

SUMMARY
Self-renewing somatic tissues depend upon the proper balance of chromatin-modifying enzymes to coordinate progenitor cell maintenance and differentiation, disruption of which can promote carcinogenesis. As a result, drugs targeting the epigenome hold significant therapeutic potential. The histone demethylase, LSD1 (KDM1A), is overexpressed in numerous cancers, including epithelial cancers; however, its role in the skin is virtually unknown. Here we show that LSD1 directly represses master epithelial transcription factors that promote differentiation. LSD1 inhibitors block both LSD1 binding to chromatin and its catalytic activity, driving significant increases in H3K4 methylation and gene transcription of these fate-determining transcription factors. This leads to both premature epidermal differentiation and the repression of squamous cell carcinoma. Together these data highlight both LSD1’s role in maintaining the epidermal progenitor state and the potential of LSD1 inhibitors for the treatment of keratinocyte cancers, which collectively outnumber all other cancers combined.
Graphical Abstract
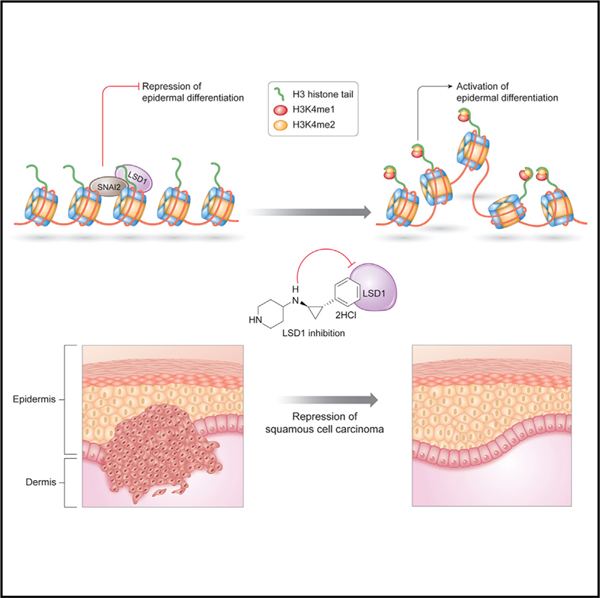
In Brief
Egolf et al. demonstrate that inhibition of the epigenetic regulator and histone demethylase, LSD1, promotes activation of the epidermal differentiation transcriptional program and, in turn, represses the invasion of cutaneous squamous cell carcinoma, one of the most common of all human cancers.
INTRODUCTION
Epigenetics encompasses the mechanisms through which gene expression and phenotypes are influenced independent of any changes to the underlying DNA sequence, and plays critical roles during development and differentiation through the intricate organization of each cell’s genome into chromatin (Atlasi and Stunnenberg, 2017). Mutations in chromatin modifiers occur in approximately 50% of all human cancers and are often associated with poor disease prognosis (Flavahan et al., 2017). By altering chromatin structure, these mutations can give rise to each of the classic hallmarks of cancer (Shen and Laird, 2013). Subsequently, considerable work has explored the use of epigenetic enzyme inhibitors to overcome tumor differentiation blocks through epigenetic reprogramming (Jin et al., 2017; Kelly and Issa, 2017). The inherently reversible nature of epigenetic marks provides additional rationale for defining the functions of chromatin modifiers in development, homeostasis, and disease, and collectively, this promise has resulted in the rapid development of numerous drugs targeting the activity of epigenetic enzymes (Shortt et al., 2017).
Epigenetics plays a particularly crucial role in self-renewing somatic epithelia, where stem cell populations must continually undergo self-renewal (Avgustinova and Benitah, 2016). A classic example of this is the epidermis, the outermost protective epithelial barrier of the skin that guards the body against external environmental damage and water loss. Through a multi-step differentiation process, epidermal progenitors (EPs) residing in the interfollicular basal stem cell layer give rise to the upper layers of the stratified epidermis (Gonzales and Fuchs, 2017). Understanding the specific transcription factors and epigenetic modifying enzymes necessary for proper regulation of the highly orchestrated transcriptional networks in normal epidermis, and how they are disrupted in epidermal cancers, may provide a unique opportunity for epigenetic therapeutic intervention.
The chromatin modifier LSD1 (KDM1A) is a histone lysine demethylase critical for organismal development and differentiation, and is frequently overexpressed in human cancers (Ding et al., 2013; Hosseini and Minucci, 2017; Li et al., 2016; Lim et al., 2010; Lv et al., 2012; Yuan et al., 2015). LSD1 acts primarily as a gene silencer by removing histone H3 lysine 4 (H3K4) mono-methylation and dimethylation (H3K4me1/2) (Shi et al., 2004; Zheng et al., 2015). In addition, in some cellular contexts, LSD1 has also been shown to demethylate H3 lysine 9 (H3K9) (Hu et al., 2008; Metzger et al., 2005), as well as non-histone targets (Huang et al., 2007; Lee et al., 2017; Nicholson and Chen, 2009; Wang et al., 2009). LSD1 is involved in repression of developmental programs and maintenance of pluripotency (Zheng et al., 2015), as well as stem cell self-renewal and cellular differentiation in myocytes, adipocytes, and during hematopoiesis (Choi et al., 2010; Musri et al., 2010; Thambyrajah et al., 2016). Despite this, the fundamental biological roles of LSD1 in the skin are virtually unknown. Here we show that pharmacologic LSD1 inhibition promotes a genome-wide loss of LSD1 binding and broad increases in H3K4 methylation and transcription at canonical epidermal differentiation-promoting transcription factors (TFs), and inhibits Ras-driven invasive neoplasia. Together, these results highlight the potential therapeutic utility of targeting epigenetic reprogramming for keratinocyte cancers (i.e., cutaneous squamous cell carcinoma [cSCC] and basal cell carcinoma [BCC]), which collectively outnumber all other human malignancies combined (Nehal and Bichakjian, 2018).
RESULTS
LSD1 Inhibition Unleashes the Epidermal Differentiation Transcriptional Program
LSD1 levels are elevated in epithelial cancers such as squamous cell carcinoma, while the opposing H3K4 histone methyltransferases, KMT2C (MLL3) and KMT2D (MLL4), display exceptionally high rates of loss-of-function mutations (Cancer Genome Atlas Network, 2015; Cancer Genome Atlas Research Network, 2014; Cancer Genome Atlas Research Network et al., 2017; Pickering et al., 2014). Together, this provides extensive rationale to understand how LSD1 functions in epithelial tissues given its potential to be inhibited with specific drugs (Cao et al., 2018; Shortt et al., 2017). To address this, we pharmacologically inhibited LSD1 in EPs using an irreversible, catalytic inhibitor of LSD1, GSK-LSD1 (Shortt et al., 2017). Consistent with the known function of LSD1 as a transcriptional repressor, after 48 h of exposure to 2 mM GSK-LSD1 or DMSO, RNA-seq differential gene expression analysis identified many more genes as upregulated (863) (Tables S1A and S1B) than downregulated (350) (Tables S1C and S1D) (Figures 1A and 1B). We observed a similar trend using a second LSD1 inhibitor, tranylcypromine (2-PCPA) (Figures 1C and S1A; Tables S1E–S1H) (Khan et al., 2013). Strikingly, treatment with either of these two distinct LSD1 inhibitors led to highly overlapping sets of transcriptional alterations genome-wide (Figures 1D and S1D; Tables S1I and S1J) from all expressed genes (Table S1K). Gene ontology (GO) analysis of the genes upregulated by GSK-LSD1 (Figure 1E), 2-PCPA (Figure S1B), or the genes overlapping between each drug highlighted genes involved in skin barrier homeostasis, keratinocyte differentiation, and cornification (Figure S1C). Genes downregulated were enriched for genes involved in extracellular structure organization, cell adhesion, and biological adhesion (Figure S1E). Numerous TFs known to be critical for epidermal progenitor differentiation were among the most highly expressed genes upon LSD1 inhibitor treatment, including OVOL2, GRHL1, NOTCH3, MAML3, MAFB, and KLF4 (Figure 1F) (Lee et al., 2014; Miyai et al., 2016; Mlacki et al., 2014; Ohashi et al., 2011; Sen et al., 2012; Watt et al., 2008). Interestingly, GLI1, the most highly downregulated transcription factor in GSK-LSD1-treated EPs (Figure 1G), is a known driver of BCC and other cancers (Epstein, 2008).
Figure 1. LSD1 Inhibitor Treatment of Epidermal Progenitors Unleashes a Pro-differentiation Transcriptional Program.

(A–C) Differentially expressed genes after 2 days of LSD1 inhibition by GSK-LSD1 (A and B) and 2-PCPA (C).
(D) Overlap between genes upregulated by 2-day GSK-LSD1 (863 genes) and 2-PCPA (1,161 genes).
(E) GO analysis of genes upregulated by 2-day GSK-LSD1 (863 genes).
(F and G) Log2 fold change values of the 50 most highly upregulated (F) and all 37 significantly downregulated (G) transcription factor-encoding genes after 2-day GSK-LSD1.
(H) Overlap between genes significantly upregulated by 2-day GSK-LSD1 (863 genes) and during in vitro epidermal differentiation (902 genes).
(I) Overlap between transcription factor-encoding genes significantly upregulated by 2-day GSK-LSD1 (93 genes) and during in vitro epidermal progenitor differentiation (103 genes).
(J) Overlap of genes significantly upregulated by 6-day GSK-LSD1 (1,678 genes) and during in vitro epidermal progenitor differentiation (902 genes).
(K) Overlap between transcription factor-encoding genes significantly upregulated by 6-day GSK-LSD1 (332 genes) and during in vitro epidermal progenitor differentiation (103 genes).
(L) transcription factor-encoding genes commonly upregulated by 2-day GSK-LSD1 and in vitro epidermal progenitor differentiation, sorted by decreasing log2 fold change between GSK-LSD1- and DMSO-treated samples.
