

Abstract
In this work, quantum mechanical methods were used to predict the microscopic and macroscopic pKa values for a set of 24 molecules as a part of the SAMPL6 blind challenge. The SMD solvation model was employed with M06–2X and different basis sets to evaluate three pKa calculation schemes (direct, vertical, and adiabatic). The adiabatic scheme is the most accurate approach (RMSE=1.40 pKa units) and has high correlation (R2=0.93), with respect to experiment. This approach can be improved by applying a linear correction to yield and RMSE of 0.73 pKa units. Additionally, we consider including explicit solvent representation and multiple lower-energy conformations to improve the predictions for outliers. Adding three water molecules explicitly can reduce the error by 2 to 4 pKa units, with respect to experiment, whereas including multiple local minima conformations does not necessarily improve the pKa prediction.
Keywords: SAMPL6, pKa, Implicit solvent, Quantum chemistry
Introduction
The use of in silico modelling in rational design has become a popular and valuable tool in current research and development for agricultural, environmental, and pharmaceutical applications, as a multifaceted technique capable of providing rapid understanding to in situ phenomena that may be difficult to measure or study[1]. Computer-aided modeling is advantageous for forecasting how a molecule may react in different environments and is heavily utilized for virtual screening and lead optimization in drug discovery as a provisional method for physicochemical and biophysical characterization, including solubility, ionization, lipophilicity, etc. While there are many computational high-throughput models for predicting physicochemical properties, challenges persist for predictions of how molecules ionize in solution. The acid dissociation constant (Ka) or its corresponding logarithmic constant (pKa), is a quantitative measure of the strength of an acid in solution in the context of acid-base reactions related to the free energy () of an acid losing a proton.
| (1) |
Many methods for predicting pKa have been designed, spanning across electronic structure theory, molecular mechanics, and machine learning approaches[2–4]. Popular QSAR-style methods have been implemented in software packages, such as ADMET Predictor (S+pKa method[5]), Epik[6], pKa Prospector[7], and ACD/pKa Percepta Platform[8]. While these empirical methods can provide instantaneous predictions, inaccuracies arise for large and flexible molecules in which steric effects and microstate conformations surpass the Hammett-Taft approach[9].
A variety of semi-empirical and quantum chemical approaches have been developed—varying by not only the level of theory, but also by the solvation model and the reaction scheme[10–13]. For semi-empirical approaches, Jensen et al. considered several combinations of semi-empirical methods and implicit solvation models to predict the pKa of 48 druglike molecules using a relative scheme [14]. From the evaluation of 6 semi-empirical methods, the AM1 and PM3 methods provided predictions within 1.4–1.6 pH units. Another study comparing the semi-empirical approaches with ab initio methods for predicting pKa values for a set of molecules containing a variety of ionizable groups, including alcohols and carboxylic acids, shows that PM6-based methods can provide predictions close to the accuracy of CBS-4B3/SMD [15].
Various ab initio methods, including CBS[16], Gaussian-n[17–20] and ccCA[21], have been applied with continuum solvation models to predict pKa values and are reported to predict pKa values as low as 0.5 pKa units from experiment, however, most of these approaches have only been employed on small molecule datasets [14, 15, 22–26]. Although wavefunction-based methods and composite ab initio methods provide high levels of accuracy, for larger molecules they are less attractive due to the computational expense, hence the interest in exploiting more approximate methods, such as electronic density-based approaches.
Density functional theory (DFT) methods are popular as they have been applied to an array of chemical applications, achieving desired accuracies for a broad range of gas phase reactions and properties [27, 28]. There are many DFT functionals and extensive assessments which illustrate that different functionals perform better for specific properties[29]. For calculations in the solution phase, DFT functionals are often used with implicit continuum models, such as CPCM[30], COSMO[31], and SMD[32] models, which are optimized for usage with modest levels of theory (smaller basis sets). Several studies employing hybrid functionals—including B3LYP, B97–1, BMK, B98, M06, and M06–2X—with the SMD model have shown that the M06–2X functional provides more accurate predictions than other functionals considered for main group element calculations, which would be expected as the SMD model was parametrized using M05–2X[26, 33]. The combination of M06–2X density functional and SMD model has been used in a recent pKa study[15], which yielded mean unsigned error of 0.9, 0.4, and 0.5 pKa units respectively for carboxylic acids, aliphatic amines, and thiols. While different groups are evaluating their methods on different datasets, it is difficult to compare the various approaches. SAMPL blind challenges provide a unique platform for designing novel approaches and assessing current methods. The need for appropriate methods for predicting of pKa was highlighted in the previous SAMPL5 challenge for predicting partition coefficients, as the ionization and tautomerization states differed in the cyclohexane and water phase. The SAMPL6 pKa challenge entails the prediction of microscopic and macroscopic pKa values divided into three sub-challenges: 1) the prediction of microscopic pKa values of associated microstates; 2) the prediction of microstate population as a function of pH ranging from 2 to 12; and 3) the macroscopic pKa. The dataset is composed of 24 drug-like fragments, each containing multiple ionization and tautomeric states.
In this work for the SAMPL6 challenge, we explored several unique approaches to predict microscopic and macroscopic pKa values. Absolute pKa values were predicted using three different calculation schemes: the direct scheme, the vertical scheme, and the adiabatic scheme. We consider multiple tactics in efforts to achieve more accurate predictions. For each scheme, we tried to improve the accuracy by (1) single point energy corrections utilizing larger basis sets; (2) including multiple conformations per microstate in the pKa calculation; (3) including explicit water molecules to stabilize neutral and charged microstates; (4) applying a linear correction to the calculated pKa values.
Methods
A source of error in pKa calculations arises from the reaction scheme used to approximate the solution phase free energy (ΔGaq). For a generic acid (HA) in water, the equilibrium of acid dissociation reaction (Ka) can be written symbolically as
| (2) |
which expresses the proton transfer from the acid to yield its conjugate base (A-) and hydronium (H3O+). For this expression, the direct thermodynamic cycle (Figure 1) is used for calculating absolute pKa values. In concentrated aqueous solutions, the expression can be simplified to the dissociation of an acid into its conjugate base (Cycle B). Previous studies comparing thermodynamic cycles with continuum solvation models highlight that the simplified expression, Cycle B, tends to be more accurate than Cycle A[24]. In Cycle A, the solution phase free energy is computed using the gas phase () and solvation free energies (). The solvation free energy of the proton, , used is −265.9 kcal/mol[34] includes the standard state correction from 1 atm to 1 M. The proton gas phase free energy ( kcal/mol) comes from the Sackur-Tetrode equation[35].
