

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a metabolic disorder due to increased accumulation of fat in the liver and in many cases to enhanced inflammation. Although the contribution of inflammation in the pathogenesis of NAFLD is well established, the cytokines that are involved and how they influence liver transformation are still poorly characterized. In addition, with other modifiers, inflammation influences NAFLD progression to liver cirrhosis and hepatocellular carcinoma, demonstrating the need to find new molecular targets with potential future therapeutic applications. We investigated gene signatures in 38 liver biopsies from patients with NAFLD and obesity who had received bariatric surgery and compared these to 10 control patients who had received a cholecystectomy, using DNA microarray technology. A subset of differentially expressed genes was then validated on a larger cohort of 103 patients who had received bariatric surgery for obesity; data were thoroughly analyzed in terms of correlations with NAFLD pathophysiological parameters. Finally, the impact of a specific cytokine, interleukin‐32 (IL32), was addressed on primary human hepatocytes (PHHs). Transcript analysis revealed an up‐regulation of proinflammatory cytokines IL32, chemokine (C‐X‐C motif) ligand 9 (CXCL9), and CXCL10 and of ubiquitin D (UBD), whereas down‐regulation of insulin‐like growth factor‐binding protein 2 (IGFBP2) and hypoxanthine phosphoribosyltransferase 1 (HPRT1) was reported in patients with NAFLD. Moreover, IL32, which is the major deregulated gene, correlated with body mass index (BMI), waist circumference, NAFLD activity score (NAS), aminotransferases (alanine aminotransferase [ALAT] and aspartate aminotransferase [ASAT]), and homeostasis model assessment of insulin resistance (HOMA‐IR) index in patients. Consistent with an instrumental role in the pathophysiology of NAFLD, treatment of control human hepatocytes with recombinant IL32 leads to insulin resistance, a hallmark metabolic deregulation in NAFLD hepatocytes. Conclusion: IL32 has a critical role in the pathogenesis of NAFLD and could be considered as a therapeutic target in patients.
Abbreviations
- AKT
protein kinase B
- ALAT
alanine aminotransferase
- ALP
alkaline phosphatase
- ASAT
aspartate aminotransferase
- BMI
body mass index
- Cont
control
- CXCL
chemokine (C‐X‐C motif) ligand
- DE
differentially expressed
- FAT10
F adjacent transcript 10
- FC
fold change
- GGT
gamma‐glutamyl transferase
- HCV
hepatitis C virus
- HDL
high‐density lipoprotein
- HOMA‐IR
homeostasis model assessment of insulin resistance
- HPRT1
hypoxanthine phosphoribosyltransferase 1
- hsCRP
highly sensitive C‐reactive protein
- IFN
interferon
- IGFBP2
insulin‐like growth factor‐binding protein 2
- IL32
interleukin‐32
- LDL
low‐density lipoprotein
- NAFLD
nonalcoholic fatty liver disease
- NAS
nonalcoholic fatty liver disease activity score
- NASH
nonalcoholic steatohepatitis
- Ob_NAFLD
obese and nonalcoholic fatty liver disease
- Ob_NASH
obese and nonalcoholic steatohepatitis
- Ob_NL
obese and normal liver
- Ob_ST
obese and hepatic steatosis
- p‐AKT
phosphorylated protein kinase B
- PHH
primary human hepatocyte
- PPAR
peroxisome proliferator‐activated receptor
- PRKCE
protein kinase C epsilon gene
- qRT‐PCR
quantitative reverse‐transcription polymerase chain reaction
- TNF
tumor necrosis factor
- UBD
ubiquitin D
- usCRP
ultrasensitive C‐reactive protein
Nonalcoholic fatty liver disease (NAFLD) is a pathologic condition characterized by enhanced accumulation of lipids in the liver and is considered by many as a common cause for liver cirrhosis development and progression to hepatocellular carcinoma.1, 2 NAFLD can be dissociated in two pathologic entities; liver steatosis, which represents the majority of cases of NAFLD (~80%), is defined by the accumulation of triglycerides in the liver, is often benign, and is associated with good prognosis and nonalcoholic steatohepatitis (NASH), which is associated with liver steatosis, inflammation, ballooning of hepatocytes, and liver fibrosis.3 A subset of patients with NASH (~30%) develops cirrhosis within 10 years, and NASH has become the second leading etiology among adults awaiting liver transplantation in the United States.3, 4, 5 Recently, it has been shown that liver fibrosis is the most important histologic feature to predict the severity of disease and long‐term outcome in patients with NAFLD.6, 7 It is worth mentioning that the presence of isolated steatosis does not preclude the progression to NASH.1 Originally, it was thought that hepatic steatosis was the first event in NAFLD followed by a second hit in which inflammation and fibrosis take place. This hypothesis has been challenged, and NASH is now viewed as a condition in which multiple pathogenic events occur simultaneously to promote liver injury.8 One major contributing factor to NAFLD is obesity,9 and weight loss is an indication for NAFLD prevention. However, patients without obesity are also susceptible to develop NAFLD but tend to have less severe disease and may have a better prognosis than patients with obesity.10 Importantly, high values of fatty liver index have been associated with enhanced insulin resistance in a large European population of middle‐aged subjects without diabetes.11 Moreover, the presence of metabolic syndrome was correlated with a high risk of NASH and particularly with a high risk of severe fibrosis among patients with NAFLD after correction for sex, age, and body mass index (BMI).12 In the last decades, many findings have contributed to identify molecules that can be targeted to stop the progression of NAFLD into cirrhosis and liver cancer. For now, a number of emerging molecular targets have been identified to potentially treat NASH. These targets include transcription factors, such as the nuclear receptors farnesoid X receptor, pregnane X receptor, peroxisome proliferator‐activated receptor alpha/beta/gamma (PPARα/β/γ), and nuclear erythroid 2 p45‐related factor 2, and several proinflammatory chemokines with the aim being to reduce inflammation, fibrogenesis, and oxidative stress and to enhance fatty acid oxidation and insulin sensitivity.13 Many of the molecules targeting either transcription factors or cytokines are currently tested in preclinical and clinical studies, and a treatment for NAFLD is eagerly awaited. Given the heterogeneity of NAFLD in patients, the treatment of NAFLD will most likely be customized to each patient based on the stage of NAFLD, and additional targets and predictive biomarkers are necessary to improve patient care.
In order to search for new molecular targets involved in NAFLD pathogenesis, we used DNA microarray chip technology to identify genes differentially expressed in liver biopsies from patients with NAFLD and obesity who had received bariatric surgery but who did not have diabetes compared to liver biopsies from patients without obesity who had received noninflammatory cholecystectomy. Surprisingly, the proportion of patients with obesity exhibiting no signs of NAFLD at histology (Ob_NL) was almost equal to that of patients with obesity with histologic hallmarks of NAFLD. We found that interleukin‐32 (IL32) expression, a proinflammatory cytokine, was robustly induced in liver samples from patients with NAFLD compared to Ob_NL and control patients without obesity. Interestingly, liver IL32 expression was significantly correlated with the NAFLD activity score (NAS), a system scoring for histolopathologic features of NAFLD developed by the NASH Clinical Research Network (CRN) and validated by the National Institute of Diabetes and Digestive and Kidney Diseases.14 IL32 also correlated with the homeostasis model assessment of insulin resistance (HOMA‐IR) index in these patients, and treatment of primary human hepatocytes (PHHs) with recombinant IL32 clearly impaired insulin signaling in these cells. Altogether, this study indicates that IL32 along with other differentially expressed genes may play an important role in the pathogenesis of NAFLD and fatty liver‐associated insulin resistance.
