

Abstract
Protein therapeutics have drastically changed the landscape of treatment for many diseases by providing a regimen that is highly specific and lacks many off target toxicities. The clinical utility of many therapeutic proteins has been undermined by the potential development of unwanted immune responses against the protein, limiting their efficacy and negatively impacting its safety profile. This review attempts to provide an overview of immunogenicity of therapeutic proteins, including immune mechanisms and factors influencing immunogenicity, impact of immunogenicity, pre-clinical screening methods, and strategies to mitigate immunogenicity.
Introduction:
Protein pharmaceuticals are one of the fastest growing class of drug molecules, including over 250 proteins used clinically for various indications. Advances in recombinant technology not only paved the way for unlimited supply of proteins, but also improved safety profiles by eliminating viral transmission that can occur with proteins isolated from natural sources. In addition, understanding the disease at the molecular level contributed to the growth of protein-based therapies by identifying key proteins that impact the severity or progression of the disease and harnessing their therapeutic potential to provide treatment. These examples include life-saving replacement therapies to treat Pompe Disease and Hemophilia A with recombinant acid alpha-glucosidase (rhGAA) 1,2 and Factor VIII (FVIII) 3, respectively, and cytokine based therapies such as interferon beta for Multiple Sclerosis 4,5. In addition, monoclonal antibody based drugs such as adalimumab (Humira), Fc-Fusion proteins such as Etanercept (Enbrel) for rheumatoid arthritis and Eloctate (Fc-FVIII) and immune checkpoint inhibitors such as pembrolizumab (Keytruda) and atezolizumab (Tecentriq), among several other antibody products have been developed.
Unfortunately, the clinical utility of many therapeutic proteins has been undermined by the potential development of unwanted immune responses against the protein. The incidence of immunogenicity of several clinically approved products has been well documented 6–8 (Tables 1 and 2), and some have failed before even reaching clinical trials. The development of antibodies can limit efficacy and negatively impact safety, hampering the clinical utility of the protein.
Table 1:
Extensive list of antibody products and immunogenicity
| Trade Name | INN | Company | Target | Type | FDA Approval | IMG Rate1 |
|---|---|---|---|---|---|---|
| ReoPro | Abciximab | Elli Lily | GPIIb/IIIa | Chimeric IgG1 Fab | 1994 | 6-44% |
| Humira | Adalimumab | Abbott | TNF-α | Human IgG1 | 2002 | 5-26% |
| Lemtrada | Alemtuzumab | Sanofi | CD-52 | Humanized IgG1 | 2014 | 1.9-8.3% |
| Praluent | Alirocumab | Sanofi | PCSK-9 | Human IgG1 | 2015 | 1.2-4.8% |
| Tecentriq | Atezolizumab | Genentech | PD-L1 | Human IgG1 | 2016 | 30-48% |
| Bavencio | Avelumab | Merck & Co. | PD-L1 | Human IgG1 | 2017 | 4.1% |
| Simulect | Basiliximab | Novartis | CD-25 | Chimeric IgG1 | 1998 | 1.2% |
| Benlysta | Belimumab | GSK | BLyS | Human IgG1 | 2011 | 0-4.8% |
| Fasenra | Benralizumab | Astrazeneca | CD-125 | Humanized IgG1 | 2017 | 13.0% |
| Avastin | Bevacizumab | Genentech | VEGF | Humanized IgG1 | 2004 | 0.6% |
| Zinplava | Bezlotuxumab | Merck & Co. | C. Difficile Toxin B | Human Monoclonal Antitoxin Antibody |
2016 | 0.0% |
| Adcetris | Brentuximab | Seattle Genetics | CD-30 | Chimeric IgG1 ADC | 2011 | 7-30% |
| Siliq | Brodalumab | Valeant Pharmaceuticals | IL-17RA | Human IgG2 | 2017 | 3.0% |
| Ilaris | Canakinumab | Novartis | IL-1ß | Human IgG1 | 2009 | 0.0% |
| Libtayo | Cemiplimab | Regeneron | PD-1 | Human IgG | 2018 | 1.3% |
| Cimzia | Certolizumab pegol | UCB | TNF-α | Humanized IgG Fab fragment | 2008 | 6-23% |
| Erbitux | Cetuximab | Eli Lilly | EGFR | Chimeric IgG1 | 2004 | 5.0% |
| Darzalex | Daratumumab | Janssen | CD-38 | Human IgG1 | 2015 | 0.0% |
| Prolia/Xgeva | Denosumab | Amgen | RANKL | Human IgG2 | 2010 | <1% |
| Dupixent | Dupilumab | Regeneron | IL-4Rα | Human IgG4 | 2017 | 7.0% |
| Imfinzi | Durvalumab | Astrazeneca | PD-L1 | Human IgG1 | 2017 | Unknown |
| Soliris | Eculizumab | Alexion Pharmaceuticals | Complement C5 | Humanized IgG2/4 | 2007 | 1-3% |
| Empliciti | Elotuzumab | BMS | SLAMF7 | Human IgG1 | 2015 | 18.5% |
| Hemlibra | Emicizumab | Genentech | Factor VIII | Humanized IgG4 | 2017 | Undetected |
| Aimovig | Erenumab | Amgen | CGRPR | Human IgG2 | 2018 | 2.6-6.2% |
| Repatha | Evolocumab | Amgen | LDL-C / PCSK9 | Human IgG2 | 2015 | 0.1% |
| Ajovy | Fremanezumab | Teva Pharmaceuticals | CGRP | Humanized IgG2 | 2018 | 0.4-1.6% |
| Emgality | Galcanezumab | Eli Lilly | CGRP | Humanized IgG4 | 2018 | 4.8-12.5% |
| Mylotarg | Gemtuzumab ozogamicin | Wyeth | CD-33 | Humanized IgG4 / toxin conjugate | 2000 | Undetected |
| Simponi | Golimumab | Janssen | TNF-α | Human IgG1 | 2009 | 2-7% |
| Trogarzo | Ibalizumab | TaiMed Biologics | CD-4 | Humanized IgG4 | 2018 | Undetected |
| Remicade | Infliximab | Janssen | TNF-α | Chimeric IgG 1 | 1998 | 10-51% |
| Vervoy | Ipilimumab | BMS | CTLA-4 | Human IgG1 | 2011 | 1.1-4.9% |
| Nucala | Mepolizumab | GSK | IL-5 | Human IgG1 | 2015 | 6.0% |
| Poteligeo | Mogamulizumab | Kyowa Kirin | CCR4 | Humanized IgG 1 | 2018 | 3.9% |
| Lumoxiti | Moxetumomab | Astrazeneca | CD-22 | Recombinant Immunotoxin | 2018 | 56.0% |
| Tysabri | Natalizumab | Biogen | VLA-4 | Humanized IgG4 | 2004 | 9-10% |
| Portrazza | Necitumumab | Eli Lilly | EGFR | Human IgG1 | 2015 | 4.1% |
| Opdivo | Nivolumab | BMS | PD-1 | Human IgG4 | 2015 | 11.2-37.8% |
| Gazyvaro | Obinutuzumab | Roche | CD-20 | Humanized IgG 1 | 2013 | 7.0% |
| Ocrevus | Ocrelizumab | Genentech | CD-20 | Humanized IgG1 | 2017 | 0.5-1.5% |
| Arzerra | Ofatumumab | GSK | CD-20 | Human IgG1 | 2009 | Undetected |
| Lartruvo | Olaratumab | Eli Lilly | PDGFR-α | Human IgG1 | 2016 | 3.5% |
| Xolair | Omalizumab | Genentech | IgE | Humanized IgG1 | 2003 | <0.1% |
| Synagis | Palivizumab | MedImmune | F-protein of RS virus | Humanized IgG1 | 1998 | 0.7% |
| Vectibix | Panitumumab | Amgen | EGFR | Human IgG2 | 2006 | 0.8-4.6% |
| Keytruda | Pembrolizumab | Merck & Co. | PD-1 | Human IgG4 | 2014 | 2.0% |
| Perjeta | Pertuzumab | Roche | HER-2 | Humanized IgG1 | 2012 | 2.8% |
| Cyramza | Ramucirumab | Eli Lilly | VEGF | Human IgG1 | 2014 | 6.0% |
| Lucentis | Ranibizumab | Genentech | VEGF-A | Humanized IgG1 Fab fragment | 2006 | 0-6% |
| Cinqair | Reslizumab | Teva Pharmaceuticals | IL-5 | Human IgG4 | 2016 | 4.8-5.4% |
| Rituxan | Rituximab | Biogen Idec | CD-20 | Chimeric IgG1 | 1997 | 1.1-11% |
| Cosentyx | Secukinumab | Novartis | IL-17a | Human IgG1 | 2015 | Undetected |
| Sylvant | Siltuximab | Janssen | cCLB8 | Chimeric IgG1 | 2014 | 0.2% |
| Ilumya | Tildrakizumab | Sun Pharma | IL-23 | Humanized IgG1 | 2018 | 6.5% |
| RoActemra | Tocilizumab | Chugai | IL-6 receptor | Humanized IgG1 | 2010 | 2.0% |
| Herceptin | Trastuzumab | Genentech | HER-2 | Humanized IgG1 | 1998 | 0.1% |
| Stelara | Ustekinumab | Janssen | IL-12/IL-23 | Human IgG1 | 2009 | 3-6% |
| Entyvio | Vedolizumab | Takeda | Integrin-α4β7 | Humanized IgG1 | 2014 | 13.0% |
Rates of Immunogenicity were derived solely from FDA licensed package inserts. In cases where IMG rates were not reported, the table was labeled as ‘Unknown’. IMG rates listed can be impacted by confounding factors such as co-medication, immune status of the patient, and detection assays used.
Table 2:
Extensive list of protein products and immunogenicity
| Trade Name | INN | Company | Target | Type | FDA Approval | IMG Rate2 |
|---|---|---|---|---|---|---|
| Activase | Alteplase | Genentech | Thrombolytic | Recombinant | 1987 | Unreported |
| Advate | Octocog Alfa | Baxter | Antihemophilic Factor VIII | Recombinant in CHO | 2003 | 20.0% |
| Amevive | Alefacept | Biogen | CD-2 inhibitor | Recombinant in CHO | 3.0% | |
| Andexxa | Portola Pharmaceuticals | Coagulation Factor Xa | Recombinant in CHO | 2018 | 17.0% | |
| Avonex | Interferon Beta-1a | Biogen | Interferon-β Mimetic | Recombinant in CHO | 1996 | 5.0% |
| Betaseron | Interferon Beta-1b | Bayer | Interferon-β Mimetic | Recombinant in E. Coli | 1993 | 45.0% |
| Bravelle | Urofollitropin | Ferring | Follicle Stimulating Hormone | Purified from Urine | 2002 | Unreported |
| Byetta | Exenatide | Astrazeneca | GLP-1 agonist | Solid Phase Peptide Chemistry | 2005 | 9-38% |
| Ceredase | Alglucerase | Genzyme | β-glucocerebrosidase Mimetic | Purified from Human Plaenta and Modified | 1994 (discontinued) | Unreported |
| Elaprase | Idursulfase | Shire | Iduronate-2-sulfatase mimetic | Recombinant in HT-1080 | 2006 | 51-68% |
| Eloctate | Bioverativ | Antihemophilic Factor VIII | Recombinant in HEK | 2014 | Unreported | |
| Epogen | Epoetin Alfa | Amgen | Erythropoietin Mimetic | Recombinant in CHO | 1989 | Unreported |
| Helixate | CSL Behring | Antihemophilic Factor VIII | Recombinant in BHK | 1993 (discontinued) | 12.5-15% | |
| Kepivance | Palifermin | Sobi | KGF Mimetic | Recombinant in E. Coli | 2004 | 2.0% |
| Koate | Grifols | Antihemophilic FactorVIII | Purified from Human Plasma | 2012 | Unreported | |
| Levemir | Insulin Detemir | Novo Nordisk | Insulin Mimetic | Recombinant in Saccharomyces Cervisiae | 2005 | 0.0% |
| Monarc-M | American Red Cross | Antihemophilic Factor VIII | Purified from Blood | 1974 | Unreported | |
| Myozyme | Alglucosidase Alfa | Genzyme | Acid Alpha-Glucosidase Mimetic | Recombinant in CHO | 2006 | 89.0% |
| Neulasta | Pegfilgrastim | Amgen | G-CSF mimetic | Recombinant in E. Coli | 2002 | 0.8% |
| Neumega | Wyeth | IL-11 Mimetic | Recombinant in E. Coli | 1997 | 1.0% | |
| NovoSeven | Novo Nordisk | Antihemophilic Factor VII | Recombinant in BHK | 1999 | Unreported | |
| Nulojix | Belatacecpt | BMS | CD-80/86 blocker | Recombinant in CHO | 2011 | 7.8% |
| Palynziq | Pegvaliase | BioMarin | Metabolizing Enzyme | Reombinant | 2018 | 100.0% |
| Proleukin | Aldesleukin | Prometheus | IL-2 Mimetic | Recombinant in E. Coli | 1992 | 66-74% |
| Pulmozyme | Dornase Alfa | Genentech | rhDNAse | Recombinant in CHO | 1993 | Unreported |
| Raplixa | The Medicines Company | Fibrin Sealant of Wounds | Purified from Human Plasma | 2015 | 2-4% | |
| ReFacto | Pfizer | Antihemophilic Factor VIII | Recombinant in CHO | 2000 | Unreported | |
| Refludan | Lepirudin | Bayer | Direct Thrombin Inhibitor | Recombinant in Yeast | 2004 (discontinued) | Unreported |
| Tanzeum | Albuglutide | GSK | GLP-1 agonist | Recombinant in Saccharomyces Cervisiae | 2014 | 5.5% |
Rates of Immunogenicity were derived solely from FDA licensed package inserts. In cases where IMG rates were not reported, the table was labeled as ‘Unknown’. IMG rates listed can be impacted by confounding factors such as co-medication, immune status of the patient, and detection assays used.
1. Mechanism of immunogenicity:
i. The Immune Response
Immune responses against therapeutic proteins can arise due to two different mechanisms; a classical immune response, or by breaking tolerance 9–11. The immunological discrimination of self and non-self-proteins is key to determining the mechanism of the immune response. In healthy individuals, the immune system maintains homeostasis between self and non-self-proteins by negative selection of self-reactive immune cells in thymus 12, and proteins that are recognized as foreign will initiate a classical immune response in patients. This response is characterized by the formation of antibodies and is typically first seen within days to weeks after administration, and is often triggered after a single injection. These types of responses are long lasting and very difficult to reverse once memory B-cells have been generated 13–15. Upon subsequent exposure to the protein, the immune system mounts a ‘secondary response,’ 13,16,17, primarily characterized by significant IgG release that greatly impacts therapy. The most well-known examples of therapeutic proteins that evoke classical immune responses are replacement therapies such as rhGAA 18, and FVIII 19,20. It also affects Mab therapeutics, as the CDR region is highly immunogenic and results in the generation of anti-idiotypic alloantibodies due to a lack of central tolerance to this region 21,22. Therapeutic proteins that are homologous to endogenous self-proteins generally do not mount a response due to established immunological tolerance, but can become immunogenic by breaking B-cell tolerance 23,24. B cell tolerance is maintained in the bone marrow mediated by negative selection of auto-reactive B cells and high BCR affinity cells are forced to undergo apoptosis 25. Through repetitive administration, tolerance can be broken to therapeutic proteins23, as in IFN-γ, IFN-β, and Erythropoietin (Epo) 26.
ii. Cellular and Molecular Mechanisms of Immunogenicity:
The cellular mechanism leading to anti-drug antibody (ADA) formation to therapeutic proteins involves two major cell types, antigen presenting cells (APC), including dendritic cells and macrophages, and T and B lymphocytes 9. Due to the high phagocytic capacity of APCs such as immature dendritic cells, administered proteins are engulfed, processed and presented in the context of major histocompatibility complex (MHC-II) to T-lymphocytes in lymph nodes 10,27. This process is also accompanied by maturation of APC by upregulation of co-stimulatory markers such as CD40, CD80 and CD86, and migration of APCs to local lymph nodes 28. With the help of secreted cytokines, APCs stimulate antigen specific T-lymphocytes that prompt the activation of B-lymphocytes and promotes their differentiation into memory B-cells and plasma cells (Figure 1 in Reference 31)29–31. Memory B-cells reside dormant until subsequent exposures to the therapeutic protein and plasma cells secrete antibodies that recognizes specific epitopes on the protein presented by APC MHC receptors 17,32. Immunogenicity is also possible through T cell independent processes in which the antigen engages B cells directly 33.