Figure 1.
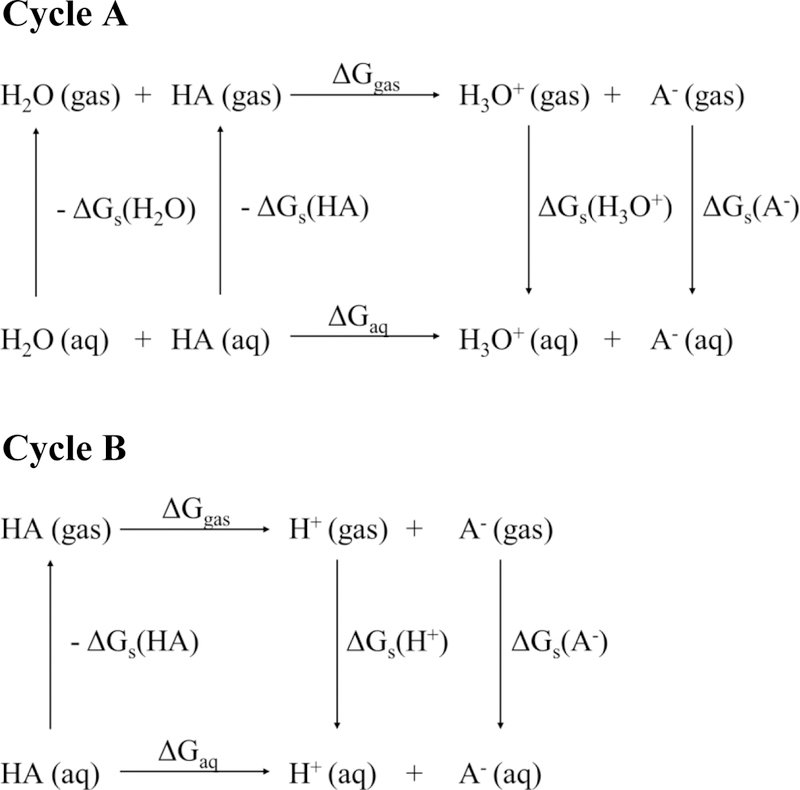
Thermodynamic cycles used for pKa calculation schemes.
| (3a) |
| (3b) |
| (3c) |
Here, we use a superscript “°” to denote the condition of 1 atm, and “*” to denote the condition of 1 M.
Calculation Schemes
In this challenge, three different schemes are used to compute the free energy foreach microstate pair. The notations and correspond to stationary points obtained from gas phase and solution phase optimizations, respectively[36].
Scheme D: Direct scheme
The direct scheme (noted Scheme D) determines the solution phase free energy without use of thermodynamic cycle.
| (4) |
In this scheme, the reaction free energy is determined by solution phase geometries. Thermal corrections to the free energy are added to the total energy to approximate . To note, all energy terms of the direct scheme are computed within the implicit solvent model. The approximation made in the direct scheme is that gas phase contributions are not needed, i.e. geometries.
In contrast, the vertical scheme (Scheme V) uses the gas phase geometry and assumes that free energy of the solute relaxing in solution phase is negligible.
Scheme V: Vertical scheme
| (5a) |
| (5b) |
In this expression, is calculated using the gas phase free energy and the solvation free energy (), which is the difference between the gas phase and solution phase total energies. Here, is determined by employing the continuum solvation approach on the gas phase structure. Thermal corrections to the gas phase free energy are used in this representation, as it is assumed that the thermal contributions in both phases are similar.
Scheme A: Adiabatic scheme
The adiabatic scheme (Scheme A) considers both the gas and solution phase geometries.
| (6a) |
| (6b) |
This scheme differs from the vertical scheme by the total energy contributions from the solute relaxed in solution, hence is determined by optimizing the molecule in solution phase. The difference between the thermal contributions in gas phase and solution phase (relaxed) can be approximated by the difference in the adiabatic and direct scheme ()
Conventionally, thermodynamic cycle is used to calculate the solution phase free energy when using continuum solvation models. The primary reason is that continuum solvation models are generally parameterized to produce accurate solvation free energies using lower levels of theory (HF or DFT with double- quality basis sets); however, by using thermodynamic cycle the solution phase free energy can be determined at different levels of theory.
Inspired by the work of J. Ho[37], we consider modifications of each scheme in hopes to obtain more accurate energetics by including single point energy corrections (augmented by “”) using larger basis sets (denoted by a superscript, ). In the D+S Scheme, the total energy term in aqueous solution is replaced with the total energy obtained with a larger basis set.
| (7) |
For the vertical and adiabatic schemes, the solvation free energies () are calculated with larger basis sets,
| (8a) |
| (8b) |
As both approaches use thermodynamic cycle, the V+S scheme and A+S schemes differ by the geometry in which the aqueous phase total energies are determined.
| (9) |
Microstate populations as a function of pH
To predict the fractional microstate populations at different pH values, we consider the following reactions from n charged microstate to m charged microstate (m < n),
while the equilibrium constant is
| (10a) |
and
| (10b) |
Therefore,
| (11) |
After applying the variable separation approach, we arrive at
| (12) |
is the partition function at specified pH value and defined as
| (13) |
Therefore, the partition function for microstate A with charge nA is
| (14) |
Note that this partition function also holds when nA < 0.
The population for microstate A with charge nA is obtained as
| (15) |
Macroscopic pKa values
To compute the macroscopic pKa values, we can use the conclusion from the derivation of the microstate population, so we have
| (16) |
Here is the Kronecker delta and then the macroscopic between the microstates with a charge of n+1 and the microstates with a charge of n is
| (17) |
QM Calculations
The initial structures of the 352 microstates were generated from the SMILES strings provided by the SAMPL6 pKa challenge using Open Babel 2.4.1[38]. Gas phase and solution phase geometry optimizations were performed using the M06–2X density functional[39]. As charged and uncharged species are represented in the molecule set, the 6–31G(d) basis set[32] are used for cationic species whereas additional diffuse functions (6–31+G(d)[40]) are included for the anionic microstates. All QM optimizations were performed with “tight” wave function and geometry convergence criteria, by using an “ultrafine” numerical quadrature as required by M06–2X functional.
To maintain consistency of the basis sets between microstate reaction pairs, duplicate calculations are carried out for neutral species using each basis set (Table 2).
Table 2.
Basis sets selection per sub-challenge.
| 6–31+G(d)/6–311++G(d,p) | 6–31G(d)/6–311G(d,p) | |
|---|---|---|
| Type I | Anionic species in the pair | Cationic species in the pair |
| Type II | ||
| Type III | Between anions or anion/neutral | Between cations or cation/neutral |
Frequencies were examined to confirm stationary points and scaled by 0.9465 and 0.9500 for methods using the 6–31G(d) and 6–31+G(d) basis sets, respectively[41]. Additional single point energy calculations for each microstate are performed using M06–2X in conjunction with 6–311G(d,p) and 6–311++G(d,p) to serve as corrections to the respective double- basis sets. Solution phase geometry optimizations and single point calculations were carried out using the SMD implicit solvation model[42]. All calculations were performed in Gaussian 16 (Rev. A.03) [43] using an Ultrafine integration grid. To improve conformational sampling, two different algorithms were considered. Per microstate, ten low-energy conformers were stochastically and systematically generated using the MOE software and compared against the optimized structures of each microstate. For microstates in which there was a large difference in the conformation, the new conformers were subjected to the aforementioned workflow.
Results & Discussion
Our method of using two basis sets is similar to the method using mixed basis set where the diffuse functions are added at the reactive center to allow improved modeling of negative ions [44]. We do not adopt using mixed basis sets because the excess electron is assumed to be delocalized over the entire molecule instead of the deprotonated atom.
Errors in pKa calculations arise from the reaction scheme in which the aqueous free energy is approximated. In this challenge, we considered several approaches for predicting absolute pKa values that differ by how free energy contributions in gas phase and solution phase are determined. Our submissions for Type I, Type II, and Type III predictions, per scheme, are listed in Table 1. To note, the calculated pKa values are reported without standard error of the mean (SEM).
Table 1.
SAMPL6 submission IDs for our approaches.
| Scheme | D | D+S | V | A | A+S |
|---|---|---|---|---|---|
| Type I | wexjs | w4z0e | arcko | wcvnu | ko8yx |
| Type II | t28dq | z0ima | yzx8f | gt0oq | i5m8f |
| Type III | y75vj | xikp8 | 5byn6 | w4iyd | ryzue |
Direct Scheme
In the direct scheme, the aqueous free energy is determined only by solution phase calculations, avoiding thermodynamic cycle. This is an attractive approach as it requires only two calculations (of each microstate pair) and would already account for solvent-induced effects since the geometries are optimized in the solution phase. From the results shown in Table 2, overall, the direct approach predicts pKa values within a mean absolute deviation (MAD) of 1.36 pKa units from the experiment. Some of the major outliers include SM01, SM06, SM14, SM23. SM18 and SM23 are suffering from the hydrogen bonding effect. These molecules can form stronger hydrogen bonding with their functional group (Phenol hydroxyl group or Aniline amino group) which is related to the macroscopic pKa, while other molecules can also suffer from the hydrogen bonding but less significantly because the hydrogen bonds being formed is much weaker. Some of these conformations were biased because the implicit solvation model cannot describe the hydrogen bonding effectively.
A previous study comparing the accuracy of the direct scheme with a low (MP2) and high (G3) level of theory, reported that use of a higher-level of theory improves the MAD with respect to experiment for carboxylic, inorganic, and cationic acids using the direct scheme from 0.4 to 0.9 pKa units [39]. Rather than using a different method, we consider improving the quality of the basis set to represent a better level of theory for this challenge. In most cases, adding additional basis functions yields poorer predictions, as great as 5.0 pKa units away from the direct scheme. This excludes SM04, SM07, SM20, SM22, and SM24, as we see that using a larger basis set yields predictions of an average of 0.5 pKa units closer to experiment (1.3 pKa units difference for SM20).
Vertical Scheme
The vertical scheme utilizes gas phase geometries and thermodynamic cycle to approximate the free energy of solvation. By contrasting the direct and vertical scheme, the difference in the gas phase contribution and solution phase contribution to the solvation free energy is highlighted. Overall, the vertical scheme provides overestimations of the pKa, yielding a MAE of 1.74 pKa units. To note, this is greater than the MAE of the direct method (This corresponds to a difference of 0.38 pKa units or 0.5 kcal/mol free energy difference distributed in the difference of the geometries). Compared to the direct scheme, the vertical scheme overestimates the pKa for SM06 and SM09. This poorer performance of the vertical scheme is surprising because this approach is common to the methods in which continuum solvation models are parameterized.
As the vertical scheme assumes the gas phase geometry, it works well for the small or rigid molecules (e.g. SM02, SM05, SM09, etc.), and we consider using larger basis sets for the solvation free energy term (Equation (3c)). In most cases, the inclusion of triple- basis sets improves the predictions by an average of 0.1 to 0.2 pKa units with respect to experiment. Cases in which the trend does not follow (in which the larger basis set yields predictions greater than that predicted using smaller basis sets), occur for polyprotic molecules, such as SM15 and SM22.
Adiabatic Scheme
Considering both optimized gas phase and solution phase structures is hypothesized to provide more accurate pKa predictions as it includes the energetic compensation for relaxing in solvent. Using the adiabatic scheme, this yields pKa values with a MAE of 1.26 pKa units. Comparing the two thermodynamic cycle-based approaches, the adiabatic scheme provides more accurate pKa than the vertical scheme for (64%) of the molecules. This highlights that the structures determined in both gas phase and solution phase are significant for determining pKa values.
Similar to the direct and vertical schemes, we examine how using a larger basis set impacts the solvation free energy. The results indicate that using a triple--level basis set on the solvation free energy term improves the pKa predictions by an average of 0.2 pKa units.
Comparison of the Schemes
Overall, the results in Table 3 illustrate a hierarchy of the different reaction schemes. Contrasting the three schemes, pKa values determined via the direct scheme and adiabatic scheme are closer to experiment than those predicted using the vertical scheme. However, this relationship only holds to the level of theory employed for each reaction scheme (in this case, using M06–2X with a double- level basis set). When applying a larger basis set to the solvation free energy calculation, the adiabatic and vertical scheme have less error (MAE is 1.10 and 1.48, respectively) with respect to experiment than the direct scheme (MAE is 1.95). The reason is that a more accurate solvation free energy can be obtained by using a larger basis set.
Table 3.
Absolute macroscopic pKa values via the direct (D), vertical (V), and adiabatic (A) schemes.
| Molecule | Experiment | Scheme |
|||||
|---|---|---|---|---|---|---|---|
| ID | pKa | D | D+S | V | V+S | A | A+S |
| SM01 | 9.53 ± 0.01 | 12.53 | 14.51 | 13.41 | 13.11 | 12.03 | 11.78 |
| SM02 | 5.03 ± 0.01 | 5.12 | 4.60 | 4.24 | 4.66 | 4.27 | 4.67 |
| SM03 | 7.02 ± 0.01 | 7.39 | 8.66 | 8.32 | 8.17 | 7.46 | 7.30 |
| SM04 | 6.02 ± 0.01 | 6.40 | 5.86 | 7.54 | 7.71 | 6.78 | 6.89 |
| SM05 | 4.59 ± 0.01 | 3.28 | 2.56 | 4.85 | 4.97 | 2.42 | 2.38 |
| SM06 | 3.03 ± 0.04 | 0.66 | −0.06 | 0.32 | 0.48 | 1.17 | 1.38 |
| SM06a | 11.74 ± 0.01 | 15.33 | 16.65 | 16.92 | 16.75 | 14.56 | 14.60 |
| SM07 | 6.08 ± 0.01 | 7.19 | 6.61 | 7.01 | 7.17 | 6.35 | 6.42 |
| SM08 | 4.22 ± 0.01 | 4.34 | 6.29 | 5.83 | 5.77 | 3.33 | 3.33 |
| SM09 | 5.37 ± 0.01 | 5.31 | 4.80 | 4.89 | 5.08 | 5.10 | 5.31 |
| SM10 | 9.02 ± 0.01 | 9.53 | 10.75 | 7.69 | 7.62 | 8.00 | 8.00 |
| SM11 | 3.89 ± 0.01 | 2.60 | 2.37 | 2.99 | 3.29 | 2.90 | 3.16 |
| SM12 | 5.28 ± 0.01 | 4.74 | 4.25 | 4.46 | 4.67 | 4.48 | 4.69 |
| SM13 | 5.77 ± 0.01 | 4.69 | 4.19 | 5.47 | 5.62 | 5.58 | 5.66 |
| SM14 | 2.58 ± 0.01 | −1.07 | −1.67 | −0.37 | −0.26 | −0.43 | −0.13 |
| SM14a | 5.30 ± 0.01 | 4.91 | 4.42 | 4.64 | 4.84 | 5.09 | 5.20 |
| SM15 | 4.70 ± 0.01 | 3.74 | 3.23 | 3.57 | 3.73 | 3.79 | 3.94 |
| SM15a | 8.94 ± 0.01 | 11.02 | 13.07 | 11.80 | 11.39 | 10.73 | 10.38 |
| SM16 | 5.37 ± 0.01 | 3.60 | 3.30 | 3.62 | 3.80 | 4.49 | 4.79 |
| SM16a | 10.65 ± 0.01 | 12.35 | 13.32 | 12.45 | 12.27 | 11.71 | 11.41 |
| SM17 | 3.16 ± 0.01 | 1.38 | 1.28 | 1.51 | 1.55 | 2.17 | 2.39 |
| SM18 | 2.15 ± 0.02 | 1.84 | 1.27 | 2.04 | 2.27 | 2.28 | 2.58 |
| SM18a | 9.58 ± 0.03 | 10.11 | 11.73 | 11.29 | 11.21 | 10.76 | 10.68 |
| SM18b | 11.02 ± 0.04 | 13.78 | 14.62 | 13.04 | 13.05 | 15.21 | 15.02 |
| SM19 | 9.56 ± 0.02 | 10.41 | 11.69 | 11.87 | 11.80 | 10.09 | 10.14 |
| SM20 | 5.70 ± 0.03 | 3.84 | 5.17 | 5.14 | 5.26 | 3.99 | 4.35 |
| SM21 | 4.10 ± 0.01 | 1.87 | 1.36 | 1.65 | 2.14 | 1.99 | 2.55 |
| SM22 | 2.40 ± 0.02 | 3.19 | 2.66 | 3.00 | 2.82 | 3.03 | 2.83 |
| SM22a | 7.43 ± 0.01 | 9.30 | 9.90 | 9.89 | 9.79 | 9.03 | 8.73 |
| SM23 | 5.45 ± 0.01 | 3.08 | 2.85 | 2.69 | 2.95 | 5.75 | 5.99 |
| SM24 | 2.60 ± 0.01 | 3.55 | 2.97 | 2.85 | 2.99 | 3.74 | 3.36 |
| MSE | −0.08 | 0.16 | 0.24 | 0.30 | 0.04 | 0.07 | |
| MAE | 1.36 | 1.95 | 1.61 | 1.48 | 1.26 | 1.10 | |
| MaxD | 3.59 | 4.98 | 5.18 | 5.01 | 4.19 | 4.00 | |
| RMSE | 1.70 | 2.38 | 1.98 | 1.85 | 1.55 | 1.40 | |
| R2 | 0.92 | 0.91 | 0.89 | 0.90 | 0.92 | 0.93 | |
pKa,2
pKa,3
Our submission to the SAMPL6 challenge (Table 1), did not include the standard state correction (which made a difference in 1.39 pKa units) and also used another value for the free energy of solvation of a proton not recommended (a difference of 0.22 pKa units); this has been corrected. These results are encouraging as the pKa predictions via the adiabatic and direct schemes correlate well with experiment, having a correlation coefficient greater than 0.9.
To confirm if the approach predicts the proper chemistry, we evaluate the different schemes on a small subset of molecules that share a similar scaffold, differing by electron donation or withdrawing groups. The molecules SM02, SM04, SM07, SM09, SM12, and SM13 share the 4-Aminoquinazoline scaffold. Ranked by acidity, SM02, SM12, and SM09 differ by substituents on the phenyl ring spanning a variance of 0.35 pKa units. The direct schemes are unable to properly determine the trend, as the predictions indicate that SM12 is more acidic than SM02 (SM12 is a Ph-Cl whereas SM02 is a Ph-CF3). In contrast, the vertical schemes rank the acidities of SM02 and SM12 correctly, however, overestimate the acidity of the SM09. This is believed to be a result of an issue with the gas phase geometry. That means using unrelaxed geometry cannot obtain accurate solvation free energy when the molecule is flexible and has different conformations in different phases. SM13 has a larger pKa and is different as it contains electronic donating groups on the quinazoline as opposed to the amino group. The direct and vertical scheme overestimate the acidity relative to SM02, SM12, and SM09. The difference between SM04 and SM07 is small quantitatively and qualitatively (0.04 pKa units). Interestingly, only the direct scheme was able to properly rank the acidities for these molecules. We also compare the microscopic pKa values with respect to experiment for these molecules and observe the same trends (Table S6).
Room for Improvement
Aside of the chosen level of theory employed, another source of error arises from the lack of explicit interactions between the solute and water, which are not accounted for in continuum solvation models. For example, functional groups such as alcohol and phenols, the ionic state may be stabilized in solution by hydrogen bonding. Including explicit water molecules with continuum solvation models, also termed microsolvation or cluster-continuum modeling, has been shown to improve pKa predictions for such issues[45]. In general, this could result in overestimation or underestimation of pKa values for acids and bases.
For example, the pKa for molecules SM01, SM15, and SM22, which may undergo deprotonation at the phenol group, are overestimated by 1.3–5.0 pKa units. As a proof of concept, we try to improve pKa predictions for SM01 by adding water molecules near the hydroxyl group. Adding one water molecule improves the prediction of the pKa by an average of 1.3 pKa units (Figure 3). By saturating the hydroxyl group with 3 water molecules, the pKa improves by an average of 3.0 pKa units (Table S3).
Figure 3.
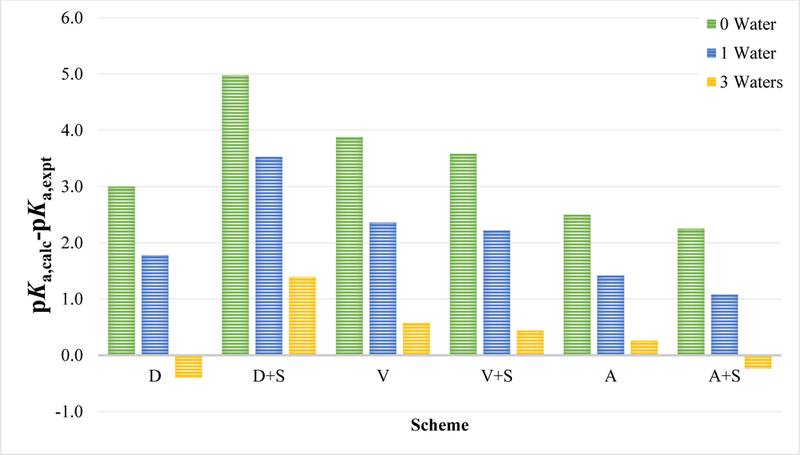
Effect of microsolvation on pKa calculations schemes for SM01.
Relative Schemes
When employing the different calculation schemes for this challenge, we only considered predicting absolute pKa values as opposed to relative pKa values. Relative schemes for calculating pKa use empirical parameters to scale or offset the solute phase free energy.
| (18) |
Using a relative scheme as an offset (A=1) to the free energy entails identifying and applying (subjectively) good reference models, which relies on chemical intuition. As this challenge includes 620 unique acid-base pairs, identifying the proper reference models proved difficult the molecules had multiple protonation sites. Alternatively, a linear regression fit can be applied to the calculated solution free energy to correct for systematic errors (e.g. concentration of water, proton solvation free energy). As this is a popular approach for calculating pKa[45, 46], we consider applying a linear regression correction to each scheme. To determine the parameters A and B, two training sets, consisting of 63 acid reactions (Table S2) and 56 bases (Table S3), were used. The linear fitting parameters determined for each scheme can be found in the Supporting Information (Table S4). For each scheme, while applying a linear regression fit does not improve the correlation (), this approach does improve the pKa predictions, with a lower MAE and RMSE than the respective absolute calculation pKa schemes (Table 4).
Table 4.
Comparison of linear regression fit macroscopic pKa values via direct (D), vertical (V), and adiabatic (A) schemes with experiment.
| Molecule ID | pKa Expt. | D | D+S | V | V+S | A | A+S |
|---|---|---|---|---|---|---|---|
| SM01 | 9.53 ± 0.01 | 9.79 | 9.89 | 8.96 | 10.07 | 9.75 | 9.73 |
| SM02 | 5.03 ± 0.01 | 5.50 | 5.41 | 5.55 | 5.21 | 4.96 | 5.16 |
| SM03 | 7.02 ± 0.01 | 7.12 | 6.81 | 6.76 | 7.39 | 7.35 | 7.36 |
| SM04 | 6.02 ± 0.01 | 6.29 | 6.21 | 7.00 | 7.11 | 6.53 | 6.57 |
| SM05 | 4.59 ± 0.01 | 4.35 | 4.12 | 5.84 | 5.41 | 3.81 | 3.71 |
| SM06 | 3.03 ± 0.04 | 2.73 | 2.46 | 2.59 | 2.62 | 3.04 | 3.08 |
| SM06a | 11.74 ± 0.01 | 11.26 | 11.01 | 11.89 | 11.94 | 11.08 | 11.22 |
| SM07 | 6.08 ± 0.01 | 6.79 | 6.68 | 7.08 | 6.77 | 6.26 | 6.27 |
| SM08 | 4.22 ± 0.01 | 5.52 | 5.56 | 5.22 | 6.30 | 5.17 | 5.25 |
| SM09 | 5.37 ± 0.01 | 5.62 | 5.53 | 5.38 | 5.47 | 5.48 | 5.57 |
| SM10 | 9.02 ± 0.01 | 8.23 | 7.91 | 6.56 | 7.25 | 7.63 | 7.72 |
| SM11 | 3.89 ± 0.01 | 3.93 | 4.00 | 4.52 | 4.36 | 4.11 | 4.20 |
| SM12 | 5.28 ± 0.01 | 5.26 | 5.19 | 5.54 | 5.22 | 5.09 | 5.18 |
| SM13 | 5.77 ± 0.01 | 5.23 | 5.15 | 5.74 | 5.81 | 5.78 | 5.79 |
| SM14 | 2.58 ± 0.01 | 1.65 | 1.44 | 2.16 | 2.16 | 2.04 | 2.13 |
| SM14a | 5.30 ± 0.01 | 5.37 | 5.30 | 5.23 | 5.33 | 5.48 | 5.50 |
| SM15 | 4.70 ± 0.01 | 4.64 | 4.54 | 4.98 | 4.64 | 4.66 | 4.70 |
| SM15a | 8.94 ± 0.01 | 9.00 | 9.13 | 8.05 | 9.18 | 9.07 | 8.99 |
| SM16 | 5.37 ± 0.01 | 4.55 | 4.58 | 4.97 | 4.68 | 5.10 | 5.24 |
| SM16a | 10.65 ± 0.01 | 9.70 | 9.26 | 8.95 | 9.64 | 9.58 | 9.53 |
| SM17 | 3.16 ± 0.01 | 3.17 | 3.31 | 3.5 | 3.28 | 3.66 | 3.72 |
| SM18 | 2.15 ± 0.02 | 3.46 | 3.30 | 4.22 | 3.73 | 3.72 | 3.84 |
| SM18a | 9.58 ± 0.03 | 8.53 | 8.42 | 8.13 | 9.09 | 9.08 | 9.14 |
| SM18b | 11.02 ± 0.04 | 10.44 | 9.94 | 9.08 | 10.04 | 11.42 | 11.44 |
| SM19 | 9.56 ± 0.02 | 8.68 | 8.40 | 8.68 | 9.40 | 8.73 | 8.86 |
| SM20 | 5.70 ± 0.03 | 5.26 | 4.98 | 5.37 | 6.03 | 5.52 | 5.79 |
| SM21 | 4.10 ± 0.01 | 3.48 | 3.36 | 4.05 | 3.65 | 3.55 | 3.82 |
| SM22 | 2.40 ± 0.02 | 4.30 | 4.18 | 4.43 | 4.07 | 4.19 | 4.00 |
| SM22a | 7.43 ± 0.01 | 8.11 | 7.46 | 7.83 | 8.36 | 8.17 | 8.11 |
| SM23 | 5.45 ± 0.01 | 4.23 | 4.30 | 4.33 | 4.15 | 5.88 | 6.00 |
| SM24 | 2.60 ± 0.01 | 4.47 | 4.43 | 4.27 | 4.18 | 4.56 | 4.37 |
| MSE | −0.04 | −0.18 | −0.06 | 0.17 | 0.11 | 0.15 | |
| MAE | 0.64 | 0.71 | 0.90 | 0.67 | 0.57 | 0.54 | |
| MaxD | 1.90 | 1.83 | 2.46 | 2.08 | 1.96 | 1.77 | |
| RMSE | 0.82 | 0.89 | 1.11 | 0.89 | 0.76 | 0.73 | |
| R2 | 0.91 | 0.90 | 0.85 | 0.90 | 0.93 | 0.93 | |
pKa2
pKa3
We believe the issue that the slope of the experimental pKa vs calculated pKa is not the expected value of 1 is due to the hydrogen bonding effects. Since the hydrogen bonding can stabilize the charged species having little effect to the neutral species, the pKa values for the bases are usually underestimated while those for the acids are usually overestimated without explicitly considering the hydrogen bonds between the solvent and the solute. The slope can approach the expected value of 1 by including explicit waters[45].
Multiple Minima Consideration
All pKa values have been determined using one conformation per microstate pair. The molecules within the SAMPL6 pKa data set are not rigid (excluding SM01 and SM22) and can adopt multiple conformations that satisfy local minima. To probe if the exclusion of multiple minima as a source of error in our pKa calculations, we generate 6 to 32 different conformations for each microstate of SM06 and re-calculate the macroscopic pKa by sequentially including the lowest energy conformations per microstate. As shown in Table 5, including multiple minima has little impact to the pKa prediction (0.1 to 0.6 pKa units). By applying the linear regression fit, the pKa predictions are closer to experiment using one conformation per microstate. Including additional conformations per microstate yields a maximum difference of 0.3 pKa unit (Table S5).
Table 5.
Macroscopic pKa values of SM06 determined as a function by microstate conformations.a
| No. Conf.b |
pKa1 |
pKa2 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D | D+S | V | V+S | A | A+S | D | D+S | V | V+S | A | A+S | |
| 1 | 0.66 | −0.06 | 0.32 | 0.48 | 1.17 | 1.38 | 15.33 | 16.65 | 16.92 | 16.75 | 14.56 | 14.60 |
| 3 | 0.67 | −0.01 | 0.30 | 0.47 | 1.17 | 1.37 | 15.49 | 16.83 | 17.21 | 17.05 | 14.89 | 14.96 |
| 5 | 0.63 | −0.03 | 0.23 | 0.40 | 1.17 | 1.42 | 15.52 | 16.92 | 17.31 | 17.16 | 15.00 | 15.09 |
| 7 | 0.61 | −0.05 | 0.30 | 0.46 | 1.19 | 1.46 | 15.55 | 16.96 | 17.39 | 17.25 | 15.09 | 15.16 |
| 9 | 0.60 | −0.05 | 0.30 | 0.46 | 1.19 | 1.48 | 15.55 | 16.96 | 17.39 | 17.29 | 15.08 | 15.19 |
| 11 | 0.62 | −0.03 | 0.30 | 0.46 | 1.19 | 1.48 | 15.55 | 16.96 | 17.39 | 17.29 | 15.09 | 15.20 |
| 13 | 0.63 | −0.02 | 0.30 | 0.46 | 1.19 | 1.48 | 15.55 | 16.96 | 17.40 | 17.29 | 15.09 | 15.20 |
| 15 | 0.63 | −0.02 | 0.30 | 0.46 | 1.19 | 1.48 | 15.55 | 16.96 | 17.40 | 17.29 | 15.09 | 15.20 |
| Range | 0.07 | 0.05 | 0.09 | 0.08 | 0.02 | 0.11 | 0.22 | 0.31 | 0.48 | 0.54 | 0.53 | 0.60 |
The experimental pKa values of SM06 are 3.03 and 11.74 for pKa1 and pKa2, respectively.
Number of conformations used per microstate to calculate the microscopic pKa.
Conclusion
In this study, three calculations schemes were used to predict the pKa of molecules in the SAMPL6 challenge. The adiabatic scheme yields more accurate pKa predictions than the direct and vertical schemes. Using a larger basis set with the adiabatic scheme yields the best results of among the other schemes, yielding an RMSE of 1.40 pKa. A combination of popular and inexpensive methods (M06–2X/Pople basis sets (6–31G(d)/6–311G(d,p) or 6–31+G(d)/6–311++G(d,p))//SMD) is being used in our approach, which means that our approach can be carried out in most popular software packages. Without additional parameterization, we have a very encouraging result with a RMSE of 1.40 and R2 of 0.93 by using different basis sets for different charged species. However, if linear correction is applied, we can improve our results to achieve a RMSE of 0.73 and R2 of 0.94. Our approach can be further improved since there are still multiple sources of error from the current method, basis set, and solvation model. In the future, we will try using different methods, basis sets, and solvation models in our approach to find out the best combination.
Supplementary Material
Figure 2.
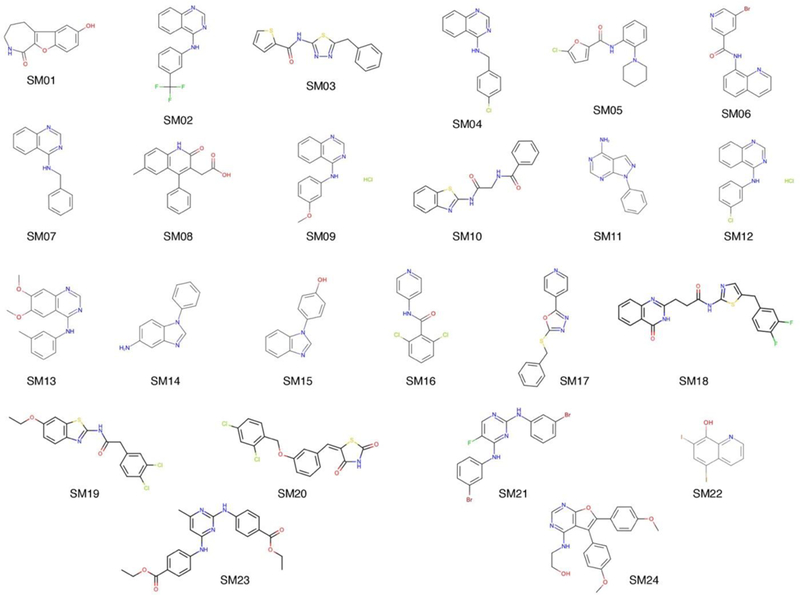
Structures of the 24 molecules in the SAMPL6 pKa challenge.
Acknowledgments
This work was supported by the intramural research program of the National Heart, Lung and Blood Institute of the National Institutes of Health and utilized the high-performance computational capabilities of the LoBoS and Biowulf Linux clusters at the National Institutes of Health (http://www.lobos.nih.gov and http://biowulf.nih.gov). The authors would also like to thank Frank C. Pickard, IV and Samarjeet Prasad for the very helpful discussion.
References
- 1.Wang Y, Xing J, Xu Y, et al. (2015) In silico ADME/T modelling for rational drug design. Q Rev Biophys 48:488–515. doi: 10.1017/S0033583515000190 [DOI] [PubMed] [Google Scholar]
- 2.Zevatskii YE, Samoilov DV. (2011) Modern methods for estimation of ionization constants of organic compounds in solution. Russ J Org Chem 47:1445–1467. doi: 10.1134/S1070428011100010 [DOI] [Google Scholar]
- 3.Seybold PG, Shields GC (2015) Computational estimation of pK a values. Wiley Interdiscip Rev Comput Mol Sci 5:290–297. doi: 10.1002/wcms.1218 [DOI] [Google Scholar]
- 4.Lee AC, Crippen GM (2009) Predicting pKa. J Chem Inf Model 49:2013–2033. doi: 10.1021/ci900209w [DOI] [PubMed] [Google Scholar]
- 5.Fraczkiewicz R, Lobell M, Goller AH, et al. (2015) Best of both worlds: Combining pharma data and state of the art modeling technology to improve in silico p K a prediction. J Chem Inf Model 55:389–397. doi: 10.1021/ci500585w [DOI] [PubMed] [Google Scholar]
- 6.Shelley JC, Cholleti A, Frye LL, et al. (2007) Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 21:681–691. doi: 10.1007/s10822-007-9133-z [DOI] [PubMed] [Google Scholar]
- 7.Software OS (2018) OpenEye Toolkits [Google Scholar]
- 8.Advanced Chemistry Development I (2015) ACD/Percepta [Google Scholar]
- 9.Wells PR (1963) Linear Free Energy Relationships. Chem Rev 63:171–219. doi: 10.1021/cr60222a005 [DOI] [Google Scholar]
- 10.Casasnovas R, Ortega-Castro J, Frau J, et al. (2014) Theoretical p K a calculations with continuum model solvents, alternative protocols to thermodynamic cycles. Int J Quantum Chem 114:1350–1363. doi: 10.1002/qua.24699 [DOI] [Google Scholar]
- 11.Ho J (2014) Predicting pKa in Implicit Solvents: Current Status and Future Directions. Aust J Chem 67:1441 . doi: 10.1071/CH14040 [DOI] [Google Scholar]
- 12.Bochevarov AD, Watson MA, Greenwood JR, Philipp DM (2016) Multiconformation, Density Functional Theory-Based p K a Prediction in Application to Large, Flexible Organic Molecules with Diverse Functional Groups. J Chem Theory Comput 12:6001–6019. doi: 10.1021/acs.jctc.6b00805 [DOI] [PubMed] [Google Scholar]
- 13.Muckerman JT, Skone JH, Ning M, Wasada-Tsutsui Y (2013) Toward the accurate calculation of pKa values in water and acetonitrile. Biochim Biophys Acta - Bioenerg 1827:882–891. doi: 10.1016/j.bbabio.2013.03.011 [DOI] [PubMed] [Google Scholar]
- 14.Jensen JH, Swain CJ, Olsen L (2017) Prediction of p K a Values for Druglike Molecules Using Semiempirical Quantum Chemical Methods. J Phys Chem A 121:699–707. doi: 10.1021/acs.jpca.6b10990 [DOI] [PubMed] [Google Scholar]
- 15.Kromann JC, Larsen F, Moustafa H, Jensen JH (2016) Prediction of pKa values using the PM6 semiempirical method. PeerJ 4:e2335 . doi: 10.7717/peerj.2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montgomery JA, Frisch MJ, Ochterski JW, Petersson GA (1999) A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J Chem Phys 110:2822–2827. doi: 10.1063/1.477924 [DOI] [Google Scholar]
- 17.Pople JA, Head-Gordon M, Fox DJ, et al. (1989) Gaussian-1 theory: A general procedure for prediction of molecular energies. J Chem Phys 90:5622–5629. doi: 10.1063/1.456415 [DOI] [Google Scholar]
- 18.Curtiss LA, Jones C, Trucks GW, et al. (1990) Gaussian‐1 theory of molecular energies for second‐row compounds. J Chem Phys 93:2537–2545. doi: 10.1063/1.458892 [DOI] [Google Scholar]
- 19.Curtiss LA, Raghavachari K, Trucks GW, Pople JA (1991) Gaussian-2 theory for molecular energies of first- and second-row compounds. J Chem Phys 94:7221–7230. doi: 10.1063/1.460205 [DOI] [Google Scholar]
- 20.Curtiss LA, Raghavach Ari K, Redfern PC, et al. (1998) Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J Chem Phys 109:7764–7776. doi: 10.1063/1.477422 [DOI] [Google Scholar]
- 21.DeYonker NJ, Cundari TR, Wilson AK (2006) The correlation consistent composite approach (ccCA): An alternative to the Gaussian-n methods. J Chem Phys 124: . doi: 10.1063/1.2173988 [DOI] [PubMed] [Google Scholar]
- 22.Ho J, Coote ML (2009) p K a Calculation of Some Biologically Important Carbon Acids - An Assessment of Contemporary Theoretical Procedures. J Chem Theory Comput 5:295–306. doi: 10.1021/ct800335v [DOI] [PubMed] [Google Scholar]
- 23.Tehan BG, Lloyd EJ, Wong MG, et al. (2002) Estimation of pKa Using Semiempirical Molecular Orbital Methods. Part 1: Application to Phenols and Carboxylic Acids. Quant Struct Relationships 21:457–472. doi: 10.1002/1521-3838(200211)21:5<457::AID-QSAR457>3.0.CO;2-5 [DOI] [Google Scholar]
- 24.Liptak MD, Shields GC (2001) Experimentation with different thermodynamic cycles used for pKa calculations on carboxylic acids using complete basis set and Gaussian-n models combined with CPCM continuum solvation methods. Int J Quantum Chem 85:727–741. doi: 10.1002/qua.1703 [DOI] [PubMed] [Google Scholar]
- 25.Liptak MD, Shields GC (2001) Accurate pKa Calculations for Carboxylic Acids Using Complete Basis Set and Gaussian-n Models Combined with CPCM Continuum Solvation Methods. J Am Chem Soc 123:7314–7319. doi: 10.1021/ja010534f [DOI] [PubMed] [Google Scholar]
- 26.Riojas AG, Wilson AK (2014) Solv-ccCA: Implicit Solvation and the Correlation Consistent Composite Approach for the Determination of p K a. J Chem Theory Comput 10:1500–1510. doi: 10.1021/ct400908z [DOI] [PubMed] [Google Scholar]
- 27.Peverati R, Truhlar DG (2014) Quest for a universal density functional: the accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos Trans A Math Phys Eng Sci 372:20120476 . doi: 10.1098/rsta.2012.0476 [DOI] [PubMed] [Google Scholar]
- 28.Mardirossian N, Head-Gordon M (2017) Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol Phys 115:2315–2372. doi: 10.1080/00268976.2017.1333644 [DOI] [Google Scholar]
- 29.Cohen AJ, Mori-Sánchez P, Yang W (2012) Challenges for Density Functional Theory. Chem Rev 112:289–320. doi: 10.1021/cr200107z [DOI] [PubMed] [Google Scholar]
- 30.Barone V, Cossi M (1998) Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A 102:1995–2001. doi: 10.1021/jp9716997 [DOI] [Google Scholar]
- 31.Klamt A, Schüürmann G (1993) COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans 2 799–805. doi: 10.1039/P29930000799 [DOI] [Google Scholar]
- 32.Francl MM, Pietro WJ, Hehre WJ, et al. (1982) Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J Chem Phys 77:3654–3665. doi: 10.1063/1.444267 [DOI] [Google Scholar]
- 33.Marenich A V, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396. doi: 10.1021/jp810292n [DOI] [PubMed] [Google Scholar]
- 34.Tissandier MD, Cowen KA, Feng WY, et al. (1998) The Proton’s Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data. J Phys Chem A 102:7787–7794. doi: 10.1021/jp982638r [DOI] [Google Scholar]
- 35.McQuarrie D (2011) Statistical Mechanics. Viva Books [Google Scholar]
- 36.Ho J, Ertem MZ (2016) Calculating Free Energy Changes in Continuum Solvation Models. J Phys Chem B 120:1319–1329. doi: 10.1021/acs.jpcb.6b00164 [DOI] [PubMed] [Google Scholar]
- 37.Ho J (2015) Are thermodynamic cycles necessary for continuum solvent calculation of pK a s and reduction potentials? Phys Chem Chem Phys 17:2859–2868. doi: 10.1039/C4CP04538F [DOI] [PubMed] [Google Scholar]
- 38.O’Boyle NM, Banck M, James CA, et al. (2011) Open Babel: An Open chemical toolbox. J Cheminform 3:1–14. doi: 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor Chem Acc 120:215–241. doi: 10.1007/s00214-007-0310-x [DOI] [Google Scholar]
- 40.Frisch MJ, Pople JA, Binkley JS (1984) Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269. doi: 10.1063/1.447079 [DOI] [Google Scholar]
- 41.Kesharwani MK, Brauer B, Martin JML (2015) Frequency and Zero-Point Vibrational Energy Scale Factors for Double-Hybrid Density Functionals (and Other Selected Methods): Can Anharmonic Force Fields Be Avoided? J Phys Chem A 119:1701–1714. doi: 10.1021/jp508422u [DOI] [PubMed] [Google Scholar]
- 42.Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396. doi: 10.1021/jp810292n [DOI] [PubMed] [Google Scholar]
- 43.Frisch MJ, Trucks GW, Schlegel HB, et al. (2016) Gaussian 16, Revision A.03. 2016 [Google Scholar]
- 44.Klicić JJ, Friesner RA, Liu SY, Guida WC (2002) Accurate prediction of acidity constants in aqueous solution via density functional theory and self-consistent reaction field methods. J Phys Chem A 106:1327–1335. doi: 10.1021/jp012533f [DOI] [Google Scholar]
- 45.Thapa B, Schlegel HB (2017) Improved p K a Prediction of Substituted Alcohols, Phenols, and Hydroperoxides in Aqueous Medium Using Density Functional Theory and a Cluster-Continuum Solvation Model. J Phys Chem A 121:4698–4706. doi: 10.1021/acs.jpca.7b03907 [DOI] [PubMed] [Google Scholar]
- 46.Klamt A, Eckert F, Diedenhofen M, Beck ME (2003) First Principles Calculations of Aqueous pKa Values for Organic and Inorganic Acids Using COSMO-RS Reveal an Inconsistency in the Slope of the pKa Scale. J Phys Chem A 107:9380–9386. doi: 10.1021/jp034688o [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.