Patients and Methods
Patient Recruitment
In order to recruit patients with NAFLD but without diabetes, patients who were morbidly obese and undergoing bariatric surgery were enrolled prospectively in the study in the Department of Hepatodigestive and Endocrine Surgery at Strasbourg University Hospital. Inclusion criteria included the following: adult men/women who were morbidly obese (BMI ≥40 kg/m2) with histologic features of NAFLD and, as the control group, adult men/women without obesity who had received surgery for noninflammatory gallstone disease. All patients had no other illness or malignancy. Exclusion criteria included viral (hepatitis B virus [HBV] or hepatitis C virus [HCV]) or autoimmune hepatitis, diabetes, alcohol consumption >20 g/day for women and >30 g/day for men, genetic hematochromatosis with the homozygote C282Y mutation, infection or inflammation at the moment of biopsy, and impaired coagulation status with platelets <60,000/mm3 and/or prothrombin rate <50%. Liver biopsies were performed for all patients who had received surgery, and histopathologic analysis of the biopsies allowed the classification of patients into four histopathologic entities as follows: controls (Cont, n = 19; mean age ± SEM, 49.5 ± 4.2 years), obese with a histologically “normal” liver without signs of steatosis (Ob_NL, n = 47; 41.5 ± 1.8 years), obese with hepatic steatosis (Ob_ST, n = 44; 40.9 ± 2.0 years), and obese with NASH (Ob_NASH, n = 12; 45.3 ± 3.5 years). The last two groups can be assimilated in the pathologic condition referred to as Ob_NAFLD, corresponding to patients with NAFLD (n = 56; 41.8 ± 1.8 years). Written informed consent was provided by all patients. This study was approved by the ethical committee board of Strasbourg University Hospital (approval number ID‐RCB: 2007‐A 00437‐46).
Biochemical Analyses
Blood samples were drawn from all fasted patients before induction of anesthesia in patients undergoing either cholecystectomy (lean control subjects) or bariatric surgery for patients with obesity (either sleeve gastrectomy or Roux‐en‐Y gastric bypass surgery). Blood samples were collected in vacutainer tubes and transported immediately to the clinical biochemistry laboratory where they were centrifuged and processed for serum measurements of standard and specialized clinical chemistry parameters (glucose, aspartate aminotransferase [ASAT], alanine aminotransferase [ALAT], gamma‐glutamyl transferase [GGT], alkaline phosphatase [ALP], total bilirubin, triglycerides, free fatty acids, total cholesterol, high‐density lipoprotein [HDL]‐cholesterol, low‐density lipoprotein [LDL]‐cholesterol, iron, ultrasensitive C‐reactive protein [usCRP] using an ADVIA 2400 clinical chemistry analyzer (Siemens Healthcare SAS, France), following the manufacturer’s instructions. Circulating insulin levels were quantified by an immunochemistry luminescence method using a COBAS 6000 analyzer (Roche Diagnostics SA, Switzerland). Ferritinemia was measured by a chemiluminescence immunoassay on an ADVIA Centaur CP analyzer (Siemens Healthcare SAS).
Gene Expression Analysis Using Microarray and Quantitative Reverse‐Transcription Polymerase Chain Reaction
We used gene expression microarray technology and quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR) to study genes that were differentially expressed in liver biopsies obtained from patients who were lean and had received a cholecystectomy (n = 10) and from patients with obesity who had received bariatric surgery. The analysis was carried out in 10 patients randomly chosen from each histopathologic group (8 patients in the NASH group; total, 38 patients). Agilent gene expression microarray chips (Reference SurePrint G3 Human GE 8x60K, Agilent Corp., France) were used to analyze genes that were differentially expressed (DE) in the liver of the four histopathologic groups (n = 10/group, 8 patients in the NASH group). To identify DE genes, we used the fold‐change rank ordering statistics method,15 which associates an f value with genes. The f values for down‐regulated genes are small (near zero), while those associated with up‐regulated genes are high (near 1). We first compared samples from Ob_NL, Ob_ST, and Ob_NASH to the Cont samples. These comparisons were performed in two steps as follows: first, comparison of groups using all data samples at a time; second, use of bootstrap analysis to select genes that were least dependent as possible to patients. In the bootstrap analysis, subsets of samples were selected from the groups in the comparison. This was repeated 100 times, and results were finally combined for each gene. The threshold f values 0.995 and 0.005 were used to select the up‐ and down‐regulated genes, respectively. The lists were combined, and unique known genes were hierarchically clustered in a heatmap in which overexpressed genes are represented in red and down‐regulated genes in blue, using Cluster 3.0 and Java Treeview software (version 1.1.6r2; http://jtreeview.sourceforge.net/). A microarray correlated genes diagram was drawn using Cytoscape 3.7.1 software (http://cytoscape.org).
For a subset of genes intercorrelated in microarray analysis, we confirmed the DE using qRT‐PCR on all complementary DNA samples (n = 122) obtained from liver biopsy‐extracted RNA, using a QIAGEN RNeasy Lipid Tissue Mini Kit (ID 74804).
Primary Hepatocyte Culture and Western Blotting
PHHs were isolated and cultured as described.16 Cells were plated 1 day before incubation with human recombinant IL32 (1 ng/mL) (CliniScience, France) for 24 hours. PHHs were then treated with human recombinant insulin (100 nM) (Sigma‐Aldrich, France) for different time points (0, 5, 10, 20, 30, and 60 minutes). Nontreated PHHs served as controls. Cells were lysed in Lysis Buffer 6 (R&D Systems), and protein concentration was determined using the Bradford assay (Bio‐Rad), following the manufacturer’s instructions. Cell lysates were run on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane using the Trans‐Blot Turbo Transfer System (Bio‐Rad). Membranes were saturated with phosphate‐buffered saline containing 5% bovine serum albumin and 0.1% tween. For protein detection, polyclonal rabbit pan‐protein kinase B (AKT) antibody (ab8805; Abcam, France), polyclonal rabbit anti‐phosphorylated (phospho‐)AKT (Thr‐308) antibody (ab38449; Abcam), and monoclonal anti‐β‐actin antibody (ab8226; Abcam) were used. IL32 protein was quantified in liver extracts with rabbit anti‐IL32 antibody (ab37158; Abcam). Protein expression was quantified using ImageJ software.
Statistical Analysis
We used analysis of variance for comparison among the four histopathologic groups analyzed, followed by Tukey’s multiple comparison test when the distribution was normal and Kruskal‐Wallis test followed by Dunn’s multiple comparison test when the distribution was not normal. Statistical significance was achieved at P < 0.05. Generated graphs and statistical regression analysis were performed using GraphPad Prism 6.0. Heatmaps for Spearman’s correlations were generated using Mev_4_8 version 10.2 (http://mev.tm4.org).
Results
Antropomorphic Data and Biochemical Parameters in Patients With Obesity and Bariatric Surgery and Patients Without Obesity who had Received a Cholecystectomy
Patients with NAFLD are usually histologically separated into patients with steatosis and patients with NASH. Patients were stratified in four main groups based on their histopathologic profile. The first group (Cont) represented the control arm of the study, corresponding to patients without obesity who had a noninflammatory cholecystectomy (n = 19). The second group (Ob_NL) was represented by patients with obesity who underwent bariatric surgery with a histologically “normal” liver (i.e., no signs of hepatic steatosis, n = 47). The third group Ob_ST (n = 44) and the fourth group Ob_NASH (n = 12) corresponded to patients with obesity who had received bariatric surgery and had histopathologic signs of NAFLD, defined by a proportion of more than 5% of hepatocytes that exhibit the presence of cytoplasmic lipid droplets with no signs of inflammation or with inflammation, respectively.17 Representative histopathologic pictures of liver biopsy sections stained with hematoxylin and eosin of the four groups are shown in Fig. 1A. BMI and waist circumference were increased in patients with obesity (Ob_NL and Ob_NAFLD) compared to the Cont group (P < 0.001; Table 1). Compared to the Cont group, fasting serum glucose levels were not changed and were slightly increased in the Ob_NL group and the Ob_NAFLD group (P < 0.05; Table 1), respectively. In addition, serum insulin levels and the HOMA‐IR index were significantly increased in the Ob_NAFLD group compared to Cont (P < 0.001) and Ob_NL (P < 0.01) groups, indicating that patients with obesity with NAFLD were insulin resistant (Table 1). The serum lipid profile indicated that total and LDL‐cholesterol levels were not changed among Ob_Cont, Ob_NL, and Ob_NAFLD, while HDL‐cholesterol was decreased in Ob_NAFLD compared with Cont (P < 0.001). In addition, compared with the Cont and Ob_NL groups, total triglycerides were increased in Ob_NAFLD (P < 0.05 and P < 0.01, respectively). Hepatic enzymes ALAT (P < 0.001), ASAT (P < 0.001), and GGT (P < 0.05) were increased in Ob_NAFLD patients compared to Ob_NL and Cont patients (Table 1). ALP, total bilirubin, iron, and transferrin saturation capacity were not changed among the three groups. Ferritin was increased in the NAFLD group compared to Ob_NL (P < 0.05) but not Cont (Table 1). Data for Ob_ST and Ob_NASH are also presented separately in Table 1, and significant statistical differences with Cont and Ob_NL are also shown for Ob_ST and Ob_NASH.