Figure 1:
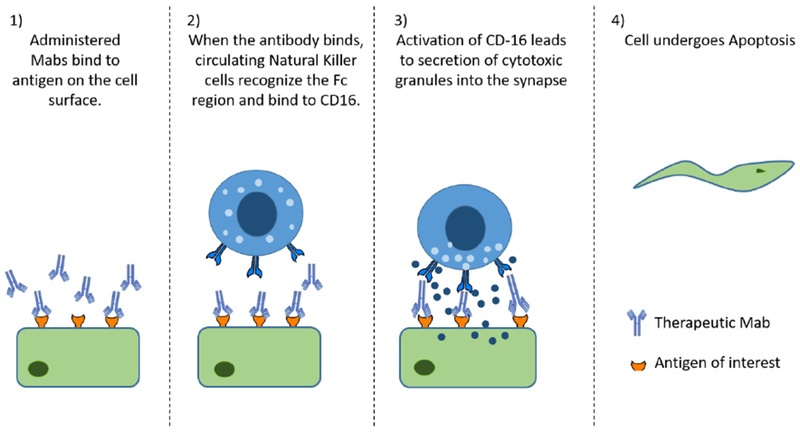
Antibody Dependent Cellular Cytotoxicity Mechanism: When Mabs bind to target antigen on the cell surface, it can lead to natural killer cells binding to the Fc region via CD16. Activation of this receptor leads to secretion of cytotoxic granules that enter the healthy cell, forcing it to undergo apoptosis.
iii. Immunological response of Therapeutic Proteins and Antibodies:
Immunogenicity of therapeutic proteins manifests as the development of ADA as well as hypersensitivity reactions 34–36. Hypersensitivity reactions can be distinguished into four types depending on the mechanism of response and effector molecule generated 37–39. Type I hypersensitivity is mediated by IgE antibodies that cross link proteins and mast cells with release of vasoactive mediators, most notably histamine, and typically manifests as anaphylaxis, rhinitis, hives and asthma 37. Type I reactions are observed within minutes to hours after administration of proteins and can typically be prevented by prophylactic administration of anti-histamines. Type I reactions can also occur independent of IgE release, involving T cells, more commonly seen with food proteins 34. In type II reactions, IgG and IgM antibodies directed against cell-surface antigens mediate cell destruction and complement activation 40. A common example of Type II hypersensitivity is blood cell incompatibility. Due to cell surface markers, blood cells from different types (A, B, O) are perceived differently upon blood transfusion and the body reacts by attacking the “invading” cells. Type II hypersensitivity is also observed with Mab products that target cell surface antigens. Upon binding of the administered Mab to cell surface antigens expressed on target or non-target tissues, the immune system can elicit antibody dependent cellular toxicity (ADCC) causing cell lysis and death 41 (Fig 1). For example, cardiotoxicity observed with trastuzumab (Anti HER-2) has been at least partially attributed to the expression of HER-2 on cardiac muscle in addition to cancerous tissue due to ADCC 42–44. Administered antibody bound to antigen can also evoke complement activation by binding to C1q and eventual deposition at the surface of the target cell, leading to complement-dependent cytotoxicity (CDC) 45,46 (Fig. 2). The design of the Fc portion of the antibody and antibody subtypes can also influence cellular cytotoxicity, as IgG2 and IgG4 show lower ADCC compared to IgG1 or IgG3, while IgG4 show no CDC effects 44,47. Type III hypersensitivity is predominantly characterized as immune-complex mediated hypersensitivity. If the amount of protein is significantly higher than ADA present, small immune complexes can form that are not effectively cleared by phagocytic cells, leading to Type III reactions due to immune complex or protein-antibody complex deposits in various tissues. Complex deposition can lead to complement activation and inflammatory responses that cause serum sickness 48. The size of the immune complexes of ADA and protein are primary determinants of type III reactions. Type IV hypersensitivity (delayed type hypersensitivity) is cell mediated, and is characterized by sensitized T helper cells that release cytokines and activate macrophages and T-cells 48.
Figure 2:

Complement Dependent Cytotoxicity: When Mabs bind to target antigen, it can deposit in healthy tissue. Circulating complement can bind to the immune complex and trigger complement activation, leading to cell death.
Based on clinical observation, about 63% of hypersensitivity reactions generated against Mabs are type I reactions, 13% experience cytokine release, while about 3% are delayed type IV reactions 34. Immune related toxicities against Mabs, particularly immune modulating Mabs, arise from two mechanisms: first, target binding in healthy tissues as described and secondly, enhanced pharmacology, attenuating the activity of target molecules on the cells 35. Recently, immune related adverse reactions have been documented for immune checkpoint inhibitors (anti CTLA-4, anti PDL −1 and anti PD1) 49,50, which manifests as inflammation involving multiple organ systems, including the gastrointestinal tract, endocrine system, skin and liver and to a lesser degree the, central nervous system, cardiovascular and pulmonary systems. These immune related adverse reactions can present at any time within first few weeks after treatment initiation up to months after therapy has been stopped. The pathophysiology underlying the toxicity is not fully understood, however it is likely to be related to the role that immune checkpoints play in maintaining immunologic homeostasis. Currently, steroid and immunosuppressive treatment is recommended for patients who have developed these adverse immune related reactions.
2. Impact of Immunogenicity on Efficacy
ADA responses against therapeutic proteins can be separated into two distinct classes; neutralizing and binding antibodies. Neutralizing antibodies recognize regions within therapeutic proteins that are critical for its biological activity, abrogating its activity directly. For example, about 30% of severe Hemophilia A patients who take recombinant FVIII as replacement, develop neutralizing antibodies, referred as “inhibitors” and in these inhibitor positive patients, hemostatic efficacy is lost 19. Similarly, 89-100% of Pompe Disease patients who are treated with recombinant human acid alpha-glucosidase (rhGAA), develop antibodies against rhGAA 51,52. Once a sustained immune response is established, the efficacy of rhGAA therapy is lost and no safe clinical options are available. In contrast, binding antibodies interact with the protein and can alter the pharmacokinetics of the protein, indirectly impacting its efficacy by reducing overall systemic exposure. During dose escalation clinical trials of emicizumab, a bi-specific FVIII mimetic, one out of six patients developed ADA that resulted in rapid clearance of the protein, indirectly impacting hemostatic efficacy 53. Similarly, long-term use of adalimumab resulted in development of ADA in about 28% of patients and it was associated with lower adalimumab concentration and poorer clinical outcomes 54,55.
i. PK/PD Modeling of Immunogenicity:
The use of pharmacokinetic and pharmacodynamic (PK/PD) approaches to assess the impact of immunogenicity on efficacy has recently gained considerable interest. As discussed, binding antibodies can impact PD indirectly by altering the PK of the protein, often by increasing the clearance 56,57. An increasing number of studies have incorporated the effect of immunogenicity to develop PK models that describes the impact of antibodies on absorption, disposition, metabolism and elimination (ADME) of therapeutic proteins 58. A few model designs have been used; models that include immunogenicity as a covariate, models that include ADA concentration as a continuous variable, and models that include immunogenicity as a supplementary clearance (Fig 3). Zhou et al. modeled immunogenicity as a covariate and found that it has a significant impact on golimumab clearance 58. Alternatively, the effect of ADA can be modeled as a separate clearance pathway, leading to direct elimination from the circulation as opposed to proteolytic degradation in tissue. Deng et al. developed a PK model for adalimumab, incorporating ADA as a factor increasing clearance 59. Casteele et al developed a non-linear mixed effects model to describe certolizumab PK in a Crohn’s disease patient and modeled ADA concentration as a continuous variable60. ADA can also be modeled as a categorical variable, as done by Rosario et al61, in which ADA is modeled based on if it is present or absent. This type of model however does not account varying levels of ADA in patients. Another consideration is the incorporation of a lag component in the model to account for the time it takes for ADA to develop. Vicini et al. modeled immunogenicity as a clearance pathway, and included a series of delay compartments through which the antibody must pass before entering the central compartment 62. Gomez-Mantilla has published an excellent review surveying the field of incorporating ADA in PK models 63.
Figure 3:

A Two compartment model (1) depicting the impact of ADA on the PK of a protein. Protein is administered into the central compartment (C1) where it can distribute into the tissue compartment (C2). CL1 and CL2 depict clearance from the central compartment and tissue compartment, respectively. The left side depicts immune complex formation. ADA in the central compartment (ADA) can bind to protein to form immune complexes (ADA+P) that can be directly eliminated via degradation pathways (CL3), complementing CL1 and CL2. The second model (2) has the same basic structure, with the addition of Tlag component (ADAL), accounting for the time it takes to initiate an immune response. ADA begins as hypothetical bolus dose before passing through a series of compartments before reaching the central compartment.
Very few studies have been published that integrate ADA response using PK/PD approaches, which is critical to completely delineate the impact of immunogenicity on efficacy. Such PK/PD integration also requires consideration of the direct effect ADA have on the PD of therapeutic proteins. We recently proposed an integrated model to define the impact of inhibitor development against FVIII in Hemophilia A where the fraction unbound of FVIII was elucidated from PK analysis and integrated with a direct response model to determine inhibitor effects on PD 64.
ii. Measurement of Anti-drug Antibodies
Measurement of ADA in patients is important in clinical trials as a requirement for new drug approvals. Different assays can have an impact on the perceived immunogenicity rates and severity, as assays must be sensitive enough to detect low levels of ADA and be able to differentiate between the therapeutic protein and the ADA. Additionally, characterizing and comparing methods is often difficult in most cases because human ADA standards are not available to determine exact concentrations. Challenges of ADA measurement include the selectivity of the assay for ADA and not drug product, the presence of false positive and negatives of several assays, and a lack of standardization across experiments makes it difficult to compare immunogenicity of different products tested differently. It is likely that early phase 3 clinical trials under or overestimated immunogenicity in protein products due to assay limitations in detection and differentiation between drug and ADA. Assay selection is key to determining the patients’ clinical outcome and this is no better shown than with adalimumab and TNF blockers. Bartelds showed that ADA against adalimumab directly correlates with patient outcomes and estimated immunogenicity rate using a radioimmunoassay65. Schouwenburg et al compared an antigen binding test and a pH shift anti-idiotypic binding test (PIA) and showed that the drug tolerant PIA assay better correlated with clinical outcomes of adalimumab treatment66. Bartelds, Shouwenburg, and Hart published a novel assay to detect anti-adalimumab antibodies which showed better detection of immunogenicity than previous assays67.
a). Bridging Assays
Bridging assays are one of the most commonly used assays in industry to detect ADA. In bridging immunoassays, the drug is labeled and ADA will form a bridge between two labeled drug molecules that can be detected by the label. These assays are useful in detecting all antibody classes, and can be used in any sample, as the ADA will always bind to the labeled drug. Recently, the Gyrolab platform has been used to develop binding assays for ADA detection68. It offers advantages such as automation and smaller volume sizes that can eliminate the use of costly resources and human error.
Unfortunately, there are some limitations to bridging assays. First, soluble antigen in plasma can cause drug molecules to bridge, resulting in false positives69. Secondly, drug interference is a major issue that can result in the determination of false negatives due to drug tolerance, seemingly decreasing the rate of immunogenicity. Drug tolerance is the amount of free drug that can be present in a sample that doesn’t interfere with the detection of ADA. If too much free drug is present in a sample, the drug can conceal the presence of low level ADA in the sample. Finally, in select cases, pre-existing antibodies can interfere with bridging immunoassays70. One example is rheumatoid factor, which can bind to the Fc region on antibodies, and result in false positives71.
b). Ligand Binding Assays
Ligand binding assays (LBA) are commonly used methods that are used to detect binding of a target. Shibata et al. compared three types of LBA to detect anti-EPO antibodies according to World Health Organization guidelines72. In their paper, they compared surface plasmon resonance, electrochemiluminescence, and biolayer interferometry. Electrochemiluminescence is a method in which light is measured as a result of chemical and electrical reactions. Biolayer interferometry is an optical method that measures light interference on a protein surface. As was hypothesized, each assay differed in their ability to detect ADA, confirming that assay selection is highly important in detecting ADA.
c). Protein Specific Assays
In some cases, protein specific assays can be used to measure ADA concentrations. In the case of FVIII, the Bethesda Assay can be used to specifically measure neutralizing anti-FVIII antibodies in concentration of BU/mL, which allows for better comparison of inhibitory ADA between studies73. In addition, FVIII is one of the few proteins where a standard is available to measure ADA by ELISA. ESH8 is an anti-FVIII antibody that can be used to generate a standard curve and is used to measure total FVIII ADA. Dong et al. developed an assay specific for anti-PEG antibodies in which biotin-PEG is conjugated to magnetic beads and anti-PEG antibodies bound to the bead is measured using a sensor that detects changes in size of the complex74. Previous assays for anti-PEG antibodies generally had high background, which is eliminated in this specific method because only the complex is detected by the sensor.
d). Drug Tolerant Assays
The presence of the drug in the sample can cause issues when detecting ADA, so drug-tolerant assays have been developed to overcome these problems and allow for better quantification of ADA. Several labs have conducted studies comparing drug tolerant assays in the detection of adalimumab ADA, looking at pH shift idiotype antigen binding, acid dissociation assays, temperature shift assays, and electrochemoluminescence assays75,76. They concluded that all assays correlated well with the amount of ADA, however the rate of immunogenicity ranged from 51-66%, with many of the issues coming from low ADA patients. Increasing the sensitivity of these assays will best identify ADA-positive samples. Mikulskis et al. developed a solution ELISA drug tolerant assay to detect ADA against monoclonal antibodies after comparing it to several other moethods77.
e). ELISA
ELISAs can also be used to detect ADA by coating the plate with the protein and incubating samples to measure ADA bound to the plate. This method has been used in our lab in conjunction with the Frey method to measure anti-rhGAA antibodies78,79. The utility of ELISAs can be limited because standards are not available for all ADA. Modified methods such as reciprocal dilutions, or optical density may be used in the place of actual concentration, however it makes it difficult to compare results between studies and between labs.
f). Other
In addition to the discussed methods, other less common methods are also used. Immune-PCR is an extension of bridging assays, used in special cases with high drug interference, in which the complex is labeled with biotin that is detected using an anti-biotin antibody conjugated to DNA80. Using PCR, the DNA can be quantified to assess ADA levels. Immuno-LC/MS can also be used to detect ADA in plasma81. Using a bridging assay, human ADA were identified and specific proteolytic cleavage patterns were identified to segregate Ig into subclasses. Before using immune-LC/MS, samples must be enriched, either by tagging the drug with biotin, or spiking excess drug into the sample to saturate ADA binding. Qu et al have also investigated methods of using LC/MS to detect antibody therapeutics, which could also be used to detect ADA82.
g). Limitations of ADA detection
There are two major complications in measuring ADA in patient samples. The first is interference from the drug and the second is assay sensitivity. Interference from circulating drug has the ability to complicate detection and removing free drug can improve these assays. The most common method to remove free drug is acid dissociation, which enriches the ADA and removes drug from the sample so that only the ADA is being detected. Secondly, some studies incorporate a wash-out period to allow circulating drug to leave the system, however this is limited in multi-dose studies. An easy, efficient method to reduce drug interference is by diluting the sample to reduce overall drug concentration so it doesn’t interfere. Sensitivity issues have been well documented and can be present in any assay. It is important to improve sensitivity to prevent the presence of false negatives that may bring down the overall immunogenicity rate.
Further, most labs have developed their own methods for ADA detection, and a reliable standardized method is not available. Standard curve generation is difficult without a standard ADA for each drug, making it difficult to compare results between studies. In addition, with many different assays available, it is not uncommon that studies may use different methods to quantify ADA of the same protein. These issues may be present in older phase 3 clinical studies in which assay sensitivity was not high, resulting in numerous false negatives and making the detection of ADA difficult, lowering overall immunogenicity.
The selection of an appropriate assay is also critical when discussing PK studies. When measuring the impact on immunogenicity on PK, it is important to accurately quantify ADA and the concentration of protein in the sample. Using an appropriate assay can influence parameters, especially in the terminal phase where sensitivity of the assay is key when determining concentration83. It can also make it difficult to compare PK profiles between studies if different assays are used.
iii. Clinical Management and Monitoring ADA
The clinical management of patients with immunogenicity is very challenging and depends on the clinical condition of the patient and the disease. In patients with Hemophilia A, after inhibitors develop, bypass therapies are available, such as NovoSeven, an activated FVIIa product, emicizumab, a bi-specific antibody that mimics FVIII by binding to FIX and FX, or activated prothrombin complex concentrate. To reverse inhibitor development, immune tolerance induction (ITI) strategies using high dose FVIII over a period of time desensitize the immune system to FVIII and induce antigen specific tolerance. The exact mechanism is still the subject of research, however it has been used successfully for decades, and various dosing regimens have been used with success to induce ITI84,85. Unfortunately, ITI approaches are very expensive, costing nearly a million dollars and is not effective in all patients. Patients who develop sustained ADA against rhGAA in the treatment of Pompe Disease have no alternative clinical options. Due to the dire situation, immunosuppressive regimens of rituximab, methotrexate and IVIG (to be discussed later) are attempted that are very costly and globally suppress the immune system, leaving the patient susceptible to infection86. The use of immune checkpoint inhibitors is complicated by immune related adverse events, as discussed (IRAE). A team approach is necessary to manage IRAE as it is a complicated process because patients must receive supportive care for management of these incidences, and careful monitoring is necessary to reduce toxicity. Management of IRAE is summarized by Amin et al 87. Based on these experiences, it is clear that clinical management of ADA is challenging as most treatments are complicated, prohibitively expensive, can be ineffective in some patients, and may expose the patient to significant risk. Prophylactic prevention of ADA development is desirable, and may be accomplished using low dose immunosuppressants or oral nanoparticles that will be discussed in detail below.
For many protein therapeutics, monitoring ADA can provide life and cost saving benefits. Routine monitoring of ADA development can be effective at improving clinical outcomes, as well as reducing the cost of therapy. Laine et al. used data collected in Finland of adalimumab and infliximab drug trough concentrations and ADA development to develop a model that shows testing patients for ADA is cost-saving in real life clinical practice88. Vincent et al. also proposed a model where drug levels and ADA measurements can be used to predict the best biologics to use in chronic inflammatory diseases89. The ABIRISK Consortium did a retrospective analysis of ADA in patients treated with IFN-β and natalizumab and found that immunogenicity testing in patients would reduce the potential number of ADA positive patients and reduce the amount of money lost treating patients that are not receiving benefit90.
