

Abstract
Nitrogen-containing heterocycles such as quinoline, quinazolinones and indole are scaffolds of natural products and have broad biological effects. During the last years those structures have been intensively synthesized and modified to yield new synthetic molecules that can specifically inhibit the activity of dysregulated protein kinases in cancer cells. Herein, a series of newly synthesized isoquinolinamine (FX-1 to 8) and isoindoloquinazolinone (FX-9, FX-42, FX-43) compounds were evaluated in regards to their anti-leukemic potential on human B- and T-acute lymphoblastic leukemia (ALL) cells. Several biological effects were observed. B-ALL cells (SEM, RS4;11) were more sensitive against isoquinolinamine compounds than T-ALL cells (Jurkat, CEM). In SEM cells, metabolic activity decreased with 10 μM up to 26.7% (FX-3), 25.2% (FX-7) and 14.5% (FX-8). The 3-(p-Tolyl) isoquinolin-1-amine FX-9 was the most effective agent against B- and T-ALL cells with IC50 values ranging from 0.54 to 1.94 μM. None of the tested compounds displayed hemolysis on erythrocytes or cytotoxicity against healthy leukocytes. Anti-proliferative effect of FX-9 was associated with changes in cell morphology and apoptosis induction. Further, influence of FX-9 on PI3K/AKT, MAPK and JAK/STAT signaling was detected but was heterogeneous. Functional inhibition testing of 58 kinases revealed no specific inhibitory activity among cancer-related kinases. In conclusion, FX-9 displays significant antileukemic activity in B- and T-ALL cells and should be further evaluated in regards to the mechanisms of action. Further compounds of the current series might serve as templates for the design of new compounds and as basic structures for modification approaches.
Keywords: Acute lymphoblastic leukemia, Heterocycles, Isoquinolinamine, Isoindoloquinazolinone, Apoptosis, Kinase inhibitor
INTRODUCTION
Heterocyclic molecules act as highly functionalized scaffolds and have received considerable attention due to their diverse biological activity. Quinoline, quinazolinones and indole are well known nitrogen containing heterocyclic structures and occur in several natural products such as plants, fungi or marine organisms. It was demonstrated that compounds with those backbones possess anti-malarial (Vandekerckhove and D’hooghe, 2015), anti-bacterial (Naeem et al., 2016), anti-fungal (Musiol et al., 2010), anti-inflammatory (Mukherjee and Pal, 2013) or anti-tumoral activity (Mantu et al., 2016).
Therefore, the interest in such molecules has risen constantly and various procedures were established and evaluated for their synthesis and modification. Over the years several structural modifications were described and numerous hybrids were generated. Thereby, novel fused heterocyclic molecules with enhanced biological activity were identified which are applicable for the treatment of different diseases including cancer (Akhtar et al., 2017).
However, their specific modes of action are divers and a wide range of biological and pharmacological properties have been reported. Famous quinoline analogs such as actinomycin D, doxorubicin or mitoxantrone induce cytotoxicity through DNA intercalation or by the inhibition of topoisomerase II (Musiol, 2017). Further, indol(Yang et al., 2017), quinolone- (Solomon and Lee, 2011) as well as quinazolinones-(Hameed et al., 2018) based structures can act as kinase inhibitors by directly binding to ATP binding site of enzymes.
Newly generated compounds are often evaluated in regards of their potential application as pharmaceuticals. Early screening strategies for the initial characterization are established and usually performed with in vitro cell-based models or in silico tools. Besides conventional testing of standard cellular and molecular markers the identification of addressed functional proteins as kinases is key to elucidate a compounds acting mechanism (Martell et al., 2013; Cozza, 2017; Eastman, 2017). Ideally, the selectivity of new kinase inhibitors should be functionally evaluated at protein level for their potential to inhibit kinase-catalysed phosphotransfer of ATP to a substrate (Yanamandra et al., 2015).
A large number of FDA-approved kinase inhibitors such as Gefitinib (Kazandjian et al., 2016), Idelalisib (Miller et al., 2015) and Midostaurin (Stansfield and Pollyea, 2017) are characterized by quinolone, quinazolinones or indole backbones (Fig. 1). These compounds specifically inhibit kinase activity of epidermal growth factor receptor, isoform p110δ of the phosphoinositide 3-kinase (PI3K) as well as FMS-like tyrosine kinase 3 receptor and have been successfully introduced for the treatment of cancers including leukemia. Moreover, numerous heterocyclic pharmaceuticals with similar scaffolds are currently being evaluated in diverse preclinical and clinical studies (Paubelle et al., 2017; Wei and Tiong, 2017; Gao et al., 2018; Li et al., 2018). Previously, we and others have demonstrated that quinoline derivative CX-4945 is a potent inhibitor of casein kinase 2 (CK2) displaying anti-proliferative potential against acute lymphoblastic leukemia (ALL) cells by downregulating PI3K/AKT pathway activity (Buontempo et al., 2014; Richter et al., 2017). CK2 is a serine threonine kinase which phosphorylates a variety of substrates across numerous pathways including PI3K/AKT, JAK/STAT and NFkB. CK2 is frequently dysregulated in hematological neoplasia including acute B- and T-cell lymphoblastic leukemia (Piazza et al., 2012).
Fig. 1.

Chemical structures. (A) Heterocyclic isoquinoline, quinazolinones and indole backbones. (B) FDA-approved kinase inhibitors with quinoline (Gefitinib), quinazinolone (Idelalisib) or indole (Midostaurin) scaffolds.
Here, a series of newly synthesized fused heterocycle compounds (isoquinolinamines and isoindoloquinazolinones derivates) were evaluated for the first time in an attempt to evaluate molecules for their anti-proliferative potential against B- and T-ALL cells. The biological activity of the compounds was studied using cell viability assays and kinase activity screening methods. Our results reveal that isoquinolinamine FX-9 is the most active candidate and exhibits the most potent activity in B- and T-ALL cells.
MATERIALS AND METHODS
Compounds
Quinoline and quinazolinones molecules (Table 1, 2) were synthesized during the original synthetic methodologies studies (Shen et al., 2015, 2016; Feng and Wu, 2016). The purity of each compound was greater 99% and confirmed by gas chromatography-mass spectrometry and nuclear magnetic resonance (1H-NMR and 13C-NMR). The analytic data and NMR spectra were published in the synthesis manuscripts (Shen et al., 2015, 2016; Feng and Wu, 2016).
Table 1.
Structures of isoindoloquinazolinones
| ID | Structure | Chemical formula | Compound name | MW |
|---|---|---|---|---|
| FX-1 |
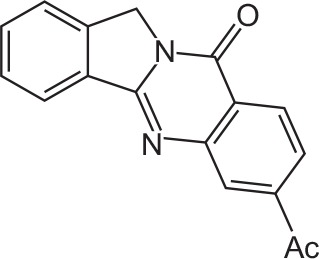
|
C17H12N2O2 | 7-Acetylisoindolo[1,2-b]quinazolin-10(12H)-one | 276.3 |
| FX-2 |
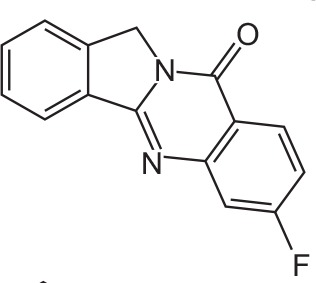
|
C15H9FN2O | 7-Fluoroisoindolo[1,2-b]quinazolin-10(12H)-one | 252.3 |
| FX-3 |
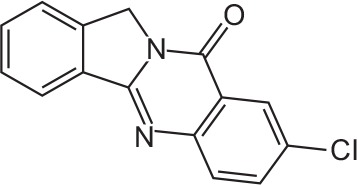
|
C15H9CIN2O | 8-Chloroisoindolo[1,2-b]quinazolin-10(12H)-one | 268.7 |
| FX-4 |
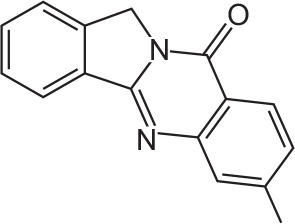
|
C16H12N2O | 7-Methylisoindolo[1,2-b]quinazolin-10(12H)-one | 248.3 |
| FX-5 |
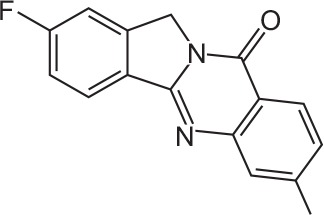
|
C16H11FN2O | 2-Fluoro-7-methylisoindolo[1,2-b]quinazolin-10(12H)-one | 266.3 |
| FX-6 |
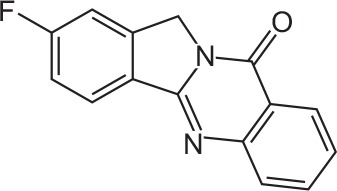
|
C15H9FN2O | 2-Fluoroisoindolo[1,2-b]quinazolin-10(12H)-one | 252.3 |
| FX-7 |
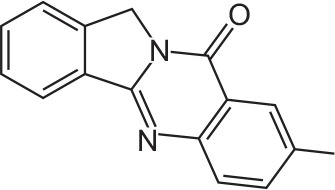
|
C16H12N2O | 8-Methylisoindolo[1,2-b]quinazolin-10(12H)-one | 248.3 |
| FX-8 |
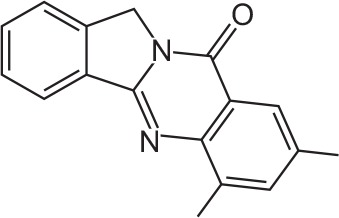
|
C17H14N2O | 6,8-Dimethylisoindolo[1,2-b]quinazolin-10(12H)-one | 262.3 |
Table 2.
Structures of isoquinolinamine
| ID | Structure | Chemical formula | Compound name | MW |
|---|---|---|---|---|
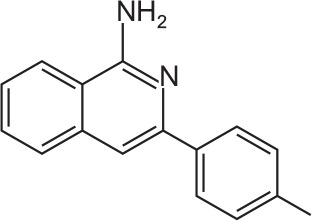
| FX-9 |
|
C16H14N2 | 3-(p-Tolyl)isoquinolin-1-amine | 234.3 |
| FX-42 |
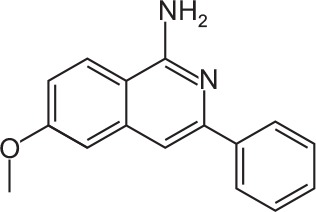
|
C16H14N2O | 6-Methoxy-3-phenylisoquinolin-1-amine | 250.3 |
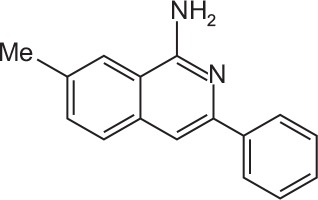
| FX-43 |
|
C16H14N2 | 7-Methyl-3-phenylisoquinolin-1-amine | 234.3 |
All compounds were dissolved in dimethyl sulfoxide (DMSO, Merck KGaA, Darmstadt, Germany) to a 10 mM stock solution and stored at −20°C. Working solutions were prepared freshly for experimental use.
Cell lines and cell culture experiments
The human ALL cell lines SEM and RS4;11 (both B-ALL) as well as Jurkat and CEM (both T-ALL) were purchased from DSMZ (Braunschweig, Germany) and cultured according to the manufacturer’s protocol. Media were supplemented with 10% heat-inactivated fetal bovine serum (Biochrom, Berlin, Germany) and 100 μg/ml penicillin and streptomycin (Biochrom, Berlin, Germany). Cells were maintained in a 5% CO2 humidified atmosphere at 37°C.
All compounds were preliminary evaluated for their anti-metabolic activities against ALL cell lines using WST-1 assay (Roche, Mannheim, Germany) as previously described (Kretzschmar et al., 2014). The assay is based on the reduction of the tetrazolium salt WST-1 to soluble formazan by mitochondrial dehydrogenases of the cells. The amount of formazan dye directly correlates to the number of metabolically active cells. Cells were seeded in 96-well plates (5×104 cells/well in triplicates) in 150 μl medium and compounds were added with a final concentration of 5 μM or 10 μM. Control cells were cultured in their medium containing DMSO in the same concentration as present in the compound exposed cells. Metabolic activity was assessed after an incubation time of 72 h. Then, 15 μl WST-1 were added to each well and incubated for additional 3 h (37°C, 5% CO2). Absorbance was measured on Glomax plate reader (Promega, Mannheim, Germany) at a wave length at 450 nm and a reference wave length at 620 nm. Metabolic activity was expressed as a percentage of DMSO-treated controls (=100%).
In addition, anti-leukemic activity of a representative compound was further evaluated on B- and T-All cell lines for up to 72 h with concentrations ranging from 0.5–5.0 μM. Proliferation was assessed by counting viable cells using trypan blue (Merck KGaA) dye exclusion. Apoptosis was analyzed by Annexin V FITC (BD Biosciences, Heidelberg, Germany) and propidium iodide PI (Sigma Aldrich, St. Louis, MO, USA) staining as previously described (Kretzschmar et al., 2014). Measurement of early apoptotic (Annexin V-FITC+ and PI–) and late apoptotic and necrotic cells (Annexin V FITC+ and PI+) was performed using flow cytometry on FACSCalibur instrument (Becton Dickinson, Heidelberg, Germany) and data were analyzed by CellQuest software (Becton Dickinson). Cell morphology was evaluated with May Grünwald Giemsa staining as previously described (Kretzschmar et al., 2014). Cells were analyzed by Evos XL Core light microscope (Life technologies, Darmstadt, Germany).
Hemolysis assay
Hemolytic activity of selected isoquinoline compounds was determined by hemoglobin release from whole blood cells as previously described (Maletzki et al., 2014). Briefly, whole blood of healthy donors (n=6) was seeded in 96-well plates and incubated with 5 μM FX-3, FX-7, FX-8 or FX-9 for 120 min. Positive control cells (=maximum lysis) were treated with 1% Sodium Dodecyl Sulfate (SDS, Merck KGaA). Following the incubation period, cell-free supernatants were transferred into a new 96-well plate and absorption was measured on Glomax plate reader (Promega) at 540 nm (reference wave length: 690 nm). Hemolytic activity was quantified according to the following formula: % hemolysis=((OD540 nm sample−OD540 nm buffer)/OD540 nm max−OD540 nm buffer))×100.
Cell viability assay on human leukocytes
Effects on leukocyte viability were analyzed by calcein-acetoxymethylester (Calcein-AM; Invitrogen, Darmstadt, Germany) staining. Human leukocytes from whole blood of six healthy donors were seeded in 96-well plates (1×105 cells/well in triplicates) after ficoll density centrifugation and incubated with 5 μM FX-3, FX-7, FX-8 or FX-9 for up to 72 h. DMSO-treated cells served as living cell control. Medium was removed after 48 h or 72 h, respectively. Calcein-AM (4 mM) was added and incubated for 30 min (37°C, 5% CO2). Analysis was performed on Glomax plate reader (Promega) at an excitation wavelength of 485 nm (emission 535 nm). For estimation of cell viability, the relative fluorescence intensities of Calcein-AM-stained DMSO-treated cells (=live control) were set to 100% and fluorescence intensities of samples were calculated. The fluorescence intensity is proportional to the number of viable cells.
CK2 kinase activity assay
Enzyme activity was determined with ADP-Glo-Kinase assay according to the manufacturer’s protocol (Promega). Briefly, FX-9 (0.1–5 μM), CX-4945 (0.1–5 μM) or DMSO was incubated with 50 ng recombinant human CK2α1 (Promega), 2.5 μg CK2α1 substrate (Promega), 10 μM ATP for 60 min at room temperature. The kinase reaction was terminated by ADP-Glo Reagent to deplete remaining ATP. In a finally step a conversion of ADP to ATP was performed with kinase detection reagent to generate luminescence using a luciferase/luciferin reaction. Luminescence was measured with the Glomax 96 well plate reader (Promega). The emitted light correlates with the amount of ADP generated in the kinase assay which is indicative of kinase activity.
Kinase activity screening
Kinase activity was determined using Eurofins’ KinaseProfilerTM Service (Diversity Panel, #50-015KP, Eurofins, Dundee, UK). Enzymatic activity was measured via substrate phosphorylation. A panel of 58 human kinases were evaluated at the Km for ATP and FX-9 was tested at 2.5 μM. Results are expressed as a percentage of the mean kinase activity in the positive control samples. The positive control value is considered to be 100%, and all test samples were measured in relation to this value. Values between 30% and 70% were defined as being partial hits, while results below 30% were considered as a strong hit.
Western Blot
Cells (1–5×106) were lysed using RIPA buffer (Cell Signaling, Danvers, MA, USA) and ultra sound exposure. Proteins were separated on Midi gels (Bio-Rad, Munich, Germany), blotted onto a PVDF membrane (Bio-Rad) using Trans-Blot® TurboTM Transfer System (Bio-Rad, 2.5 A, 25 V, 10 min), blocked in LI-COR blocking buffer (LI-COR Biotechnology, Lincoln, NE, USA) and detected via LI-COR Odyssey Imaging System (LICOR Biotechnology) and Image Studio Lite software (LI-COR Biotechnology). Antibodies are listed in Supplementary Table 1.
Statistical analysis
Results within each experiment were described using mean and standard deviation. Significance between control and treated was calculated using two tailed student’s t-test, Excel software, version 2010 (Microsoft Deutschland GmbH, Munich, Germany). A p-value <0.05 was considered to be significant. Calculation of IC50 values was performed using GNUPLOT package, version 5.2 (SourceForge Media, La Jolla, CA, USA).
RESULTS
Biological activities of isoindoloquinazolinones
Chemical structures of eight newly synthesized isoindoloquinazolinones (FX-1-8) are given in Table 1. Anti-metabolic effects of each compound (5 μM, 10 μM) were analyzed on two B- and two T-ALL cell lines after 72 h exposure time (Table 3). Three compounds (FX-3, FX-7 and FX-8) exhibited anti-leukemic potential in B-ALL cell lines indicated by a reduction of metabolic activity of more than 50% compared to DMSO-treated control cells. Here, strongest effects were observed with a concentration of 10 μM in SEM cells. Metabolic activities were reduced up to 26.7% (FX-3), 25.2% (FX-7) or 14.5% (FX-8). At a concentration of 10 μM FX-2 exhibited strongest effects in T-ALL cells. Metabolic activity decreased to 77.2% in CEM as well as to 54.3% in Jurkat cells.
Table 3.
Effects of isoindoloquinazolinone compounds on metabolic activity
| ID | SEM | RS4;11 | CEM | Jurkat | ||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| 5 μM | 10 μM | 5 μM | 10 μM | 5 μM | 10 μM | 5 μM | 10 μM | |
| FX-1 | 107.3% ± 4.7 | 97.7% ± 7.3 | 99.3% ± 2.9 | 94.4% ± 4.1 | 87.1% ± 4.5 | 83.9% ± 6.6 | 93.4% ± 12.1 | 68.5 ± 14.8 |
| FX-2 | 100.2% ± 3.1 | 69.8% ± 2.7 | 97.3% ± 1.2 | 79.8% ± 0.9 | 86.3% ± 4.9 | 77.2% ± 3.0 | 81.3% ± 13.8 | 54.3% ± 11.9 |
| FX-3 | 32.0% ± 3.0 | 26.7% ±4.3 | 73.8% ± 0.3 | 40.2% ± 3.7 | 76.2% ± 2.4 | 86.0% ± 4.9 | 57.9% ± 13.9 | 77.0% ± 1.2 |
| FX-4 | 96.8% ± 1.0 | 64.9% ± 3.5 | 91.2% ± 4.3 | 85.9% ± 4.5 | 101.8% ± 2.9 | 90.6% ± 2.5 | 90.4% ± 1.8 | 86.6% ± 0.8 |
| FX-5 | 91.7% ± 4.2 | 74.7% ± 5.1 | 91.4% ± 5.8 | 86.7% ± 7.1 | 101.0% ± 1.8 | 95% ± 3.1 | 94.0% ± 4.5 | 96.5% ± 2.1 |
| FX-6 | 72.1% ± 3.9 | 51.4% ± 7.6 | 76.6% ± 3.5 | 67.1% ± 11.5 | 93.2% ± 1.6 | 107.1% ± 4.3 | 104.6% ± 1.8 | 97.5% ± 7.2 |
| FX-7 | 52.1% ± 7.1 | 25.2% ± 3.7 | 61.3% ± 11.3 | 49.4% ± 7.6 | 96.1% ± 5.5 | 85.0% ± 3.6 | 91.4% ± 9.3 | 84.3% ± 10.3 |
| FX-8 | 23.3% ± 4.6 | 14.5% ± 2.2 | 61.5% ± 8.7 | 55.8% ± 10.0 | 97.0% ± 4.5 | 85.0% ± 1.9 | 95.2% ± 16.3 | 92.5% ± 14.0 |
Values represent metabolic activity expressed as mean ± standard deviation of one experiment performed in triplicates.
Biological activities of isoquinolinamines
Chemical structures of newly synthesized isoquinolinamines (FX-9, FX-42, FX-43) are displayed in Table 2. Anti-metabolic effects of the compounds were assessed in SEM and Jurkat cells after 72 h exposure time (Table 4). FX-9 exhibited the strongest effect against both cell lines. Herein, metabolic activities were reduced up to 1.2% in SEM and up to 10.4% in Jurkat (both 5 μM). Exposure of 10 μM FX-42 decreased metabolic activity in SEM cells to 24.5% and in Jurkat to 32.4%. No significant effects on metabolic activities in ALL cells were induced by FX-43.
Table 4.
Effects of isoquinolinamine compounds on metabolic activity
| ID | SEM | Jurkat | ||
|---|---|---|---|---|
|
|
|
|||
| 5 μM | 10 μM | 5 μM | 10 μM | |
| FX-9 | 1.2% ± 0.3 | 0.6% ± 0.2 | 10.4% ± 0.6 | 13.6% ± 2.3 |
| FX-42 | 66.7% ± 0.4 | 24.5% ± 0.3 | 88.4% ± 12.6 | 32.4% ± 1.0 |
| FX-43 | 94.7% ± 2.7 | 94.3% ± 0.6 | 106.9% ± 6.7 | 103.6% ± 1.3 |
Values represent metabolic activity expressed as mean ± standard deviation of one experiment performed in triplicates.
FX compounds exert no hemolytic and leukotoxic effects
In order to examine if the observed effects were specific for leukemia cells whole blood or leukocytes from six healthy donors were incubated with 5 μM FX-3,-7,-8 or -9 (Fig. 2). Hemolytic activity of FX-compounds was determined by hemoglobin release from whole blood cells after 120 min incubation (Fig. 2A). Minimal hemolysis with values below 20% was induced compared to the positive control (=maximal lysis). Viability of leukocytes was analyzed using a calcein-based assay. Exposure of FX-compounds for up to 72 h did not influence cell viability significantly compared to DMSO-treated cells (Fig. 2B). The amount of viable cells was above than 90% in all samples.
Fig. 2.

Hemolysis and lymphotoxicity assays. (A) Hemolytic activity of FX-compounds (5 μM) was determined by hemoglobin release from whole blood cells after 120 min incubation. Positive controls (=maximum lysis) were treated with 1% SDS. (B) Leukocytes were incubated in the presence of FX-compounds (5 μM) for a period of 48 and 72 h. Viability was analyzed using Calcein-AM and quantified in comparison to DMSO treated controls, which were set to be =100%. Results show data of six different healthy donors. Experiments were performed in triplicates. Values are given as the mean x-fold increase ± standard deviation.
FX-9 influences morphology and induces apoptosis
In addition, we selected FX-9 as best candidate and characterized the compound in more detail in regards to its anti-proliferative potential on B- and T-ALL cells. Therefore, cells were exposed to concentrations ranging from 0.1–5.0 μM over 72 h. A dose dependent effect of FX-9 on proliferation was observed in ALL cells (Fig. 3A). FX-9 inhibited cell proliferation with a 50% inhibitory concentration (IC50) ranging from 0.54 μM in RS4;11 cells to 1.94 μM in CEM cells (Table 5). B-ALL cell lines showed a significant inhibition of proliferation starting at a concentration of 0.5 μM. Proliferation was significantly reduced up to 69.5% in SEM as well as 60.0% in RS4;11 compared to control cells (=100%). In T-ALL cell lines proliferation was significantly reduced starting at a concentration of 2.5 μM. At this concentration proliferation was inhibited in Jurkat up to 16.3% and in CEM up to 24.1% compared to control cells (100%).
Fig. 3.

FX-9 inhibits cell proliferation and induces changes on ALL cell morphology. B- and T-ALL cells were exposed to FX-9 and DMSO for up to 72 h to evaluate cell proliferation, morphology and cell death. (A) FX-9 significantly inhibited cell proliferation of B- and T-ALL cells after 72 h. (B) Cellular morphology was assessed by May-Grünwald staining. Results are exemplary displayed for RS4;11 and CEM after 48 h. Cytomorphology was altered in both cell lines. FX-9 treatment induced an extensive cytoplasmic vacuolization (red arrow), karyopyknosis (black arrow), karyorrhexis (green arrow) and karyolysis (blue arrow). (C) The amount of apoptotic and necrotic cells (dead cells) in B- and T-ALL cell lines was analyzed using annexin V and propidium iodid staining after 72 h. FX-9 significantly induced apoptosis in all cell lines. (D) Western blot revealed that apoptosis was induced via caspase 3 pathway as indicated by an increased amount of cleaved caspase 3 protein in FX-9 treated SEM and RS4;11 cells after 48 h. Results are displayed as mean ± standard deviation of three independent experiments. Asterisk represents a statistically significance difference between FX-9 and DMSO treated cells with a *p<0.05.
Table 5.
IC50 values of FX-9 based on proliferation
| SEM | RS4;11 | Jurkat | CEM | |
|---|---|---|---|---|
| IC50 [μM] | 1.46 ± 0.01 | 0.54 ± 0.02 | 1.67 ± 0.03 | 1.94 ± 0.03 |
Values represent mean ± standard deviation of three separate experiments performed in triplicates.
To evaluate possible changes on cellular morphology cells were exposed to 1.0 μM or 2.5 μM of FX-9 for 48 h and were analyzed with light microscopy after May Grunwald staining (Fig. 3B). Cytomorphology was altered in B- and T-ALL cell lines as exemplary shown for RS4;11 and CEM cells. Exposure to 1.0 μM FX-9 led to an increased amount of vacuoles and karyopyknosis in RS4;11. Extensive cytoplasmic vacuolization, karyopyknosis, and karyolysis were induced with 2.5 μM FX-9 in RS4;11 as well as in CEM cells.
To examine whether FX-9 induces apoptosis we performed flow cytometry analyses using Annexin V and propidium iodide staining. Apoptosis is expressed as the total amount of early apoptotic and late apoptotic and necrotic cells. A dose dependent effect of FX-9 on cell death was observed in B-and T-ALL cells after 72 h of drug exposure (Fig. 3C, Supplementary Fig. 1). FX-9 significantly induced apoptosis in ALL cell lines compared to DMSO-treated cells. Apoptosis increased with 5 μM FX-9 in SEM up to 75.9% vs. 7.4% (DMSO), in RS4;11 up to 84.6% vs. 11.7% (DMSO), in Jurkat up to 48.7% vs. 18.7% (DMSO) as well as in CEM up to 90.4% vs. 7.3% (DMSO).
To further explore whether cells die via apoptosis, we examined caspase 3 expression using western blot. Apoptosis induction by caspase 3 was analyzed in SEM and RS4;11 cells and results are presented in Fig. 3D. After incubation for 48 h incubation an increased amount of cleaved caspase 3 was induced by 2.5 μM and 5.0 μM FX-9.
FX-9 influences PI3K/AKT and MAPK signaling
In order to characterize the effects of FX-9 on protein level western blot analysis was performed. In detail, PI3K/AKT, JAK/STAT and MAPK signaling were analyzed in SEM and RS4;11 cells (Fig. 4). After incubation of FX-9 for up to 48 h the expression of pAKT, pSTAT3, pERK and p-p38 was modified. Strongest effects were detectable at 48 h FX-9 exposure. In SEM cells FX-9 reduced pAKT, p-p38 and pSTAT3 level whereas in RS4;11 cells a dose dependent increase of pERK occurred.
Fig. 4.

FX-9 influences JAK/STAT, MAPK and PI3K/AKT pathway. SEM and RS4;11 cells were exposed to DMSO, 2.5 μM or 5.0 μM FX-9 for 48 h. For quantification, Image Studio Lite software (Version 5.2, LI-COR Biotechnology) was used to determine signal intensities of the protein bands. Protein bands were normalized to the GAPDH band of the respective sample and the ratio of phosphorylated forms to the corresponding total protein signal intensities was calculated. Values are expressed as ratio. DMSO control is considered as value 1 and samples were measured in relation to the control. (A) FX-9 decreased levels of p-STAT3 in SEM cells and increased pERK protein level in RS4;11 (B) FX-9 reduced levels of p-AKT and p-p38 in SEM cells. Exemplary results for SEM and RS4;11 cells are displayed. GAPDH was used as loading control.
FX-9 does not inhibit CK2 activity
FX-9 is a quinolinamine analog and displays chemical structure similarities to CX-4945 a known inhibitor of the CK2 enzyme. In order to investigate if FX-9 induced effects are mediated via CK2 inhibition we analyzed CK2 enzyme activity using a luminescent ADP detection system (Fig. 5). In the presence of 10 μM FX-9 activity of CK2 was not changed significantly. In contrast, CK2 enzyme activity decreased significantly with 1 μM CX-4945 up to 3.4 ± 0.3% compared to DMSO (100%).
Fig. 5.

FX-9 does not inhibit CK2 activity. Recombinant human CK2 was incubated with CK2α1 substrate, ATP and increasing concentrations of FX-9 or CX-4945. Reduction of CK2 enzyme activity was induced with 0.1 μM CX-4945. No changes of CK2 enzyme activity were observed with FX-9. Results are displayed as mean and standard deviation of three technical replicates of one experiment.
In addition, the activities of further kinases were analyzed in the presence of FX-9 using a radiometric-based assay. Therefore, a commercially designed panel of human kinases associated with MAPK, PI3K/AKT or cell cycle pathway was selected. Activities were determined against human kinases at their Km for ATP and with 2.5 μM FX-9. Results of this screening are summarized in Table 6. Exposure of 2.5 μM FX-9 did not influence the activity of any of the selected kinases.
Table 6.
Results of kinase activity screening
| Kinase | ATP Km (μM) | Kinase activity (% control) | SD |
|---|---|---|---|
| Abl | 45 | 96 | 2 |
| ALK | 200 | 112 | 10 |
| AMPKα1 | 45 | 98 | 1 |
| ASK1 | 200 | 101 | 1 |
| Aurora-A | 15 | 92 | 0 |
| CaMKI | 200 | 100 | 4 |
| CDK1/cyclinB | 45 | 97 | 1 |
| CDK2/cyclinA | 45 | 90 | 9 |
| CDK6/cyclinD3 | 200 | 103 | 5 |
| CDK7/cyclinH/MAT1 | 90 | 91 | 0 |
| CDK9/cyclin T1 | 45 | 86 | 3 |
| CHK1 | 90 | 94 | 1 |
| CK1γ1 | 15 | 98 | 5 |
| CK2α2 | 10 | 95 | 9 |
| c-RAF | 120 | 92 | 1 |
| DRAK1 | 10 | 81 | 7 |
| eEF-2K | 15 | 86 | 4 |
| EGFR | 10 | 101 | 3 |
| EphA5 | 10 | 98 | 5 |
| EphB4 | 10 | 83 | 5 |
| Fyn | 70 | 97 | 7 |
| GSK3β | 15 | 88 | 5 |
| IGF-1R | 200 | 89 | 10 |
| IKKα | 10 | 91 | 1 |
| IRAK4 | 200 | 95 | 1 |
| JAK2 | 45 | 111 | 4 |
| KDR | 90 | 83 | 3 |
| LOK | 120 | 101 | 4 |
| Lyn | 70 | 87 | 9 |
| MAPKAP-K2 | 90 | 87 | 6 |
| MEK1 | 10 | 90 | 2 |
| MLK1 | 45 | 84 | 3 |
| Mnk2 | 120 | 90 | 11 |
| MSK2 | 45 | 89 | 1 |
| MST1 | 90 | 87 | 2 |
| mTOR | 70 | 100 | 2 |
| NEK2 | 120 | 106 | 0 |
| p70S6K | 15 | 85 | 4 |
| PAK2 | 90 | 88 | 1 |
| PDGFRβ | 200 | 99 | 1 |
| Pim-1 | 90 | 99 | 0 |
| PKA | 10 | 92 | 0 |
| PKBα | 155 | 98 | 4 |
| PKCα | 45 | 99 | 1 |
| PKCθ | 15 | 96 | 7 |
| PKG1α | 90 | 98 | 11 |
| Plk3 | 70 | 99 | 2 |
| PRAK | 15 | 79 | 4 |
| ROCK-I | 70 | 93 | 4 |
| Rse | 45 | 92 | 3 |
| Rsk1 | 45 | 95 | 5 |
| SAPK2a | 90 | 98 | 2 |
| SRPK1 | 10 | 94 | 4 |
| TAK1 | 45 | 87 | 1 |
| PI3 Kinase (p110b/p85a) | 200 | 97 | 1 |
| PI3 Kinase (p120g) | 100 | 98 | 2 |
| PI3 Kinase (p110d/p85a) | 200 | 100 | 1 |
| PI3 Kinase (p110a/p85a) | 200 | 101 | 1 |
Human kinases were evaluated at the Km for ATP and FX-9 was tested at 2.5 μM. Kinase activities are expressed as a percentage of the mean kinase activity in the positive control samples (=100%) Data interpretation of kinase activity: values <30% means a strong hit; values between 30% and 70% means a partial hit. Values >70% means no hit.
DISCUSSION
The aim of the current study was to evaluate the biological activity of eleven newly synthesized isoindoloquinazolinones (FX-1 to FX-8) and isoquinolinamines (FX-9, FX-42, FX-43) compounds against human ALL cells. The FX compounds are nitrogen containing heterocyclic structures with quinoline, quinazolinones or indole scaffolds that have been recently synthesized (Shen et al., 2015, 2016; Feng and Wu, 2016). Those core structures have been extensively examined in various neoplastic cells types and demonstrated anti-proliferative activity (He et al., 2003; Dadashpour and Emami, 2018; Mphahlele et al., 2018). Here, we studied for the first time the efficacy of these novel synthesized small molecules in ALL cells.
ALL is a hematological neoplasia characterized by clonal accumulation of B or T lymphoblastic precursor cells. The outcome of ALL patients depends on disease-related or patient-related factors including age, genetic alterations or immuno-phenotype (Terwilliger and Abdul-Hay, 2017). Therefore, the identification of new agents which have anti-leukemic potential is still of interest.
In the present study, two B-ALL cell lines (SEM and RS4;11) and two T-ALL cell lines (Jurkat, CEM) with different cell doubling times and genetic alterations were selected as cell line-based models to evaluate the FX-compounds. These cell lines represent ALL subtypes associated with a poor clinical outcome. The B-ALL cells are characterized by mixed lineage leukemia (MLL) gene rearrangement [t(4;11)] (Stumpel et al., 2009) while T-ALL cell lines are characterized by complex aberrant tetraploid karyotypes and numerous genetic mutations (Schneider et al., 1977; Moore et al., 1985). Furthermore, all cell lines exhibit aberrantly activated PI3K/AKT (Schult et al., 2010; Cani et al., 2015) and MAPK (Cani et al., 2015) signaling pathway.
Our results demonstrated that the metabolic activity in B-ALL cells was more reduced after isoindoloquinazolinones exposure than in T-ALL cell lines. Best effects were induced with FX-3, FX-7 and FX-8. Here, SEM cells were more sensitive against these FX-compounds than RS4;11 cells. The metabolic activity was more reduced with FX-8. Interestingly, either FX-3, FX-7 nor FX-8 showed biological activity in T-ALL cell lines Jurkat and CEM. Our results revealed that these three compounds exhibit a subtype specific anti-leukemic potential.
The compounds FX-1 to FX-8 are isoindolo[1;2-b]quinazolin-10 molecules which are modified with aryl halide (Cl, F) or functional groups such as acetyl or methyl moieties. The addition of a methyl group or fluoride to isoindoloquinazolinones appears to influences the biological activity in B-ALL cells. Thereby, the position and the number of methyl groups are potentially of importance as demonstrated by FX-4, FX-7 and FX-8. The addition of chlor or a acetyl group to the isoindoloquinazolinones molecule (FX-1, FX-3) had no impact on the metabolic activity of ALL cells. Karki et al (2009) also demonstrated that modification of quinoxaline structures influences antileukemic cell effects. The authors showed that cytostatic activity was dependent on substituents and their position. They identified that quinoxaline derivatives with methyl or halogen groups at position 4th or at 9th exhibited strong anticancer activity.
Further, biological activity of three isoquinolinamines (FX-9, FX-42, FX-43) was evaluated against ALL cells, exemplarily on SEM and Jurkat cells. Here, metabolic activity decreased dose dependently with FX-9 and FX-42 in both cell lines and FX-9 exhibited strongest effects. No effect on metabolic activity was observed with FX-43.
The compounds FX-9, FX-42 and FX-43 are characterized by an isoquinolinamine core structure which is linked to a modified phenyl group [(3-8p-tolyl), 6-methyox-3phenyl or 7-methyl-3-phenyl). Our first screening by analyzing metabolic activity revealed that FX-9 was the most active compound against B- and T-ALL cells. Therefore, this compound was selected to characterize the biological effect in more detail.
Interestingly, FX-9-induced effects on healthy blood cells could be excluded. Here, neither cytotoxicity nor hemolytic activity was inducible. Further, our results indicated that FX-9 mediated anti-metabolic effects in ALL cells were associated with inhibition of cell proliferation and morphological changes. Cytoplasmic vacuolization, karyopyknosis and karyolysis after FX-9 exposure on B- and T-ALL cells occurred and were distinct signs for apoptosis (Hotchkiss et al., 2009). By further analyses of phosphatidylserine expression on cell surface using Annexin V staining as well as the detection of cleaved caspase 3 protein demonstrated that FX-9 is an apoptosisinducing compound for ALL cells.
FX-9 is a fused ring structure with two heterocycle molecules (isoquinoline amine, phenyl ring). The quinoline structure is often present in numerus kinase inhibitors (Solomon and Lee, 2011). Furthermore, it has been demonstrated that isoquinoline modified structures have a high affinity to DNA (Musiol, 2017).
One of such quinolin-based molecule is CX-4945. CX-4945 blocks CK-2 activity and potently downregulates PI3K/AKT, JAK/STAT and NFkB signaling (Chon et al., 2015). Gefitinib also has a quinoline moiety. Gefitinib is a selective inhibitor of the epidermal growth factor receptor (EGFR) which prevents subsequent activation of the downstream signaling cascades that stimulate cell proliferation, differentiation, survival, and migration (Rahman et al., 2014). Both drugs are approved as type I inhibitors and ATP competitors that binds to the active conformation of the kinase in the ATP pocket (Cozza et al., 2012; Wu et al., 2015).
We hypothesized that FX-9 mediated effects on ALL cells can also be explained by potent inhibition of dysregulated signaling. We and others have shown that ALL cells can be efficiently targeted by specific inhibition of PI3K, mTOR or CK2 (Schult et al., 2012; Neri et al., 2014; Buontempo et al., 2017). In fact, our results showed that FX-9 did not directly inhibit CK2 enzyme activity as we had hypothesized. However, changes in the expression of key signaling proteins like pAKT, pSTAT3, pERK or p-p38 have been detected but the modulation of the proteins were heterogeneous between SEM and RS4;11 cells. In SEM cells FX-9 reduced pAKT and pSTAT3 signaling whereas in RS4;11 cells a dose dependent increase of pERK occurred. Both cell lines exhibited different basal level of pERK and pSTAT3. Therefore it is possible that signaling varies through activation of feedback loops after FX-9 exposure.
Additionally, we aimed to find a specific target for FX-9 by performing a kinase activity screening on a panel of 58 key kinases for cancer. Our results showed that none of the tested enzymes were inhibited in their activity. Based on these results we cannot definitely exclude that FX-9 is a kinase inhibitor. The human genome encodes 568 protein kinases and most of them are associated with cancer initiation and progression (Ardito et al., 2017). In our study we only analyzed the activity of 58 kinases.
However, it has also been shown that the bioactivity of quinoline-based structures is the result of DNA binding and the subsequent inhibition of DNA-dependent enzymes for examples topoisomerases (Musiol, 2017). Therefore, FX-9 associated cytotoxicity might also be explained by DNA intercalation and this has to be clarified in future studies.
In conclusion, a series of newly synthesized isoindoloquin-azolinone and isoquinolinamine compounds have been analyzed for the first time in regards to their antileukemic activity. The results demonstrated that the compound 3-(p-tolyl) isoquinolin-1-amine named as FX-9 showed the strongest anti-proliferative potential towards ALL cells and displayed no cytotoxicity against healthy blood cells. Based on these results further research should be carried out to investigate the detailed mechanisms of action. In addition, compounds from this series might serve as templates for the future design of new compounds and as basic structure for modification approaches.
Acknowledgments
This work was supported by FORUN grant of Rostock University Medical Center as part of the InterAKT initiative.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
- Akhtar MJ, Yar MS, Khan AA, Ali Z, Haider MR. Recent advances in the synthesis and anticancer activity of some molecules other than nitrogen containing heterocyclic moeities. Mini Rev Med Chem. 2017;17:1602–1632. doi: 10.2174/1389557516666161031121639. [DOI] [PubMed] [Google Scholar]
- Ardito F, Giuliani M, Perrone D, Troiano G, Lo Muzio L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (review) Int J Mol Med. 2017;40:271–280. doi: 10.3892/ijmm.2017.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buontempo F, Orsini E, Martins LR, Antunes I, Lonetti A, Chiarini F, Tabellini G, Evangelisti C, Melchionda F, Pession A, Bertaina A, Locatelli F, McCubrey JA, Cappellini A, Barata JT, Martelli AM. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: targeting the unfolded protein response signaling. Leukemia. 2014;28:543–553. doi: 10.1038/leu.2013.349. [DOI] [PubMed] [Google Scholar]
- Buontempo F, McCubrey JA, Orsini E, Ruzzene M, Cappellini A, Lonetti A, Evangelisti C, Chiarini F, Evangelisti C, Barata JT, Martelli AM. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia. 2017;32:1–10. doi: 10.1038/leu.2017.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani A, Simioni C, Martelli AM, Zauli G, Tabellini G, Ultimo S, McCubrey JA, Capitani S, Neri LM. Triple Akt inhibition as a new therapeutic strategy in T-cell acute lymphoblastic leukemia. Oncotarget. 2015;6:6597–6610. doi: 10.18632/oncotarget.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon HJ, Bae KJ, Lee Y, Kim J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front Pharmacol. 2015;6:70. doi: 10.3389/fphar.2015.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozza G. The development of CK2 inhibitors: from traditional pharmacology to in silico rational drug design. Pharmaceuticals. 2017;10:26. doi: 10.3390/ph10010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozza G, Pinna LA, Moro S. Protein kinase CK2 inhibitors: a patent review. Expert Opin Ther Pat. 2012;22:1081–1097. doi: 10.1517/13543776.2012.717615. [DOI] [PubMed] [Google Scholar]
- Dadashpour S, Emami S. Indole in the target-based design of anticancer agents: a versatile scaffold with diverse mechanisms. Eur J Med Chem. 2018;150:9–29. doi: 10.1016/j.ejmech.2018.02.065. [DOI] [PubMed] [Google Scholar]
- Eastman A. Improving anticancer drug development begins with cell culture: misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget. 2017;8:8854–8866. doi: 10.18632/oncotarget.12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J-B, Wu X-F. Potassium tert-butoxide-promoted synthesis of 1-aminoisoquinolines from 2-methylbenzonitriles and benzonitriles under catalyst-free conditions. Adv Synth Catal. 2016;358:2179–2185. doi: 10.1002/adsc.201600169. [DOI] [Google Scholar]
- Gao X, Cen L, Li F, Wen R, Yan H, Yao H, Zhu S. Oral administration of indole substituted dipyrido[2, 3-d]pyrimidine derivative exhibits anti-tumor activity via inhibiting AKT and ERK1/2 on hepatocellular carcinoma. Biochem Biophys Res Commun. 2018;505:761–767. doi: 10.1016/j.bbrc.2018.09.120. [DOI] [PubMed] [Google Scholar]
- Hameed A, Al-Rashida M, Uroos M, Ali SA, Arshia, Ishtiaq M, Khan KM. Quinazoline and quinazolinone as important medicinal scaffolds: a comparative patent review (2011–2016) Expert Opin Ther Pat. 2018;28:281–297. doi: 10.1080/13543776.2018.1432596. [DOI] [PubMed] [Google Scholar]
- He L, Chang HX, Chou TC, Savaraj N, Cheng CC. Design of antineoplastic agents based on the “2-phenylnaphthalene-type” structural pattern - synthesis and biological activity studies of 11H-indolo[3.2-c]quinoline derivatives. Eur. J. Med. Chem. 2003;38:101–107. doi: 10.1016/S0223-5234(02)01420-4. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361:1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki SS, Hazare R, Kumar S, Bhadauria VS, Balzarini J, De Clercq E. Synthesis, anticancer and cytostatic activity of some 6H-indolo[2,3-b]quinoxalines. Acta Pharm. 2009;59:431–440. doi: 10.2478/v10007-009-0040-9. [DOI] [PubMed] [Google Scholar]
- Kazandjian D, Blumenthal GM, Yuan W, He K, Keegan P, Pazdur R. FDA approval of gefitinib for the treatment of patients with metastatic EGFR mutation-positive non-small cell lung cancer. Clin Cancer Res. 2016;22:1307–1312. doi: 10.1158/1078-0432.CCR-15-2266. [DOI] [PubMed] [Google Scholar]
- Kretzschmar C, Roolf C, Langhammer T-S, Sekora A, Pews-Davtyan A, Beller M, Frech MJ, Eisenlöffel C, Rolfs A, Junghanss C. The novel arylindolylmaleimide PDA-66 displays pronounced antiproliferative effects in acute lymphoblastic leukemia cells. BMC Cancer. 2014;14:71. doi: 10.1186/1471-2407-14-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Luo M, Cai B, Haroon-Ur-Rashid, Huang M, Jiang J, Wang L, Wu L. Design, synthesis, biological evaluation and structure-activity relationship of sophoridine derivatives bearing pyrrole or indole scaffold as potential antitumor agents. Eur J Med Chem. 2018;157:665–682. doi: 10.1016/j.ejmech.2018.08.021. [DOI] [PubMed] [Google Scholar]
- Maletzki C, Klier U, Marinkovic S, Klar E, Andrä J, Linnebacher M. Host defense peptides for treatment of colorectal carcinoma - a comparative in vitro and in vivo analysis. Oncotarget. 2014;5:4467–4479. doi: 10.18632/oncotarget.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantu D, Antoci V, Moldoveanu C, Zbancioc G, Mangalagiu II. Hybrid imidazole (benzimidazole)/pyridine (quinoline) derivatives and evaluation of their anticancer and antimycobacterial activity. J Enzyme Inhib Med Chem. 2016;31:96–103. doi: 10.1080/14756366.2016.1190711. [DOI] [PubMed] [Google Scholar]
- Martell RE, Brooks DG, Wang Y, Wilcoxen K. Discovery of novel drugs for promising targets. Clin Ther. 2013;35:1271–1281. doi: 10.1016/j.clinthera.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Miller BW, Przepiorka D, de Claro RA, Lee K, Nie L, Simpson N, Gudi R, Saber H, Shord S, Bullock J, Marathe D, Mehrotra N, Hsieh LS, Ghosh D, Brown J, Kane RC, Justice R, Kaminskas E, Farrell AT, Pazdur R. FDA approval: idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin Cancer Res. 2015;21:1525–1529. doi: 10.1158/1078-0432.CCR-14-2522. [DOI] [PubMed] [Google Scholar]
- Moore DE, Weise K, Zawydiwski R, Thompson EB. The karyotype of the glucocorticoid-sensitive, lymphoblastic human T-cell line CCRF-CEM shows a unique deleted and inverted chromosome 9. Cancer Genet Cytogenet. 1985;14:89–94. doi: 10.1016/0165-4608(85)90219-5. [DOI] [PubMed] [Google Scholar]
- Mphahlele M, Mmonwa M, Aro A, McGaw L, Choong Y. Synthesis, biological evaluation and molecular docking of novel indole-aminoquinazoline hybrids for anticancer properties. Int J Mol Sci. 2018;19:2232. doi: 10.3390/ijms19082232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Pal M. Medicinal chemistry of quinolines as emerging anti-inflammatory agents: an overview. Curr Med Chem. 2013;20:4386–4410. doi: 10.2174/09298673113209990170. [DOI] [PubMed] [Google Scholar]
- Musiol R. An overview of quinoline as a privileged scaffold in cancer drug discovery. Expert Opin Drug Discov. 2017;12:583–597. doi: 10.1080/17460441.2017.1319357. [DOI] [PubMed] [Google Scholar]
- Musiol R, Serda M, Hensel-Bielowka S, Polanski J. Quinoline-based antifungals. Curr Med Chem. 2010;17:1960–1973. doi: 10.2174/092986710791163966. [DOI] [PubMed] [Google Scholar]
- Naeem A, Badshah S, Muska M, Ahmad N, Khan K. The current case of quinolones: synthetic approaches and antibacterial activity. Molecules. 2016;21:268. doi: 10.3390/molecules21040268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri LM, Cani A, Martelli AM, Simioni C, Junghanss C, Tabellini G, Ricci F, Tazzari PL, Pagliaro P, McCubrey JA, Capitani S. Targeting the PI3K/Akt/mTOR signaling pathway in B-precursor acute lymphoblastic leukemia and its therapeutic potential. Leukemia. 2014;28:739–748. doi: 10.1038/leu.2013.226. [DOI] [PubMed] [Google Scholar]
- Paubelle E, Zylbersztejn F, Thomas X. The preclinical discovery of vosaroxin for the treatment of acute myeloid leukemia. Expert Opin Drug Discov. 2017;12:747–753. doi: 10.1080/17460441.2017.1331215. [DOI] [PubMed] [Google Scholar]
- Piazza F, Manni S, Ruzzene M, Pinna LA, Gurrieri C, Semenzato G. Protein kinase CK2 in hematologic malignancies: reliance on a pivotal cell survival regulator by oncogenic signaling pathways. Leukemia. 2012;26:1174–1179. doi: 10.1038/leu.2011.385. [DOI] [PubMed] [Google Scholar]
- Rahman AFMM, Korashy HM, Kassem MG. Gefitinib. Profiles Drug Subst Excip Relat Methodol. 2014;39:239–264. doi: 10.1016/B978-0-12-800173-8.00005-2. [DOI] [PubMed] [Google Scholar]
- Richter A, Roolf C, Sender S, Kong W, Knübel G, Sekora A, Gladbach YS, Hamed M, Fuellen G, Vollmar B, Jeremias I, Panse JP, Escobar HM, Junghanss C. Casein kinase II inhibition by CX-4945 and epigenetic modulation by decitabine demonstrate significant antiproliferative activity as single agents as well as in combination in acute b-lymphoblastic leukemia cells. Blood. 2017;130:3887. [Google Scholar]
- Schneider U, Schwenk HU, Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int. J Cancer. 1977;19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- Schult C, Dahlhaus M, Ruck S, Sawitzky M, Amoroso F, Lange S, Etro D, Glass A, Fuellen G, Boldt S, Wolkenhauer O, Neri LM, Freund M, Junghanss C. The multikinase inhibitor Sorafenib displays significant antiproliferative effects and induces apoptosis via caspase 3, 7 and PARP in B- and T-lymphoblastic cells. BMC Cancer. 2010;10:560. doi: 10.1186/1471-2407-10-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schult C, Dahlhaus M, Glass A, Fischer K, Lange S, Freund M, Junghanss C. The dual kinase inhibitor NVP-BEZ235 in combination with cytotoxic drugs exerts anti-proliferative activity towards acute lymphoblastic leukemia cells. Anticancer Res. 2012;32:463–474. [PubMed] [Google Scholar]
- Shen C, Man NYT, Stewart S, Wu X-F. Palladium-catalyzed dicarbonylative synthesis of tetracycle quinazolinones. Org Biomol Chem. 2015;13:4422–4425. doi: 10.1039/C5OB00368G. [DOI] [PubMed] [Google Scholar]
- Shen C, Spannenberg A, Wu X-F. Palladium-catalyzed carbonylative four-component synthesis of thiochromenones: the advantages of a reagent capsule. Angew Chemie Int Ed. 2016;55:5067–5070. doi: 10.1002/anie.201600953. [DOI] [PubMed] [Google Scholar]
- Solomon VR, Lee H. Quinoline as a privileged scaffold in cancer drug discovery. Curr Med Chem. 2011;18:1488–1508. doi: 10.2174/092986711795328382. [DOI] [PubMed] [Google Scholar]
- Stansfield LC, Pollyea DA. Midostaurin: a new oral agent targeting FMS-like tyrosine kinase 3-mutant acute myeloid leukemia. Pharmacother J Hum Pharmacol Drug Ther. 2017;37:1586–1599. doi: 10.1002/phar.2039. [DOI] [PubMed] [Google Scholar]
- Stumpel DJPM, Schneider P, van Roon EHJ, Boer JM, de Lorenzo P, Valsecchi MG, de Menezes RX, Pieters R, Stam RW. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood. 2009;114:5490–5498. doi: 10.1182/blood-2009-06-227660. [DOI] [PubMed] [Google Scholar]
- Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7:e577. doi: 10.1038/bcj.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove S, D’hooghe M. Quinoline-based anti-malarial hybrid compounds. Bioorg Med Chem. 2015;23:5098–5119. doi: 10.1016/j.bmc.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Wei AH, Tiong IS. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood. 2017;130:2469–2474. doi: 10.1182/blood-2017-08-784066. [DOI] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Yanamandra M, Mitra S, Giri A. Development and application of PI3K assays for novel drug discovery. Expert Opin Drug Discov. 2015;10:171–186. doi: 10.1517/17460441.2015.997205. [DOI] [PubMed] [Google Scholar]
- Yang D, Tong D, Zhang Q, Wang Y, Sun J, Zhang F, Zhao G. Design, synthesis, and evaluation of novel Akt1 inhibitors based on an indole scaffold. Chem Biol Drug Des. 2017;90:791–803. doi: 10.1111/cbdd.13000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.