Figure 1.
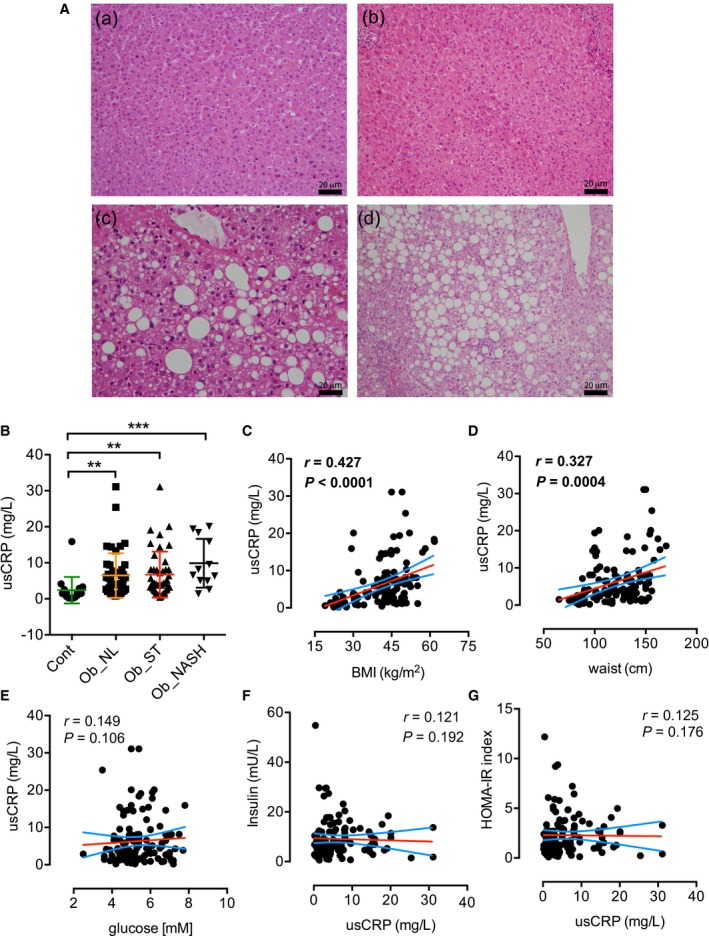
Histology and usCRP measurement in patients who received bariatric surgery compared with patients who underwent cholecystectomy. (A) Histopathologic representative pictures of hematoxylin and eosin staining of liver sample paraffin‐embedded sections from (a) control patients who received a cholecystectomy, (b) Ob_NL, (c) Ob_ST, and (d) Ob_NASH patients who received bariatric surgery (scale bar, 20 micrometers). (B) usCRP immunoanalytic measurement in patients with obesity who received bariatric surgery, with NAFLD (Ob_ST and Ob_NASH) and without NAFLD (Ob_NL), compared with control patients who received a cholecystectomy; usCRP was significantly correlated to BMI (C) and waist circumference (D) but not with glucose (E), insulin (F), and HOMA‐IR index (G). Results are expressed as mean ± SEM; **P < 0.01, ***P < 0.001.
Table 1.
Antropomorphic and Biochemical Analyses From Patients With Obesity who Received BARIATRIC SURGERY AND THEIR Controls Without Obesity
| Cont | Ob_NL | Ob_ST | Ob_NASH | Ob_NAFLD | |
|---|---|---|---|---|---|
| n | 19 | 47 | 44 | 12 | 56 |
| Age (years) | 49.5 ± 4.2 | 41.5 ± 1.8 | 40.9 ± 2.0 | 45.3 ± 3.5 | 41.8 ± 1.8 |
| BMI (kg/m2) | 24.7 ± 0.6 | 42.6 ± 0.9§ | 45.3 ± 1.0§ | 46.2 ± 2.6§ | 45.5 ± 1.0§ |
| Waist (cm) | 84.1 ± 2.4 | 121.4 ± 2.9§ | 131.8 ± 3.2§ | 129.6 ± 5.6§ | 131.3 ± 2.7§ |
| Glucose (mM) | 5.0 ± 0.2 | 5.1 ± 0.1 | 5.7 ± 0.1*/† | 5.8 ± 0.3 | 5.7 ± 0.1*/† |
| Insulin (mU/L) | 5.3 ± 0.8 | 7.5 ± 0.9 | 11.7 ± 1.4†/* | 10.6 ± 2.3* | 11.4 ± 1.2‡/† |
| HOMA‐IR index | 1.2 ± 0.2 | 1.8 ± 0.2 | 3.1 ± 0.4‡/† | 2.7 ± 0.5* | 2.98 ± 0.3‡/† |
| Total chol (mmol/L) | 4.4 ± 0.3 | 4.1 ± 0.2 | 4.1 ± 0.1 | 4.0 ± 0.2 | 4.1 ± 0.1 |
| LDL‐chol (mmol/L) | 2.6 ± 0.2 | 2.5 ± 0.1 | 2.4 ± 0.1 | 2.2 ± 0.2 | 2.4 ± 0.1 |
| HDL‐chol (mmol/L) | 1.2 ± 0.1 | 1.0 ± 0.04† | 0.8 ± 0.03‡/* | 0.9 ± 0.05 | 0.8 ± 0.02‡/* |
| TG (mmol/L) | 1.4 ± 0.2 | 1.4 ± 0.1 | 2.1 ± 0.2*/* | 1.9 ± 0.2 | 2.06 ± 0.1†/† |
| FFA (mmol/L) | 0.7 ± 0.1 | 0.8 ± 0.04 | 0.8 ± 0.04 | 0.7 ± 0.07 | 0.8 ± 0.04 |
| ALAT (U/L) | 24.7 ± 6.0 | 29.8 ± 3.4 | 54.6 ± 4.5§/§ | 66.9 ± 20.8†/* | 57.2 ± 5.6§/§ |
| ASAT (U/L) | 24.6 ± 4.0 | 27.3 ± 2.3 | 39.2 ± 2.9‡/† | 49.5 ± 14.3* | 41.4 ± 3.8§/‡ |
| GGT (U/L) | 26.9 ± 6.2 | 39.2 ± 7.0 | 40.4 ± 3.7* | 68.9 ± 23.3* | 46.5 ± 5.8†/* |
| ALP (U/L) | 54.5 ± 4.8 | 68.3 ± 5.9 | 58.1 ± 2.4 | 69.5 ± 13.0 | 60.6 ± 3.4 |
| Total bilirubin (μmol/L) | 9.2 ± 1.2 | 9.8 ± 0.6 | 8.8 ± 0.5 | 14.2 ± 4.7 | 9.9 ± 1.1 |
| Iron (μmol/L) | 14.7 ± 1.6 | 11.9 ± 0.8 | 13.6 ± 0.7 | 14.0 ± 1.6 | 13.7 ± 0.7 |
| Ferritin (μg/L) | 139.3 ± 30.9 | 111.5 ± 17.9 | 179.2 ± 20.3ns/* | 142.7 ± 31.5 | 171.4 ± 17.3ns/* |
| TSC (%) | 30.3 ± 3.3 | 25.3 ± 1.9 | 28.5 ± 1.6 | 27.0 ± 2.7 | 28.2 ± 1.4 |
Symbols before the slash (/) represent comparison with the control group; those after the slash (/) represent comparison with the Ob_NL group. Results are expressed as mean ± SEM.
P < 0.05,
P < 0.01,
P < 0.001,
P < 0.0001.
Abbreviations: Chol, cholesterol; FFA, free fatty acid; ns, not significant; TSC, transferrin saturation capacity.
Several groups have studied CRP status in individuals with obesity, yet the association with insulin resistance remains elusive. In this study, usCRP was significantly increased in the serum of patients with obesity and bariatric surgery (i.e., Ob_NL, Ob_ST and Ob_NASH patients; Fig. 1B) compared to control lean patients and correlated positively with BMI and waist circumference in patients (Fig. 1C,D, respectively). This result was not surprising given that obesity is characterized by metabolic low‐grade and chronic inflammation, yet the values of usCRP were not as high as in the case of an acute inflammatory response. However, usCRP was not correlated with glucose, insulin, or HOMA‐IR index in our cohort of patients (Fig. 1E‐G, respectively). In summary, the inflammatory acute‐phase response protein usCRP was not correlated with insulin resistance in our cohort of patients.