3. Factors influencing immunogenicity:
Several factors can influence the immunogenicity of therapeutic proteins and can be classified as patient, product, or treatment related factors. The duration of treatment, route, and frequency of administration impact the immunogenicity of the protein 91. It is commonly accepted that subcutaneous administration of therapeutic proteins, while more user friendly, is more immunogenic than intravenous administration, likely due to migratory potential of cutaneous dendritic cells 92,93. As discussed, prolonged, chronic therapies are often more immunogenic and frequent administration has also been shown to increase immunogenicity 94.
Patient related factors that can influence immunogenicity include the immune status of the patient and polymorphisms of the MHC receptor. The MHC (or HLA in humans) is highly polymorphic 95 and several different alleles have been identified for MHC II with many different subunits, such as DP, DM, DOA, DOB, DQ and DR 96–98. These receptor subtypes differ in binding affinity for epitopes and therefore, inter-patient differences in MHC subtypes can influence the generation of an immune response. Immune status can also greatly influence a patient’s immune response to the protein, as autoimmune patients often respond more strongly to therapeutic proteins than immunocompromised patients do.
Alterations in the actual protein product can have a significant effect on their immunogenic potential. The presence of immunogenic epitopes recognized by MHC-II 99, aggregates in the final product 100, and post translation modification such as glycosylation all influence the immunogenicity of protein products. The link between aggregates and immunogenicity is currently the subject of intense research 101,102 . The nature of aggregates that evoke an immune response are not clear, as there are several aggregates with distinct molecular and biophysical characteristics that have been identified that differ in their ability to engage the immune system. To get a better understanding of the link between aggregation and immunogenicity, a classification system has been formulated to organize aggregates based on size, reversibility, structure, covalent modification and morphology 103,104. For example, the presence of sub visible particles and native like aggregates has been shown to evoke a strong immune response and may be partially complicit in breaking tolerance due to the repeated exposure to immunogenic epitopes 105,106. Additionally, it has been shown that native-like aggregates of IFN-α are much more immunogenic than non-native aggregates and aggregates that lack a native epitope 107,108. Studies performed by our group using Factor VIII also showed that native-like aggregates of FVIII are more highly immunogenic when administered in a murine model of Hemophilia A 107. In addition to aggregation, oxidation of proteins and aggregates in formulations have also been shown to influence immunogenicity. Jiskoot et al. showed that oxidation-mediated aggregation resulted in increased immunogenicity of IFN-β109. Hermeling et al also showed that oxidized aggregates are more immunogenic than native protein108.
Recombinant proteins can be produced in bacterial cells, such as E. Coli, or mammalian cells, such as CHO cells. Proteins expressed in bacteria do not undergo post translational modifications such as glycosylation, while proteins from mammalian cells do, which can result in different immunogenic profiles. A classic example of the influence of glycosylation can be seen in Interferon-β products. The first product developed, Betaseron, is produced in E. Coli cells and contained no glycosylation. Avonex was developed after, and is produced in CHO cells with recombinant DNA technology. Between the two products, Betaseron has much higher immunogenicity than Avonex, at 35% versus 5%, respectively 110. The difference in immunogenicity can be partially attributed to the changes in glycosylation patterns of these products that can lead to aggregation. Glycosylation patters on Mabs can also influence their toxicity profile in the body. Most Mabs in the clinic are IgG class and contain a glycosylation site at amino acid 297, and occasionally in the Fab region as well. Fucose structures on the Mab reduce IgG binding to the Fc receptor, reducing ADCC, while less terminally bound galactose reduced CDC activity 111. Several very informative review articles are available regarding the impact of aggregation and glycosylation on immunogenicity 6,100–102,112
4. Prediction of immunogenicity using preclinical studies:
Prediction of clinical immunogenicity using preclinical approaches can be useful when assessing new biologic products. Bococizumab, a Mab in development to lower LDL levels in the body by inhibiting PCSK9 was recently discontinued after phase III clinical trials, citing higher immunogenicity incidence rates causing reduced efficacy of the Mab over time as compared to similar products in its class. 113,114. A bioengineered version of Factor VIIa (rFVIIa) was discontinued from further clinical development after phase III clinical trials, due to the detection of unwanted immunogenicity. A post hoc analysis of the bioengineered rFVIIa proposed to modify neo-epitopes not present on rFVIIa, demonstrating the value of rigorous preclinical screening 115. The ability to predict such immune responses during preclinical screening could reduce drug attrition and also lead to selection of better protein candidates that have been specifically engineered to lower immunogenicity 115. Unfortunately, most pre-clinical immunogenicity models are limited in their ability to predict the incidence rate of immunogenicity, but have indispensable value in assessing relative immunogenicity between similar products, rank ordering the immunogenicity of multiple molecules and formulations, and elucidating the mechanism of immune response to further our ability to design less immunogenic proteins. Preclinical studies in vivo are commonly used to screen immunogenicity of therapeutic proteins and the relative immunogenicity between different products. In vitro methods such as epitope mapping, HLA affinity, and T cell proliferation assays are used to assess the immunogenic potential of proteins and identify immunogenic regions. In silico programs have been developed that focus on epitope mapping of the proteins three dimensional structure.
a). In Vivo Immunogenicity Assessment:
Generally speaking, any human or humanized protein for therapeutic use is going to be immunogenic when administered to an animal, thus results should be interpreted with caution. However, mouse models have made significant contributions to biomedical research, including our understanding of the immune response against antigens and foreign proteins. The ease of breeding and housing, and manipulation of their genome system for specific applications are key reasons for their utility in biomedical research.
i. Mouse Models:
Mouse models have been used to predict immunogenicity, compare relative immunogenicity between products and elucidating immune mechanisms 116–120. In an experiment by Jiang et al, they compared the relative immunogenicity of several types of FVIII products to determine if their new FVIII fc fusion was less immunogenic than the currently used products 121. It was found that mice treated with rFVIIIFc had significantly lower antibody response as compared to B-Domain deleted FVIII or full length FVIII, which is confirmed by clinical observation of Eloctate (rFVIII-Fc) having much lower immunogenicity 122–124. In a study by Lu et al, BALC/c mice were used to compare the relative immunogenicity of DNA vaccines by different routes of administration and showed that intramuscular administration was the least immunogenic 125. Studies from our lab have used mouse models to show that administration of FVIII or rhGAA complexed with phosphatidylserine based liposomes is significantly less immunogenic than administration of the free protein alone 31,78,126. B6-129 mice with the GAA gene knocked out 127 have been used as a model to assess immunogenicity of rhGAA 78,128.
With increasing interest regarding factors influencing immunogenicity, mouse models have also been used to compare the immunogenicity of aggregated products. Using a mouse model of Hemophilia A, our lab showed native aggregates are relatively more immunogenic than monomeric protein and non-native aggregates 129. Jiskoot et al. showed that the aggregated forms of insulin were more immunogenic than the commercial products 130. Braun et al. performed a study with IFN-α which showed the formulation became significantly more immunogenic after the formation of aggregates, which occurs in nearly all marketed IFN-α products 131.
Another important use of mouse models is to provide mechanistic insight into immune system activity. Adoptive transfer and T cell proliferation studies can be performed to assess the role of T and B cells in protein immunogenicity. Adoptive transfer studies performed by our group suggest a role for CD4+CD25+ T cells in suppressing an immune response against FVIII. T cells were transferred from mice that were tolerized to FVIII into recipient mice, which then exhibited significantly lower ADA development 31. A recent study by van Beers et al. showed that immunological memory did not develop after administration of IFN-β, despite the development of anti-IFN-β antibodies 132. This has also been seen clinically in patients that have switched interferon products due to immunogenicity. Anti-IFN antibodies have been known to be cross reactive, however when switching products, patients do not see an increase in antibody level 5,133.
ii. HLA Transgenic Mice:
Due to physiological differences, animal MHC receptors don’t directly mimic human HLA receptors 134. HLA and MHC genes are some of the most polymorphic genes, making it difficult to assess protein immunogenicity due to high inter-subject variability in HLA/MHC expression. 96,97,135. To combat this, transgenic (Tg) mouse models have been developed that mimic a human subject and are tolerized to a specific protein. 136,137. The resulting mouse will tolerate the therapeutic protein of interest, and the development of immunogenicity will result from the breaking of self-tolerance, as opposed to a classical immune response to foreign antigens. Transgenic mouse models have been developed for a wide range of therapeutic proteins, such as IFN-β 136,138, human growth hormone 139, insulin 140,141, and FVIII 137.
One of the earliest uses of transgenic tolerant mice investigated the immunogenicity of human insulin and insulin analogues. Ottesen et al showed that transgenic mice are tolerant of human insulin and did not produce antibodies 141. Incidentally, they also found that increasing the number of protein modifications correlated with an increase in ADA. Beers et al utilized a transgenic mouse model tolerant to human IFN-β to investigate immunogenicity towards IFN-β products. They showed that Betaferon was the only product tested that broke immune tolerance in transgenic mice. In addition, it had the highest immunogenicity amongst all products tested 138, which agrees with other reports of IFN-β immunogenicity 142. Transgenic murine models are useful, however development of the model is a labor intensive process, and new models must be developed for each therapeutic protein of interest. Some transgenic mouse models are provided in Table 3.
Table 3:
Transgenic Mouse Models
| Protein of Interest | Reference |
|---|---|
| Tissue Plasminogen Activator | Stewart et al. 248 |
| Insulin | Ottensen et al. 141 |
| Interferon-β | Hermeling et al. 136 |
| Human Growth Hormone | Sweetser et al. 139 |
| Factor VIII | Fakharzadeh er al. 137 |
| Factor VIIa | Lenk et al. 249 |
| Transforming Growth Factor-β | Carrier et al. 250 |
| Acid Alpha-glucosidase | Khanna et al. 251 |
NOD scid gamma (NSG) mice are highly immunocompromised and can be transfected to investigate the human immune system in an in vivo model. NSG mice are deficient in most immune cells; T and B cells, dendritic cells, macrophages and natural killer cells, as well as complement and cytokine signaling. The Jackson Laboratories pioneered the development of the NSG model, and have developed humanized NSG mouse models143–145 . CD34+ humanized mice are engrafted with cord blood derived hematopoietic stem cells to develop a functional immune system in the mouse, displaying normal T cell and inflammatory function. To this point, NSG mice have primarily been used to investigate the pathology of infectious diseases in a humanized mouse model, but used properly, these mice could provide insight into the mechanism of therapeutic protein immunogenicity.
iii. Higher Order Animal Models:
Higher order animal species may have better clinical utility for predicting immunogenicity due to a higher degree of homology between human and non-human primate proteins and more similarities in immune mechanism. Rhesus monkeys and chimpanzees have been used to examine the relative immunogenicity of human growth hormone (HGH) and tissue plasminogen activator (tPA). Zwickl et al. investigated immunogenicity of HGH in rhesus monkeys and found that their results correlated well with clinical observation in regards to relative immunogenicity, however the predictive power was lessened when trying to assess incidence rate of immunogenicity 146. In a separate study, this group also investigated the impact of immunogenicity of a marketed tPA analog compared to native monkey tPA. They concluded that antibody development is not more significant against the tPA analog and these low antibody levels do not impact its enzymatic behavior, suggesting immunogenicity does not pose a great risk to clinical treatment 147.
In addition to relative immunogenicity, primate models can also be used to investigate the impact neo-epitopes may have on protein immunogenicity. Zwickl again used rhesus monkeys to investigate the impact of neo-epitopes on the immunogenicity of insulin. Monkeys were administered human insulin, LysPro insulin and porcine insulin and the development of antibodies was assessed. Their results showed that alterations present in modified human insulin (LysPro insulin) does not give rise to neo-epitopes in monkeys, as only 1 monkey developed low levels of anti-insulin antibodies 148. These results are corroborated by clinical observation showing human and LysPro insulin have similar incidence rates of immunogenicity 149.
Canine models have also occasionally been utilized to assess immunogenicity. In studies performed by Finn et al, they assessed the immunogenicity of modified Factor IX in a dog model of Hemophilia B. They showed there was no antibody formation against FIX-R338L even after treatment with wild type FIX 150,151. In a separate study be Randolph et al, they investigated whether administration of recombinant canine erythropoietin was immunogenic in dogs and found there were no immunogenicity risks 152.
iv. Limitations of Animal Models:
Despite their uses, animal models still possess significant hurdles impacting their usefulness in predicting immunogenicity due to the lack of genetic diversity in mice and basic differences in immune mechanisms. Factors influencing immunogenicity even in humans are not fully understood, and variability between human and animal systems makes prediction difficult. The strain of mouse used to develop a model, or investigate immunogenicity can also have a large impact on their usefulness. C57BL/6 mice and Balb/c mice differ greatly in their immunologic systems and can respond very differently to stimulation by proteins 153–155. Generally, Balb/c mice have higher cytokine release and T cell function as compared to C57BL/6, while C57BL/6 mice generally have higher natural killer cell activity 155,156. In addition, both strains of mice respond differently to pro- and anti- inflammatory cytokines 156. Differences between humans and mice exist in Toll-like receptors, antibody subsets and the balance of leukocyte subsets that make it difficult to directly correlate human and murine immune responses.
As with murine models, higher order animal species are still ineffective at predicting immunogenicity incidence rates. Gunn et al performed a study looking at the immunogenicity of interleukin-3 (IL-3) and found that all primates formed antibodies, while clinically, few patients form antibodies against IL-3 157. Additionally, primates are not an effective model for assessing immunogenicity of monoclonal antibodies. In a study by van Meer et al. they attempted to assess the incidence rate and impact of immunogenicity of several approved mAb products in monkeys and compared it to available clinical data 158. They found that only 59% of the mAbs tested had comparable immunogenicity between monkeys and humans, while 30% over-predicted and 11% under-predicted.
Laboratory animals commonly used for pre-clinical immunogenicity testing are bred in hyper hygienic conditions and lack exposure to pathogens that strengthen the basal activity of the immune system 159. In contrast, humans are constantly exposed to various pathogens in everyday life that keep the immune system well maintained. Therefore, the physiological microbiome may not be representative of a natural human immune system. Nevertheless, the use of pre-clinical models is indispensable in terms of comparing relative immunogenicity and elucidating the mechanism of immune responses in studies that are not ethically possible or practical in humans.
b. In vitro Immunogenicity Assessment:
Compared to in vivo testing, in vitro systems are typically less time consuming and more cost effective. In vitro assays can be used to investigate the cellular mechanism of the immune response, identify immunogenic epitopes on the proteins, and evaluate MHC affinity, T cell proliferation, and immunogenic effects of the whole protein. In theory, in vitro scaling can be used effectively with screening tools to investigate the mechanistic basis by which the immune system responds to foreign antigens, as well as identifying key immunogenic epitopes present on the protein.
i. Epitope Mapping:
Epitope mapping is a robust method that can identify immunogenic epitopes of a protein by individually analyzing peptide fragments of the protein 160,161. Therapeutic proteins are broken down into overlapping amino acid chains, and peptides are exposed to immune cells. After exposure, immunogenicity can be assessed by measuring cytokines and surface markers that indicate an inflammatory immune response has occurred. This exhaustive method was used by Herzog and colleagues to map the immunogenic T-cell epitopes of rhGAA 162. Mice were immunized with free GAA protein after which the splenocytes were extracted and exposed to amino acid chains developed from the full length protein. Using an ELISpot assay, peptides were assessed for IFN-γ secretion and immunogenic epitopes were identified 162. Hamze et al mapped infliximab and rituximab to identify CD4 T cell epitopes. Using lymphocytes from healthy donors, they identified nine T cell epitopes present in the variable region of both antibodies that were able to stimulate PBMC to secrete a variety of cytokines163. Epitope mapping has been performed for additional proteins, such as FVIII 164 and AAV2 165. Epitope mapping from the full protein is highly labor intensive, and in silico programs are often used to identify regions that may be immunogenic to narrow down epitope candidates.
