

Abstract
Internalization of a variety of different heptahelical G protein-coupled receptors has been shown to be influenced by a number of different structural determinants of the receptors, including the carboxyl terminus. To investigate the role of the carboxyl terminus of cholecystokinin (CCK) receptors in receptor internalization, the rat wild type (WT) CCK-A receptor (WT CCKAR) and the rat WT CCK-B receptor (WT CCKBR) were truncated after amino acid residue 399 (CCKAR Tr399) and 408 (CCKBR Tr408), thereby deleting the carboxyl-terminal 45 and 44 residues, respectively. All WT and mutant CCK receptors were stably expressed in NIH/3T3 cells. Internalization of the CCKAR Tr399 was not significantly different from the WT CCKAR. In contrast, internalization of the CCKBR Tr408 was decreased to 26% compared with the WT CCKBR internalization of 92%. The mutation of all 10 serine and threonine residues (as potential phosphorylation sites) in the carboxyl terminus of the CCKBR to alanines (mutant CCKBR ΔS/T) could account for the majority of this effect (39% internalization). All mutant receptors displayed similar ligand binding characteristics, G protein coupling, and signal transduction as their respective WT receptors, indicating that the carboxyl termini are not necessary for these processes. Thus, internalization of the CCKBR, unlike that of the CCKAR, depends on the carboxyl terminus of the receptor. These results suggest that, despite the high degree of homology between CCKAR and CCKBR, the structural determinants that mediate the interaction with the endocytic pathway reside in different regions of the receptors.
Many G protein-coupled receptors (GPCRs)1 undergo internalization (or sequestration) to intracellular sites within minutes after agonist exposure. Internalized receptors are either degraded in lysosomes (1) or undergo resensitization and reinsertion into the plasma membrane (2–4). The relationship between internalization and desensitization is unsettled for most GPCRs and may vary between different receptors and different cell systems. For some receptors, receptor internalization seems to be a major factor in the acute desensitization process that dampens the receptor-mediated cellular response to agonist stimulation (5, 6). For other receptors, receptor internalization makes very little (7) or no (8, 9) contribution to receptor desensitization, arguing that these two processes are unrelated.
The search for the structural basis of GPCRs that couples to the endocytic machinery has revealed a variety of different structural determinants for different receptors that influence receptor internalization. Although for many receptors the carboxyl terminus has been shown to influence receptor internalization (5, 8, 10–18), a variety of other structural determinants including areas within the second (19) and third (20) intracellular loop and a conserved NPXnY motif near the seventh transmembrane domain (21) have been found to be critical for internalization for different GPCRs. However, a universal structural element that mediates internalization has not been identified and may not exist. The signaling mechanism that allows the agonist-occupied receptor to couple to the endocytic machinery and to internalize is unknown for the majority of the GPCRs. For some receptors, however, receptor phosphorylation has been suggested to be implicated in receptor internalization because the mutation of potential serine and threonine phosphorylation sites caused the loss of receptor internalization (5, 10, 15, 17). Only recently, functional studies on the β2-adrenergic receptor have demonstrated a mechanism in which phosphorylation of the carboxyl terminus of the receptor leads to receptor internalization by enhancing the affinity for β-arrestin, an adaptor molecule between the receptor and clathrin-coated vesicles (22, 23).
Cholecystokinin (CCK) receptors belong to the superfamily of GPCRs (24). There are two types of CCK receptors, which have nearly 50% amino acid homology and can be distinguished on the basis of their affinities to the agonists cholecystokinin octapeptide (CCK-8) and gastrin. The cholecystokinin type A receptor (CCKAR) binds CCK-8 with much greater affinity than gastrin, whereas the cholecystokinin type B receptor (CCKBR) binds CCK-8 and gastrin nearly equally well. The signal transduction mechanisms are similar for both the CCKAR and the CCKBR. Both receptors couple to a pertussis toxin-insensitive G protein and elicit the production of inositol phosphates and diacylglycerol (24).
Stimulation of the CCKAR promotes enzyme secretion in the pancreas, smooth muscle contraction in the gallbladder, and regulation of satiety in selective areas of the central and peripheral nervous systems. The CCKBR is present in the stomach where it mediates acid secretion from parietal cells and is also found throughout the central nervous system where it regulates dopamine release and anxiety (24).