Gene Signature of Ob_NAFLD Patients Revealed Involvement of Specific Inflammatory Cytokines
To better understand the molecular changes that occur in the liver of patients with obesity in the context of NAFLD, we performed microarray analysis in 10 patients randomly chosen from each group (8 patients in the NASH group). We identified 846 genes that were significantly DE in patients with obesity compared to control samples (fold change and f values are presented in Supporting Table S1). Of these, 595 genes were DE in the Ob_NL group, 395 genes in the Ob_ST group, and 243 genes in the Ob_NASH group. To identify DE genes that may be more relevant biologically, we set a fold‐change threshold of 2 for up‐regulated genes and 0.5 (−2‐fold) for down‐regulated genes (i.e., 0.5 ≥ fold change ≥2). Hence, we identified 144 DE genes in patients with obesity (Ob_NL, Ob_ST, or Ob_NASH). The list of DE genes (with 0.5 ≥ fold change ≥2) in each group is summarized in Fig. 2C and Supporting Table S2. Hierarchical clustering of the identified genes was represented in a heatmap in which genes are classified in rows and patients in columns (Fig. 2A; Supporting Fig. S2 for clarity). As expected, Cont patients were clustered together except 1 Ob_NL patient clustered with this group. Ob_NASH and Ob_ST patients clustered mainly together (except 1 Ob_NL clustering with this group), forming a fairly homogeneous molecular group named NAFLD that could be easily distinguished from the Cont patients who were lean (Fig. 2A). However, using this approach the NAFLD group was not separated into two clear independent groups, in line with the difficulty to histopathologically classify patients with fatty liver disease in either the steatosis or NASH entity. Indeed, 2/8 Ob_NASH patients clustered with the Ob_ST group (Fig. 2A). Venn diagrams (http://bioinfogp.cnb.csic.es/tools/venny/) presented show the number of genes specifically DE in each group and the ones that are commonly DE among the three groups.
Figure 2.
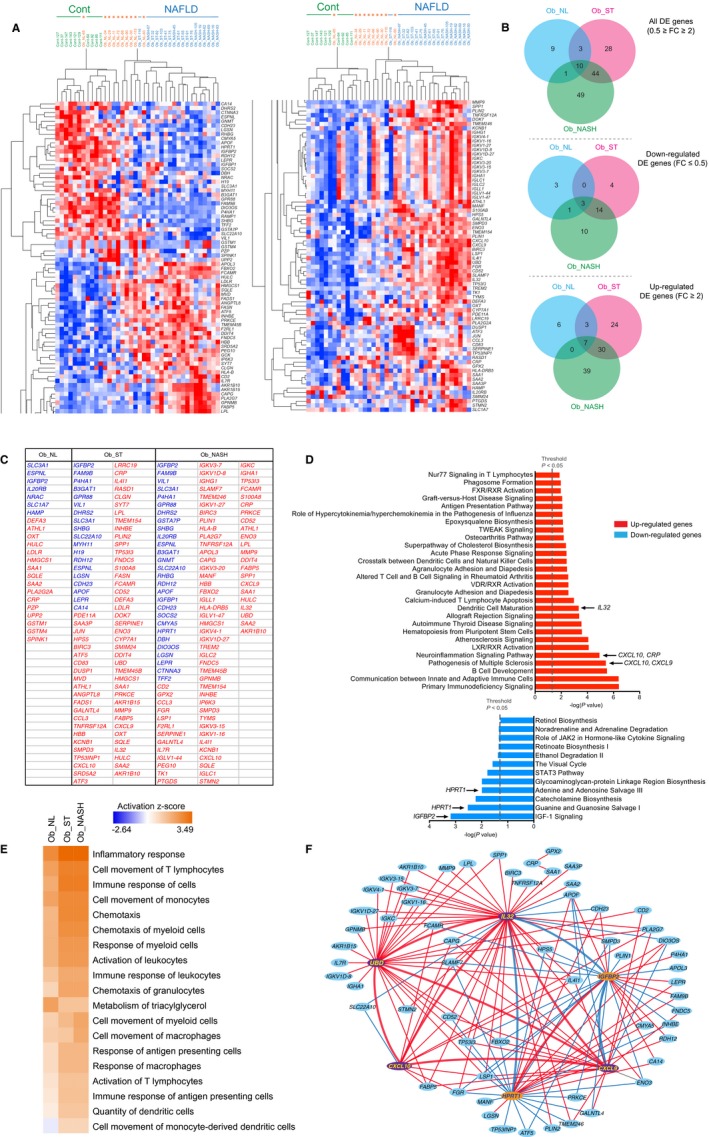
Representation and pathway analysis of DE gene expression dataset. (A) Hierarchical cluster analysis of microarray DE genes in liver biopsies from patients with obesity who had bariatric surgery and control patients without obesity who had a cholecystectomy. DE genes are represented with FC ≥2 and ≤0.5. Overexpressed genes are in red, and down‐regulated genes are in blue. Rows and columns represent genes and patients, respectively. (B) Venn representation of the DE genes. The number of genes commonly DE between the Ob_NL, Ob_ST, and OB_NASH groups are indicated as well as those specific to each group. (C) List of DE genes in the three groups of patients Ob_NL, Ob_ST, and Ob_NASH compared to the control group (0.5 ≥ FC ≥ 2). (D) Functional analysis (IPA) of differentially regulated genes between patients with steatohepatitis and their controls. Regarding up‐regulated genes, the immune response and inflammatory pathways involving the proinflammatory genes CXCL9, CXC10, and IL32 are among the most significantly enriched pathways. IL32 is particularly enriched in the dendritic cell maturation pathway. For down‐regulated genes, IGFBP2 is annotated in the IGF‐1 signaling pathway, the most significantly enriched pathway. HPRT1 is annotated in the second and fourth most enriched nucleotide salvage pathways. The dashed line indicates the Fisher exact test P value threshold set at 0.05. (E) Heatmap of IPA disease and biological function comparison analysis between Ob_NL, Ob_ST, and Ob_NASH liver samples. The intensity of the orange color indicates the functions that are overrepresented in a given condition based on genes that are DE, whereas the intensity of the blue color reflects the functions that are underrepresented. The activation z score represents the bias in gene regulation that predicts that a biological function is overrepresented or underrepresented in a given condition. (F) Gene‐to‐gene correlation between microarray DE genes. Positive correlations ≥0.6 are indicated with red lines and negative correlations ≤−0.6 are indicated with blue lines. Abbreviations: FC, fold change; IPA, Ingenuity Pathway Analysis.
Functional gene enrichment analysis of up‐regulated genes in the Ob_NASH patients showed that inflammation‐associated pathways and those associated with the adaptive and innate immune response, such as the pathogenesis multiple sclerosis pathway (Fisher’s exact test, P = 3.9E–06) in which the proinflammatory chemokine (C‐X‐C motif) ligand 9 (CXCL9) and CXCL10 genes are annotated or the dendritic cell maturation pathway (Fisher’s exact test, P = 4.7E–04) in which a gene encoding a less well‐characterized inflammatory cytokine, namely IL32, is annotated, were also enriched (Fig. 2D). This analysis supports the notion that the up‐regulation of the inflammatory genes CXCL9, CXCL10, and IL32 may play important roles in the pathogenesis of NASH. Interestingly, in Ob_ST patients, both CXCL9 and CXCL10 were annotated in the pathogenesis of the multiple sclerosis pathway, which is the second most enriched pathway (Fisher’s exact test, P = 3.1E–06), whereas IL32 was not annotated in the most significant enriched pathways (Fisher’s exact test, P = 0.02), suggesting that IL32 might play a more prominent role in NASH than in steatosis. The full list of enriched pathways and their annotated genes in the obese NAFLD bariatric groups (Ob_ST and Ob_NASH) regarding both down‐ and up‐regulated genes is summarized in Supporting Table S3. Moreover, a comparison using the Ingenuity Pathway Analysis bioinformatics tool (http://qiagenbioinformatics.com/products/ingenuity-pathway-analysis) showed that several biological functions linked to the inflammatory response, chemotaxis, and cell movement of immune cells are predicted to be activated in Ob_ST liver samples, and some of them even more in Ob_NASH patients as is the case for the movement of myeloid cells and macrophages (Fig. 2E). This analysis also revealed that the metabolism of triglycerides is predicted to be much less activated in liver samples of Ob_NAFLD patients compared to Ob_NL patients, in line with the observed decrease in the expression of the fatty acid oxidation‐inducing gene PPARα (Supporting Fig. S3) in Ob_NAFLD samples and not in Ob_NL samples.
