

Abstract
Studies have shown that tumors commonly exhibit normal or enhanced respiration in addition to glycolytic metabolism. In this issue, Courtney et al. (2018) report a reduction in mitochondrial function in kidney cancer patients and thus a classic “Warburg Effect” that further illustrates the heterogeneity of human cancer metabolism.
Nearly a century ago, Otto Warburg and colleagues observed that tumors exhibit significantly elevated rates of glucose uptake, even in the presence of oxygen. This observation has been foundational to our understanding of cancer metabolism and has provided the basis for modern tumor detection and monitoring via 18-fluoro-2-deoxyglucose position emission tomography (FDG-PET) (Liberti and Locasale, 2016). After many iterations over the years, Warburg’s hypothesis, now referred to as the Warburg effect or aerobic glycolysis, was conceptually defined as a switch from oxidative glucose metabolism involving respiration in the mitochondria to fermentation from glycolysis that generates lactate as the primary energy source.
However, even within the initial work from Warburg and others it was found that many cancers undergo respiration. Indeed, in recent years, with the advent of in vivo analysis of cancer metabolism, it has been shown that non-small cell lung (NSCLC) tumors (Hensley et al., 2016), liver cancers (Yuneva et al., 2012), as well as primary and metastatic brain tumors (Maher et al., 2012) exhibit significant and even elevated levels of complete glucose oxidation, as indicated by stable isotope tracing of mouse tumors or human patients infused with 13C-glucose. Certain cancers remain dependent on mitochondrial function in vivo (Liu et al., 2016; Weinberg and Chandel, 2015), while others specifically require aerobic glycolysis (Liberti et al., 2017). Notably, these phenomena have been attributed to oncogene-induced metabolic programming, which has been primarily characterized in cell culture, although intriguing evidence from in vivo studies suggests that the environment may play an equal, if not larger, role to these genetic alterations in shaping metabolic state (Davidson et al., 2016; Yuneva et al., 2012). Overall, these observations have led to an evolved understanding of the Warburg effect, with the current view being that tumors acquire the ability to undergo enhanced aerobic glycolysis while simultaneously maintaining, if not also upregulating, mitochondrial metabolism.
Findings reported in this issue of Cell Metabolism (Courtney et al., 2018) now provide some of the first experimental evidence in human patients that the metabolic switch proposed by Warburg indeed occurs in certain contexts. Five patients exhibiting primary clear cell renal cell carcinoma (ccRCC) were infused with 13C-glucose before surgery as conducted previously in NSCLC and brain tumor patients. Surprisingly, it was found that this cancer subtype exhibited a remarkably different metabolic profile compared to those found in other types of patient tumors. While the ccRCC tumors demonstrated increased labeling of glycolytic metabolites (consistent with previous observations in other tumors), an accompanying reduction in labeling of TCA metabolites was also observed compared to normal kidney tissue. Furthermore, ccRCC tumors exhibited a reduced fraction of labeled acetyl-coAand nearly undetectable labeled glutamine, indicating significantly reduced pyruvate dehydrogenase (PDH) and glutamine synthetase enzyme activity. As a final measure, [1,2-13C]-acetate labeling data collected from one ccRCC patient further indicated low TCA cycle turnover independent of PDH activity. These findings define a metabolic phenotype characterized by reduced utilization of glucose oxidation in favor of aerobic glycolysis, in line with the classic view of the Warburg effect (Figure 1).
Figure 1. Human Kidney Tumors Exhibit a Distinct Metabolic Profile from Other Tumor Types.
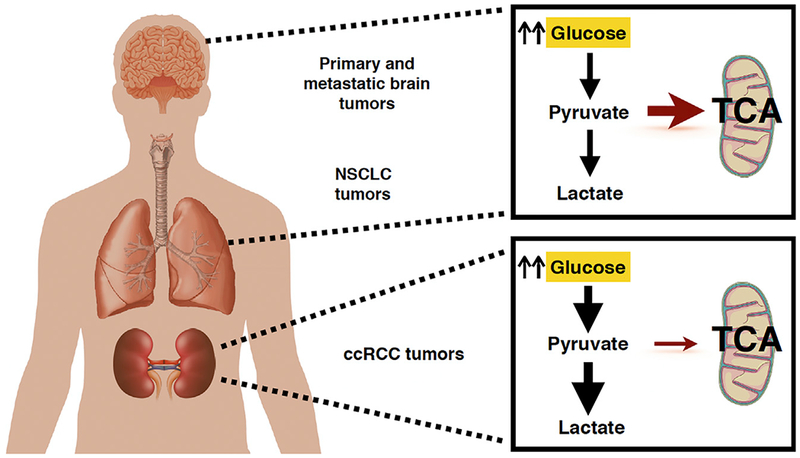
Patient isotope tracing of human brain and lung tumors using 13C-glucose has shown increased glucose uptake and lactate production while maintaining significant glucose oxidation and respiration in the mitochondria. Human ccRCC kidney tumors, however, display markedly different metabolic programming, using glycolysis as a primary energy source with a decrease in mitochondrial respiration, in line with a classic Warburg effect.
These observations are particularly interesting upon consideration of the genetic features of this cancer subtype, as the majority of ccRCCs are driven by loss of the tumor suppressor VHL gene. This gene encodes an E3 ubiquitin ligase component that is known to promote degradation of the hypoxia inducible factors HIF-1α and HIF-2α, resulting in ccRCC having a pseudo-hypoxic state characterized by increased HIF accumulation and activity due to VHL loss. Given the well-characterized transcriptional role of HIF-1α in regulating the Warburg effect, it has been proposed that metabolic dysfunction may be the most prominent characteristic of these tumors. As the results of this study provide some of the first direct evidence with isotope tracers in human patients to support this notion, it is tempting to thus conclude that the hard-wired induction of this pseudo-hypoxic state may be sufficient to promote the canonical Warburg effect.
While the results of this important study are very insightful, there are some important factors to take into account when interpreting the data. Variability across and within patient tumors is large, creating significant limitations in extrapolating generalities from relatively small sample sizes. The current study, albeit limited in power, presents what is currently feasible in human studies. Additionally, there currently is a lack of consensus on how to interpret metabolic flux from isotope labeling data and, unfortunately, in many cases the interpretation can significantly differ depending on the type of model and analysis used. Furthermore, it remains to be determined whether the observed metabolic phenotype is genetically hard-wired or a result of the hypoxic, glucose-deprived tumor environment found in ccRCC.
Nevertheless, these findings clearly demonstrate a greater complexity of mitochondrial function in patient tumors than what has been previously observed. Moving forward, it will be interesting to see whether this decrease in glucose oxidation is a property inherent to tumors lacking the VHL gene, the cell of origin in ccRCC, or a consequence of a metabolic adaptation to a kidney-specific tissue environment. It seems likely that this metabolic phenotype will be observed in other types of cancer, given the common presence of downregulation of respiratory genes in tumors (Reznik et al., 2017).
It will be interesting to understand how this decrease in respiration may influence the tumor microenvironment and host-tumor interactions compared to tumors that retain relatively normal rates of cellular respiration. For example, immune cell populations and their effector functions have different requirements for mitochondrial metabolism; if the decreased respiration is environmentally driven, this would be predicted to also affect the immune cell infiltrates and their corresponding effector functions. As methodology for in vivo isotope tracing, in addition to metabolic imaging approaches, continues to be refined and more broadly applied, the underlying mechanisms that define the heterogeneity of cancer cell metabolism in humans will undoubtedly become better understood. Finally, several ongoing investigations in humans are aiming to target mitochondrial metabolism in tumors. The findings in this current and subsequent future studies will hopefully inform these clinical investigations.
ACKNOWLEDGMENTS
Support from the National Institutes of Health (R01CA193256 to J.W.L. and F31CA224973 to S.M.S.) and the American Cancer Society (129832-RSG-16-214-01-TBE to J.W.L.) is gratefully acknowledged.
REFERENCES
- Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, Wimberly J, McNeil SS, Kapur P, Lotan Y, et al. (2018). Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab 28, this issue, 793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, and Locasale JW (2016). The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci 41, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, et al. (2017). A Predictive Model for Selective Targeting of the Warburg Effect through GAPDH Inhibition with a Natural Product. Cell Metab 26, 648–659.e8, e648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Romero IL, Litchfield LM, Lengyel E, and Locasale JW (2016). Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab 24, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher EA, Marin-Valencia I, Bachoo RM, Mashimo T, Raisanen J, Hatanpaa KJ, Jindal A, Jeffrey FM, Choi C, Madden C, et al. (2012). Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed 25, 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznik E, Wang Q, La K, Schultz N, and Sander C (2017). Mitochondrial respiratory gene expression is suppressed in many cancers. eLife 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, and Chandel NS (2015). Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol 11, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Matés JM, Alonso FJ, Wang C, Seo Y, et al. (2012). The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]