(M) The 25 most differentially regulated transcription factor-encoding genes commonly upregulated by 6-day GSK-LSD1 and in vitro epidermal progenitor differentiation, sorted by decreasing log2 fold change between GSK-LSD1- and DMSO-treated samples.
(N) Increased expression of human genes that define the gene ontology term ‘‘keratinocyte differentiation’’ (GO:0030216) upon in vitro epidermal progenitor differentiation, after 2- or 6-day GSK-LSD1.
We next asked whether the LSD1-inhibited epidermal progenitor transcriptome overlapped with EPs differentiated in vitro (Toufighi et al., 2015). We compared the gene expression profile of differentiated EPs (Figure S1F; Tables S1L–S1O) with that of LSD1 inhibitor-treated EPs and found a significant intersection for both upregulated (Figure 1H) and downregulated genes (Figure S1G), including key upregulated (Figures 1I and 1L) or downregulated (Figures S1H and S1I) established epidermal differentiation TFs. Intersection between the TFs upregulated by LSD1 inhibition and TFs deemed as critical for epidermal pro-genitor differentiation (Klein et al., 2017) yielded a restricted number of TFs including all three members of the Grainyhead-like family of TFs (GRHL1, GRHL2, and GRHL3), together with OVOL2, NOTCH3, and KLF4 (Figures S1J and S1K).
Consistent with our initial results, exposure to 6 days of GSK-LSD1 at the same dose (2 mM) led to similar, but even more pro-found transcriptional changes than those at 48 h or 2 days, including more upregulated genes (1,678) than downregulated genes (1,377) (Figure S2A; Tables S1P–S1Q). Analysis of genes commonly upregulated or downregulated by 2 or 6 days of GSK-LSD1 revealed highly significant overlap between the two RNA-seq datasets (Figures S2B and S2C), with upregulated genes enriched for genes involved in epidermal differentiation, cornification, and keratinization (Figure S2G), whereas genes commonly downregulated were enriched for genes contributing to extracellular matrix organization, collagen fibril organization, and heterotypic cell-cell adhesion (Figure S2H). In addition, there was an even more significant overlap between 6-day GSK-LSD1-treated EPs and in-vitro-differentiated EPs than those treated for 2 days with GSK-LSD1, both at the gene (Figures 1J and S2D) and transcription factor (Figures 1K, S2E, and S2F) level. These changes include numerous TFs known as key determinants of epidermal progenitor differentiation, such as NOTCH3, OVOL1, ZNF750, GRHL1, and GRHL3 (Figure 1M). We next investigated the expression of the 288 genes that define the GO term ‘‘keratinocyte differentiation’’ (GO:003216) and found that although 2-day exposure to GSK-LSD1 resulted in a moderate increase in expression of epidermal progenitor differentiation genes, 6-day exposure to GSK-LSD1 triggered a dramatic increase in their expression and included numerous established differentiation genes (i.e., DSG1, SPRR1B, PI3, KLK13, KRT1, and CDSN) (Figures 1N).
Finally, we treated EPs with siRNAs against LSD1 to determine whether genetic knockdown recapitulated our results with the pharmacological LSD1 inhibitors. After 72 h of siRNA treatment, LSD1 protein abundance was significantly reduced (Figure S2I). RNA sequencing (RNA-seq) demonstrated that although the changes in the transcriptional landscape were not as broad as the inhibitors, genes significantly upregulated by siLSD1 treatment were again enriched for genes involved in epidermal cornification and keratinocyte differentiation (Figures S2J and S2K). The majority of upregulated genes overlapped with genes upregulated by GSK-LSD1 treatment (Figure S2L), and those overlapping genes were enriched for epidermal cornification and differentiation (Figure S2M). Even though GRHL1 and GRHL3 were again upregulated, it did not quite reach statistical significance, although several known targets of these epidermal differentiation TFs were significantly increased in gene expression, such as KRT80, IVL, and DSG1 (Figure S2N). Together, these data link LSD1 to the transcriptional repression of differentiation genes and TFs in EPs. Furthermore, these differentiation gene expression programs can be unleashed with inhibition of LSD1.
LSD1 Inhibition Prevents LSD1 Binding at SNAI2-Repressed Epidermal Differentiation Genes
Having found LSD1 inhibitors induce a differentiation-related transcriptional program in EPs, we hypothesized that LSD1 may be enriched at the regulatory elements of key epidermal progenitor differentiation genes. To test this, we mapped LSD1 binding genome-wide using chromatin immunoprecipitation followed by sequencing [ChIP-seq]) in EPs treated with DMSO or GSK-LSD1 (2 mM). Irreversible LSD1 inhibitors, such as GSK-LSD1, have been reported to prevent binding of LSD1 to the genome in addition to inhibiting LSD1’s catalytic activity (Maiques-Diaz et al., 2018). Consistent with this, GSK-LSD1 treatment dramatically reduced LSD1 binding to chromatin genome-wide. Differential binding analysis identified 1,432 lost LSD1 binding sites with GSK-LSD1 (Table S2A), whereas 1,174 LSD1 sites were maintained (Table S2B), and only 14 sites demonstrated increased binding (Table S2C) (Figures 2A–2E). Lost LSD1 peaks were enriched at promoters, defined here as sequences located up to 1 kb upstream of a gene’s transcriptional start site, thus also encompassing local enhancer regions. Together, this suggests that LSD1 is preferentially lost from regulatory elements upon LSD1 inhibition (Figure 2F). In line with this, analysis of the genes nearest to each GSK-LSD1 lost LSD1 peak showed that the majority (71%) were associated with protein-coding genes (Figure S3A).
Figure 2. LSD1 Inhibition Prevents LSD1 Binding to Epidermal Differentiation Genes and SNAI2 Binding Sites.

(A) ChIP-seq demonstrates dramatic reduction in LSD1 peaks with LSD1 inhibition.
(B) Comparison of fragments per kilobase mapped (FPKM) normalized LSD1 binding intensities in DMSO- or GSK-LSD1-treated EPs.
(C and D) LSD1 binding intensities at shared, GSK-LSD1 lost, or GSK-LSD1-gained LSD1 sites (1 kb apart peak center) by heatmap (C) and violin plot (D).
(E) Average profiles of LSD1 binding at shared, GSK-LSD1 lost, and GSK-LSD1-gained sites. Solid lines represent LSD1 binding with DMSO, whereas dotted lines represent LSD1 binding with GSK-LSD1.
(F) Distribution of LSD1 binding sites at shared, GSK-LSD1 lost, and GSK-gained LSD1 sites (numbers = log2 enrichment).
(G) Top de novo motifs associated with GSK-LSD1 lost sites.
(H and I) Overlap between genes (H) and transcription factor-encoding genes (I) upregulated by GSK-LSD1 and associated with GSK-LSD1 lost LSD1 sites.
(J) Log2 fold changes (left) and LSD1 binding intensities (right) for GSK-LSD1 upregulated TFs with GSK-LSD1 lost LSD1 sites.
(K) Overlap of GSK-LSD1 lost LSD1 sites and SNAI2 binding sites (GSE55421) (Mistry et al., 2014).
(L) Overlap of LSD1 peaks common to DMSO- and GSK-LSD1-treated samples and SNAI2 peaks.
(M) LSD1 immunoprecipitation (IP) pulls down SNAI2 by coIP.
(N and O) LSD1 and SNAI2 shared target genes that lose LSD1 binding and increase in expression, HES5 (N) and CLDN1 (O).
(P and Q) LSD1 bound genes that do not lose LSD1 binding or increase in expression, HIST1H4C (P) and ZNF687 (Q), highlighting absence of SNAI2 binding.
We next integrated these data with our RNA-seq results to identify genes that had upregulated gene expression and loss of LSD1 binding upon LSD1 inhibition. A total of 146 genes (Figure 2H), of which 23 were TFs (Figures 2I), were both upregulated by LSD1 inhibition and associated with LSD1 lost sites. Importantly, TFs critical for epidermal progenitor differentiation (i.e., GRHL1, GRHL3, NOTCH3, KLF4) were among the 146 genes (Figure 2J). This suggests that the majority of the transcriptional effects resulting from LSD1 inhibition are driven secondarily through LSD1’s direct role in regulating expression of key TFs. There was no significant overlap between LSD1 binding sites that were maintained and genes upregulated (Figure S3B) or downregulated (Figure S3C) by GSK-LSD1. GO analysis of the 146 overlapping genes again demonstrated a strong enrichment of terms involved in epidermal differentiation and cornification (Figure S3D), whereas those shared sites between DMSO- and GSK-LSD1-treated EPs did not (Figure S3E).
We then evaluated transcription factor binding motifs that were enriched at LSD1 binding sites lost upon GSK-LSD1 treatment. This identified Fra1 (FOSL1) and SNAI2 (Slug) as the top two most highly enriched motifs at these sites (Figure 2G), both known critical regulators of keratinocyte differentiation (Eckert et al., 2013; Klein et al., 2017; Mistry et al., 2014). SNAI2, acting through its SNAG domain, has been reported to recruit LSD1 to target genes in mammary, colon, and neuroblastoma cancer cells in vitro (Ferrari-Amorotti et al., 2013). Because the primary function of SNAI2 in epidermal progenitors is transcriptional repression of keratinocyte differentiation (Mistry et al., 2014), we next performed co-immunoprecipitation (coIP) experiments using full-length endogenous LSD1 and SNAI2 proteins from human EPs and found that LSD1 and SNAI2 indeed do directly interact in this context (Figures 2M and S3L). Furthermore, transcriptional changes induced by LSD1 inhibition significantly overlap with those seen with SNAI2 inhibition (Figures S3G–S3J), including the upregulation of genes involved in epidermal differentiation and cornification (Figure S3K). Collectively, these observations suggest that inhibiting LSD1 in EPs may block LSD1/SNAI2 binding and induce differentiation via activation of SNAI2-repressed target genes.
