presence of gadolinium in a clinical sample can cause an artefactually elevated selenium result.56,57
In general, double charged interferences in ICP-MS are quite rare, and are not a significant concern in most biological applications. Clinical laboratories usually monitor double charged ion formation using a tuning solution containing cerium or barium. The percentage of double charged ions (% Ce2+/Ce+ or Ba2+/Ba+) is measured daily during instrument tuning and suitability checks. The acceptance criteria is usually <2–3% double charged ions.24
Polyatomic Interference
The most problematic type of spectroscopic interference is from polyatomic ions. Polyatomic ions form in the high-temperature plasma, either due to incomplete atomisation or from recombination reactions during the extraction of ions into the mass spectrometer.16 These ions may be derived from the sample matrix, reagents used for sample preparation, plasma gases (argon) or entrained atmospheric gases. For example, in samples containing chloride (or where hydrochloric acid is used during sample preparation), chlorine oxide (35Cl16O) and argon chloride (40Ar35Cl) are formed in the plasma. These ions share the same m/z ratio as vanadium (51V) and arsenic (75As) respectively. The presence of chloride may therefore lead to erroneous results for these analytes. A few other common polyatomic ions (and the analytes for which they interfere) are listed in Table 3. Elements in the fourth row (period 4) of the periodic table are particularly prone to polyatomic interference, as these masses (approximately 40–82 amu) frequently overlap with common types of polyatomic ions such as oxides, argides and chlorides.
Table 3.
Common polyatomic interferences in biological samples and the resolution required to resolve these interferences by HR-ICP-MS (adapted from ref. 80).
| Analyte | Interfering ion | Resolution required (M/ΔM) |
|---|---|---|
| 75As+ | 40Ar35Cl+ | 7773 |
| 59Co16O+ | 11498 | |
| 52Cr+ | 40Ar12C+ | 2376 |
| 36Ar16O+ | 2367 | |
| 78Se+ | 40Ar38Ar+ | 9970 |
| 80Se+ | 40Ar40Ar+ | 9521 |
| 40Ar40Ca+ | 9295 | |
| 51V+ | 35Cl16O+ | 2573 |
| 34S16OH+ | 1452 |
A convenient way to monitor polyatomic ion formation in ICP-MS is to measure the degree of oxide formation which is thought to be proportional to other matrix-based interferences. A tuning solution containing cerium is often analysed because it forms a strong oxide bond and therefore has a high oxide formation rate. An oxide value (CeO+/Ce+: i.e. m/z 156/140) of <3% is considered acceptable.
Tailing Interference
An often overlooked source of spectroscopic interference in ICP-MS is that arising from spectral overlap from an adjacent mass. The magnitude of this type of interference is dependent on the abundance sensitivity of the mass analyser. In biological fluids such as blood, this type of interference can be seen in the analysis of manganese. Manganese has a solitary isotope at a mass of 55 amu, which is bordered on either side by isotopes of iron (54Fe and 56Fe). The concentration of iron in blood is roughly four orders of magnitude higher than that of manganese, therefore the mass peaks of iron may contribute to the manganese signal, causing a falsely high result which could mask manganese deficiency. This type of interference may also affect isotope ratio measurements.58,59
Non-Spectroscopic Interference
Non-spectroscopic interferences may be broadly divided into matrix effects and instrument drift. Both may cause analytical error if they are not appropriately corrected.
Matrix Effects
Matrix effects can be defined as an enhancement or, more commonly, suppression of an analyte signal due to properties or constituents of the sample matrix. These effects are thought to arise from a complicated interplay of various mechanisms which occur in nearly all components of the instrument.60 A few of these mechanisms are discussed below.
Sample Introduction Effects
In the sample introduction system, the delivery of sample aerosol to the plasma is highly dependent on the size distribution of aerosol droplets and the aerosol transport efficiency. Changes in physical and chemical properties which affect aerosol characteristics will therefore affect the measured analyte signal. Sample viscosity, vapour pressure, surface tension, ionic strength and acidity all play an important role.18 Changes which increase the delivery of sample to the plasma, such as a reduction in sample viscosity, and an increase in vapour pressure will lead to signal enhancement, provided the plasma is not overloaded.61 Overloading the plasma with solvent can have a cooling effect which will decrease ionisation, leading to signal suppression. This is usually only a concern with high vapour pressure organic solvents which are not routinely analysed in clinical laboratories. Laboratories are more likely to encounter highly viscous samples such as joint fluids which may cause signal suppression if not appropriately diluted before analysis.
Samples with a high ionic strength may experience significant matrix effects in the sample introduction system as a result of two separate and diametric mechanisms.62 An increase in ionic strength decreases sample volatility which decreases the efficiency of aerosol transport to the plasma; however high ionic strength also promotes a process known as coulomb fission of aerosol droplets in the spray chamber.62 Coulomb fission creates smaller droplets which are more efficiently transported to the ICP, and this may counteract any signal suppression arising from the aforementioned decrease in volatility.63
These effects are also dependent on the temperature of the spray chamber and the design of the sample introduction system, including the type of nebuliser and spray chamber. Therefore the effect of a given sample matrix is often unpredictable. In fact, the most significant source of matrix effects in ICP-MS occurs downstream of the sample introduction system, in the plasma and interface/ion optics region of the instrument.
