

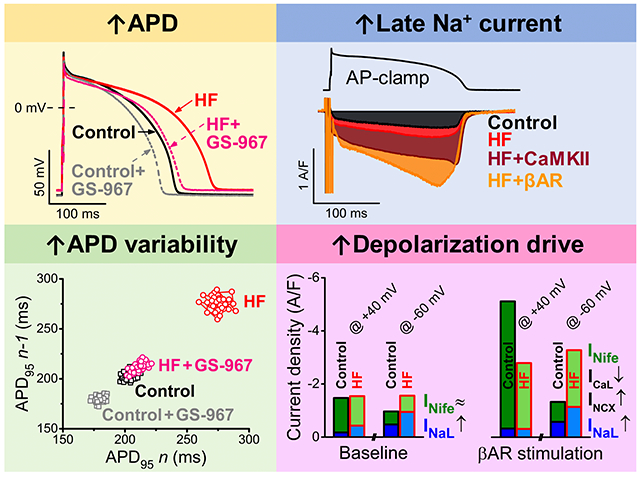
Abstract
Background:
Heart failure (HF) is characterized by electrophysiological remodeling resulting in increased risk of cardiac arrhythmias. Previous reports suggest that elevated inward ionic currents in HF promote action potential (AP) prolongation, increased short-term variability of AP repolarization (STV) and delayed afterdepolarizations. However, the underlying changes in late Na+, L-type Ca2+, and Na+/Ca2+ exchanger currents (INaL, ICaL and INCX, respectively) are often measured in nonphysiological conditions (square pulse voltage-clamp, slow pacing rates, exogenous Ca2+ buffers).
Methods:
We measured the major inward currents and their Ca2+- and β-adrenergic dependence under physiological AP-clamp in rabbit ventricular myocytes in chronic pressure/volume overload-induced HF (versus age-matched control).
Results:
AP duration and STV were increased in HF, and importantly, inhibition of INaL decreased both parameters to the control level. INaL was slightly increased in HF versus control even when intracellular Ca2+ was strongly buffered. But under physiological AP-clamp with normal Ca2+ cycling, INaL was markedly upregulated in HF versus control (dependent largely on Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity). β-adrenergic stimulation (often elevated in HF), further enhanced INaL. ICaL was decreased in HF when Ca2+ was buffered, but CaMKII-mediated Ca2+-dependent facilitation upregulated physiological ICaL to the control level. Furthermore, ICaL response to β-adrenergic stimulation was significantly attenuated in HF. Inward INCX was upregulated at phase 3 of AP in HF when assessed by combining experimental data and computational modeling.
Conclusions:
Our results suggest that CaMKII-dependent upregulation of INaL in HF significantly contributes to AP prolongation and increased STV, which may lead to increased arrhythmia propensity, and is further exacerbated by adrenergic stress.
Keywords: electrophysiology, heart failure, L-type calcium current, calcium/calmodulin-dependent protein kinase II, late sodium current, Ion Channels/Membrane Transport
Graphical Abstract
Introduction
Heart failure (HF) is characterized by increased risk for cardiac arrhythmias. Arrhythmogenic alterations in the ventricular action potential (AP), including early and delayed afterdepolarizations,1–4 AP duration (APD) prolongation,5, 6 and increased beat-to-beat variability of APD6–8 have been extensively studied in failing ventricular cardiomyocytes. Alterations in membrane potential stability have been associated with extensive ionic remodeling, including enhanced Na+/Ca2+ exchanger current (INCX),9, 10 increased late Na+ current (INaL),11, 12 and reduced repolarization reserve.13, 14 In line with the electrophysiology results, the protein expression level of Na+/Ca2+ exchanger (NCX) was also found to be increased in HF,9 whereas that of several K+ channels were decreased.15 But in the case of Na+ channels both the protein level and peak current density were decreased,12, 16 suggesting that the increased INaL in HF results from altered regulation and gating. Accordingly, Ca2+-calmodulin-dependent protein kinase II (CaMKII), was found to be centrally involved in INaL upregulation in HF.17–19
However, INaL in previous HF studies was typically measured in non-physiologic conditions (square-pulse voltage-clamp, slow stimulation rates, strong buffering of intracellular Ca2+ concentration ([Ca2+]i), and room temperature), which might have masked some important aspects of INaL regulation. Indeed, INaL was found to be larger in AP-clamped healthy guinea-pig and rabbit myocytes than the tiny INaL found in conventional voltage-clamp studies.20, 21 β-adrenergic receptor (βAR) stimulation can further enhance INaL (via both CaMKII-dependent and independent mechanisms19, 21, 22) and increased sympathetic activity is commonly observed in HF. Consequently, enhanced INaL in HF may play a critical role in APD prolongation leading to cardiac arrhythmias and Na+ overload.23 Accordingly, inhibition of either INaL or CaMKII was found to exert beneficial effects in HF.18, 24 Unlike upregulated INCX and INaL, L-type Ca2+ current (ICaL) was found to be unchanged or slightly decreased in previous studies in HF. 4, 25, 26 However, because βAR signaling, Ca2+ and CaMKII all regulate ICaL,27–29 prior voltage-clamp studies could easily misestimate changes in physiological ICaL or INaL that occur during the AP in HF.
The present study measures APD, INaL, ICaL and INCX in HF versus control rabbit hearts during physiologic APs, with or without βAR activation and also assesses the involvement of CaMKII. We performed AP-clamp recordings with physiologic ionic composition, pacing rate, [Ca2+]i and temperature. We hypothesized that Ca2+ transients and increased CaMKII activity under these conditions significantly enhance inward currents in HF leading to arrhythmogenic alterations in AP morphology. We used a previously well-characterized chronic nonischemic HF rabbit model (combined volume and pressure overload), which is also arrhythmogenic.4, 9, 14, 30 APs and major inward ionic currents were measured under current- and AP-clamp respectively in ventricular myocytes using specific blockers of INaL (GS-967) and ICaL (nifedipine). Because under physiological conditions our nifedipine-sensitive current includes both ICaL and some inward INCX (because nifedipine inhibits Ca2+ transients that modulate INCX), we included computational modeling to better clarify the distinct profiles of ICaL and INCX under AP-clamp. Modulation of ICaL and INaL by Ca2+, CaMKII, and βAR stimulation (ie, pathophysiological settings characteristic of HF) was also investigated to assess the regulatory changes in the net depolarizing drive during the plateau and repolarization phases of the AP.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. All animal handling and laboratory procedures were in accordance with the approved protocols of the local Institutional Animal Care and Use Committee confirming to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (8th edition, 2011).
Arrhythmogenic Rabbit Nonischemic HF Model
HF was induced in New Zealand White rabbits (all male, 2.5–3 kg, 3–4 months old) by aortic insufficiency and 4 weeks later by aortic constriction as previously described.9 Data here was obtained from 12 HF rabbits and 8 age-matched control rabbits. HF progression was monitored periodically by echocardiography and myocytes were isolated when left ventricular end-systolic dimension exceeded 1.4 cm (at ≈2.5 years of age). HF animals versus control hearts were near twice the heart weight/body weight (5.27±0.61 versus 2.67±0.10 g/kg; P<0.01), exhibited ≈40% larger left ventricular end-diastolic diameter (2.31±0.10 versus 1.61±0.08 cm; P<0.001), ≈50% larger left ventricular end-systolic diameter (1.63±0.08 versus 1.08±0.05 cm; P<0.001), evidence of pulmonary congestion (lung weight, 19.13±2.29 versus 14.11±0.40 g; P<0.05), and abdominal ascites fluid accumulation, all similar to our prior studies on this rabbit model.4, 9, 14 Enzymatic isolation of cardiomyocytes from the midmyocardial region of the left ventricular free wall was performed as previously described.14
Electrophysiology
Isolated cells were transferred to a temperature-controlled plexiglass chamber (Cell Microsystems) and continuously superfused with a modified, bicarbonate-containing Tyrode’s solution containing (in mmol/L): NaCl 124, NaHCO3 25, KCl 4, CaCl2 1.2, MgCl2 1, HEPES 10, Glucose 10, with pH=7.4. APs and underlying ionic currents were recorded in whole-cell configuration of patch-clamp technique. Electrodes were fabricated from borosilicate glass (World Precision Instruments) with tip resistances of 2 to 2.5 MΩ when filled with internal solution containing (in mmol/L): K-aspartate 110, KCl 25, NaCl 5, Mg-ATP 3, HEPES 10, cAMP 0.002, phosphocreatine-K2 10, and EGTA 0.01, with pH=7.2. This composition preserved physiological myocyte Ca2+ transient and contraction.20, 31, 32 Electrodes were connected to the input of an Axopatch 200B amplifier (Axon Instruments), with outputs digitized at 50 kHz using Digidata 1440A A/D card (Molecular Devices) under software control (pClamp 10). Series resistance (on cell) was typically 3 to 5 MΩ and was compensated by 85%. Experiments were discarded when the series resistance was high or increased substantially (>10%) during experiments. Reported AP voltages are already corrected to the liquid junction potentials. All experiments were conducted at 37±0.1 C.
APs were recorded in current-clamp experiments where cells were stimulated with depolarizing pulses (1.5× the threshold amplitude, 2 ms duration) delivered via patch pipette at pacing frequencies from 1 to 5 Hz. After reaching steady-state (3 min at each frequency), 50 consecutive APs were recorded to measure average behaviour. Then pacing was returned to 1 Hz and the selective INaL inhibitor GS-967 (1 µmol/L) was added to perfusate. When GS-967 effects stabilized (typically within 3 min) AP recordings at different pacing frequencies were repeated. AP duration at 95% of repolarization (APD95) was determined. Series of 50 consecutive APs were analyzed to estimate short-term variability of APD95 (STV) according to the following formula: STV=Σ(│APDn+1–APDn│)/[(nbeats–1)×√2], where APDn and APDn+1 indicate the durations of the nth and (n+1)th APs, and nbeats denotes the total number of consecutive beats analyzed.33 Changes in STV are presented as Poincaré plots of 50 consecutive APD95.
AP-clamp experiments were conducted as previously described.14, 34, 35 Briefly, the basic steps are as follows: (1) Record the cell’s steady-state AP under I-clamp (self AP-clamp) or choose a previously recorded typical AP (canonical AP-clamp). (2) Apply this AP onto the cell as voltage command under V-clamp at a given pacing frequency. The net current output (reference current) should reach steady-state and be stable over time. (3) Isolate the current of interest by using its specific blocker to remove it from the net current output (compensation current). (4) The current of interest is obtained by subtraction: Drug-sensitive current=Reference current–Compensation current. (5) Next, isolate the second current of interest by applying the second channel blocker, when it reaches steady-state another compensation current is recorded, and the second current of interest can be determined again by subtraction. Figure I in the Data Supplement shows a representative example. Under AP-clamp, all ionic currents were recorded as difference currents after each specific blocker had reached its steady-state effect (~3 min perfusion). 60 consecutive traces were recorded (to evaluate the stability) and averaged in each case before applying a drug (reference current) and 3-min after drug application (compensation current). 1 µmol/L GS-967 and 10 µmol/L nifedipine were used to measure INaL and ICaL (including some INCX), respectively. As previously validated,21 the GS-967-sensitive current recorded in our conditions is an excellent selective measure of INaL. Experiments were performed both when Ca2+ cycling was preserved (Physiol) and when [Ca2+]i was buffered below the diastolic level using 10 mmol/L 1,2-bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid (BAPTA) in the internal solution (BAPTAi) to assess [Ca2+]i-sensitivity of these currents under AP-clamp. To test the effect of CaMKII, cells were pretreated for 2 hours before experiment with the specific CaMKII inhibitor, autocamtide-2-related inhibitory peptide (AIP, cell-permeable myristoylated form, 1 µmol/L). Both the perfusion and pipettes solutions were also supplemented with AIP. In experiments examining the effect of βAR stimulation, isoproterenol (ISO, 3–300 nmol/L) was applied on AP-clamped cells. After ISO reached a steady-state effect (≈2 min), blockers were added sequentially to the perfusion solution to measure INaL and ICaL. All AP-clamp experiments were performed in Tyrode solution supplemented with selective inhibitors of K+ and Cl– currents (5 mmol/L 4-aminopyridine for Ito, 1 µmol/L E-4031 for IKr, 1 µmol/L HMR-1556 for IKs, 300 µmol/L BaCl2 for IK1, 100 nmol/L apamin for IKCa, and 30 µmol/L CaCCinh-A01 for IClCa). Experiments were excluded from analysis if significant rundown of ICaL was observed (in periodic tests) or if membrane current did not reach steady-state.
Conventional square pulse voltage-clamp experiments to measure the biophysical parameters of ICaL was performed using pipette solution containing 5 mmol/L EGTA and 2.1 mmol/L CaCl2 (free [Ca2+]i=100 nmol/L using the MaxChelator software), and in the presence of selective K+ and Cl– channel inhibitors in the bath (as above) and Na+ was replaced by Li+ to inhibit NCX. ICaL was measured using a 500 ms long voltage steps from holding potential of –80 mV to test potentials (between –40 and +20 mV) every 5 s (0.2 Hz stimulation) with a 50-ms pre-step to –40 mV to inactivate Na+ channels. To investigate Ca2+/CaMKII-dependence of ICaL, a 100-ms long depolarization pulse to 0 mV were used in every 0.5 s (2 Hz stimulation).
Ion currents were normalized to cell capacitance, determined in each cell using short (10 ms) hyperpolarizing pulses from −10 mV to −20 mV. Cell capacitance was 144.4±1.2 pF in age-matched controls (118 cells/8 animals) versus 194.1±3.3 pF in HF (158 cells/12 animals) using 2-sample Student’s t test, P<0.001.
Chemicals and reagents were purchased from Sigma-Aldrich, if not specified otherwise. E-4031 and HMR-1556 were from Tocris Bioscience. GS-967 was from Gilead.
Computational Modeling and Simulation
In silico experiments were performed using our recently updated rabbit ventricular myocyte model36 that integrates detailed descriptions of membrane electrophysiology, Ca2+ and Na+ handling,37 protein kinase A and CaMKII signalling pathways,38 and myofilament contraction.39 This model describes changes in CaMKII activity during each heartbeat, resulting in dynamic functional modulation of CaMKII phosphorylation targets (L-type Ca2+ channels, ryanodine receptors and phospholamban). These effects are enhanced in HF, where CaMKII expression and activation is increased (and as in prior work we elevated CaMKII content to 6-fold).38 We updated our model to account for HF-induced remodeling, based on our new data here and our previous HF model (including two-fold increase in NCX maximal transport rate, and altered sarcoplasmic reticulum (SR) Ca2+ release and reuptake).40 Based on our novel ICaL observations here, we shifted steady-state activation (5 mV negative) but left steady-state inactivation unchanged in HF. We also reduced ICaL maximal conductance (GCaL) by 20% in HF, resulting in the unaltered peak ICaL that we observed in control versus HF myocytes (Table and Figure II in the Data Supplement).
We used our updated cellular models to simulate AP-clamp experiments at 2 Hz pacing in control and HF myocytes with physiologic Ca2+ handling (Figure III in the Data Supplement; exhibiting reduced Ca2+ transients in HF) and/or with CaMKII inhibition (simulated by clamping fractional phosphorylation of CaMKII targets to the levels predicted without pacing). We applied the same AP trace used in wet AP-clamp experiments as the voltage-command. All simulations were performed in MATLAB (The MathWorks, Natick, MA, USA) using the stiff ordinary differential equation solver ode15s. Model code is available for download at: https://somapp.ucdmc.ucdavis.edu/Pharmacology/bers/ or http://elegrandi.wixsite.com/grandilab/downloads.
Statistical Analysis
Data are presented as Mean±SEM. The number of cells in each experimental group was reported as n=number of cells/number of animals. Statistical significance of differences was tested by paired Student’s t test or analysis of variance (ANOVA) with Bonferroni posttest as appropriate using Origin2016 software. Differences were deemed significant if P<0.05.
RESULTS
Frequency-Dependent Changes in AP Shape and Effect of Late Na+ Current Inhibition in HF
Figure 1 shows representative APs and group analysis in HF and age-matched control myocytes before and after treatment with the selective INaL inhibitor GS-967 (1 µM). Baseline APD at 95% repolarization (APD95) was longer in HF versus control at 1 Hz pacing (269.5±17.5 versus 199.2±7.7 ms; P<0.001) (Figure 1A and 1B). GS-967 decreased APD95 in HF by 22% (to 211.6±6.8 ms, NS), and also decreased APD95 in control, but by only 13% (to 174.2±5.9 ms; P<0.001). At faster pacing rates APD95 converged for HF and control, and similarly for the +GS-967 treatment curves (Figure 1B). Resting membrane potential (RMP) was slightly depolarized (by ≈4 mV, Figure 1C) in HF consistent with reduced IK1 in HF.4, 5, 14 AP amplitude (APA) was significantly lower (by 8–10 mV) in HF independent of GS-967 treatment. Similarly, the maximum rate of rise (dV/dtmax) during AP upstroke was also decreased by ≈25% in HF (Figure 1D). These effects are likely to be at least partly attributed to lower Na+ channel availability (with the more positive diastolic Vm), but elevated intracellular [Na+] ([Na+]i) and altered Na+ channel expression (both known to occur in HF)41–43 could also be involved. These effects were similar upon acute INaL inhibition with GS-967. The maximum repolarization rate (–dV/dtmax) during AP phase 3 was significantly slower in HF (by ≈25%), and INaL block partially restored –dV/dtmax and limited the difference between control and HF (≈15%). Collectively, these data are consistent with peak INa amplitude in HF being normal except for slight reduction in availability associated with the slightly depolarized diastolic Vm, and smaller APA and dV/dtmax, but enhanced INaL that contributes to APD prolongation and slowed repolarization.
Figure 1.

Frequency-dependent effects of late Na+ current inhibition on action potential (AP) in heart failure (HF). A Representative APs recorded at 1 Hz steady-state pacing in HF and age-matched control before and after treatment with the selective late Na+ current inhibitor GS-967 (1 µmol/L). B Frequency-dependence of AP duration measured at 95% of repolarization (APD95). C Resting membrane potential (RMP) was slightly more positive in HF in line with decreased AP amplitude (APA) at 1 Hz pacing. GS-967 (GS) had no effect on either AP parameters. D Maximal rate of rise (dV/dtmax) and maximal rate of phase 3 repolarization (–dV/dtmax) were significantly decreased in HF compared to control. GS increased –dV/dtmax already in control but even more in HF. E Representative Poincare plots of 50 consecutive APD95 values at 1 Hz pacing. F Frequency-dependent short-term variability of APD95 (STV). STV was increased at low pacing frequencies in HF, which was decreased with GS to the control level. Columns and bars represent mean±SEM. n refers to cells/animals measured in each group. Paired and unpaired Student’s t tests following analysis of variance (ANOVA). *P<0.05, **P<0.01, ***P<0.001 versus control; †P<0.05, ††P<0.01 versus control+GS-967.
Figure 1E and 1F show that HF myocytes also exhibited higher short-term variability of APD95 (STV) at 1 and 2 Hz, in HF versus control (4.80±0.51 versus 3.21±0.21 ms, respectively, 1 Hz pacing; P<0.01). Importantly, GS-967 treatment in HF decreased not only mean APD95 but also STV to control values (3.19±0.46 ms; NS). GS-967 also decreased STV at all pacing rates in control (2.10±0.13 ms; P<0.001).
INaL Magnitude and Dynamics Changes Under AP in HF
The AP data indicate increased depolarization drive during the AP plateau and repolarization phases in HF versus control myocytes. Thus, we studied the 3 major inward plateau currents (INaL, ICaL, and INCX) under physiological AP-clamp. We did not measure the fast Na+ current due to technical limitations (peak INa overlaps the capacitive transient under physiological AP-clamp), but altered peak INa can contribute to pathological excitability in HF.44 The reduced dV/dtmax observed (Figure 1D) would be consistent with reduced Na+ current availability and peak INa in HF. GS-967-sensitive INaL and nifedipine-sensitive current were recorded from the same myocyte using AP-clamp sequential dissection21, 35 (Figure I in the Data Supplement) with either preserved [Ca2+]i cycling (Physiol) or with [Ca2+]i buffered with 10 mmol/L BAPTA in the pipette (BAPTAi). Involvement of CaMKII was also tested using the selective CaMKII inhibitory peptide AIP. A previously recorded typical rabbit ventricular AP was used as voltage command in all AP-clamp experiments (ie, a canonical AP-clamp analogous to the mean for 2 Hz where HF and control APD95 were similar).14 Because HF myocytes were larger (≈35% increase in cell capacitance) all reported currents are normalized to the corresponding cell capacitance.
INaL was measured as GS-967-sensitive current under AP-clamp (Figure 2; See Reference 21 and Figure 1 therein for additional controls and technical details).21 The density of INaL increased during AP repolarization as the driving force for Na+ influx increased, achieving peak density during phase 3 repolarization of the AP (at ≈–50 mV in both control and HF). Importantly, peak INaL density was 82% higher in HF versus control myocytes in our physiological condition (–0.93±0.03 versus –0.51±0.01 A/F, respectively; P<0.001; Figure 2A and 2D). The INaL I-V analysis reveals that INaL is increased during the entire AP without change in Vm-dependence (Figure 2A, bottom). Buffering [Ca2+]i with BAPTAi did not alter peak INaL density in control (–0.54±0.02 A/F; NS; Figure 2E), but decreased peak INaL by 23% in HF (–0.72±0.03 A/F; P<0.001; Figure 2B). Nevertheless, INaL density and integral charge were still larger in HF versus control myocytes with BAPTAi (Figure 2E and 2F). AIP pretreatment (to inhibit CaMKII) decreased INaL in HF to the level of untreated control (–0.54±0.02 A/F; NS; Figure 2C and 2D). However, AIP also decreased INaL in control (–0.37±0.01 A/F; P<0.001; Figure 2C and 2D), such that INaL was still higher in HF versus control after AIP treatment (as for BAPTAi). We conclude that INaL is elevated in HF under all conditions studied, that basal INaL at 2 Hz pacing with Ca2+ transients, is partly dependent upon CaMKII activity, and that large INaL increase in HF is substantially CaMKII-dependent.
Figure 2.

Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent upregulation of late Na+ current (INaL) in heart failure (HF) under action potential (AP)-clamp. INaL was measured as GS-967 (1 µM)-sensitive current in HF and age-matched control. AP-clamp using a prerecorded typical AP (shown above) was applied at 2 Hz pacing. A INaL traces (mean±SEM) recorded under preserved [Ca2+]i cycling (physiol). INaL was significantly increased already during the early plateau phase of the AP and it achieved a nearly doubled peak density during phase 3 repolarization in HF cells having Ca2+ transients. Current-voltage relationship under AP-clamp is shown below. B INaL traces (mean±SEM) recorded under buffered [Ca2+]i using 10 mmol/L BAPTA in the pipette (BAPTAi). Buffering [Ca2+]i significantly reduced INaL peak density in HF. C INaL traces (mean±SEM) recorded in cells pretreated with the specific CaMKII inhibitor AIP (autocamtide-2-related inhibitory peptide; 1 µmol/L). AIP reduced INaL in HF to the untreated control level; however, AIP also decreased INaL in control. D Peak INaL density was significantly upregulated in HF under AP, partially by a CaMKII-dependent acute effect on INaL. E INaL density measured at the mid-plateau of the AP. F Net charges carried by INaL under AP in HF and age-matched control. Columns and bars represent mean±SEM. n refers to cells/animals measured in each group. Analysis of variance (ANOVA) with Bonferroni posttest, *P<0.05, **P<0.01, ***P<0.001. Ctl indicates control. G-I Simulated time courses of INaL under AP-clamp in control and HF obtained with physiol, BAPTAi and CaMKII inhibition conditions.
We used this data to update our most recent computational model of the rabbit ventricular myocyte.36 The basal value of INaL maximal conductance (GNaL) was scaled to 0.0527 mS/µF to reflect the peak INaL observed during the AP in control myocytes with physiologic Ca2+ handling. Inactivation of INaL was set to 600 ms as previously.45 GNaL was modeled as a function of chronic HF-induced remodeling (not influenced by acute CaMKII inhibition, ie, 50% increase in GNaL in failing versus nonfailing myocytes -guided by data in AIP-treated cells, Figure 2C) and simulated CaMKII activation reproduced the increase in peak INaL observed with physiologic Ca2+ handling versus CaMKII inhibition in both failing and nonfailing conditions, as previously done.46 GNaL (in mS/µF) was calculated as:
where HFremodeling indicates the absence/presence of chronic HF-induced remodeling (0 and 1, respectively), and PNaVs (ranging from 0 to 1) is the fraction of phosphorylated Na+ channels, modeled as previously described (Figure IV in the Data Supplement).46 The in silico AP-clamp experiments quantitatively reproduced the experimental data on the role of physiological Ca2+ transients and CaMKII activity in upregulating INaL in control and more strongly in HF (Figure 2G through 2I).
Nifedipine-sensitive Inward Current Changes in HF (ICaL and INCX)
Next, we measured nifedipine-sensitive current (INife) under AP-clamp (Figure 3). Under physiological conditions, nifedipine inhibits ICaL, and consequently abolishes Ca2+ transients. Note that INife was recorded when other Ca-sensitive currents (eg, IKs, IK(Ca) and ICl(Ca)) were pharmacologically inhibited (see Methods). Thus, the measured INife is a composite current containing ICaL and the inward shift in INCX that is driven by elevated [Ca]i. Peak INife density in the early plateau phase of the AP (at ≈+35 mV in both control and HF) was unaltered in HF versus control under physiological condition (Figure 3A and 3D). However, INife was slightly increased in HF during the AP plateau and terminal repolarization phases (Figure 3A and 3E), potentially due to either less Ca2+-dependent inactivation (CDI) of ICaL in HF (due to reduced Ca2+ transients)4, 9, enhanced Ca2+/CaMKII-dependent facilitation (CDF), altered Ca2+ channel subunit composition, or alternatively, but less likely41 to more inward INCX. In contrast, when INife was recorded with BAPTAi (ie, without Ca2+ transient and inward INCX), peak INife density (more exclusively ICaL) under AP-clamp was significantly decreased in HF (Figure 3B and 3D), but BAPTA also abolished the small hump near terminal repolarization, consistent with loss of expected inward INCX at this time (note superimposition of INife versus Vm curve between –80 and –40 mV). In BAPTAi the INife decay was still slower in HF versus control, despite low CDI expected in both cases (Figure 3B and 3E). When CaMKII was inhibited, the INife density and integrated charge movement were reduced versus Physiol for both HF and control (Figure 3C, 3D, and 3F). Since the peak inward INife is likely dominated by peak ICaL, this might reflect the involvement of basal CaMKII activity in maintaining physiological peak ICaL when Ca2+ transients and CaMKII are functional (Figure 3A and 3D).
Figure 3.

Nifedipine-sensitive current in heart failure (HF) under action potential (AP)-clamp. The L-type Ca2+ current and the inward Na+/Ca2+ exchange current under action potential (AP) were measured as a composite nifedipine-sensitive current (INife) in HF and age-matched control. AP-clamp using a prerecorded typical AP (shown above) was applied at 2 Hz pacing before and after application of 10 µmol/L nifedipine (Nife). A INife traces (mean±SEM) recorded under preserved [Ca2+]i cycling (physiol). INife was increased during the mid-plateau and the late-plateau phases of the AP in HF. Current-voltage relationship under AP-clamp is shown below. INife traces (mean±SEM) recorded under buffered [Ca2+]i using 10 mmol/L BAPTA in the pipette (BAPTAi). Buffering [Ca2+]i significantly reduced INife peak density in HF. C INife traces (mean±SEM) recorded in cells pretreated with the specific CaMKII (Ca2+/calmodulin-dependent protein kinase II) inhibitor AIP (autocamtide-2-related inhibitory peptide; 1 µmol/L). AIP slightly reduced INife peak density in HF. D Peak INife density was significantly upregulated in HF under AP by CaMKII. E INife density measured at the mid-plateau of the AP. F Net charges carried by INife under AP in HF and age-matched control. Columns and bars represent mean±SEM. n refers to cells/animals measured in each group. Analysis of variance (ANOVA) with Bonferroni posttest, *P<0.05, **P<0.01, ***P<0.001.
Changes in the Biophysical Properties of L-type Ca2+ Current in HF
Next, we measured the biophysical parameters of ICaL using conventional square-pulse voltage-clamp and [Ca2+]i buffered to 100 nmol/L by inclusion of 5 mmol/L EGTA in the pipette solution. The peak density of ICaL and the I-V relationship were not significantly different (Figure 4A), but the steady-state activation curve of ICaL was shifted slightly (4.7 mV) to more negative potentials in HF versus control (Figure 4B). The inactivation time constants of ICaL (obtained by biexponential fits of ICaL decay) were slightly prolonged in HF versus control, consistent with slower CDI in HF (Figure 4D and 4E), but Vm-dependence of inactivation (Figure 4C) and the recovery from inactivation kinetics (Figure 4F) were unchanged.
Figure 4.

Biophysical properties of L-type Ca2+ current (ICaL) in heart failure (HF). A Current-voltage relationship of ICaL peak density in HF and age-matched control. ICaL was measured in the presence of 5 mmol/L EGTA ([Ca2+]i=100 nmol/L) in the pipette. B Steady-state activation of ICaL was shifted by 5 mV to more negative potentials in HF. C Steady-state inactivation of ICaL was unaltered in HF. D Representative ICaL traces elicited by depolarization pulses to 0 mV (voltage protocol shown in the inset). E Decay time constants (τfast, τslow) of ICaL were slightly increased in HF. F ICaL recovery from inactivation was unchanged in HF. G Representative ICaL traces under preserved [Ca2+]i cycling (physiol). ICaL was elicited with depolarization pulse to 0 mV at 2 Hz. Inset shows significantly increased decay time constants but unaltered ICaL amplitude in HF. H Representative ICaL traces under buffered [Ca2+]i using 10 mmol/L BAPTA in the pipette (BAPTAi). Inset shows that the amplitude of the fast ICaL decay was significantly reduced in HF versus control, whereas τfast was still slightly increased in HF. I Representative ICaL traces following specific CaMKII inhibition with AIP (1 µmol/L). Inset shows the decay time constants of ICaL and the corresponding amplitudes. The inactivation time course of ICaL was fitted by a biexponential function. Columns and bars represent mean±SEM. n refers to cells/animals measured in each group. Analysis of variance (ANOVA) with Bonferroni posttest, *P<0.05, **P<0.01, ***P<0.001.
We also analyzed ICaL in the pipette conditions used for AP-clamp studies (Physiol, BAPTAi, and CaMKII inhibition). Peak ICaL density elicited by a step pulse to 0 mV was unaltered in HF versus control with preserved [Ca2+]i cycling (Figure 4G). This contrasts with the diminished peak ICaL observed when either [Ca2+]i is clamped very low (BAPTAi) or CaMKII is inhibited (Figure 4H and 4I), both conditions where CaMKII-dependent CDF is suppressed. We infer that Ca2+-dependent CaMKII upregulates peak ICaL (via CDF) in HF to the level of control cells (when measured in physiological conditions; compare Figure 4G through 4I).
CDI of ICaL was, as expected, stronger under physiological [Ca2+]i versus when [Ca2+]i was buffered by EGTA or BAPTA (Figure 4G versus 4E and 4H). However, the τ of inactivation was significantly slowed in HF versus control (Figure 4G, inset), consistent with known smaller Ca2+ transients in HF and thus less CDI.4, 9 Using 10 mmol/L BAPTA in the pipette (BAPTAi) should eliminate both CDI as well as CDF (Figure 4H). Indeed, BAPTAi significantly slowed the τ of inactivation (versus Physiol or EGTA; Figure 4H and 4I) but τfast was still slightly slower in HF versus control (Figure 4H, inset). When CaMKII was inhibited with AIP (Figure 4I) the inactivation τ values were more like those in physiological buffer (Figure 4G). The slowed ICaL inactivation in HF was still observable in BAPTA, consistent with part of that effect being independent of CDI, and may reflect some PKA-dependent effect in HF myocytes.47, 48
Computer Models Help to Report Physiological ICaL and INCX during the AP in HF
To help delineate the relative contributions of ICaL and INCX to INife under AP-clamp, we used our rabbit ventricular myocyte model36 with ICaL properties tuned to those measured in Figure 4 (for details see Methods, Table and Figure II in the Data Supplement). Figure 5 shows how in silico experiments can inform the distinct profiles of ICaL and INCX under AP-clamp in control and HF cells under physiologic Ca2+ transients (Figure III in the Data Supplement). Figure 5A and 5B shows calculated ICaL and INa/Ca in physiological conditions, which have the expected overall shapes.10, 37, 41, 49, 50After ICaL activation and the start of SR Ca2+ release, CDI causes ICaL decline to a plateau that can increase slightly as AP repolarization causes an increase in the Ca2+ driving force, until terminal repolarization deactivates ICaL. INCX is initially outward (Ca2+ influx) driven by the rapid AP depolarization. But once SR Ca2+ release occurs, the high submembrane [Ca2+]i drives a first peak of inward INCX which declines as [Ca2+]i falls, but is followed by a second peak during terminal repolarization (driven by voltage) until the [Ca2+]i reaches the diastolic level. In HF the ICaL waveform is similar, although CDI is slowed. The higher [Na+]i and smaller Ca2+ transient in HF shifts the INCX waveform outward during the plateau, and the higher NCX expression levels cause the larger inward INCX tail upon final repolarization.10, 41
Figure 5.

Simulated time courses of L-type Ca2+ current (ICaL) and Na+/Ca2+ exchanger current (INCX) under action potential (AP)-clamp in heart failure (HF). Simulated ICaL and INCX have been obtained with our updated rabbit ventricular myocyte model that integrates detailed descriptions of electrophysiology, Ca2+ and Na+ handling, PKA and CaMKII signaling, and myofilament contraction. A-B Simulated ICaL and INCX under AP-clamp at 2 Hz pacing in control (left) and HF (right) with physiological Ca2+ handling. C-D Simulated ICaL and INCX after nifedipine (Nife) treatment (simulated assuming complete block of ICaL) under AP-clamp at 2 Hz pacing in control and HF. E-F Nifedipine-sensitive current (INife) expressed as sum of the changes in simulated ICaL and INCX before and after nifedipine treatment in control and HF.
Figure 5C and 5D shows how ICaL and INCX are expected to change following 3-min of nifedipine exposure, which blocks ICaL and SR Ca2+ release. Without ICaL or SR Ca2+ release the AP drives outward INCX (Ca2+ influx) throughout much of the AP, and then that same amount of Ca2+ which entered via outward INCX is extruded via inward INCX during repolarization. Higher NCX expression in HF myocytes makes inward and outward INCX larger in HF. The residual INCX during nifedipine exposure must be taken into account when inferring the predicted INife shown in Figure 5E and 5F. The shape of the predicted INife is similar to the INife recorded in vitro under AP-clamp in cardiomyocytes (Figure 3A). This analysis also explains why the inward INCX tail at terminal repolarization is less prominent than expected in the measured INife traces in Figure 3A and 3C. While this simulation analysis cannot extract absolute ICaL and INCX waveforms that occur during the AP in control versus HF myocytes, Figure 5A provides a qualitative estimate of likely changes that occur in ICaL and INCX during the HF AP.
Altered β-Adrenergic Response of Inward Currents in HF
Elevated sympathetic tone is often reported in HF in conjunction with altered responses to βAR activation. We therefore tested the effects of acute βAR stimulation on INaL and INife in HF versus control myocytes using AP-clamp. Because downstream effects of βAR stimulation are mediated both by protein kinase A (PKA) and CaMKII and the activities of these kinases are known to be altered in HF, ionic currents were measured again with physiological preserved [Ca2+]i cycling and with heavily buffered [Ca2+]i.
Figure 6A shows INaL measured after βAR agonist isoproterenol (ISO, 10 nM) treatment. ISO increased INaL in both control and HF myocytes, but by a much larger percent in control (by 110% versus 40%; Figure 6C). Nevertheless, the resulting INaL with ISO (and its integral) was still larger in HF. Buffering [Ca2+]i reduced the effect of ISO on INaL, indicating the involvement of Ca2+ and/or CaMKII (as well as PKA) in mediating the ISO effect. Again, the effect on control was larger than in HF (70% versus 30%). Moreover, in BAPTAi the ISO-induced INaL peak density was not significantly different between control and HF. That is consistent with Ca2+- or CaMKII-dependence being involved with ISO-induced higher INaL in HF versus control.
Figure 6.

Altered response of inward currents to β-adrenergic receptor (βAR) stimulation in heart failure (HF). The late Na+ current (INaL) and the nifedipine-sensitive current (INife) were recorded after 2-min pretreatment with βAR agonist isoproterenol (ISO, 10 nmol/L). A INaL traces (mean±SEM) under AP-clamp at 2 Hz pacing measured with preserved [Ca2+]i cycling (physiol) and [Ca2+]i buffering using 10 mmol/L BAPTA in the pipette (BAPTAi) after ISO pretreatment in HF and age-matched control. Current-voltage relationship under AP-clamp is shown below. B INife traces (mean±SEM) under AP-clamp after ISO pretreatment in HF and age-matched control. C Upregulation of INaL peak and net charge induced by ISO, which was reduced with BAPTAi. D Robust increase in INife after ISO stimulation, which was reduced in BAPTAi, indicating a Ca2+-dependent pathway in mediating the response of βAR stimulation on INife (besides the classical protein kinase A effect). HF cells exhibited significantly reduced response of INife after ISO stimulation both with and without [Ca2+]i buffering. Symbols and bars represent mean±SEM. n refers to cells measured in each group, and the cells in each group came from 3 to 6 individual animals. Analysis of variance (ANOVA) with Bonferroni posttest, *P<0.05, **P<0.01, ***P<0.001.
INife also increased after ISO treatment, as expected for the known effects of βAR stimulation on ICaL and Ca2+ transient amplitude (which drives inward INCX; Figure 6B). Again, the ISO-induced increase in INife was smaller in HF. This blunted βAR response in HF was present with both physiological Ca2+ cycling and BAPTAi conditions, but for INife the ISO-induced increase for BAPTAi (2.4-fold in control versus 2-fold in HF) was only slightly smaller than for physiological conditions (3.3-fold in control versus 2.2-fold increase in HF; Figure 6D).
Because the ISO effects on both INaL and INife were blunted in HF versus control, we tested whether this was due to decreased ISO-sensitivity or limited maximal response. ISO concentrations between 3 and 300 nM were applied in HF and control (Figure 7). Steady-state contracting AP-clamped myocytes in HF were unstable at higher ISO concentrations, so these experiments were performed with 10 mmol/L BAPTA in the pipette (BAPTAi). ISO increased INaL in both control and HF, but the half maximal [ISO] (EC50) was slightly higher in HF (16.1±1.4 versus 10.5±1.3 nM; P<0.05; Figure 7A). However, the maximal ISO-induced INaL density was not different in control versus HF (Figure 7A). ICaL was also markedly increased after ISO application with similar EC50 (≈12 nM) in control and HF. However, the maximal ICaL response after ISO stimulation was only half as much in HF versus control (4.6- versus 2.7-fold increase in peak ICaL; Figure 7B). This indicates unchanged ISO-sensitivity, but weaker potency in raising ICaL. The blunted ISO response of ICaL in HF may limit Ca2+ transients and inotropy in HF during sympathetic activity.
Figure 7.

β-adrenergic receptor (βAR) responsiveness of inward currents in heart failure (HF). Dose-response effect of isoproterenol (ISO) on late Na+ current (INaL) and L-type Ca2+ current (ICaL) peak densities under AP-clamp at 2 Hz pacing rate. Pipette solution contained 10 mmol/L BAPTA (BAPTAi). A INaL measured as GS-967-sensitive current significantly increased after ISO application. INaL sensitivity (EC50, half maximal effective concentration) to ISO was slightly reduced in HF, and INaL exhibited similar maximal response in HF than in control despite the increased basal INaL in HF. B ICaL measured as nifedipine-sensitive current was markedly increased after ISO treatment; however, the magnitude of the response was significantly blunted in HF with no change in ISO-sensitivity. EC50 values, Hill coefficients and maximum responses were determined by fitting data to the Hill equation, indicated by solid lines. Symbols and bars represent mean±SEM. n refers to the number of cells measured in each group, and the cells in each group came from 3 to 6 individual animals. Analysis of variance (ANOVA) with Bonferroni posttest, *P<0.05, **P<0.01, ***P<0.001.
Relative Contributions of Inward Currents to AP Plateau and Phase 3 Repolarization
The dynamic interplay of time-and voltage-dependent activation and inactivation of inward currents governs the AP waveform. Each inward current has a unique profile and magnitude during the cardiac AP. The relative contributions of major inward currents (INaL, INife≈ICaL+INCX) during the AP plateau and repolarization in control and in HF are shown in Figure 8, compared at different Vm (+40, –20, and –60 mV), and shown as integrated charge movement during the AP (Figure 8, insets). INife early in the AP (phase 1, at +40 mV) predominantly represents ICaL, whereas late during terminal repolarization (at –60 mV) is mainly inward INCX (during physiological Ca2+ transients).
Figure 8.

Relative contribution of inward currents to net depolarizing current in heart failure (HF). Relative contributions and magnitudes of the major inward currents (INaL and INife) are compared in distinct phases of the action potential (AP) repolarization process in HF to those in age-matched control. Magnitude of INife at the early repolarization phase (phase 1) of the AP (at +40 mV) predominantly represents ICaL, whereas the magnitude of INife during the terminal repolarization (at –60 mV) is generated predominantly by the inward INCX (under physiological conditions). A INaL and INife traces measured in control and HF under AP-clamp at 2 Hz pacing without using any Ca2+ buffer or β-adrenergic receptor (βAR) agonist. Mean traces and SEM are shown. B When [Ca2+]i cycling is preserved, upregulation of INaL increased the net inward current during phase 3 of AP. C βAR stimulation using isoproterenol (ISO, 10 nmol/L) significantly upregulated not only ICaL but also INaL and INCX. However, HF cells were hyporesponsive to ISO-induced stimulation (ie, both ICaL and INaL increased in a smaller extent than in control), thus net inward current at +40 mV (predominantly ICaL) was significantly decreased in HF compared with control. Despite hyporesponsiveness, the net inward current during AP terminal repolarization was still significantly increased in HF because of the upregulated INaL and INCX. D CaMKII inhibition using the specific inhibitory peptide AIP largely diminished the difference between control and HF by reducing the upregulated INaL in HF. E Buffering [Ca2+]i (BAPTAi) eliminated the inward INCX and significantly decreased the extent of INaL enhancement in HF limiting the increase in net inward current. F Under βAR stimulation in BAPTAi, HF cells exhibited slightly decreased ICaL, but similar INaL density compared to control. The contributions of these inward currents to total net charge are shown in insets. Columns and bars represent mean±SEM. Statistics and n numbers are shown in Figures 2 through 6.
Total inward current during AP phase 1 (early repolarization) was unaltered in HF versus control in physiologic conditions, but had greater INaL contribution in HF (Figure 8A and 8B). Later during AP repolarization (–20 and –60 mV) total inward current was higher in HF, mainly due to increased INaL (Figure 8B). In control myocytes, ISO increased both inward currents, but the smaller ISO-induced increases of ICaL in HF reduced ICaL throughout the AP (Figure 8C). Total inward current in HF was reduced early (+40 mV) but increased progressively later in the AP (–20 and –60 mV) which could promote failure of repolarization and EADs. Despite βAR hyporesponsiveness, INaL and INCX were still higher in HF and contributed to that increased total inward current late in the AP (with ISO).
In contrast to physiological conditions, CaMKII inhibition limited both INaL and INife increases in HF and abolished differences in late AP total inward current between HF and control (Figure 8D versus 8B). Similarly, using strong [Ca2+]i buffering also reduced INaL and INife in HF, and total inward current was unaltered early but still slightly increased late during the AP in HF versus control (Figure 8E). ISO did not change total inward current during phase 3 of the AP in the presence of BAPTAi (presumably because inward INCX is prevented; Figure 8F).
Discussion
Arrhythmogenic AP Alterations in HF and the Role of Increased Late Na+ Current
HF-induced ionic remodeling causes characteristic changes in ventricular AP profile in our rabbit HF model including APD prolongation, reduction of phase 1 and phase 3 repolarization rates, depolarized resting membrane potential and reduced AP upstroke velocity under steady-state pacing (see Figure 1A through 1D), which mostly agree with literature data on various animal HF models and human HF.4–7, 18, 25, 26, 51 Moreover, temporal variability of AP repolarization (characterized by STV) was also increased in HF (Figure 1E and 1F), further enhancing the arrhythmogenic substrate.6–8 Delayed afterdepolarizations are frequently reported in HF,3, 4 and we have shown previously increased DADs that depended on CaMKII-mediated SR Ca2+ leak after cessation of pacing bouts in this HF model.14,52, 53
The HF-related arrhythmogenic alterations in ventricular AP were more pronounced at slow pacing rates than at fast pacing rates (Figure 1) in line with previous experimental14, 54 and clinical data.55 Importantly, selective INaL inhibition in HF reduced both APD and STV to control values at all pacing rates. The rate-dependence of APD prolongation and increase in STV associated with enhanced INaL in HF can be explained by the following mechanisms: (1) INaL availability may decrease at faster pacing rates.56 (2) Amplitude of Ca2+ transients may decrease at faster pacing rates in HF cells unlike healthy control, altering the magnitude of Ca2+-dependent ionic currents and INCX.57 (3) The delayed rectifier K+ currents that counterbalance INaL during AP plateau and phase 3 are also increased in HF under physiological conditions at faster pacing rates,14 and the increase of those K+ currents has also been shown to be Ca2+/CaMKII-dependent.14
In line with our results in HF rabbits, it has been shown that both the INaL inhibitor ranolazine and AIP exert potent antiarrhythmic effects in a transverse aortic constriction (pressure overload) induced HF model in mice18 as well as in failing human myocytes.24 Inhibition of the enhanced INaL prevented APD prolongation and STV enhancement in our rabbit HF model, thus it reduced the substrate for arrhythmias. Moreover, enhanced INaL can also contribute to increased Na+ loading and (via NCX) Ca2+ loading, and contribute to increased occurrence of afterdepolarizations that triggers arrhythmias in HF.23 Furthermore, enhanced Na+ current and spontaneous SR Ca2+ leak can contribute to increased STV.33, 58 Of note, the INaL maximal conductance used in the mathematical model to fit our AP-clamp current data is ~10-fold larger than previously estimated in rabbit myocyte experiments using a different experimental approach (1-day cell culture, square voltage pulses to −20 mV, abolished Ca2+ cycling).45 Physiologically dynamic Ca2+-dependent CaMKII activation and AP shape may contribute to these differences which underlines the importance of using physiological AP-clamp to measure ionic currents.
Changes in Inward Currents in HF that Shape the AP
Electrophysiological remodeling in HF leads to arrhythmogenic alterations in AP morphology that involves changes in the expression and regulation of multiple ion channels.26 Experimental data detailing such changes have been integrated in computational models to understand mechanistically how arrhythmogenesis occurs in HF.40, 59 However, ionic currents were usually recorded under nonphysiologic conditions, which may mask modulation by Ca2+ transients and CaMKII on ionic currents. This regulation may be even more impactful under pathological states including HF (where CaMKII-activity is increased).14, 52, 60 Accordingly, we have shown that Ca2+ transient, CaMKII, and βAR stimulation regulates K+ currents under physiological AP-clamp, and importantly, that this regulation was significantly altered in HF.14 Importantly, Ca2+/CaM and CaMKII are also known to regulate both Na+ and Ca2+ channels. Ca2+/CaM binding to the IQ motif of these channels is known to enhance CDI that is evident, especially for ICaL,61, 62 but a similar mechanism has been shown for fast INa and INaL.63, 64 CDI serves as a feedback mechanism preventing cellular Ca2+ overload.29 In contrast, CaMKII increases both ICaL (via CDF)27–29 and INaL.17, 64
INaL represents a non- or slowly-inactivating component of Na+ current that persists throughout the AP plateau and phase 3, as shown in Figure 2. The gating mechanism(s) contributing to INaL have been extensively studied and may include the early burst and late scattered openings, non-equilibrium gating and steady-state “window” current (overlap between the activation and inactivation curves); however, the details are still not fully resolved.65 INaL enhancement in HF has been demonstrated in different animal models and human;11, 12 however, we found an even more pronounced increase in INaL under physiological AP-clamp than previous reports of smaller INaL under nonphysiological voltage-clamp conditions.11, 17, 19, 45, 56 We showed that INaL upregulation in HF was largely Ca2+-dependent (Figure 2), in line with the increased CaMKII activity in HF that upregulates INaL.17–19 However, INaL was still increased in HF following cytosolic Ca2+ buffering with BAPTA, potentially reflecting an increased basal PKA-dependent phosphorylation of Na+ channels or remodeling with increased expression of neural Na+ channel isoforms in HF.42, 43
ICaL is the main inward current during the AP plateau in ventricular cardiomyocytes, as shown in Figure 3. ICaL amplitude was unchanged in HF under physiological conditions or using the slow Ca2+ buffer EGTA (which does not eliminate subsarcolemmal [Ca2+] changes, Figure 4). In contrast, ICaL was slightly decreased in HF when measured with BAPTAi and AIP, indicating a key role of CaMKII in maintaining ICaL in HF. ICaL decay was significantly slower in HF. This might be explained by reduced CDI (because of reduced Ca2+ transient amplitude), enhanced CDF (via increased CaMKII activity), and/or altered Ca2+ channel subunit composition. Increased expression of auxiliary β2 subunit is known to occur in HF66 and it has been shown to reduce the rate of ICaL inactivation, shift activation to more negative potentials, and significantly diminishes PKA response, all in agreement with our data.67–69
The difference in nifedipine-sensitive inward current measured with and without [Ca2+]i buffering suggests enhanced INCX, in agreement with previous reports.9 INCX kinetics during the rabbit ventricular AP during Ca2+ transients was previously measured in an elegant study of Weber et al.49 Here, we could not explicitly separate INCX from INife (Figure 3), but our measurements of ICaL alone (Figure 4) and in silico models (Figure 5) were consistent with the upregulation of NCX previously reported in this HF model. Our modeling provided quantitative estimates of INCX dynamics under AP-clamp in control and HF (Figure 5). Importantly, the increased INCX peak during phase 3 of AP repolarization in HF may contribute to EADs and APD prolongation. However, the precise contribution of INCX to arrhythmogenic AP alterations in HF requires further studies.
β-Adrenergic Stimulation-Induced Changes in Inward Currents in HF
Increased sympathetic activity but blunted β-adrenergic response are hallmarks of HF and significantly contribute to contractile dysfunction, alterations in Ca2+ handling system, and arrhythmogenesis.4, 14, 52, 70, 71
ICaL, INaL. and INCX are all known to be increased in ventricular myocytes after βAR stimulation. Downstream signaling mediating βAR effects on Na+ and Ca2+ channels involves both PKA and CaMKII; however, there is still debate on the exact molecular mechanisms and phosphorylation sites.72, 73 We found that ICaL peak was upregulated mainly by PKA during ISO stimulation (by comparing physiological and BAPTAi conditions), whereas INaL was upregulated by PKA and CaMKII in an almost equal manner both in control and HF (Figure 6). Importantly, the ISO-induced increases in ICaL and INaL were limited in HF both in physiological and BAPTAi conditions (Figure 6). While the ISO EC50 for INaL activation was slightly increased in HF, the eventual maximal INaL density was the same in HF versus age-matched control, in part because of the increased basal INaL in HF (Figure 7A). On the other hand, the ISO EC50 for ICaL activation was not altered in HF, but the maximal effect was ≈40% smaller in HF versus age-matched control (Figure 7B) similar to prior report in this HF model.4 This blunted βAR response could be due to reduced number of β1ARs,70 lower local cAMP levels,71 or altered phosphatases and phosphosdiesterases in HF.52
ICaL, INaL, and INCX reach their peak density sequentially during the AP, and their relative contributions to net inward current also dynamically change during the AP time course, as demonstrated previously in pig51 and guinea-pig20, 31 cardiomyocytes. In this study we demonstrated their detailed contribution in a chronic pressure/volume overload-induced HF rabbit model (Figure 8). The enhanced INaL and INCX increase the demand on the repolarizing K+ currents, that are also remodeled in HF.13, 14 The balance between these depolarizing and repolarizing currents in HF is shifted during phase 3 of the AP so as to slow repolarization and prolong APD.51
Conclusions
We measured INaL under physiologic recording conditions (AP-clamp with physiologic ionic composition, pacing rate, [Ca2+]i and temperature) and demonstrated a key role INaL enhancement in HF in APD prolongation and increased STV, especially at slow heart rates. INaL increase and its consequences may be exacerbated upon sympathetic activation. Our results agree with previous studies showing beneficial effects of INaL inhibition in HF and point to INaL as a therapeutic target to reduce the risk of arrhythmias in HF.18, 24 However, it should be considered that selective INaL inhibition decreased APD, albeit more modestly, in control. On the other hand, hyporesponsiveness of ICaL to βAR stimulation limits Ca2+ entry and that can aggravate the contractile deficit in HF. Our data demonstrate the importance of utilizing physiological recording conditions when measuring ionic currents, especially in cardiac pathologies associated with altered [Ca2+]i handing and CaMKII activity, in order to improve our understanding of electrophysiological remodeling and mechanistic bases of arrhythmogenesis in HF.
Supplementary Material
What is Known:
Significant ion channel remodeling occurs in heart failure (HF) which results in prolongation of the cardiac action potential (and QT interval in ECG) and increases the risk of arrhythmias.
Enhanced late Na+ current and upregulation of Na+/Ca2+ exchanger (NCX) have been reported in HF.
What the Study Adds:
CaMKII-dependent and beta-adrenergic upregulation of late Na+ current in HF enhances the net depolarization drive and significantly contributes to arrhythmogenic action potential alterations in HF.
Reveal CaMKII-dependent facilitation but β-adrenergic hypo-responsiveness of L-type Ca2+ current in HF.
Acknowledgments:
We thank Dr William T. Ferrier and Linda Talken for performing the surgical procedures, and Lynette M. Mendoza for performing echocardiography. We also thank Logan R.J. Bailey, Johanna M. Borst, Austen J. Lucena, Maura Ferrero, Matthew L. Stein, Ian P. Palmer, and Maximilien Bergman for their help in animal care and cell isolation.
Sources of Funding: This work was supported by grants from the National Institute of Health P01-HL080101 and R01-HL030077 (Dr Bers), R01-HL142282 (Drs Bers and Bossuyt), OT2-OD023848, OT2-OD026580 and R01-HL131517, R01-HL141214 (Dr Grandi), R01-HL090880 (Drs Izu and Chen-Izu), R01-HL123526 (Dr Chen-Izu), and K99-HL138160 (Dr Morotti); the American Heart Association 14GRNT20510041 (Dr Chen-Izu), and 15SDG24910015 (Dr Grandi); the Hungarian Scientific Research Fund OTKA101196 (Dr Bányász); the Heart Rhythm Society post-doctoral fellowship 16OA9HRS (Dr Morotti); and the UC Davis School of Medicine Dean’s Fellow award (Dr Grandi).
Footnotes
Disclosures: Dr Luiz Belardinelli is a former employee of Gilead Sciences, Inc, which is the patent holder of GS-967. Current affiliation of Dr Belardinelli is InCarda Therapeutics, Inc. (Brisbane, CA, USA). The authors declare that they have no conflict of interest.
References:
- 1.Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels AP, Wellens HJ, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: Time to revise current concepts. Cardiovasc Res. 2000;46:376–392. [DOI] [PubMed] [Google Scholar]
- 2.Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verkerk AO, Veldkamp MW, Baartscheer A, Schumacher CA, Klopping C, van Ginneken AC, Ravesloot JH. Ionic mechanism of delayed afterdepolarizations in ventricular cells isolated from human end-stage failing hearts. Circulation. 2001;104:2728–2733. [DOI] [PubMed] [Google Scholar]
- 4.Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. [DOI] [PubMed] [Google Scholar]
- 5.Kaab S, Nuss HB, Chiamvimonvat N, O’Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–273. [DOI] [PubMed] [Google Scholar]
- 6.Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90:2534–2539. [DOI] [PubMed] [Google Scholar]
- 7.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piccirillo G, Magri D, Matera S, Magnanti M, Torrini A, Pasquazzi E, Schifano E, Velitti S, Marigliano V, Quaglione R, Barilla F. Qt variability strongly predicts sudden cardiac death in asymptomatic subjects with mild or moderate left ventricular systolic dysfunction: A prospective study. Eur Heart J. 2007;28:1344–1350. [DOI] [PubMed] [Google Scholar]
- 9.Pogwizd SM, Qi M, Yuan WL, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 1999;85:1009–1019. [DOI] [PubMed] [Google Scholar]
- 10.Weber CR, Piacentino V 3rd, Houser SR, Bers DM. Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation. 2003;108:2224–2229. [DOI] [PubMed] [Google Scholar]
- 11.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. [DOI] [PubMed] [Google Scholar]
- 12.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. [DOI] [PubMed] [Google Scholar]
- 13.Beuckelmann DJ, Nabauer M, Erdmann E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ Res. 1993;73:379–385. [DOI] [PubMed] [Google Scholar]
- 14.Hegyi B, Bossuyt J, Ginsburg KS, Mendoza LM, Talken L, Ferrier WT, Pogwizd SM, Izu LT, Chen-Izu Y, Bers DM. Altered repolarization reserve in failing rabbit ventricular myocytes: Calcium and beta-adrenergic effects on delayed- and inward-rectifier potassium currents. Circ Arrhythm Electrophysiol. 2018;11:e005852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure - a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98:1383–1393. [DOI] [PubMed] [Google Scholar]
- 16.Rivaud MR, Agullo-Pascual E, Lin X, Leo-Macias A, Zhang M, Rothenberg E, Bezzina CR, Delmar M, Remme CA. Sodium channel remodeling in subcellular microdomains of murine failing cardiomyocytes. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, Sag CM, Hund TJ, Mohler PJ, Belardinelli L, Hasenfuss G, Maier LS, Sossalla S. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J Mol Cell Cardiol. 2013;61:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ, Hund TJ. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation. 2012;126:2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horvath B, Banyasz T, Jian Z, Hegyi B, Kistamas K, Nanasi PP, Izu LT, Chen-Izu Y. Dynamics of the late Na(+) current during cardiac action potential and its contribution to afterdepolarizations. J Mol Cell Cardiol. 2013;64:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hegyi B, Banyasz T, Izu LT, Belardinelli L, Bers DM, Chen-Izu Y. Beta-adrenergic regulation of late Na(+) current during cardiac action potential is mediated by both PKA and CaMKII. J Mol Cell Cardiol. 2018;123:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda JJ, Lee H, Shibata EF. Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ Res. 1992;70:199–207. [DOI] [PubMed] [Google Scholar]
- 23.Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS, Shryock JC. Cardiac late Na(+) current: Proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm. 2015;12:440–448. [DOI] [PubMed] [Google Scholar]
- 24.Sag CM, Mallwitz A, Wagner S, Hartmann N, Schotola H, Fischer TH, Ungeheuer N, Herting J, Shah AM, Maier LS, Sossalla S, Unsold B. Enhanced late ina induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J Mol Cell Cardiol. 2014;76:94–105. [DOI] [PubMed] [Google Scholar]
- 25.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. [DOI] [PubMed] [Google Scholar]
- 26.Nattel S, Maguy A, Le Bouter S, Yeh YH. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–456. [DOI] [PubMed] [Google Scholar]
- 27.Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca2+/calmodulin-dependent protein-kinase mediates Ca2+-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ Res. 1994;75:854–861. [DOI] [PubMed] [Google Scholar]
- 28.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bers DM, Morotti S. Ca(2+) current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front Pharmacol. 2014;5:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoeker GS, Hanafy MA, Oster RA, Bers DM, Pogwizd SM. Reduced arrhythmia inducibility with calcium/calmodulin-dependent protein kinase ii inhibition in heart failure rabbits. J Cardiovasc Pharmacol. 2016;67:260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banyasz T, Horvath B, Jian Z, Izu LT, Chen-Izu Y. Profile of L-type Ca(2+) current and Na(+)/Ca(2+) exchange current during cardiac action potential in ventricular myocytes. Heart Rhythm. 2012;9:134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horvath B, Vaczi K, Hegyi B, Gonczi M, Dienes B, Kistamas K, Banyasz T, Magyar J, Baczko I, Varro A, Seprenyi G, Csernoch L, Nanasi PP, Szentandrassy N. Sarcolemmal Ca(2+)-entry through L-type Ca(2+) channels controls the profile of Ca(2+)-activated Cl(−) current in canine ventricular myocytes. J Mol Cell Cardiol. 2016;97:125–139. [DOI] [PubMed] [Google Scholar]
- 33.Szentandrassy N, Kistamas K, Hegyi B, Horvath B, Ruzsnavszky F, Vaczi K, Magyar J, Banyasz T, Varro A, Nanasi PP. Contribution of ion currents to beat-to-beat variability of action potential duration in canine ventricular myocytes. Pflugers Arch. 2015;467:1431–1443. [DOI] [PubMed] [Google Scholar]
- 34.Chen-Izu Y, Izu LT, Hegyi B, Bányász T. Recording of ionic currents under physiological conditions: Action potential-clamp and ‘onion-peeling’ techniques In: Jue T, ed. Modern Tools of Biophysics. New York, NY: Springer; New York; 2017:31–48. [Google Scholar]
- 35.Banyasz T, Horvath B, Jian Z, Izu LT, Chen-Izu Y. Sequential dissection of multiple ionic currents in single cardiac myocytes under action potential-clamp. J Mol Cell Cardiol. 2011;50:578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartos DC, Morotti S, Ginsburg KS, Grandi E, Bers DM. Quantitative analysis of the Ca2+ -dependent regulation of delayed rectifier K+ current IKs in rabbit ventricular myocytes. J Physiol. 2017;595:2253–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca(2+) handling. Biophys J. 2010;99:2038–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Negroni JA, Morotti S, Lascano EC, Gomes AV, Grandi E, Puglisi JL, Bers DM. Beta-adrenergic effects on cardiac myofilaments and contraction in an integrated rabbit ventricular myocyte model. J Mol Cell Cardiol. 2015;81:162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shannon TR, Wang F, Bers DM. Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys J. 2005;89:4096–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. [DOI] [PubMed] [Google Scholar]
- 42.Huang B, El-Sherif T, Gidh-Jain M, Qin D, El-Sherif N. Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. J Cardiovasc Electrophysiol. 2001;12:218–225. [DOI] [PubMed] [Google Scholar]
- 43.Mishra S, Reznikov V, Maltsev VA, Undrovinas NA, Sabbah HN, Undrovinas A. Contribution of sodium channel neuronal isoform Na(v)1.1 to late sodium current in ventricular myocytes from failing hearts. J Physiol. 2015;593:1409–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clancy CE, Chen-Izu Y, Bers DM, Belardinelli L, Boyden PA, Csernoch L, Despa S, Fermini B, Hool LC, Izu L, Kass RS, Lederer WJ, Louch WE, Maack C, Matiazzi A, Qu Z, Rajamani S, Rippinger CM, Sejersted OM, O’Rourke B, Weiss JN, Varro A, Zaza A. Deranged sodium to sudden death. J Physiol. 2015;593:1331–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morotti S, Edwards AG, McCulloch AD, Bers DM, Grandi E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J Physiol. 2014;592:1181–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schroder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RH, Weil J, Herzig S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 1998;98:969–976. [DOI] [PubMed] [Google Scholar]
- 48.Findlay I Voltage- and cation-dependent inactivation of L-type Ca2+ channel currents in guinea-pig ventricular myocytes. J Physiol. 2002;541:731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weber CR, Piacentino V, Ginsburg KS, Houser SR, Bers DM. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ Res. 2002;90:182–189. [DOI] [PubMed] [Google Scholar]
- 50.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and ca transient. J Mol Cell Cardiol. 2010;48:112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hegyi B, Bossuyt J, Griffiths LG, Shimkunas R, Coulibaly Z, Jian Z, Grimsrud KN, Sondergaard CS, Ginsburg KS, Chiamvimonvat N, Belardinelli L, Varro A, Papp JG, Pollesello P, Levijoki J, Izu LT, Boyd WD, Banyasz T, Bers DM, Chen-Izu Y. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc Natl Acad Sci U S A. 2018;115:E3036–E3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. [DOI] [PubMed] [Google Scholar]
- 53.Curran J, Brown KH, Santiago DJ, Pogwizd S, Bers DM, Shannon TR. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. J Mol Cell Cardiol. 2010;49:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vermeulen JT, McGuire MA, Opthof T, Coronel R, de Bakker JM, Klopping C, Janse MJ. Triggered activity and automaticity in ventricular trabeculae of failing human and rabbit hearts. Cardiovasc Res. 1994;28:1547–1554. [DOI] [PubMed] [Google Scholar]
- 55.Davey PP, Barlow C, Hart G. Prolongation of the QT interval in heart failure occurs at low but not at high heart rates. Clin Sci (Lond). 2000;98:603–610. [PubMed] [Google Scholar]
- 56.Wu L, Ma J, Li H, Wang C, Grandi E, Zhang P, Luo A, Bers DM, Shryock JC, Belardinelli L. Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation. 2011;123:1713–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. SR calcium handling and calcium after-transients in a rabbit model of heart failure. Cardiovasc Res. 2003;58:99–108. [DOI] [PubMed] [Google Scholar]
- 58.Johnson DM, Heijman J, Bode EF, Greensmith DJ, van der Linde H, Abi-Gerges N, Eisner DA, Trafford AW, Volders PG. Diastolic spontaneous calcium release from the sarcoplasmic reticulum increases beat-to-beat variability of repolarization in canine ventricular myocytes after beta-adrenergic stimulation. Circ Res. 2013;112:246–256. [DOI] [PubMed] [Google Scholar]
- 59.Gomez JF, Cardona K, Trenor B. Lessons learned from multi-scale modeling of the failing heart. J Mol Cell Cardiol. 2015;89:146–159. [DOI] [PubMed] [Google Scholar]
- 60.Hegyi B, Bers DM, Bossuyt J. CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol. 2019;127:246–259. [DOI] [PubMed] [Google Scholar]
- 61.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. [DOI] [PubMed] [Google Scholar]
- 62.Grandi E, Morotti S, Ginsburg KS, Severi S, Bers DM. Interplay of voltage and Ca-dependent inactivation of L-type Ca current. Prog Biophys Mol Biol. 2010;103:44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sarhan MF, Tung CC, Van Petegem F, Ahern CA. Crystallographic basis for calcium regulation of sodium channels. Proc Natl Acad Sci U S A. 2012;109:3558–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: Similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen-Izu Y, Shaw RM, Pitt GS, Yarov-Yarovoy V, Sack JT, Abriel H, Aldrich RW, Belardinelli L, Cannell MB, Catterall WA, Chazin WJ, Chiamvimonvat N, Deschenes I, Grandi E, Hund TJ, Izu LT, Maier LS, Maltsev VA, Marionneau C, Mohler PJ, Rajamani S, Rasmusson RL, Sobie EA, Clancy CE, Bers DM. Na+ channel function, regulation, structure, trafficking and sequestration. J Physiol. 2015;593:1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hullin R, Matthes J, von Vietinghoff S, Bodi I, Rubio M, D’Souza K, Friedrich Khan I, Rottlander D, Hoppe UC, Mohacsi P, Schmitteckert E, Gilsbach R, Bunemann M, Hein L, Schwartz A, Herzig S. Increased expression of the auxiliary beta(2)-subunit of ventricular L-type Ca(2)+ channels leads to single-channel activity characteristic of heart failure. PloS One. 2007;2:e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, Alvania RS, Johns DC, Marban E, Yue DT. Novel functional properties of Ca(2+) channel beta subunits revealed by their expression in adult rat heart cells. J Physiol. 2002;541:435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miriyala J, Nguyen T, Yue DT, Colecraft HM. Role of CaVbeta subunits, and lack of functional reserve, in protein kinase A modulation of cardiac CaV1.2 channels. Circ Res. 2008;102:e54–64. [DOI] [PubMed] [Google Scholar]
- 69.Beetz N, Hein L, Meszaros J, Gilsbach R, Barreto F, Meissner M, Hoppe UC, Schwartz A, Herzig S, Matthes J. Transgenic simulation of human heart failure-like L-type Ca2+-channels: Implications for fibrosis and heart rate in mice. Cardiovasc Res. 2009;84:396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Desantiago J, Ai X, Islam M, Acuna G, Ziolo MT, Bers DM, Pogwizd SM. Arrhythmogenic effects of beta2-adrenergic stimulation in the failing heart are attributable to enhanced sarcoplasmic reticulum Ca load. Circ Res. 2008;102:1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barbagallo F, Xu B, Reddy GR, West T, Wang Q, Fu Q, Li M, Shi Q, Ginsburg KS, Ferrier W, Isidori AM, Naro F, Patel HH, Bossuyt J, Bers D, Xiang YK. Genetically encoded biosensors reveal pka hyperphosphorylation on the myofilaments in rabbit heart failure. Circ Res. 2016;119:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Katchman A, Yang L, Zakharov SI, Kushner J, Abrams J, Chen BX, Liu G, Pitt GS, Colecraft HM, Marx SO. Proteolytic cleavage and PKA phosphorylation of alpha1C subunit are not required for adrenergic regulation of CaV1.2 in the heart. Proc Natl Acad Sci U S A. 2017;114:9194–9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Herren AW, Weber DM, Rigor RR, Margulies KB, Phinney BS, Bers DM. CaMKII phosphorylation of Na(V)1.5: Novel in vitro sites identified by mass spectrometry and reduced S516 phosphorylation in human heart failure. J Proteome Res. 2015;14:2298–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.