Epitope spreading can complicate the prediction of immunogenic epitopes on a protein. During a typical immune response, T cell activation is dependent on recognition of immunodominant epitopes, however epitope spreading results in the recognition of sub-dominant or cryptic epitopes 166. Epitope spreading can occur as a result of altered endocytic process, variable T cell interaction, or mutations in B cell processing, which causes alterations in epitope recognition 167. Epitope spreading has been implicated in autoimmune conditions, as it can result in continual tissue damage as the immune system recognizes different epitopes. In a study by Bernard et al, they showed that after immunization with MOG protein, the ADA target changed and new immunogenic MOG epitopes were identified 168. As a result of epitope spreading, continual tissue damage occurs as the next wave of autoreactive T cells invades with a different epitope target. The unpredictability of epitope spreading is extremely difficult to identify in pre-clinical models, and while immunodominant epitopes can be predicted, it may be more difficult to predict sub-dominant epitopes that arise later on.
ii. MHC Binding Assays:
MHC binding assays can be used to detect high affinity peptides and are often used in conjunction with epitope mapping to identify protein regions that are at a high risk of immunogenicity 169–171. Griswold et al 172 describe a high throughput method to perform a peptide MHC binding assay that allows for the rapid analysis of up to 90 peptides on a single plate using a mechanized protocol. Peptides of interest are incubated at various doses with control peptides and soluble MHC proteins to assess affinity. Higher affinity peptides bind more strongly to MHC, resulting in an increased likelihood that a T cell will recognize the epitope170,173–175 ProImmune has developed a peptide binding assay that can be used to provide MHC binding data and identify potentially immunogenic epitopes 176.
iii. T-Cell Dependent Assays:
Upon stimulation by an immunogenic protein, T cells proliferate and release cytokines to exert their effect. Typically, T cell proliferation is measured through the use of radiolabeling with thymidine 177 or fluorescent labeling dyes such as carboxyfluorescein succinimidyl ester (CFSE) 178,179. CFSE is a fluorescent dye that can be used to assess repeated T-cell divisions 179 and is rapidly replacing radiolabeling cells due to less hazardous waste and simplicity. CFSE staining can also be accompanied with phenotypic staining to distinguish different subtypes of proliferating T cells. Karle et al. and Rubic-Schneider et al assessed T cell proliferation using thymidine to determine the immunogenicity of secukinumab and EPO aggregates, respectively 180,181
In addition to proliferation, cytokine release can also be measured to determine immunogenicity. Assaying the type and degree of cytokine activity can give general information about the immune response that is occurring in these T cells. Commonly, ELISA and ELISpot methods are used to measure cytokines such as IL-2 and I FN-γ released by T cells in vitro 174,182. ELISpot assays are highly sensitive methods that can be used to determine the number of cells that secrete specific cytokines. Developed by Czerkinsky et al in 1983, ELISpot assays are more sensitive than ELISA assays and are typically more effective in quantifying the cytokine response 183. Cytokines can also be detected by flow cytometry and can performed in conjunction with T cell phenotyping to classify the type of T cell response that is occurring 184,185. Peripheral Blood Mononuclear Cell (PBMC) assays can provide insight into the potential immune response without testing in humans using preparations that contain several types of immune cells 169,186, better mimicking in vivo immune systems. PBMC preparations contain several different cell types, including macrophages, monocytes, and lymphocytes, and can include cells from both the adaptive and innate immune systems. PBMC from healthy human donors were used to assess the immunogenicity of aggregated antibodies by assessing cytokine secretion and T cell proliferation in a study by Joubert et al 187.
iv. Innate Immune Screening:
Given the increasing evidence of interplay between the innate and adaptive arms of the immune response, using innate cell systems for in vitro screening may improve the predictive power of pre-clinical screening. Ahmadi et al used PBMC preparations in which CD8 reactive T cells were removed to model the innate response, and showed excellent correlation between in vitro testing and clinical immunogenicity seen with Mabs 188. Innate lymphoid cells (ILCs) may be useful as an in vitro system but has not yet been explored in the context of immunogenicity of therapeutic proteins 189. In vitro innate system cell lines transfected with toll-like receptors in combination with human and mouse macrophages have been successfully used to screen for impurities in therapeutic protein preparations 190.
v. B cell Epitope Mapping:
B cells recognize discontinuous epitopes composed of amino acid sequences that are in close proximity in the protein’s three dimensional conformation that more closely mimic the native protein 191,192. As a result, the prediction of B cell epitopes is much more difficult than two dimensional epitopes recognized by T cells192,193. In B cell epitope mapping, immunogenic regions of the protein are identified and modifications can be made to the protein to remove these sections. There are two types of epitopes that can be recognized by antibody-producing B cells; structural and functional epitopes 192. Structural epitopes are generally larger, around 16-22 amino acids, and comprise the amino acids on the protein that come in contact with the antibody. Functional epitopes are shorter 3-5 amino acid chains that influence the affinity of the therapeutic protein and the antibody 192,193.
Currently, the most accurate method of predicting structural epitopes is through the use of X-ray crystallography to identify epitopes on the protein that bind to the antibody. X-ray crystallography allows for the identification of precise sequences that bind to the antibody and can provide information on strength of binding as well 194,195. Nuclear Magnetic Resonance (NMR) can also be used to identify structural epitopes without forming crystals, however it is limited to use with small proteins and peptides 194,196. In addition to the preferred methods listed above, electron microscopy can be used to examine structural epitopes on larger antigens such as viral capsids. For example, electron microscopy was used to identify structural epitopes on the surface of the Human Papilloma Virus 16 capsid and showed that antibody binding results in conformational changes 197,198.
Functional epitopes are commonly determined by antigen fragmentation, competitive binding or modification testing. Fragmentation assays are performed to assess whether or not an antibody binds to the epitope fragment of interest 199,200. Primarily a qualitative approach, western blots or ELISAs are commonly used to assess whether the antibody will bind to the epitope fragment. Positive binding interactions indicate the peptide fragment may be immunogenic. Competitive binding assays can be used to determine if multiple antibodies can bind to epitopes on a protein at the same time 201, providing information on the number of potentially immunogenic epitopes on a therapeutic protein. An easy way to determine if a specific sequence is immunogenic is to modify the amino acid chain and assess its binding to ADA. Modification testing relies on mutagenesis of amino acid residues to assess their impact on binding of the antibody to the immunogenic sequence 202. By mutating individual amino acids, one can identify detrimental residues, and can replace them if it does not abrogate activity of the protein.
vi. Limitations of in vitro immunogenicity assessment:
One of the main issues predicting immunogenicity in vitro is attempting to replicate the immune system ex vivo. The immune system is a complex system of T-cells, B-cells dendritic cells, and macrophages, and this cannot be easily replicated in in vitro systems. It is difficult to predict the interplay between two dimensional epitopes and the three dimensional protein structure and how APC will digest the protein to present epitopes via MHC. It is likely there is variability in the way the proteins are processed by immune cells in vivo versus in vitro that is undetected by in vitro assays. Synthesized peptide sequences may not contain post-translational modifications that are essential to the protein 203,204 which would lead to altered signaling, potentially resulting in false negatives or false positives 204. Even if immunogenic epitopes are detected, they can only be modified if they do not interfere with protein activity. As with many systems, genetic diversity is an issue that is difficult to replicate. Patient-derived PBMC from various genetic backgrounds are a possibility, however a very large pool of donors would be required to adequately capture HLA polymorphisms to investigate.
Several well-known factors influencing immunogenicity such as route of administration, frequency, and dose are very difficult to predict in vitro 7,91. Unpredictable treatment factors also can emerge that influence immunogenicity and are impossible to predict. One example is Eprex, an Epo analog that was used to increase red blood cell growth in anemic patients. Initial treatment with Eprex was successful, however some patients being treated with Eprex started showing higher incidence of red cell aplasia 205,206. It was determined that the issue was antibody mediated and treatment with Eprex was becoming less effective. Initial investigations suggested that organic compounds leached from the rubber stopper of the pre-filled syringes was the most probable cause of immunogenicity 205,206, however further testing has concluded that tungsten present during the manufacturing process may be the root cause of Eprex aggregation181,207. Small factors like these are likely to go undetected during preclinical testing and there is little chance of predicting such occurrences.
c. In Silico Predictions:
Due to technological advances in proteomics and sequence mapping, epitope databases have been constructed that provide a library of existing epitopes and methods of prediction. Several databases exist such as IEDB, Epitome, and SEDB 208–210, that provide information on two dimensional epitopes that are recognized by T cells. The linear binding interface between epitopes and MHC make it easier to predict as opposed to B cell epitopes that are influenced by protein folding. Table 4 highlights several key databases that have been developed.
Table 4:
In Silico Epitope Databases
Several programs and webpages have also been developed that can be used to analyze molecules and predict immunogenic epitopes on potential therapeutic proteins. Soria-Guerra et al reviewed several programs that can be used for in silico epitope prediction 211. These various programs range in MHC subtype predictions and methods, and some programs have the ability to simulate amino acid substitutions to assess changes in immunogenicity. MHCPred is an algorithm developed by Doytchinova that can be used to predict immunogenicity of epitope sequences 212,213 Users can input the amino acid sequence and assess its MHC binding potential to various alleles. EpiMatrix was developed by EpiVax to predict allele specific binding of therapeutic proteins to MHCII receptors, and has been improved to also assess binding at the T-cell receptor interface 169,214. Table 5 summarizes some of the algorithms that have been developed for peptide-MHC binding.
Table 5:
Commonly Used Algorithms and Programs for Epitope Prediction
Epitope prediction can be applied to most therapeutic proteins, however predictions must be assessed in conjunction with in vitro methods. Predictions should also be combined with activity assessment to ensure altering immunogenic epitopes does not impact protein activity. In silico methods could be improved with greater ability to predict peptide-MHC affinity, which drives epitope immunogenicity. It is also difficult to predict peptide affinity for the T-cell receptor, as the TCR recognizes three dimensional structure. As described above, HLA polymorphisms make it extremely difficult to predict population-wide immunogenic epitopes. Algorithms may be able to identify epitopes that are most immunogenic, but patients may still experience immunogenicity due to alternative epitopes that are presented by their individual APC. Post translational modifications are also difficult to simulate in silico.
The use of quantitative structure activity relationship (QSAR) models to improve safety and reduce immunogenicity of protein pharmaceuticals is gaining momentum to predict and reduce immunogenic regions of proteins. This approach attempts to employ0 structural, statistical, and machine based learning to connect structural features to immunogenicity and activity, Generating an integrated data set of protein structures from databanks, pre-clinical and clinical outcomes could be a first step in designing less immunogenic proteins and formulations. Similar QSAR models have been used to predict skin sensitivity to several compounds 215. This approach could also be utilized to predict immunogenicity of protein pharmaceuticals. For example, Kumar et al utilized computational tools to delineate the relationship between epitopes and aggregates using a structure based approach216,217.
5. Strategies to mitigate immunogenicity of therapeutic proteins:
Strategies are being developed that are aimed to prevent as well as reverse an established ADA response by (i) inducing immunological tolerance, (ii) modifying the product or (iii) formulation, (iv) modulation of the immune system using small molecule drugs, and (v) manipulating T and B cells to reverse established immune responses. The reversal of an established immune response is very difficult, often requiring lymphocyte deletion, and have been met with limited success, thus prophylactic regimens to induce tolerance to therapeutic proteins are preferred.
i. Immune Tolerance Induction:
Immune tolerance induction is a method of presenting an antigen in a way that will prevent or reverse the development of ADA in an antigen specific manner. Immunologically, Tregs have shown to have a critical role in immune tolerance induction through TGF-β mediated pathways 218. Our group is developing a method to induce tolerance to various therapeutic proteins by co-administering the antigen with phosphatidylserine (PS) based liposomes. PS is a naturally occurring phospholipid present on the inner leaflet of cells. Upon cellular apoptosis, PS flips to the outer membrane and acts as an “eat me” signal to phagocytic cells that engulf and digest cellular debris without the initiation of an immune response. We have shown that co-administration of PS with various therapeutic proteins and antigens (including rhGAA, FVIII and ovalbumin) can induce durable tolerance and prevent the development of antibodies 78,126 (unpublished data). Additionally, we have shown this method to be antigen specific, as rechallenge with an unrelated antigen showed a robust immune response 31. Sule et al. showed that dendritic cells conditioned with anti-inflammatory cytokines IL-10 and TGF-β were able to suppress antibody development in Hemophilia A (HA) mice 219. In their study, they transferred DC that were treated with IL-10 and TGF-β into HA mice and then challenged the mice with free protein. They also showed that this method is antigen specific, as challenge with human serum albumin resulted in a normal immune response. Zhang and Scott have developed a method to induce immunologic tolerance to FVIII, rhGAA, and AAV8 using Poly lactic-co-glycolic acid (PLGA) based nanoparticles containing rapamycin 128,220,221. These studies showed that administration of antigen with their nanoparticle can significantly reduce antibody development in mouse models.
Another method of inducing immunological tolerance is hepatic gene therapy. In this method, viral vectors containing the gene sequence of the protein of interest are administered and incorporated into hepatocytes, which produce the protein of interest. Work done by Herzog first showed that immune tolerance can be induced to Factor IX by adeno-associated virus (AVV-FIX) gene delivery to the liver 222. Mice that were administered AAV-FIX showed no antibody development and substantially lower T cell responses in vitro. They showed this method was antigen specific and induced the proliferation of regulatory CD4+ T cells. Gene therapy for the treatment of Hemophilia A and B is quickly gaining interest in the industry and the FDA.
ii. Protein Modifications:
Directly modifying the protein may lead to a decrease in immunogenicity by modifying immunogenic regions identified on the protein. Reducing impurities, aggregates and host cell associated proteins have further improved the safety of therapeutic proteins. Using protein engineering approaches and in silico tools, deimmunization and epitope removal strategies have been employed to design less immunogenic therapeutic proteins, which were employed to engineer a less immunogenic form of FVIII. Pratt et al. identified key immunogenic epitopes on the FVIII protein and assess amino acid substitutions that would reduce immunogenicity, while maintaining procoagulant activity. Using in vitro T cell proliferation assays and MHC binding assays, they determined that substitutions at the 2196 amino acid of full length FVIII reduced T cell proliferation while maintaining activity, illustrating the ability of in vitro methods to develop a less immunogenic FVIII 223. Additionally, Pastan et al. developed a less immunogenic anti-CD25 immunotoxin by identifying and eliminating immunogenic T cell epitopes. After amino acid modification, they showed that the T cell response was reduced by 74.8%, while maintaining cell cytotoxicity 224. One strategy that has been shown to reduce clinical immunogenicity for Mabs is to increase the human content in the antibody. One example showed that switching the construct of the antibody OKT3 from murine to chimeric, humanized, or fully human drastically reduced the incidence rate of immunogenicity 225. Anne de Groot reviewed several potential mechanisms to bioengineer less immunogenic proteins in her review 226.
iii. Formulation Related Changes:
Modifying the formulation to decrease aggregation may also help to reduce therapeutic protein immunogenicity. Our lab has shown that the presence of O-Phospho-L-serine (OPLS) prevents the formation of FVIII aggregates by occupying the lipid binding region that is susceptible to aggregation in aqueous formulations 227. Senga and Honda showed that the inclusion of a small peptide AF.2A1 in Mab formulations will bind to the Fc region of non-native IgG conformations. Once bound, the formulation can be separated to remove Mab bound to AF.2A1 and the resulting formulation will be devoid of aggregates228. AF.2A1 peptide beads can be used to rid a formulation of aggregates, while OPLS can be used to prevent the formation of aggregates in FVIII formulations.
iv. Immune Modulatory Drugs
Small molecules drugs can also be used to help patients better tolerate therapeutic proteins by modulating the immune response primarily through immunosuppression. Commonly used immunosuppressive drugs are methotrexate, rapamycin, bortezomib, and cyclophosphamide. Methotrexate (MTX) is an anti-cancer drug that is commonly used to treat certain types of cancer and autoimmune diseases, such as breast cancer, leukemia, rheumatoid arthritis, and Crohn’s disease. MTX is used to treat autoimmune diseases due to its ability to suppress T cell activation and down-regulate B cell activity. Kishnani et al. and Richards et al. developed low dose MTX regimens for use in Infantile Pompe Disease patients to induce tolerance to administered rhGAA 79,229 and shown clinically to prevent immunogenicity. Maini et al. showed that the administration of MTX with the anti-TNF antibody infliximab reduced antibody development and patient response improved 230. Rapamycin is a small molecule macrolide that is commonly used to prevent transplant rejection by inhibiting the activation of T and B cells via the mTOR receptor 231,232. As mentioned, Scott and Zhang used rapamycin in their PLGA nanoparticle to induce tolerance. Herzog et al. used rapamycin in combination with FVIII delivered orally to induce tolerance to FVIII in Hemophilia A mice 233. Bortezomib is a small molecule drug that inhibits proteasome activity, preventing the degradation of pro-apoptotic signals, leading to cell death. It has been used in Infantile Pompe Disease patients along with MTX and RTX to reverse high sustained ADA and restore efficacy of therapy 234. Cyclophosphamide is primarily used as a chemotherapeutic agent to kill rapidly proliferating cells but at low doses has been recognized to have immunomodulatory effects through its influence on dendritic cell homeostasis and cytokine secretion 235. Historically, cyclophosphamide has been shown to induce tolerance to FVIII when given as a pre-treatment 236,237 Investigative therapies also include cyclophosphamide/steroid combinations to induce tolerance in FVIII inhibitor positive patients 238 and has also been tested for use in preventing ADA formation in the treatment of Pompe Disease to no avail 239. Azathioprine is an immunosuppressive drug that can be given in combination with infliximab to improve the treatment of Crohn’s Disease240. It was suggested by Colombel et al that the improvement may be partially attributed to reducing the immunogenicity of infliximab241. Additionally, azathioprine was used in combination with infliximab or adalimumab to reduce ADA frequency by 47%242.
v. Lymphocyte Manipulation to Reverse ADA development
In addition to tolerance induction strategies, methods have also been investigated which manipulate and eliminate lymphocytes to prevent or reverse an immune response against the therapeutic protein (Fig. 4). These approaches can impact ADA development, but also compromise the immune system of the patient, leaving them susceptible to infections. Kishnani et al. developed an immunosuppressive regimen that showed the ability to stop and reverse the development of antibodies against GAA in infantile Pompe Disease patients 229,243, comprised of rituximab (RTX), MTX, and gammaglobulins. While this method was able to stop antibody development, it also caused a significant reduction in CD20 positive B cells during the course of RTX and MTX administration that did not recover until after treatment with RTX and MTX was stopped. Anti-CD3 antibodies have been used with success in preventing antibody development against rhGAA 244 and FVIII 245 in mice by altering cytokine production to favor a Th1 response, however they were not able to modulate ADA development long term, likely due to the generation of new lymphocyte cells that are reactive to the therapeutic protein. Active tolerance is not being induced with the use of immunosuppressive drugs, therefore is only maintained for one cell lifespan.