Plasma Effects
A number of sample-specific factors may affect analyte ionisation in the plasma. Relatively high levels of easily-ionised elements in the sample such as sodium and potassium tend to shift the thermal ionisation equilibrium of the analyte toward the neutral atom, thereby decreasing the instrument signal.64 On the other hand, the presence of carbon in the sample matrix has the opposite effect, enhancing analyte ionisation (at least for elements with a high ionisation potential). The mechanism here is thought to be charge transfer reactions in the plasma between carbon ions (C+) and the analyte. This effect has been reported for numerous elements, most notably arsenic, selenium, mercury and tellurium; all of which have a high ionisation potential (but still lower than that of carbon). The magnitude of carbon-induced signal enhancement varies between different instrument manufacturers and sources of carbon; up to five-fold increases in signal have been observed for arsenic and selenium.6,65–67 Laboratories often take advantage of this effect to increase analytical sensitivity. For example, organic solvents such as isopropanol or methanol are often added to sample diluent solutions or mobile phases for speciation methods.8,65 In addition to enhancing sensitivity, this also has a matrix-matching effect for the standards and samples.
Space-Charge Effects
The most significant source of matrix effect in ICP-MS is the space-charge effect which occurs in the interface and ion optics of the instrument. Space-charge is caused by the mutual electrostatic repulsion between positively-charged ions in the ion beam. Although the gas extracted from the plasma through the sample cone is essentially neutral, the large reduction in pressure contributes to a diffusion of electrons away from the ion beam, which consequently develops a net positive charge.68 Positively-charged ions repel each other, causing the ion beam to broaden, thereby reducing transmission of ions to the detector. This effect is mass dependent; light analytes have lower kinetic energy (lower inertia) and are therefore more severely affected than higher mass analytes.69 Heavy matrix elements also cause a higher degree of suppression compared to an equimolar amount of a lower mass matrix element.69 This intimate connection between mass and space-charge highlights the importance of selecting an internal standard with a similar mass to the analyte.
Instrument Drift
Over time, dissolved solids in samples may deposit in the nebuliser and/or interface cones. Solid deposition on the cool tip of the interface cones reduces ion transmission into the mass spectrometer by occluding the orifice, leading to signal suppression. The degree of occlusion increases with time, causing a characteristic downward drift in signal, which will cause artefactually low results if not corrected.3,70 As discussed previously, changes in room temperature (which may affect the spray chamber temperature) can also lead to instrument drift; therefore well-controlled air-conditioning in the laboratory is essential.
Reduction and Elimination of Spectroscopic Interference
A number of strategies have been employed to reduce or eliminate spectroscopic interference in ICP-MS. These techniques may target the pre-analytical, analytical or post-analytical stage of laboratory testing.
Sample Pre-Treatment
Polyatomic interference may arise from reagents used for sample preparation. For example, hydrochloric acid is occasionally used during sample preparation to stabilise mercury in solution.5 However, as stated previously, this will cause spectroscopic interference on vanadium and arsenic. This interference can be avoided by simply selecting an alternate reagent for sample preparation such as ammonium pyrolidinedithiocarbamate (which can also be used to stabilise mercury).8
Matrix Removal
In hyphenated ICP-MS applications (such as HPLC-ICP-MS), in addition to separating different chemical species of an element, the chromatographic separation can also be used to separate analytes from spectroscopic interference. For example, ion chromatography coupled to ICP-MS is often used for arsenic speciation. By optimising the chromatographic conditions, interference from 40Ar35Cl can be eliminated by ensuring that chloride elutes at a different retention time to organic and inorganic arsenicals in the sample.71
Isotope Selection
A simple approach to avoiding spectroscopic interference is to select an interference-free isotope of the analyte. Most elements in the periodic table have at least one isotope that is free from isobaric overlap with another element, however many of these isotopes suffer from either polyatomic or double charged interference. It is therefore not always possible to select an isotope that is free from interference. Another factor to consider when selecting isotopes during method development is the relative isotopic abundance. Low abundance isotopes will clearly yield lower analytical sensitivity, and may therefore be unsuitable.
Correction Equation
Mathematical equations can be used to correct for some cases of spectroscopic interference in ICP-MS.72 This involves estimating the level of interference by measuring a secondary isotope of the interfering ion, and back-calculating the interference based on known isotope abundance ratios. For example, in samples containing chloride, the interference of 40Ar35Cl on arsenic at m/z 75 can be estimated by measuring 40Ar37Cl at m/z 77 and using the chlorine isotope ratio (35Cl:37Cl, 75.8%:24.2%) to back-calculate 40Ar35Cl. The equation becomes more complicated if the sample contains selenium which will contribute to the signal at m/z 77. Alternatively, a correction factor can be experimentally-determined.6 Most instrument software packages are able to perform these calculations automatically. The adequacy of a correction equation can be evaluated by analysing an ‘interference check solution’ containing known concentrations of interfering elements (e.g. chloride). Logically, the accuracy of the corrected result will also depend on the relative signals of the analyte and interferent.