To address possible crosstalk between the different genes found in our molecular analysis, we applied Pearson’s correlation analysis. Six genes were highly intercorrelated, including IL32, CXCL9, CXCL10, ubiquitin D (UBD)/F adjacent transcript 10 (FAT10), insulin‐like growth factor‐binding protein 2 (IGFBP2) and hypoxanthine phosphoribosyltransferase 1 (HPRT1). The last three genes are: i) UBD, a tumor necrosis factor alpha (TNFα)‐inducible gene mediating nuclear factor kappa B (NF‐κB) activation18 and highly up‐regulated in HCC19; ii) IGFBP2, a leptin‐regulated gene with antidiabetic effects20; and iii) HPRT1, the gene encoding the enzyme hypoxanthine phosphoribosyltransferase 1, which plays a central role in the generation of purine nucleotides through the purine salvage pathway. HPRT1 deficiency has been shown to cause Lesch‐Nyhan syndrome with hyperuricemia and gout21 (Fig. 2F). These genes form a hexagram in which the proinflammatory genes (IL32, CXCL9, and CXCL10) and UBD are positively intercorrelated. IGFBP2 and HPRT1 were positively intercorrelated and negatively correlated with IL32 (Fig. 2F). Altogether, our analysis showed that the up‐regulation of specific proinflammatory cytokines, particularly IL32, is part of the molecular signature of metabolic‐induced liver disease.
Large‐Scale Analysis of Inflammatory Gene Expression and Possible Link With Insulin Resistance
To validate our microarray data, we used qRT‐PCR to assess the relative expression of IL32, CXCL9, CXCL10, UBD/FAT10, IGFBP2, and HPRT1 in the entire patient cohort (n = 122). Consistently, compared to control patients, IL32 expression was increased 2.5‐fold in the NAFLD group (P < 0.001) while a moderate but not statistically significant increase in IL32 expression was observed in Ob_NL patients (1.6‐fold; Fig. 3A). The expressions of CXCL9 and CXCL10 were also significantly increased in the Ob_NAFLD group (1.9‐fold and 1.9‐fold; P < 0.001 and P < 0.05, respectively), while the change in expression was significant only for CXCL9 and not for CXCL10 in the Ob_NL group (1.9‐fold and 1.1‐fold, respectively; Fig. 3C,D). The decreased expression of IGFBP2 and HPRT1 was confirmed in all patients with obesity with a more pronounced decrease in the NAFLD group (0.5‐fold, P < 0.001 and 0.7‐fold, P < 0.01, respectively; Fig. 3E,F). Finally, the expression of UBD/FAT10 was significantly increased in patients with NAFLD, and this elevation was more pronounced in patients with NASH (3.3‐fold, P < 0.01; Fig. 4B). Importantly, consistent with gene expression analysis, IL32 protein level was increased in liver samples of patients with NAFLD (Fig. 5A).
Figure 3.
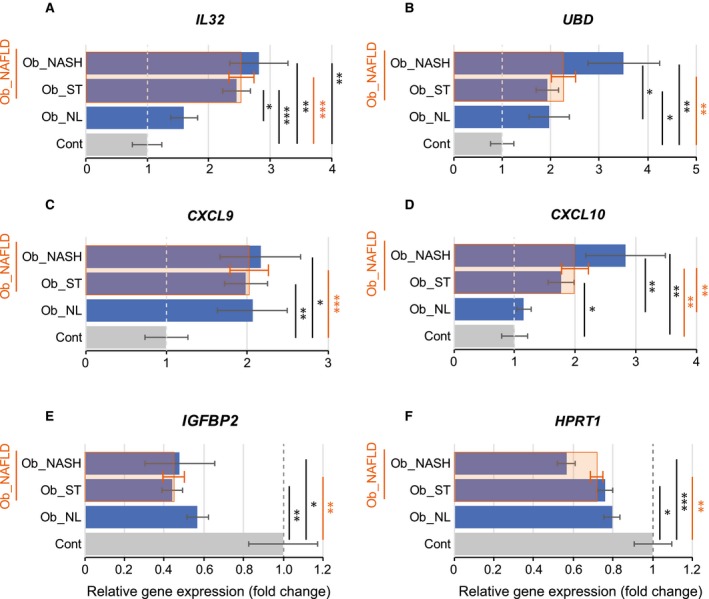
qRT‐PCR expression profile of the most intercorrelated genes found in microarray analysis. (A) IL32, (B) UBD, (C) CXCL9, and (D) CXCL10 were significantly up‐regulated in patients with NAFLD, whereas (E) IGFBP2 and (F) HPRT1 were significantly down‐regulated compared to controls. Results are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 4.
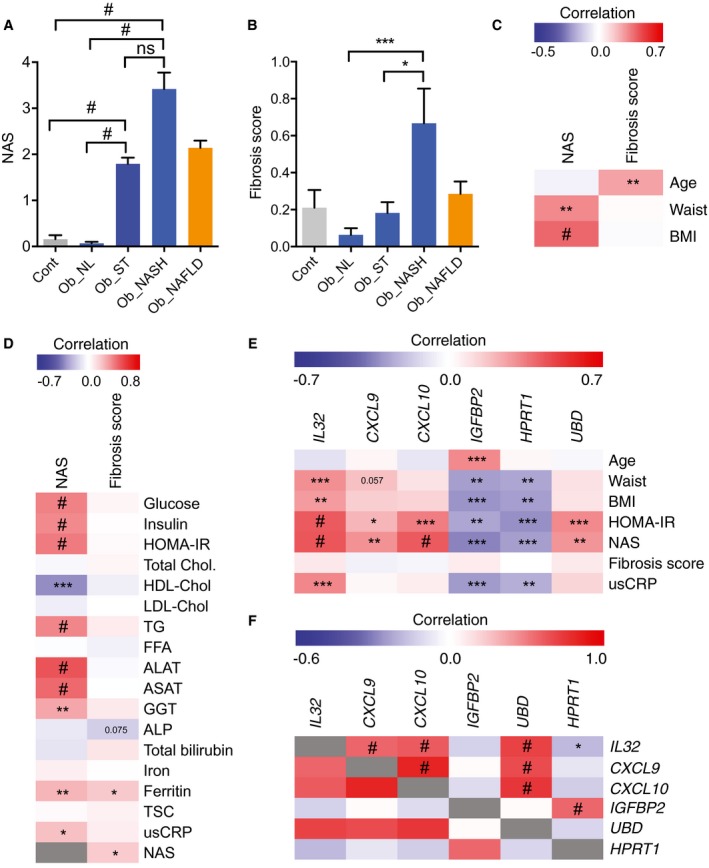
NAFLD activity score, fibrosis score and correlation with biochemical parameters and NAFLD molecular signature. (A) NAS variation in control patients without obesity who received a cholecystectomy and in patients who received bariatric surgery in Ob_NL, Ob_ST, Ob_NASH, and Ob_NAFLD (Ob_ST + Ob_NASH) groups. (B) Fibrosis score variation in Ob_NL, Ob_ST, Ob_NASH, and Ob_NAFLD compared to controls. (C) Heatmap of the Spearman correlation of NAS and fibrosis score relative to age, waist circumference, and BMI in our cohort of patients. (D) Heatmap of the Spearman correlation of NAS and fibrosis score relative to biochemical parameters in our cohort of patients. (E) Heatmap of the Spearman correlation of the DE genes IL32, CXCL9, CXCL10, IGFBP2, HPRT1, and UBD in liver samples relative to age, waist circumference, BMI, HOMA‐IR index, NAS, fibrosis score, and usCRP in our cohort of patients. (F) Heatmap of gene‐to‐gene correlation analysis of the qRT‐PCR profile for the most intercorrelated genes found by microarray analysis. IL32 correlated positively and significantly with CXCL9, CXCL10, and UBD and correlated negatively with HPRT1. The down‐regulated genes IGFBP2 and HPRT1 were strongly and positively intercorrelated. The intensities of the red and blue color reflect the degree of positive and negative correlation, respectively. The gray color is the correlation of each parameter to itself (r = 1). Results are expressed as mean ± SEM; #P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. Abbreviations: FFA, free fatty acid; TG, triglyceride; TSC, transferrin saturation capacity.