Figure 4:
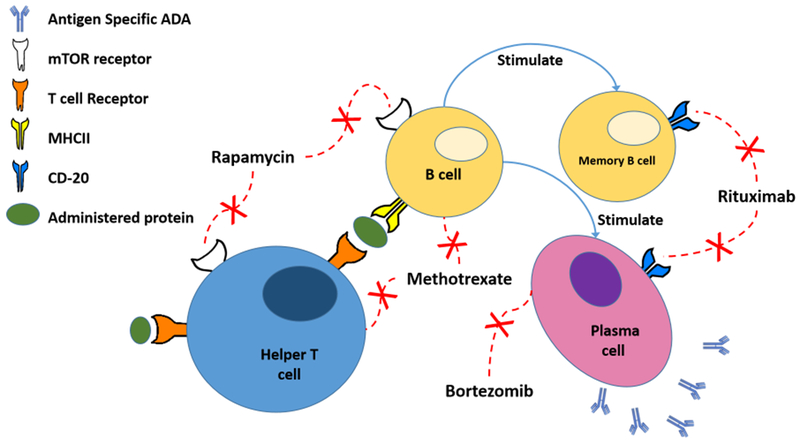
Effect of various immunomodulatory drugs on the immune system. Rapamycin inhibits T and B cell activation through the mTOR pathway. Rituximab blocks CD20, killing CD20 expressing B cells. Methotrexate inhibits DNA synthesis and kills rapidly dividing cells. Bortezomib inhibits proteasomes, promoting apoptosis in rapidly dividing cells.
Scott et al. engineered FVIII expressing cytotoxic T cell that could selectively target and kill FVIII-specific B cells. They engineered immunodominant epitopes on the FVIII protein that were expressed as antigen receptors on the surface of cytotoxic T cells, which bound to and specifically killed FVIII reactive B cells in vivo and in vitro 246. In the interest of B cell targeting, Brettschneider et al. fused the C2 domain of FVIII to endotoxin A of pseudomonas aeruginosa which selectively targeted FVIII specific memory B cells and eliminated them 247.
There are a variety of methods being assessed to induce immunological tolerance and the clinical translatability of these methods is still being investigated. Antigen specific tolerance is the desired method, as patients would not be susceptible to secondary infection. There is also a degree of risk assessment required in these tolerance induction strategies. In some cases, such as Pompe Disease, antibodies develop in nearly all patients, leaving the patient without relief and no other clinical options. In this case, administering an immunosuppressive regimen may be a more acceptable option, whereas in other diseases, the incidence of immunogenicity is lower, and patients can transition to another product. Mitigation strategies are still under development, however there are several promising methods being investigated.
Conclusion
Clinical immunogenicity of therapeutic proteins is a growing issue that threatens to undermine the development of potent protein drugs. Understanding the mechanism of the immune response and factors influencing immunogenicity of therapeutic proteins will allow us to design safer drugs and prevent immunogenicity. Engineering proteins that are inherently less immunogenic, or developing methods to prevent or reverse an established immune response to therapeutic proteins can help solve the major issue of immunogenicity. Several methods are available that can attempt to predict immunogenicity of therapeutic proteins, but there is still large debate over the usefulness of these predictive tools. As was described, in vivo models are most useful for relative immunogenicity ranking, but the prediction of immunogenicity incidence rate is something that is very difficult to perform in models due to basic differences between human and animal immune systems. In vitro models are useful in predicting if the protein will interact with MHC molecules, however these systems do not expose the protein to the complete array of factors that can influence immunogenicity in human patients. In silico models are rapidly being developed and can be useful in predicting immunogenic epitopes, but need to be combined with in vitro testing to ensure the predictions are useful. Despite their limitations, preclinical methods for predicting immunogenicity are instrumental to drug development. Immunogenicity of therapeutic proteins is a clinical challenge, but success in overcoming it would improve the safety and efficacy of several lifesaving therapies. Several mitigation strategies are being actively pursued to not only prevent, but reverse an established immune response.
Acknowledgements:
This work was financially supported by National Institutes of Health R01 grant HL-70227 to Dr. Sathy V. Balu-Iyer. The opinions expressed in this review are those of the authors and do not represent the opinion or position of SUNY University at Buffalo, the FDA, NIH, or any other corporate entity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A 2001. Recombinant human acid &agr;-glucosidase enzyme therapy for infantile glycogen storage disease type II: Results of a phase I/II clinical trial. Genetics in Medicine 3(2):132–138. [PubMed] [Google Scholar]
- 2.Klinge L, Straub V, Neudorf U, Schaper J, Bosbach T, Görlinger K, Wallot M, Richards S, Voit T 2005. Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscular Disorders 15(1):24–31. [DOI] [PubMed] [Google Scholar]
- 3.Lusher JM, Arkin S, Abildgaard CF, Schwartz RS 1993. Recombinant Factor VIII for the Treatment of Previously Untreated Patients with Hemophilia A -- Safety, Efficacy, and Development of Inhibitors. New England Journal of Medicine 328(7):453–459. [DOI] [PubMed] [Google Scholar]
- 4.Rudick RA, Ransohoff RM, Lee JC, Peppler R, Yu M, Mathisen PM, Tuohy VK 1998. In vivo effects of interferon beta-1a on immunosuppressive cytokines in multiple sclerosis. Neurology 50(5):1294–1300. [DOI] [PubMed] [Google Scholar]
- 5.Herndon RM, Rudick RA, Munschauer FE III, Mass MK, Salazar AM, Coats ME, Labutta R, Richert JR, Cohan SL, Genain C 2005. Eight-year immunogenicity and safety of interferon beta-1a-Avonex® treatment in patients with multiple sclerosis. Multiple Sclerosis Journal 11(4):409–419. [DOI] [PubMed] [Google Scholar]
- 6.Baker M, Reynolds HM, Lumicisi B, Bryson CJ 2010. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self/nonself 1(4):314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schellekens H, Jiskoot W. 2013. Immunogenicity of therapeutic proteins Pharmaceutical biotechnology, ed.: Springer; p 133–141. [Google Scholar]
- 8.Schellekens H 2002. Immunogenicity of therapeutic proteins: Clinical implications and future prospects. Clinical Therapeutics 24(11):1720–1740. [DOI] [PubMed] [Google Scholar]
- 9.Xing Y, Hogquist KA 2012. T-cell tolerance: central and peripheral. Cold Spring Harbor perspectives in biology 4(6):a006957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janeway CA, Travers P, Walport M, Shlomchik M 2017. Immunobiology: the immune system in health and disease. 2005 New York: Garland Science; 6. [Google Scholar]
- 11.Shishido SN, Varahan S, Yuan K, Li X, Fleming SD 2012. Humoral innate immune response and disease. Clinical immunology (Orlando, Fla) 144(2):142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robey a E Fowlkes BJ 1994. Selective Events in T Cell Development. Annual Review of Immunology 12(1):675–705. [DOI] [PubMed] [Google Scholar]
- 13.Ademokun AA, Dunn-Walters D. 2001. Immune Responses: Primary and Secondary eLS, ed.: John Wiley & Sons, Ltd. [Google Scholar]
- 14.Letendre P 2010. Primary and Secondary Immune Responses. ed.: University of Alberta. [Google Scholar]
- 15.Inaba K, Witmer MD, Steinman RM 1984. Clustering of dendritic cells, helper T lymphocytes, and histocompatible B cells during primary antibody responses in vitro. The Journal of Experimental Medicine 160(3):858–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motta I, Truffa-Bachi P. Annales d’immunologie, 1980, pp 223–227. [PubMed] [Google Scholar]
- 17.Guan SG, Qi AS 1995. Contributions of memory B cells to secondary immune response. Bulletin of mathematical biology 57(5):713–731. [DOI] [PubMed] [Google Scholar]
- 18.Kishnani PS, Goldenberg PC, DeArmey SL, Heller J, Benjamin D, Young S, Bali D, Smith SA, Li JS, Mandel H 2010. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Molecular genetics and metabolism 99(1):26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee C, Kessler C, Varon D, Martinowitz U, Heim M, Jacquemin M, SAINT-REMY JM 1998. Factor VIII immunogenicity. Haemophilia: State of the Art 4(4):552–557. [DOI] [PubMed] [Google Scholar]
- 20.Lollar P, Healey JF, Barrow RT, Parker ET. 2001. Factor Viii Inhibitors In Monroe DM, Hedner U, Hoffman MR, Negrier C, Savidge GF, White GC, editors. Hemophilia Care in the New Millennium, ed., Boston, MA: Springer US; p 65–73. [Google Scholar]
- 21.Harding FA, Stickler MM, Razo J, DuBridge RB 2010. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs 2(3):256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K 2011. Engineering the variable region of therapeutic IgG antibodies. mAbs 3(3):243–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schellekens H 2003. Immunogenicity of therapeutic proteins. Nephrology Dialysis Transplantation 18(7):1257–1259. [DOI] [PubMed] [Google Scholar]
- 24.Dalum I, Jensen MR, Hindersson P, Elsner HI, Mouritsen S 1996. Breaking of B cell tolerance toward a highly conserved self protein. The Journal of Immunology 157(11):4796–4804. [PubMed] [Google Scholar]
- 25.Nemazee D 2017. Mechanisms of central tolerance for B cells. Nature reviews Immunology 17(5):281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.KROMMINGA A, SCHELLEKENS H 2005. Antibodies against Erythropoietin and Other Protein-Based Therapeutics: An Overview. Annals of the New York Academy of Sciences 1050(1):257–265. [DOI] [PubMed] [Google Scholar]
- 27.Busch DH, Pamer EG 1999. T cell affinity maturation by selective expansion during infection. Journal of Experimental Medicine 189(4):701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujii S-i, Liu K, Smith C, Bonito AJ, Steinman RM 2004. The Linkage of Innate to Adaptive Immunity via Maturing Dendritic Cells In Vivo Requires CD40 Ligation in Addition to Antigen Presentation and CD80/86 Costimulation. The Journal of Experimental Medicine 199(12):1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wakelin D, Denham DA. 1983. The immune response Trichinella and trichinosis, ed.: Springer; p 265–308. [Google Scholar]
- 30.Coffman RL, Seymour BW, Lebman DA, Hiraki DD, Christiansen JA, Shrader B, Cherwinski HM, Savelkoul HF, Finkelman FD, Bond MW 1988. The role of helper T cell products in mouse B cell differentiation and isotype regulation. Immunological reviews 102(1):5–28. [DOI] [PubMed] [Google Scholar]
- 31.Gaitonde P, Ramakrishnan R, Chin J, Kelleher RJ, Bankert RB, Balu-Iyer SV 2013. Exposure to Factor VIII Protein in the Presence of Phosphatidylserine Induces Hypo-responsiveness toward Factor VIII Challenge in Hemophilia A Mice. Journal of Biological Chemistry 288(24):17051–17056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.den Haan JMM, Arens R, van Zelm MC 2014. The activation of the adaptive immune system: Cross-talk between antigen-presenting cells, T cells and B cells. Immunology Letters 162(2, Part B):103–112. [DOI] [PubMed] [Google Scholar]
- 33.Janeway CA Jr, Travers P, Walport M, Shlomchik MJ 2001. B-cell activation by armed helper T cells. [Google Scholar]
- 34.Isabwe GAC, Garcia Neuer M, de las Vecillas Sanchez L, Lynch D-M, Marquis K, Castells M 2018. Hypersensitivity reactions to therapeutic monoclonal antibodies: Phenotypes and endotypes. Journal of Allergy and Clinical Immunology 142(1):159–170.e152. [DOI] [PubMed] [Google Scholar]
- 35.Brennan PJ, Bouza TR, Hsu FI, Sloane DE, Castells MC 2009. Hypersensitivity reactions to mAbs: 105 desensitizations in 23 patients, from evaluation to treatment. Journal of Allergy and Clinical Immunology 124(6):1259–1266. [DOI] [PubMed] [Google Scholar]
- 36.Capanoglu M, Dibek Misirlioglu E, Azkur D, Vezir E, Guvenir H, Gunduz M, Toyran M, Civelek E, Kocabas CN 2016. IgE-Mediated Hypersensitivity and Desensitisation with Recombinant Enzymes in Pompe Disease and Type I and Type VI Mucopolysaccharidosis. International archives of allergy and immunology 169(3):198–202. [DOI] [PubMed] [Google Scholar]
- 37.Buelow Becky JR. 2015. Immediate Hypersesnsitivity Reactions. ed., Medscape.com: Medscape. [Google Scholar]
- 38.Hou W, Xu G, Wang H. 2011. CHAPTER 1 - Basic immunology and immune system disorders In Hou W, Xu G, Wang H, editors. Treating Autoimmune Disease with Chinese Medicine, ed., Edinburgh: Churchill Livingstone; p 1–12. [Google Scholar]
- 39.Kindt TJ, Goldsby RA, Osborne BA, Kuby J. 2007. Kuby immunology. ed.: Macmillan. [Google Scholar]
- 40.King TC. 2007. 2 - Inflammation, Inflammatory Mediators, and Immune-Mediated Disease In King TC, editor Elsevier’s Integrated Pathology, ed., Philadelphia: Mosby; p 21–57. [Google Scholar]
- 41.Cochrane AMG, Thomson AD, Moussouros A, Eddleston ALWF, Williams R 1976. ANTIBODY-DEPENDENT CELL-MEDIATED (K CELL) CYTOTOXICITY AGAINST ISOLATED HEPATOCYTES IN CHRONIC ACTIVE HEPATITIS. The Lancet 307(7957):441–444. [DOI] [PubMed] [Google Scholar]
- 42.Keefe DL 2002. Trastuzumab-associated cardiotoxicity. Cancer 95(7):1592–1600. [DOI] [PubMed] [Google Scholar]
- 43.De Korte M, De Vries E, Lub-de Hooge M, Jager P, Gietema J, van der Graaf W, Sluiter W, van Veldhuisen D, Suter T, Sleijfer DT 2007. 111Indium-trastuzumab visualises myocardial human epidermal growth factor receptor 2 expression shortly after anthracycline treatment but not during heart failure: a clue to uncover the mechanisms of trastuzumab-related cardiotoxicity. European Journal of Cancer 43(14):2046–2051. [DOI] [PubMed] [Google Scholar]
- 44.Brennan FR, Morton LD, Spindeldreher S, Kiessling A, Allenspach R, Hey A, Mϋller P, Frings W, Sims J 2010. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. mAbs 2(3):233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pawluczkowycz AW, Beurskens FJ, Beum PV, Lindorfer MA, van de Winkel JG, Parren PW, Taylor RP 2009. Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. The Journal of Immunology:jimmunol. 0900632. [DOI] [PubMed] [Google Scholar]
- 46.Dall’Ozzo S, Tartas S, Paintaud G, Cartron G, Colombat P, Bardos P, Watier H, Thibault G 2004. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer research 64(13):4664–4669. [DOI] [PubMed] [Google Scholar]
- 47.Michaelsen TE, Aase A, Norderhaug L, Sandlie I 1992. Antibody dependent cell-mediated cytotoxicity induced by chimeric mouse-human IgG subclasses and IgG3 antibodies with altered hinge region. Molecular Immunology 29(3):319–326. [DOI] [PubMed] [Google Scholar]
- 48.Movat HZ. 1971. Inflammation, Immunity, and Hypersensitivity. ed.: Medical Department, Harper & Row. [Google Scholar]
- 49.Postow MA, Sidlow R, Hellmann MD 2018. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. New England Journal of Medicine 378(2):158–168. [DOI] [PubMed] [Google Scholar]
- 50.Hargadon KM, Johnson CE, Williams CJ 2018. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. International immunopharmacology 62:29–39. [DOI] [PubMed] [Google Scholar]
- 51.Lumizyme [Package Insert] Genzyme Corporation CM, 2011. ed. [Google Scholar]
- 52.Myozyme [Package Insert] Genzyme Corporation CM, 2007. [Google Scholar]
- 53.Shima M, Hanabusa H, Taki M, Matsushita T, Sato T, Fukutake K, Kasai R, Yoneyama K, Yoshida H, Nogami K 2017. Long-term safety and efficacy of emicizumab in a phase 1/2 study in patients with hemophilia A with or without inhibitors. Blood Advances 1(22):1891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.West R, Zelinkova Z, Wolbink G, Kuipers E, Stokkers P, Van Der Woude C 2008. Immunogenicity negatively influences the outcome of adalimumab treatment in Crohn’s disease. Alimentary pharmacology & therapeutics 28(9):1122–1126. [DOI] [PubMed] [Google Scholar]
- 55.Karmiris K, Paintaud G, Noman M, Magdelaine–Beuzelin C, Ferrante M, Degenne D, Claes K, Coopman T, Van Schuerbeek N, Van Assche G 2009. Influence of trough serum levels and immunogenicity on long-term outcome of adalimumab therapy in Crohn’s disease. Gastroenterology 137(5):1628–1640. [DOI] [PubMed] [Google Scholar]