Optimisation of Instrument Settings
The formation of spectroscopic interferences such as oxides and double charged ions can be minimised by optimising instrument operating conditions such as RF power, nebulisation gas flow rate and the sampling position within the plasma (the latter can be adjusted by changing the distance between the torch and the sample cone).63 By using low RF power and high nebulisation gas flow rate, the energy of the plasma can be substantially reduced, generating a so-called cool (or cold) plasma. By operating the ICP in cool plasma conditions, the formation of argon-based interferences such as ArO+ and Ar+ is significantly reduced.73,25 Cool plasma is a double-edged sword, however; the lower-energy plasma also exacerbates matrix effects and reduces ionisation of analytes with a high ionisation potential. For this reason, cool plasma has traditionally been used for analysis of low matrix samples such as deionised water and mineral acids used in the manufacture of semiconductors.25
High-Resolution ICP-MS
High-resolution instruments such as magnetic sector-ICP-MS are capable of separating most cases of spectroscopic interference. Magnetic sector instruments are capable of operating at a resolution of approximately 10,000 (M/ΔM), which is sufficient to resolve many of the isobars listed in Table 3. The utility of high resolution is exemplified in the analysis of chromium in blood, which is often monitored along with cobalt in patients who have received a metal-on-metal hip arthroplasty.74,75 In samples containing carbon, chromium suffers polyatomic interference from 40Ar12C (chromium has a mass of 51.9405 amu, and 40Ar12C has a mass of 51.9624 amu). The resolution required to separate these ions is approximately 2376, hence a magnetic sector mass analyser is able to distinguish these ions.7 In fact, these ions have been successfully resolved using a double-focusing magnetic sector ICP-MS in medium-resolution mode (resolution 3400).7 High-resolution instruments are significantly more expensive than quadrupole instruments, however, and are therefore mainly used for research purposes.
Collision and Reaction Gases
The most popular method to reduce spectroscopic interference is via collision or reaction with a gas in an enclosed cell positioned immediately before the quadrupole. This cell is known as a collision or reaction cell (CRC). It consists of a quadrupole (or higher order multipole such as an octopole) enclosed in a cell that can be pressurised with gas. Inert or reactive gases (or a combination of both) are introduced into the cell to induce collisions with ions generated in the ICP. These collisions may reduce spectroscopic interference by either a chemical reaction process or by reducing the kinetic energy of polyatomic ions.
Reaction Mode
A reactive gas in the CRC may chemically resolve isobaric ions by one of three mechanisms.
On-mass detection: the interfering ion reacts with the gas, forming a new polyatomic ion with a different mass to the analyte. The analyte can then be measured free of interference.
Off-mass detection: the analyte reacts with the gas, forming a product ion with a different mass. The analyte can then be measured indirectly using the product ion mass.
Neutralisation: the interfering ion is neutralised via a charge exchange reaction with the gas.
A number of reactive gases have been utilised for reaction mode ICP-MS. These include ammonia (NH3), methane (CH4), hydrogen (H2), oxygen (O2), nitrous oxide (N2O), nitrogen oxide (NO), carbon dioxide (CO2), fluoromethane (CH3F), sulfur hexafluoride (SF6) and carbon disulphide (CS2).76–80 A disadvantage of reactive gases is they are non-specific and will not eliminate all polyatomic ions, which may preclude multi-element analysis.
Kinetic Energy Discrimination
When an inert gas such as helium is used in the CRC, ions generated in the ICP undergo collisions with gas molecules causing a reduction in kinetic energy. Analyte ions are also affected, although to a lesser extent than polyatomic ions. This is because polyatomic ions have a larger cross-sectional area, therefore they undergo more collisions. Lower energy polyatomic ions can be readily filtered out by applying an appropriate voltage difference between the CRC and the analyser quadrupole, the analyte ions having sufficient energy to surmount this energy barrier.81 Although analyte ions also lose kinetic energy due to collisions in the CRC, the dramatic reduction in polyatomic interference results in much higher signal-to-background ratio for the analyte, and therefore lower detection limits. This process is called kinetic energy discrimination (KED). Unfortunately, KED is effective only for polyatomic ions; isobaric elements and double charged isobars are not removed.
Most modern ICP-MS instruments are equipped with a CRC, although the design varies somewhat between manufacturers. Analytik Jena instruments utilise a so-called integrated collision reaction cell (iCRC); hydrogen or helium gas is directly injected into the plasma through the skimmer cone, thereby removing interfering ions before they reach the ion optics. The mechanism of interference removal is similar to CRC technology.
Tandem Mass Spectrometry
A relatively new development in ICP-MS is the introduction of triple-quadrupole (tandem mass spectrometry) instruments. These instruments are equipped with an additional quadrupole positioned upstream of the CRC. This quadrupole simplifies reaction chemistry in the cell by permitting only certain ions (with a particular m/z) to enter, allowing more controlled and predictable reaction processes to occur. This configuration is useful in a number of clinical applications, such as the measurement of titanium in blood.82 Titanium suffers interference from several isobaric elements and polyatomic ions (namely 48Ca+, 31P17O+ and 31P16O1H+), which can be obviated using a mass-shift method with ammonia in the CRC.82 However, in this particular example, the product ion is itself subject to interference from isotopes of other elements. Using triple-quadrupole instruments, this type of interference at the product ion mass can be excluded by the first quadrupole. Although titanium has no discernible biological role, it is often used in surgical reconstruction applications and metallic knee or hip arthroplasties. Elevated blood concentrations of titanium can indicate premature failure of the joint. Therefore accurate quantification is of considerable clinical interest.