This prompted us to query publicly available SNAI2 ChIP-seq data from EPs (Mistry et al., 2014). Consistent with our motif analysis, overlap between SNAI2 peaks and GSK-LSD1 lost sites indicates that GSK-LSD1 lost sites are significantly associated with SNAI2 binding sites (Figure 2K). These included genes involved in epidermal differentiation, such as HES5, CLDN1, CLDN7, and PPL (Figures 2N, 2O, S3M, and S3N) (Furuse et al., 2002; Sevilla et al., 2007). In contrast, shared DMSO-and GSK-LSD1-treated LSD1 peak regions did not overlap significantly with SNAI2 peaks (Figure 2L), nor did these genes increase in expression (Figures 2P and 2Q). Furthermore, transcription factor binding motifs associated with LSD1 sites shared between DMSO- and GSK-LSD1-treated samples included different transcription factor binding motifs, e.g., Nkx2–5, Vdr, and Smad3–1 (Figure S3F). Together these data indicate that GSK-LSD1 both inhibits LSD1 catalytic activity and blocks a significant proportion of LSD1 binding to its targets, and that these LSD1 lost sites overlap with SNAI2 transcription factor binding sites. We also demonstrate that LSD1 and SNAI2 directly interact, collectively suggesting that LSD1 is required for SNAI2-mediated transcriptional repression, and that LSD1 inhibitors may block this repressive function.
LSD1 Inhibition Increases H3K4 Methylation at Epidermal Differentiation Genes and Transcription Factors
Because loss of LSD1 binding at SNAI2-repressed epidermal progenitor differentiation genes correlated with an upregulation of these genes, we hypothesized that LSD1 inhibitors may enrich the regulatory elements of these genes with H3K4 methylation, leading to their activation. To test this, we mapped H3K4me1 and H3K4me2 genome-wide in DMSO- or GSK-LSD1-treated EPs, and observed a global increase of 15% in H3K4 monomethylated regions and approximately 6% in H3K4 dimethylated regions (Figure 3A). GSK-LSD1 appeared to have a more drastic effect on differential H3K4me2 (974 regions) (Table S2E) than H3K4me1 enrichment (138 regions) (Table S2D) (Figures 3B, 3D, and 3F). Similar to the LSD1 binding sites, the increases in H3K4me1 and H3K4me2 were enriched at promoter regions in GSK-LSD1-treated samples (Figure 3C). Consistent with the known roles of H3K4me1 at enhancers and H3K4me2 at promoters, respectively, H3K4me2 was more enriched at promoter regions in comparison with H3K4me1 (Figure S4A).
Figure 3. LSD1 Inhibition Drives Genome-wide Increases of H3K4me1 and H3K4me2 in Epidermal Progenitors.

(A) Number of H3K4me1/me2 peaks in DMSO- or GSK-LSD1-treated EPs in combined replicates (n = 2).
(B) Number of GSK-LSD1-gained or lost H3K4me1 and H3K4me2 regions.
(C) Distribution of GSK-LSD1-gained H3K4me1/me2 regions. Bolded numbers equal log2 enrichment.
(D) Average profile plots of H3K4me1 and H3K4me2 binding at GSK-LSD1-gained H3K4me1 or H3K4me2 regions in DMSO- (blue lines) or GSK-LSD1-treated EPs (red lines).
(E) Overlap between GSK-LSD1 lost LSD1 sites (1,432 peaks and 1,301 unique genes), GSK-LSD1-gained H3K4me1 regions (138 regions and 138 unique genes), and GSK-LSD1-gained H3K4me2 regions (974 regions and 941 unique genes).
(F) H3K4me1 and H3K4me2 binding occupancy at GSK-LSD1-gained H3K4me1 (left, blue) or H3K4me2 (right, green) regions in DMSO- or GSK-LSD1- treated EPs.
(G) Average profiles of LSD1, H3K4me1, and H3K4me2 binding at LSD1 peaks shared between DMSO- and GSK-LSD1-treated EPs (left panel), at GSK-LSD1 lost LSD1 peaks (center panel) and at GSK-LSD1 lost LSD1 peaks associated with GSK-LSD1 upregulated genes (right panel).
(H and I) University of California, Santa Cruz (UCSC) genome browser demonstrating representative epidermal differentiation TFs, NOTCH3 (H) and GRHL3 (I), that display overlapping SNAI2 and LSD1 peaks, lost LSD1 binding with GSK-LSD1, and increases in H3K4me1 and/or H3K4me2 and concomitant gene expression.
We next intersected GSK-LSD1 lost LSD1 binding sites with GSK-LSD1-gained H3K4me1 and H3K4me2 regions and found 24 genes that were associated with lost LSD1 binding and increases in both H3K4me1 and H3K4me2 (Figure 3E; Table S2L), including OVOL2 and NOTCH3 (Figure 3H). A total of 151 genes gained only H3K4me2 (Figures 3E and 3I; Table S2J), and 17 genes gained only H3K4me1 (Figures 3E and S4C; Table S2I). There were some genes that gained H3K4me1 and/or H3K4me2 that were not bound by LSD1 in either DMSO- or GSK-LSD1-treated EPs (Tables S2G and S2H), as well as genes that lost LSD1 binding with GSK-LSD1 treatment but did not demonstrate any significant changes in H3K4 methylation (Figure S4D; Table S2F). Collectively, these data suggest that LSD1 inhibitor treatment had larger effects on increasing H3K4me2 than H3K4me1 (Figure 3E). Average profile plots confirmed specific increases of H3K4 methylation surrounding both total LSD1 lost sites and LSD1 lost sites associated with genes upregulated by GSK-LSD1 (Figures 3G and S4B). In contrast, maintained LSD1 sites did not exhibit these increases in H3K4 methylation (Figure 3G, center and right compared with left). SNAI2-target genes with lost LSD1 binding, increased H3K4me1 and H3K4me2, and upregulated gene expression following GSK-LSD1 treatment included the major pro-epithelial differentiation TFs, NOTCH3 and GRHL3 (Figures 3H and 3I).
Altogether, these results highlight how pharmacological inhibition of LSD1 enzymatic activity in proliferating EPs impaired LSD1 binding at a set of genes implicated in epidermal progenitor differentiation and triggered their increased expression along with associated increases in H3K4me1 and H3K4me2. Further-more, our results suggest that the portion of LSD1 cistrome affected by pharmacological inhibition of its enzymatic activity might be driven by transcriptional repressors known to play important roles in inhibiting cell differentiation and maintaining the self-renewal potential of epidermal progenitors (i.e., FRA1 and SNAI2) (Eckert et al., 2013; Klein et al., 2017; Mistry et al., 2014).
LSD1 Inhibition Promotes Epidermal Differentiation and Represses Squamous Cell Carcinoma
We next wanted to test how LSD1 inhibition functionally affected epidermal progenitor differentiation and growth. First, we validated the upregulation of NOTCH3, GRHL3, KLF4, and AP2-g (TFAP2C) following GSK-LSD1 treatment (Figure 4A). We then assessed how proliferating epidermal progenitor cell behavior and fate may be altered by activation of these pro-differentiation epithelial TFs. GSK-LSD1 treatment significantly reduced epidermal progenitor cell growth as compared with DMSO (Figure 4B). Next, we used three-dimensional (3D) human organotypic (OTC) skin models to assess the effects of LSD1 inhibition on epidermal behavior (Simpson et al., 2010). In contrast with DMSO-treated controls, which yielded a relatively normal stratified epidermis, GSK-LSD1-treated EPs showed regions of sporadic cornification, the end product of terminal differentiation within the epidermis (Figure 4C), consistent with our data from two-dimensional (2D) cultures that indicated that LSD1 inhibitors activated epidermal differentiation programs.
Figure 4. LSD1 Inhibition Promotes Differentiation and Represses Squamous Cell Carcinoma.

(A) NOTCH3, GRHL3, KLF4, and AP2-g (TFAP2C) are upregulated in GSK-LSD1-treated EPs.
(B) GSK-LSD1-treated EPs display significantly reduced growth compared with DMSO.
(C) GSK-LSD1-treated 3D human OTCs on collagen rafts prematurely cornify (arrows) as compared with DMSO.
(D) GSK-LSD1-treated oncogenic 3D human OTCs on human dermis prematurely cornify as compared with DMSO (H&E).
(E and F) GSK-LSD1-treated (2, 10, or 20 mM) oncogenic 3D human OTCs established on human dermis display significantly less epidermal area (E) and dermal protrusions (F) than DMSO. Error bars represent SDs.
(G) IF of oncogenic 3D OTCs for Krt14 (red), Collagen VII (green), or DAPI (blue) (original magnification 320; scale bar, 50 mm).
(H) Overlap of genes upregulated by GSK-LSD1 in EPs and genes downregulated in cSCC compared with normal skin (top).
(I) LSD1 expression negatively correlates with expression of epithelial differentiation TFs in HNSCC data from TCGA.
We then hypothesized that activating differentiation with LSD1 inhibitors may inhibit keratinocyte cancers. To test this, we utilized a model of human cSCC in which proliferating EPs are engineered to express two medically relevant oncodrivers, CDK4 (R24C) and tamoxifen-induced mutant H-RAS (G12V) (Lazarov et al., 2002), which are sufficient to convert normal OTC epidermis into invasive cSCC (Ridky et al., 2010). Similar to our results in EPs, GSK-LSD1 treatment of the engineered cSCC-like cells upregulated the expression of the same pro-epidermal differentiation TFs as compared with DMSO (Figure S4E). In the invasive cSCC OTC system, DMSO-treated control OTCs produced a thickened epidermis and invasive projections into the dermis consistent with previous results (Figures 4D and 4G) (Ridky et al., 2010). In contrast, OTCs treated with GSK-LSD1 at 2, 10, or 20 mM exhibited significantly reduced epidermal area and dermal protrusions, and displayed less invasive phenotypes (Figures 4D–4G). Consistent with this, these cultures also showed earlier flattening and squamatization of the epidermis, as well as expression of differentiation markers like involucrin (IVL) (Figure S4F).