Figure 5.
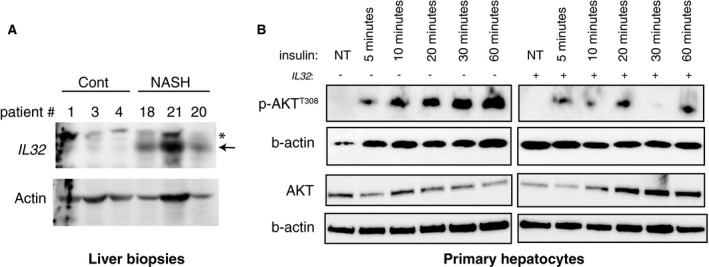
IL32 protein expression in liver biopsies from Cont and Ob_NASH patients and effect of recombinant IL32 on insulin signaling in PHHs. (A) Immunoblots of protein extracts from patients with NASH and control liver biopsies showing an increase in IL32 expression in NASH. (B) Recombinant IL32 treatment induces decreased insulin signaling in protein lysates from PHHs as demonstrated by the robust decrease in AKTT308 phosphorylation after insulin stimulation. Abbreviations: Cont, controls; NT, nontreated; *, non specific.
IL32 Gene Expression Levels Correlate With NAS
In order to understand the biological relevance of the change in gene expression of the above‐mentioned genes, we calculated NAS14 and the fibrosis score (METAVIR) for all patients in the study. We found a significant increase in NAS in Ob_ST and Ob_NASH patients but not in Ob_NL patients (Fig. 4A); however, the fibrosis score was increased in Ob_NASH patients compared to Ob_ST and Ob_NL patients (Fig. 4B). NAS was significantly and positively correlated with waist circumference and BMI and not with the age of patients, whereas the fibrosis score was positively correlated with age but not with waist circumference and BMI (Fig. 4C). Furthermore, we analyzed the potential correlation of NAS and fibrosis score with classical biochemical parameters. NAS correlated significantly and positively with glucose, insulin, HOMA‐IR, triglyceride levels, ALAT, ASAT, GGT, ferritin, and usCRP and negatively with HDL‐cholesterol (Fig. 4D). Fibrosis score correlated with ferritin and NAS score (Fig. 4D), suggesting that hepatic steatosis contributes to fibrosis development. We then correlated the expression levels of the six genes analyzed by qRT‐PCR with age and with clinical, histologic, and biological parameters, such as waist circumference, BMI, HOMA‐IR, NAS, fibrosis score, and usCRP. Only IGFBP2 transcript levels correlated positively with age. IL32 correlated positively with waist circumference, BMI, HOMA‐IR, NAS, and usCRP. CXCL9, CXCL10, and UBD positively correlated with HOMA‐IR and NAS, suggesting that these genes contribute to hepatic steatosis and insulin resistance. IGFBP2 and HPRT1 negatively correlated with waist circumference, BMI, HOMA‐IR, NAS, and usCRP. None of the genes correlated with the fibrosis score in our cohort of patients (Fig. 4E).
In line with our previous correlation on microarray data, IL32 expression was positively correlated with CXCL9, CXCL10, and UBD/FAT10 and negatively correlated with HPRT1 (Fig. 4F). CXCL9 was positively correlated with CXCL10, and UBD was correlated with both CXCL9 and CXCL10 (Fig. 4F). Altogether, our large‐scale analysis confirmed the critical role of inflammatory cytokines, in particular IL32, in liver injury in NAFLD and particularly in patients with NASH. As such, these analyses further support our conclusion that increased IL32, CXCL9, CXCL10, and UBD levels and decreased IGFBP2 and HPRT1 expression represent a molecular signature of insulin resistance and liver injury in patients with obesity with NAFLD.
IL32 Supplementation Promotes Insulin Resistance in PHHs
Following the observation that IL32 protein levels were increased in patients with NASH (Fig. 5A), we aimed to address the direct impact of IL32 on hepatocytes. We treated PHH cells with recombinant IL32. PHHs were treated with recombinant IL32 (1 ng/mL) for 24 hours before a time‐course exposition to insulin (100 nM) during 5, 10, 20, 30, and 60 minutes. The ratio of AKT phosphorylation (p‐AKTT308)/AKT was determined in IL32‐treated and untreated PHHs after insulin addition as an indicator of insulin signaling pathway activation. As expected, in the control condition (without IL32 treatment), treatment of PHHs with insulin induced an increase in p‐AKT/AKT ratio with time, with a maximum increase at 60 minutes (Fig. 5B). Pretreatment of PHHs with IL32 yielded a robust reduction in p‐AKT/AKT ratio 30 minutes after insulin addition, demonstrating that IL32 contributes to insulin resistance and blunts insulin receptor signaling (Fig. 5B).
Discussion
Here, we identified IL32, a proinflammatory cytokine known to play a role in host defense, inflammatory diseases, and cancer (reviewed in Joosten et al.22), as a novel factor clearly associated with NAFLD and insulin resistance in patients with obesity who underwent bariatric surgery. IL32 expression was significantly increased in the liver of patients with NAFLD, and this elevation was more pronounced in patients with NASH. Similar to IL32, the expression of two other proinflammatory chemokines were enhanced, namely CXCL9 and CXCL10. Furthermore, we observed a positive correlation between liver expression of IL32 and CXCL9 and CXCL10, supporting solid crosstalk among these cytokines in NASH pathophysiology. The expression of CXCL9 and CXCL10 has been reported to be increased in the liver and serum of patients infected with HCV23 as was the expression of IL32 in patients infected with HCV24 and HBV.25 Increased intrahepatic transcript level of CXCL10 (also known as interferon γ [IFNγ]‐induced protein 10) has been associated with intralobular inflammation, and CXCL9 (also known as monokine induced by IFNγ) was shown to correlate with the grade of liver inflammation.26 Furthermore, CXCL10 has been recently shown to mediate macrophage‐associated inflammation in a murine model of NASH.27 These two cytokines are notoriously known to be activated by IFNγ,28, 29, 30 a molecule with immune regulatory functions secreted by T helper 1 (Th1) inflammatory cells in the context of virus‐induced and metabolic inflammation.31, 32, 33, 34, 35
How is IL32 produced in the context of NAFLD and particularly in NASH? It is well established that myeloid and lymphoid cells infiltrate adipose tissue and liver as a result of excessive nutrient load.34, 36 One possibility is that IL32 can be produced following the activation of Th1 cells. Indeed, the activation of these cells is known to produce IFNγ and TNFα in the context of obesity and oxidative stress‐induced liver inflammation, which translates into the promotion of the classical M1 inflammatory‐dominated response.34, 35 IL32 was initially discovered as a protein that is produced following the activation of both natural killer cells and T cells.37 Later, IL32 was shown to stimulate the production of several proinflammatory cytokines, including TNFα, and to activate typical cytokine signaling pathways of NF‐κB.38 Reversely, TNFα and IFNγ were shown to activate IL32 production in various pathologic contexts,38, 39, 40 underpinning the presence of an autoperpetuating loop between IL32 and TNFα to sustain inflammation. UBD/FAT10 was also significantly correlated with IL32, CXCL9, and CXCL10 in our cohort of patients. UBD is an IFNγ‐ and TNFα‐inducible ubiquitin‐like protein with a putative role in immune response.41, 42 An IFN sequence response element is present on the promoter of the UBD/FAT10 gene, which responds to synergistic stimulation by TNFα and IFNγ to induce Mallory‐Denk body (MDB)‐like aggresomes.43 It was reported that UBD deletion prevents the formation of MDBs through the maintenance of the physiological 26S proteasome.44 Overexpression of both IL32 and UBD in the liver of patients with NAFLD and obesity strongly suggests that these two molecules participate in the immune response and hence contribute to alterations in cell proteostasis; this ultimately leads to liver injury in patients with NAFLD, especially in patients with NASH in whom IL32 and UBD up‐regulation was more pronounced.