- 56.Chirmule N, Jawa V, Meibohm B 2012. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. The AAPS journal 14(2):296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sailstad J, Amaravadi L, Clements-Egan A, Gorovits B, Myler H, Pillutla R, Pursuhothama S, Putman M, Rose M, Sonehara K 2014. A white paper–consensus and recommendations of a global harmonization team on assessing the impact of immunogenicity on pharmacokinetic measurements. The AAPS journal 16(3):488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu ZH, Lee H, Vu T, Hu C, Yan H, Baker D, Hsu B, Pendley C, Wagner C, Davis HM, Zhou H 2010. Population pharmacokinetics of golimumab in patients with ankylosing spondylitis: impact of body weight and immunogenicity. Int J Clin Pharmacol Ther 48(9):596–607. [DOI] [PubMed] [Google Scholar]
- 59.Ng CM, Loyet KM, Iyer S, Fielder PJ, Deng R 2014. Modeling approach to investigate the effect of neonatal Fc receptor binding affinity and anti-therapeutic antibody on the pharmacokinetic of humanized monoclonal anti-tumor necrosis factor-α IgG antibody in cynomolgus monkey. European Journal of Pharmaceutical Sciences 51:51–58. [DOI] [PubMed] [Google Scholar]
- 60.Vande Casteele N, Mould DR, Coarse J, Hasan I, Gils A, Feagan B, Sandborn WJ 2017. Accounting for Pharmacokinetic Variability of Certolizumab Pegol in Patients with Crohn’s Disease. Clinical Pharmacokinetics 56(12):1513–1523. [DOI] [PubMed] [Google Scholar]
- 61.Rosario M, Dirks NL, Gastonguay MR, Fasanmade AA, Wyant T, Parikh A, Sandborn WJ, Feagan BG, Reinisch W, Fox I 2015. Population pharmacokinetics-pharmacodynamics of vedolizumab in patients with ulcerative colitis and Crohn’s disease. Alimentary Pharmacology & Therapeutics 42(2):188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen X, Hickling T, Kraynov E, Kuang B, Parng C, Vicini P 2013. A Mathematical Model of the Effect of Immunogenicity on Therapeutic Protein Pharmacokinetics. The AAPS Journal 15(4):1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gómez-Mantilla JD, Trocóniz IF, Parra-Guillén Z, Garrido MJ 2014. Review on modeling anti-antibody responses to monoclonal antibodies. Journal of Pharmacokinetics and Pharmacodynamics 41(5):523–536. [DOI] [PubMed] [Google Scholar]
- 64.Shetty KA, Kosloski MP, Mager DE, Balu-Iyer SV 2015. Soy Phosphatidylinositol Containing Nanoparticle Prolongs Hemostatic Activity of B-Domain Deleted Factor VIII in Hemophilia A Mice. Journal of Pharmaceutical Sciences 104(2):388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bartelds GM, Krieckaert CM, Nurmohamed MT, et al. 2011. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA 305(14):1460–1468. [DOI] [PubMed] [Google Scholar]
- 66.van Schouwenburg PA, Krieckaert CL, Rispens T, Aarden L, Wolbink GJ, Wouters D 2013. Long-term measurement of anti-adalimumab using pH-shift-anti-idiotype antigen binding test shows predictive value and transient antibody formation. Annals of the Rheumatic Diseases 72(10):1680–1686. [DOI] [PubMed] [Google Scholar]
- 67.van Schouwenburg PA, Bartelds GM, Hart MH, Aarden L, Wolbink GJ, Wouters D 2010. A novel method for the detection of antibodies to adalimumab in the presence of drug reveals “hidden” immunogenicity in rheumatoid arthritis patients. Journal of Immunological Methods 362(1):82–88. [DOI] [PubMed] [Google Scholar]
- 68.Eriksson C, Agaton C, Kange R, Sundberg M, Nilsson P, Ek B, Uhlen M, Gustafsson M, Hober S 2006. Microfluidic analysis of antibody specificity in a compact disk format. Journal of proteome research 5(7):1568–1574. [DOI] [PubMed] [Google Scholar]
- 69.Carrasco-Triguero M, Mahood C, Milojic-Blair M, Amaya C, Ruppel J, Hong K, Yi JH, Kaur S 2012. Overcoming soluble target interference in an anti-therapeutic antibody screening assay for an antibody-drug conjugate therapeutic. Bioanalysis 4(16):2013–2026. [DOI] [PubMed] [Google Scholar]
- 70.Eriksson S, Halenius H, Pulkki K, Hellman J, Pettersson K 2005. Negative interference in cardiac troponin I immunoassays by circulating troponin autoantibodies. Clinical chemistry 51(5):839–847. [DOI] [PubMed] [Google Scholar]
- 71.Myler H, Felix T, Zhu J, Hruska M, Piccoli SP 2014. Measuring biotherapeutics with endogenous counterparts and pre-existing antibodies: an interferon case study. Bioanalysis 6(8):1113–1122. [DOI] [PubMed] [Google Scholar]
- 72.Shibata H, Nishimura K, Miyama C, Tada M, Suzuki T, Saito Y, Ishii-Watabe A 2018. Comparison of different immunoassay methods to detect human anti-drug antibody using the WHO erythropoietin antibody reference panel for analytes. J Immunol Methods 452:73–77. [DOI] [PubMed] [Google Scholar]
- 73.Verbruggen H, Novakova I, Wessels J, Boezeman J, Berg Mvd, Mauser-Bunschoten E 1995. The Nijmegen modification of the Bethesda assay for factor VIII: C inhibitors: improved specificity and reliability. [PubMed]
- 74.Dong H, Mora JR, Brockus C, Chilewski SD, Dodge R, Merrifield C, Dickerson WM, DeSilva B 2015. Development of a Generic Anti-PEG Antibody Assay Using BioScale’s Acoustic Membrane MicroParticle Technology. The AAPS journal 17(6):1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bloem K, van Leeuwen A, Verbeek G, Nurmohamed MT, Wolbink GJ, van der Kleij D, Rispens T 2015. Systematic comparison of drug-tolerant assays for anti-drug antibodies in a cohort of adalimumab-treated rheumatoid arthritis patients. J Immunol Methods 418:29–38. [DOI] [PubMed] [Google Scholar]
- 76.Hart MH, de Vrieze H, Wouters D, Wolbink G-J, Killestein J, de Groot ER, Aarden LA, Rispens T 2011. Differential effect of drug interference in immunogenicity assays. Journal of Immunological Methods 372(1):196–203. [DOI] [PubMed] [Google Scholar]
- 77.Mikulskis A, Yeung D, Subramanyam M, Amaravadi L 2011. Solution ELISA as a platform of choice for development of robust, drug tolerant immunogenicity assays in support of drug development. J Immunol Methods 365(1–2):38–49. [DOI] [PubMed] [Google Scholar]
- 78.Schneider JL, Balu-Iyer SV 2016. Phosphatidylserine Converts Immunogenic Recombinant Human Acid Alpha-Glucosidase to a Tolerogenic Form in a Mouse Model of Pompe Disease. Journal of pharmaceutical sciences 105(10):3097–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Joseph A, Munroe K, Housman M, Garman R, Richards S 2008. Immune tolerance induction to enzyme-replacement therapy by co-administration of short-term, low-dose methotrexate in a murine Pompe disease model. Clinical & Experimental Immunology 152(1):138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Spengler M, Adler M, Jonas A, Niemeyer CM 2009. Immuno-PCR assays for immunogenicity testing. Biochem Biophys Res Commun 387(2):278–282. [DOI] [PubMed] [Google Scholar]
- 81.Chen LZ, Roos D, Philip E 2016. Development of Immunocapture-LC/MS Assay for Simultaneous ADA Isotyping and Semiquantitation. J Immunol Res 2016:7682472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.An B, Zhang M, Qu J 2014. Towards Sensitive and Accurate Analysis of Antibody Biotherapeutics by LC/MS. Drug Metabolism and Disposition:dmd.114.058917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marini JC, Sendecki J, Cornillie F, Popp JW, Black S, Blank M, Gils A, Van Stappen T, Hamann D, Rispens T, Thérien L, Chun K, Shankar G 2017. Comparisons of Serum Infliximab and Antibodies-to-Infliximab Tests Used in Inflammatory Bowel Disease Clinical Trials of Remicade®. The AAPS Journal 19(1):161–171. [DOI] [PubMed] [Google Scholar]
- 84.DIMICHELE DM, HOOTS WK PIPE SW RIVARD GE, SANTAGOSTINO E 2007. International workshop on immune tolerance induction: consensus recommendations1. Haemophilia 13(s1):1–22. [DOI] [PubMed] [Google Scholar]
- 85.Reipert BM, Van Den Helden PMW, Schwarz H-P, Hausl C 2007. Mechanisms of action of immune tolerance induction against factor VIII in patients with congenital haemophilia A and factor VIII inhibitors. British Journal of Haematology 136(1):12–25. [DOI] [PubMed] [Google Scholar]
- 86.Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M, Jones SA, Olson R, White A, Wells C, Bali D, Case LE, Young SP, Rosenberg AS, Kishnani PS 2012. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genetics In Medicine 14:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Villadolid J, Amin A 2015. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Translational lung cancer research 4(5):560–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Laine J, Jokiranta TS, Eklund KK, Väkeväinen M, Puolakka K 2016. Cost-effectiveness of routine measuring of serum drug concentrations and anti-drug antibodies in treatment of rheumatoid arthritis patients with TNF-α blockers. Biologics : targets & therapy 10:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vincent FB, Morand EF, Murphy K, Mackay F, Mariette X, Marcelli C 2013. Antidrug antibodies (ADAb) to tumour necrosis factor (TNF)-specific neutralising agents in chronic inflammatory diseases: a real issue, a clinical perspective. Annals of the Rheumatic Diseases 72(2):165–178. [DOI] [PubMed] [Google Scholar]
- 90.Link J, Ramanujam R, Auer M, Ryner M, Hässler S, Bachelet D, Mbogning C, Warnke C, Buck D, Hyldgaard Jensen PE, Sievers C, Ingenhoven K, Fissolo N, Lindberg R, Grummel V, Donnellan N, Comabella M, Montalban X, Kieseier B, Soelberg Sørensen P, Hartung H-P, Derfuss T, Lawton A, Sikkema D, Pallardy M, Hemmer B, Deisenhammer F, Broët P, Dönnes P, Davidson J, Fogdell-Hahn A, Consortium A 2017. Clinical practice of analysis of anti-drug antibodies against interferon beta and natalizumab in multiple sclerosis patients in Europe: A descriptive study of test results. PloS one 12(2):e0170395–e0170395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schellekens H 2005. Factors influencing the immunogenicity of therapeutic proteins. Nephrology Dialysis Transplantation 20(suppl_6):vi3–vi9. [DOI] [PubMed] [Google Scholar]
- 92.Segura E, Valladeau-Guilemond J, Donnadieu M-H, Sastre-Garau X, Soumelis V, Amigorena S 2012. Characterization of resident and migratory dendritic cells in human lymph nodes. The Journal of experimental medicine 209(4):653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Haniffa M, Gunawan M, Jardine L 2015. Human skin dendritic cells in health and disease. Journal of dermatological science 77(2):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ross C, Clemmesen KM, Svenson M, Soelberg Sørensen P, Koch-Henriksen N, Lange Skovgaard G, Bendtzen K 2000. Immunogenicity of interferon-β in multiple sclerosis patients: Influence of preparation, dosage, dose frequency, and route of administration. Annals of Neurology 48(5):706–712. [PubMed] [Google Scholar]
- 95.Lawlor DA, Ward FE, Ennis PD, Jackson AP, Parham P 1988. HLA-A and B polymorphisms predate the divergence of humans and chimpanzees. Nature 335:268. [DOI] [PubMed] [Google Scholar]
- 96.Li D, Sun K, Zhao Y, Lin A, Li S, Jiang Y, Feng J 2017. Polymorphism in the major histocompatibility complex (MHC class II B) genes of the Rufous-backed Bunting (Emberiza jankowskii). PeerJ 5:e2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Williams TM 2001. Human Leukocyte Antigen Gene Polymorphism and the Histocompatibility Laboratory. The Journal of molecular diagnostics : JMD 3(3):98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kropshofer H, Hämmerling GJ, Vogt AB 1999. The impact of the non-classical MHC proteins HLA-DM and HLA-DO on loading of MHC class II molecules. Immunological reviews 172(1):267–278. [DOI] [PubMed] [Google Scholar]
- 99.Arnold PY, La Gruta NL, Miller T, Vignali KM, Adams PS, Woodland DL, Vignali DA 2002. The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. The Journal of Immunology 169(2):739–749. [DOI] [PubMed] [Google Scholar]
- 100.Ratanji KD, Derrick JP, Dearman RJ, Kimber I 2014. Immunogenicity of therapeutic proteins: Influence of aggregation. Journal of Immunotoxicology 11(2):99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang W, Singh SK, Li N, Toler MR, King KR, Nema S 2012. Immunogenicity of protein aggregates—concerns and realities. International Journal of Pharmaceutics 431(1–2):1–11. [DOI] [PubMed] [Google Scholar]
- 102.Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, Topp EM 2016. Immunogenicity of therapeutic protein aggregates. Journal of pharmaceutical sciences 105(2):417–430. [DOI] [PubMed] [Google Scholar]
- 103.Narhi LO, Schmit J, Bechtold-Peters K, Sharma D 2012. Classification of Protein Aggregates1. Journal of pharmaceutical sciences 101(2):493–498. [DOI] [PubMed] [Google Scholar]
- 104.Joubert MK, Luo Q, Nashed-Samuel Y, Wypych J, Narhi LO 2011. Classification and characterization of therapeutic antibody aggregates. Journal of Biological Chemistry:jbc. M110 160457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJA, Middaugh CR, Winter G, Fan Y-X, Kirshner S, Verthelyi D, Kozlowski S, Clouse KA, Swann PG, Rosenberg A, Cherney B 2009. Overlooking subvisible particles in therapeutic protein products: Gaps that may compromise product quality. Journal of Pharmaceutical Sciences 98(4):1201–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Barnard JG, Singh S, Randolph TW, Carpenter JF 2011. Subvisible Particle Counting Provides a Sensitive Method of Detecting and Quantifying Aggregation of Monoclonal Antibody Caused by Freeze-Thawing: Insights Into the Roles of Particles in the Protein Aggregation Pathway. Journal of Pharmaceutical Sciences 100(2):492–503. [DOI] [PubMed] [Google Scholar]
- 107.Pisal DS, Kosloski MP, Middaugh CR, Bankert RB, Balu-iyer SV 2012. Native-Like Aggregates of Factor VIII Are Immunogenic in von Willebrand Factor Deficient and Hemophilia a Mice. Journal of Pharmaceutical Sciences 101(6):2055–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hermeling S, Schellekens H, Maas C, Gebbink MF, Crommelin DJ, Jiskoot W 2006. Antibody response to aggregated human interferon alpha2b in wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J Pharm Sci 95(5):1084–1096. [DOI] [PubMed] [Google Scholar]
- 109.van Beers MMC, Sauerborn M, Gilli F, Brinks V, Schellekens H, Jiskoot W 2011. Oxidized and Aggregated Recombinant Human Interferon Beta is Immunogenic in Human Interferon Beta Transgenic Mice. Pharmaceutical Research 28(10):2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rudick RA, Goelz SE 2011. Beta-interferon for multiple sclerosis. Experimental Cell Research 317(9):1301–1311. [DOI] [PubMed] [Google Scholar]
- 111.Liu L 2015. Antibody Glycosylation and Its Impact on the Pharmacokinetics and Pharmacodynamics of Monoclonal Antibodies and Fc-Fusion Proteins. Journal of Pharmaceutical Sciences 104(6):1866–1884. [DOI] [PubMed] [Google Scholar]
- 112.Rosenberg AS 2006. Effects of protein aggregates: an immunologic perspective. The AAPS journal 8(3):E501–E507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ridker PM, Tardif J-C, Amarenco P, Duggan W, Glynn RJ, Jukema JW, Kastelein JJ, Kim AM, Koenig W, Nissen S 2017. Lipid-reduction variability and antidrug-antibody formation with bococizumab. New England Journal of Medicine 376(16):1517–1526. [DOI] [PubMed] [Google Scholar]
- 114.Fazio S, Robertson DG, Joh T, Wan H, Riel T, Forgues P, Baum CM, Garzone PD, Gumbiner B 2017. Effects of 12 weeks of treatment with intravenously administered bococizumab, a humanized monoclonal antibody blocking proprotein convertase subtilisin/kexin type 9, in hypercholesterolemic subjects on high-dose statin. Cardiovascular therapeutics. [DOI] [PubMed] [Google Scholar]
- 115.Lamberth K, Reedtz-Runge SL, Simon J, Klementyeva K, Pandey GS, Padkjær SB, Pascal V, León IR, Gudme CN, Buus S 2017. Post hoc assessment of the immunogenicity of bioengineered factor VIIa demonstrates the use of preclinical tools. Science translational medicine 9(372):eaag1286. [DOI] [PubMed] [Google Scholar]
- 116.De Groot AS, McMurry J, Moise L 2008. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Current opinion in pharmacology 8(5):620–626. [DOI] [PubMed] [Google Scholar]
- 117.De Groot AS, Moise L 2007. Prediction of immunogenicity for therapeutic proteins: state of the art. Current Opinion in Drug Discovery and Development 10(3):332. [PubMed] [Google Scholar]
- 118.De Groot AS, Scott DW 2007. Immunogenicity of protein therapeutics. Trends in immunology 28(11):482–490. [DOI] [PubMed] [Google Scholar]
- 119.Brinks V, Jiskoot W, Schellekens H 2011. Immunogenicity of Therapeutic Proteins: The Use of Animal Models. Pharmaceutical Research 28(10):2379–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shankar G, Shores E, Wagner C, Mire-Sluis A 2006. Scientific and regulatory considerations on the immunogenicity of biologics. Trends in Biotechnology 24(6):274–280. [DOI] [PubMed] [Google Scholar]
- 121.Krishnamoorthy S, Liu T, Drager D, Patarroyo-White S, Chhabra ES, Peters R, Josephson N, Lillicrap D, Blumberg RS, Pierce GF, Jiang H 2016. Recombinant factor VIII Fc (rFVIIIFc) fusion protein reduces immunogenicity and induces tolerance in hemophilia A mice. Cellular immunology 301:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ing M, Gupta N, Teyssandier M, Maillère B, Pallardy M, Delignat S, Lacroix-Desmazes S 2016. Immunogenicity of long-lasting recombinant factor VIII products. Cellular Immunology 301:40–48. [DOI] [PubMed] [Google Scholar]
- 123.Young G, Mahlangu J, Kulkarni R, Nolan B, Liesner R, Pasi J, Barnes C, Neelakantan S, Gambino G, Cristiano L 2015. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia A. Journal of Thrombosis and Haemostasis 13(6):967–977. [DOI] [PubMed] [Google Scholar]
- 124.Mahlangu J, Powell JS, Ragni MV, Chowdary P, Josephson NC, Pabinger I, Hanabusa H, Gupta N, Kulkarni R, Fogarty P 2014. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood 123(3):317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang S, Zhang C, Zhang L, Li J, Huang Z, Lu S 2008. The relative immunogenicity of DNA vaccines delivered by the intramuscular needle injection, electroporation and gene gun methods. Vaccine 26(17):2100–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ramani K, Miclea RD, Purohit VS, Mager DE, Straubinger RM, Balu-Iyer SV 2008. Phosphatidylserine containing liposomes reduce immunogenicity of recombinant human factor VIII (rFVIII) in a murine model of hemophilia A. Journal of Pharmaceutical Sciences 97(4):1386–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Raben N, Danon M, Gilbert A, Dwivedi S, Collins B, Thurberg B, Mattaliano R, Nagaraju K, Plotz P 2003. Enzyme replacement therapy in the mouse model of Pompe disease. Molecular genetics and metabolism 80(1):159–169. [DOI] [PubMed] [Google Scholar]
- 128.Lim H-H, Yi H, Kishimoto TK, Gao F, Sun B, Kishnani PS 2017. A pilot study on using rapamycin-carrying synthetic vaccine particles (SVP) in conjunction with enzyme replacement therapy to induce immune tolerance in Pompe disease. Molecular genetics and metabolism reports 13:18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Purohit VS, Middaugh CR, Balasubramanian SV 2006. Influence of aggregation on immunogenicity of recombinant human Factor VIII in hemophilia A mice. Journal of Pharmaceutical Sciences 95(2):358–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Torosantucci R, Brinks V, Kijanka G, Halim LA, Sauerborn M, Schellekens H, Jiskoot W 2014. Development of a Transgenic Mouse Model to Study the Immunogenicity of Recombinant Human Insulin. Journal of Pharmaceutical Sciences 103(5):1367–1374. [DOI] [PubMed] [Google Scholar]
- 131.Braun A, Kwee L, Labow MA, Alsenz J 1997. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon alpha (IFN-alpha) in normal and transgenic mice. Pharm Res 14(10):1472–1478. [DOI] [PubMed] [Google Scholar]
- 132.van Beers MMC, Sauerborn M, Gilli F, Brinks V, Schellekens H, Jiskoot W 2010. Aggregated Recombinant Human Interferon Beta Induces Antibodies but No Memory in Immune-Tolerant Transgenic Mice. Pharmaceutical Research 27(9):1812–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Perini P, Facchinetti A, Bulian P, Massaro AR, Pascalis DD, Bertolotto A, Biasi G, Gallo P 2001. Interferon-beta (INF-beta) antibodies in interferon-beta1a-and interferon-beta1b-treated multiple sclerosis patients. Prevalence, kinetics, cross-reactivity, and factors enhancing interferon-beta immunogenicity in vivo. European cytokine network 12(1):56–61. [PubMed] [Google Scholar]
- 134.Ploegh HL, Orr HT, Strominger JL 1981. Major histocompatibility antigens: The human (HLA-A,-B,-C) and murine (H-2K, H-2D) class I molecules. Cell 24(2):287–299. [DOI] [PubMed] [Google Scholar]
- 135.Messaoudi I, Patiño JAG, Dyall R, LeMaoult J, Nikolich-Žugich J 2002. Direct Link Between MHC Polymorphism, T Cell Avidity, and Diversity in Immune Defense. Science 298(5599):1797–1800. [DOI] [PubMed] [Google Scholar]
- 136.Hermeling S, Jiskoot W, Crommelin D, Bornss C, Schellekens H 2005. Development of a Transgenic Mouse Model Immune Tolerant for Human Interferon Beta. Pharmaceutical Research 22(6):847–851. [DOI] [PubMed] [Google Scholar]
- 137.Fakharzadeh SS, Zhang Y, Sarkar R, Kazazian HH 2000. Correction of the coagulation defect in hemophilia A mice through factor VIII expression in skin. Blood 95(9):2799–2805. [PubMed] [Google Scholar]
- 138.van Beers MMC, Sauerborn M, Gilli F, Hermeling S, Brinks V, Schellekens H, Jiskoot W 2010. Hybrid transgenic immune tolerant mouse model for assessing the breaking of B cell tolerance by human interferon beta. Journal of Immunological Methods 352(1):32–37. [DOI] [PubMed] [Google Scholar]
- 139.Sweetser DA, Hauft SM, Hoppe PC, Birkenmeier EH, Gordon JI 1988. Transgenic mice containing intestinal fatty acid-binding protein-human growth hormone fusion genes exhibit correct regional and cell-specific expression of the reporter gene in their small intestine. Proceedings of the National Academy of Sciences 85(24):9611–9615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Selden RF, Skośkiewicz MJ, Howie KB, Russell PS, Goodman HM 1986. Regulation of human insulin gene expression in transgenic mice. Nature 321(6069):525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ottesen JL, Nilsson P, Jami J, Weilguny D, Dϋhrkop M, Bucchini D, Havelund S, Fogh JM 1994. The potential immunogenicity of human insulin and insulin analogues evaluated in a transgenic mouse model. Diabetologia 37(12):1178–1185. [DOI] [PubMed] [Google Scholar]
- 142.Bertolotto A, Malucchi S, Sala A, Orefice G, Carrieri P, Capobianco M, Milano E, Melis F, Giordana M 2002. Differential effects of three interferon betas on neutralising antibodies in patients with multiple sclerosis: a follow up study in an independent laboratory. Journal of Neurology, Neurosurgery, and Psychiatry 73(2):148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, Watanabe T, Akashi K, Shultz LD, Harada M 2005. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 106(5):1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R 2005. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174(10):6477–6489. [DOI] [PubMed] [Google Scholar]
- 145.Shultz LD, Brehm MA, Bavari S, Greiner DL 2011. Humanized mice as a preclinical tool for infectious disease and biomedical research. Annals of the New York Academy of Sciences 1245(1):50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zwickl CM, Cocke KS, Tamura RN, Holzhausen LM, Brophy GT, Bick PH, Wierda D 1991. Comparison of the immunogenicity of recombinant and pituitary human growth hormone in rhesus monkeys. Fundamental and applied toxicology : official journal of the Society of Toxicology 16(2):275–287. [DOI] [PubMed] [Google Scholar]
- 147.Zwickl CM, Hughes BL, Piroozi KS, Smith HW, Wierda D 1996. Immunogenicity of tissue plasminogen activators in rhesus monkeys: antibody formation and effects on blood level and enzymatic activity. Fundamental and applied toxicology : official journal of the Society of Toxicology 30(2):243–254. [DOI] [PubMed] [Google Scholar]
- 148.Zwickl CM, Smith HW, Zimmermann JL, Wierda D 1995. Immunogenicity of biosynthetic human LysPro insulin compared to native-sequence human and purified porcine insulins in rhesus monkeys immunized over a 6-week period. Arzneimittel-Forschung 45(4):524–528. [PubMed] [Google Scholar]
- 149.Fineberg NS, Fineberg SE, Anderson JH, Birkett MA, Gibson RG, Hufferd S 1996. Immunologic effects of insulin lispro [Lys (B28), Pro (B29) human insulin] in IDDM and NIDDM patients previously treated with insulin. Diabetes 45(12):1750–1754. [DOI] [PubMed] [Google Scholar]
- 150.Finn JD, Simioni P, Iacobelli N, Zhou S, Nichols TC, High KA, Arruda VR 2009. FIX-R338L (FIX Padua) as a Successful Alternative for the Treatment of Canine Severe Hemophilia B. Blood 114(22):694–694. [Google Scholar]
- 151.Finn JD, Nichols TC, Svoronos N, Merricks EP, Bellenger DA, Zhou Z, Simioni P, High KA, Arruda VR 2012. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Randolph JF, Stokol T, Scarlett JM, MacLeod JN 1999. Comparison of biological activity and safety of recombinant canine erythropoietin with that of recombinant human erythropoietin in clinically normal dogs. American journal of veterinary research 60(5):636–642. [PubMed] [Google Scholar]
- 153.Kolaczkowska E, Chadzinska M, Seljelid R, Plytycz B 2001. Strain differences in some immune parameters can be obscured by circadian variations and laboratory routines: studies of male C57BL/6J, Balb/c and CB6 F1 mice. Laboratory animals 35(1):91–100. [DOI] [PubMed] [Google Scholar]
- 154.Jiang X, Shen C, Yu H, Karunakaran KP, Brunham RC 2010. Differences in innate immune responses correlate with differences in murine susceptibility to Chlamydia muridarum pulmonary infection. Immunology 129(4):556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Engler H, Zawatzky R, Kirchner H, Armerding D 1982. Experimental infection of inbred mice with herpes simplex virus. IV. Comparison of interferon production and natural killer cell activity in susceptible and resistant adult mice. Archives of virology 74(4):239–247. [DOI] [PubMed] [Google Scholar]
- 156.Karp JD, Cohen N, Moynihan JA 1994. Quantitative differences in interleukin-2 and interleukin-4 production by antigen-stimulated splenocytes from individually- and group-housed mice. Life Sciences 55(10):789–795. [DOI] [PubMed] [Google Scholar]
- 157.Gunn H 1997. Immunogenicity of Recombinant Human Interleukin-3. Clinical Immunology and Immunopathology 83(1):5–7. [DOI] [PubMed] [Google Scholar]
- 158.van Meer PJK, Kooijman M, Brinks V, Gispen-de Wied CC, Silva-Lima B, Moors EHM, Schellekens H 2013. Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. mAbs 5(5):810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tao L, Reese TA 2017. Making Mouse Models That Reflect Human Immune Responses. Trends in Immunology 38(3):181–193. [DOI] [PubMed] [Google Scholar]
- 160.Ressing ME, Sette A, Brandt RM, Ruppert J, Wentworth PA, Hartman M, Oseroff C, Grey HM, Melief CJ, Kast WM 1995. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A*0201-binding peptides. The Journal of Immunology 154(11):5934–5943. [PubMed] [Google Scholar]
- 161.Sachse K, Helbig JH, Lysnyansky I, Grajetzki C, Mϋller W, Jacobs E, Yogev D 2000. Epitope Mapping of Immunogenic and Adhesive Structures in Repetitive Domains of Mycoplasma bovis Variable Surface Lipoproteins. Infection and Immunity 68(2):680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nayak S, Sivakumar R, Cao O, Daniell H, Byrne BJ, Herzog RW 2012. Mapping the T helper cell response to acid α-glucosidase in Pompe mice. Molecular genetics and metabolism 106(2):189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hamze M, Meunier S, Karle A, Gdoura A, Goudet A, Szely N, Pallardy M, Carbonnel F, Spindeldreher S, Mariette X, Miceli-Richard C, Maillère B 2017. Characterization of CD4 T Cell Epitopes of Infliximab and Rituximab Identified from Healthy Donors. Frontiers in Immunology 8(500). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.JONES TD, PHILLIPS WJ, SMITH BJ, BAMFORD CA, NAYEE PD, BAGLIN TP, GASTON JSH, BAKER MP 2005. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. Journal of Thrombosis and Haemostasis 3(5):991–1000. [DOI] [PubMed] [Google Scholar]
- 165.Moskalenko M, Chen L, van Roey M, Donahue BA, Snyder RO, McArthur JG, Patel SD 2000. Epitope Mapping of Human Anti-Adeno-Associated Virus Type 2 Neutralizing Antibodies: Implications for Gene Therapy and Virus Structure. Journal of Virology 74(4):1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Vanderlugt CL, Miller SD 2002. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nature Reviews Immunology 2:85. [DOI] [PubMed] [Google Scholar]
- 167.Cornaby C, Gibbons L, Mayhew V, Sloan CS, Welling A, Poole BD 2015. B cell epitope spreading: mechanisms and contribution to autoimmune diseases. Immunol Lett 163(1):56–68. [DOI] [PubMed] [Google Scholar]
- 168.Bernard CCA, Johns TG, Slavin A, Ichikawa M, Ewing C, Liu J, Bettadapura J 1997. Myelin oligodendrocyte glycoprotein: a novel candidate autoantigen in multiple sclerosis. Journal of Molecular Medicine 75(2):77–88. [DOI] [PubMed] [Google Scholar]
- 169.Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS 2013. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clinical Immunology 149(3, Part B):534–555. [DOI] [PubMed] [Google Scholar]
- 170.Lazarski CA, Chaves FA, Jenks SA, Wu S, Richards KA, Weaver JM, Sant AJ 2005. The Kinetic Stability of MHC Class II:Peptide Complexes Is a Key Parameter that Dictates Immunodominance. Immunity 23(1):29–40. [DOI] [PubMed] [Google Scholar]
- 171.Buus S, Sette A, Colon SM, Miles C, Grey HM 1987. The relation between major histocompatibility complex (MHC) restriction and the capacity of Ia to bind to immunogenic peptides. Science 235:1353–1359. [DOI] [PubMed] [Google Scholar]
- 172.Salvat R, Moise L, Bailey-Kellogg C, Griswold KE 2014. A High Throughput MHC II Binding Assay for Quantitative Analysis of Peptide Epitopes. Journal of Visualized Experiments : JoVE (85):51308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Antunes DA, Vieira GF, Rigo MM, Cibulski SP, Sinigaglia M, Chies JA 2010. Structural allele-specific patterns adopted by epitopes in the MHC-I cleft and reconstruction of MHC: peptide complexes to cross-reactivity assessment. PloS one 5(4):e10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tanguay S, Killion JJ 1994. Direct comparison of ELISPOT and ELISA-based assays for detection of individual cytokine-secreting cells. Lymphokine and cytokine research 13(4):259–263. [PubMed] [Google Scholar]
- 175.Van Der Burg SH, Visseren M, Brandt R, Kast WM, Melief C 1996. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide complex stability. The Journal of Immunology 156(9):3308–3314. [PubMed] [Google Scholar]
- 176.ProImmune. 2006. ProImmune REVEAL Mouse Class II MHC-Peptide Binding Assays. ed.
- 177.Fujita S, Shimada M, Nakamura T 1966. H3-Thymidine autoradiographic studies on the cell proliferation and differentiation in the external and the internal granular layers of the mouse cerebellum. Journal of Comparative Neurology 128(2):191–207. [DOI] [PubMed] [Google Scholar]
- 178.Fulcher D, Wong S 1999. Carboxyfluorescein succinimidyl ester-based proliferative assays for assessment of T cell function in the diagnostic laboratory. Immunology and cell biology 77(6):559–564. [DOI] [PubMed] [Google Scholar]
- 179.Lyons AB 1999. Divided we stand: tracking cell proliferation with carboxyfluorescein diacetate succinimidyl ester. Immunology and Cell Biology 77(6):509–515. [DOI] [PubMed] [Google Scholar]
- 180.Karle A, Spindeldreher S, Kolbinger F 2016. Secukinumab, a novel anti-IL-17A antibody, shows low immunogenicity potential in human in vitro assays comparable to other marketed biotherapeutics with low clinical immunogenicity. mAbs 8(3):536–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Rubic-Schneider T, Kuwana M, Christen B, Aßenmacher M, Hainzl O, Zimmermann F, Fischer R, Koppenburg V, Chibout S-D, Wright TM, Seidl A, Kammϋller M 2017. T-cell assays confirm immunogenicity of tungsten-induced erythropoietin aggregates associated with pure red cell aplasia. Blood Advances 1(6):367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Karlsson AC, Martin JN, Younger SR, Bredt BM, Epling L, Ronquillo R, Varma A, Deeks SG, McCune JM, Nixon DF 2003. Comparison of the ELISPOT and cytokine flow cytometry assays for the enumeration of antigen-specific T cells. Journal of immunological methods 283(1):141–153. [DOI] [PubMed] [Google Scholar]
- 183.Czerkinsky CC, Nilsson L-Å, Nygren H, Ouchterlony Ö, Tarkowski A 1983. A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. Journal of immunological methods 65(1–2):109–121. [DOI] [PubMed] [Google Scholar]
- 184.Maecker HT. 2004. Cytokine flow cytometry Flow Cytometry Protocols, ed.: Springer; p 95–107. [Google Scholar]
- 185.Jung T, Schauer U, Heusser C, Neumann C, Rieger C 1993. Detection of intracellular cytokines by flow cytometry. Journal of immunological methods 159(1–2):197–207. [DOI] [PubMed] [Google Scholar]
- 186.Kleiveland CR. 2015. Peripheral Blood Mononuclear Cells In Verhoeckx K, Cotter P, López-Expósito I, Kleiveland C, Lea T, Mackie A, Requena T, Swiatecka D, Wichers H, editors. The Impact of Food Bioactives on Health: in vitro and ex vivo models, ed., Cham: Springer International Publishing; p 161–167. [PubMed] [Google Scholar]
- 187.Joubert MK, Hokom M, Eakin C, Zhou L, Deshpande M, Baker MP, Goletz TJ, Kerwin BA, Chirmule N, Narhi LO, Jawa V 2012. Highly Aggregated Antibody Therapeutics Can Enhance the in Vitro Innate and Late-stage T-cell Immune Responses. The Journal of biological chemistry 287(30):25266–25279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ahmadi M, Bryson CJ, Cloake EA, Welch K, Filipe V, Romeijn S, Hawe A, Jiskoot W, Baker MP, Fogg MH 2015. Small Amounts of Sub-Visible Aggregates Enhance the Immunogenic Potential of Monoclonal Antibody Therapeutics. Pharmaceutical Research 32(4):1383–1394. [DOI] [PubMed] [Google Scholar]
- 189.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE 2013. Innate lymphoid cells—a proposal for uniform nomenclature. Nature Reviews Immunology 13(2):145. [DOI] [PubMed] [Google Scholar]
- 190.Haile LA, Puig M, Kelley-Baker L, Verthelyi D 2015. Detection of Innate Immune Response Modulating Impurities in Therapeutic Proteins. PLOS ONE 10(4):e0125078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kirpach J, Muller CP. 2001. Epitopes eLS, ed.: John Wiley & Sons, Ltd. [Google Scholar]
- 192.Ponomarenko JV, Van Regenmortel MH 2009. B cell epitope prediction. Structural bioinformatics:849–879. [Google Scholar]
- 193.Potocnakova L, Bhide M, Pulzova LB 2016. An Introduction to B-Cell Epitope Mapping and In Silico Epitope Prediction. Journal of Immunology Research 2016:6760830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Haste Andersen P, Nielsen M, Lund O 2006. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Science 15(11):2558–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Padavattan S, Flicker S, Schirmer T, Madritsch C, Randow S, Reese G, Vieths S, Lupinek C, Ebner C, Valenta R 2009. High-affinity IgE recognition of a conformational epitope of the major respiratory allergen Phl p 2 as revealed by X-ray crystallography. The Journal of Immunology 182(4):2141–2151. [DOI] [PubMed] [Google Scholar]
- 196.Ding W, Huang X, Yang X, Dunn JJ, Luft BJ, Koide S, Lawson CL 2000. Structural identification of a key protective B-cell epitope in lyme disease antigen OspA1. Journal of molecular biology 302(5):1153–1164. [DOI] [PubMed] [Google Scholar]
- 197.Chen XS, Garcea RL, Goldberg I, Casini G, Harrison SC 2000. Structure of small virus-like particles assembled from the L1 protein of human papillomavirus 16. Molecular cell 5(3):557–567. [DOI] [PubMed] [Google Scholar]
- 198.White WI, Wilson SD, Palmer-Hill FJ, Woods RM, Ghim S-j, Hewitt LA, Goldman DM, Burke SJ, Jenson AB, Koenig S 1999. Characterization of a major neutralizing epitope on human papillomavirus type 16 L1. Journal of virology 73(6):4882–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Morris GE. 1996. Epitope Mapping by Chemical Fragmentation In Morris GE, editor Epitope Mapping Protocols, ed., Totowa, NJ: Humana Press; p 121–127. [DOI] [PubMed] [Google Scholar]
- 200.Davidson HW, Watts C 1989. Epitope-directed processing of specific antigen by B lymphocytes. The Journal of Cell Biology 109(1):85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kessler JH, Benckhuijsen WE, Mutis T, Melief CJ, van der Burg SH, Drijfhout JW 2004. Competition-Based Cellular Peptide Binding Assay for HLA Class I. Current protocols in immunology:18.12. 11–18.12. 15. [DOI] [PubMed] [Google Scholar]
- 202.Cunningham BC, Wells JA 1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244(4908):1081–1085. [DOI] [PubMed] [Google Scholar]
- 203.Beevers L 1982. Post-translational modifications Nucleic Acids and Proteins in Plants I, ed.: Springer; p 136–168. [Google Scholar]
- 204.Walsh G, Jefferis R 2006. Post-translational modifications in the context of therapeutic proteins. Nature biotechnology 24(10):1241–1252. [DOI] [PubMed] [Google Scholar]
- 205.Pang J, Blanc T, Brown J, Labrenz S, Villalobos A, Depaolis A, Gunturi S, Grossman S, Lisi P, Heavner GA 2007. Recognition and Identification of UV-absorbing Leachables in EPREX® Pre-filled Syringes: An Unexpected Occurrence at a Formulation—Component Interface. PDA Journal of Pharmaceutical Science and Technology 61(6):423–432. [PubMed] [Google Scholar]
- 206.Boven K, Stryker S, Knight J, Thomas A, van Regenmortel M, Kemeny DM, Power D, Rossert J, Casadevall N 2005. The increased incidence of pure red cell aplasia with an Eprex formulation in uncoated rubber stopper syringes. Kidney international 67(6):2346–2353. [DOI] [PubMed] [Google Scholar]
- 207.Seidl A, Hainzl O, Richter M, Fischer R, Böhm S, Deutel B, Hartinger M, Windisch J, Casadevall N, London GM, Macdougall I 2012. Tungsten-induced denaturation and aggregation of epoetin alfa during primary packaging as a cause of immunogenicity. Pharmaceutical research 29(6):1454–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.2018. Immune Epitope Database and Analysis Resource. ed., www.iedb.org: National Institute of Allergy and Infectious Diseases. [Google Scholar]
- 209.Schlessinger A, Ofran Y, Yachdav G, Rost B 2006. Epitome: database of structure-inferred antigenic epitopes. Nucleic Acids Research 34(Database issue):D777–D780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sharma O, Das A, Krishna R, Kumar MS, Mathur P 2012. Structural Epitope Database (SEDB): a web-based database for the epitope, and its intermodular interaction along with the tertiary structure information. J Proteomics Bioinform 5:084–089. [Google Scholar]
- 211.Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S 2015. An overview of bioinformatics tools for epitope prediction: implications on vaccine development. Journal of biomedical informatics 53:405–414. [DOI] [PubMed] [Google Scholar]
- 212.Guan P, Hattotuwagama CK, Doytchinova IA, Flower DR 2006. MHCPred 2.0. Applied bioinformatics 5(1):55–61. [DOI] [PubMed] [Google Scholar]
- 213.Guan P, Doytchinova IA, Zygouri C, Flower DR 2003. MHCPred: a server for quantitative prediction of peptide–MHC binding. Nucleic acids research 31(13):3621–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.De Groot AS, Jesdale BM, Szu E, Schafer JR, Chicz RM, Deocampo G 1997. An interactive Web site providing major histocompatibility ligand predictions: application to HIV research. AIDS research and human retroviruses 13(7):529–531. [DOI] [PubMed] [Google Scholar]
- 215.Alves VM, Capuzzi SJ, Muratov EN, Braga RC, Thornton TE, Fourches D, Strickland J, Kleinstreuer N, Andrade CH, Tropsha A 2016. QSAR models of human data can enrich or replace LLNA testing for human skin sensitization. Green Chemistry 18(24):6501–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Agrawal NJ, Kumar S, Wang X, Helk B, Singh SK, Trout BL 2011. Aggregation in protein-based biotherapeutics: Computational studies and tools to identify aggregation-prone regions. Journal of Pharmaceutical Sciences 100(12):5081–5095. [DOI] [PubMed] [Google Scholar]
- 217.Kumar S, Singh SK, Wang X, Rup B, Gill D 2011. Coupling of aggregation and immunogenicity in biotherapeutics: T-and B-cell immune epitopes may contain aggregation-prone regions. Pharmaceutical research 28(5):949. [DOI] [PubMed] [Google Scholar]
- 218.Chen M-L, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, Khazaie K 2005. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proceedings of the National Academy of Sciences of the United States of America 102(2):419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sule G, Suzuki M, Guse K, Cela R, Rodgers JR, Lee B 2012. Cytokine-Conditioned Dendritic Cells Induce Humoral Tolerance to Protein Therapy in Mice. Human Gene Therapy 23(7):769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Zhang A-H, Rossi RJ, Yoon J, Wang H, Scott DW 2016. Tolerogenic nanoparticles to induce immunologic tolerance: Prevention and reversal of FVIII inhibitor formation. Cellular Immunology 301:74–81. [DOI] [PubMed] [Google Scholar]
- 221.Meliani A, Boisgerault F, Hardet R, Marmier S, Collaud F, Ronzitti G, Leborgne C, Verdera HC, Sola MS, Charles S 2018. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nature communications 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Mingozzi F, Liu Y-L, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, Arruda VR, High KA, Herzog RW 2003. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. The Journal of Clinical Investigation 111(9):1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ettinger RA, Liberman JA, Gunasekera D, Puranik K, James EA, Thompson AR, Pratt KP 2018. FVIII proteins with a modified immunodominant T-cell epitope exhibit reduced immunogenicity and normal FVIII activity. Blood Advances 2(4):309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mazor R, Kaplan G, Park D, Jang Y, Lee F, Kreitman R, Pastan I 2017. Rational design of low immunogenic anti CD25 recombinant immunotoxin for T cell malignancies by elimination of T cell epitopes in PE38. Cellular Immunology 313:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Adair JR, Athwal DS, Bodmer MW, Bright SM, Collins AM, Pulito VL, Rao PE, Reedman R, Rothermel AL, Xu D, et al. 1994. Humanization of the murine anti-human CD3 monoclonal antibody OKT3. Human antibodies and hybridomas 5(1–2):41–47. [PubMed] [Google Scholar]
- 226.De Groot AS, Martin W 2009. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clinical Immunology 131(2):189–201. [DOI] [PubMed] [Google Scholar]
- 227.Miclea RD, Purohit VS, Balu-Iyer SV 2007. O-phospho-L-serine, multi-functional excipient for B domain deleted recombinant factor VIII. The AAPS Journal 9(2):E251–E259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Senga Y, Honda S 2018. Suppression of Aggregation of Therapeutic Monoclonal Antibodies during Storage by Removal of Aggregation Precursors Using a Specific Adsorbent of Non-Native IgG Conformers. Bioconjugate chemistry 29(10):3250–3261. [DOI] [PubMed] [Google Scholar]
- 229.Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M, Jones SA, Olson R, White A, Wells C 2012. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genetics in medicine 14(1):135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Maini RN, Breedveld FC, Kalden JR, Smolen JS, Davis D, MacFarlane JD, Antoni C, Leeb B, Elliott MJ, Woody JN, Schaible TF, Feldmann M 1998. Therapeutic efficacy of multiple intravenous infusions of anti-tumor necrosis factor α monoclonal antibody combined with low-dose weekly methotrexate in rheumatoid arthritis. Arthritis & Rheumatism 41(9):1552–1563. [DOI] [PubMed] [Google Scholar]
- 231.Wicker LS, Boltz RC Jr, Matt V, Nichols EA, Peterson LB, Sigal NH 1990. Suppression of B cell activation by cyclosporin A, FK506 and rapamycin. European journal of immunology 20(10):2277–2283. [DOI] [PubMed] [Google Scholar]
- 232.Groth CG, Bäckman L, Morales J-M, Calne R, Kreis H, Lang P, Touraine J-L, Claesson K, Campistol JM, Durand D 1999. SIROLIMUS (RAPAMYCIN)-BASED THERAPY IN HUMAN RENAL TRANSPLANTATION: Similar Efficacy and Different Toxicity Compared with Cyclosporine1, 2. Transplantation 67(7):1036–1042. [DOI] [PubMed] [Google Scholar]
- 233.Moghimi B, Sack BK, Nayak S, Markusic DM, Mah CS, Herzog RW 2011. Induction of tolerance to factor VIII by transient co-administration with rapamycin. Journal of Thrombosis and Haemostasis 9(8):1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Banugaria SG, Prater SN, McGann JK, Feldman JD, Tannenbaum JA, Bailey C, Gera R, Conway RL, Viskochil D, Kobori JA, Rosenberg AS, Kishnani PS 2012. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: lessons learned from Pompe disease. Genetics In Medicine 15:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sistigu A, Viaud S, Chaput N, Bracci L, Proietti E, Zitvogel L 2011. Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Seminars in Immunopathology 33(4):369–383. [DOI] [PubMed] [Google Scholar]
- 236.Nilsson IM, Berntorp E, Zettervall O 1988. Induction of Immune Tolerance in Patients with Hemophilia and Antibodies to Factor VIII by Combined Treatment with Intravenous IgG, Cyclophosphamide, and Factor VIII. New England Journal of Medicine 318(15):947–950. [DOI] [PubMed] [Google Scholar]
- 237.Mayumi H, Umesue M, Nomoto K 1996. Cyclophosphamide-induced immunological tolerance: an overview. Immunobiology 195(2):129–139. [DOI] [PubMed] [Google Scholar]
- 238.Green D, Rademaker AW, Briët E 1993. A prospective, randomized trial of prednisone and cyclophosphamide in the treatment of patients with factor VIII autoantibodies. Thrombosis and haemostasis 69(05):753–757. [PubMed] [Google Scholar]
- 239.Banugaria SG, Patel TT, Mackey J, Das S, Amalfitano A, Rosenberg AS, Charrow J, Chen Y-T, Kishnani PS 2012. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Molecular genetics and metabolism 105(4):677–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Anderson PJ 2005. Tumor necrosis factor inhibitors: Clinical implications of their different immunogenicity profiles. Seminars in Arthritis and Rheumatism 34(5, Supplement 1):19–22. [DOI] [PubMed] [Google Scholar]
- 241.Colombel JF, Sandborn WJ, Reinisch W, Mantzaris GJ, Kornbluth A, Rachmilewitz D, Lichtiger S, D’Haens G, Diamond RH, Broussard DL, Tang KL, van der Woude CJ, Rutgeerts P 2010. Infliximab, Azathioprine, or Combination Therapy for Crohn’s Disease. New England Journal of Medicine 362(15):1383–1395. [DOI] [PubMed] [Google Scholar]
- 242.Garcês S, Demengeot J, Benito-Garcia E 2013. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: a systematic review of the literature with a meta-analysis. Annals of the Rheumatic Diseases 72(12):1947–1955. [DOI] [PubMed] [Google Scholar]
- 243.Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS 2009. Elimination of antibodies to recombinant enzyme in Pompe’s disease. The New England journal of medicine 360(2):194–195. [DOI] [PubMed] [Google Scholar]
- 244.Ohashi T, Iizuka S, Shimada Y, Higuchi T, Eto Y, Ida H, Kobayashi H 2012. Administration of Anti-CD3 Antibodies Modulates the Immune Response to an Infusion of α-glucosidase in Mice. Molecular Therapy 20(10):1924–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Waters B, Qadura M, Burnett E, Chegeni R, Labelle A, Thompson P, Hough C, Lillicrap D 2009. Anti-CD3 prevents factor VIII inhibitor development in hemophilia A mice by a regulatory CD4+ CD25+-dependent mechanism and by shifting cytokine production to favor a Th1 response. Blood 113(1):193–203. [DOI] [PubMed] [Google Scholar]
- 246.Parvathaneni K, Scott DW 2018. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Advances 2(18):2332–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Brettschneider K, Schmidt A, Kahle J, Orlowski A, Stichel D, Schwabe D, Konigs C Elimination of factor VIII-specific B cells by immunotoxins composed of a single factor VIII domain fused to Pseudomonas exotoxin A. Journal of Thrombosis and Haemostasis 0(0). [DOI] [PubMed] [Google Scholar]
- 248.Stewart TA, Hollingshead PG, Pitts SL, Chang R, Martin LE, Oakley H 1989. Transgenic mice as a model to test the immunogenicity of proteins altered by site-specific mutagenesis. Molecular biology & medicine 6(4):275–281. [PubMed] [Google Scholar]
- 249.Lenk C, Unterthurner S, Schuster M, Weiller M, Antoine G, Malisauskas M, Scheiflinger F, Schwarz HP, de la Rosa M, Reipert BM 2013. Development of a transgenic mouse model with immune tolerance for human coagulation factor VIIa. Pharm Res 30(11):2855–2867. [DOI] [PubMed] [Google Scholar]
- 250.Carrier Y, Yuan J, Kuchroo VK, Weiner HL 2007. Th3 Cells in Peripheral Tolerance. I. Induction of Foxp3-Positive Regulatory T Cells by Th3 Cells Derived from TGF-β T Cell-Transgenic Mice. The Journal of Immunology 178(1):179–185. [DOI] [PubMed] [Google Scholar]
- 251.Khanna R, Flanagan JJ, Feng J, Soska R, Frascella M, Pellegrino LJ, Lun Y, Guillen D, Lockhart DJ, Valenzano KJ 2012. The Pharmacological Chaperone AT2220 Increases Recombinant Human Acid α-Glucosidase Uptake and Glycogen Reduction in a Mouse Model of Pompe Disease. PLoS ONE 7(7):e40776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Huang J, Honda W 2006. CED: a conformational epitope database. BMC Immunology 7(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Toseland CP, Clayton DJ, McSparron H, Hemsley SL, Blythe MJ, Paine K, Doytchinova IA, Guan P, Hattotuwagama CK, Flower DR 2005. AntiJen: a quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data. Immunome Research 1:4–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Saha S, Bhasin M, Raghava GP 2005. Bcipep: A database of B-cell epitopes. BMC Genomics 6(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Duquesnoy RJ, Marrari M, Sousa LCDdM, Barroso JRPdM, Aita KMdSU, da Silva AS, do Monte SJH 2013. 16th IHIW: A Website for Antibody-Defined HLA Epitope Registry. International Journal of Immunogenetics 40(1):54–59. [DOI] [PubMed] [Google Scholar]
- 256.Doytchinova IA, Guan P, Flower DR 2006. EpiJen: a server for multistep T cell epitope prediction. BMC Bioinformatics 7(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Bhasin M, Raghava GP 2003. Prediction of promiscuous and high-affinity mutated MHC binders. Hybridoma and hybridomics 22(4):229–234. [DOI] [PubMed] [Google Scholar]
- 258.Dönnes P, Elofsson A 2002. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinformatics 3(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Darnell S, Riese M 2012. Precise Predictions of Linear B Cell Epitopes in Protean 3D.
- 260.Yao B, Zhang L, Liang S, Zhang C 2012. SVMTriP: a method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PloS one 7(9):e45152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Moise L, Gutierrez AH, Bailey-Kellogg C, Terry F, Leng Q, Abdel Hady KM, VerBerkmoes NC, Sztein MB, Losikoff PT, Martin WD, Rothman AL, De Groot AS 2013. The two-faced T cell epitope: examining the host-microbe interface with JanusMatrix. Human vaccines & immunotherapeutics 9(7):1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]