We next hypothesized that the expression of the pro-differentiation transcriptional programs upregulated by LSD1 inhibition would be suppressed in human patient cSCCs. To test this, we queried published gene expression data from human patient cSCCs (Chitsazzadeh et al., 2016). Indeed, there was significant overlap between those genes suppressed in human cSCC and those genes upregulated by LSD1 inhibition in EPs after 2 days of GSK-LSD1 (Figures 4H). Finally, we examined publicly available gene expression data from The Cancer Genome Atlas (TCGA) for Head and Neck Squamous Cell Carcinoma (HNSCC) given that HNSCC has been shown to be the most transcriptionally similar cancer to cSCC (Chitsazzadeh et al., 2016), and because cSCC gene expression data are currently unavailable in TCGA. Notably, LSD1 is commonly overexpressed and associated with poor prognosis and survival in HNSCC (Alsaqer et al., 2017). Intriguingly, we found that LSD1 expression was significantly negatively correlated with the expression of the pro-epithelial differentiation genes GRHL1, MAML3, NOTCH3, NOTCH1, and CLDN4 in HNSCC (Figures 4I and S4G), in accordance with our evidence that LSD1 is a direct repressor of these major pro-differentiation genes and TFs. Together, these data underscore the pro-differentiation gene expression effects of LSD1 inhibitors in both EPs and cSCC models, and highlight their potential as a pro-differentiation therapy in invasive SCC.
DISCUSSION
Self-renewing somatic tissues such as the skin rely on precise epigenetic changes in order to orchestrate the dramatic transcriptional changes that occur during the transition from early stem-like EPs into fully differentiated cells (Avgustinova and Benitah, 2016). Epigenetic dysregulation disrupts differentiation and, in turn, can promote disease such as cancer. Therefore, the ability to potentially reverse disrupted epigenetics through therapies targeting chromatin regulators is an active area of pharmaceutical research (Shortt et al., 2017).
Given its direct accessibility, the skin is more amenable to use of potentially broad-based inhibitors of chromatin modifiers given the ability to avoid systemic delivery and side effects through topical delivery systems. Furthermore, keratinocyte cancers, made up of BCC and cSCC, outnumber all other human malignancies combined and are increasing in incidence with the aging of the population (Nehal and Bichakjian, 2018). Although generally treatable with surgery, cSCC in particular displays increased rates of metastasis and death in the aged and immunocompromised (Nehal and Bichakjian, 2018). Interestingly, cSCC has been shown to share common mutational and transcriptional underpinnings with all other forms of SCC, and this ‘‘pan-squamous’’ group of cancers displays the highest rates of mutations in epigenetic modifiers (Campbell et al., 2018; Chitsazzadeh et al., 2016; Dotto and Rustgi, 2016). Together, this underscores the critical importance of obtaining an in-depth mechanistic understanding of how these major epigenetic regulators function in both epithelial homeostasis and disease.
Given that LSD1 is elevated across numerous human cancers and can be targeted with relatively specific inhibitors (Shortt et al., 2017), we set out to define the role of LSD1 in the skin. We present evidence that LSD1’s main function within the epidermis is to actively maintain EPs in their more basal stem cell state through the direct repression of major TFs driving epithelial differentiation. Inhibition of LSD1 results in a dramatic reduction of LSD1 binding genome-wide at known SNAI2-repressed canonical epithelial differentiation genes and TFs (Mistry et al., 2014), and is coupled with broad increases in H3K4 methylation, as well as the expression of these genes. Functionally this results in reduced proliferation of EPs, as well as reduced growth and invasion in a model of cSCC (Ridky et al., 2010).
The enrichment of LSD1 binding at SNAI2 target genes, as well as the direct interaction of LSD1 and SNAI2 in EPs, strongly suggest that LSD1 and SNAI2 might cooperate to maintain a more stem cell-like state by repressing the epidermal differentiation program. This finding, together with the well-established function of SNAI2 as a critical regulator of epithelial mesenchymal transition (EMT) in various cancer contexts (Casas et al., 2011), suggests that LSD1 inhibition may work through diverse mechanisms beyond just promoting differentiation to prevent cancer progression. Collectively, these observations underscore the exciting potential for LSD1 inhibitors to treat cSCC.
Notably, an earlier study of ZNF750, a transcription factor involved in epidermal differentiation, demonstrated an interaction between LSD1 and ZNF750 (Boxer et al., 2014). Upon examining the effects of LSD1 inhibition on a panel of 80 ZNF750-regulated genes, this study suggested that LSD1 may play a role in the repression of progenitor genes. However, our study has employed more comprehensive and unbiased genome-wide and functional approaches, and through these methods, we are able to show that, on balance, LSD1’s major role in the epidermis is actually to repress differentiation genes, because LSD1 inhibition leads to a genome-wide loss of LSD1 binding, increased H3K4 methylation, and increased gene expression of canonical epidermal differentiation genes.
Future studies will be needed to test the ability of LSD1 inhibitors, and particularly the potential of topically delivered LSD1 inhibitors, to treat cSCC in vivo. Along these lines, recent evidence has also highlighted the ability of LSD1 inhibitors to synergize with immunotherapies such as inhibitors of PD-1 (Sheng et al., 2018), suggesting even further potential when used in combination with other therapies that have demonstrated efficacy (Migden et al., 2018). More broadly, this study also suggests that a deeper mechanistic understanding of chromatin regulators in epithelial tissues may lead to novel therapeutic opportunities for a range of cutaneous diseases. For instance, psoriasis and acne, two diseases that affect broad segments of the population (Lim et al., 2017), have also been shown to benefit from therapies thought to work through pro-differentiation mechanisms (Beckenbach et al., 2015).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian C. Capell (capellb@pennmedicine.upenn.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Normal Human Epidermal Keratinocyte Isolation and Culture
Primary epidermal progenitors were isolated from de-identified discarded neonatal human foreskin obtained by Core B of the Penn Skin Biology and Diseases and Resource-based Center. Foreskin was incubated for 12 h at 4°C in 2.4 U/mL Dispase II. Sterile forceps were used to separate the underlying dermis. The epidermal sheet was transferred to a 60-mm tissue culture plate, incubated in 0.25% trypsin for 10 min at 37°C, and then neutralized with 1 mL of fetal bovine serum (FBS). Sterile forceps were used to scrape the epidermal sheet against the dish to dissociate cells. The suspension was passed through a 40-mm strainer and then centrifuged at 200 g for 5 min. The cell pellet was resuspended in 5 mL keratinocyte medium (described next). Epidermal progenitors were cultured in a 50:50 mix of 1 × keratinocyte–SFM supplemented with human recombinant epidermal growth factor and bovine pituitary extract combined with medium 154 supplemented with human keratinocyte growth supplement and 1% 10,000 U/mL penicillin–streptomycin at 37°C.
Human Skin Organoids
Collagen Rafts
The 3D organotypic human skin cultures were performed as described previously(Simpson et al., 2010). Briefly, J2 3T3 fibroblasts were grown in DMEM + 10% FBS. Cells were released from culture plates using 0.25% trypsin for 5 min at 37°C, resuspended in DMEM + 10% FBS, and counted using a hemacytometer to determine the volume needed to obtain 0.75 million to 1 million fibro-blasts per organotypic culture. The required volume was centrifuged in a 50-mL sterile conical tube at 200 g for 5 min, and the supernatant was removed. The fibroblast cell pellet was resuspended in 1/10 the final required volume (2 mL per culture) of 10 × collagen resuspension buffer (1.1 g of NaHCO3 plus 2.39 g of HEPES in 50 mL of 0.05 N NaOH) and held on ice. One-tenth the final volume of 10 × DMEM (Sigma) was then added, and the cells were mixed by vigorous pipetting. Purified high-concentration rat tail collagen I (Corning) was added and diluted with sterile dH2O to a final concentration of 4 mg per milliliter of the final volume. NaOH (0.05 N) was added to a pH of ~7. The collagen–fibroblast slurry was mixed by inverting, and then 2 mL was pipetted into the upper chamber of a sixwell transwell insert (Corning) placed within a deep-well six-well tissue culture plate (Corning). The fibroblast– collagen matrices were allowed to polymerize for 60 min at 37°C. Next, the matrices were submerged in DMEM + 10% FBS and placed overnight at 37°C. The next day, NHEKs were trypsinized, resuspended in 38 DMEM + 10% FBS, counted to collect 1 million cells per culture, and centrifuged at 200 g for 5 min, and the supernatant was discarded. The NHEK pellet was resuspended in E-medium supplemented with 5 ng/mL EGF (Sigma) to a volume of 2 mL per culture. The DMEM was removed from both the upper and lower chambers of the transwell plates containing the collagen–fibroblast matrices. Two milliliters of NHEKs (1 million cells) was seeded atop each matrix in the upper transwell chamber, and 14 mL of E-medium with 5 ng/mL EGF was added to the bottom chamber. The cultures were placed overnight at 37°C. The next day, the medium was aspirated from both the top and bottom chambers of the transwell. To place the NHEK monolayers at an air–liquid interface and induce stratification, 10 mL of E-medium (without EGF supplementation) was added only to the bottom chamber of the transwell, and the cultures were grown for up to 12 d at 37°C and fed 10 mL of Emedium every other day. To generate protein lysates, the transwell apparatus was removed from the plate, and the organotypic culture was separated from the underlying matrix using sterile forceps. The culture was transferred into urea sample buffer (8 M urea, 1% SDS, 10% glycerol, 60 mM Tris, 5% β-mercaptoethanol at pH 6.8) and dissolved by vigorous pipetting using a 25-gauge needle and a 1-mL syringe. Organotypic cultures were prepared for routine histology by submerging the culture in 10% neutral-buffered formalin for 24–48 h.