Reduction and Elimination of Non-Spectroscopic Interference
ICP-MS as a technique has made significant strides in resolving spectroscopic interference, however non-spectroscopic interference remains somewhat problematic. Calibration strategies such as internal standardisation, the method of standard additions and isotope dilution have already been discussed. Additional strategies to attenuate or correct for these effects are discussed below, however none are without drawbacks.
Dilution
Matrix effects are dependent on the absolute concentration of matrix components, therefore sample dilution (either during sample preparation or performed online) will reduce the severity of matrix effects.83 Sample dilution also reduces instrument drift arising from deposition of solids in the nebuliser and interface cones. As discussed earlier, a TDS level of <0.2% is often recommended.
Matrix-Analyte Separation
Numerous sample pre-treatment techniques have been employed to affect matrix-analyte separation in a wide range of industries. This includes precipitation, liquid-liquid extraction, solid-phase extraction, anodic stripping voltammetry and hydride generation. In a clinical biochemistry context, hydride generation is perhaps the most common of such techniques. A borohydride reducing agent is used to selectively transform the analyte to a volatile vapour of the corresponding hydride, thereby separating the analyte from the sample matrix. Hydride-forming elements include mercury, arsenic, antimony, selenium and tin.84 In addition to separating the analyte from the sample matrix (which reduces both spectroscopic and non-spectroscopic interference), this technique also provides enhanced sensitivity.85
Conclusion
ICP-MS is a highly sensitive analytical technique for the determination of trace elements of clinical interest in biological fluids. ICP-MS offers numerous features which make it particularly attractive for the clinical laboratory. These include: high sensitivity, wide linear dynamic range, wide elemental coverage, multi-element capability, high sample throughput and simple sample preparation. Although mass spectrometry is highly specific, scientists and clinicians should be cognisant of the potential for interference, and analytical factors which may affect the accuracy of reported results. Initial capital expenditure (instrument cost and laboratory set-up) and ongoing operating costs (argon supply) are not insignificant, and laboratories must assess all these factors when selecting a method for a clinical application.
Acknowledgments
The authors would like to thank Dr Lee Price, Gertruida Pool, Greg Ward and Robert Flatman for reviewing the draft manuscript.
Footnotes
Competing Interests: None declared.
References
- 1.Geldmacher-von Mallinckrodt M, Meissner D. General aspects of the role of metals in clinical chemistry. In: Seiler HG, Sigel A, Sigel H, editors. Handbook on metals in clinical and analytical chemistry. New York: Marcel Dekker, Inc; 1994. pp. 13–29. [Google Scholar]
- 2.Jaishankar M, Tseten T, Anbalagan N, Mathew BB, Beeregowda KN. Toxicity, mechanism and health effects of some heavy metals. Interdiscip Toxicol. 2014;7:60–72. doi: 10.2478/intox-2014-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Houk RS, Fassel VA, Flesch GD, Svec HJ, Gray AL, Taylor CE. Inductively Coupled Argon Plasma as an Ion Source for Mass Spectrometric Determination of Trace Elements. Anal Chem. 1980;52:2283–9. [Google Scholar]
- 4.McShane WJ, Pappas RS, Paschal D. Analysis of total arsenic, total selenium and total chromium in urine by inductively coupled plasma-dynamic reaction cell-mass spectrometry. J Anal At Spectrom. 2007;22:630–5. [Google Scholar]
- 5.Rodushkin I, Engström E, Stenberg A, Baxter DC. Determination of low-abundance elements at ultra-trace levels in urine and serum by inductively coupled plasma-sector field mass spectrometry. Anal Bioanal Chem. 2004;380:247–57. doi: 10.1007/s00216-004-2742-7. [DOI] [PubMed] [Google Scholar]
- 6.Choe K-Y, Gajek R. Determination of trace elements in human urine by ICP-MS using sodium chloride as a matrix-matching component in calibration. Anal Methods. 2016;8:6754–63. [Google Scholar]
- 7.Case CP, Ellis L, Turner JC, Fairman B. Development of a routine method for the determination of trace metals in whole blood by magnetic sector inductively coupled plasma mass spectrometry with particular relevance to patients with total hip and knee arthroplasty. Clin Chem. 2001;47:275–80. [PubMed] [Google Scholar]
