

Abstract
Protein-protein interactions (PPIs) play a crucial role in various biological processes. To better comprehend the pathogenesis and treatments of various diseases, it is necessary to learn the detail of these interactions. However, the current experimental method still has many false-positive and false-negative problems. Computational prediction of protein-protein interaction has become a more important prediction method which can overcome the obstacles of the experimental method. In this work, we proposed a novel computational domain-based method for PPI prediction, and an SVM model for the prediction was built based on the physicochemical property of the domain. The outcomes of SVM and the domain-domain score were used to construct the prediction model for protein-protein interaction. The predicted results demonstrated the domain-based research can enhance the ability to predict protein interactions.
1. Introduction
Protein commonly consists of one or more submolecule parts, which are termed as domain. Domain is a structural or functional module of protein, and it is usually evolutionarily conserved units. Differential association of domains provides a way to create new functions for organisms [1]. The interactions between domains can help locate a protein at a specific subcellular site, which recognize protein posttranslational modification or participate in signal transduction. The interactions can also regulate the enzymatic activity, vigor, and substrate specificity [2]. Recently, many comprehensive studies about domain have been conducted. For example, PDZ domain, which was found in various proteins, including protein tyrosine phosphatase and nitric oxide synthase, plays an important role in regulating protein-protein interactions, protein targets, and protein complex formations [3]. The PB1 domain exists in many signaling proteins involved in the multiple signaling pathway, including the mitogen-activated protein kinase pathway [4] and cellular polarity pathways [5]. Proteins containing the PB1 domain have a close relationship to the occurrence of cancer, such as breast cancer and lung cancer. More and more findings indicate that abnormalities in the domain can lead to various diseases. Therefore, it holds an important practical significance for the domain-based drug design and disease treatment in clinical research, such as arteriosclerosis and cancer. Domain-based studies might help to understand the molecular mechanisms of human diseases, to develop appropriate disease models, and to provide tools for diagnosis.
Domain-based prediction has provided a new perspective for the study of protein-protein interactions (PPIs). PPIs play a crucial role in biological processes, including immune response, signal transduction, and the occurrence and development of disease. Usually, there are two methods predicting protein-protein interactions, experimental method and computational method. Experimental techniques identifying protein-protein interactions are the earliest research methods, including yeast two-hybrid (Y2H) [6], tandem affinity purification (TAP) [7], co-immunoprecipitation (Co-IP) [8], and other techniques. However, high- and low-throughput experimental techniques have some constraints on manpower and material, and experimental results often have high false positives and false negatives. Thus, computational methods have been developed for PPI prediction. The classification of computational method is mainly based on its different features. The commonly used features are protein sequence, protein evolutionary, three-dimensional structure, and domain information. Currently, sequence-based methods have achieved some good prediction results [9–18]. You et al. [19] considered the sequence order and dipeptide information of the protein primary sequence and proposed a matrix-based representation of protein sequence, which is used as the input information of an SVM. However, the sequence-based approaches only use the sequential information, and the 3D structure information was ignored. It is generally believed that protein interactions are mediated by some their specific domain interactions [20], so the domain-based method is widely used in recent years.
Wojcik and Schachter have developed an interacted domain pair profile method to predict protein-protein interactions. They applied their method to predict an interaction map of Escherichia coli [21]. Kim et al. have proposed a statistical scoring system, based on the interacting domain pairs from InterPro, to measure the interaction probability between domains and to represent protein-protein interactions [22]. Hayashida et al. have used conditional random field to predict PPIs based on mutual information between residues of domain-domain interactions [23]. Kamada et al. have used domain features with support vector regression (SVR) and relevance vector machine (RVR) to predict the strengths of PPIs [24]. Singhal and Resat have applied the InterDom (the interacting domain database) domain-domain interaction scores as the feature information. They developed a multiparameter optimization method—DomainGA—which used the obtained score to predict the interactions between proteins [25]. Zhang et al. have also used the domain-domain interaction scores. His method used DDI confidence probabilities to calculate the confidence probability of the predicted PPI [26].
Currently, the features that domain-based methods used just contain the domain co-occurrence relationships or the proportion of an important domain. The domain information is not completely considered. The domain interactions, which are crucial to the understanding of biomolecule interactions, also provide a global view of the protein-protein interaction network. In order to effectively utilize the information of the domain, we proposed a new domain-based method to predict protein-protein interactions.
In this paper, we both considered the physicochemical property of domain and the domain-domain interaction score. The physicochemical property of domain was used as the SVM feature to construct the DDI prediction model. Finally, the DDI prediction model is combined with the domain-domain interaction score to construct the PPI prediction model.
2. Materials and Methods
2.1. Proposed Method
The flow chart of our method is given in Figure 1.
Figure 1.
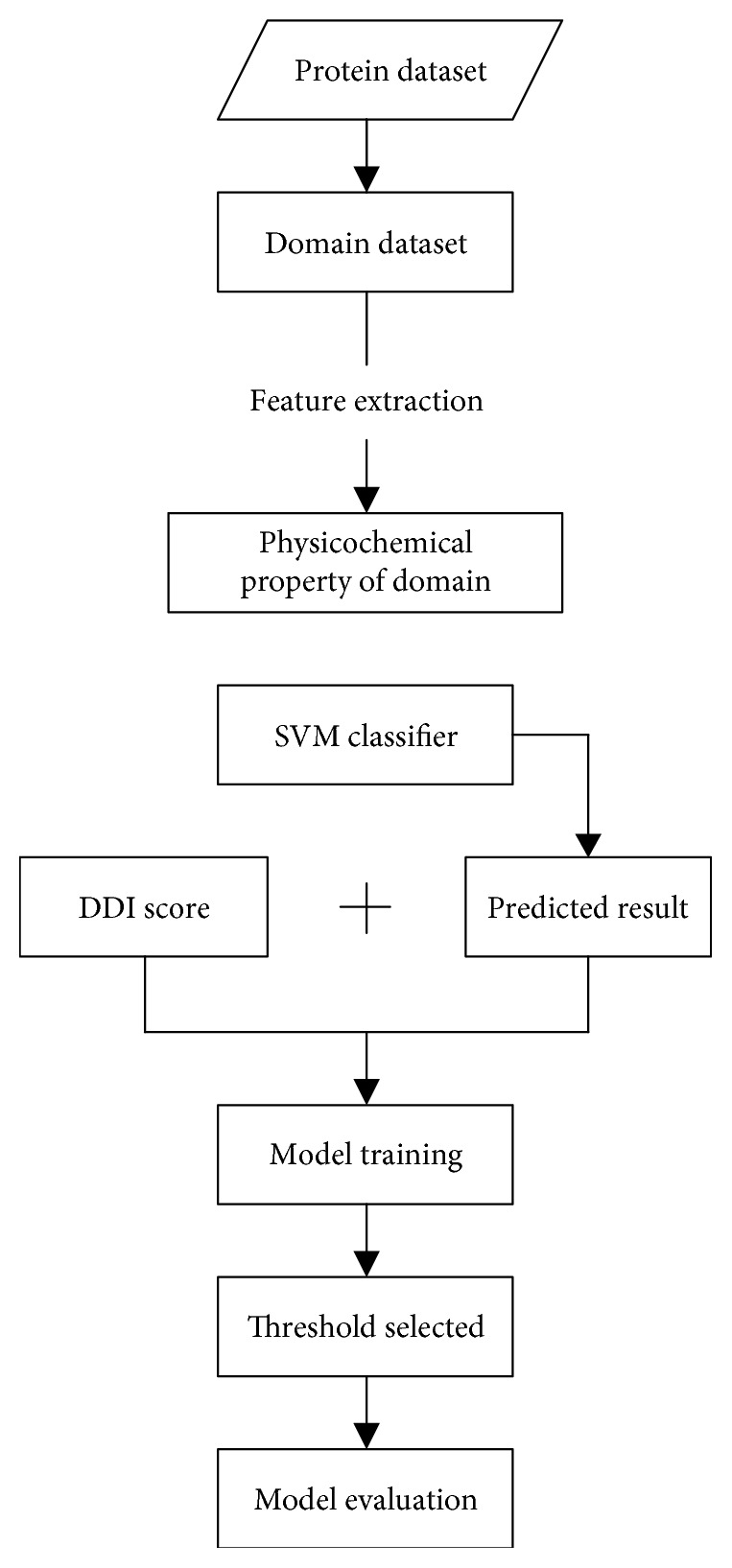
Flow chart of the method.
2.2. Datasets
2.2.1. Protein Dataset
The positive protein-protein interaction data were collected from the interacting adhesome protein-protein. It can be obtained on the website of The Adhesome: A Focal Adhesion Network (http://www.adhesome.org/) [27, 28]. Xiao-Yong et al.'s noninteraction dataset, where any protein pair does not have sequence identity higher than 25% [29], was used for obtaining the negative PPI data. Pan's dataset was commonly used in protein-protein interaction studies [30, 31].
2.2.2. Domain Dataset
We used the protein database mentioned above as our source database to extract the domain of its protein. The domains of protein and sequence information of these domains were obtained from the Pfam database (version 32.0 http://pfam.xfam.org/). We constructed the corresponding domain-domain pairs. Meanwhile, interacting and noninteracting domain pairs were chosen in the InterDom database (interacting domains http://interdom.lit.ofg.sg/) and 3did database (https://3did.irbbarcelona.org/index.php). The InterDom database had a set of confidence scores of DDIs which used 1.5 as the cutoff of false-positive and nonfalse-positive prediction [32]. The interacting domain-domain was selected where the Interdom score is greater than 1.5. Noninteracting DDIs are not available in the two domain level databases which we used above.
The positive protein dataset contained 427 positive PPI, and we constructed 1040 positive DDI from it. There were 403 noninteracting protein pairs, in the negative protein dataset, and we constructed 1040 negative DDI from it. The Interdom score was used in our protein-protein predicting model. But the domain-domain interaction score was not available for the negative domain dataset. So, we set up a score as a background noise to the negative DDI, which was chosen from the Interdom score in the positive domain dataset. 1040 values were ranged from small to large, and the 20% position value of 1.74 was selected.
2.3. Feature Extraction
The physicochemical property of domain pairs was used as features of our method. The domain and the corresponding sequence information were downloaded from the Pfam database. According to the sequence information of the domain, the physicochemical property of the domain can be obtained with the online tools ProtParam (https://web.expasy.org/protparam/) and ProtComp (https://www.expasy.org/). ProtParam can calculate various physicochemical parameters for a given protein [33]. ProtComp can predict the subcellular localization of animal/fungi proteins (version 9.0 http://www.softberry.com/berry.phtml?group=programs&subgroup=proloc&topic=protcompan). Thelocation of a protein in a cell has a close relationship to its biological function [34]. The detailed calculated parameter for ProtParam is listed in Table 1.
Table 1.
Parameters for ProtParam.
| Molecular weight | Aliphatic index |
| Atom composition | Instability index |
| Theoretical pI | Ext. coefficient (1) |
| Number of amino acids | Ext. coefficient (2) |
| Amino acid composition (20) | Estimated half-life |
| Total number of negatively charged residues (Asp + Glu) | Extinction coefficients |
| Total number of positively charged residues (Arg + Lys) | Grand average of hydropathicity (gravy) |
ProtComp calculated the weight of each position from ten positions and chose the most accurate one. To numerically represent the feature of the domain-domain pairs, ten domain location's information was encoded into numbers as shown in Table 2.
Table 2.
Encoded information for the location.
| Location | Code |
|---|---|
| Nuclear | 1 |
| Plasma membrane | 2 |
| Extracellular | 3 |
| Cytoplasmic | 4 |
| Mitochondrial | 5 |
| Endoplasmic reticulum | 6 |
| Peroxisomal | 7 |
| Lysosomal | 8 |
| Golgi | 9 |
| Vacuolar | 10 |
In order to reduce the interference of correlation factors, we carried out a correlation analysis for these features. Finally, ten meaningful physicochemical property features were picked out. They were amino acid numbers, theoretical pI, total number of negatively charged residues, total number of positively charged residues, total number of atoms, Ext. coefficient 1, instability index, aliphatic index, grand average of hydropathicity, and the domain location.
To reduce the impact of large differences in values between various features on results, we did normalized processing for these features according to Mapminmax function. Equation (1) is defined as follows:
| (1) |
The specific value of twenty physicochemical properties for domain was listed in Supplementary Tables and . Finally, the feature of the DDI was a 20-dimensional eigenvector.
2.4. Classification
There are numerous machine-learning techniques for predicting protein-protein interactions. Support vector machine (SVM) is the usual technique for classification and regression [35, 36]. In recent years, it has been widely used in bioinformatic researches and has made outstanding performances [30, 31, 37–41]. In this paper, SVM was used to design the classifier. The domain pairs class label was set +1 for interacting pairs and 0 for noninteracting pairs. The kernel function plays an important role in nonlinear classification. In this paper, the RBF kernel was chosen as the kernel function. The optimal parameters c and g were 9.1896 and 3.0314, which were optimized by the grid search method for SVM classifiers. The fivefold cross-validation method indicates that the data are randomly divided into five equal parts. One part is used as a testing set in turn, and the other four parts are used as a training test. It can effectively prevent the overfitting problem. At the same time, our results have been counted at least five times until the results are relatively stable.
The software libsvm 3.23 (http://www.csie.ntu.edu.tw/∼cjlin/libsvm/) was employed in this work.
2.5. Assessment of Prediction System
In order to evaluate the prediction performance of our approach, the following six measurements: accuracy (Acc), sensitivity (SN), specificity (SPE), precision (Pre), Matthews correlation coefficient (MCC), and F1 score values were used. Their mathematical description is defined as follows:
| (2) |
where TP (the true positive value) is the number of interactions predicted correctly; TN (the true negative value) is the number of noninteraction pairs predicted correctly; and FN (the false negative value) and FP (the false positive value) are the number of interactions incorrectly predicted as noninteractions and noninteracting proteins incorrectly as interactions.
3. Results and Discussion
This section is divided into four parts: the first part is the intermediate result of the prediction of the domain-domain interaction, the second part is the result of protein prediction, the third part is the comparison of different methods, and the last part is the limitations of our model.
3.1. Results of DDIs
We used the physicochemical property of domain to build the SVM prediction model. To evaluate the robustness of our method and to reduce impact of data independence, fivefold cross validation was used to ensure the reliability of the results. The SVM calculation was run five times. The result of domain-domain interaction prediction is shown in Table 3.
Table 3.
Performances of result for five-time predictions with SVM.
| Time | Acc (%) | SN (%) | SPE (%) | Pre (%) | F 1 (%) | MCC (%) |
|---|---|---|---|---|---|---|
| 1 | 94.62 | 95.19 | 94.04 | 94.11 | 94.65 | 89.24 |
| 2 | 94.13 | 94.23 | 94.04 | 94.05 | 94.14 | 88.27 |
| 3 | 94.95 | 95.00 | 94.90 | 94.91 | 94.95 | 89.90 |
| 4 | 95.24 | 95.38 | 95.10 | 95.11 | 95.25 | 90.48 |
| 5 | 94.52 | 94.42 | 94.62 | 94.61 | 94.51 | 89.04 |
From Table 3, we can see that the DDI prediction model achieved an acceptable performance. The highest prediction accuracy was 95.24%. The average prediction accuracy was 94.69%. Two indicators, the F1 and MCC, can better evaluate the overall performance of the classifier. The average value of F1 was 94.54%, and the MCC was 89.39%. These results show that the domain's physicochemical properties are effective feature information for domain-domain interaction.
3.2. Results of PPIs
The domain-domain interaction score in the Interdom database and DDI predicted label results were used to build a protein-protein prediction model. In order to reduce the numerical difference between the domain-domain score, the value was obtained by the following algorithm:
| (3) |
In which λmn represented the Interdom score of m domain and n domain pair and the Smax represented the maximum score of domain-domain in our database. Pij represented the DDI-predicted label results, which was a probability score that the interacting domain-domain we predicted to the total theoretical domain pairs in a protein pair. Pij was defined by using the following equation:
| (4) |
Num_predicted was the number of predicted domain pairs with our model for one certain protein pair. Num_DDI was the theoretical number of all domain-domain pairs in the same protein pair.
In this section, we assumed that domain-domain interactions were independent [42]. We estimated the probability of each PPI by the following equation:
| (5) |
Grid algorithm N ∗ N is used to find the optimal parameters A and B. We set the value of N from 0 to 0.6 by 0.1. A total of 49 uniform lattices trained the protein sets. In order to evaluate the results of the training, we set ten thresholds from 0.1 to 0.55, with an interval of 0.05. The values of TP, TN, FP, FN, the false negative rate (fn), the false positive rate (fp), ACC, and SN were calculated. These evaluation indexes are described in detail in Section 2.5. The specific algorithm for fn and fp was as follows:
| (6) |
The results of protein-protein interacting possibility were compared by the accuracy and ROC curves with AUC scores. Finally, the parameters A and B with high accuracy and large ACU area were selected. The final equation was as follows:
| (7) |
In order to select the optimal threshold, we used formula (7) to train the parameter for the protein-protein dataset. The result is shown in Figure 2.
Figure 2.
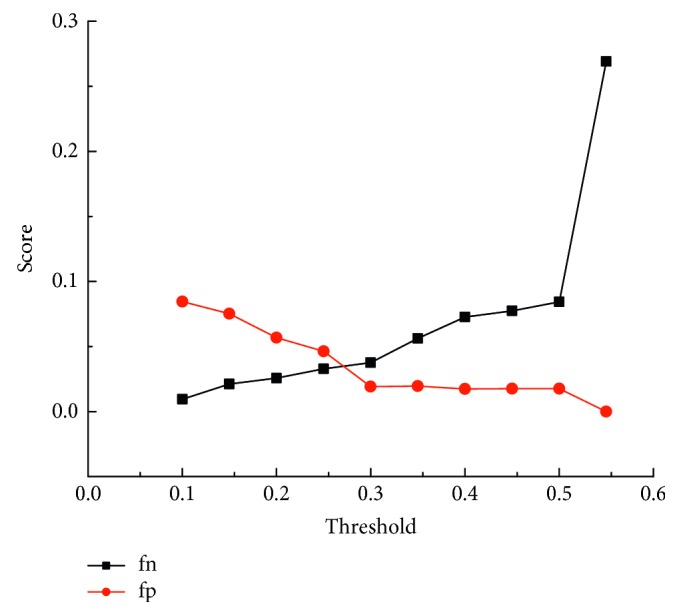
fn and fp of the predicted result.
The X-axis represented different thresholds, and the Y-axis represented the values of fn and fp. The suitable threshold was determined according to two principles: (1) fn and fp should be as small as possible and (2) fn and fp should be as equal as possible. Therefore, we chose 0.26 as the optimal threshold. To obtain a more accurate threshold, we calculated the protein training results of three thresholds that are 0.25, 0.26, and 0.27. We also calculated the AUC (the area under the ROC curve). The results showed the same result in Table 4, so we chose 0.26 as the optimal threshold.
Table 4.
Prediction results based on three thresholds.
| Threshold | TP | TN | FP | FN | Acc (%) | SN (%) | SPE (%) | Pre (%) | MCC (%) | F 1 (%) | AUC (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.25 | 413 | 383 | 20 | 14 | 95.90 | 96.72 | 95.04 | 95.38 | 91.81 | 96.00 | 91.92 |
| 0.26 | 413 | 383 | 20 | 14 | 95.90 | 96.72 | 95.04 | 95.38 | 91.81 | 96.00 | 91.92 |
| 0.27 | 413 | 383 | 20 | 14 | 95.90 | 96.72 | 95.04 | 95.38 | 91.81 | 96.00 | 91.92 |
3.3. Comparison with Different Prediction Methods
To demonstrate the prediction performance, we compared our method with other SVM-based methods. In order to compare more accurately, we chose the different studies which not only used Pan's database but also used SVM classifier. The results are shown in Table 5.
Table 5.
Results of comparison with different methods.
| Method | Acc (%) | SN (%) | Pre (%) | MCC (%) |
|---|---|---|---|---|
| This paper | 95.9 | 96.72 | 95.04 | 91.81 |
| Zhang1 | 82.11 | 80.4 | 84.73 | 80.07 |
| SP-SVM2 | 70 | 66 | 72 | — |
| LDA-SVM3 | 69 | 63 | 72 | — |
| PSEAAC_SVM4 | 68 | 63 | 70 | — |
| Yunus5 | 93.45 | 89.29 | 89.84 | 85.71 |
As shown in Table 5, among different methods, the performance of our method achieved the best result. This suggests that our method based on domain to predict protein-protein interactions is relatively successful.
3.4. Limitations of Our Model
Although the accuracy of our method is acceptable, there are still some limitations for our model to be used widely. For example, the number of our dataset and the physiochemical property are small, and in future work, we plan to test our model on a bigger dataset with more features. For our approach, independent software and online tools development work are still in progress.
4. Conclusions
In this paper, we proposed a new domain-based method to predict protein-protein interaction. We used the domain's physicochemical property and interaction score to construct the protein interaction-predicting model. The predicted result, which achieved a good performance, indicates that our method is relatively successful. The physicochemical property of the domain as features for PPI prediction is of great significance. Applying our approach to large dataset and finding more effective feature information for predicting PPI will be part of our future work. Furthermore, our methods can be used for the prediction of new PPIs, and the result could provide some reference significance for dealing with related bioinformatics problems.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant no. 31870932), Natural Science Foundation of Shanxi (Grant no. 201801D121232), Program for the Innovative Talents of Higher Learning Institutions of Shanxi, and 131Talents Project of Shanxi Province.
Data Availability
The physicochemical property of the domain and corresponding protein data used to support the findings of this study are included within the supplementary information files (Supplementary Tables and ).
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Supplementary Materials
Supplementary Table S1: 1040 positive DDI. Supplementary Table S2: 1040 negative DDI.
References
- 1.Bitard-Feildel T., Kemena C., Greenwood J. M., Bornberg-Bauer E. Domain similarity based orthology detection. BMC Bioinformatics. 2015;16(1):p. 154. doi: 10.1186/s12859-015-0570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawson T., Nash P. Protein–protein interactions define specificity in signal transduction. Genes & Development. 2000;14(9):1027–1047. [PubMed] [Google Scholar]
- 3.Dong H., O’Brien R. J., Fung E. T., Lanahan A. A., Worley P. F., Huganir R. L. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386(6622):279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura K., Uhlik M. T., Johnson N. L., Hahn K. M., Johnson G. L. PB1 domain-dependent signaling complex is required for extracellular signal-regulated kinase 5 activation. Molecular and Cellular Biology. 2006;26(6):2065–2079. doi: 10.1128/mcb.26.6.2065-2079.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joberty G., Petersen C., Gao L., Macara I. G. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nature Cell Biology. 2000;2(8):531–539. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- 6.Fields S., Song O.-k. A novel genetic system to detect protein-protein interactions. Nature. 1989;340(6230):245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 7.Puig O., Caspary F., Rigaut G., et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24(3):218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 8.Fiil B. K., Qiu J.-L., Petersen K., Petersen M., Mundy J. Coimmunoprecipitation (co-IP) of nuclear proteins and chromatin immunoprecipitation (ChIP) from Arabidopsis. Cold Spring Harbor Protocols. 2008;2008(10):p. pdb.prot5049. doi: 10.1101/pdb.prot5049. [DOI] [PubMed] [Google Scholar]
- 9.Bock J. R., Gough D. A. Predicting protein-protein interactions from primary structure. Bioinformatics. 2001;17(5):455–460. doi: 10.1093/bioinformatics/17.5.455. [DOI] [PubMed] [Google Scholar]
- 10.Shen J., Zhang J., Luo X., et al. Predicting protein-protein interactions based only on sequences information. Proceedings of the National Academy of Sciences. 2007;104(11):4337–4341. doi: 10.1073/pnas.0607879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y., Wang J., Yang Z., Deng N. Sequence-based protein-protein interaction prediction via support vector machine. Journal of Systems Science and Complexity. 2010;23(5):1012–1023. doi: 10.1007/s11424-010-0214-z. [DOI] [Google Scholar]
- 12.Dong Q., Zhou S., Liu X. Prediction of protein protein interactions from primary sequences. International Journal of Data Mining and Bioinformatics. 2010;4(2):p. 211. doi: 10.1504/ijdmb.2010.032151. [DOI] [PubMed] [Google Scholar]
- 13.Shi M.-G., Xia J.-F., Li X.-L., Huang D.-S. Predicting protein-protein interactions from sequence using correlation coefficient and high-quality interaction dataset. Amino Acids. 2010;38(3):891–899. doi: 10.1007/s00726-009-0295-y. [DOI] [PubMed] [Google Scholar]
- 14.Pirogova E., Vojisavljevic V., Cosic I. Prediction of protein active and/or binding site using time-frequency analysis: application to ras oncogene proteins. Proceedings of the ISSNIP Biosignals and Biorobotics Conference: Biosignals and Robotics for Better and Safer Living (BRC); January 2012; Manaus, Brazil. [Google Scholar]
- 15.Cao J., Kuang C., Liu X., Yao Y., He P., Dai Q. A new prediction strategy for protein structural classes using protein sequence-structure features. Journal of Computational and Theoretical Nanoscience. 2015;12(10):3023–3027. doi: 10.1166/jctn.2015.4076. [DOI] [Google Scholar]
- 16.Ding Y., Tang J., Guo F. Identification of protein-protein interactions via a novel matrix-based sequence representation model with amino acid contact information. International Journal of Molecular Sciences. 2016;17(10):p. 1623. doi: 10.3390/ijms17101623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L., Luo Q., Xiao W., et al. A machine-learning approach for predicting palmitoylation sites from integrated sequence-based features. Journal of Bioinformatics and Computational Biology. 2016;15(1) doi: 10.1142/s0219720016500256.1650025 [DOI] [PubMed] [Google Scholar]
- 18.Wang T., Li L., Huang Y.-A., Zhang H., Ma Y., Zhou X. Prediction of protein-protein interactions from amino acid sequences based on continuous and discrete wavelet transform features. Molecules. 2018;23(4):p. 823. doi: 10.3390/molecules23040823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You Z. H., Li J., Gao X., et al. Detecting protein-protein interactions with a novel matrix-based protein sequence representation and support vector machines. BioMed Research International. 2015;2015:9. doi: 10.1155/2015/867516.867516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou K. C., Maggiora G. M. Domain structural class prediction. Protein Engineering Design and Selection. 1998;11(7):523–538. doi: 10.1093/protein/11.7.523. [DOI] [PubMed] [Google Scholar]
- 21.Wojcik J., Schachter V. Protein-protein interaction map inference using interacting domain profile pairs. Bioinformatics. 2001;17(1):S296–S305. doi: 10.1093/bioinformatics/17.suppl_1.s296. [DOI] [PubMed] [Google Scholar]
- 22.Kim W. K., Park J., Suh J. K. Large scale statistical prediction of protein-protein interaction by potentially interacting domain (PID) pair. Genome Informatics. 2002;13:42–50. [PubMed] [Google Scholar]
- 23.Hayashida M., Kamada M., Song J., Akutsu T. Conditional random field approach to prediction of protein-protein interactions using domain information. BMC Systems Biology. 2011;5(1):1–9. doi: 10.1186/1752-0509-5-s1-s8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamada M., Sakuma Y., Hayashida M., Akutsu T. Prediction of protein-protein interaction strength using domain features with supervised regression. Scientific World Journal. 2014;2014:7. doi: 10.1155/2014/240673.240673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singhal M., Resat H. A domain-based approach to predict protein-protein interactions. BMC Bioinformatics. 2007;8(1):1–19. doi: 10.1186/1471-2105-8-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X., Jiao X., Song J., Chang S. Prediction of human protein-protein interaction by a domain-based approach. Journal of Theoretical Biology. 2016;396:144–153. doi: 10.1016/j.jtbi.2016.02.026. [DOI] [PubMed] [Google Scholar]
- 27.Ronen Z. B., Shalev I., Avi M. A., Ravi I., Benjamin G. Functional atlas of the integrin adhesome. Nature Cell Biology. 2007;9(8):858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winograd-Katz S. E., Fässler R., Geiger B., Legate K. R. The integrin adhesome: from genes and proteins to human disease. Nature Reviews Molecular Cell Biology. 2014;15(4):273–288. doi: 10.1038/nrm3769. [DOI] [PubMed] [Google Scholar]
- 29.Xiao-Yong P., Ya-Nan Z., Hong-Bin S. Large-scale prediction of human protein-protein interactions from amino acid sequence based on latent topic features. Journal of Proteome Research. 2010;9(10):4992–5001. doi: 10.1021/pr100618t. [DOI] [PubMed] [Google Scholar]
- 30.Shao-Wu Z., Li-Yang H., Ting-He Z. Prediction of protein-protein interaction with pairwise kernel support vector machine. International Journal of Molecular Sciences. 2014;15(2):3220–3233. doi: 10.3390/ijms15023220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Göktepe Y. E., Kodaz H. Prediction of protein-protein interactions using an effective sequence based combined method. Neurocomputing. 2018;303:68–74. doi: 10.1016/j.neucom.2018.03.062. [DOI] [Google Scholar]
- 32.Ng S.-K., Zhuo Z., Soon-Heng T., Kui L. InterDom: a database of putative interacting protein domains for validating predicted protein interactions and complexes. Nucleic Acids Research. 2003;31(1):251–254. doi: 10.1093/nar/gkg079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker J. M. The Proteomics Protocols Handbook. Totowa, NJ, USA: Humana Press; 2005. [Google Scholar]
- 34.Xu H., Yan S., Dai Q., He P.-A., Liao B., Yao Y.-H. Protein subcellular location prediction based on pseudo amino acid composition and PSI-blast profile. Journal of Computational and Theoretical Nanoscience. 2015;12(10):3756–3762. doi: 10.1166/jctn.2015.4272. [DOI] [Google Scholar]
- 35.Cortes C., Vapnik V. Support-vector networks. Machine Learning. 1995;20(3):273–297. [Google Scholar]
- 36.Vapnik V. N. The Nature of Statistical Learning Theory. Berlin, Germany: Springer; 1995. [Google Scholar]
- 37.Li L., Yu S., Xiao W., et al. Sequence-based identification of recombination spots using pseudo nucleic acid representation and recursive feature extraction by linear kernel SVM. BMC Bioinformatics. 2014;15(1):p. 340. doi: 10.1186/1471-2105-15-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L., Cui X., Yu S., et al. PSSP-RFE: accurate prediction of protein structural class by recursive feature extraction from PSI-BLAST profile, physical-chemical property and functional annotations. PLoS One. 2014;9(3) doi: 10.1371/journal.pone.0092863.e92863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xi M., Sun J., Liu L., Fan F., Wu X. Cancer feature selection and classification using a binary quantum-behaved particle swarm optimization and support vector machine. Computational and Mathematical Methods in Medicine. 2016;2016:9. doi: 10.1155/2016/3572705.3572705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiang Q., Liao B., Li X., et al. Subcellular localization prediction of apoptosis proteins based on evolutionary information and support vector machine. Artificial Intelligence in Medicine. 2017;78:41–46. doi: 10.1016/j.artmed.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Mustaqeem A., Anwar S. M., Majid M. Multiclass classification of cardiac arrhythmia using improved feature selection and SVM invariants. Computational and Mathematical Methods in Medicine. 2018;2018:10. doi: 10.1155/2018/7310496.7310496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deng M., Mehta S., Sun F., Chen T. Inferring domain-domain interactions from protein-protein interactions. Genome Research. 2002;12(10):1540–1548. doi: 10.1101/gr.153002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: 1040 positive DDI. Supplementary Table S2: 1040 negative DDI.
Data Availability Statement
The physicochemical property of the domain and corresponding protein data used to support the findings of this study are included within the supplementary information files (Supplementary Tables and ).