Both CCKARs and CCKBRs have been shown to undergo ligand-induced internalization in pancreatic acini (25) or transfected NIH 3T3 cells (26), respectively. They have also been shown to undergo desensitization in their native cells (27–29), but the impact of receptor internalization in this process is unclear. The CCKAR is phosphorylated predominantly (>95%) in the third intracellular loop within seconds after agonist stimulation (30), in part by protein kinase C and in part by another heparin-inhibitable enzyme, probably a G protein receptor kinase (31). The pattern of ligand-induced phosphorylation of the CCKBR has not been previously demonstrated.
In this study, we investigated the structural determinants that mediate internalization of CCK receptors. Deletion of the 44 carboxyl-terminal amino acid residues profoundly decreased internalization of the CCKBR. Interestingly, a deletion of the corresponding amino acids in the CCKAR did not affect internalization. The mutation of all serine and threonine residues in the carboxyl terminus to alanines decreased internalization of the CCKBR to almost the same degree as truncation of the carboxyl terminus.
EXPERIMENTAL PROCEDURES
Materials
NIH/3T3 cells were obtained from the American Type Culture Collection (Rockville, MD). Dulbecco’s modified Eagle’s medium (DMEM), calf serum, trypsin-EDTA, and aminoglycoside G418 were from Life Technologies, Inc. (Gaithersburg, MD). Sulfated cholecystokinin octapeptide (CCK-8) was purchased from Research Plus, Inc. (Bayonne, NJ). 125I-Bolton-Hunter-labeled CCK-8 (125I-BH-CCK-8) (2200 Ci/mmol) was obtained from NEN Life Science Products. 45Ca2+ (10–40 mCi/mg Ca2+) and [γ−32P]ATP, triethylammonium salt (6000 Ci/mmol) were from Amersham Life Science, Inc. Potassium thiocyanate, sodium fluoride, aluminum chloride, and EGTA were purchased from Sigma. Bovine serum albumin (BSA) was from ICN Biomedicals Inc. (Aurora, OH). Hydrofluor was from National Diagnostics, Inc. (Atlanta, GA).
Mutant CCK Receptor Construction
The wild type (WT) rat CCKAR and CCKBR were subcloned into the vector pCDL/SRa containing a neomycin resistance gene (32). Truncated receptor mutants were created by introducing a stop codon using the polymerase chain reaction. The WT CCKAR was truncated after amino acid residue 399 (CCKAR Tr399), and the WT CCKBR was truncated after amino acid residue 408 (CCKBR Tr408) (see Fig. 1). All serine and threonine residues in the carboxyl terminus were mutated to alanines by site-directed mutagenesis (Muta-Gene® phagemid in vitro mutagenesis kit, Bio-Rad) for both the CCKAR and the CCKBR and are referred to as CCKAR ΔS/T and CCKBR ΔS/T (see Fig. 1). All mutations were confirmed by DNA sequence analysis using the dsDNA cycle sequencing system (Life Technologies, Inc.).
FIG. 1. Amino acid models of the rat CCKAR and the rat CCKBR illustrating the receptor mutations.