- 8.McShane WJ, Pappas RS, Wilson-McElprang V, Paschal D. A rugged and transferable method for determining blood cadmium, mercury, and lead with inductively coupled plasma-mass spectrometry. Spectrochim Acta Part B At Spectrosc. 2008;63:638–44. [Google Scholar]
- 9.Bocca B, Mattei D, Pino A, Alimonti A. Uncertainty evaluation in the analysis of biological samples by sector field inductively coupled plasma mass spectrometry. Part A: Measurements of Be, Cd, Hg, Ir, Pb, Pd, Pt, Rh, Sb, U, Tl and W in human serum. Rapid Commun Mass Spectrom. 2010;24:2363–9. doi: 10.1002/rcm.4650. [DOI] [PubMed] [Google Scholar]
- 10.Pappas RS. Sample Preparation Problem Solving for Inductively Coupled Plasma-Mass Spectrometry with Liquid Introduction Systems I. Solubility, Chelation, and Memory Effects. Spectroscopy (Springf) 2012;27:20–31. [PMC free article] [PubMed] [Google Scholar]
- 11.Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, et al. The human serum metabolome. PLoS One. 2011;6:e16957. doi: 10.1371/journal.pone.0016957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonnen R, Kol R, Laichter Y, Marcus P, Halicz L, Lorber A, et al. Determination of Uranium in Human Hair by Acid Digestion and FIAS-ICPMS. J Radioanal Nucl Chem. 2000;243:559–62. [Google Scholar]
- 13.Rodrigues JL, Nunes JA, Batista BL, de Souza SS, Barbosa F., Jr A fast method for the determination of 16 elements in hair samples by inductively coupled plasma mass spectrometry (ICP-MS) with tetramethylammonium hydroxide solubilization at room temperature. J Anal At Spectrom. 2008;23:992–6. [Google Scholar]
- 14.Dombovári J, Varga Z, Becker JS, Mátyus J, Kakuk G, Papp L. ICP-MS determination of trace elements in serum samples of healthy subjects using different sample preparation methods. At Spectrosc. 2001;22:331–5. [Google Scholar]
- 15.Vanhaecke F, Resano M, Moens L. Electrothermal vaporisation ICP-mass spectrometry (ETV-ICP-MS) for the determination and speciation of trace elements in solid samples – A review of real-life applications from the author’s lab. Anal Bioanal Chem. 2002;374:188–95. doi: 10.1007/s00216-002-1338-3. [DOI] [PubMed] [Google Scholar]
- 16.Templeton DM. Inductively coupled plasma-atomic emission spectroscopy (ICP-AES) and inductively coupled plasma-mass spectrometry (ICP-MS) In: Seiler HG, Sigel A, Sigel H, editors. Handbook on metals in clinical and analytical chemistry. New York: Marcel Dekker, Inc; 1994. pp. 167–80. [Google Scholar]
- 17.Jakubowski N, Feldmann I, Stuewer D. Analytical improvement of pneumatic nebulization in ICP-MS by desolvation. Spectrochim Acta Part B At Spectrosc. 1992;47:107–18. [Google Scholar]
- 18.Sharp BL. Pneumatic nebulisers and spray chambers for inductively coupled plasma spectrometry. A review. Part 2. Spray chambers. J Anal At Spectrom. 1988;3:939–63. [Google Scholar]
- 19.Novak P, Zuliani T, Milačič R, Ščančar J. Development of an analytical procedure for the determination of polybrominated diphenyl ethers in environmental water samples by GC–ICP-MS. Anal Chim Acta. 2014;827:64–73. doi: 10.1016/j.aca.2014.04.020. [DOI] [PubMed] [Google Scholar]
- 20.Navas-Acien A, Francesconi KA, Silbergeld EK, Guallar E. Seafood intake and urine concentrations of total arsenic, dimethylarsinate and arsenobetaine in the US population. Environ Res. 2011;111:110–8. doi: 10.1016/j.envres.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitt JJ. Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry. Clin Biochem Rev. 2009;30:19–34. [PMC free article] [PubMed] [Google Scholar]
- 22.Chan SK, Van Hoven RL, Montaser A. Generation of a helium inductively coupled plasma in a low-gas-flow torch. Anal Chem. 1986;58:2342–3. [Google Scholar]
- 23.Jorabchi K, McCormick R, Levine JA, Liu H, Nam S, Montaser A. High efficiency nebulization for helium inductively coupled plasma mass spectrometry. Spectrochim Acta Part B At Spectrosc. 2006;61:945–50. [Google Scholar]
- 24.Pupyshev AA, Semenova EV. Formation of doubly charged atomic ions in the inductively coupled plasma. Spectrochim Acta Part B At Spectrosc. 2001;56:2397–418. [Google Scholar]
- 25.Niu H, Houk RS. Fundamental aspects of ion extraction in inductively coupled plasma mass spectrometry. Spectrochim Acta Part B At Spectrosc. 1996;51:779–815. [Google Scholar]
- 26.Farnsworth PB, Spencer RL. Ion sampling and transport in inductively coupled plasma mass spectrometry. Spectrochim Acta Part B At Spectrosc. 2017;134:105–22. [Google Scholar]
- 27.Trace Analyses in Metal Matrices Using the ELAN DRC II. Perkin Elmer application note. [Accessed 25 June 2019]. https://www.perkinelmer.com/CMSResources/Images/44-74108APP_ELANDRCIIMetalMatrices.pdf.