Our analysis showed that recombinant IL32 caused a significant decrease in insulin signaling in PHHs, underscoring a potential causal role in insulin resistance. Whether this effect is direct or indirect needs further clarification. Mechanistically, IL32‐mediated insulin resistance could be due to the feed‐forward loop involving IL32 and TNFα. The latter has been shown to inhibit insulin signaling through c‐Jun N‐terminal kinase 1‐mediated Ser307 phosphorylation of insulin receptor substrate 1 (IRS1).45 The second possibility is that IL32 could promote protein kinase C epsilon (PKCε) (encoded by the PKCε gene (PRKCE)) activation, which is known to suppress insulin signaling through reduced insulin‐stimulated IRS2 tyrosine phosphorylation.46 In line with this, our microarray data set showed a ~3‐fold increase in PRKCE transcripts in the liver of patients with NAFLD compared to controls and a positive correlation between liver IL32 expression and PRKCE (r = 0.607).
Most interestingly, the expression of IL32, CXCL9, CXCL10, and UBD correlated with NAS and the HOMA‐IR index, while IGFBP2 and HPRT1 correlated negatively with these parameters. NAS was primarily designed by the NASH CRN as a system of scoring histopathologic features of NAFLD during clinical trials.14 Although early studies have used threshold values of NAS ≥5 as a surrogate for the histologic diagnosis of NASH, a recent study showed that NAS <4 is not a synonym of a benign histology.47 This was indeed our case as only 2 patients with NASH had NAS ≥5 while 10 patients with NASH had NAS ≤5. Consistent with results from the study by Brunt et al.47 in which higher values of NAS were associated with increased levels of ALAT and ASAT, we observed a significant and positive correlation of NAS with hepatic aminotransferases, a hallmark for the severity of liver injury. The fact that the NAFLD molecular signature (increased IL32, CXCL9, CXCL10, and UBD and decreased IGFBP2 and HPRT1) was correlated with NAS as well as aminotransferases (e.g., IL32 correlates with ALAT and ASAT; P = 0.002 and P = 0.006, respectively) strongly suggests that these genes contribute to the pathogenesis of NASH. The reason why this molecular signature was not correlated with fibrosis lies in the possibility that the fibrosis was not extensive enough in our cohort of patients, or due to the lower number of patients with NASH (n = 12) compared to those with steatosis (n = 44) in whom few patients had a fibrosis score of 1, or simply that these genes are not associated with fibrosis in the context of metabolic‐induced liver injury. Nevertheless, the expression of these genes remains to be verified in liver biopsies of patients with NAFLD in whom a Fibroscan or Fibrotest has revealed a high fibrosis score compared to patients with NAFLD with a low fibrosis score. Obviously, much effort is needed to understand how these genes interact and the signaling pathway network activated in order to achieve insulin resistance and liver injury.
Finally, our analysis reported a positive correlation between usCRP and BMI and between usCRP and waist circumference but did not reveal any correlation between usCRP with HOMA‐IR. In line with this, whether usCRP is correlated to insulin resistance remains controversial in the literature. Indeed, Hossain et al.48 reported a positive correlation between highly sensitive CRP (hsCRP) and HOMA‐IR (P = 0.03) in 140 patients with prediabetes, yet these patients were not obese (BMI, 26.04 ± 4.51 kg/m2). Another study showed that hsCRP correlated with BMI and waist to hip ratio but not with HOMA‐IR in West Africans while it was correlated with all three parameters in an African‐American population.49 Finally, Namburi et al.50 showed that hsCRP was correlated with BMI and insulin but not with HOMA‐IR in children and adolescents with obesity. Although, we observed a positive correlation between liver IL32 expression and usCRP, it seems that these two proteins may share common inflammatory pathways but not a pathway interfering with the insulin signaling pathway. Altogether, we believe that although usCRP is associated with obesity and NAFLD, it is not a reliable marker of insulin resistance in patients with obesity who are prediabetic.
In summary, our large‐scale analysis identifies new protagonists in the pathogenesis of NAFLD. Mainly, we showed a robust increase in liver expression of proinflammatory IL32 that was significantly correlated with NAS, aminotransferases, and insulin resistance in patients with obesity who underwent bariatric surgery. Moreover, IL32 supplementation on PHHs impairs insulin signaling, supporting a causal effect of IL32 in the pathogenesis of liver insulin resistance. Nevertheless, molecular mechanisms governing the interplay between these proteins to promote liver injury, insulin resistance, and hepatic liver disease progression remain to be dissected. Given that IL32 has been shown to be involved in hepatitis‐induced liver damage, it is plausible that activation of the TNFα–IL32–IFNγ–UBD axis may be a general mechanism leading to liver injury. We propose that inhibition of IL32, for example, using neutralizing antibodies in patients with NAFLD, may prove efficient in the treatment of metabolic liver disease.
Supporting information
Acknowledgment
We thank Dr. S. Bayer and Dr. C. Metzger from the Unité de Coordination de la Biologie des Essais Cliniques, Hôpitaux Universitaires de Strasbourg, for their help in patient blood sample management and technical assistance. We acknowledge the Centre de Ressources Biologiques, Strasbourg, France, for the management of patient‐derived liver tissues. We are indebted to Dr. K. Hnia for his input and critical reading of the manuscript and to Professor J. Auwerx who initiated this project. We thank the Hôpitaux Universitaires de Strasbourg for promoting this study and particularly the Fonds Regional de Coopération pour la Recherche (Grand Est‐Alsace Champagne‐Ardenne Lorraine) for their grant to the project Pathological Obesity and Metabolic Aging (OMAGE).
Supported by the Programme Hospitalier de Recherche Clinique‐regional (number 3966‐2007 to Hôpitaux Universitaires de Strasbourg).
Trial Registration clinicaltrials.gov, Identifier: NCT00844779.