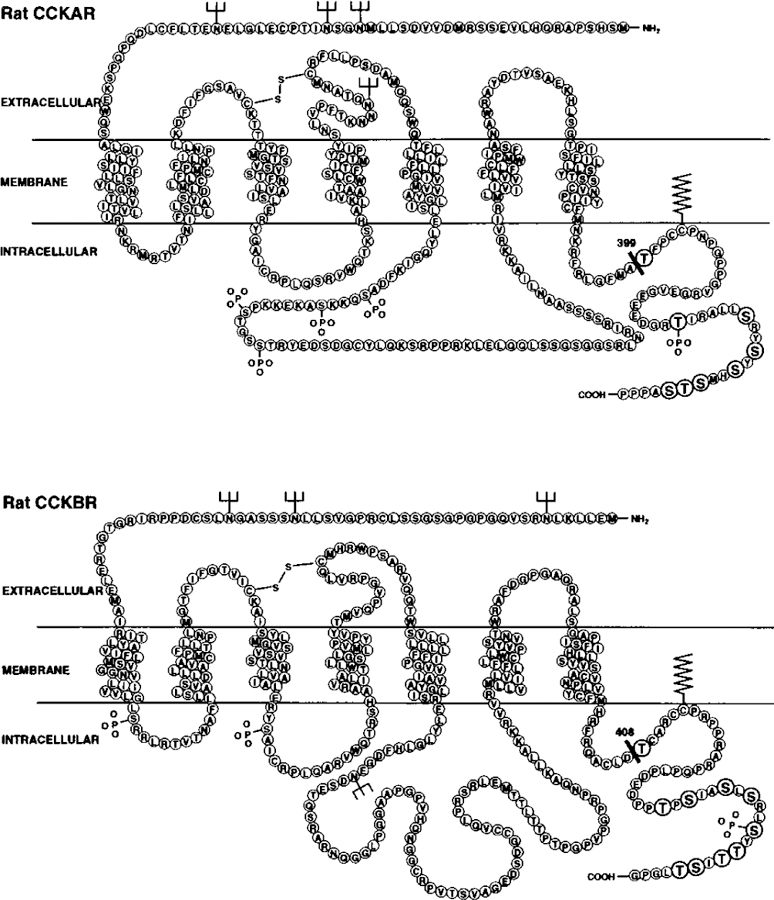
The models show the predicted amino acid sequence of the WT CCKAR (upper panel) and the WT CCKBR (lower panel). The mutant CCKAR Tr399 was constructed by truncation of the carboxyl terminus after amino acid residue 399, as indicated by the black bar. The mutant CCKAR ΔS/T was constructed by mutation of all eight serine and threonine residues in the carboxyl terminus (indicated by the enlarged and embolded circles) to alanines. In parallel, the mutant CCKBR Tr408 was constructed by truncation of the carboxyl terminus after amino acid residue 408 as indicated by the black bar. The mutant CCKBR ΔS/T was constructed by mutation of all 10 serine and threonine residues in the carboxyl terminus (indicated by the enlarged and embolded circles) to alanines.
Stable Expression of CCK Receptors in NIH/3T3 Cells
All WT and mutant receptors were stably transfected into NIH/3T3 cells by electroporation (500 microfarads, 0.25 kV, Gene Pulser®, Bio-Rad) of 2 × 107 cells in a volume of 0.25 ml with 20 µg of the linearized recombinant vector containing the respective WT or mutant receptor cDNA in the presence of 500 µg/ml salmon sperm DNA as a carrier. Cell clones stably expressing the receptors were then selected for G418 resistance (250 µg of G418/ml) and 125I-BH-CCK-8 binding. Cells were maintained in DMEM, 10% calf serum/G418 (250 µg/ml) at 37 °C in a 6% CO2 atmosphere.
Radioligand Binding Displacement Studies
Transfected NIH/3T3 cells were plated in 24-well tissue culture plates and assayed the following day for radioligand binding. For binding displacement studies, cells were incubated for 90 min with 50 pM 125I-BH-CCK-8 in the absence or presence of increasing concentrations of unlabeled CCK-8 in DMEM, 0.1% BSA at 37 °C (0.5 ml final volume). Nonspecific binding was defined as total binding in the presence of 1 µM unlabeled CCK-8 and was always <15% that of total binding. After termination of the binding reaction by washing the cells two times with phosphate-buffered saline (PBS; 8.1 mM NaH2PO4, 1.5 mM KH2PO4, 138 mM NaCl, 2.7 mM KCl, pH 7.4) containing 4% BSA at room temperature, cells were solubilized with 1 ml of 1% SDS, and radioactivity was detected in a Packard Autogamma counter (Packard Instrument Co). The half-maximal inhibition of binding (IC50) was determined with the nonlinear curve fitting computer program ALLFIT (33).
Radioligand Stripping Studies
Cells were prepared as mentioned above and then incubated with 50 pM 125I-BH-CCK-8 at 37 °C for various time intervals between 5 and 120 min. At the times indicated, cells were rapidly washed two times with PBS, 4% BSA and subjected to 0.5 M KSCN for 10 min at room temperature. Cell-associated radioactivity was measured after solubilizing the cells with 1 ml of 1% SDS. In all cases, parallel incubations were performed in the presence of 1 µM unlabeled CCK-8 to determine nonsaturable binding at each time point. Internalized receptor was defined as cell-associated radioactivity after stripping and expressed as the percent of the total binding.