- 28.Hu J, Deng D, Liu R, Lv Y. Single nanoparticle analysis by ICPMS: a potential tool for bioassay. J Anal At Spectrom. 2018;33:57–67. [Google Scholar]
- 29.Miller PE, Denton MB. The quadrupole mass filter: Basic operating concepts. J Chem Educ. 1986;63:617. [Google Scholar]
- 30.Ying JF, Douglas DJ. High resolution inductively coupled plasma mass spectra with a quadrupole mass filter. Rapid Commun Mass Spectrom. 1996;10:649–52. [Google Scholar]
- 31.Kościelniak P. Non-linear robust regression procedure for calibration in flame atomic absorption spectrometry. Anal Chim Acta. 1993;278:177–87. [Google Scholar]
- 32.Jamari NLA, Dohmann JF, Raab A, Krupp EM, Feldmann J. Novel non-targeted analysis of perfluorinated compounds using fluorine-specific detection regardless of their ionisability (HPLC-ICPMS/MS-ESI-MS) Anal Chim Acta. 2019;1053:22–31. doi: 10.1016/j.aca.2018.11.037. [DOI] [PubMed] [Google Scholar]
- 33.Miyake Y, Yamashita N, So MK, Rostkowski P, Taniyasu S, Lam P, et al. Trace analysis of total fluorine in human blood using combustion ion chromatography for fluorine: A mass balance approach for the determination of known and unknown organofluorine compounds. J Chromatogr A. 2007;1154:214–21. doi: 10.1016/j.chroma.2007.03.084. [DOI] [PubMed] [Google Scholar]
- 34.Montiel J, Grindlay G, Gras L, de Loos-Vollebregt M, Mora J. The influence of the sample introduction system on signals of different tin compounds in inductively coupled plasma-based techniques. Spectrochim Acta Part B At Spectrosc. 2013;81:36–42. [Google Scholar]
- 35.Martínez-Sierra JG, Sanz FM, Espilez PH, Santamaria-Fernandez R, Gayón JMM, Alonso JIG. Evaluation of different analytical strategies for the quantification of sulfur-containing biomolecules by HPLC-ICP-MS: Application to the characterisation of 34S-labelled yeast. J Anal At Spectrom. 2010;25:989–97. [Google Scholar]
- 36.Amayo KO, Petursdottir A, Newcombe C, Gunnlaugsdottir H, Raab A, Krupp EM, et al. Identification and quantification of arsenolipids using reversed-phase HPLC coupled simultaneously to high-resolution ICPMS and high-resolution electrospray MS without species-specific standards. Anal Chem. 2011;83:3589–95. doi: 10.1021/ac2005873. [DOI] [PubMed] [Google Scholar]
- 37.Vanhaecke F, Vanhoe H, Dams R, Vandecasteele C. The use of internal standards in ICP-MS. Talanta. 1992;39:737–42. doi: 10.1016/0039-9140(92)80088-u. [DOI] [PubMed] [Google Scholar]
- 38.Finley-Jones HJ, Molloy JL, Holcombe JA. Choosing internal standards based on a multivariate analysis approach with ICP(TOF)MS. J Anal At Spectrom. 2008;23:1214–22. [Google Scholar]
- 39.Collie JT, Massie JR, Jones OA, Morrison PD, Greaves RF. A candidate reference method using ICP-MS for sweat chloride quantification. Clin Chem Lab Med. 2016;54:561–7. doi: 10.1515/cclm-2015-0506. [DOI] [PubMed] [Google Scholar]
- 40.Bechlin MA, Fortunato FM, Ferreira EC, Gomes Neto JA, Nóbrega JA, Donati GL, et al. Bismuth as a general internal standard for lead in atomic absorption spectrometry. Anal Chim Acta. 2014;831:24–30. doi: 10.1016/j.aca.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 41.Bechlin MA, Ferreira EC, Gomes Neto JA, Ramos JC, Borges DLG. Contributions on the Use of Bismuth as Internal Standard for Lead Determinations Using ICP-Based Techniques. J Braz Chem Soc. 2015;26:1879–86. [Google Scholar]
- 42.Harvey RR, Virji MA, Edwards NT, Cummings KJ. Comparing plasma, serum and whole blood indium concentrations from workers at an indium-tin oxide (ITO) production facility. Occup Environ Med. 2016;73:864–7. doi: 10.1136/oemed-2016-103685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palmer CD, Lewis ME, Geraghty CM, Barbosa F, Parsons PJ. Determination of lead, cadmium and mercury in blood for assessment of environmental exposure: A comparison between inductively coupled plasma–mass spectrometry and atomic absorption spectrometry. Spectrochim Acta Part B At Spectrosc. 2006;61:980–90. [Google Scholar]
- 44.Praamsma ML, Arnason JG, Parsons PJ. Monitoring Mn in whole blood and urine: a comparison between electrothermal atomic absorption and inorganic mass spectrometry. J Anal At Spectrom. 2011;26:1224–32. [Google Scholar]
- 45.Heitland P, Köster HD. Fast, simple and reliable routine determination of 23 elements in urine by ICP-MS. J Anal At Spectrom. 2004;19:1552–8. [Google Scholar]
- 46.Gajek R, Barley F, She J. Determination of essential and toxic metals in blood by ICP-MS with calibration in synthetic matrix. Anal Methods. 2013;5:2193–202. [Google Scholar]
- 47.Beauchemin D. Online Standard Addition Method with ICPMS Using Flow Injection. Anal Chem. 1995;67:1553–7. [Google Scholar]
- 48.Huang C, Beauchemin D. Direct multielemental analysis of human serum by ICP-MS with on-line standard addition using flow injection. J Anal At Spectrom. 2003;18:951–2. [Google Scholar]
- 49.Andrén H, Rodushkin I, Stenberg A, Malinovsky D, Baxter D. Sources of mass bias and isotope ratio variation in multi-collector ICP-MS: optimization of instrumental parameters based on experimental observations. J Anal At Spectrom. 2004;19:1217–24. [Google Scholar]
- 50.Xie Q, Kerrich R. Isotope ratio measurement by hexapole ICP-MS: mass bias effect, precision and accuracy. J Anal At Spectrom. 2002;17:69–74. [Google Scholar]
- 51.Haldimann M, Zimmerli B, Als C, Gerber H. Direct determination of urinary iodine by inductively coupled plasma mass spectrometry using isotope dilution with iodine-129. Clin Chem. 1998;44:817–24. [PubMed] [Google Scholar]
- 52.Haldimann M, Baduraux M, Eastgate A, Froidevaux P, O’Donovan S, Von Gunten D, et al. Determining picogram quantities of uranium in urine by isotope dilution inductively coupled plasma mass spectrometry. Comparison with α-spectrometry. J Anal At Spectrom. 2001;16:1364–9. [Google Scholar]
- 53.Baxter DC, Rodushkin I, Engström E, Klockare D, Waara H. Methylmercury Measurement in whole blood by isotope-dilution GC-ICPMS with 2 sample preparation methods. Clin Chem. 2007;53:111–6. doi: 10.1373/clinchem.2007.072520. [DOI] [PubMed] [Google Scholar]
- 54.Keyes WR, Turnlund JR. Determination of molybdenum and enriched Mo stable isotope concentrations in human blood plasma by isotope dilution ICP-MS. J Anal At Spectrom. 2002;17:1153–6. [Google Scholar]
- 55.Joffe P, Thomsen HS, Meusel M. Pharmacokinetics of gadodiamide injection in patients with severe renal insufficiency and patients undergoing hemodialysis or continuous ambulatory peritoneal dialysis. Acad Radiol. 1998;5:491–502. doi: 10.1016/s1076-6332(98)80191-8. [DOI] [PubMed] [Google Scholar]
- 56.Ryan JB, Grant S, Walmsley T, Florkowski CM, George PM. Falsely elevated plasma selenium due to gadolinium contrast interference: a novel solution to a preanalytical problem. Ann Clin Biochem. 2014;51:714–6. doi: 10.1177/0004563214529262. [DOI] [PubMed] [Google Scholar]
- 57.Walter A, Nelms S, Harrington CF, Taylor A. Interference of gadolinium on the measurement of selenium in human serum by inductively coupled plasma-quadrupole mass spectrometry. Ann Clin Biochem. 2011;48:176–7. doi: 10.1258/acb.2010.010030. [DOI] [PubMed] [Google Scholar]
- 58.Gray PJ, Zhang L, Xu H, McDiarmid M, Squibb K, Centeno J. Determination of 236U/238U and 235U/238U isotope ratios in human urine by inductively coupled plasma mass spectrometry. Microchem J. 2012;105:94–100. [Google Scholar]
- 59.Boulyga S, Klötzli U, Prohaska T. Improved abundance sensitivity in MC-ICP-MS for determination of 236U/238U isotope ratios in the 10−7 to 10−8 range. J Anal At Spectrom. 2006;21:1427–30. [Google Scholar]
- 60.Kim Y-S, Kawaguchi H, Tanaka T, Mizuike A. Non-spectroscopic matrix interferences in inductively coupled plasma-mass spectrometry. Spectrochim Acta Part B At Spectrosc. 1990;45:333–9. [Google Scholar]
- 61.Browner RF, Boorn AW, Smith DD. Aerosol transport model for atomic spectrometry. Anal Chem. 1982;54:1411–9. [Google Scholar]
- 62.Fraser MM, Beauchemin D. Evidence supporting the occurrence of Coulomb fission during conventional sample introduction in inductively coupled plasma mass spectrometry. J Anal At Spectrom. 2009;24:469–75. [Google Scholar]
- 63.Agatemor C, Beauchemin D. Matrix effects in inductively coupled plasma mass spectrometry: a review. Anal Chim Acta. 2011;706:66–83. doi: 10.1016/j.aca.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 64.Olivares JA, Houk RS. Suppression of analyte signal by various concomitant salts in inductively coupled plasma mass spectrometry. Anal Chem. 1986;58:20–5. [Google Scholar]
- 65.Larsen EH, Stürup S. Carbon-enhanced inductively coupled plasma mass spectrometric detection of arsenic and selenium and its application to arsenic speciation. J Anal At Spectrom. 1994;9:1099–105. [Google Scholar]
- 66.Kovačevič M, Goessler W. Direct introduction of volatile carbon compounds into the spray chamber of an inductively coupled plasma mass spectrometer: Sensitivity enhancement for selenium. Spectrochim Acta Part B At Spectrosc. 2005;60:1357–62. [Google Scholar]
- 67.Gajek R, Choe K-Y. Determination of ultra-trace elements in human plasma or serum by ICP-MS using sodium in the presence of carbon as a single calibration matrix-match component. J Anal At Spectrom. 2015;30:1142–53. [Google Scholar]