Organotypic Culture
Split-thickness human skin was obtained from SBDRC. Skin was washed in PBS containing 5% penicillin-streptomycin and incubated at 37°C for 10 days in PBS with penicillin-steptomycin, where PBS was changed every 2 days. The epidermis was separated from the dermis and discarded. The dermis was washed with PBS and incubated at 4°C for 6 to 12 weeks, with PBS changed every 2 days. For assembly of organotypic tissue, the dermis was cut into 1 cm2 square pieces. The cut dermis was elevated onto a sterilized annular dermal support tissue culture device in a manner that the basement membrane was oriented upward(Duperret et al., 2015; Ridky et al., 2010). 50 uL Matrigel was added to the bottom (non-basement membrane) side of the dermis to block any openings in the dermis due to hair follicles and incubated at 37°C for 30 minutes until solidified. Primary human keratinocytes with CDK4 R24C and ER-H-RAS G12V overexpression were seeded onto the basement membrane side at a density of 8×105 cells in a total volume of 80 µL. Keratinocyte growth medium (KGM) was added below the dermal support device allowing the basement membrane side to be exposed to air. KGM consists of 3:1 mixture of DMEM:Ham’s F12, supplemented with 10% FBS, adenine (1.8 × 10–4 M), hydrocortisone (0.4 µg/ml), insulin (5 µg/ml), cholera toxin (1 × 10–10 M), EGF (10 ng/ml), transferrin (5 µg/ml), and triido-l-thyronine (1.36 ng/ml). KGM medium was changed every other day, along with any 4-hydroxytamoxifen (100 nM in 100% EtOH) to activate H-RAS. Organotypic tissue was harvested after 14 days, fixed in 10% formalin overnight, and placed in 70% EtOH for paraffin embedding.
Retroviral Transduction
Phoenix cells were used for retrovirus production containing ER-H-RAS G12V and CDK4 R24C constructs(Duperret et al., 2015). Phoenix cells containing either construct were plated in a 6-well plate at approximately 40% confluency. 24 hours after plating, cell culture medium was replaced, and cells were moved to 32°C for optimal virus production. Primary human keratinocytes were transduced at 30% confluence. Cells were spun at 1000 rpm for 1 hour at room temperature and keratinocyte growth medium was replaced after 1 hour of incubation at 37°C. Primary human keratinocytes were first transduced with CDK4 R24C and then two days later were transduced with ER-H-RAS G12V. Overexpression of both vectors was confirmed with western blot; for ER-H-RAS, 100 nM 4-hydroxytamoxifen was added for 72 hours before collecting cells for western blot and was confirmed via downstream activation of p-ERK. Keratinocytes containing both CDK4 and ER-H-RAS were frozen and thawed for each experiment.
METHOD DETAILS
2D Keratinocyte Culture Treatments
LSD1 Inhibitor –
For LSD1 inhibitor experiments, epidermal progenitors were treated with LSD1 inhibitors GSK-LSD1 or 2-PCPA. For 48 hour experiments, cells were treated twice with inhibitor at 0 and 24 hours before harvesting. For 6d experiments, cells were treated once daily with inhibitor at 0, 24, 48, 72, 96, and 120 hours before harvesting at 144 hours.
Differentiation Media –
For differentiation experiments, NHEKs were cultured in medium containing 1.22 mM calcium chloride for 48 h and then harvested.
siRNAs-
For siRNA experiments, primary human epidermal progenitors were treated with either siRNAs against LSD1 or a control siRNA at a dose of 500 nM. Cells were harvested 72 hours after transfection.
RNA-sequencing
Total RNA was extracted using RNeasy kit following the manufacturer’s instructions. All RNA-seq libraries were prepared using the NEBNext poly(A) mRNA magnetic isolation module followed by NEBNext Ultra Directional RNA library preparation kit for Illumina. Library quality was checked by Agilent BioAnalyzer 2100 and libraries were quantified using the Library Quant Kit for Illumina. Libraries were then sequenced using a NextSeq500 platform (75-base-pair (bp) single-end reads). All RNA-seq was aligned using RNA STAR(Dobin et al., 2013) under default settings to Homo sapiens University of California at Santa Cruz (UCSC) hg19 (RefSeq and Gencode gene annotations). FPKM (fragments per kilobase per million mapped fragments) generation and differential expression analysis were performed using DESeq2(Love et al., 2014). Statistical significance was obtained using an adjusted p value (padj) generated by DESeq2 of less than 0.05. All RNA-seq experiments were performed in triplicate. For Figure 1N, the list of human genes that defines the gene ontology term ‘‘Keratinocyte differentiation’’ (GO:0030216) was filtered to retrieve genes that have a positive log2 fold change and an adjusted p value ≤ 0.05 in the epidermal differentiation RNA-seq dataset, and then further filtered to keep genes that have sufficient expression to assess differential gene expression in both 2d and 6d GSK-LSD1 RNA-seq datasets (i.e., baseMean > = 10), and then ordered by decreasing log2 fold change in the epidermal differentiation RNA-seq dataset.
ChIP-sequencing
ChIP-seq was performed as described previously(Capell et al., 2016; Lin-Shiao et al., 2018). Briefly, keratinocytes cultured in 10-cm2 dishes were fixed in 1% formaldehyde for 5 min, and fixation was quenched with the addition of glycine to 125 mM for an additional 5 min. Cells were harvested by scraping from plates and washed twice in 1 × PBS before storage at –80°C. ChIP extracts were sonicated for 15 minutes in a Covaris sonicator. All ChIPs were performed using 500 µg of extract and 2 µg of antibody per sample. Thirty microliters of Protein G Dynabeads was used per ChIP. ChIP DNA was also used to make sequencing libraries using NEBNext Ultra DNA library preparation kit for Illumina. Library quality was checked by Agilent BioAnalyzer 2100 and libraries were quantified using the Library Quant Kit for Illumina. Libraries were then sequenced using a NextSeq500 33 platform (75-bp, single-end reads). After sequencing, all data were demultiplexed from the raw reads using Illumina’s BCL2FASTQ from BaseSpace. Further ChIP-seq analysis described below. Antibodies used for ChIP-seq include anti-LSD1, anti-Histone 3 (monomethyl K4), and anti-Histone 3 (dimethyl K4) as described in the KRT.
ChIP-sequencing Data Processing
ChIP-seq reads were aligned to human reference genome (hg19) using Bowtie2 V2–1.0(Langmead and Salzberg, 2012). Only uniquely mapped reads were considered for further analysis. Aligned reads form biological replicates were pooled together. For LSD1 ChIP-seq analysis, tag directories were generated for pooled DMSO treated samples and for pooled GSK-LSD1-treated samples using HOMER v4.10.1(Heinz et al., 2010), allowing a maximum of 1 tag per base pair to remove PCR duplicates and using the given fragment length. Peaks were called for pooled vehicle-treated samples and for pooled GSK-LSD1-treated samples using HOMER v4.10.1, using the corresponding inputs and default parameters. Visualization tracks were generated using HOMER v4.10.1, allowing a maximum of 1 tag per base pair to remove PCR duplicates, normalizing experiments to 10 millions total tags and subtracting the corresponding input (-tbp 1, -i -subtract) and visualized using UCSC Genome Browser(Kent et al., 2002). For the differential analysis of LSD1 peaks, DMSO-treated and GSK-LSD1-treated peak sets were concatenated, then sorted and merged using BEDtools v2.27.1(Quinlan, 2014; Quinlan and Hall, 2010), allowing a maximum distance of 100 base pairs (bp) to merge peaks. The resulting peak file was gene annotated and reads were counted in each tag directories and normalized to fragments per kilobase mapped (FPKM) using HOMER v4.10.1. To retrieve high confidence peaks, the peak file was filtered to retrieve peaks with a read count greater than 2.5 FPKM in both condition (e.g., DMSO-treated or GSK-LSD1-treated). Differential peaks were defined as GSK-LSD1-lost if the read counts in DMSO-treated samples were at least 2 times higher than in GSK-LSD1-treated samples. Identically, peaks were defined as GSK-LSD1 gained if the read counts in GSK-LSD1-treated samples were at least 2 times higher than in DMSO-treated samples. Heatmaps and average profile plots were generated by annotating the corresponding peaks sets using HOMER v4.10.1 and custom R 34 scripts. Analysis of transcription factor motifs at each peak set was performed using HOMER v4.10.1 and a fragment size of 200 bp, scaling sequence logos by information content. Gene ontology analyses of DMSO / GSKLSD1-shared LSD1 sites, GSK-LSD1-lost LSD1 sites and GSK-LSD1-gained LSD1 sites were performed using HOMER gene ontology annotation, using the default genome size (2e9) and the default genome background. SNAI2 ChIPseq reads were retrieve form GEO GSE55421 and processed as described above, filtering out peaks with a FPKM lower than 2.5. Overlap between sets of LSD1 peaks and SNAI2 peaks was assessed by merging peak files, using HOMER v4.10.1, allowing a maximum merging distance of 1000 base pairs. For H3K4me1 and H3K4me2 ChIP-seq analysis, broad peaks were called in each condition on pooled biological replicates using macs2 - v2.1.1.20160309 using a broad-cutoff of 0.05 and default parameters(Zhang et al., 2008). Differential peaks were identified using macs2 bdgdiff default parameters. The resulting peak file was merged using BEDtools v2.27.1, in order to stitch together differential peaks up to 5 kilobases (kb) apart. Genomic distribution, average profile plots, heatmaps and visualization tracks of H3K4me1 and H3K4me2 in DMSO-treated samples and in GSK-LSD1-treated samples as well as differential H3K4me1 and H3K4me2 regions were retrieved by annotating the corresponding sets of peaks using HOMER v4.10.1, as described above. Overlaps between GSK-LSD1-gained H3K4me1, GSK-LSD1 gained H3K4me2 regions and GSKLSD1-lost LSD1 sites were assessed on peak-associated genes by associating those peaks with the nearest gene’s transcriptional start site using HOMER v4.10.1 and a custom R script.
Transcription Factor Analyses
The list of human transcription factors representing 2,765 transcription factor-encoding genes was derived from (Lambert et al., 2018).
Growth Curve Measurements
210,000 epidermal progenitors were seeded on a 10 cm surface area on day 0. Cells were treated with DMSO or GSK-LSD1 (2 µM) the following day and retreated at the same time a day later. On day 3, cell number was measured with a Countess automated cell counter (Life Technologies) following standard procedures and default parameter settings. 210,000 cells were plated back into non-drug media. The following day, the drug regiment proceeded as before with cell number being counted every 2 days for a total of 12 days.