Laser Scanning Confocal Microscopy
NIH/3T3 cells expressing WT or mutant CCK receptors were plated in Nunc cover glass chamber slides and assayed the following day. For internalization studies, cells were treated with Rhodamine Green-conjugated CCK-8 (RG-CCK-8) for 15 min at 37 °C, labeled with the cell surface marker Rhodamine B-conjugated concanavalin A (RB-conA) for 2 min at 4 °C, and observed under a laser scanning confocal microscope (Zeiss inverted LSM 410). RG-CCK-8 was excited using a 488-nm argon/krypton laser, and emitted fluorescense was detected through a 515–540-nm band-pass filter. Fluorescence of RB-conA was excited with a 568-nm helium/neon laser, and fluorescence was detected with a 590-nm band-pass filter. All observations were performed using a pinhole of 40, a × 63 oil immersion lens, and an electronic zoom of 3.2 to yield a 2016-fold magnification.
Binding Studies in the Presence of Aluminum Fluoride
NIH/3T3 cells expressing WT or mutant CCK receptors were scraped out of the tissue culture flasks and resuspended at a concentration of 106 cells/ml in DMEM, 0.1% BSA. Following a 30-min preincubation of the cells with 30 mM NaF and 10 µM AlCl3 at 37 °C, 125I-BH-CCK-8 was added to a concentration of 50 pM in a final volume of 0.5 ml and allowed to bind for 90 min at 37 °C. Samples of 0.2 ml of the cell suspension were transferred into 1 ml of PBS, 4% BSA. Cell-associated 125I-BH-CCK-8 was separated from free radioligand by centrifugation (1 min, 13,000 × g) and subsequently washed and recentrifuged two times with 1 ml of PBS, 4% BSA. Gamma radioactivity associated with the pelleted cells was determined in a Packard Autogamma counter. Control experiments were performed as described above either using 30 mM NaCl in place of NaF or in the absence of NaF and AlCl3. For all experiments, nonsaturable binding was determined in the presence of 1 µM CCK-8. Results are expressed as percent of total binding of transfected cells in the absence of NaF and AlCl3.
Measurement of 45Ca2+ Efflux
Similar to a previously described method (34, 35), cells were plated in 24-well tissue culture plates and loaded with 45Ca2+ for 12–16 h with a concentration of 8 µCi/ml. After washing the cells seven times with DMEM containing 3 mM EGTA at room temperature, cells were stimulated with increasing concentrations of unlabeled CCK-8 at 37 °C in a final volume of 0.5 ml. After a 2-min stimulation, 0.1 ml of the supernatant was sampled for 45Ca2+ efflux into Hydrofluor, and radioactivity was measured using a Packard 2500 TR liquid scintillation analyzer. The half-maximal stimulation (EC50) was calculated with the ALLFIT program (33).
RESULTS
To investigate the role of the carboxyl terminus in the internalization of cholecystokinin receptors, we initially truncated the CCKAR after amino acid residue 399 (mutant CCKAR Tr399) and the CCKBR after residue 408 (mutant CCKBR Tr408), thereby deleting the last 45 and 44 residues of the carboxyl terminus, respectively (Fig. 1). These truncation sites were chosen to eliminate all 8 potential phosphorylation sites in the carboxyl terminus of the CCKAR and all 10 potential phosphorylation sites in the carboxyl terminus of the CCKBR. To examine a possible specific involvement of potential phosphorylation sites, all serine and threonine residues in the carboxyl terminus were mutated to alanines, producing the mutants CCKAR ΔS/T and CCKBR ΔS/T (Fig. 1).
NIH/3T3 cells were stably transfected with either WT or mutant CCK receptor subcloned into the pCDL/SRa vector containing the neomycin resistance gene. At least five G418-resistant clones were assayed for 125I-BH-CCK-8 radioligand binding for each WT and mutant transfection. Cell clones with similar receptor densities (receptors/cell = 87 × 103 for the WT CCKAR, 291 × 103 for the CCKAR ΔS/T, 69 × 103 for the CCKAR Tr399, 66 × 103 for the WT CCKBR, 86 × 103 for the CCKBR ΔS/T, and 26 × 103 for the CCKBR Tr408) were chosen for further studies. 125I-BH-CCK-8 binding competition curves demonstrated specific high affinity binding of CCK-8 for all WT and mutant CCKARs and CCKBRs (Fig. 2). All receptors had a similar affinity for CCK-8 with IC50s between 0.37 ± 0.11 and 0.7 ± 0.02 nM for CCKAR constructs (Fig. 2, left panel) and 0.42 ± 0.05 and 0.53 ± 0.05 nM for CCKBR constructs (Fig. 2, right panel).