Present address for Federico Costantino is Groupe Hospitalier St‐Vincent, Clinique Sainte‐Anne, Strasbourg France; Cosimo Callari is Department of Surgery, Ospedale Buccheri La Ferla Fatebenefratelli, 90123 Palermo, Italy; Jacopo D’Agostino is Pole Obésité Etang de Berre, Clinique de Martigues, Martigues, France; Stefan Paveliu is Clinique St‐George, Nice, France; Monica Gualtierotti is Chirurgia Generale Oncologica e Mininvasiva, Ospedale Niguarda Cà Granda, Milano, Italy; Mirjam B. Zeisel is Inserm U1052, CNRS UMR 5286, Centre Léon Bérard, Cancer Research Center of Lyon, Université de Lyon, Lyon, France.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332:1519‐1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med 2017;377:2063‐2072. [DOI] [PubMed] [Google Scholar]
- 3. Rinella ME, Sanyal AJ. Management of NAFLD: a stage‐based approach. Nat Rev Gastroenterol Hepatol 2016;13:196‐205. [DOI] [PubMed] [Google Scholar]
- 4. Argo CK, Northup PG, Al‐Osaimi AM, Caldwell SH. Systematic review of risk factors for fibrosis progression in non‐alcoholic steatohepatitis. J Hepatol 2009;51:371‐379. [DOI] [PubMed] [Google Scholar]
- 5. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547‐555. [DOI] [PubMed] [Google Scholar]
- 6. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stål P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology 2015;61:1547‐1554. [DOI] [PubMed] [Google Scholar]
- 8. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014;59:713‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koppe SW. Obesity and the liver: nonalcoholic fatty liver disease. Transl Res 2014;164:312‐322. [DOI] [PubMed] [Google Scholar]
- 10. Leung JC, Loong TC, Wei JL, Wong G, Chan AW, Choi PC, et al. Histological severity and clinical outcomes of nonalcoholic fatty liver disease in nonobese patients. Hepatology 2017;65:54‐64. [DOI] [PubMed] [Google Scholar]
- 11. Gastaldelli A, Kozakova M, Højlund K, Flyvbjerg A, Favuzzi A, Mitrakou A.; RISC Investigators . Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology 2009;49:1537‐1544. [DOI] [PubMed] [Google Scholar]
- 12. Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003;37:917‐923. [DOI] [PubMed] [Google Scholar]
- 13. Musso G, Cassader M, Gambino R. Non‐alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov 2016;15:249‐274. [DOI] [PubMed] [Google Scholar]
- 14. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 15. Dembélé D, Kastner P. Fold change rank ordering statistics: a new method for detecting differentially expressed genes. BMC Bioinformatics 2014;15:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 2011;17:589‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab 2005;288:E462‐E468. [DOI] [PubMed] [Google Scholar]
- 18. Gong P, Canaan A, Wang B, Leventhal J, Snyder A, Nair V, et al. The ubiquitin‐like protein FAT10 mediates NF‐kappaB activation. J Am Soc Nephrol 2010;21:316‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee CG, Ren J, Cheong IS, Ban KH, Ooi LL, Yong Tan S, et al. Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene 2003;22:2592‐2603. [DOI] [PubMed] [Google Scholar]
- 20. Hedbacker K, Birsoy K, Wysocki RW, Asilmaz E, Ahima RS, Farooqi IS, et al. Antidiabetic effects of IGFBP2, a leptin‐regulated gene. Cell Metab 2010;11:11‐22. Erratum in: Cell Metab 2010;11:239. [DOI] [PubMed] [Google Scholar]
- 21. Rossiter B, Caskey CT. Hypoxanthine‐guanine phosphoribosyltransferase deficiency: Lesch‐Nyhan syndrome and gout In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. Molecular and Metabolic Bases of Inherited Disease. 8th ed New York, NY: McGraw‐Hill; 1995:1679‐1706. [Google Scholar]
- 22. Joosten LA, Heinhuis B, Netea MG, Dinarello CA. Novel insights into the biology of interleukin‐32. Cell Mol Life Sci 2013;70:3883‐3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larrubia JR, Benito‐Martínez S, Calvino M, Sanz‐de‐Villalobos E, Parra‐Cid T. Role of chemokines and their receptors in viral persistence and liver damage during chronic hepatitis C virus infection. World J Gastroenterol 2008;14:7149‐7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moschen AR, Fritz T, Clouston AD, Rebhan I, Bauhofer O, Barrie HD, et al. Interleukin‐32: a new proinflammatory cytokine involved in hepatitis C virus‐related liver inflammation and fibrosis. Hepatology 2011;53:1819‐1829. [DOI] [PubMed] [Google Scholar]
- 25. Xu Q, Pan X, Shu X, Cao H, Li X, Zhang K, et al. Increased interleukin‐32 expression in chronic hepatitis B virus‐infected liver. J Infect 2012;65:336‐342. [DOI] [PubMed] [Google Scholar]
- 26. Apolinario A, Majano PL, Alvarez‐Pérez E, Saez A, Lozano C, Vargas J, et al. Increased expression of T cell chemokines and their receptors in chronic hepatitis C: relationship with the histological activity of liver disease. Am J Gastroenterol 2002;97:2861‐2870. [DOI] [PubMed] [Google Scholar]
- 27. Tomita K, Freeman BL, Bronk SF, LeBrasseur NK, White TA, Hirsova P, et al. CXCL10‐mediates macrophage, but not other innate immune cells‐associated inflammation in murine nonalcoholic steatohepatitis. Sci Rep 2016;6:28786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Amichay D, Gazzinelli RT, Karupiah G, Moench TR, Sher A, Farber JM. Genes for chemokines MuMig and Crg‐2 are induced in protozoan and viral infections in response to IFN‐gamma with patterns of tissue expression that suggest nonredundant roles in vivo. J Immunol 1996;157:4511‐4520. [PubMed] [Google Scholar]
- 29. Sgadari C, Farber JM, Angiolillo AL, Liao F, Teruya‐Feldstein J, Burd PR, et al. Mig, the monokine induced by interferon‐gamma, promotes tumor necrosis in vivo. Blood 1997;89:2635‐2643. [PubMed] [Google Scholar]
- 30. Basset L, Chevalier S, Danger Y, Arshad MI, Piquet‐Pellorce C, Gascan H, et al. Interleukin‐27 and IFNγ regulate the expression of CXCL9, CXCL10, and CXCL11 in hepatitis. J Mol Med (Berl) 2015;93:1355‐1367. [DOI] [PubMed] [Google Scholar]
- 31. Farrar MA, Schreiber RD. The molecular cell biology of interferon‐gamma and its receptor. Annu Rev Immunol 1993;11:571‐611. [DOI] [PubMed] [Google Scholar]
- 32. Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark‐Lewis I, et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T‐lymphocytes. J Exp Med 1996;184:963‐969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN‐γ‐inducible protein 10 (IP‐10; CXCL10)‐deficient mice reveal a role for IP‐10 in effector T cell generation and trafficking. J Immunol 2002;168:3195‐3204. [DOI] [PubMed] [Google Scholar]
- 34. Brestoff JR, Artis D. Immune regulation of metabolic homeostasis in health and disease. Cell 2015;161:146‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014;59:886‐897. [DOI] [PubMed] [Google Scholar]
- 36. Dali‐Youcef N, Mecili M, Ricci R, Andres E. Metabolic inflammation: connecting obesity and insulin resistance. Ann Med 2013;45:242‐253. [DOI] [PubMed] [Google Scholar]
- 37. Dahl CA, Schall RP, He HL, Cairns JS. Identification of a novel gene expressed in activated natural killer cells and T cells. J Immunol 1992;148:597‐603. [PubMed] [Google Scholar]
- 38. Kim SH, Han SY, Azam T, Yoon DY, Dinarello CA. Interleukin‐32: a cytokine and inducer of TNFalpha. Immunity 2005;22:131‐142. [DOI] [PubMed] [Google Scholar]
- 39. Netea MG, Azam T, Lewis EC, Joosten LA, Wang M, Langenberg D, et al. Mycobacterium tuberculosis induces interleukin‐32 production through a caspase‐ 1/IL‐18/interferon‐gamma‐dependent mechanism. PLoS Medicine 2006;3:e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heinhuis B, Koenders MI, van Riel PL, van de Loo FA, Dinarello CA, Netea MG, et al. Tumour necrosis factor alpha‐driven IL‐32 expression in rheumatoid arthritis synovial tissue amplifies an inflammatory cascade. Ann Rheum Dis 2011;70:660‐667. [DOI] [PubMed] [Google Scholar]
- 41. Raasi S, Schmidtke G, de Giuli R, Groettrup M. A ubiquitin‐like protein which is synergistically inducible by interferon‐gamma and tumor necrosis factor‐alpha. Eur J Immunol 1999;29:4030‐4036. [DOI] [PubMed] [Google Scholar]
- 42. Bates EE, Ravel O, Dieu MC, Ho S, Guret C, Bridon JM, et al. Identification and analysis of a novel member of the ubiquitin family expressed in dendritic cells and mature B cells. Eur J Immunol 1997;27:2471‐2477. [DOI] [PubMed] [Google Scholar]
- 43. Oliva J, Bardag‐Gorce F, Lin A, French BA, French SW. The role of cytokines in UbD promoter regulation and Mallory‐Denk body‐like aggresomes. Exp Mol Pathol 2010;89:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. French SW, French BA, Oliva J, Li J, Bardag‐Gorce F, Tillman B, et al. FAT10 knock out mice livers fail to develop Mallory‐Denk bodies in the DDC mouse model. Exp Mol Pathol 2012;93:309‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature 2002;420:333‐336. [DOI] [PubMed] [Google Scholar]
- 46. Samuel VT, Liu Z‐X, Wang A, Beddow SA, Geisler JG, Kahn M, et al. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 2007;117:739‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander‐Tetri BA; NASH Clinical Research Network (CRN) . Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology 2011;53:810‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hossain IA, Akter S, Bhuiyan FR, Shah MR, Rahman MK, Ali L. Subclinical inflammation in relation to insulin resistance in prediabetic subjects with nonalcoholic fatty liver disease. BMC Res Notes 2016;9:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Doumatey AP, Lashley KS, Huang H, Zhou J, Chen G, Amoah A, et al. Relationships among obesity, inflammation, and insulin resistance in African Americans and West Africans. Obesity (Silver Spring) 2010;18:598‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Namburi RP, Ponnala AR, Karthik TS, Rani PR, Maheshwari R. A study on metabolic variables and its association with high sensitive C‐reactive protein in obese children and adolescents. Indian J Endocrinol Metab 2013;17(Suppl. 1):S360‐S362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials