

Abstract
Macrophages are important mediators of inflammation and tissue remodeling. Recent insights into the heterogeneity of macrophage subpopulations has renewed interest in their functional diversity in homeostasis and disease. Additionally, their plasticity enables them to perform a variety of functions in response to changing tissue contexts, such as those imposed by aging. These qualities make macrophages particularly intriguing cells given their dichotomous role in protecting against, or accelerating diseases of the cardiovascular system and the eye, two tissues particularly susceptible to the effects of aging. Here, we review novel perspectives on macrophage biology as informed by recent studies detailing the diversity of macrophage identity and function, as well as mechanisms influencing macrophage behavior that might offer opportunities for new therapeutic strategies.
Keywords: Macrophage, heterogeneity, plasticity, aging, disease, eye, heart
New insights into macrophage lineage, function, and plasticity in health and disease
In recent years, it has become increasingly clear that the differential effects of mammalian macrophages during homeostasis and disease are due to their diverse identities and divergent activation states. Furthermore, the different subpopulations of macrophages in a particular tissue undergo dynamic shifts in response to perturbations within these populations and changing tissue microenvironments, as seen in aging. Renewed interest in these aspects of macrophage origin, function, and plasticity has led to the identification of mechanisms responsible for this complexity. In particular, recent studies combining lineage tracing, single-cell transcriptomics, and morphologic and molecular analysis have elucidated the role of diverse macrophage populations in mediating various aspects of development, steady-state function and pathogenesis in the human cardiovascular system and the eye, two organ systems that commonly develop pathology with increased age (Key Table). In this review, we discuss the differential homeostatic and pathogenic functions of various macrophage populations as defined by lineage diversity, molecular expression and phenotypic activation, and their contributions in mouse models of myocardial injury, atherosclerosis, photoreceptor degeneration, and age-related macular degeneration. These novel mechanistic insights demonstrate the possibility of targeting macrophage identity and plasticity as potential therapeutic strategies for diseases where macrophages play a crucial role.
Key Table: Putative functional roles of diverse macrophage populations in the mammalian heart and eye.
Inferences are primarily based on studies in mice with minor contributions from human studies. Ref: references. CNV: choroidal neovascularization
| Macrophage Population |
Origin | Role in Heart | Ref |
|---|---|---|---|
|
Ccr2−
Resident Macrophage |
• Yolk sac progenitor • Fetal liver progenitor • Maintained through local proliferation |
• Promotes coronary development Promotes cardiac regeneration • Facilitates electrical conduction • Inhibits cardiac monocyte recruitment |
[4, 15-17, 101] |
|
Ccr2+
Resident Macrophage |
• Bone marrow derived monocytes | • Promotes cardiac inflammatory response • Promotes monocyte and neutrophil recruitment |
[15, 17, 20, 21] |
|
Recruited
Macrophage |
• Bone marrow derived monocytes | • Phagocytosis/Efferocytosis • Leukocyte recruitment to the heart • Oxidative damage • Promotes inflammatory response • Promotes angiogenesis |
[15, 17, 18, 20-22, 103] |
| Macrophage Population |
Origin | Role in Eye | Ref |
| Microglia | • Yolk sac progenitor | • Retinal neural circuit development • Phagocytosis during photoreceptor degeneration (possibly protective or pathogenic) • Release of inflammatory cytokines in photoreceptor degeneration |
[34, 35, 37, 38] |
|
Recruited
Macrophage |
• Bone marrow derived monocytes | • Possible promotion of photoreceptor degeneration • Inhibition of angiogenesis (young macrophages) • Promotion of angiogenesis, inflammation (aged macrophages) |
[40, 41, 44, 45] |
|
Recruited M1-like Macrophage |
• Bone marrow derived monocytes | • Possible pathogenic development in early AMD phenotypes • Possible pathogenic development in CNV • Possible protective function against CNV |
[83, 85, 86] |
|
Recruited M2-like Macrophage |
• Bone marrow derived monocytes | • Possible pathogenic development in CNV • Possible protective function against CNS |
[82, 83, 86, 87] |
Functions of resident and recruited macrophages
More than a decade ago, the dominant belief was that macrophages in adult mammalian tissues were simply derived from circulating monocytes [1, 2]. While initially controversial, it is now accepted that tissue-resident macrophages are not derived from infiltrating monocytes, but are an independent and self-replenishing population derived from embryonic hematopoietic progenitors (Box 1) [3–6]. Tissue resident and recruited macrophages are not only derived from distinct origins in development, but also have distinct gene expression and functional profiles.
A long-standing paradigm has been to group macrophages into an arbitrary M1-M2 dichotomy (see Glossary) where M1 macrophages represent the classical inflammatory population stimulated by interferon gamma (IFNγ), microbial stimuli such as lipopolysaccharide (LPS), and/or pro-inflammatory cytokines, while M2 macrophages have been associated with tissue repair and wound healing, stimulated by specific cytokines including interleukin 4 (IL-4) [7, 8]. While tissue resident macrophages would traditionally be classified under the M2 population and recruited infiltrating monocyte-derived macrophages thought to be predominantly inflammatory M1-like cells, we and others have recently begun to understand that this dichotomy represents an over-simplification of the roles of both resident and recruited macrophages. Both M1 and M2 macrophages can be divided into many heterogeneous populations based on expression profiles and functions, as discussed later in this review [9, 10].
Multiple roles of resident and recruited macrophages in the heart
In many cases, the functions and expression profiles of tissue resident macrophages are directly related to the organs in which they reside [11]. Even within a single organ, tissue resident macrophages represent a highly heterogeneous population with varying function [5, 11–14]. For example, within both the mouse and human heart, tissue resident macrophages can be distinguished by the expression of C-C motif chemokine receptor 2 (CCR2). CCR2 expression is typically associated with macrophages derived from infiltrating Ly6Chi monocytes, which maintain CCR2 expression after differentiation [15]. Under steady state conditions, CCR2+ macrophages are maintained by gradual monocyte input and are established as a small, long-lived resident population (from here on referred to as “CCR2+ resident macrophages”). These cells have robust inflammatory potential and function to recruit monocytes and neutrophils following tissue injury, resulting in collateral tissue injury and poor outcomes in numerous models of cardiac disease. In contrast, CCR2- resident macrophages represent the major resident macrophage population, are embryonically derived, have reduced inflammatory activity, and are involved in various processes -- including those traditionally associated with M2 macrophages -- comprising coronary development, steady-state electrical conduction, and tissue repair (Figure 1) [15–17].
Figure 1: Origin and Function of Cardiac Macrophages in Mice and Humans.
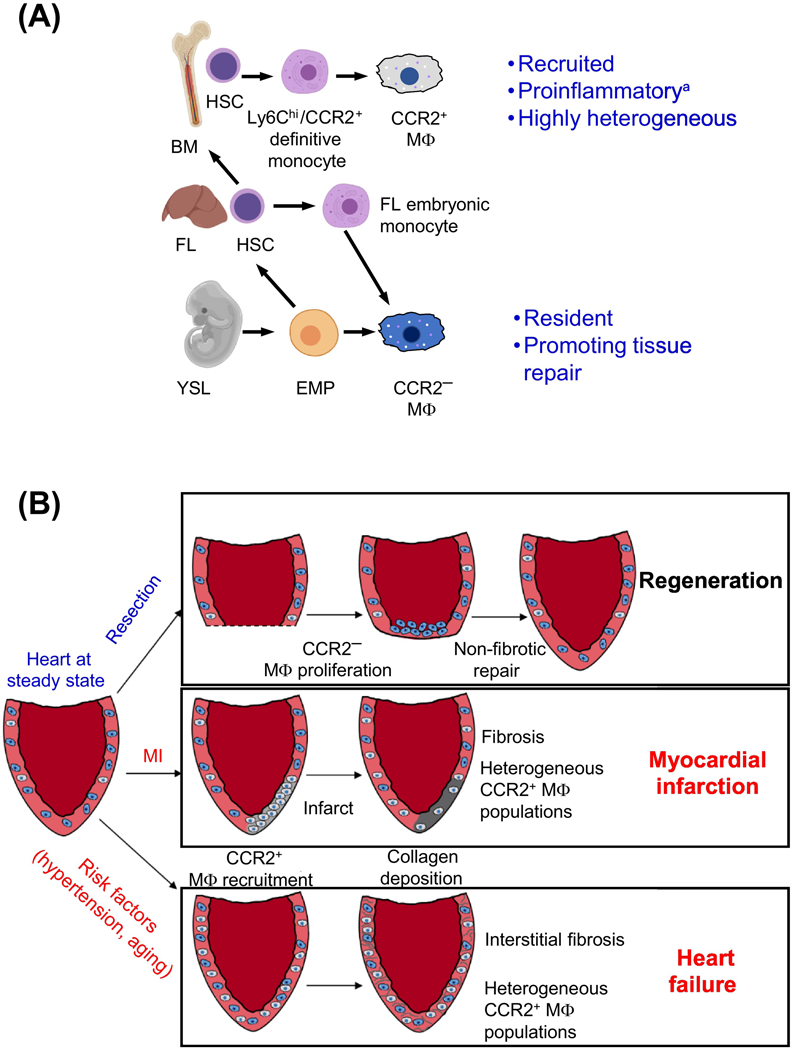
(a) CCR2+ macrophages are derived from recruited Ly6chi/CCR2+ monocytes that develop from definitive hematopoietic stem cells with origins in the fetal liver and bone marrow. CCR2- macrophages differentiate from erythro-myeloid progenitors (EMPs) that are specified in the yolk sac as well as hematopoietic stem cells from the fetal liver. Fetal liver monocytes, which are derived from a population of yolk syncytial layer EMPs, remain a poorly understood population. (b) Models of macrophage roles in heart regeneration and disease are shown. In hearts with regenerative capacity, such as neonatal mice and lower vertebrates, injury/resection results in local proliferation of CCR2- macrophages at the site of injury, promoting non-fibrotic repair. Following myocardial infarction in adult murine hearts without regenerative capacity, resident CCR2- macrophages die and monocyte derived macrophages, which retain CCR2 expression, are recruited to the infarct where they promote inflammation and stimulate collagen deposition resulting in localized fibrosis at the site of injury. In the case of heart failure, CCR2- macrophages are recruited to the failing heart but are not localized to a single site, resulting in formation of interstitial fibrosis that stiffens the ventricular wall, decreasing cardiac function. Radiolabeled probes have also demonstrated that CCR2+ cells are recruited to acute MI sites and failing hearts in humans. *While recruited monocyte derived macrophages represent predominantly pro-inflammatory macrophages, a smaller population of recruited macrophages demonstrate wound-healing properties in contrast to the majority of the recruited population. FL – Fetal Liver, BM – Bone Marrow, HSC – Hematopoietic Stem Cell, YSL – Yolk Syncytial Layer, EMP – Erythro-Myeloid Progenitor
While infiltrating monocyte-derived macrophages are predominately thought to display M1-like inflammatory behaviors, these cells may also acquire M2-like anti-inflammatory phenotypes [21]. Alternative activation of recruited macrophages through interleukin 13 (IL-13) can result in cardioprotective anti-inflammatory effects in myocarditis. In mice infected with coxsackievirus B3, introduction of recombinant IL-13 rescued ejection fraction and reduced the overall area of inflammation via M2 macrophage polarization [18]. Additionally, a subset of recruited macrophages have been shown to take on a less inflammatory and pro-wound-healing function in mice following myocardial infarction (MI), as evidenced by the failure of undertaking debris clearance and reverse remodeling [19–21]. Outside the heart, recruited macrophages have also demonstrated tissue repair activity in mice during late stages of inflammation in the kidney following injury, based on the expression of pro-wound-healing markers such as Ccl17 and Igf1 [22]. Other recent studies have further reported the heterogeneous nature of recruited macrophages following tissue injury, highlighting the wide array of functions monocyte-derived macrophages may acquire [15, 17]. Furthermore, in mice, lineage tracing has shown that blood monocytes can also be recruited to fulfill the role of resident macrophages in a variety of tissues, suggesting that these recruited macrophages are plastic, and likely capable of taking on steady-state homeostatic roles within tissues that have been depleted of resident macrophages via targeted knockout approaches [23–25]. Together, these studies demonstrate the highly plastic and heterogeneous nature of macrophages, both resident and recruited, highlighting their differentiation and activation functions as potential therapeutic ‘targets’.
Single-cell transcriptomics reveal macrophage subpopulations with diverse functions in cardiac or other contexts
Recent next-generation sequencing tools such as single-cell RNA sequencing (scRNA-Seq) have allowed for in-depth analysis of macrophages and confirm the idea of a network of macrophage complexity and heterogeneity that has been put forth in recent years [10, 26]. In the past year, numerous studies have utilized this technique and discovered significant diversity in the expression profile of both resident and recruited macrophages under steady-state and disease conditions in tissues such as the heart, liver, lung, and aorta in both humans and mouse models [15, 27–31].
A recent study using scRNA-Seq in the murine steady-state heart demonstrated that multiple populations of macrophages could be observed to exhibit varying functions based on gene expression [31]. Clusters were identified based on similar gene expression enrichment and characterized by pathways enriched within the cluster. One cluster, enriched for Timd4 and lymphatic vessel endothelial hyaluronan receptor 1 (Lyve1) expression has also been shown to harbor high expression of genes associated with regeneration, angiogenesis, and endocytosis by gene pathway analysis [31]. Additional clusters marked by high Ccr2 and interferon-stimulated gene (ISG) expression were identified as exhibiting more inflammatory components in these populations than clusters with enriched Timd4 and Lyve1 expression; these data were based on gene pathway enrichment of classical inflammatory pathways such as interleukin 12 (IL-12), IFNγ and type I IFN responses [31].
Another study analyzed the influence of CCR2- resident and CCR2+ resident macrophages on recruited macrophage heterogeneity following ischemia reperfusion (IR) injury, to model re-perfused myocardial infarction [15]. Authors used CD169-DTR mice to globally ablate the CCR2- resident population, and CCR2-DTR to globally ablate the CCR2+ resident macrophage population. The authors took advantage of the rapid turnover of circulating CCR2+ monocytes to selectively target CCR2+ resident macrophages. Several days after diphtheria toxin treatment, CCR2+ monocytes had fully recovered, while the CCR2+ resident macrophage population remained depleted. At 4 days post-injury, monocytes expressing lymphocyte antigen 6C2 (Ly6c2) and Plac8 were readily identified within the heart, indicating continued presence of recruited monocytes. Additionally, 6 clusters/populations of macrophages with significantly different gene expression profiles were identified via unsupervised clustering and differential expression analysis. These populations, distinguished by enriched expression of Lyve1 and Timd4, IFN-induced protein with tetratricopeptide repeats 3 (Ifit3), arginase 1 (Arg1), integrin subunit beta 7 (Itgb7), alanyl aminopeptidase (Anpep), and C-C motif chemokine receptor like 2 (Ccrl2) expressed varying amounts of reparative (eg. Fn1, Arg1, Egr1) and inflammatory (eg. Il1b, Irf7, Cxcl10) genetic markers. The discrete nature of these populations was confirmed by immunostaining and each was found to have distinct morphology and localization within the infarcted heart. Additionally, loss of either resident or recruited macrophages resulted in a shift in the populations of the analyzed macrophages. Depletion of inflammatory CCR2+ resident macrophages prior to IR injury resulted in a reduction in the inflammatory Ifit3+ recruited population and expansion of the Itgb7+ population relative to controls, while depletion of reparative CCR2- resident macrophages resulted in increased numbers of Arg1+ and Ccrl2+ macrophages and reduced Lyve1+ macrophages [15].
In this study, the observation that highly inflammatory cells were CCR2+ highlights the potential to analyze patients with chronic or acute myocardial injury for recruitment of CCR2+ monocytes and macrophages following MI, as a candidate indicator of potential outcomes, but further studies are warranted. A recent study in mice and humans using a CCR2-specific probe in conjunction with positron emission tomography (PET) demonstrated this possibility. Specifically, while a small population of resident CCR2+ macrophages could be identified even in uninjured hearts, the increase in visualized CCR2 signal after myocardial infarction in mice was dramatic and could be easily distinguished from baseline. Additionally, this probe was capable of detecting CCR2+ monocytes and macrophages in humans post-myocardial infarction and in chronic ischemic cardiomyopathy, thus providing proof of concept for the development of putative biomarkers and targeted treatments based on CCR2 [32].
Additional studies performed in other tissues also highlight this heterogeneity of macrophages and enforce the need to further study the dynamics of macrophage differentiation and functions across tissues. A study in the mouse lung following partial pneumonectomy identified multiple populations of macrophages, including an enrichment of Arg1+ macrophages compared to sham-operated mice [27]. Similar to findings in the heart, when CCR2 was knocked out, lineage tracing demonstrated that fewer macrophages were recruited to the site of injury. However, in contrast to observations in the heart, lineage tracing demonstrated that recruitment of CCR2+ monocytes was associated with significantly better recovery and regeneration of tissue following pneumonectomy, as lung tissue failed to adequately regenerate in absence of CCR2. This supported the notion that recruited macrophages can have distinct functions in different tissues [27]. Additionally, a recent study using scRNA-Seq demonstrated the existence of multiple populations of resident and recruited macrophages in mouse lung airspace under healthy and acute inflammatory conditions; these macrophage populations exhibited distinct transcriptional profiles relevant to the inflammatory process [33]. As in the cardiac study [32], these macrophage populations were identified using unsupervised clustering and analysis of classical inflammatory and reparative markers [33]. Similar studies in atherosclerosis in mice and steady-state human liver, among other tissues, have found similar results: macrophages have been grouped into multiple and distinct populations using unsupervised clustering based on gene expression profiles; based on known phenotypic marker genes coupled to experimental analysis relying on the amelioration or exacerbation of disease phenotypes, such macrophage populations were grouped to reflect distinct presumed functions, [28, 29]. Collectively, these single cell studies reinforce the highly heterogeneous nature of macrophage populations -- both resident and recruited -- demonstrating the importance of characterizing macrophages beyond the arbitrary M1-M2 paradigm so as to enable the development of indicators of disease outcomes and/or putative targeted therapies.
Macrophage lineage diversity in the eye
As in the cardiovascular system and elsewhere, multiple populations of macrophages with distinct origins, function in the eye under healthy maintenance, or diseased states (Figure 2). Microglia of the murine central nervous system (CNS) are a long-lived, self-replenishing tissue-resident population of mononuclear phagocytes derived from the yolk sac [34]. Recent lineage tracing in mice has also confirmed the yolk sac origin of retinal microglia [35]. Additionally, retinal microglia and their equivalents elsewhere in the CNS have many features in common: They both contribute to synaptic circuit formation during development and exhibit ramified morphology under steady-state conditions [34]. However, adult retinal microglia exhibit distinctive spatial organization, stratifying within the synaptic plexiform layers of the retina (Figure 2) [34]. A recent study in mice using scRNA-Seq suggested that nearly all mononuclear phagocytes isolated from steady-state retina consist of microglia, as evidenced from molecular profiling using unsupervised clustering and expression of resting microglia markers, including Tmem119 [36].
Figure 2: Murine Retinal Macrophage Lineage and Activation in Health and Disease.
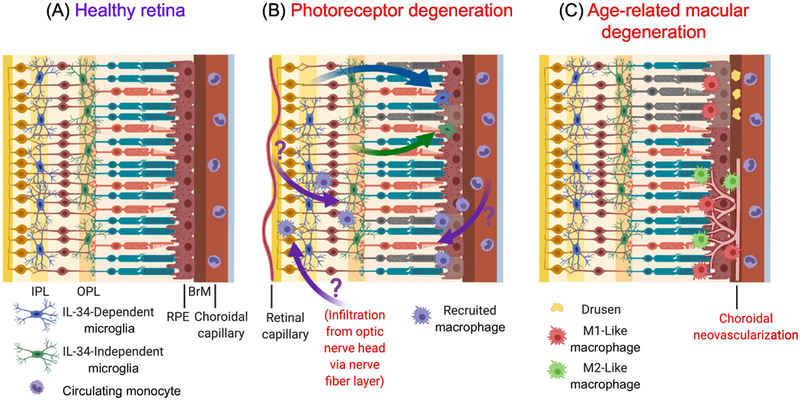
(a) In the healthy retina, microglia stratify within the outer plexiform layer (OPL) and inner plexiform layer (IPL) and mediate homeostatic functions. Different retinal microglial populations occupy different niches, with IL-34-dependent microglia residing within the IPL and IL-34-independent microglia occupying the OPL. Monocytes circulate within choroidal and retinal capillaries, separated from the retina by the blood-retina barrier. (b) In light-induced photoreceptor degeneration or retinitis pigmentosa, microglia migrate to the photoreceptor layer and subretinal space to engulf photoreceptor debris (green and blue arrows), accompanied by a shift in their gene expression and functional profile. Resident microglia from both the IPL and OPL contribute to this responding population, with conflicting evidence about their role in promoting or preventing photoreceptor and retinal pigment epithelium (RPE) demise. Controversy exists over whether monocyte-derived recruited macrophages can infiltrate the retina via various vascular sources and contribute to photoreceptor degeneration (purple arrows). (c) In dry age-related macular degeneration (AMD), lipid-rich deposits (drusen) accumulate in Bruch’s membrane (BrM) or underneath the retina, causing inflammation resulting in RPE and photoreceptor death. Experimental models suggest that these phenotypes are mediated by the inflammatory effects of M1-like macrophages. Wet AMD is characterized by choroidal neovascularization (CNV), the growth of new vessels from the choroid through the BrM and RPE, and into the subretinal space. Mouse models of CNV have yielded results implicating both M1-like and M2-like macrophages in protective and pathogenic roles [82, 83, 85–87].
The relative contribution of resident microglia and monocyte-derived recruited macrophages in the retina under disease contexts remains unclear. Notably, in the absence of pathology, immunostaining of IBA1 in the retina suggests that murine microglia are restricted to the retinal synaptic plexiform layers and are not found among the photoreceptors in the outer retina; however, in different models of retinitis pigmentosa (RP), murine microglia have been shown to appear in the outer retina and to affect photoreceptor loss -- hypothesized to be due to phagocytosis of photoreceptor proteins released during degeneration [34, 35, 37].
Until recently, studies have suggested that microglia play a pathogenic role in mediating photoreceptor loss. For example, ex vivo live imaging has shown that Cx3cr1-driven GFP-labeled microglia contribute to photoreceptor death in the rd10 mouse model of RP by direct phagocytosis of rod photoreceptors, as well as by expressing IL-1β, which in turn can promote photoreceptor apoptosis (as evidenced from terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and decreased thickness of the photoreceptor-containing outer nuclear layer (ONL) relative to controls) [37]. Moreover, pharmacologic inhibition of microglial activation by cyclic Arg-Gly-Asp-Phe-Val peptide (cRGD) or minocycline, can decrease microglial phagocytosis and cytokine secretion relative to treatment with the inactive analog of cRGD, cRAD, concurrent with increased preservation of photoreceptor morphology, decreased photoreceptor TUNEL, and preservation of photoreceptor function (as assessed by electroretinography and optokinetic responses) [37, 38]. However, a recent study demonstrated a protective function of microglia in removing degenerating photoreceptor debris and preservation of retinal pigment epithelium (RPE) by depleting microglia in a light-induced damage model of acute photoreceptor degeneration (using mice with a Rpe65L450M/+ mutation conferring susceptibility to light damage), and in a model of chronic RP RhoP23H/WT mice [35]. When retinal microglia migrated from the plexiform layers to the subretinal space in response to light damage, single-cell RNA-Seq analysis demonstrated a shift in gene expression in these cells from steady state immune-response function,s to a protective phenotype (including cell recognition, adhesion, lipid metabolism, and antioxidant functions); this demonstrated plasticity of retinal microglia undergoing programmatic changes from homeostatic to disease response functions [35, 39]. However, to this date, the role of microglia in the eye remains quite ambivalent.
Studies also offer conflicting views on the role of recruited monocytes in exacerbating or ameliorating photoreceptor degeneration. One study in the rd10 model of RP reported that ablation of monocyte recruitment by deletion of Ccr2 partially rescued retinal structure, photoreceptor morphology, and function, as measured by electroretinography [40]. In contrast, others suggest a lesser role of monocyte-derived macrophages in photoreceptor degeneration, noting the absence of recruited monocytes to the subretinal space or ONL, as well as a lack of phagocytic activity [35, 37]. In a separate Arrestin-1 knockout mouse model of rapid light-induced photoreceptor degeneration, in vivo imaging and scRNA-Seq showed early infiltration of monocytes from retinal vessels and from the optic nerve head relative to baseline, followed by microglial migration to the photoreceptor layer after light exposure [36, 41]. However, elimination of monocyte recruitment by intravitreal injection of neutralizing antibody to C-C motif chemokine ligand 2 (CCL2), or genetic ablation of Müller glia-derived CCL2, did not alleviate photoreceptor degeneration in this model, suggesting that infiltrating monocytes might not exacerbate photoreceptor degeneration [41].
In the eye, these studies also suggest remarkable heterogeneity of both resident and recruited macrophages (at steady-state and under disease conditions), where distinct populations of macrophages might harbor significantly different functions. The roles of seemingly similar macrophages (based on expression profiles) might even be defined depending on the specific tissue and the health of that tissue. As such, there is much interest in obtaining a greater understanding of the dynamics between resident and recruited macrophages, as well as among specific subsets within each population. Investigation of monocyte/macrophage dynamics within specific tissues under different conditions—including the mechanisms by which different macrophage populations acquire their detrimental fates—might allow for the development of putative treatments to treat conditions linked to ocular inflammation.
Macrophage alterations in the pathogenesis of age-related diseases
Recent insights into the mechanisms driving macrophage function have highlighted the role of organismal aging and its contributions to shifting macrophages from a disease-protective state to a disease-promoting phenotype (Box 2). In mammalian hearts, past the neonatal period, macrophages lose their regenerative capacity and instead, promote inflammation and fibrosis following myocardial injury [42]. Moreover, aged macrophages acquire a foam cell phenotype and can promote atherosclerotic plaque formation and plaque angiogenesis [43]. This phenotype portends eventual plaque rupture that can lead to vascular catastrophe in the form of MI and stroke. In the eye, macrophages can promote pathogenic ocular neovascularization that causes hemorrhage and fibrosis in the retina in diseases such as age-related macular degeneration (AMD)—the leading cause of blindness in the elderly in the industrialized world [44, 45]. Additionally, macrophage aging and phenotypic plasticity can also affect other age-associated pathologic processes, including insulin resistance and neurodegeneration (extensively reviewed elsewhere [46, 47]). Here, we discuss diverse macrophage populations and phenotypes mediating various aspects of age-related cardiovascular and ocular diseases.
Macrophages in cardiac disease and regeneration
Macrophages have long been known to play a role in myocardial wound healing and recovery following cardiac injury, secreting a complex plethora of cytokines, growth factors and extracellular matrix proteins that ultimately regulate the quality of tissue repair. Tissue-resident macrophages have a strong presence in the heart -- even at steady-state -- and constitute approximately 8 percent of non-cardiomyocytes in the mouse heart; these perform homeostatic functions, including immune surveillance, promotion of angiogenesis, and suppression of inflammation [48–50]. Additionally, macrophages also play an important role in cardiac regeneration in species where this phenomenon occurs. Model organisms such as zebrafish and salamander are capable of regenerating large portions of the heart even when fully resected from embryonic through adult stages [51, 52]. Mammalian hearts also demonstrate an ability for robust regeneration at early neonatal stages, as supported by numerous studies in mice and anecdotal evidence in humans [42, 53–55]. In each of the studied model organisms, macrophages play a crucial role in heart regeneration by enabling the beneficial remodeling of the extracellular matrix and the neovascularization of the wound [56–58].
In contrast, after the brief neonatal period, mammalian hearts are incapable of functional regeneration and can develop fibrotic lesions at the site of injury that are deleterious to cardiac function [42]. In mice, neonatal hearts have demonstrated an expansion of embryonic-derived resident macrophages which can promote cardiac recovery after injury [56]. Although also present in adult hearts, this embryonic-derived population does not expand after injury and instead, is replaced by infiltrating pro-inflammatory monocyte-derived macrophages lacking reparative capacity [14]. In mouse MI, embryonic-derived resident tissue macrophages begin to accumulate at the site of injury within 5 minutes, but die rapidly within the infarct [59–61]. Within sites of injury, resident macrophages are largely replaced by infiltrating monocyte-derived macrophages drawn to the injury site by CCL2 and CCL7, along with a population of inflammatory neutrophils peaking within one day after injury [62]. Resident macrophages continue to be maintained through proliferation outside of the infarct [62–64]. Infiltrating Ly6Chi monocytes and derived macrophages become the predominant leukocyte population within the injured mouse heart between 2 and 5 days post-infarction before gradually returning to steady-state numbers [65–67]. Throughout aging, macrophages in the heart also shift to constituting a population of recruited pro-inflammatory macrophages, which may contribute to poorer outcomes in the event of cardiac injury, relative to earlier stages of development [5, 49, 68] (Box 2).
Additionally, in mice, self-renewing embryonic-derived CCR2- macrophages can promote beneficial healing following MI, while recruited CCR2+ pro-inflammatory macrophages can promote increased fibrosis and be detrimental to wound healing [15, 31]. Researchers have observed similar effects in models of heart failure. In a murine cardiac pressure overload state induced by transverse aortic constriction (TAC), CCL2, CCL7, and CCL12 ligands, which bind CCR2, have been shown to become upregulated in cells of the heart, including endothelial cells, smooth muscle cells, and fibroblasts, along with resident immune cells [69]. This, in turn, has resulted in an increase in Ly6ChiCCR2+ circulating monocytes and CCR2+ macrophage recruitment into the heart relative to controls. Moreover, antibody-mediated CCR2 inhibition early in pressure overload has resulted in reduced monocyte numbers and macrophage recruitment, reduced fibrosis and hypertrophy, as well as improvement in systolic function compared to untreated mice [69]. An additional study further supported the detrimental role of recruited CCR2+ macrophages in heart failure with preserved ejection fraction (HFpEF), a condition currently lacking effective therapies [70]. Similar to the TAC pressure overload model, mice with reduced diastolic function due to hypertension or advanced age exhibited increased numbers of circulating monocytes and recruitment of macrophages relative to healthy mice[71]. Specifically, in this murine model of diastolic dysfunction, recruited macrophages produced IL-10, which stimulated fibroblasts to increase collagen production, thus resulting in increased interstitial fibrosis between cardiomyocytes, and further perpetuating the heart failure phenotype (attenuated by an anti-IL-10 neutralizing antibody) [71]. Consistent with other emerging evidence that populations of macrophages are not constrained to their traditional functional roles [15, 25, 27], a recent study in mice showed that both classically inflammatory recruited macrophages and resident macrophages expressed interferon regulatory factor 3 (IRF3) in response to free double-stranded DNA released from damaged and dying cardiomyocytes [72]. This in turn promoted type I IFN production, further contributing to the inflammatory cascade, and revealing a novel function of macrophages that primarily phagocytose tissue without releasing inflammatory signals [72]. All of these studies demonstrate that macrophages can respond to varying types of injury and have different responses dependent on the type of tissue injury that has occurred. Consequently, future studies are needed to elucidate the mechanisms driving these functions.
Potentially relevant to functional regeneration of the heart following injury is the timing of the immune response. In species such as salamander and zebrafish, the response to cardiac injury is much more rapid than in mammals, and the switch from inflammatory to reparatory function of the immune system occurs much sooner, based on RNA sequencing at multiple time points after injury. Specifically, evidence suggests that a rapid response and repression of inflammation is key to the reparative function of macrophages because when this function is delayed using clodronate administration, the formation of scar tissue can prevent macrophages -- normally entering the tissue after acute inflammation -- to repair the injured tissue [57, 58, 73]. Indeed, lineage tracing in mice has demonstrated that there is a shift from reparative embryonic-derived macrophages towards recruited pro-inflammatory macrophages during aging [68]. An additional study suggests that mice exhibit age-related increases in proinflammatory gene expression, including Ccl4 and Cx3Cl1 [74]. Yet, despite the detrimental nature of inflammation in cardiac repair, phagocytosis and efferocytosis are also critical functions of macrophages in the positive response to injury. Specifically, depletion of either macrophages, or phagocytotic genes in macrophages, after MI in mice, can result in an inability to remove dead cells and cellular debris from dying cardiomyocytes; this also results in impaired remodeling, reduced cardiac function, and greater risk of cardiac wall rupture, relative to mice with intact macrophage populations. Macrophage deletion with clodronate or knockout of Class A macrophage scavenger receptor (SR-A) caused an increase in the frequency of ventricular wall rupture, ventricular wall thickening and remodeling, and reduced survival compared to control mice after cardiac injury [65, 75–77]. Moreover, loss of macrophage expression of vascular endothelial growth factor A (Vegfa) also resulted in reduced cardiac function as a result of poor revascularization during injury repair in mice relative to controls; VEGFA staining and numbers of vascular endothelial cell nuclei were reduced in cryoinjured hearts when macrophages were depleted with clodronate compared to untreated injured controls [76]. Conversely, a drastic increase in the numbers of monocytes and macrophages can be detrimental to the healing of an infarct in mouse models [78, 79]. These results suggest that a delicate balance between pro and anti-inflammatory functions of monocytes and macrophages in the heart is necessary for optimal injury healing.
Additional diseases of the cardiovascular system, such as atherosclerosis, have also been shown to depend on immune components (Box 3). Taken together, because macrophages can play various roles in cardiovascular tissues, it may be possible to target their recruitment or proliferation in cardiac diseases, thus aiming to improve pathology and/or promote tissue regeneration. However, robust testing is still warranted to advance in this front.
Conflicting roles of macrophage populations in age-related macular degeneration
The role of macrophages has garnered increasing interest in the pathogenesis of human age-related macular degeneration (AMD), although the protective versus pathogenic roles of divergently activated macrophages, as well as diverse lineages, remains unknown. AMD begins with a dry stage characterized by cholesterol-rich deposits such as drusen, and can progress to either an atrophic form, or a wet, neovascular form, characterized by growth of new vessels termed choroidal neovascularization (CNV), CNV can cause blindness from hemorrhaging and fibrosis [80]. Macrophage early response to injury is inflammatory, and in the following days, this phenotype shifts towards anti-inflammatory, fibrosis-promoting, and pro-angiogenic functions, as well as wound healing [81]. Accordingly, investigators have found these trends in macrophage behavior in models of AMD. Specifically, laser injury-induced CNV constitutes a murine model often used to study angiogenesis and inflammation in neovascular AMD. Early during the injury response (within 1–3 days post-laser injury), M1-like macrophages predominate in the posterior segment of the eye (consisting of the retina, retinal pigment epithelium, choroid and sclera), as evidenced from increased expression of Mi-associated markers (iNOS, TNFα, IL-6, CXCL10, and CXCL11) relative to controls; this is followed by decreased expression of these markers by day 7 post-laser injury, while expression of alternatively activated M2 markers (ARG1, CD163, CD206) increases through days 1–3, peaking at day 7, and remaining high up to day 21 [82]. Flow cytometry classification of macrophages as M1-like (CD11b+CD80+) or M2-like (CD11b+CD206+) confirmed these observations, reporting an early increase and variable decline in the M1-like macrophage population, followed by a later rise and sustained presence of M2-like macrophages in the posterior segment of the eye after laser injury [83]. These temporal dynamics in macrophage polarization may describe an early M1 -like inflammatory role of macrophages followed by M2-like tissue remodeling and angiogenic functions during the development of CNV lesions [84].
It remains unclear whether pro-inflammatory and anti-inflammatory macrophages function in a protective or pathogenic role in either early or late time points after ocular injury. Some argue for a pathogenic role of inflammatory macrophages in early disease by ablating M1-like macrophages and demonstrating amelioration of disease phenotypes [85]. For instance, subretinal macrophages in mice injected with carboxyethylpyrrole-modified mouse serum albumin (CEP-MSA)—recapitulating some pathologic features of dry AMD (including damage to Bruch’s membrane, RPE and photoreceptor death)—have been found to express M1-associated (Tnf, Il12, Il6, Il1b and Ccl2) but not M2-associated (Il10) markers [85]. Elimination of macrophage recruitment via Ccr2 KO ameliorated the development of AMD disease phenotypes after CEP-MSA injection, demonstrating a role of M1-like inflammatory macrophages in the development of early AMD disease phenotypes (RPE dysfunction and loss) [85]. Others have demonstrated a possible role of M1-like macrophages in mediating the later phenotype of neovascularization [86]. Specifically, in both laser-induced CNV and an oxygen-induced model of retinopathy of prematurity, removal of Il17a by genetic knockout or neutralizing antibody, reduced the M1 population and increased the M2 population, and caused reduced neovascularization in both disease models, relative to controls. By contrast, adding recombinant IL-17A intravitreously resulted in an increased M1 population relative to controls, suggesting that either M1 macrophages might be pathogenic, or M2 macrophages might be protective in ocular neovascularization in these models [86]. Moreover, in vitro, IL-17A-treated RAW264.7 macrophages expressed higher M1-like markers as well as increased VEGF relative to untreated cells; this treatment also caused increased VEGF receptor (VEGFR½) transcription in cultured human umbilical vein endothelial cells (HUVEC) and increased HUVEC proliferation relative to controls, suggesting a possible direct role of M1-like macrophages in promoting pathologic angiogenesis [86], although this remains to be further tested.
Others attribute pathologic neovascularization to the action of alternatively activated M2-like macrophages later during the healing process [82, 83, 87]. For example, in the laser-injury mouse model of CNV described above, the late rise of the M2-like population from day 3 post-injury, sustained up to 21 days, concurs with earlier studies of the laser CNV model showing that neovascularization begins on day 3 and peaks at day 7 after laser injury [82, 83]. Another study using the laser CNV mouse model reported a pathogenic role of late-responding M2 macrophages, where the M2 population was decreased upon inhibition of Rho-associated protein kinase 1 and 2 (ROCK1 and ROCK2) or ROCK2 alone, relative to controls, concomitant with a decrease in the extent of CNV development [83]. Furthermore, this study showed that intravitreal injection of M2 macrophages or M2 cytokines (IL-4, IL-10, IL-13) worsened CNV relative to controls, while injection of M1 macrophages or M1 cytokines (IFNγ and LPS) ameliorated CNV; this suggested a pathogenic role for M2 macrophages in CNV development (at least in the laser CNV model), as well as a potential protective role for M1 macrophages [83]. Additionally, inhibition of M2 polarization by doxycycline administration also attenuated laser-induced CNV [87]. Evidently, further testing will be needed to elucidate these ambiguous roles. Nevertheless, despite limitations in our mechanistic understanding of the roles of macrophages in pathogenesis or protection in the eye, modulation of macrophage activation by ROCK inhibition is already being explored clinically with potential promise for macrophage-and age-mediated diseases such as AMD (Box 4) [88–94].
At this time, the role and even the presence of M1-like versus M2-like macrophages in AMD remains unclear. Models of AMD pathogenesis that can account for the data presented above seem to suggest that both M1-and M2-like macrophages might be pathogenic. It may be possible that the pathogenesis of AMD involves an early inflammatory, M1-mediated process, followed by a subsequent M2-mediated aberrant healing response. This model in turn suggests a concept of inflammation where both M1-like and M2-like macrophage populations could contribute to mediating the chronic inflammatory environment of AMD. It is also possible that macrophage activation and plasticity allows both classical and alternatively activated macrophages to orchestrate tissue inflammation and angiogenesis simultaneously, although the relative contribution of each macrophage subset might change with each phase of the disease. Additionally, alterations in macrophage phenotype due to aging may cause them to promote AMD pathogenesis through changes in their lipid handling functions (Box 5). Further investigations will be needed to test these hypotheses.
Concluding Remarks
In summary, it is clear that macrophages display great diversity in their lineage, phenotype, and plasticity. Given their presence in multiple tissues and their role in a multitude of disease contexts, the divergent characteristics of macrophages complicate their interaction with their environment and their subsequent role in preventing or exacerbating disease. The field is just beginning to gain a nuanced catalog of the extent of macrophage diversity, as well as the beginning of a molecular mechanistic understanding of their phenotypes. Recent advances in understanding the gene expression and function of macrophage subpopulations in the cardiovascular system and the eye may inform mechanistic studies on the pathogenic or protective roles of these populations, paving the way for translational research based on macrophage modulation (see Outstanding Questions). Many challenges persist in the study of macrophage function. In particular, there is a need for improved characterization of macrophage ontogeny and phenotypic activation beyond the M1/M2 designation, which may be addressed by greater utilization of single cell molecular profiling strategies such as scRNA-Seq. Greater nuance in macrophage population characterization, combined with improved disease models, will further resolve current controversies in the disease-promoting or ameliorating roles of diverse aging macrophage populations. Despite the numerous challenges, studying the diverse functions of macrophages offers great potential for understanding the divergent physiologic roles and disease courses mediated by this seemingly singular cell type. A precise understanding of these differences will be critical to developing precision therapeutics targeting macrophage-mediated pathogenic mechanisms.
Box 1: Diversity of macrophage origins in mice and humans
Yolk-sac blood islands appear in mouse embryos at approximately E7.5 and in humans at approximately day 18–19 estimated gestational age with the first macrophages appearing quickly thereafter [95–97]. These earliest primitive hematopoietic progenitors are capable of becoming erythrocytes or macrophages only. Additional progenitor cells from the yolk syncytial layer arise at E8.5 in mice and 4–6 weeks of gestation in humans and are unique bipotential progenitors, known as erythro-myeloid progenitors (EMPs), capable of giving rise to erythroid and myeloid cells only [6, 98, 99]. Macrophages derived from the initial wave of EMPs are unique in that i) they are thought not to pass through a monocytic intermediary state and ii) depend on certain specific transcription factors; notably, they are independent of Myb but are dependent on Pu.1, in contrast to hematopoietic stem cells that give rise to definitive monocytes which are dependent on both Myb and Pu.1 [100]. While this differential requirement of Myb and Pu.1 was examined in mice, the high homology between mammalian and human hematopoiesis suggests that a similar requirement occurs in humans. A later wave of EMPs will traffic to the fetal liver and differentiate into extra-embryonic derived monocytes that differ from definitive monocytes by lack of a macrophage colony-stimulating factor 1 receptor (CSF1R) requirement for development (as evidenced by assessments of murine hematopoiesis) [101]. Subsets of these yolk syncytial layer derived EMPs and fetal liver derived monocytes, in both humans and mice, will spread through the circulatory system to developing organs and become major contributors to tissue-resident macrophages [4, 30, 101, 102].
In contrast, circulating or infiltrating monocytes are derived from hematopoietic stem cells which colonize the bone marrow during development [103]. Definitive monocytes are divided into two major populations; i) infiltrating monocytes transmigrating from circulation into tissue in response to specific signals and are identified by Ly6chi and CD14+ expression in mice and humans respectively, and represent the population that directly contributes to recruited macrophages in tissues [104]; ii) monocytes exhibiting Ly6clo expression in mice and CD16+ expression in humans and which are primarily responsible for patrolling vessel walls and for endothelial support. These monocytes are derived from Ly6chi monocytes in response to signaling, including Delta-Like 1 (DLL1) and upon upregulation of transcription factor NR4A3, although other sources and signaling pathways deriving Ly6clo monocytes are likely [105, 106]. Infiltrating monocytes demonstrate rapid turnover in circulation and will infiltrate tissues through extravasation in response to injury and inflammation, begin to express inflammatory markers themselves, and can differentiate into macrophages and dendritic cells.
Box 2. Age-associated metabolic changes influencing macrophage priming and function
The mechanisms governing plasticity along the spectrum of the so-called M1-like and M2-like macrophages are beginning to be elucidated, suggesting that age-associated metabolic changes might be important in influencing macrophage activation, and in promoting the pathogenic activity of aged macrophages. For instance, murine macrophages polarized towards a pro-inflammatory, M1-like phenotype display a predominantly glycolytic metabolism, whereas anti-inflammatory, tissue-remodeling M2-like macrophages show dependence on higher oxidative metabolism [107]. Thus, the description of aging as a state of ‘chronic inflammation’ is broadly consistent with the observed mitochondrial dysfunction across tissues with aging (including in macrophages), although emerging evidence suggests that M2-like macrophages might also contribute to age-related inflammation in a noncanonical manner, by expression of senescence markers p16INK4a and ß-galactosidase [108–110]. Supporting the notion that macrophages can rely on mitochondrial respiration to acquire an anti-inflammatory M2-like phenotype, recent mouse work has demonstrated that induction of M1-like polarization can result in the elimination of mitochondria (as quantified by decreased mitochondrial mass visualized with MitoTracker) and a decrease in mitochondrial respiration (measured via extracellular flux analysis) relative to controls, thereby preventing M1-like macrophages from repolarizing to an M2-like phenotype; this in turn, has suggested a loss of plasticity towards an anti-inflammatory phenotype, as in the case of mitochondrial dysfunction seen with aging [111, 112]. Additional work has shown that maintaining murine macrophage mitochondrial respiration and M2-like polarization requires the de novo synthesis of nicotinamide mononucleotide (NMN), precursor to nicotinamide adenine dinucleotide (NAD+, a multifunctional cofactor decreased systemically in aging) [113]. Loss of de novo NMN synthesis by genetic or pharmacologic blockade of the kynurenine pathway resulted in decreased mitochondrial respiration (measured via extracellular flux analysis) and increased secretion of M1-like cytokines such as IL-1β and IL-18 relative to controls; this has suggested that decreased de novo NMN synthesis in aged macrophages might be driving their inflammatory phenotype [113]. Consequently, age-associated metabolic changes can influence and limit the ability of macrophages to switch phenotypes; it remains to be seen whether these changes indeed underlie the ability of aged macrophages to promote disease in the cardiovascular system and the eye.
Box 3: Atherosclerosis: A vascular disease with significant cardiac implications
Atherosclerosis – the build-up of lipoproteins in the walls of arteries resulting in plaques – has a robust immune component that is tightly linked to the heart. Circulating monocytes respond to signals from macrophages in arterial plaques to extravasate into the tissue. After infiltration, monocytes differentiate into macrophages which then begin to phagocytose LDL cholesterol that has been deleteriously modified due to oxidative stress within the arterial walls [114, 115]. These macrophages continue to proliferate within arterial plaques [116, 117]. Additionally, these accumulated macrophages, when loaded with cholesterol, are retained within the plaque due to limited mobility which perpetuates the inflammatory environment of the atheroma [118]. These cholesterol-rich foam cells (a hallmark of atherosclerosis) will eventually succumb to apoptosis, and, combined with the reduced ability of remaining macrophages to clear them through efferocytosis, will release the accumulated cholesterol as well as cell death signals, further propagating the inflammatory environment within the atheroma [43, 119, 120]. Antithetically, M2-like macrophages, required for plaque resolution, are derived from Ly6chi circulating macrophages long thought to be highly inflammatory. These macrophages are capable of clearance of inflammation in humans and mice when driven to M2 polarization with IL-13 [121, 122]. MI and atherosclerosis are also tightly linked diseases. Destabilized plaques can dislodge from the arterial wall resulting in MI. The occurrence of MI also stimulates increased monocyte production, which, in addition to being recruited to the site of cardiomyocyte injury, are recruited to the plaque, furthering the progression of atherosclerosis [123].
The confluence of events that lead to advanced atherosclerosis present multiple potential targets for promoting the resolution of disease. A recent active Phase 3 clinical trial, the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS; a randomized, double-blind, placebo-controlled, event-driven interventional trial of quarterly subcutaneous canakinumab in the prevention of recurrent cardiovascular events among stable post-myocardial infarction patients with elevated hsCRP; NCT01327846)I used targeted blocking of IL-1β to reduce inflammation. The primary outcome was time to first occurrence of major adverse cardiovascular event, and the results demonstrated efficacy in high-risk atherosclerosis patients in reducing the rates of MI and stroke without reducing lipid concentrations relative to placebo treatment [124]. This trial, while also presenting negative interactions involving infection due to immunosuppression, highlights the potential for macrophage and inflammatory targeted treatments in the resolution of cardiovascular disease. Additional studies have demonstrated that similar to their role in MI, CCR2+ macrophages might also be detrimental in the case of atherosclerosis, further accentuating the potential for targeting CCR2 in predicting outcomes and developing treatments [125, 126], although this warrants further investigation.
Box 4: Therapeutic modulation of macrophage activation
Recent mechanistic insights raise the possibility of modulation of macrophage activation as potential treatment strategies for ocular diseases. Inhibition of either ROCK½ or ROCK2 alone in experimental CNV in mice demonstrated reduction of CNV by decreasing M2 polarization [83]. As such, the potential of ROCK inhibitors in modulating macrophage activation are being examined for the treatment of various diseases in translational and clinical studies.
The ROCK inhibitor fasudil has already been approved in some countries for cerebral vasospasm [88, 89]. Ripasudil and netarsudil, ROCK inhibitors delivered as ophthalmic solutions, are approved for lowering intraocular pressure in glaucoma and ocular hypertension [90, 91]. Other compounds such as Y-27632 are undergoing multiple clinical trials for diseases of corneal endothelial cells, thought to mediate their therapeutic effects via modulation of the cytoskeleton, cell junction, or cell proliferation [88, 92]. The multiple putative mechanisms of action of ROCK inhibitors have also been theorized for the treatment of a variety of retinal diseases including AMD [89].
Fasudil has also been shown to reduce the severity of experimental autoimmune encephalomyelitis (EAE; mouse model of multiple sclerosis), concurrent with a shift from M1-like to M2-like macrophages, as well as decrease in inflammatory cytokine secretion by microglia relative to controls [94]. It has also ameliorated the disease phenotype in the superoxide dismutase 1 (SOD1) mouse model of amyotrophic lateral sclerosis (ALS) relative to controls; this has led to an ongoing (recruiting), randomized, placebo-controlled Phase 2 clinical trial of fasudil in patients with ALS, measuring as primary endpoints, safety and tolerability (NCT03792490)II [93]. The putative therapeutic mechanism in the EAE and SOD1 models contrasts with those reported to reduce M2 macrophage numbers in the amelioration of CNV [83]; this highlights the differential effects of ROCK inhibition on macrophages of varying lineages and occupying diverse microenvironments. The possibility of controlling macrophage phenotypic activation to increase the number of reparative or tissue resident-like macrophages underscores the importance of connecting single-cell observations with mechanistic studies for a more precise understanding of monocyte differentiation.
Box 5. Macrophage lipid metabolism and AMD pathogenesis
In both mice and humans, certain microRNAs have been implicated in the regulation of CNV in AMD by modulating macrophage lipid metabolism via different mechanisms. Impaired cholesterol trafficking and reverse cholesterol transport are key defects in the biogenesis of drusen, characteristic of early, dry AMD [80]. Moreover, genome wide association studies have identified polymorphisms in the cholesterol efflux transporter ATP binding cassette subfamily A member 1 (ABCA1) as being associated with AMD risk in humans [127, 128]. Increased miR-33 expression in peritoneal macrophages of 18 months-old aged mice (compared to 3 months-old young mice) can reduce macrophage ABCA1 expression and likely disrupt cholesterol trafficking from the eye; indeed, this phenomenon has been shown to promote CNV in animal models [44, 80]. Recently, conditional knockout of ABCA1 and ABCG1 from macrophages caused drusen deposition and photoreceptor neurodegeneration leading to vision loss in mice [129]. However, increased miR-150 expression can affect macrophage lipid metabolism via regulation of stearoyl-CoA desaturase-2, thus promoting CNV in the murine laser-induced CNV model. Thus, it is possible that miR-150 could serve as a putative biomarker to distinguish patients with physiologically aging macrophages from those with AMD risk-associated macrophages [130]. However, this possibility still requires robust testing.
Outstanding Questions
What are the specific roles of the subpopulations of resident and recruited macrophages in various adult tissues under steady state and disease conditions in mice and humans? What are the interactions between these populations that dictate their overall functions in tissue homeostasis and disease development or prevention?
How do macrophages acquire a detrimental fate in various tissue contexts relevant to disease, and is this fate influenced by environmental cues or cell-intrinsic factors? What are points of intervention in the path leading towards detrimental macrophages that might be exploited for the development of putative treatments in human diseases involving inflammation and regeneration?
What are the relative contributions of each metabolic factor to macrophage polarization and plasticity? How do overarching programmatic metabolic changes such as those occurring with aging converge to determine macrophage phenotype?
What differences in microenvironments in young versus aged tissues contribute to the establishment of protective or pathogenic macrophages? For example, why do murine injured neonatal hearts regenerate with proliferation of embryonic-derived resident macrophages, while adult hearts recruit inflammatory, fibrogenic monocyte-derived macrophages, despite the continued presence of resident macrophages?
What can explain the seemingly conflicting results concerning the protective or pathogenic roles of M1-like and M2-like macrophage populations in various eye injury mouse models? What can single-cell transcriptomics do to resolve ambiguity in macrophage population and subpopulation-specific effects on these disease phenotypes, as well as clarify the relative contributions of microglia versus recruited macrophages in the retina?
Highlights
Recent studies combining single-cell transcriptomics, imaging, and functional assays provide mechanistic validation of prior observations indicating that murine macrophage heterogeneity contributes to divergent roles in healthy tissue and during disease response
Disease models in mice have identified subpopulations of macrophages with characteristic gene expression and behavior profiles that ameliorate or exacerbate disease phenotypes in the cardiovascular system and the eye
Analysis of multiple macrophage populations can reveal molecular mechanisms of macrophage plasticity and phenotype switching (or lack thereof), in response to context-dependent tissue changes
Mechanistic insights from basic research has spurred numerous translational and clinical studies exploring the measurement or modulation of human macrophage function and identity in improving disease outcomes
Acknowledgments
This work was supported by NIH Grants R01 EY019287 06A1 (RSA), R01 HL138466 (KJL), R01 HL139714 (KJL), R21 EY026707 02 (RSA), K08 HL123519 (KJL), Vision Core Grant P30 EY02687, Institutional National Research Service Award 5 T32 EY013360 from the National Eye Institute (ZW), 5 T32 HL13463502 Training in Integrative and Systems Biology of Cardiovascular Disease (ALK), Glenn Foundation for Medical Research Award (RSA), Starr Foundation (RSA), Kuzma Family Gift Fund (RSA), Jeffrey Fort Innovation Fund (RSA), Edward N. and Della L. Thome Foundation Award (RSA), the Vitreoretinal Surgery Foundation (ZW), Burroughs Welcome Fund (1014782) (KJL), Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CHII-2015-462 and CH-II-2017-628) (KJL), Foundation of Barnes-Jewish Hospital (8038-88) (KJL), and an unrestricted grant from Research to Prevent Blindness, Inc., New York, NY to the Department of Ophthalmology, Washington University School of Medicine, St. Louis, MO. Figures were created with BioRender.com
Glossary
- AMD
blinding disease characterized by atrophic dry AMD (neurodegenerative phenotype involving lipid drusen deposition, inflammation and photoreceptor degeneration) or neovascular wet AMD (causing proliferative, pathogenic angiogenesis in the choroid)
- Arrestin-1
protein that deactivates rhodopsin. Loss of function results in excess phototransduction, causing photoreceptors to become susceptible to light-induced degeneration.
- Atheroma
Plaque comprising accumulated lipids, large numbers of macrophages, dead cells and scar tissue in pathologic arterial walls.
- CCL2
primary ligand of CCR2, or monocyte chemoattractant protein (MCP-1). Promotes monocyte, T-cell, and dendritic cell recruitment.
- CCR2
chemokine receptor for monocyte chemoattractant protein (MCP-1/CCL2) that promotes monocyte chemotaxis and recruitment resulting in inflammation.