Immunoblotting
Cell were washed twice with PBS and lysed with RIPA buffer (Sigma, Cat# R0278) supplemented with Halt protease and phosphatase inhibitors (Thermo Fisher, Cat# 78440). Lysates were incubated at 4°C for 15 min, sheared with a 25 g needle, and then pelleted at 15 000 rpm for 10 min at 4°C. Supernatants were quantified using the Bradford Assay (Quick Start Bradford 1X Dye Reagent from Bio-Rad, Cat# 500–0205). Samples were separated by electrophoresis in 4%–20% SDS/PAGE gels with 20 µg per lane, transferred to PVDF membrane, and blotted with antibodies. Secondary horseradish peroxidase-conjugated secondary antibodies (Santa Cruz) and Amersham ECL Prime Western Blotting Detection Reagents (GE Healthcare, Cat# RPN2232) were used for detection. Antibodies used for immunoblotting include anti-GRHL3-C12, anti-NOTCH3-D11B8, anti-KLF4, anti-AP-2γ, anti-LSD1, and anti-Slug as described in the KRT.
Co-immunoprecipitation Experiments
Co-immunoprecipitations experiments were performed as previously described(Dou et al., 2015). Briefly, 30 µL of magnetic Protein G Dynabeads were washed twice in 1 mL BSA 0.5%, resuspended in 250 µL BSA 0.5% and conjugated for 1h to 2h at 4°C under rotation with 1 µg of antibodies against either LSD1, SNAI2, or IgG as a negative control. About 500,000 proliferating epidermal progenitors cells were harvested from a 10 cm culture plate at 50% confluence and lysed for 1h at 4°C under rotation in 250 µL of immunoprecipitation buffer (IP buffer: 20 mM Tris pH 7.5, 134 mM NaCl, 1 mM CaCl2, 1% NP-40, 10% 36 glycerol, supplemented with freshly made 1 mM MgCl2, 1:100 Halt protease and phosphatase inhibitor cocktail (Thermo Fisher, Cat# 78440) and benzonase at 12.5 U ml-1. Benzonase is critical for the efficient release of chromatin-bound proteins to the supernatant and MgCl2 is critical for its activity. Cell lysates were centrifuged at top speed for 10 minutes. 500 µg cell lysate were incubated overnight at 4°C with antibody conjugated magnetic beads previously washed 3 times with 1 mL BSA 0.5%. Immunoprecipitates were collected using a magnet, washed four to five times with IP buffer devoid of MgCl2, protease/phosphatase inhibitors and benzonase, then boiled with NuPage loading dye and analyzed by western blotting. Antibodies used for Co-IP include anti-LSD1, anti-Slug, and anti-IgG as described in the KRT.
Immunofluorescence
3D organotypic skin cultures were processed for histological examination by Core A of the Penn Skin Biology and Disease Resource-based Center. Tissue slides were exposed to xylene and ethanol and then treated with Targeting Unmasking Fluid (Pan Path, Cat# Z000R.0000) to deparaffinize the tissues. Sections were incubated in BlockAid Blocking Solution (Thermo Fisher, Cat# B10710) for 2 h at 37°C and then incubated O/N in primary antibody. Following secondary antibody incubation and washes, the sections were mounted with ProLong Gold with DAPI (Thermo Fisher, Cat# P36935). The slides were observed and representative images captured using a Keyence BZ-X700 Series All in One Fluorescent Microscope and a Nikon Eclipse microscope. Antibodies used for IF include anti-KRT14, anti-Collagen VII, anti-IVL, and anti-Filaggrin as described in the KRT. All IF figures appear at 20x magnification and are marked by scale bars equal to 50 µm.
Human Skin Organoid LSD1 Inhibitor Assays
Collagen Rafts
Human skin organotypics established on a collagen raft were treated with DMSO or GSK-LSD1 every 48 hours starting on day 0 and harvested after 7 days.
Devitalized Human Dermis
KGM media containing 4-hydroxytamoxifen (100 nM in 100% EtOH) and DMSO or GSK-LSD1 was replaced daily and organotypic tissue was harvested after 12 days.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental Design
The number of experimental replicates are detailed in the figure legends and represent independent biological experiments. Experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Statistical Analysis
All statistical tests for experiments and the corresponding P values are detailed in the figure legends. P values less than 0.05 are considered statistically significant and higher levels of significance are noted in the figures and figure legends. Statistical details for specific experiments are described in further detail below. For gene overlaps, statistical significance of overlaps between lists of genes or list of unique genes associated with ChIP-seq peaks were assessed using the R package GeneOverlap which relies on a Fisher’s exact test to calculate the statistical significance of the overlap. The union of genes or transcription factors expressed in proliferating epidermal progenitors upon GSK-LSD1 treatment or 2-PCPA-treatment was used as a background (14,104 genes and 2,134 transcription factors, respectively). Statistical analyses performed for all RNA-seq and ChIP-seq data are detailed in the method details. For RNA-seq data, genes were considered significantly up- or downregulated if the adjusted p value generated by DESeq2 for that gene was less than 0.05. Gene ontology analyses were either performed using PANTHER(Mi et al., 2019) or HOMER as stated in figure legends using the indicated background genes as a reference list. PANTHER gene ontology overrepresentation tests rely on the use of a Fisher’s exact test together with a Benjamini-Hochberg False Discovery Rate (FDR) correction. HOMER gene ontology analyses were performed using HOMER’s annotatePeaks.pl script. Enrichment of gene ontology terms is calculated assuming a cumulative hypergeometric distribution. Genomic annotation of LSD1 and H3K4me1/me2 binding sites and their corresponding enrichment (log2) were calculated using HOMER annotatePeaks.pl scripts. Transcription factor binding motifs at LSD1 binding sites and their respective statistical significance were computed using HOMER findMotifsGenome.pl script which uses ZOOPS scoring (zero or one occurrence per sequence) coupled with the hypergeometric enrichment calculations (or binomial) to determine motif enrichment. For transcriptional correlation plots, P values were retrieved from TCGA. For epidermal area and protrusion quantification, four representative images (as seen in Figure 4D) of each biological replicate for each condition (DMSO versus LSD1 inhibitor treatments) were used to determine the average epidermal area or number of protrusions of an individual sample. These values were utilized in a one-way ANOVA and a post hoc Tukey’s multiple comparisons test to determine significance between groups using GraphPad Prism 7.04. Epidermal area was determined using ImageJ(Schneider et al., 2012). For the growth curve measurement assay, a 2-tailed Student’s t test was used to determine significance using GraphPad Prism 7.04.
DATA AND CODE AVAILABILITY
The accession number for all data is GEO: GSE133766. The accession number for the ChIP-seq data reported in this paper is GEO: GSE133560. The accession number for the two RNA-seq datasets reported in this paper are GEO: GSE133737 and GSE133738. Cutsom R scripts used in this study are available upon request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Notch3 (D11B8) | Cell Signaling | Cat# 5276 RRID: AB_10560515 |
| Goat polyclonal anti-Klf4 | R&D | Cat# AF3640. RRID: AB_2130224 |
| Rabbit polyclonal anti-AP-2γ | Cell Signaling | Cat# 2320. RRID: AB_2202287 |
| Mouse monoclonal anti-Grhl3 (C12) | Santa Cruz | Cat# sc398838. RRID: N/A |
| Rabbit polyclonal anti-Kdm1a | Abcam | Cat# ab17721. RRID: AB_443964 |
| Rabbit polyclonal anti-Histone 3 (monomethyl K4) | Abcam | Cat# ab8895. RRID: AB_306847 |
| Rabbit polyclonal anti-Histone 3 (dimethyl K4) | Abcam | Cat# ab7766. RRID: ABB_2560996 |
| Mouse monoclonal anti-Keratin 14 | Abcam | Cat# ab7800. RRID: N/A |
| Rabbit polyclonal anti-Collagen VII | Millipore | Cat# 234192. RRID: AB_211739 |
| Mouse monoclonal anti-Involucrin (SY5) | Abcam | Cat# ab68. RRID: AB_305656 |
| Rabbit polyclonal anti-Filaggrin | Abcam | Cat# ab81468. RRRID: AB_1640512 |
| Rabbit polyclonal anti-IgG | Abcam | Cat# ab46540. RRID: AB_2614925 |
| Rabbit monoclonal anti-Slug (C19G7) | Cell Signaling | Cat# 9585. RRID: AB_2239535 |
| Rabbit monoclonal anti-CDK4 | Cell Signaling | Cat# 12790 |
| Rabbit monoclonal anti-p-ERK | Cell Signaling | Cat# 4370P |
| Bacterial and Virus Strains | ||
| Retrovirus: LZRS-ER-H-RAS G12V | Duperret et al., 2015; PMID 26359297 | N/A |
| Retrovirus: LZRS-Cdk4 R24C | Duperret et al., 2015; PMID 26359297 | N/A |
| Biological Samples | ||
| Primary epidermal progenitors (neonatal human epidermal keratinocytes, NHEKs) | Penn Skin Biology and Diseases Resources-based Center (SBDRC) | https://dermatology.upenn.edu/sbdrc/ |
| NHEKs transfected with CDK4 R24C and ER-H-RAS G12V expression | Penn Skin Biology and Diseases Resources-based Center (SBDRC) and Duperret et al., 2015; PMID 26359297 | N/A |
| Primary Normal Adult Human Dermis | Penn Skin Biology and Diseases Resources-based Center (SBDRC) | https://dermatology.upenn.edu/sbdrc/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| LSD1 inhibitor; GSK-LSD1 | Cayman Chemical | Cat# 16439 |
| LSD1 inhibitor; 2-PCPA | BPS Bioscience | Cat# 27305 |
| 4-hydroxytamoxifen | Sigma-Aldrich | Cat# H7904–5MG |
| Critical Commercial Assays | ||
| RNeasy Kit | QIAGEN | Cat# 74106 |
| NEBNext Poly(A) mRNA magnetic isolation module | New England Biolabs | Cat# E7490S |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7420L |
| NEBNext Oligos for Illumina (Index Primer Set 1) | New England Biolabs | Cat# E7335L |
| NEBNext Oligos for Illumina (Index Primer Set 2) | New England Biolabs | Cat# E7500S |
| NEBNext Library Quantification Kit for Illumina | New England Biolabs | Cat# E7630L |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645L |