- 68.Hill SJ, Fisher A, Liezers M. Plasma Generation, Ion Sampling and Focusing. In: Nelms SM, editor. ICP Mass Spectrometry Handbook. Oxford: Blackwell Publishing; 2005. pp. 1–25. [Google Scholar]
- 69.Gillson GR, Douglas DJ, Fulford JE, Halligan KW, Tanner SD. Nonspectroscopic interelement interferences in inductively coupled plasma mass spectrometry. Anal Chem. 1988;60:1472–4. [Google Scholar]
- 70.Douglas DJ, Kerr LA. Study of solids deposition on inductively coupled plasma mass spectrometry samplers and skimmers. J Anal At Spectrom. 1988;3:749–52. [Google Scholar]
- 71.Sheppard BS, Shen W-L, Caruso JA, Heitkemper DT, Fricke FL. Elimination of the argon chloride interference on arsenic speciation in inductively coupled plasma mass spectrometry using ion chromatography. J Anal At Spectrom. 1990;5:431–5. [Google Scholar]
- 72.Raut NM, Huang L-S, Aggarwal SK, Lin K-C. Mathematical correction for polyatomic isobaric spectral interferences in determination of lanthanides by inductively coupled plasma mass spectrometry. J Chin Chem Soc. 2005;52:589–97. [Google Scholar]
- 73.Wollenweber D, Straßburg S, Wünsch G. Determination of Li, Na, Mg, K, Ca and Fe with ICP-MS using cold plasma conditions. Fresenius J Anal Chem. 1999;364:433–7. [Google Scholar]
- 74.Afolaranmi GA, Tettey J, Meek RMD, Grant MH. Release of Chromium from Orthopaedic Arthroplasties. Open Orthop J. 2008;2:10–8. doi: 10.2174/1874325000802010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hart AJ, Sabah SA, Bandi AS, Maggiore P, Tarassoli O, Sampson B, et al. Sensitivity and specificity of blood cobalt and chromium metal ions for predicting failure of metal-on-metal hip replacement. J Bone Joint Surg Br. 2011;93:1308–13. doi: 10.1302/0301-620X.93B10.26249. [DOI] [PubMed] [Google Scholar]
- 76.Jones DR, Jarrett JM, Tevis DS, Franklin M, Mullinix NJ, Wallon KL, et al. Analysis of whole human blood for Pb, Cd, Hg, Se, and Mn by ICP-DRC-MS for biomonitoring and acute exposures. Talanta. 2017;162:114–22. doi: 10.1016/j.talanta.2016.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Batista BL, Rodrigues JL, Nunes JA, Souza VC, Barbosa F., Jr Exploiting dynamic reaction cell inductively coupled plasma mass spectrometry (DRC-ICP-MS) for sequential determination of trace elements in blood using a dilute-and-shoot procedure. Anal Chim Acta. 2009;639:13–8. doi: 10.1016/j.aca.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 78.Gong Z-S, Jiang X-H, Sun C-Q, Tian Y-P, Guo G-H, Zhang Y-Z, et al. Determination of 21 elements in human serum using ICP-MS with collision/reaction cell. Int J Mass Spectrom. 2017;423:20–6. [Google Scholar]
- 79.Bolea-Fernandez E, Balcaen L, Resano M, Vanhaecke F. Potential of methyl fluoride as a universal reaction gas to overcome spectral interference in the determination of ultratrace concentrations of metals in biofluids using inductively coupled plasma-tandem mass spectrometry. Anal Chem. 2014;86:7969–77. doi: 10.1021/ac502023h. [DOI] [PubMed] [Google Scholar]
- 80.D’Ilio S, Violante N, Majorani C, Petrucci F. Dynamic reaction cell ICP-MS for determination of total As, Cr, Se and V in complex matrices: still a challenge? A review. Anal Chim Acta. 2011;698:6–13. doi: 10.1016/j.aca.2011.04.052. [DOI] [PubMed] [Google Scholar]
- 81.Yamada N. Kinetic energy discrimination in collision/reaction cell ICP-MS: Theoretical review of principles and limitations. Spectrochim Acta Part B At Spectrosc. 2015;110:31–44. [Google Scholar]
- 82.Balcaen L, Bolea-Fernandez E, Resano M, Vanhaecke F. Accurate determination of ultra-trace levels of Ti in blood serum using ICP-MS/MS. Anal Chim Acta. 2014;809:1–8. doi: 10.1016/j.aca.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 83.Barros AI, Pinheiro FC, Amaral CDB, Lorençatto R, Nóbrega JA. Aerosol dilution as a simple strategy for analysis of complex samples by ICP-MS. Talanta. 2018;178:805–10. doi: 10.1016/j.talanta.2017.10.024. [DOI] [PubMed] [Google Scholar]
- 84.Welz B. Atomic Absorption Spectrometry. In: Seiler HG, Sigel A, Sigel H, editors. Handbook on metals in clinical and analytical chemistry. New York: Marcel Dekker, Inc; 1994. pp. 86–105. [Google Scholar]
- 85.Matoušek T, Wang Z, Douillet C, Musil S, Stýblo M. Direct Speciation Analysis of Arsenic in Whole Blood and Blood Plasma at Low Exposure Levels by Hydride Generation-Cryotrapping-Inductively Coupled Plasma Mass Spectrometry. Anal Chem. 2017;89:9633–7. doi: 10.1021/acs.analchem.7b01868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Košler J, Sylvester PJ. Present Trends and the Future of Zircon in Geochronology: Laser Ablation ICPMS. Rev Mineral Geochem. 2003;53:243–75. [Google Scholar]