

Abstract
X-linked carriers of chronic granulomatous disease (CGD) may become phenotypically affected if substantial skewing from lyonisation occurs. We describe a 73-year-old female carrier with an overt CGD phenotype due to skewed lyonisation, complicated by macrophage activation syndrome (MAS)/haemophagocytic lymphohistiocytosis (HLH) secondary to Burkholderiacepacia complex septicaemia that was successfully treated with a combination of three antibiotics, an antifungal, granulocyte colony stimulating factor, intravenous immune globulin (IVIG) and ciclosporin. Fully phenotypic immunodeficiency is possible in X-linked CGD carriers when skewed lyonisation occurs, rendering such patients to all the same sequelae of CGD such as MAS/HLH. MAS/HLH should be thoroughly excluded when evaluating ‘cepacia syndrome’ in non-CGD patients.
Keywords: immunology, pneumonia (respiratory medicine)
Background
Chronic granulomatous disease (CGD) is a rare, monogenic condition with an incidence of approximately 1 in 250 000 live births and is heterogeneous in phenotype.1 It is due to a defective nicotinamide-adenine dinucleotide-phosphate oxidase enzyme complex: NADPH. NADPH oxidase is essential for neutrophil respiratory (oxidative) burst. It catalyses the first reaction in the respiratory burst pathway, allowing the generation of superoxide oxygen anions (O2 −) from molecular oxygen (O2).1 2 The clinical hallmarks of the disease include recurrent gram positive bacterial abscesses, gram negative bacterial (including Burkholderia) sepsis and recurrent invasive fungal infections.3 4 NADPH is a large enzymatic complex with multiple components; genetic defects in the genes that encode any of the subunits can give rise to CGD.5 Most of the genes responsible are inherited in an autosomal recessive fashion, except for CYBB which encodes the gp91phox component; this gene is present on the X chromosome, making inheritance of this defect X-linked. X-linked carriers for CGD are prone to immune dysregulation and immunodeficiency if utilisation of the X chromosome that carries the aberrant CYBB mutation is preferentially selected, a process known as skewed lyonisation.6
Macrophage activation syndrome (MAS)/haemophagocytic lymphohistiocytosis (HLH) secondary to Burkholderia cepacia complex infection is a rare, but well-recognised complication of CGD.7–9 MAS/HLH is characterised by increased and circuitous stimulating cytokine production by macrophages and lymphocytes, including natural killer cells and cytotoxic T lymphocytes, which results in a hyperinflammatory state leading to cytopenias, fevers, rash, hepatosplenomegaly and end-organ damage. Treatment of MAS/CGD involves a combination of targeted therapy to remove the precipitant, as well as controlling inflammation with immunomodulation and immunosuppression.10
B. cepacia complex is a group of closely, phylogenetically related phytopathogen bacteria that are uncommonly associated with disease in the otherwise healthy human. It can cause a severe and almost universally fatal infection in patients with cystic fibrosis (CF); this has previously been described as the ‘cepacia syndrome’.11 B. cenocepacia is associated with the greatest risk of ‘cepacia syndrome’ out of all the members of the B. cepacia complex. Generally, CF patients are colonised with B. cepacia for an asymptomatic period before the ‘cepacia syndrome’ occurs, although the timing between colonisation and infection is variable.12 The pathophysiology of the ‘cepacia syndrome’ is incompletely understood. Furthermore, only few cases of successfully managed ‘cepacia syndrome’ exist.13–17
MAS/HLH due to B. cepacia septicaemia in an X-linked carrier of CGD with skewed lyonisation has not been described previously. We detail such a case that was successfully managed and compare this with the interventions used successfully in treating ‘cepacia syndrome’ in CF.
Case presentation
A 73-year-old acutely unwell woman, who was previously known to be an X-linked CGD carrier with a distant history of polymyalgia rheumatica (PMR), was evaluated by our service. Six months prior to admission, she had had multiple recurrent lower respiratory tract infections. Each time she managed as an outpatient with oral antibiotics, although she required hospital admission for parenteral antibiotics on two occasions in 5 weeks prior to her acute deterioration. She had been diagnosed with a cryptogenic right-sided pneumonia with high fevers; this was despite exhaustive investigation on each occasion for a microbial pathogen, including two bronchoscopies. The latter episode was also managed with oral glucocorticosteroids (prednisone, 15 mg daily).