| High Sensitivity DNA Chips | Agilent Technologies | Cat# 5067–4626 |
| Deposited Data | ||
| All datatsets form this study | This study | GEO: GSE133766 |
| RNA-seq NHEK LSD1 inhibitors | This study | GEO: GSE133737 |
| RNA-seq NHEK Differentiation versus Proliferation | This study | GEO: GSE133738 |
| ChIP-seq NHEK LSD1 | This study | GEO: GSE133560 |
| Experimental Models: Cell Lines | ||
| J2 3T3 Fibroblasts | Kerafast | Cat# EF3003 |
| Phoenix cells-AMPHO | ATCC | Cat# CRL-3213 |
| Oligonucleotides | ||
| LSD1 targeted siRNA: SMARTpool: Accell KDM1A siRNA | Dharmacon | Cat# E-009223–00-0005 |
| Non-targeting control siRNA: Accell Non-Targeting Control siRNA | Dharmacon | Cat# D-001910–01-20 |
| Recombinant DNA | ||
| ER-H-RAS G12V | Duperret et al., 2015; PMID 26359297 | N/A |
| Cdk4 R24C | Duperret et al., 2015; PMID 26359297 | N/A |
| Software and Algorithms | ||
| STAR | Dobin et al., 2013; PMID 23104886 | https://doi.org/10.1093/bioinformatics/bts635 |
| DESeq2 | Love et al., 2014; PMID 25516281 | https://doi.org/10.1186/s13059–014-0550–8 |
| Bowtie2 V2–1.0 | PMID 22388286 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| HOMER v4.10.1 | Heinz et al., 2010; PMID 20513432 | https://doi.org/10.1016/j.molcel.2010.05.004 |
| http://homer.ucsd.edu/homer/ | ||
| BEDtools v2.27.1 | Quinlan and Hall 2010; PMID 20110278. | https://bedtools.readthedocs.io/en/latest/# |
| Quinlan, 2014; PMID 25199790 | https://doi.org/10.1093/bioinformatics/btq033 | |
| https://doi.org/10.1002/0471250953.bi1112s47 | ||
| macs2 v2.1.1.20160309 | Zhang et al., 2008; PMID 18798982 | https://github.com/taoliu/MACS |
| R version 3.5.0 | https://www.r-project.org/ | |
| R Studio version 1.1.383 | https://www.rstudio.com/ | |
| ImageJ | Schneider et al., 2012; PMID 22930834 | https://imagej.nih.gov/ij/ |
| NextSeq500 | https://www.illumina.com/systems/sequeseque-platforms/nextseq.html | |
| Illumina BaseSpace | https://www.illumina.com/products/by-type/informatics-products/basespace-sequence-hub.html | |
| UCSC Genome Brower | Kent et al., 2002; PMID 12045153 | https://genome.ucsc.edu |
| PANTHER | Mi et al., 2019; PMID 30804569 | https://doi.org/10.1093/nar/gky1038 |
| http://www.pantherdb.org | ||
| Other | ||
| Keratinocyte-SFM (1X) | GIBCO | Cat# 10724–011 |
| Medium 154 | GIBCO | Cat# M-154–500 |
| Human Keratinocyte Growth Supplement (100X) | GIBCO | Cat# S-001–5 |
| Supplements For Keratinocytes –SFM | GIBCO | Cat# 37000–015 |
| Protein G Dynabeads | Thermo Fisher | Cat# 10004D |
| Benzonase Nuclease | Sigma | Cat# E1014–5KU |
| DMEM high glucose 4.5 g/L | Mediatech | Cat# MT10–013-CV |
| Ham’s F-12 | Lonza | Cat# 12–615F |
| Fetal Bovine Serum | Life Technologies | Cat# 16000–044 |
| Adenine hydrochloride hydrate | Sigma-Aldrich | Cat# A9795–5G |
| Hydrocortisone | Sigma-Aldrich | Cat# H4001–1G |
| Insulin from bovine pancreas | Sigma-Aldrich | Cat# I1882–100MG |
| Cholera Toxin from Vibrio cholerae | Sigma-Aldrich | Cat# C8052–0.5MG |
| EGF Human Recombinant | Invitrogen | Cat# PHG0311L |
| 3,3ʹ,5 Triido-L-thyronine | Sigma-Aldrich | Cat# T2877–100MG |
| Transferrin human | Sigma-Aldrich | T8158–100MG |
| Matrigel Matrix | Corning | Cat# 354234 |
Highlights.
LSD1 represses master epidermal transcription factors that promote differentiation
LSD1 inhibition activates the epidermal differentiation transcriptional program
LSD1 inhibition represses invasion in a model of cutaneous squamous cell carcinoma
ACKNOWLEDGMENTS
Research here was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) (grant K08AR070289), the Damon Runyon Cancer Foundation, the Dermatology Foundation, and the Penn-Wistar SPORE in Skin Cancer (funded by NCI grant P50CA174523), all to B.C.C., as well as the Penn Skin Biology and Diseases Resource-based Center (NIAMS grant 1P30AR069589–01). C.L.S. was supported by the Dermatology Foundation and the National Psoriasis Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.058.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alsaqer SF, Tashkandi MM, Kartha VK, Yang YT, Alkheriji Y, Salama A, Varelas X, Kukuruzinska M, Monti S, and Bais MV (2017). Inhibition of LSD1 epigenetically attenuates oral cancer growth and metastasis. Oncotarget 8, 73372–73386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlasi Y, and Stunnenberg HG (2017). The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet 18, 643–658. [DOI] [PubMed] [Google Scholar]
- Avgustinova A, and Benitah SA (2016). Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol 17, 643–658. [DOI] [PubMed] [Google Scholar]
- Beckenbach L, Baron JM, Merk HF, Löffler H, and Amann PM (2015). Retinoid treatment of skin diseases. Eur. J. Dermatol 25, 384–391. [DOI] [PubMed] [Google Scholar]
- Boxer LD, Barajas B, Tao S, Zhang J, and Khavari PA (2014). ZNF750 interacts with KLF4 and RCOR1, KDM1A, and CTBP1/2 chromatin regulators to repress epidermal progenitor genes and induce differentiation genes. Genes Dev 28, 2013–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Yau C, Bowlby R, Liu Y, Brennan K, Fan H, Taylor AM, Wang C, Walter V, Akbani R, et al. (2018). Genomic, pathway network, and immunologic features distinguishing squamous carcinomas. Cell Rep 23, 194–212.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2015). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network; Analysis Working Group: Asan University; BC Cancer Agency; Brigham and Women’s Hospital; Broad Institute; Brown University; Case Western Reserve University; Dana-Farber Cancer Institute; Duke University; Greater Poland Cancer Centre; Harvard Medical School; Institute for Systems Biology; KU Leuven; Mayo Clinic; Memorial Sloan Kettering Cancer Center; National Cancer Institute; Nationwide Children’s Hospital; Stanford University; University of Alabama; University of Michigan; University of North Carolina; University of Pittsburgh; University of Rochester; University of Southern California; University of Texas MD Anderson Cancer Center; University of Washington; Van Andel Research Institute; Vanderbilt University; Washington University; Genome Sequencing Center: Broad Institute; Washington University in St. Louis; Genome Characterization Centers: BC Cancer Agency; Broad Institute; Harvard Medical School; Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University; University of North Carolina; University of Southern California Epigenome Center; University of Texas MD Anderson Cancer Center; Van Andel Research Institute; Genome Data Analysis Centers: Broad Institute; Brown University; Harvard Medical School; Institute for Systems Biology; Memorial Sloan Kettering Cancer Center; University of California Santa Cruz; University of Texas MD Anderson Cancer Center; Biospecimen Core Resource: International Genomics Consortium; Research Institute at Nationwide Children’s Hospital; Tissue Source Sites: Analytic Biologic Services; Asan Medical Center; Asterand Bioscience; Barretos Cancer Hospital; BioreclamationIVT; Botkin Municipal Clinic; Chonnam National University Medical School; Christiana Care Health System; Cureline; Duke University; Emory University; Erasmus University; Indiana University School of Medicine; Institute of Oncology of Moldova; International Genomics Consortium; Invidumed; Israelitisches Krankenhaus Hamburg; Keimyung University School of Medicine; Memorial Sloan Kettering Cancer Center; National Cancer Center Goyang; Ontario Tumour Bank; Peter MacCallum Cancer Centre; Pusan National University Medical School; Ribeiraõ Preto Medical School; St. Joseph’s Hospital &Medical Center; St. Petersburg Academic University; Tayside Tissue Bank; University of Dundee; University of Kansas Medical Center; University of Michigan; University of North Carolina at Chapel Hill; University of Pittsburgh School of Medicine; University of Texas MD Anderson Cancer Center; Disease Working Group: Duke University; Memorial Sloan Kettering Cancer Center; National Cancer Institute; University of Texas MD Anderson Cancer Center; Yonsei University College of Medicine; Data Coordination Center: CSRA Inc.; Project Team: National Institutes of Health (2017). Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Collings CK, Morgan MA, Marshall SA, Rendleman EJ, Ozark PA, Smith ER, and Shilatifard A (2018). An Mll4/COMPASS-Lsd1 epigenetic axis governs enhancer function and pluripotency transition in embryonic stem cells. Sci. Adv 4, eaap8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS, et al. (2016). MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev 30, 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, and Yang J (2011). Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res 71, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitsazzadeh V, Coarfa C, Drummond JA, Nguyen T, Joseph A, Chilukuri S, Charpiot E, Adelmann CH, Ching G, Nguyen TN, et al. (2016). Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun 7, 12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Jang H, Kim H, Kim ST, Cho EJ, and Youn HD (2010). Histone demethylase LSD1 is required to induce skeletal muscle differentiation by regulating myogenic factors. Biochem. Biophys. Res. Commun 401, 327–332. [DOI] [PubMed] [Google Scholar]