- CD169-DTR
Diphtheria toxin receptor (DTR) under the CD169/Sialoadhesin promoter. Cells expressing DTR become sensitive to diphtheria toxin and undergo cell death when exposed, allowing for targeted cell ablation. CD169 is expressed on CCR2− but not CCR2+ macrophages.
- CNV
Growth of new vessels from the choroid and extending into the subretinal space or underneath; can cause hemorrhaging and fibrosis leading to blindness.
- Drusen
Lipid-rich deposits underneath the retina or RPE that can cause inflammation and damage to photoreceptors and the RPE.
- Efferocytosis
clearance of dead and/or dying cells by macrophages and other phagocytes.
- Foam cells
-
Fat-laden phagocytic cells; a hallmark of atherosclerotic disease.
Contribute to the instability of arterial plaques and the progression of disease.
- Heart Failure with Preserved Ejection Fraction
A form of heart failure in which ejection fraction is unaltered as compared to healthy hearts, characterized by diastolic dysfunction.
- Lymphatic vessel endothelial hyaluronan receptor 1
An integral membrane protein capable of binding hyaluronic acid. Macrophages expressing LYVE1 degrade collagen, promoting tissue repair.
- M1-M2 Dichotomy
classical grouping of macrophages into one of two broad classes based on activation and function; M1 macrophages are classically activated by IFN or LPS and promote inflammation. M2 macrophages are alternatively activated by cytokines such as IL-10 or IL-13 and promote wound healing and tissue repair.
- Optokinetic response
Eye movements comprised of smooth pursuit and saccades allowing the eye to follow a moving object or pattern. Used clinically and experimentally as an assay of visual acuity.
- Pressure Overload
pathological state in which a chamber of the heart must contract against higher than normal afterload in order to eject blood.
- Rd10 RP mouse model
Mice carrying a mutation in rod cGMP phosphodiesterase 6B (Pde6b) exhibiting progressive loss of photoreceptors.
- Retinal pigment epithelium
Single layer of pigmented cells underneath the retina that provides metabolic support to photoreceptors, as well as forming a component of the blood-retina barrier, susceptible to inflammatory damage.
- Retinitis pigmentosa
Blinding disease characterized by progressive degenerative loss of photoreceptors due to a genetic mutation.
- Retinopathy of prematurity
Abnormal blood vessel development in the retina due to excess oxygen supplementation of premature infants.
- Rho-associated protein kinase 1 and 2
A family of kinases mediating the effects of Rho GTPases with a range of cellular functions.
- RhoP23H/WT model of RP
Mice carrying a P23H mutation in the rhodopsin gene that causes progressive photoreceptor degeneration.
- Transverse Aortic Constriction
experimental method for inducing pressure overload to the left ventricle of the heart in rodents.
- Unsupervised Clustering
Analysis used in single cell RNA sequencing combining linear (Principal Component Analysis – PCA) and non-linear (t-Distributed Stochastic Neighbor Embedding – tSNE) reduction algorithms to identify groups of cells with similar gene expression profiles from large datasets.
Footnotes
Resources
This trial is listed in www.clinicaltrials.gov:https://clinicaltrials.gov/ct2/show/NCT01327846
This trial is listed in www.clinicaltrials.gov:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Landsman L et al. (2007) Distinct differentiation potential of blood monocyte subsets in the lung. J Immunol 178 (4), 2000–7. [DOI] [PubMed] [Google Scholar]
- 2.van Furth R et al. (1972) The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ 46 (6), 845–52. [PMC free article] [PubMed] [Google Scholar]
- 3.Hashimoto D et al. (2013) Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38 (4), 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ginhoux F et al. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330 (6005), 841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epelman S et al. (2014) Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40 (1), 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez Perdiguero E et al. (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518 (7540), 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3 (1), 23–35. [DOI] [PubMed] [Google Scholar]
- 8.Mantovani A et al. (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25 (12), 677–86. [DOI] [PubMed] [Google Scholar]
- 9.Yang J et al. (2014) Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nahrendorf M and Swirski FK (2016) Abandoning M1/M2 for a Network Model of Macrophage Function. Circ Res 119 (3), 414–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gautier EL et al. (2012) Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 13 (11), 1118–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yona S et al. (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38 (1), 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies LC et al. (2013) Tissue-resident macrophages. Nat Immunol 14 (10), 986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavine KJ et al. (2014) Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A 111 (45), 16029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bajpai G et al. (2019) Tissue Resident CCR2-and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ Res 124 (2), 263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hulsmans M et al. (2017) Macrophages Facilitate Electrical Conduction in the Heart. Cell 169 (3), 510-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bajpai G et al. (2018) The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 24 (8), 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang H et al. (2017) Interleukin-13 reduces cardiac injury and prevents heart dysfunction in viral myocarditis via enhanced M2 macrophage polarization. Oncotarget 8 (59), 99495–99503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cihakova D et al. (2008) Interleukin-13 protects against experimental autoimmune myocarditis by regulating macrophage differentiation. Am J Pathol 172 (5), 1195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nahrendorf M and Swirski FK (2013) Monocyte and macrophage heterogeneity in the heart. Circ Res 112 (12), 1624–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujiu K et al. (2014) Cardioprotective function of cardiac macrophages. Cardiovasc Res 102 (2), 232–9. [DOI] [PubMed] [Google Scholar]
- 22.Lin SL et al. (2009) Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol 183 (10), 6733–43. [DOI] [PubMed] [Google Scholar]
- 23.van de Laar L et al. (2016) Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 44 (4), 755–68. [DOI] [PubMed] [Google Scholar]
- 24.Scott CL et al. (2016) Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun 7, 10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Misharin AV et al. (2017) Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214 (8), 2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue J et al. (2014) Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40 (2), 274–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lechner AJ et al. (2017) Recruited Monocytes and Type 2 Immunity Promote Lung Regeneration following Pneumonectomy. Cell Stem Cell 21 (1), 120–134 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cochain C et al. (2018) Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res 122 (12), 1661–1674. [DOI] [PubMed] [Google Scholar]
- 29.MacParland SA et al. (2018) Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 9 (1), 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stremmel C et al. (2018) Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat Commun 9 (1), 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dick SA et al. (2019) Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20 (1), 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heo GS et al. (2019) Molecular Imaging Visualizes Recruitment of Inflammatory Monocytes and Macrophages to the Injured Heart. Circ Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mould KJ et al. (2019) Single cell RNA sequencing identifies unique inflammatory airspace macrophage subsets. JCI Insight 4 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silverman SM and Wong WT (2018) Microglia in the Retina: Roles in Development, Maturity, and Disease. Annu Rev Vis Sci 4, 45–77. [DOI] [PubMed] [Google Scholar]
- 35.O’Koren EG et al. (2019) Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 50 (3), 723–737 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ronning KE et al. (2019) Molecular profiling of resident and infiltrating mononuclear phagocytes during rapid adult retinal degeneration using single-cell RNA sequencing. Sci Rep 9 (1), 4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao L et al. (2015) Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med 7 (9), 1179–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng B et al. (2014) Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J Neurosci 34 (24), 8139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin JB and Apte RS (2019) Visualizing the Heterogeneity of Retinal Microglia. Immunity 50 (3), 544–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo C et al. (2012) Knockout of ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp Eye Res 104, 39–47. [DOI] [PubMed] [Google Scholar]
- 41.Karlen SJ et al. (2018) Monocyte infiltration rather than microglia proliferation dominates the early immune response to rapid photoreceptor degeneration. J Neuroinflammation 15 (1), 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam NT and Sadek HA (2018) Neonatal Heart Regeneration. Circulation 138 (4), 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore KJ et al. (2018) Macrophage Trafficking, Inflammatory Resolution, and Genomics in Atherosclerosis: JACC Macrophage in CVD Series (Part 2). J Am Coll Cardiol 72 (18), 2181–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sene A et al. (2013) Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab 17 (4), 549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura R et al. (2015) IL10-driven STAT3 signalling in senescent macrophages promotes pathological eye angiogenesis. Nat Commun 6, 7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olefsky JM and Glass CK (2010) Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72, 219–46. [DOI] [PubMed] [Google Scholar]
- 47.Costantini E et al. (2018) The Role of Immunosenescence in Neurodegenerative Diseases. Mediators Inflamm 2018, 6039171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinto AR et al. (2016) Revisiting Cardiac Cellular Composition. Circ Res 118 (3), 400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heidt T et al. (2014) Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res 115 (2), 284–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lavine KJ et al. (2018) The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J Am Coll Cardiol 72 (18), 2213–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poss KD et al. (2002) Heart regeneration in zebrafish. Science 298 (5601), 2188–90. [DOI] [PubMed] [Google Scholar]
- 52.Becker RO et al. (1974) Regeneration of the ventricular myocardium in amphibians. Nature 248 (5444), 145–7. [DOI] [PubMed] [Google Scholar]
- 53.Haubner BJ et al. (2016) Functional Recovery of a Human Neonatal Heart After Severe Myocardial Infarction. Circ Res 118 (2), 216–21. [DOI] [PubMed] [Google Scholar]
- 54.Porrello ER et al. (2011) Transient regenerative potential of the neonatal mouse heart. Science 331 (6020), 1078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Porrello ER and Olson EN (2014) A neonatal blueprint for cardiac regeneration. Stem Cell Res 13 (3 Pt B), 556–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aurora AB et al. (2014) Macrophages are required for neonatal heart regeneration. J Clin Invest 124 (3), 1382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lai SL et al. (2017) Reciprocal analyses in zebrafish and medaka reveal that harnessing the immune response promotes cardiac regeneration. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Godwin JW et al. (2017) Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regen Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jung K et al. (2013) Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ Res 112 (6), 891–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leuschner F et al. (2012) Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med 209 (1), 123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen B et al. (2019) The Functional Heterogeneity of Resident Cardiac Macrophages in Myocardial InjuryCCR2(+) Cells Promote Inflammation, Whereas CCR2(−) Cells Protect. Circ Res 124 (2), 183–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Honold L and Nahrendorf M (2018) Resident and Monocyte-Derived Macrophages in Cardiovascular Disease. Circ Res 122 (1), 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li W et al. (2016) Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shiraishi M et al. (2016) Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest 126 (6), 2151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nahrendorf M et al. (2007) The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204 (12), 3037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hilgendorf I et al. (2014) Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res 114 (10), 1611–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sager HB et al. (2015) Targeting Interleukin-1beta Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation 132 (20), 1880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Molawi K et al. (2014) Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med 211 (11), 2151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patel B et al. (2018) CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl Sci 3 (2), 230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iliesiu AM and Hodorogea AS (2018) Treatment of Heart Failure with Preserved Ejection Fraction. Adv Exp Med Biol 1067, 67–87. [DOI] [PubMed] [Google Scholar]
- 71.Hulsmans M et al. (2018) Cardiac macrophages promote diastolic dysfunction. J Exp Med 215 (2), 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.King KR et al. (2017) IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med 23 (12), 1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Godwin JW et al. (2013) Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A 110 (23), 9415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yabluchanskiy A et al. (2016) Myocardial Infarction Superimposed on Aging: MMP-9 Deletion Promotes M2 Macrophage Polarization. J Gerontol A Biol Sci Med Sci 71 (4), 475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frantz S et al. (2013) Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J 27 (3), 871–81. [DOI] [PubMed] [Google Scholar]
- 76.van Amerongen MJ et al. (2007) Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol 170 (3), 818–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsujita K et al. (2007) Targeted deletion of class A macrophage scavenger receptor increases the risk of cardiac rupture after experimental myocardial infarction. Circulation 115 (14), 1904–11. [DOI] [PubMed] [Google Scholar]
- 78.Panizzi P et al. (2010) Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol 55 (15), 1629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cochain C et al. (2012) The chemokine decoy receptor D6 prevents excessive inflammation and adverse ventricular remodeling after myocardial infarction. Arterioscler Thromb Vasc Biol 32 (9), 2206–13. [DOI] [PubMed] [Google Scholar]
- 80.Sene A and Apte RS (2014) Eyeballing cholesterol efflux and macrophage function in disease pathogenesis. Trends Endocrinol Metab 25 (3), 107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spiller KL et al. (2014) The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 35 (15), 4477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang Y et al. (2016) Macrophage polarization in experimental and clinical choroidal neovascularization. Sci Rep 6, 30933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zandi S et al. (2015) ROCK-isoform-specific polarization of macrophages associated with age-related macular degeneration. Cell Rep 10 (7), 1173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kelly J et al. (2007) Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J Clin Invest 117 (11), 3421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cruz-Guilloty F et al. (2013) Infiltration of proinflammatory m1 macrophages into the outer retina precedes damage in a mouse model of age-related macular degeneration. Int J Inflam 2013, 503725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhu Y et al. (2016) Interleukin-17A neutralization alleviated ocular neovascularization by promoting M2 and mitigating M1 macrophage polarization. Immunology 147 (4), 414–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.He L and Marneros AG (2014) Doxycycline inhibits polarization of macrophages to the proangiogenic M2-type and subsequent neovascularization. J Biol Chem 289 (12), 8019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Okumura N et al. (2017) Application of Rho Kinase Inhibitors for the Treatment of Corneal Endothelial Diseases. J Ophthalmol 2017, 2646904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamaguchi M et al. (2017) Rho-Kinase/ROCK as a Potential Drug Target for Vitreoretinal Diseases. J Ophthalmol 2017, 8543592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaneko Y et al. (2016) Effects of K-115 (Ripasudil), a novel ROCK inhibitor, on trabecular meshwork and Schlemm’s canal endothelial cells. Sci Rep 6, 19640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kahook MY et al. (2019) Long-term Safety and Ocular Hypotensive Efficacy Evaluation of Netarsudil Ophthalmic Solution. Am J Ophthalmol. [DOI] [PubMed] [Google Scholar]
- 92.Okumura N et al. (2013) The ROCK inhibitor eye drop accelerates corneal endothelium wound healing. Invest Ophthalmol Vis Sci 54 (4), 2493–502. [DOI] [PubMed] [Google Scholar]
- 93.Tonges L et al. (2014) Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 62 (2), 217–32. [DOI] [PubMed] [Google Scholar]
- 94.Liu C et al. (2013) Targeting the shift from M1 to M2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS One 8 (2), e54841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takahashi K et al. (1989) Differentiation, maturation, and proliferation of macrophages in the mouse yolk sac: a light-microscopic, enzyme-cytochemical, immunohistochemical, and ultrastructural study. J Leukoc Biol 45 (2), 87–96. [DOI] [PubMed] [Google Scholar]
- 96.Tavian M and Peault B (2005) Embryonic development of the human hematopoietic system. Int J Dev Biol 49 (2–3), 243–50. [DOI] [PubMed] [Google Scholar]
- 97.Tavian M et al. (1999) Emergence of intraembryonic hematopoietic precursors in the pre-liver human embryo. Development 126 (4), 793–803. [DOI] [PubMed] [Google Scholar]
- 98.Mass E et al. (2016) Specification of tissue-resident macrophages during organogenesis. Science 353 (6304). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McGrath KE et al. (2015) Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep 11 (12), 1892–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schulz C et al. (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336 (6077), 86–90. [DOI] [PubMed] [Google Scholar]
- 101.Hoeffel G et al. (2015) C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42 (4), 665–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hoeffel G et al. (2012) Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 209 (6), 1167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hettinger J et al. (2013) Origin of monocytes and macrophages in a committed progenitor. Nat Immunol 14 (8), 821–30. [DOI] [PubMed] [Google Scholar]
- 104.Jakubzick C et al. (2013) Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity 39 (3), 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Auffray C et al. (2007) Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317 (5838), 666–70. [DOI] [PubMed] [Google Scholar]
- 106.Gamrekelashvili J et al. (2016) Regulation of monocyte cell fate by blood vessels mediated by Notch signalling. Nat Commun 7, 12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Galvan-Pena S and O’Neill LA (2014) Metabolic reprograming in macrophage polarization. Front Immunol 5, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sun N et al. (2016) The Mitochondrial Basis of Aging. Mol Cell 61 (5), 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.van Beek AA et al. (2019) Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends in Immunology 40 (2), 113–127. [DOI] [PubMed] [Google Scholar]
- 110.Pence BD and Yarbro JR (2018) Aging impairs mitochondrial respiratory capacity in classical monocytes. Experimental Gerontology 108, 112–117. [DOI] [PubMed] [Google Scholar]
- 111.Van den Bossche J et al. (2016) Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep 17 (3), 684–696. [DOI] [PubMed] [Google Scholar]
- 112.Esteban-Martinez L et al. (2017) Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J 36 (12), 1688–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Minhas PS et al. (2019) Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat Immunol 20 (1), 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kzhyshkowska J et al. (2012) Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 217 (5), 492–502. [DOI] [PubMed] [Google Scholar]
- 115.Kunjathoor VV et al. (2002) Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 277 (51), 49982–8. [DOI] [PubMed] [Google Scholar]
- 116.Katsuda S et al. (1993) Human atherosclerosis. IV. Immunocytochemical analysis of cell activation and proliferation in lesions of young adults. Am J Pathol 142 (6), 1787–93. [PMC free article] [PubMed] [Google Scholar]
- 117.Robbins CS et al. (2013) Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 19 (9), 1166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Randolph GJ (2008) Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr Opin Lipidol 19 (5), 462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jerome WG (2006) Advanced atherosclerotic foam cell formation has features of an acquired lysosomal storage disorder. Rejuvenation Res 9 (2), 245–55. [DOI] [PubMed] [Google Scholar]
- 120.Feng B et al. (2003) The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol 5 (9), 781–92. [DOI] [PubMed] [Google Scholar]
- 121.Cardilo-Reis L et al. (2012) Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol Med 4 (10), 1072–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nathan C and Ding A (2010) Nonresolving inflammation. Cell 140 (6), 871–82. [DOI] [PubMed] [Google Scholar]
- 123.Dutta P et al. (2012) Myocardial infarction accelerates atherosclerosis. Nature 487 (7407), 325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ridker PM et al. (2017) Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med 377 (12), 1119–1131. [DOI] [PubMed] [Google Scholar]
- 125.Bot I et al. (2017) A novel CCR2 antagonist inhibits atherogenesis in apoE deficient mice by achieving high receptor occupancy. Sci Rep 7 (1), 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nahrendorf M and Swirski FK (2017) Cholesterol, CCR2, and monocyte phenotypes in atherosclerosis. Eur Heart J 38 (20), 1594–1596. [DOI] [PubMed] [Google Scholar]
- 127.Neale BM et al. (2010) Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci U S A 107 (16), 7395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen W et al. (2010) Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci U S A 107 (16), 7401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ban N et al. (2018) Impaired monocyte cholesterol clearance initiates age-related retinal degeneration and vision loss. JCI insight 3 (17), e120824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lin JB et al. (2018) Macrophage microRNA-150 promotes pathological angiogenesis as seen in age-related macular degeneration. JCI insight 3 (7), e120157. [DOI] [PMC free article] [PubMed] [Google Scholar]