She eventually deteriorated with respiratory compromise with increasing shortness of breath, headaches, scalp tenderness and fevers despite an increased dose of prednisone at 25 mg/day. Given her history of PMR, a temporal artery biopsy was collected, but revealed no diagnostic features of vasculitis. A lumbar puncture was performed, although cerebrospinal fluid analysis was bland. An initial CT chest showed prominent right middle lobe consolidation (figure 1), although no further pathogens were isolated by further bronchoscopy and bronchial wash.
Figure 1.

Axial slice of CT chest with extensive right-sided consolidation.
Subsequent blood cultures, however, were positive for B. cepacia complex and her antibiotics were later changed to a combination of three antimicrobials: ceftazidime (2 g two times per day) and trimethoprim/sulfamethoxazole (5+25 mg/kg four times a day) given intravenously and tobramycin (160 mg two times per day) given via the nebulised route. Treatment dose voriconazole (300 mg two times per day) was also given intravenously. It was prescribed empirically as a fungal chest infection could not be reasonably excluded. Other antibiotics, such as piperacillin/tazobactam, meropenem, azithromycin and doxycyclin, were given in different combinations prior to the diagnosis of B. cepacia as the causative pathogen, but rationalised quickly thereafter. In addition to antimicrobial medications, she was also administered granulocyte colony stimulating factor (filgrastim 300 µg alternate days) in an attempt to increase her total (including phenotypically normal) neutrophil count.
Despite the increased antibiotics, her shortness of breath and fevers acutely deteriorated into frank septic shock requiring admission to the intensive care unit. She was able to be managed with high flow nasal oxygen and intravenous vasopressor support.
Investigations
Suspicion of MAS was considered given her acutely inflammatory deterioration (C reactive protein 299.7 mg/L, Reference Range (RR) <5, erythrocyte sedimentation rate 106 mm in 1 hour (RR <35), increasing transaminases (alanine transaminase 126 U/L RR 5–55, aspartate transaminase (AST) 197 U/L RR 5–55, respectively) along with unexplained thrombocytopenia and non-haemolytic anaemia (nadir platelet count and haemoglobin were 83×109/L (RR 150−400×109/L) and 75 g/L (120–150 g/L), respectively); a screening serum ferritin returned at 10 903 µg/L (RR 20–300) and she was also found to have new onset hypertriglyceridaemia at 2.3 mmol/L (RR <2.0). A bone marrow aspirate revealed a hypercellular marrow with reactive changes and prominent haemophagocytosis (figure 2).
Figure 2.

Bone marrow aspirate with haemophagocytosis.
Differential diagnosis
MAS/HLH due to B. cenocepacia septicaemia, secondary to symptomatic CGD due to lyonisation.
Treatment
Considering her overwhelming sepsis and primary immunodeficiency, her MAS/HLH was treated with an amended protocol; intravenous immune globulin (IVIG; 150 g, equivalent to 2 g/kg of body weight) was administered in divided daily doses over 5 days, pulsed intravenous methylprednisolone (1 g daily for three doses) followed by oral prednisone at 50 mg/day on a prolonged taper. She also received ciclosporin at a conservative dose of 1.5 mg/kg/day; it was adjusted based on trough and 2-hour post-dosing serum levels.
Outcome and follow-up
The patient was discharged from hospital 3 weeks after treatment of her sepsis, with her immunosuppression (prednisone and ciclosporin) weaned judiciously in conjunction with careful monitoring of her inflammatory markers. Her transaminases, CRP and ferritin were normalised over the next 3 months (figure 3).
Figure 3.
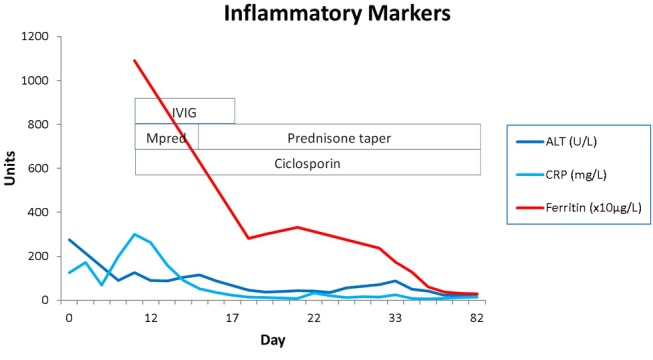
Inflammatory marker trend with treatment. ALT, Alanine transaminases; CRP, C reactive protein; IVIG, intravenous immunoglobulin; Mpred, methylprednisone.
Five years after her admission, she maintains health and independence and lives with her husband in the community. She is maintained on antimicrobial prophylaxis.
Discussion
The underlying condition of the patient was diagnosed at the age of 33 in 1975, after which she had had two male children who died of overwhelming pyogenic sepsis at 9 and 14 months of age. She has a daughter who is also an X-linked carrier for CGD. Despite many decades of good health, she had been referred to our service in 2012 for evaluation of several (largely mild) lower respiratory tract infections, as well as persistent mediastinal lymphadenopathy. The latter had been biopsied to reveal non-necrotising granulomatous infiltrate and was thought to have been ‘sarcoidosis’. Testing requested by our service at that time confirmed her genetic status, with a mutated CYBB gene (figure 4).
Figure 4.

Sequencing of CYBB. Pathological deletion of adenine at position 764, resulting in reading frameshift and early termination (c.764hetdelA → p.Lys255Argfs) (A).
Nitroblue tetrazolium (NBT) and dihydrorhodamine (DHR) flow cytometry chemiluminescence testing revealed two distinct populations of neutrophils; only 2% was considered as having an adequate respiratory burst on phorbol myristate acetate (PMA) stimulation, consistent with an extremely skewed lyonisation (figure 5). Recommendation at that time was for long-term antimicrobial prophylaxis; however, she declined this avenue and had subsequently been lost to follow-up until her current presentation.
Figure 5.

Dihydrorhodamine flow cytometric evaluation. (A) Control neutrophil population unstimulated. (B) Control neutrophil population after stimulation with PMA. (C) Patient neutrophil population unstimulated. (D) Patient neutrophil population after stimulation with PMA.
MAS/HLH, although well recognised as a complication of CGD, is rare. There have been 14 cases to date describing MAS/HLH in CGD patients,7 18–21 with four of them appearing to be triggered by a member of the B. cepacia complex. Case reports of female X-linked carriers with a mutated CYBB gene, who have become immunodeficient through skewed lyonisation, have also previously been described,6 22–24 although our case is the first documented case of an elderly female carrier of X-linked CGD with profound skewing of lyonisation resulting in susceptibility to B. cepacia with triggering of MAS/HLH.
There are five cases of confirmed ‘cepacia syndrome’ in CF patients whereby management resulted in survival. The management of each of these cases is outlined in table 1.
Table 1.
Successfully treated cases of ‘cepacia syndrome’ in patients with cystic fibrosis
| Author | Age (years) | Gender | Pre-existing comorbidity | Pre-existing immunosuppression | Organism | Systemic antibiotics | Nebulised antibiotics | Additional treatment |
| Grimwood et al 13 | 10 | F | – | – | Burkholderia cenocepacia | Ceftazidime, trimethoprim-sulfamethoxazole, tobramycin | Tobramycin | Recombinant human DNase |
| Kazachkov et al 16 | 11 | F | – | – | B. cepacia | Ceftazidime, meropenem, chloramphenicol, trimethoprim-sulfamethoxazole | – | Methylprednisolone |
| Weidmann et al 15 | 31 | F | En-bloc liver–pancreas transplant | Mycophenolate mofetil, tacrolimus | B. cenocepacia | Meropenem, trimethorpim-sulfamethoxazole, temocillin | Meropenem Tobramycin |
– |
| Nash et al 17 | 24 | M | Bilateral lung transplant | Tacrolimus, azathioprine (temporarily withheld during episode of sepsis), prednisone | B. cenocepacia | Ticarcillin-clavulanate, meropenem, amikacin, colistin | Amikacin Colistin | – |
| 24 and 30 (same patient with two different episodes) | F | Bilateral lung transplant | Ciclosporin, azathioprine, prednisone | B. cenocepacia | 1st episode—Ceftazidime, tobramycin, chloramphenicol (oral) 2nd episode —Meropenem, ciprofloxacin (oral), azithromycin (oral) and doxycycline (oral) |
Tobramycin | Surgical debridement | |
| Gilchrist et al 14 | 38 | M | – | – | B. cenocepacia | Tobramycin, meropenem, trimethoprim-sulfamethoxazole, choramphenicol | – | Oral prednisolone, ciclosporin |
NADPH is a large enzymatic complex that straddles the membrane and cytosol of the secondary granules of neutrophils. It is composed of the membrane-bound cytochrome b558 (consisting of two proteins, gp91phox and p22phox) which is associated with the cytosolic components p47phox, p67phox, p40phox, as well as Rho-GTPase.3 5 Mutations in CYBB (the gene that encodes gp91phox) account for approximately two-thirds of cases of CGD, with the remaining cases attributed to mutations in the genes responsible for p47phox, p40phox, p22phox and p67phox.5
CYBB is inherited in an X-linked fashion. Mutations of the other genes are inherited in an autosomal recessive manner. Approximately 30% of CYBB mutations are de novo.25 Being X-linked, mutations in CYBB universally affects men with a diagnosis usually being reached in early infancy. The overall prognosis has historically been poor, although prophylactic antimicrobials increases life expectancy markedly. Bone marrow stem cell transplantation is a curative option for some patients.4 5
Carriers of a mutated CYBB are prone to two well-described consequences: immune dysregulation and extreme lyonisation. Immune dysregulation is common in carriers and can manifest as a paradoxically increased propensity to autoimmunity.23 26 The patients are at much higher risk of systemic diseases such as systemic lupus erythematosus. Additionally, granulomatous involvement of multiple organs can occur, with biopsies having a similar appearance to sarcoid.26 Our patient had both of these phenomena: steroid responsive polymyalgia rheumatica and previously confirmed mediastinal granulomatous lymphadenopathy. We also suspected large vessel vasculitis, although temporal artery biopsy was non-diagnostic.
Skewed lyonisation can also occur with advancing age, so that the preferred X-chromosome selected by neutrophil progenitors harbours the mutated CYBB. Infections typical of CGD do not occur generally in carriers until lyonisation results in less than 5%–10% neutrophils that are respiratory burst competent.23 Our patient had previously been a confirmed heterozygous X-linked CYBB (gp91phox) mutant carrier, with DHR flow cytometry testing revealing only 2% of neutrophils capable of an adequate respiratory burst. Additionally, she had had a mediastinal lymph node biopsy consistent with granulomatous infiltrate that was thought, retrospectively, to be due to her CGD carrier status. Her stably deranged LFTs, while not invasively investigated, were in keeping with granulomatous liver infiltrate well described in carriers of CGD.26
MAS and HLH are conditions that probably lie on a spectrum of the same clinical entity. They are characterised by a hyperinflammatory state, with defined clinical and laboratory criteria.10 By convention, MAS as a diagnosis is used when the clinical syndrome presents secondary to an infection or underlying autoimmune disease, while HLH is reserved when the primary defect is genetic (such as defects of granule trafficking within natural killer cells or other cytotoxic lymphocytes) or malignant, although there is no universally accepted consensus and both terms are frequently used interchangeably.
In CF, ‘cepacia syndrome’ is uncommon but rapidly fatal sequelae secondary to colonisation and septicaemia from members of the B. cepacia complex. The original description11 and subsequent case reports describe a state of uncontrolled hyperinflammation preceded by B. cepacia bacteraemia that almost universally results in death. Only a select few case reports to date have documented successful treatment of cepacia syndrome (table 1); one report aggressively treated the B. cepacia with a combination of antibiotics alone.13 Another utilised antibiotics and high dose intravenous glucocorticosteroids.16 A third reported case was successfully managed with aggressive antibiotics, however, that patient also had pre-existing dual immunosuppression with mycophenolate mofetil and tacrolimus (a calcineurin inhibitor) for a previous en-bloc liver–pancreas transplant.15 Likewise, other cases of B. cepacia sepsis in CF also had calcineurin inhibitor-dependent immunosuppression for prevention of solid organ transplantation rejection as part of background therapy.17 The most recent published case documents successful treatment of cepacia syndrome with combination antibiotics, oral glucocorticosteroids and ciclosporin as a novel treatment without a history of previous transplantation.14 Although full biochemical and clinical examination is not documented in these cases, some of the pathophysiology of ‘cepacia syndrome’ may potentially explained by unrecognised MAS/HLH; the cornerstone of treatment of MAS/HLH is aggressive eradication of the triggering event (such as infection or malignancy) if possible, glucocorticosteroids, high dose IVIG and ciclosporin with or without etoposide. Confirmed ‘MAS/HLH’ in CF patients secondary to B. cepacia has not been described in the literature to our knowledge, although cases due to other organisms exist.27 28
To our knowledge, this is the first report of MAS/HLH secondary to B. cepacia in an elderly patient with confirmed X-linked CGD carrier-status with a full pathological phenotype due to an extreme of lyonisation. Aggressive control of B. cepacia infection with multiple antibiotics and management of MAS/HLH with high dose IVIG and ciclosporin immunosuppression were effective in this patient. In a non-CGD setting, such as in CF, ‘cepacia syndrome’ should be investigated with attempt to exclude MAS, as the latter has clearer therapeutic options.
Learning points.
Female carriers of X-linked chronic granulomatous disease (CGD) can become symptomatic through progressive lyonisation.
Burkholderia cepacia septicaemia and macrophase activation syndrome/haemophagocytic lymphohistiocytosis (MAS/HLH) are rare complications of CGD and can occur in female carriers.
MAS/HLH should be considered a differential diagnosis in non-CGD cases of ‘cepacia syndrome’.
Acknowledgments
The authors' sincerest gratitude goes to the staff of the Immunology Laboratory at The Children’s Hospital at Westmead; Reta Nambiar and Lou Gacis (performing and reporting the NBT and the DHR testing), Alvin Benig (processing of samples) with further comments and interpretation by Dr Melanie Wong. The authors are also indebted to Penelope Motum and Vinay Vanguru (Department of Haematology, Liverpool Hospital) for reporting and providing the photographs of the bone marrow aspirate.
Footnotes
Contributors: NU acted as the main author. KK provided editorial assistance and review. AW was responsible for CYBB gene sequencing validation and performance/interpretation of all neutrophil function testing.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Patient consent for publication: Obtained.
References
- 1. Agarwal S. Chronic granulomatous disease. J Clin Diagn Res 2015;9:SD01–2. 10.7860/JCDR/2015/12139.5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cachat J, Deffert C, Hugues S, et al. Phagocyte NADPH oxidase and specific immunity. Clin Sci 2015;128:635–48. 10.1042/CS20140635 [DOI] [PubMed] [Google Scholar]
- 3. Segal BH, Veys P, Malech H, et al. Chronic granulomatous disease: lessons from a rare disorder. Biol Blood Marrow Transplant 2011;17:S123–31. 10.1016/j.bbmt.2010.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Åhlin A, Fasth A. Chronic granulomatous disease–conventional treatment vs. hematopoietic stem cell transplantation: an update. Curr Opin Hematol 2015;22:41–5. 10.1097/MOH.0000000000000097 [DOI] [PubMed] [Google Scholar]
- 5. Seger RA. Chronic granulomatous disease: recent advances in pathophysiology and treatment. Neth J Med 2010;68:334–40. [PubMed] [Google Scholar]
- 6. Lun A, Roesler J, Renz H. Unusual late onset of X-linked chronic granulomatous disease in an adult woman after unsuspicious childhood. Clin Chem 2002;48:780–1. [PubMed] [Google Scholar]
- 7. Hisano M, Sugawara K, Tatsuzawa O, et al. Bacteria-associated haemophagocytic syndrome and septic pulmonary embolism caused by Burkholderia cepacia complex in a woman with chronic granulomatous disease. J Med Microbiol 2007;56:702–5. 10.1099/jmm.0.47071-0 [DOI] [PubMed] [Google Scholar]
- 8. van Montfrans JM, Rudd E, van de Corput L, et al. Fatal hemophagocytic lymphohistiocytosis in X-linked chronic granulomatous disease associated with a perforin gene variant. Pediatr Blood Cancer 2009;52:527–9. 10.1002/pbc.21851 [DOI] [PubMed] [Google Scholar]
- 9. Parekh C, Hofstra T, Church JA, et al. Hemophagocytic lymphohistiocytosis in children with chronic granulomatous disease. Pediatr Blood Cancer 2011;56:460–2. 10.1002/pbc.22830 [DOI] [PubMed] [Google Scholar]
- 10. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–31. 10.1002/pbc.21039 [DOI] [PubMed] [Google Scholar]
- 11. Isles A, Maclusky I, Corey M, et al. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr 1984;104:206–10. 10.1016/S0022-3476(84)80993-2 [DOI] [PubMed] [Google Scholar]
- 12. Dobbin CJ, Soni R, Jelihovsky T, et al. Cepacia syndrome occurring following prolonged colonisation with Burkholderia cepacia . Aust N Z J Med 2000;30:288–9. 10.1111/j.1445-5994.2000.tb00828.x [DOI] [PubMed] [Google Scholar]
- 13. Grimwood K, Kidd TJ, Tweed M. Successful treatment of cepacia syndrome. J Cyst Fibros 2009;8:291–3. 10.1016/j.jcf.2009.04.002 [DOI] [PubMed] [Google Scholar]
- 14. Gilchrist FJ, Webb AK, Bright-Thomas RJ, et al. Successful treatment of cepacia syndrome with a combination of intravenous cyclosporin, antibiotics and oral corticosteroids. J Cyst Fibros 2012;11:458–60. 10.1016/j.jcf.2012.04.002 [DOI] [PubMed] [Google Scholar]
- 15. Weidmann A, Webb AK, Dodd ME, et al. Successful treatment of cepacia syndrome with combination nebulised and intravenous antibiotic therapy. J Cyst Fibros 2008;7:409–11. 10.1016/j.jcf.2008.02.005 [DOI] [PubMed] [Google Scholar]
- 16. Kazachkov M, Lager J, LiPuma J, et al. Survival following Burkholderia cepacia sepsis in a patient with cystic fibrosis treated with corticosteroids. Pediatr Pulmonol 2001;32:338–40. 10.1002/ppul.1127 [DOI] [PubMed] [Google Scholar]
- 17. Nash EF, Coonar A, Kremer R, et al. Survival of Burkholderia cepacia sepsis following lung transplantation in recipients with cystic fibrosis. Transpl Infect Dis 2010;12:551–4. 10.1111/j.1399-3062.2010.00525.x [DOI] [PubMed] [Google Scholar]
- 18. Akagi K, Kawai T, Watanabe N, et al. A case of macrophage activation syndrome developing in a patient with chronic granulomatous disease-associated colitis. J Pediatr Hematol Oncol 2014;36:e169–72. 10.1097/MPH.0b013e31828e5dae [DOI] [PubMed] [Google Scholar]
- 19. Álvarez-Cardona A, Rodríguez-Lozano AL, Blancas-Galicia L, et al. Intravenous immunoglobulin treatment for macrophage activation syndrome complicating chronic granulomatous disease. J Clin Immunol 2012;32:207–11. 10.1007/s10875-011-9616-5 [DOI] [PubMed] [Google Scholar]
- 20. Valentine G, Thomas TA, Nguyen T, et al. Chronic granulomatous disease presenting as hemophagocytic lymphohistiocytosis: a case report. Pediatrics 2014;134:e1727–30. 10.1542/peds.2014-2175 [DOI] [PubMed] [Google Scholar]
- 21. Sirinavin S, Techasaensiri C, Pakakasama S, et al. Hemophagocytic syndrome and Burkholderia cepacia splenic microabscesses in a child with chronic granulomatous disease. Pediatr Infect Dis J 2004;23:882–4. 10.1097/01.inf.0000137565.23501.03 [DOI] [PubMed] [Google Scholar]
- 22. Sarwar G, de Malmanche T, Rassam L, et al. Chronic granulomatous disease presenting as refractory pneumonia in late adulthood. Respirol Case Rep 2015;3:54–6. 10.1002/rcr2.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rösen-Wolff A, Soldan W, Heyne K, et al. Increased susceptibility of a carrier of X-linked chronic granulomatous disease (CGD) to Aspergillus fumigatus infection associated with age-related skewing of lyonization. Ann Hematol 2001;80:113–5. 10.1007/s002770000230 [DOI] [PubMed] [Google Scholar]
- 24. Anderson-Cohen M, Holland SM, Kuhns DB, et al. Severe phenotype of chronic granulomatous disease presenting in a female with a de novo mutation in gp91-phox and a non familial, extremely skewed X chromosome inactivation. Clin Immunol 2003;109:308–17. 10.1016/j.clim.2003.08.002 [DOI] [PubMed] [Google Scholar]
- 25. Roos D, de Boer M. Molecular diagnosis of chronic granulomatous disease. Clin Exp Immunol 2014;175:139–49. 10.1111/cei.12202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Battersby AC, Cale AM, Goldblatt D, et al. Clinical manifestations of disease in X-linked carriers of chronic granulomatous disease. J Clin Immunol 2013;33:1276–84. 10.1007/s10875-013-9939-5 [DOI] [PubMed] [Google Scholar]
- 27. Casciaro R, Cresta F, Favilli F, et al. Macrophage activation syndrome induced by A/H1N1 influenza in cystic fibrosis. Pediatr Pulmonol 2014;49:E10–12. 10.1002/ppul.22778 [DOI] [PubMed] [Google Scholar]
- 28. Şişmanlar Eyüboğlu T, Aslan AT, Ramaslı Gursoy T, et al. Macrophage activation syndrome due to Nocardia spp in a pediatric patient with cystic fibrosis. Pediatr Pulmonol 2019;54:E10–12. 10.1002/ppul.24262 [DOI] [PubMed] [Google Scholar]