- Ding J, Zhang ZM, Xia Y, Liao GQ, Pan Y, Liu S, Zhang Y, and Yan ZS (2013). LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. Br. J. Cancer 109, 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotto GP, and Rustgi AK (2016). Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 29, 622–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duperret EK, Dahal A, and Ridky TW (2015). Focal-adhesion-independent integrin-av regulation of FAK and c-Myc is necessary for 3D skin formation and tumor invasion. J. Cell Sci 128, 3997–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert RL, Adhikary G, Young CA, Jans R, Crish JF, Xu W, and Rorke EA (2013). AP1 transcription factors in epidermal differentiation and skin cancer. J. Skin Cancer 2013, 537028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein EH (2008). Basal cell carcinomas: attack of the hedgehog. Nat. Rev. Cancer 8, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari-Amorotti G, Fragliasso V, Esteki R, Prudente Z, Soliera AR, Cattelani S, Manzotti G, Grisendi G, Dominici M, Pieraccioli M, et al. (2013). Inhibiting interactions of lysine demethylase LSD1 with snail/slug blocks cancer cell invasion. Cancer Res 73, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Gaskell E, and Bernstein BE (2017). Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, and Tsukita S (2002). Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J. Cell Biol 156, 1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales KAU, and Fuchs E (2017). Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 43, 387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini A, and Minucci S (2017). A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 9, 1123–1142. [DOI] [PubMed] [Google Scholar]
- Hu Q, Kwon YS, Nunez E, Cardamone MD, Hutt KR, Ohgi KA, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, and Fu XD (2008). Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc. Natl. Acad. Sci. USA 105, 19199–19204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, and Berger SL (2007). p53 is regulated by the lysine demethylase LSD1. Nature 449, 105–108. [DOI] [PubMed] [Google Scholar]
- Jin X, Jin X, and Kim H (2017). Cancer stem cells and differentiation therapy. Tumour Biol 39, 1010428317729933. [DOI] [PubMed] [Google Scholar]
- Kelly AD, and Issa JJ (2017). The promise of epigenetic therapy: reprogramming the cancer epigenome. Curr. Opin. Genet. Dev 42, 68–77. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MN, Suzuki T, and Miyata N (2013). An overview of phenylcyclopropylamine derivatives: biochemical and biological significance and recent developments. Med. Res. Rev 33, 873–910. [DOI] [PubMed] [Google Scholar]
- Klein RH, Lin Z, Hopkin AS, Gordon W, Tsoi LC, Liang Y, Gudjonsson JE, and Andersen B (2017). GRHL3 binding and enhancers rearrange as epidermal keratinocytes transition between functional states. PLoS Genet 13, e1006745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The Human Transcription Factors. Cell 172, 650–665. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q, Fang M, Tao S, Green CL, and Khavari PA (2002). CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat. Med 8, 1105–1114. [DOI] [PubMed] [Google Scholar]
- Lee B, Villarreal-Ponce A, Fallahi M, Ovadia J, Sun P, Yu QC, Ito S, Sinha S, Nie Q, and Dai X (2014). Transcriptional mechanisms link epithelial plasticity to adhesion and differentiation of epidermal progenitor cells. Dev. Cell 29, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Park JH, Choi HJ, Won HY, Joo HS, Shin DH, Park MK, Han B, Kim KP, Lee TJ, et al. (2017). LSD1 demethylates HIF1a to inhibit hydroxylation and ubiquitin-mediated degradation in tumor angiogenesis. Oncogene 36, 5512–5521. [DOI] [PubMed] [Google Scholar]
- Li X, Li T, Chen D, Zhang P, Song Y, Zhu H, Xiao Y, and Xing Y (2016). Overexpression of lysine-specific demethylase 1 promotes androgen-independent transition of human prostate cancer LNCaP cells through activation of the AR signaling pathway and suppression of the p53 signaling pathway. Oncol. Rep 35, 584–592. [DOI] [PubMed] [Google Scholar]
- Lim S, Janzer A, Becker A, Zimmer A, Schu€le R, Buettner R, and Kirfel J (2010). Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31, 512–520. [DOI] [PubMed] [Google Scholar]
- Lim HW, Collins SAB, Resneck JS Jr., Bolognia JL, Hodge JA, Rohrer TA, Van Beek MJ, Margolis DJ, Sober AJ, Weinstock MA, et al. (2017). The burden of skin disease in the United States. J. Am. Acad. Dermatol 76, 958–972.e2. [DOI] [PubMed] [Google Scholar]
- Lin-Shiao E, Lan Y, Coradin M, Anderson A, Donahue G, Simpson CL, Sen P, Saffie R, Busino L, Garcia BA, et al. (2018). KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev 32, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv T, Yuan D, Miao X, Lv Y, Zhan P, Shen X, and Song Y (2012). Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 7, e35065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiques-Diaz A, Spencer GJ, Lynch JT, Ciceri F, Williams EL, Amaral FMR, Wiseman DH, Harris WJ, Li Y, Sahoo S, et al. (2018). Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep 22, 3641–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, and Schüle R (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439. [DOI] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, and Thomas PD (2019). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc 14, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, Chung CH, Hernandez-Aya L, Lim AM, Chang ALS, et al. (2018). PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med 379, 341–351. [DOI] [PubMed] [Google Scholar]
- Mistry DS, Chen Y, Wang Y, Zhang K, and Sen GL (2014). SNAI2 controls the undifferentiated state of human epidermal progenitor cells. Stem Cells 32, 3209–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyai M, Hamada M, Moriguchi T, Hiruma J, Kamitani-Kawamoto A, Watanabe H, Hara-Chikuma M, Takahashi K, Takahashi S, and Kataoka J (2016). Transcription Factor MafB Coordinates Epidermal Keratinocyte Differentiation. J. Invest. Dermatol 136, 1848–1857. [DOI] [PubMed] [Google Scholar]
- Mlacki M, Darido C, Jane SM, and Wilanowski T (2014). Loss of Grainy head-like 1 is associated with disruption of the epidermal barrier and squamous cell carcinoma of the skin. PLoS ONE 9, e89247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musri MM, Carmona MC, Hanzu FA, Kaliman P, Gomis R, and Párrizas M (2010). Histone demethylase LSD1 regulates adipogenesis. J. Biol. Chem 285, 30034–30041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehal KS, and Bichakjian CK (2018). Update on Keratinocyte Carcinomas. N. Engl. J. Med 379, 363–374. [DOI] [PubMed] [Google Scholar]
- Nicholson TB, and Chen T (2009). LSD1 demethylates histone and non-histone proteins. Epigenetics 4, 129–132. [DOI] [PubMed] [Google Scholar]
- Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H, Kalman RA, Nakagawa M, Darling DS, Basu D, et al. (2011). A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res 71, 6836–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, Tsai KY, Curry JL, Tetzlaff MT, Lai SY, et al. (2014). Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res 20, 6582–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR (2014). BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinformatics 47, 11.12.1–11.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridky TW, Chow JM, Wong DJ, and Khavari PA (2010). Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat. Med 16, 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen GL, Boxer LD, Webster DE, Bussat RT, Qu K, Zarnegar BJ, Johnston D, Siprashvili Z, and Khavari PA (2012). ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev. Cell 22, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevilla LM, Nachat R, Groot KR, Klement JF, Uitto J, Djian P, Määttä A, and Watt FM (2007). Mice deficient in involucrin, envoplakin, and periplakin have a defective epidermal barrier. J. Cell Biol 179, 1599–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, and Laird PW (2013). Interplay between the cancer genome and epigenome. Cell 153, 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu PH, et al. (2018). LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 174, 549–563.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, and Shi Y (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. [DOI] [PubMed] [Google Scholar]
- Shortt J, Ott CJ, Johnstone RW, and Bradner JE (2017). A chemical probe toolbox for dissecting the cancer epigenome. Nat. Rev. Cancer 17, 160–183. [DOI] [PubMed] [Google Scholar]
- Simpson CL, Kojima S, and Getsios S (2010). RNA interference in keratinocytes and an organotypic model of human epidermis. Methods Mol. Biol 585, 127–146. [DOI] [PubMed] [Google Scholar]
- Thambyrajah R, Mazan M, Patel R, Moignard V, Stefanska M, Marino-poulou E, Li Y, Lancrin C, Clapes T, Möröy T, et al. (2016). GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol 18, 21–32. [DOI] [PubMed] [Google Scholar]
- Toufighi K, Yang JS, Luis NM, Aznar Benitah S, Lehner B, Serrano L, and Kiel C (2015). Dissecting the calcium-induced differentiation of human primary keratinocytes stem cells by integrative and structural network analyses. PLoS Comput. Biol 11, e1004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, et al. (2009). The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet 41, 125–129. [DOI] [PubMed] [Google Scholar]
- Watt FM, Estrach S, and Ambler CA (2008). Epidermal Notch signalling: differentiation, cancer and adhesion. Curr. Opin. Cell Biol 20, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, Li Z, Qi B, Zhang W, Cheng J, and Wang Y (2015). High expression of the histone demethylase LSD1 associates with cancer cell proliferation and unfavorable prognosis in tongue cancer. J. Oral Pathol. Med 44, 159–165. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YC, Ma J, Wang Z, Li J, Jiang B, Zhou W, Shi X, Wang X, Zhao W, and Liu HM (2015). A Systematic Review of Histone Lysine-Specific Demethylase 1 and Its Inhibitors. Med. Res. Rev 35, 1032–1071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for all data is GEO: GSE133766. The accession number for the ChIP-seq data reported in this paper is GEO: GSE133560. The accession number for the two RNA-seq datasets reported in this paper are GEO: GSE133737 and GSE133738. Cutsom R scripts used in this study are available upon request.