

Abstract
Two methods for the synthesis of bis(imidazol-2-ylidene)pyridine iron dialkyl complexes, (CNC)Fe(CH2SiMe3)2, have been developed. The first route consists of addition of two equivalents of LiCH2SiMe3 to the iron dihalide complex, (CNC)FeBr2, while the second relies on addition of the free CNC ligand to readily-prepared (py)2Fe(CH2SiMe3)2 (py = pyridine). With aryl-substituted CNC ligands, octahedral complexes of the type (ArCNC)Fe(CH2SiMe3)2(N2) (ArCNC = bis(arylimidazol-2-ylidene)pyridine) were isolated, where the dinitrogen ligand occupies the site trans to the pyridine of the CNC-chelate. In contrast, the alkyl-substituted variant, (tBuACNC)Fe(CH2SiMe3)2 (tBuACNC = 2,6-(tBu-imidazol-2-ylidene)2pyridine) was isolated as the five-coordinate compound lacking dinitrogen. Exposure of the (ArCNC)Fe(CH2SiMe3)2(N2) derivatives to an H2 atmosphere resulted in formation of the corresponding iron hydride complexes (ArCNC)FeH4. These compounds catalyzed hydrogen isotope exchange between the deuterated benzene solvent and H2, generating isotopologues and isotopomers of (ArCNC)Fe(Hn)(D4-n) (n = 0–4). When (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) (3,5-Me2MesCNC = 2,6-(2,4,6-Me3-C6H2-imidazol-2-ylidene)2-3,5-Me2-pyridine) was treated successively with H2 and then N2, the corresponding reduced dinitrogen complex (3,5-Me2MesCNC)Fe(N2)2 was isolated. The same product was also obtained following addition of pinacolborane to (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2).
Graphical Abstract
INTRODUCTION
The oxidative addition of non-polar bonds such as H–H, B–H, Si–H and C–H is a fundamental substrate activation step in organometallic transformations1 and poses a fundamental challenge for catalysis with earth-abundant first row transition metals.2 One strategy for enabling this reactivity with iron and cobalt is to introduce pincer ligands with strong σ-donating phosphines or N-heterocyclic carbenes to enforce a strong ligand field and promote formation of low-spin complexes prone to two-electron reactivity.2a,c Examples of concerted, two-electron oxidative addition with both iron and cobalt are now well documented.2
Aryl-substituted bis(arylimidazol-2-ylidene)pyridine iron dinitrogen compounds, [(ArylCNC)Fe(N2)2] originally synthesized by Danopoulos and coworkers,3,4 are exemplary of the strong-field ligand strategy and promote the oxidative addition of C–H,3a Si–H,3b and H–H bonds.3c These complexes have also been shown to be highly active pre-catalysts for the hydrogenation of hindered, essentially unactivated olefins.5 More recently, a variant with saturated arylimidazolines, (H4-arylCNC)Fe(N2)2 (H4-arylCNC = bis(arylimidazolin-2-ylidene)pyridine), (H4-arylCNC)Fe(N2)2 (H4-arylCNC = bis(arylimidazolin-2-ylidene)pyridine) was synthesized and shown to be an effective catalyst for hydrogen isotope exchange (HIE) and the tritiation of active pharmaceutical ingredients.6 The catalyst exhibits both broad functional group tolerance and high HIE activity in polar solvents such as N-methyl pyrrolidone (NMP) and exhibits sterically-driven site selectivity that is complementary to state-of-the-art iridium catalysts by ignoring directing groups and being governed by steric effects.
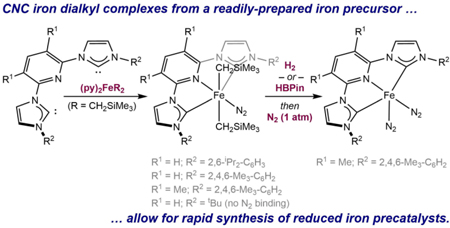
One challenge associated with these compounds is their synthesis and extreme air- and moisture-sensitivity (Scheme 1a). Typical routes to the dinitrogen complexes involve reduction of iron(II) dihalide precursors with strong reducing agents, typically sodium amalgam or sodium naphthalenide. The success and efficiency of this reaction also proved highly dependent on the nature of the aryl substituents as iron complexes bearing smaller groups required stoichiometric sodium and catalytic naphthalene for reduction to the corresponding iron bis(dinitrogen) complex.5 No examples of iron bis(dinitrogen) complexes with N-alkyl rather than N-aryl substituents on the carbene are known. Another limitation of the current synthetic routes is the reliance on Fe[N(SiMe3)2]2 as the iron precursor for preparation of the iron dihalides. This material is a low-melting, green solid that is highly air sensitive and requires distillation for purification.7 Alternative synthetic routes that rely on more accessible precursors that are compatible with a range of ligand substitution patterns are therefore valuable and of interest.
Scheme 1.

Synthesis of (CNC)Fe Precatalysts from a) (CNC)Fe dihalide complexes and b) (CNC)Fe dialkyl complexes
Iron dialkyl complexes, (CNC)Fe(CH2SiMe3)2 (CNC = bis(imidazol-2-ylidene)pyridine) are attractive targets for isolable catalyst precursors due to their potential synthetic accessibility and activation by straightforward treatment with H2, silanes or boranes.8 The commercial availability of LiCH2SiMe3 and the synthetic accessibility of (pyridine)2Fe(CH2SiMe3)2 reported by Cámpora9 also motivated exploration of routes utilizing these reagents. Previous reports from our laboratory have demonstrated the utility of both iron and cobalt dialkyl complexes of this type as precatalysts for alkene hydrogenation, hydroboration, hydrosilylation, HIE and C(sp2/3)–H borylation reactions.8 Here, we describe the synthesis and characterization of a family of (CNC)Fe(CH2SiMe3)2 complexes, their propensity to coordinate dinitrogen as a function of N-heterocyclic carbene substituent, and their reactivity with hydrogen.
RESULTS AND DISCUSSION
Our studies commenced with the preparation of dialkyl derivatives of [(iPrCNC)Fe], as this compound is the most studied in its class. Dialkylation of the iron dihalide, (iPrCNC)FeBr2 was initially explored with the addition of two equivalents of LiCH2SiMe3 in diethyl ether at −35 ºC. A color change to dark violet was observed and filtration, followed by recrystallization from pentane at −35 ºC furnished (iPrCNC)Fe(CH2SiMe3)2(N2) (1-Ns2(N2)) as a diamagnetic violet solid in 61% isolated yield (Scheme 2). An alternative route was inspired by the work of Cámpora, who demonstrated that addition of tridentate, aryl-substituted pyridine(diimine) ligands to (py)2Fe(CH2SiMe3)2 resulted in pyridine displacement and formation of the five-coordinate iron dialkyl complexes.9 To explore analogous chemistry with the iPrCNC chelate, the free carbene was prepared by treatment of the bis(imidazolium) salt iPrCNC(HBr)2 with KHMDS (HMDS = N(SiMe3)2).10 Addition of the resulting free chelate to a readily-generated solution of (py)2Fe(CH2SiMe3)2 in pentane at −35 ºC resulted in a dark violet solution, which upon concentration and filtration, resulted in isolation of 1-Ns2(N2) in 67% yield based on iPrCNC.
Scheme 2.

Two Methods for the Synthesis of (iPrCNC)Fe(CH2SiMe3)2(N2).
The 1H NMR spectrum of 1-Ns2(N2) in benzene-d6 is consistent with a C2v symmetric complex with resolved coupling of the pyridine protons and the methylene groups of the neosilyl ligand detectable at –1.11 ppm. The carbene carbon atoms give rise to a signal in the 13C NMR spectrum at 223.1 ppm, consistent with other (CNC)Fe complexes.3–6,11 The 13C resonances of the alkyl carbons directly bound to iron, however, were not observed by 13C NMR spectroscopy.
Single crystals of 1-Ns2(N2) suitable for X-ray diffraction were obtained from a concentrated pentane solution at –35 °C. The solid-state structure of 1-Ns2(N2) is best described as an idealized octahedral complex with the two alkyl ligands oriented trans to each other, although slightly deviated from linearity (C40–Fe1–C33 = 163.26(14)°, Figure 1). The coordination site trans to the pyridine is occupied by a dinitrogen ligand, the coordination of which is maintained in solution as judged by IR spectroscopy (νN2 (C6D6) = 2129 cm−1). The bite of the CNC chelate results in a deviation of the C(11)-Fe(1)-C(10) angle from linearity (C11–Fe1–C10 = 159.68(15)°) despite a yaw angle of 15.7° (obtained from (N1-C11-Fe1)-(N2-C11-Fe1))/2). Having two strong-field, mutually trans X-type ligands and a neutral ligand trans to the pyridine in the CNC-plane around the iron center is a general structural feature of this class of complexes as it has also been observed in related iron dihydride and silyl–hydride complexes.3b,c The zero-field, solid-state 57Fe Mößbauer spectrum of 1-Ns2(N2) was measured at 80 K and exhibited a doublet with an isomer shift (δ) of 0.13 mm/s and a quadrupole splitting (|ΔEQ|) of 1.71 mm/s, consistent with a low-spin iron(II) complex (Figure 2a).12
Figure 1.

Solid-state molecular structure of (iPrCNC)Fe(CH2SiMe3)2(N2) (1-Ns2(N2)) at 30% probability ellipsoids. Hydrogen atoms and one aryl substituent are omitted for clarity. Selected bond distances (Å) and angles (deg): C10–Fe1 1.936(3), C11–Fe1 1.933(3), N3–Fe1 1.877(3), N6–Fe1 1.829(3), C33–Fe1 2.137(3), C40–Fe1 2.135(3); N6–Fe1–N3 177.39(13), N6–Fe1–C11 100.79(14); N3–Fe1–C11 79.80(14), N6–Fe1–C10 99.50(14), N3–Fe1–C10 79.88(13), C11–Fe1–C10 159.68(15), C40–Fe1–C33 163.26(14).
Figure 2.

Zero-field 57Fe Mößbauer spectra of (a) (iPrCNC)Fe(CH2SiMe3)2(N2) (1-Ns2(N2)), (b) (MesCNC)Fe(CH2SiMe3)2(N2) (2-Ns2(N2)), (c) (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) (3-Ns2(N2)), (d) (tBuACNC)Fe(CH2SiMe3)2 (4-Ns2) at 80 K in the solid state. Simulated parameters (blue lines) are (a) δ = 0.13 mm/s, |ΔEQ| = 1.71 mm/s; (b) δ = 0.13 mm/s, |ΔEQ| = 1.62 mm/s; (c) δ = 0.11 mm/s, |ΔEQ| = 1.90 mm/s; (d) δ = 0.13 mm/s, |ΔEQ| = 3.89 mm/s.
For the synthesis of other iron dialkyl complexes of this type, the route involving pyridine displacement from (py)2Fe(CH2SiMe3)2 was preferred because it obviates the use of Fe[N(SiMe3)2]2. While the yields using the two routes are similar, the iron dialkyl precursor obviates the distillation of an air-sensitive iron precursor. Addition of the free pyridine dicarbene pincer MesCNC to a pentane solution of (py)2Fe(CH2SiMe3)2 followed by filtration furnished (MesCNC)Fe(CH2SiMe3)2(N2) (2-Ns2(N2), MesCNC = 2,6-(2,4,6-Me3-C6H2-imidazol-2-ylidene)2pyridine) as a violet-brown solid in 72% yield (Scheme 3). Using a similar procedure, the related 3,5-dimethyl pyridine substituted iron dialkyl dinitrogen complex (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) (3-Ns2(N2), 3,5-Me2MesCNC = 2,6-(2,4,6-Me3-C6H2-imidazol-2-ylidene)2 3,5-Me2-pyridine) was isolated as a brown solid in 90% yield (Scheme 3). The dimethyl substituents at the 3- and 5-positions of the central pyridine ring were originally reported by Danopoulos and coworkers to prevent deleterious metalation at those sites and generally gave improved yields.3a
Scheme 3.
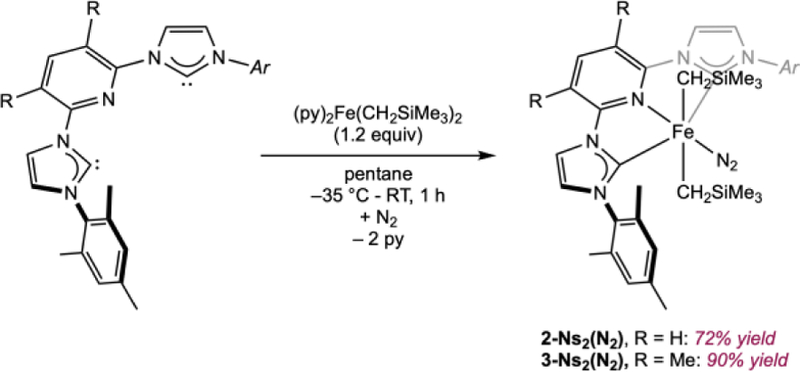
Synthesis of (MesCNC)Fe(CH2SiMe3)2(N2) and (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2)
Both diamagnetic iron complexes 2-Ns2(N2) and 3-Ns2(N2) were characterized by a combination of NMR, IR and Mößbauer spectroscopies. 1H and 13C NMR spectra exhibit features similar to 1-Ns2(N2), with the exception that the carbene carbons bound to iron were not located by 13C NMR spectroscopy. Dinitrogen coordination was confirmed by benzene solution IR spectroscopy with strong bands located at 2126 cm−1 and 2118 cm−1 for 2-Ns2(N2) and 3-Ns2(N2), respectively. The zero-field 57Fe Mößbauer spectra for both complexes exhibit parameters as expected for octahedral iron complexes in a strong ligand field (δ = 0.13 mm/s; |ΔEQ| = 1.62 mm/s (2-Ns2(N2); δ = 0.11 mm/s; |ΔEQ| = 1.90 mm/s (3-Ns2(N2)) (Figure 2b, c).12 Both the infrared and Mößbauer spectroscopic data support a more electron-donating pincer upon introduction of methyl groups in the 3,5-positions of the central pyridine ring. The solid-state structure of 3-Ns2(N2) was determined by X-ray diffraction of single crystals obtained from a pentane/Et2O solution at –35 °C, confirming the idealized octahedral geometry of the complex with trans alkyl ligands (Figure 3).
Figure 3.

Solid-state molecular structure of (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) (3-Ns2(N2)) at 30% probability ellipsoids. Hydrogen atoms and one aryl substituent are omitted for clarity. Selected bond distances (Å) and angles (deg): C11–Fe1 1.8969(14), C10–Fe1 1.8983(15), C32–Fe1 2.1587(14), C36–Fe1 2.1256(15), Fe1–N6 1.8241(13), Fe1–N3 1.8822(12); N6–Fe1–C11 99.29(6), N3–Fe1–C11 80.69(6), N6–Fe1–C10 99.56(6), N3–Fe1–C10 80.57(6), C11–Fe1–C10 160.77(7), N6–Fe1–N3 178.53(5), C36–Fe1–C32 169.46(6).
With a reliable synthetic method in hand, variants with alkyl substituents on the imidazol-2-ylidenes of the CNC-ligands were targeted (Scheme 4). Addition of tBuACNC (tBuACNC = 2,6-(tBu-imidazol-2-ylidene)2pyridine) to a pentane solution of (py)2Fe(CH2SiMe3)2 followed by filtration furnished a brown diamagnetic solid in 77% yield identified as (tBuACNC)Fe(CH2SiMe3)2 (4-Ns2). In contrast to the aryl-substituted derivatives described above, no infrared band assignable to a dinitrogen stretch was observed in the benzene solution infrared spectrum. The 1H NMR spectrum of 4-Ns2 exhibited signals for the methylene groups of the neosilyl ligand at 8.34 ppm, as identified by a cross-peak between the methylene group and the carbon atom of the SiMe3-group in the 1H-13C HMBC (heteronuclear multiple bond correlation) NMR spectrum. The triplet assigned to the 4-pyridine proton is shifted significantly downfield to 11.01 ppm in comparison to 1-Ns2(N2) (7.18 ppm) and 2-Ns2(N2) (7.11 ppm). Although the signal for this carbon was not directly observed by 13C NMR spectroscopy, the HSQC (HSQC = heteronuclear single quantum coherence) NMR spectrum exhibited a cross peak at 81.7 ppm. A signal for the carbene carbon was again not observable. Although 4-Ns2 is diamagnetic, the anomalous NMR chemical shifts are likely a result of population of low-lying triplet state.
Scheme 4.
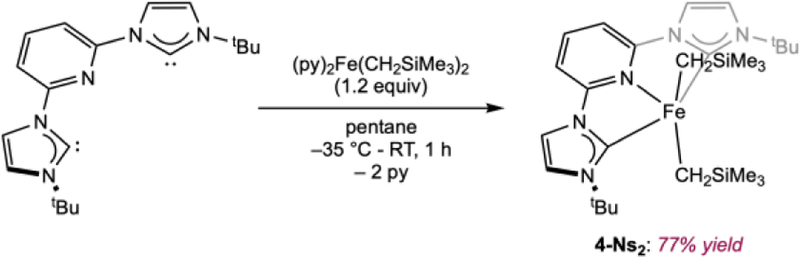
Synthesis of (tBuACNC)Fe(CH2SiMe3)2
Single crystals of 4-Ns2 suitable for X-ray diffraction were obtained from a diethyl ether solution stored at –35 °C and a representation of the solid-state structure is depicted in Figure 4. The coordination environment around the iron is between square pyramidal and trigonal bipyramidal with the two neosilyl ligands in a mutually trans disposition (C20–Fe1–C24 = 148.63(7)°) and in plane with the two carbene ligands. The tBu-groups shield the sixth coordination site, likely preventing coordination of dinitrogen and resulting in the isolation of 4-Ns2 as a five-coordinate, 16-electron complex.
Figure 4.

Solid-state molecular structure of (tBuACNC)Fe(CH2SiMe3)2 (4-Ns2) at 30% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (deg): C10–Fe1 1.9467(17), C11–Fe1 1.9392(17), C20–Fe1 2.0633(16), C24–Fe1 2.0716(17), Fe1–N3 1.8205(14); N3–Fe1–C11 81.53(6), N3–Fe1–C10 81.55(6), C11–Fe1–C10 163.03(7), C20–Fe1–C24 148.63(7).
The diamagnetism of 4-Ns2 is noteworthy as other five-coordinate iron dialkyl complexes with tridentate pincer ligands usually adopt a trigonal bipyramidal geometry and have either S =1 or 2 ground states.8j-l,9,13 To our knowledge, an octahedral coordination environment has thus far been a prerequisite for diamagnetism among iron dialkyl complexes.14 The higher anisotropy of the electron density around the Fe(II) center this coordination environment confers has implications for Mößbauer spectroscopy. The zero-field 57Fe Mößbauer spectrum of 4-Ns2 exhibits a doublet with an isomer shift of 0.13 mm/s and a quadrupolar splitting of 3.89 mm/s, the latter being significantly higher than observed for the octahedral, N2-containing complexes as well as trigonal bipyramidal, paramagnetic iron bis(neosilyl) complexes (Figure 3d).8j Similar behavior was also observed with square-planar, ferrous complexes when compared to their tetrahedral counterparts.15
Synthesis of (ACNC)Fe(CH2SiMe3)2 analogs with smaller alkyl substituents on the imidazol-2-ylidene were unsuccessful as addition of MeACNC (MeACNC = 2,6-(Me-imidazol-2-ylidene)2pyridine) to (py)2Fe(CH2SiMe3)2 yielded a black solid, which was insoluble in benzene-d6 and did not exhibit any assignable resonances in the 1H NMR spectrum in THF-d8. Iron dialkyl complexes with the pincer containing saturated N-heterocyclic carbenes were not obtained using this method as deprotonation of (H4-iPrCNC)(HBr)2 and (H4-MesCNC)(HBr)2 produced a complex mixture of unidentified products, likely due to the decreased stability of free imidazolin-2-ylidenes when compared to their unsaturated counterparts.16
With the various (CNC)Fe dialkyl complexes in hand, reactivity studies were conducted, specifically focused on the generation of reduced or hydride complexes, with potential application as hydrogenation and HIE catalysts.3c,5,6 First, ligand-induced reductive elimination of the alkyl ligands was targeted.14a,17 Exposure of a diethyl ether solution of 1-Ns2(N2) to a carbon monoxide atmosphere did not yield the reductive elimination product (iPrCNC)Fe(CO)23b,11 but resulted in clean substitution of the dinitrogen ligand (Scheme 5). The diamagnetic iron product, (iPrCNC)Fe(CH2SiMe3)2(CO) (1-Ns2(CO)) was isolated in 81% yield following recrystallization from a Et2O/pentane mixture and was stable under an N2 atmosphere. As expected, both the 1H and 13C NMR spectra exhibited similar features to 1-Ns2(N2). The zero-field, solid-state 57Fe Mößbauer spectrum exhibited a doublet with an isomer shift of –0.03 mm/s and a quadrupole splitting of 0.65 mm/s. Coordination of CO was also corroborated by infrared spectroscopy in toluene solution (ν(CO) = 1919 cm−1).
Scheme 5.

Synthesis of (iPrCNC)Fe(CH2SiMe3)2(CO)
Single crystals suitable of 1-Ns2(CO) for X-ray diffraction were obtained from a pentane solution at –35 °C and a representation of the solid-state structure presented in Figure 5 establishes an idealized octahedral geometry nearly identical to 1-Ns2(N2) with the CO ligand trans to the pyridine and two alkyl ligands mutually trans. While the Fe-carbene and Fe-alkyl bond lengths are similar to those in 1-Ns2(N2), the Fe-pyridine bond length is significantly longer (1.925(2) Å in 1-Ns2(CO), 1.829(3) Å in 1-Ns2(N2)), likely a result of the stronger trans influence of the CO ligand as compared to N2.
Figure 5.

Solid-state molecular structure of (iPrCNC)Fe(CH2SiMe3)2(CO) (1-Ns2(CO)) at 30% probability ellipsoids. Hydrogen atoms and one aryl substituent are omitted for clarity. Selected bond distances (Å) and angles (deg): C44–Fe1 1.730(3), N3–Fe1 1.925(2), C10–Fe1 1.931(3), C11–Fe1 1.915(3), C36–Fe1 2.137(2), C40–Fe1 2.154(3); C44–Fe1–C11 100.45(12), C44–Fe1–N3 178.25(11), C11–Fe1–N3 79.81(10), C44–Fe1–C10 100.14(12), C11–Fe1–C10 159.37(12), N3–Fe1–C10 79.64(10), C36–Fe1–C40 170.16(11).
Addition of the stronger field ligand, carbon monoxide to 4-Ns2 overcomes the apparent steric penalty associated with formation of a six-coordinate complex as diamagnetic 4-Ns2(CO) was isolated. CO coordination was confirmed by the observation of a strong band centered at 1919 cm−1 in the benzene-d6 solution infrared spectrum. The 1H NMR spectrum of 4-Ns2(CO) also differs slightly from 4-Ns2 with the methylene groups of the neosilyl ligand (−1.34 ppm) and 4-pyridine (7.10 ppm) protons significantly shifted upfield, comparable to the spectroscopic features observed with 1-Ns2(CO).
Given the performance of [(CNC)Fe] complexes in catalytic alkene hydrogenation and HIE chemistry,6 hydrogenolysis of the iron dialkyl complexes was studied (Scheme 6a). Exposure of a benzene-d6 solution of 1-Ns2(N2) to 4 atm of H2 resulted in a color change from violet to brown. Monitoring the progress of the reaction by 1H NMR spectroscopy revealed formation of SiMe4 along with a new diamagnetic product with upfield resonances between −8 and −10 ppm, consistent with formation of iron hydrides (Scheme 6b). In analogy to spectroscopic features reported for (H4-iPrCNC)Fe(N2)2 under similar reaction conditions,3c these resonances were assigned to the isotopologues of (iPrCNC)FeH4, generated from hydrogenolysis of the iron alkyl ligands and subsequent HIE with the benzene-d6 solvent. Performing a similar procedure with 2-Ns2(N2) and 3-Ns2(N2) also produced 1H NMR signals consistent with formation of isotopologues and isotopomers of the corresponding iron dihydride-dihydrogen complexes.3c Exposure of 4-Ns2 to dihydrogen also liberated SiMe4, but no signals assignable to an organometallic iron compound were observed.
Scheme 6.

(a) Reaction of (CNC)Fe(CH2SiMe3)2(N2) complexes with H2 in the Presence of C6D6 and (b) High-Field Excerpts of the Corresponding 1H NMR Spectra
Attention was also devoted to the synthesis of reduced iron dinitrogen complexes, (CNC)Fe(N2)2 using the dialkyl derivatives as precursors. Such routes would obviate harsh and hazardous alkali metal reduction reactions. The aryl-susbtituted compound, 3-Ns2(N2) was chosen as a representative example (Scheme 7). Stirring a suspension of 3-Ns2(N2) in pentane successively under 4 atm of H2 followed by 1 atm of N2 resulted in the isolation of (3,5-Me2MesCNC)Fe(N2)2 (3-(N2)2) as a diamagnetic, brown solid in 96% yield. The same complex was also obtained in 94% isolated yield following addition of 1.2 equiv of HBPin (Pin = pinacolato) to a pentane suspension of 3-Ns2(N2). When this procedure was conducted in a sealed NMR tube in benzene-d6, concomitant formation of a 1:1 mixture of TMS and PinBCH2SiMe3 was observed by 1H NMR spectroscopy, supporting initial generation of an iron alkyl-hydride or alkyl-boryl complex and subsequent reductive elimination. Excess HBPin was detected in the reaction mixture by 1H NMR spectroscopy, indicating that HBPin does not undergo irreversible oxidative addition to the reduced iron product. This observation is also supported by the near quantitative isolated yield of 3-Ns2(N2). Addition of HBPin to a benzene-d6 solution of 4-Ns2 produced no reaction over the course of one hour at ambient temperature. Heating the mixture to 70 ºC induced loss of SiMe4 and formation of PinBCH2SiMe3 but no defined organometallic product could be identified by 1H NMR spectroscopy.
Scheme 7.
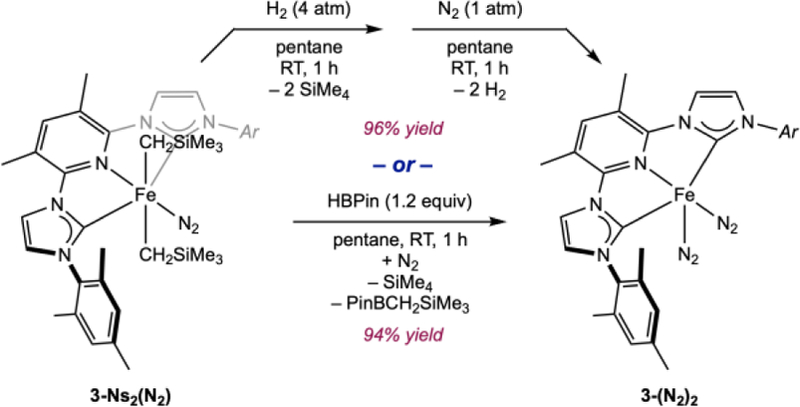
Synthesis of (3,5-Me2MesCNC)Fe(N2)2
The benzene solution infrared spectrum of 3-(N2)2 displayed four strong bands assigned as N2 stretches, the relative intensities of which were concentration dependent. Under dilute conditions (~0.007 M), the bands at 2094 cm−1 and 2022 cm−1 were dominant and assigned to the monomeric, five-coordinate bis(dinitrogen) complex, (3,5-Me2MesCNC)Fe(N2)2 (Figure 6a). Recording the infrared data in the solid state (KBr pellet) also produced four bands, likely due to the presence of both monomer and dimer ([(3,5-Me2MesCNC)Fe(N2)]2(µ2-N2)). These features are reminiscent of (MesCNC)Fe(N2)2, where both monomer and dimer were observed in the crystal lattice.5 The equilibrium process was also detected by 1H NMR spectroscopy as seven broad singlets were observed at ambient temperature while the 13C NMR spectrum needed to be recorded in dilute solution (<0.007 M) to observe the signals with the carbene carbons detected at 205.1 ppm. Mößbauer spectroscopy was used to assess the purity and identity of the obtained 3-(N2)2 complex, which exhibited a clean doublet with an isomer shift of 0.29 mm/s and quadrupole splitting of 0.55 mm/s, very similar to the features observed for (iPrCNC)Fe(N2)2 (Figure 6b).11
Figure 6.

Benzene solution IR spectra of (3,5-Me2MesCNC)Fe(N2)2 3-(N2)2 in C6D6 at low (0.007 M upper spectrum) and high (0.03 M, lower spectrum) concentration. (b) Zero-field 57Fe Mößbauer spectrum of (3,5-Me2MesCNC)Fe(N2)2 at 80 K in the solid state. Simulated parameters (blue line) are δ = 0.29 mm/s, |ΔEQ| = 0.55 mm/s.
CONCLUDING REMARKS
A synthetic method has been developed to access a series of (CNC)Fe dialkyl complexes. With aryl-substituted N-heterocyclic carbenes on the pincer, dinitrogen coordinates to the position trans to the pyridine of the chelate to form diamagnetic, idealized octahedral low-spin iron(II) complexes. The dinitrogen ligand is labile and in one instance was displaced by CO to from the corresponding carbonyl complex. The synthetic route to iron dialkyl complexes was also compatible with iron complexes bearing tert-butyl rather than aryl substituents on the carbene portion of the pincer. The resulting iron dialkyl complex is five-coordinate and diamagnetic but exhibits NMR spectroscopic features to suggest contributions from a paramagnetic excited state. Hydrogenation of the aryl-substituted iron dialkyl complexes generated isotopologues and isotopomers of the resulting iron hydrides, demonstrating activity in catalytic hydrogen isotope exchange with benzene-d6. The iron bis(dinitrogen) complex, (3,5-Me2MesCNC)Fe(N2)2 was synthesized from (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) by successive reaction with H2 and N2 or reaction with HBPin in the presence of N2. This straightforward method to access reduced (CNC)Fe complexes and in situ generation of (CNC)Fe hydride complexes circumvents the use of Fe[N(SiMe3)2]2, metallic reductants and isolation of very sensitive dinitrogen complexes and permits a higher variability of the ligand substituents. It will therefore allow a more rapid assessment of catalytic activity in hydrogenation and HIE, and generally facilitate reactivity studies of (CNC)Fe complexes. Studies along these lines are currently underway in our laboratory.
EXPERIMENTAL SECTION
General Considerations.
All air- and moisture-sensitive manipulations were carried out using vacuum line, Schlenk and cannula techniques or in an MBraun inert atmosphere (nitrogen) dry box unless otherwise noted. All glassware was stored in a pre-heated oven prior to use. The solvents used for air- and moisture-sensitive manipulations were dried and deoxygenated using literature procedures.18 Hydrogen gas was purchased from Airgas National Welders and passed through a column of MnO2 supported on vermiculite and 3 Å molecular sieves prior to use on a Schlenk manifold. HBpin and KHMDS were purchased from Sigma Aldrich and used as received. LiCH2SiMe3 (1.0 m in pentane) was purchased from Sigma Aldrich, concentrated, recrystallized from pentane at –35 °C and dried under vacuum to obtain LiCH2SiMe3 as a colorless crystalline solid which was stored at –35 °C. The following compounds were prepared according to literature procedures: 2,6-diiodo-3,5-dimethylpyridine,19 1-mesityl-1H-imidazole,20 iPrCNC(HBr)2,21 MesCNC(HBr)2,21 tBuACNC(HBr)2,22 (py)4FeCl2.23
1H NMR spectra were recorded on either Bruker AVANCE 300 or 500 spectrometers operating at 300.13 MHz, and 500.46 MHz, respectively. All 1H and 13C NMR chemical shifts are reported in ppm relative to SiMe4 using the 1H (chloroform-d: 7.26 ppm; benzene-d6: 7.16 ppm) and 13C (chloroform-d: 77.16 ppm; benzene-d6: 128.06 ppm) chemical shifts of the solvent as a standard. 1H NMR data for diamagnetic compounds are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, sept = septet, br = broad, m = multiplet), coupling constants (Hz), integration, assignment. Elemental analyses were performed at Robinson Microlit Laboratories, Inc., in Ledgewood, NJ. Infrared spectroscopy was conducted on a Thermo-Nicolet iS10 FT-IR spectrometer calibrated with a polystyrene standard. For some iron dinitrogen complexes a satisfactory elemental analysis could not be achieved, which is likely due to the highly sensitive nature of these compounds. The identity and purity of the bulk material can however adequately be assessed by the combination of NMR and Mößbauer spectroscopies.
Zero-field 57Fe Mößbauer spectra were recorded on a SEE Co. Mößbauer spectrometer (MS4) at 80 K in constant acceleration mode. 57Co/Rh was used as the radiation source. WMOSS software24 was used for the quantitative evaluation of the spectral parameters (least-squares fitting to Lorentzian peaks). The temperature of the sample was controlled by a Janis Research Co. CCS-850 He/N2 cryostat within an accuracy of 0.3 K. Isomer shifts were determined relative to α-iron at 298 K.
Single crystals suitable for X-ray diffraction were coated with polyisobutylene oil in a drybox, transferred to a nylon loop and then quickly transferred to the goniometer head of a Bruker SMART APEX DUO diffractometer equipped with a molybdenum X-ray tube (λ = 0.71073 Å) and a Cu X-ray tube (λ = 1.54178 Å). Preliminary data revealed the crystal system. The data collection strategy was optimized for completeness and redundancy using the Bruker COSMO software suite. The space group was identified, and the data were processed using the Bruker SAINT+ program and corrected for absorption using SADABS. The structures were solved using direct methods (SHELXS) completed by subsequent Fourier synthesis and refined by full-matrix least-squares procedures.
Preparation of 3,5-Me2MesCNC(HI)2.
A glass ampoule was filled with 2,6-diiodo-3,5-dimethylpyridine (1.44 g, 4.0 mmol, 1.0 equiv) and 1-mesityl-1H-imidazole (2.24 g, 12 mmol, 3.0 equiv). The ampoule was attached to a Schlenk line, evacuated, and then flame sealed. The sealed ampoule was placed in an oven at 200 °C for 7 days, allowed to cool to RT and opened. The solid residue was taken up in CH2Cl2 and the resulting mixture was concentrated to a volume of ca. 5 mL. Et2O (ca. 400 mL) were added and the resulting precipitate was collected through filtration and dried to yield 2.84 g (97% yield) of 3,5-Me2MesCNC(HI)2 as a sandy solid. 1H NMR (500 MHz, CDCl3, 25 ºC): δ 10.73 (t, J = 1.6 Hz, 2H, 2-imidazolium H), 8.87 (t, J = 1.8 Hz, 2H, 4/5-imidazolium H), 7.98 (s, 1H, p-pyr H), 7.35 (t, J = 1.8 Hz, 2H, 4/5-imidazolium H), 7.02 (s, 4H, Ar-H), 2.62 (s, 6H, m-pyr-(CH3)2), 2.33 (s, 6H, 4-Ar-CH3), 2.16 (s, 12H, 2,6-Ar-(CH3)2). 13C{1H} NMR (126 MHz, CDCl3, 25 ºC): δ 147.6 (p-pyr C), 142.3 (m-pyr C), 141.8 (4-Ar C), 138.3 (2-imidazolium C), 134.0 (2,6-Ar C), 131.7 (o-pyr-C), 130.3 (1-Ar C), 130.2 (3,5-Ar C), 124.9 (4/5-imidazolium C), 123.2 (4/5-imidazolium C), 21.2 (4-Ar-CH3), 18.3 (m-pyr-(CH3)2), 18.1 (2,6-Ar-(CH3)2)
Preparation of iPrCNC.
iPrCNC was prepared using a slightly modified literature procedure.10,25 In a N2-filled glovebox a 100 mL round bottom flask was charged with iPrCNC(HBr)2 (347 mg, 0.5 mmol, 1.0 equiv) and THF (ca. 10 mL) and the resulting suspension was cooled in the cold well (–196 °C) for 20 min. A 20 mL scintillation vial was charged with KHMDS (210 mg, 1.05 mmol, 2.1 equiv) and THF (ca. 5 mL) and the resulting suspension was cooled in the freezer (–35 °C) for 20 min. The cold solution of KHMDS in THF was added to the cold suspension of MesCNC(HBr)2 and the resulting mixture was allowed to warm to RT while being stirred for 1 h. All volatiles were removed in vacuo and the residue was triturated in pentane (ca. 20 mL). All volatiles were removed in vacuo and the residue was taken up in toluene (ca. 30 mL). The mixture was filtered through celite (eluent: toluene) and all volatiles were removed from the filtrate in vacuo to yield 264 mg (99% yield) of iPrCNC as an off-white solid. 1H NMR (400 MHz, C6D6, 25 ºC): δ 8.44 (d, J = 8.0 Hz, 2H), 8.06 (d, J = 1.7 Hz, 2H), 7.26 (dd, J = 8.4, 7.1 Hz, 2H), 7.16 – 7.11 (m, 4H), 7.07 (t, J = 8.0 Hz, 1H), 6.62 (d, J = 1.7 Hz, 2H), 2.91 (hept, J = 6.9 Hz, 4H), 1.20 (d, J = 6.8 Hz, 12H), 1.11 (d, J = 6.9 Hz, 12H). The characterization data matched previously reported data.10
Preparation of MesCNC.
MesCNC was prepared in an analogous fashion to iPrCNC using MesCNC(HBr)2 (609 mg, 1.0 mmol, 1.0 equiv) and KHMDS (419 mg, 2.1 mmol, 2.1 equiv). This procedure yielded 430 mg (96% yield) of MesCNC as an off-white solid. 1H NMR (300 MHz, C6D6, 25 ºC): δ 8.54 (d, J = 8.0 Hz, 2H), 8.12 (d, J = 1.8 Hz, 2H), 7.11 (t, J = 8.0 Hz, 1H), 6.81 (s, 4H), 6.46 (d, J = 1.7 Hz, 2H), 2.15 (s, 6H), 2.13 (s, 12H). The characterization data matched previously reported data.25
Preparation of 3,5-Me2MesCNC.
3,5-Me2MesCNC was prepared in an analogous fashion to iPrCNC using 3,5-Me2MesCNC(HI)2 (731 mg, 1.0 mmol, 1.0 equiv) and KHMDS (419 mg, 2.1 mmol, 2.1 equiv). This procedure yielded 470 mg (99% yield) of 3,5-Me2MesCNC as an off-white solid. 1H NMR (400 MHz, C6D6, 25 ºC): δ 7.70 (d, J = 1.7 Hz, 2H, 4/5-imidazolylidene H), 7.12 (s, 1H, p-pyr H), 6.80 (s, 4H, Ar-H), 6.43 (d, J = 1.7 Hz, 2H, 4/5-imidazolylidene H), 2.73 (s, 6H, m-pyr-(CH3)2), 2.15 (s, 6H, 4-Ar-CH3), 2.12 (s, 12H, 2,6-Ar-(CH3)2). 13C{1H} NMR (101 MHz, C6D6, 25 ºC): δ 221.3 (2-imidazolylidene C), 148.9 (o-pyr C), 146.6 (p-pyr C), 139.1 (Ar C), 137.4 (Ar C), 135.5 (Ar C), 129.2 (3,5-Ar C), 124.9 (m-pyr C), 119.98 (4/5-imidazolylidene C), 119.96 (4/5-imidazolylidene C), 21.0 (4-Ar-CH3), 20.5 (m-pyr-(CH3)2), 18.2 (2,6-Ar-(CH3)2).
Preparation of tBuACNC.
tBuACNC was prepared in an analogous fashion to iPrCNC using tBuACNC(HBr)2 (485 mg, 1.0 mmol, 1.0 equiv) and KHMDS (419 mg, 2.1 mmol, 2.1 equiv). This procedure yielded 270 mg (83% yield) of tBuACNC as a yellow-orange solid. 1H NMR (400 MHz, C6D6, 25 ºC): δ 8.51 (d, J = 8.0 Hz, 2H, m-pyr H), 8.07 (d, J = 1.9 Hz, 2H, 4/5-imidazolylidene H), 7.18 (t, J = 8.0 Hz, 1H, p-pyr H), 6.77 (d, J = 1.8 Hz, 2H, 4/5-imidazolylidene H), 1.49 (s, 18H, C(CH3)3). 13C{1H} NMR (101 MHz, C6D6, 25 ºC): δ 216.2 (2-imidazolylidene C), 153.1 (o-pyr C), 140.3 (p-pyr C), 116.6 (4/5-imidazolylidene C), 116.1 (4/5-imidazolylidene C), 111.0 (m-pyr C), 56.5 (C(CH3)3), 31.2 (C(CH3)3).
Preparation of (iPrCNC)Fe(CH2SiMe3)2(N2).
In a N2-filled glovebox a 20 mL scintillation vial was charged with (iPrCNC)FeBr2 (500 mg, 0.67 mmol, 1.0 equiv) and Et2O (ca. 5 mL) and the resulting slurry was cooled in the freezer (–35 °C) for 20 min. Another 20 mL scintillation vial was charged with LiCH2SiMe3 (125 mg, 1.33 mmol, 2.0 equiv) and Et2O (ca. 10 mL) and the resulting solution was likewise cooled in the freezer (–35 °C) for 20 min. The cold solution of LiCH2SiMe3 was added dropwise to the cold and rapidly stirred slurry of (iPrCNC)FeBr2 and the resulting mixture was allowed to warm to RT while being stirred for 3 h. All volatiles were removed in vacuo, the residue was extracted with pentane, and the resulting mixture was filtered through Celite to give a purple solution. All volatiles were removed in vacuo and the residue recrystallized from pentane at –35 °C to yield 325 mg (61% yield) of (iPrCNC)Fe(CH2SiMe3)2(N2) as a violet solid. Single crystals suitable for X-ray diffraction were grown from a concentrated solution in pentane at –35 °C. IR (C6D6): νNN = 2129 cm-1. 1H NMR (400 MHz, C6D6, 25 ºC): δ 7.23 (d, J = 2.1 Hz, 2H, 4/5-imidazolylidene H), 7.18 (t, J = 9.2 Hz, 2H, p-Ar H), 7.11 (d, J = 7.6 Hz, 4H, m-Ar H), 7.00 (t, J = 7.8 Hz, 1H, p-pyr H), 6.75 (d, J = 2.1 Hz, 2H, 4/5-imidazolylidene H), 6.61 (d, J = 7.8 Hz, 2H, m-pyr H), 3.28 (sept, J = 6.8 Hz, 4H, CH(CH3)2), 1.39 (d, J = 6.7 Hz, 12H, CH(CH3)2), 1.08 (d, J = 6.8 Hz, 12H, CH(CH3)2), −0.50 (s, 18H, CH2Si(CH3)3), −1.11 (s, 4H, CH2Si(CH3)3). 13C{1H} NMR (101 MHz, C6D6, 25 ºC): δ 223.1 (2-imidazolylidene C), 153.6 (o-pyr C), 147.3 (Ar C), 136.7 (Ar C), 129.9 (p-Ar C), 126.6 (4/5-imidazolylidene C), 126.4 (p-pyr C), 124.0 (m-Ar C), 112.8 (4/5-imidazolylidene C), 100.00 (m-pyr C), 28.6 (CH(CH3)2), 27.1 (CH(CH3)2), 23.0 (CH(CH3)2), 2.8 (CH2Si(CH3)3), −15.0 (CH2Si(CH3)3, located by HSQC analysis).
Alternative Preparation of (iPrCNC)Fe(CH2SiMe3)2(N2).
In a N2-filled glovebox a 20 mL scintillation vial was charged with (py)4FeCl2 (260 mg, 0.59 mmol, 1.2 equiv) and pentane (ca. 5 mL) and the resulting suspension was cooled in the freezer (–35 °C) for 20 min. Another 20 mL scintillation vial was charged with LiCH2SiMe3 (113 mg, 1.20 mmol, 2.46 equiv) and pentane (ca. 5 mL) and the resulting solution was likewise cooled in the freezer (–35 °C) for 20 min. The cold solution of LiCH2SiMe3 was added to the cold suspension of (py)4FeCl2 and the resulting mixture was allowed to warm to RT while being stirred for 1–2 h. The resulting violet reaction mixture was filtered through Celite (eluent: pentane) and all volatiles were removed in vacuo. The residue was taken up in pentane (ca. 5 mL) and the resulting solution containing (py)2Fe(CH2SiMe3)2 was cooled in the freezer (–35 °C) for 20 min. A 100 mL round bottom flask was charged with iPrCNC (260 mg, 0.49 mmol, 1.0 equiv) and cold pentane (ca. 30 mL, –35 °C). The cold solution containing (py)2Fe(CH2SiMe3)2 was added to the cold suspension of MesCNC and the resulting mixture was allowed to warm to RT while being stirred for 1 h. All volatiles were removed in vacuo from the resulting violet reaction mixture and the remaining solid was washed with cold pentane (3 × 3 mL) and dried under vacuum to yield 260 mg (67% yield) of (iPrCNC)Fe(CH2SiMe3)2(N2) as a violet solid. All characterization data matched the material obtained above.
Preparation of (MesCNC)Fe(CH2SiMe3)2(N2).
In a N2-filled glovebox a 20 mL scintillation vial was charged with (py)4FeCl2 (511 mg, 1.15 mmol, 1.2 equiv) and pentane (ca. 10 mL) and the resulting suspension was cooled in the freezer (–35 °C) for 20 min. Another 20 mL scintillation vial was charged with LiCH2SiMe3 (222 mg, 2.36 mmol, 2.46 equiv) and pentane (ca. 5 mL) and the resulting solution was likewise cooled in the freezer (–35 °C) for 20 min. The cold solution of LiCH2SiMe3 was added to the cold suspension of (py)4FeCl2 and the resulting mixture was allowed to warm to RT while being stirred for 1–2 h. The resulting violet reaction mixture was filtered through Celite (eluent: pentane) and all volatiles were removed in vacuo. The residue was taken up in pentane (ca. 5 mL) and the resulting solution containing (py)2Fe(CH2SiMe3)2 was cooled in the freezer (–35 °C) for 20 min. A 100 mL round bottom flask was charged with MesCNC (430 mg, 0.96 mmol, 1.0 equiv) and cold pentane (ca. 30 mL, –35 °C). The cold solution containing (py)2Fe(CH2SiMe3)2 was added to the cold suspension of MesCNC and the resulting mixture was allowed to warm to RT while being stirred for 1 h during which time a microcrystalline solid precipitated from the reaction mixture. The resulting violet suspension was concentrated to ca. 10 mL and filtered. The solid was washed with cold pentane (3 × 3 mL) and dried under vacuum to yield 487 mg (72% yield) of (MesCNC)Fe(CH2SiMe3)2(N2) as a violet-brown solid. Anal Calcd for C37H51FeN7Si2: C, 62.96; H, 7.28; N, 13.89. Found: C, 62.58; H, 6.79; N, 13.72. IR (C6D6): νNN = 2126 cm-1. 1H NMR (300 MHz, C6D6, 25 ºC): δ 7.21 (s, 2H, 4/5-imidazolylidene H), 7.11 (t, J = 7.8 Hz, 1H, p-pyr H), 6.71 (s, 4H, Ar H), 6.61 (d, J = 7.8 Hz, 2H, m-pyr H), 6.36 (s, 2H, 4/5-imidazolylidene H), 2.27 (s, 12H, 2,6-Ar-(CH3)2), 2.03 (s, 6H, 4-Ar-CH3), −0.49 (s, 18H, CH2Si(CH3)3), −0.73 (s, 4H, CH2Si(CH3)3). 13C{1H} NMR (126 MHz, C6D6, 25 ºC): δ 153.3 (o-pyr C), 138.3 (4-Ar C), 137.0 (Ar C), 136.4 (Ar C), 129.5 (3-Ar C), 126.0 (p-pyr C), 124.9 (4/5-imidazolylidene C), 113.3 (4/5-imidazolylidene C), 100.3 (m-pyr C), 21.0 (4-Ar-CH3), 19.2 (2,6-Ar-(CH3)2), 2.9 (CH2Si(CH3)3). (CH2Si(CH3)3 and 2-imidazolylidene C not located).
Preparation of (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2).
(3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) was prepared in an analogous fashion to (MesCNC)Fe(CH2SiMe3)2(N2) using 3,5-Me2MesCNC (412 mg, 0.89 mmol, 1.0 equiv), (py)4FeCl2 (460 mg, 1.04 mmol, 1.2 equiv) and LiCH2SiMe3 (201 mg, 2.13 mmol, 2.46 equiv). This procedure yielded 564 mg (90% yield) of (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) as a violet/brown solid. Single crystals suitable for X-ray diffraction were grown from a concentrated solution in Et2O/pentane at –35 °C. Anal Calcd for C39H55FeN7Si2: C, 63.82; H, 7.55; N, 13.36. Found: C, 63.49; H, 7.56; N, 12.89. IR (C6D6): νNN = 2118 cm-1. 1H NMR (500 MHz, C6D6, 25 ºC): δ 7.70 (s, 2H, 4/5-imidazolylidene H), 6.71 (s, 4H, Ar H), 6.42 (s, 1H, p-pyr H), 6.35 (s, 2H, 4/5-imidazolylidene H), 2.30 (s, 12H, 2,6-Ar-(CH3)2), 2.29 (s, 6H, m-pyr-(CH3)2), 2.03 (s, 6H, 4-Ar-CH3), −0.45 (s, 18H, CH2Si(CH3)3), −1.01 (s, 4H, CH2Si(CH3)3). 13C{1H} NMR (126 MHz, C6D6, 25 ºC): δ 150.9 (o-pyr C), 138.1 (4-Ar C), 137.0 (Ar C), 136.5 (Ar C), 134.5 (p-pyr C), 129.5 (3-Ar C), 124.1 (4/5-imidazolylidene C), 116.3 (4/5-imidazolylidene C), 111.0 (m-pyr C), 21.0 (4-Ar-CH3), 19.3 (2,6-Ar-(CH3)2), 18.7 (m-pyr-(CH3)2), 2.8 (CH2Si(CH3)3). (CH2Si(CH3)3 and 2-imidazolylidene C not located).
Preparation of (tBuACNC)Fe(CH2SiMe3)2.
(tBuACNC)Fe(CH2SiMe3)2 was prepared in an analogous fashion to (MesCNC)Fe(CH2SiMe3)2(N2) using tBuACNC (187 mg, 0.58 mmol, 1.0 equiv), (py)4FeCl2 (307 mg, 0.69 mmol, 1.2 equiv) and LiCH2SiMe3 (134 mg, 1.42 mmol, 2.46 equiv). This procedure yielded 247 mg (77% yield) of (tBuACNC)Fe(CH2SiMe3)2 as a brown solid. Single crystals suitable for X-ray diffraction were grown from a concentrated solution in Et2O at –35 °C. Anal Calcd for C27H47FeN5Si2: C, 58.57; H, 8.56; N, 12.65. Found: C, 58.37; H, 8.12; N, 12.66. 1H NMR (400 MHz, C6D6, 25 ºC): δ 11.01 (t, J = 7.6 Hz, 1H, p-pyr H), 8.34 (s, 4H, CH2Si(CH3)3), 8.26 (s, 2H, 4/5-imidazolylidene H), 8.23 (d, J = 7.8 Hz, 2H, m-pyr H), 7.89 (s, 2H, 4/5-imidazolylidene H), 1.94 (s, 18H, C(CH3)3), −0.87 (s, 18H, CH2Si(CH3)3). 13C{1H} NMR (101 MHz, C6D6, 25 ºC): δ 151.3 (o- or p-pyr C), 125.1 (4/5-imidazolylidene C), 120.5 (m-pyr C), 105.9 (4/5-imidazolylidene C), 81.7 (CH2Si(CH3)3, located by HSQC analysis), 58.4 (C(CH3)3), 35.8 (C(CH3)3), 1.6 (CH2Si(CH3)3). (2-imidazolylidene C and o- or p-pyr C not located).
Preparation of (iPrCNC)Fe(CH2SiMe3)2(CO).
In a N2-filled glovebox a 50 mL Schlenk tube with a Teflon valve was charged with (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) (125 mg, 0.16 mmol, 1.0 equiv) and Et2O (ca. 15 mL). The tube was sealed, brought outside of the glovebox and attached to a Schlenk line where the solution was frozen (–196 °C, liquid nitrogen). Following evacuation of the headspace, CO (1 atm) was admitted. The tube was sealed and allowed to warm to RT upon which a rapid color change from purple to pink occurred and the resulting suspension was vigorously stirred for 30 min. The resulting reaction mixture was filtered through celite and all volatiles were removed in vacuo. The residue was recrystallized from a Et2O/pentane mixture to yield 101 mg (81% yield) of (iPrCNC)Fe(CH2SiMe3)2(CO) as a burgundy solid. Anal Calcd for C44H63FeN5OSi2: C, 66.89; H, 8.04; N, 8.86. Found; C, 66.74; H, 7.97; N, 8.69. IR (toluene): νCO = 1919 cm-1. 1H NMR (400 MHz, C6D6, 25 ºC): δ 7.22 (t, J = 8 Hz, 2H, p-Ar H), 7.09 – 7.15 (m, 7H, 4/5-imidazolylidene H, m-Ar H, p-pyr H), 6.63 – 6.67 (m, 4H, 4/5-imidazolylidene H, m-pyr H), 3.11 (sept, J = 7 Hz, 4H, CH(CH3)2), 1.39 (d, J = 7 Hz, 12H, CH(CH3)2), 1.05 (d, J = 6.8 Hz, 12H, CH(CH3)2), −0.49 (s, 18H, CH2Si(CH3)3), −1.43 (s, 4H, CH2Si(CH3)3). 13C{1H} NMR (101 MHz, C6D6, 25 ºC): δ 227.6 (2-imidazolylidene C), 151.6 (o-pyr C), 147.0 (Ar C), 136.7 (Ar C), 130.0 (p-Ar C), 126.4 (4/5-imidazolylidene C), 124.1 (m-Ar C), 112.7 (4/5-imidazolylidene C), 100.4 (m-pyr C), 28.5 (CH(CH3)2), 27.0 (CH(CH3)2), 23.0 (CH(CH3)2), 2.9 (CH2Si(CH3)3) (CH2Si(CH3)3, CO and p-pyr C not located).
Observation of (tBuACNC)Fe(CH2SiMe3)2(CO).
In a N2-filled glovebox a 20 mL scintillation vial was charged with (tBuACNC)Fe(CH2SiMe3)2 (11 mg, 0.02 mmol, 1.0 equiv) and C6D6 (ca. 0.5 mL). The brown solution was transferred to a J-Young Tube which was sealed, brought outside of the glovebox, and attached to a Schlenk line where the solution was frozen (–196 °C, liquid nitrogen). Following evacuation of the headspace, CO (1 atm) was admitted. The tube was sealed, and the solvent was slowly melted immediately resulting in a color change to purple. The contents of the tube were analyzed by 1H, 13C NMR and IR spectroscopies. IR (C6H6): νCO = 1899 cm-1. 1H NMR (400 MHz, C6D6, 25 ºC) δ 7.10 (t, J = 8.1 Hz, 1H, p-pyr H), 7.07 (d, J = 2.3 Hz, 2H, 4/5-imidazolylidine H), 6.74 (d, J = 2.3 Hz, 2H, 4/5-imidazolylidine H), 6.57 (d, J = 7.9 Hz, 2H, m-pyr H), 1.83 (s, 18H, C(CH3)3), −0.57 (s, 18H, CH2Si(CH3)3), −1.34 (s, 4H, CH2Si(CH3)3). 13C NMR (101 MHz, C6D6, 25 ºC) δ 223.9 (2-imidazolylidine C), 150.9 (o-pyr C), 132.2 (p-pyr C), 121.4 (4/5-imidazolylidine C), 111.7 (4/5-imidazolylidine C), 101.0 (m-pyr C), 58.6 (C(CH3)3), 31.7 (C(CH3)3), 2.3 (Si(CH3)3) (CO and CH2Si(CH3)3 not located).
Preparation of (3,5-Me2MesCNC)Fe(N2)2.
In a N2-filled glovebox a 20 mL scintillation vial was charged with (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) (200 mg, 0.26 mmol, 1.0 equiv) and pentane (ca. 5 mL). HBPin (45 μL, 0.3 mmol, 1.2 equiv) was added at RT and the resulting suspension was vigorously stirred for 1 h. The resulting brown suspension was filtered and the solid was washed with pentane (3 × 3 mL) and dried under vacuum to yield 143 mg (94% yield) of (3,5-Me2MesCNC)Fe(N2)2 as a brown solid. IR (C6D6): νNN = 2094 (major), 2055 (minor), 2022 (major), 1983 (minor) cm-1. 1H NMR (500 MHz, C6D6, 25 ºC): δ 7.86 (s, 2H, 4/5-imidazolylidene H), 6.79 (s, 4H, Ar H), 6.64 (s, 1H, p-pyr H), 6.48 (s, 2H, 4/5-imidazolylidene H), 2.43 (s, 6H, m-pyr-(CH3)2), 2.27 (s, 12H, 2,6-Ar-(CH3)2), 1.99 (s, 6H, 4-Ar-CH3). 13C{1H} NMR (126 MHz, C6D6, 25 ºC): δ 205.1 (2-imidazolylidene C), 140.1 (o-pyr C), 138.3 (4-Ar C), 137.4 (Ar C), 136.8 (Ar C), 129.4 (3-Ar C), 123.1 (4/5-imidazolylidene C), 122.0 (p-pyr C), 115.1 (4/5-imidazolylidene C), 110.7 (m-pyr C), 21.1 (4-Ar-CH3), 19.1 (m-pyr-(CH3)2), 18.1 ((2,6-Ar-(CH3)2)).
Alternative Preparation of (3,5-Me2MesCNC)Fe(N2)2.
In a N2-filled glovebox a 50 mL Schlenk tube with a Teflon valve was charged with (3,5-Me2MesCNC)Fe(CH2SiMe3)2(N2) (200 mg, 0.26 mmol, 1.0 equiv) and pentane (ca. 5 mL). The tube was sealed, brought outside of the glovebox and attached to a Schlenk line where the solution was frozen (–196 °C, liquid nitrogen). Following evacuation of the headspace, H2 (1 atm) was admitted at –196 °C. The tube was sealed and allowed to warm to RT and the resulting suspension was vigorously stirred for 1 h. The tube was brought back into the glovebox and opened, and the resulting brown suspension was transferred to a 20 mL scintillation vial and vigorously stirred for 1 h under an N2 atmosphere. The resulting brown suspension was filtered and the solid was washed with pentane (3 × 3 mL) and dried under vacuum to yield 146 mg (96% yield) of (3,5-Me2MesCNC)Fe(N2)2 as a brown solid. All characterization data matched the material obtained above.
Observation of (CNC)Fe hydrides and isotopologues.
In a N2-filled glovebox a J-Young NMR-tube was charged with a solution of (CNC)Fe(CH2SiMe3)2(N2) (10 mg) in C6D6 (0.5 mL). The tube was sealed, brought outside of the glovebox and attached to a Schlenk line and the contents of the tube were frozen in liquid nitrogen. Following evacuation of the headspace H2 (1 atm) was admitted at –196 °C. The tube was sealed, warmed to RT and inverted three times and the solution immediately analyzed by 1H NMR spectroscopy.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (R01 GM121441) for financial support.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
NMR Spectra (PDF)
Accession Codes
CCDC 1916329, 1916330, 1916331 and 1916332 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: + 441223 336033.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Labinger JA “Tutorial on Oxidative Addition” Organometallics 2015, 34, 4784–4795. [Google Scholar]
- (2).a)Fürstner A “Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion” ACS Cent. Sci 2016, 2, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chirik PJ; Wieghardt K “Radical Ligands Confer Nobility on Base-Metal Catalysts” Science 2010, 327, 794–795. [DOI] [PubMed] [Google Scholar]; c) Arevalo R; Chirik PJ “Enabling two-electron pathways with iron and cobalt: From ligand design to catalytic applications.” J. Am. Chem. Soc 2019, 141, 9106–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]; For other specific examples see: c) Sun J; Luo L; Luo Y; Deng L “An NHC-silyl-NHC pincer ligand for the oxidative addition of C–H, N–H, and O–H bonds to cobalt(I) complexes.” Angew. Chem. Int. Ed 2017, 56, 2720–2724. [DOI] [PubMed] [Google Scholar]; d) Tokmic K; Markus CR; Zhu L; Fout AR “Well-defined cobalt(I) dihydrogen catalyst: Eperimental evidence for a Co(I)-Co(III) redox process in olefin hydrogenation” J. Am. Chem. Soc 2016, 138, 11907–11913. [DOI] [PubMed] [Google Scholar]
- (3).a)Danopoulos AA; Pugh D; Smith H; Saßmannshausen J “Structural and Reactivity Studies of “Pincer” Pyridine Dicarbene Complexes of Fe0: Experimental and Computational Comparison of the Phosphine and NHC Donors” Chem. Eur. J 2009, 15, 5491–5502. [DOI] [PubMed] [Google Scholar]; b) Pugh D; Wells NJ; Evans DJ; Danopoulos AA “Reactions of ‘pincer’ pyridine dicarbene complexes of Fe(0) with silanes” Dalton Trans 2009, 7189–7195. [DOI] [PubMed] [Google Scholar]; c) Yu RP; Darmon JM; Semproni SP; Turner ZR; Chirik PJ “Synthesis of Iron Hydride Complexes Relevant to Hydrogen Isotope Exchange in Pharmaceuticals” Organometallics 2017, 36, 4341–4343. [Google Scholar]
- (4).Danopoulos AA; Wright JA; Motherwell WB “Molecular N2 complexes of iron stabilised by N-heterocyclic ‘pincer’ dicarbene ligands” Chem. Commun 2005, 784–786. [DOI] [PubMed] [Google Scholar]
- (5).Yu RP; Darmon JM; Hoyt JM; Margulieux GW; Turner ZR; Chirik PJ “High-Activity Iron Catalysts for the Hydrogenation of Hindered, Unfunctionalized Alkenes” ACS Catal 2012, 2, 1760–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yu RP; D. Hesk D; Rivera N; Pelczer I; Chirik PJ “Iron-catalysed tritiation of pharmaceuticals” Nature 2016, 529, 195–199. [DOI] [PubMed] [Google Scholar]
- (7).Andersen RA; Faegri K Jr.; Green JC; Haaland A; Lappert MF; Leung WP; Rypdal K “Synthesis of bis[bis(trimethylsilyl)amido]iron(II). Structure and bonding in M[N(SiMe3)2]2 (M = manganese, iron, cobalt): two-coordinate transition-metal amides.” Inorg. Chem 1988, 27, 1782–1786. [Google Scholar]
- (8).a)Obligacion JV; Chirik PJ “Mechanistic Studies of Cobalt-Catalyzed C(sp2)−H Borylation of Five-Membered Heteroarenes with Pinacolborane” ACS Catal 2017, 7, 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]; b)Palmer WN; Chirik PJ “Cobalt-Catalyzed Stereoretentive Hydrogen Isotope Exchange of C(sp3)–H Bonds” ACS Catal 2017, 7, 5674–5678. [DOI] [PMC free article] [PubMed] [Google Scholar]; c)Palmer WN; Obligacion JV; Pappas I; Chirik PJ “Cobalt-Catalyzed Benzylic Borylation: Enabling Polyborylation and Functionalization of Remote, Unactivated C(sp3)–H Bonds” J. Am. Chem. Soc 2016, 138, 766–769. [DOI] [PubMed] [Google Scholar]; d)Obligacion JV; Semproni SP; Pappas I; Chirik PJ “Cobalt-Catalyzed C(sp2)‑H Borylation: Mechanistic Insights Inspire Catalyst Design” J. Am. Chem. Soc 2016, 138, 10645–10653. [DOI] [PubMed] [Google Scholar]; e)Palmer WN; Diao T; Pappas I; Chirik PJ “High-Activity Cobalt Catalysts for Alkene Hydroboration with Electronically Responsive Terpyridine and α-Diimine Ligands” ACS Catal 2015, 5, 622–626. [Google Scholar]; f)Obligacion JV; Semproni SP; Chirik PJ “Cobalt-Catalyzed C−H Borylation” J. Am. Chem. Soc 2014, 136, 4133–4136. [DOI] [PubMed] [Google Scholar]; g)Hoyt JM; Shevlin M; Margulieux GW; Krska SW; Tudge MT; Chirik. PJ “Synthesis and Hydrogenation Activity of Iron Dialkyl Complexes with Chiral Bidentate Phosphines” Organometallics 2014, 33, 5781–5790. [Google Scholar]; h)Friedfeld MR; Margulieux GW; Schaefer BA; Chirik. PJ “Bis(phosphine)cobalt Dialkyl Complexes for Directed Catalytic Alkene Hydrogenation” J. Am. Chem. Soc 2014, 136, 13178–13181. [DOI] [PubMed] [Google Scholar]; i)Friedfeld MR; Shevlin M; Hoyt JM; Krska SW; Tudge MT; Chirik. PJ “Cobalt Precursors for High-Throughput Discovery of Base Metal Asymmetric Alkene Hydrogenation Catalysts” Science 2013, 342, 1076–1080. [DOI] [PubMed] [Google Scholar]; j)Tondreau AM; Atienza CCH; Darmon JM; Milsmann C; Hoyt HM; Weller KJ; Nye SA; Lewis KM; Boyer J; Delis JGP; Lobkovsky E; Chirik PJ ”Synthesis, Electronic Structure, and Alkene Hydrosilylation Activity of Terpyridine and Bis(imino)pyridine Iron Dialkyl Complexes” Organometallics 2012, 31, 4886–4893. [Google Scholar]; k)Tondreau AM; Darmon JM; Wile BM; Floyd SK; Lobkovsky E; Chirik PJ “Enantiopure Pyridine Bis(oxazoline) “Pybox” and Bis(oxazoline) “Box” Iron Dialkyl Complexes: Comparison to Bis(imino)pyridine Compounds and Application to Catalytic Hydrosilylation of Ketones” Organometallics 2009, 28, 3928–3940. [Google Scholar]; l)Tondreau AM; Lobkovsky E; Chirik PJ “Bis(imino)pyridine Iron Complexes for Aldehyde and Ketone Hydrosilylation” Org. Lett 2008, 10, 2789–2792. [DOI] [PubMed] [Google Scholar]
- (9).Cámpora J; Naz AM; Palma P; Álvarez E “2,6-Diiminopyridine Iron(II) Dialkyl Complexes. Interaction with Aluminum Alkyls and Ethylene Polymerization Catalysis” Organometallics 2005, 24, 4878–4881. [Google Scholar]
- (10).Danopoulos AA; Winston S; Motherwell WB “Stable N-functionalised ‘pincer’ bis carbene ligands and their ruthenium complexes; synthesis and catalytic studies.” Chem. Commun 2002, 1376–1377. [Google Scholar]
- (11).Darmon JM; Yu RP; Semproni SP; Turner ZR Stieber SCE; DeBeer S; Chirik PJ; “Electronic Structure Determination of Pyridine N-Heterocyclic Carbene Iron Dinitrogen Complexes and Neutral Ligand Derivatives” Organometallics 2014, 33, 5423–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gütlich P; Bill E; Trautwein AX, Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications; Springer-Verlag: Berlin Heidelberg, 2011, p 85. [Google Scholar]
- (13).Fernández I; Trovitch RJ; Lobkovsky E; Chirik PJ “Synthesis of Bis(imino)pyridine Iron Di- and Monoalkyl Complexes: Stability Differences between FeCH2SiMe3 and FeCH2CMe3 Derivatives” Organometallics 2008, 27, 109–118. [Google Scholar]
- (14).a)Joannou MV; Darmon JD; Bezdek MJ; Chirik PJ “Exploring C(sp3)–C(sp3) reductive elimination from an isolable iron metallacycle” Polyhedron 2019, 159, 308–317. [Google Scholar]; b)Allen OR; Dalgarno SJ; Field LD; Jensen P; Turnbull AJ; Willis AC “Addition of CO2 to Alkyl Iron Complexes, Fe(PP)2Me2” Organometallics 2008, 27, 2092–2098. [Google Scholar]; c)Venturi C; Bellachioma G; Cardaci G; Macchioni A; Zuccaccia C “Syntheses of di-hydrocarbyl derivatives of carbonyl phosphine complexes of iron” Inorg. Chimica Acta 2005, 358, 3815–3823. [Google Scholar]; d)Girolami GS; Wilkinson G; Galas AMR; Thornton-Pett M; Hursthouse MB “Synthesis and properties of the divalent 1,2-bis(dimethylphosphino)ethane (dmpe) complexes MCl2(dmpe)2 and MMe2(dmpe)2(M = Ti, V, Cr, Mn, or Fe). X-Ray crystal structures of MCl2(dmpe)2(M = Ti, V, or Cr), MnBr2(dmpe)2, TiMe1.3Cl0.7(dmpe)2, and CrMe2(dmpe)2” Dalton Trans 1985, 1339–1348; [Google Scholar]; e)Lau W; Huffman JC; Kochi JK “Electrochemical oxidation-reduction of organometallic complexes. Effect of the oxidation state on the pathways for reductive elimination of dialkyliron complexes” Organometallics 1982, 1, 155–169. [Google Scholar]
- (15).Hawrelak EJ; Bernskoetter WH; Lobkovsky E; Yee G; Bill E; Chirik PJ “Square planar versus tetrahedral geometry in four coordinate iron(II) complexes.” Inorg. Chem 2005, 44, 3103–3111. [DOI] [PubMed] [Google Scholar]
- (16).González-Sebastián L; Chaplin AB “Synthesis and complexes of imidazolinylidene-based CCC pincer ligands” Inorg. Chimica Acta 2017, 460, 22–28. [Google Scholar]
- (17).Russell SK; Lobkovsky E; Chirik PJ “Iron-Catalyzed Intermolecular [2π + 2π] Cycloaddition.” J. Am. Chem. Soc 2011, 133, 8858–8861. [DOI] [PubMed] [Google Scholar]
- (18).Pangborn AB; Giardello MA; Grubbs RH; Rosen RK; Timmers FJ Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar]
- (19).Pugh D “2,6-Dibromo-3,5-dimethylpyridineand 2,6-diiodo-3,5-dimethylpyridine” Acta Cryst. C 2006, C62, 590–592. [DOI] [PubMed] [Google Scholar]
- (20).Liu J; Chen J; Zhao J; Zhao Y; Zhang H “A Modified Procedure for the Synthesis of 1‐Arylimidazoles” Synthesis 2003, 17, 2661–2666. [Google Scholar]
- (21).Danopoulos AA; Tulloch AAD; Winston S; Eastham G; Hursthouse MB “Chelating and ‘pincer’ dicarbene complexes of palladium; synthesis and structural studies” Dalton Trans 2003, 1009–1015. [Google Scholar]
- (22).Nielsen DJ; Cavell KJ; Skelton BW;White AH “Methyl-palladium(II) complexes of pyridine-bridged bis(nucleophilic heterocyclic carbene) ligands: Substituent effects on structure, stability, and catalytic performance” Inorganica Chim. Acta 2006, 359, 1855–1869. [Google Scholar]
- (23).Baudisch O; Hartung WH Tetrapyridino-Ferrous Chloride (Yellow Salt). Inorganic Syntheses; Booth HS, Ed.; McGraw-Hill: Ohio, 1939; 184–185. [Google Scholar]
- (24).Prisecaru I WMOSS4 Mössbauer Spectral Analysis Software, www.wmoss.org, 2009–2016.
- (25).Pugh D; Wright JA; Freeman S; Danopoulos AA “‘Pincer’ dicarbene complexes of some early transition metals and uranium.” Dalton Trans 2006, 775–778. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.