FIG. 2. Displacement of 125I-BH-CCK-8 binding to NIH/3T3 cells stably expressing WT or mutant CCK receptors.
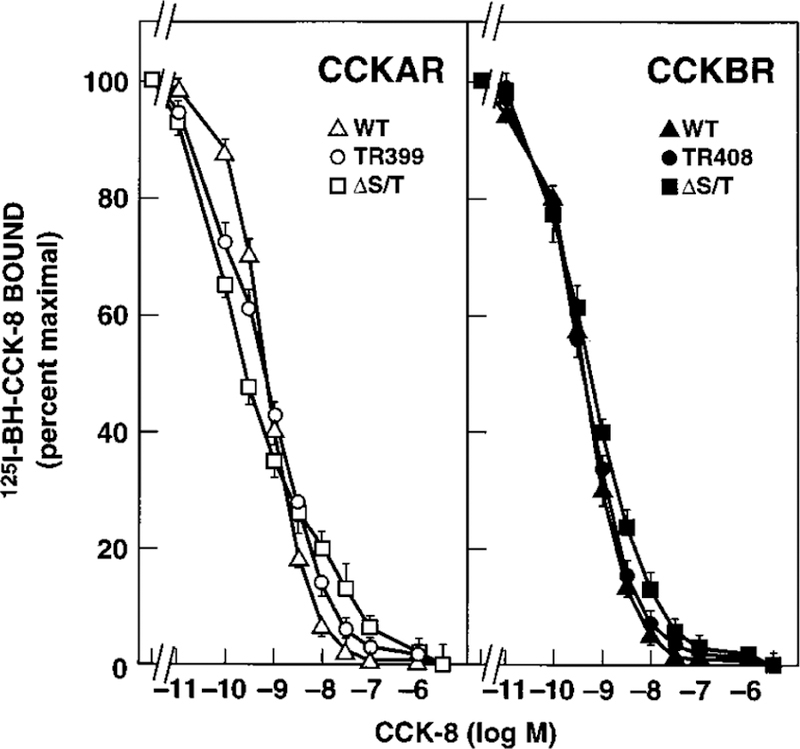
NIH/3T3 cells stably expressing the indicated WT or mutant CCKAR (left panel) or CCKBR (right panel) were incubated with 125I-BH-CCK-8 (50 pM) either alone or with the indicated concentrations of unlabeled CCK-8. Data are presented as percent of saturable binding in the absence of unlabeled ligand. Each value represents the mean ± S.E. of at least three experiments performed in duplicate.
To investigate the effect of the deletion of the carboxyl terminus or the mutation of the serine and threonine residues to alanines on receptor-mediated ligand internalization, radioligand stripping experiments were performed. NIH/3T3 cells expressing the WT CCKAR or WT CCKBR internalized radio-labeled CCK-8 rapidly and to a high degree (Fig. 3). For both WT receptors, >50% of the radioligand was internalized before 15 min. The internalization process reached a plateau after 60 min, with a maximal internalization of 89 ± 2.7% (WT CCKAR) and 92 ± 3.3% (WT CCKBR) of the radioligand at 120 min (Fig. 3). Cells expressing the CCKAR Tr399 as well as cells expressing the CCKAR ΔS/T displayed nearly the same degree and kinetics of internalization as the WT CCKAR (Fig. 3, left panel). In contrast, the internalization of 125I-BH-CCK-8 was profoundly attenuated in cells transfected with the mutant CCKBRs. At 15 min, 89% of the radioligand was stripped with KSCN for both the truncated and the ΔS/T CCKBR mutants, equivalent to 11% internalization (Fig. 3, right panel). After a 120-min incubation, only 26 ± 0.8 and 39 ± 5.3% of the radio-ligand was internalized in cells transfected with the CCKBR Tr408 and CCKBR ΔS/T, respectively (Fig. 3, right panel). There was a small, however significant, difference in internalization between CCKBR Tr408 and CCKBR ΔS/T for the later time points (90 and 120 min). To exclude that differences in internalization were due to clonal variation, KSCN-stripping experiments were also performed on a population of transiently transfected COS-7 cells with similar results. To further demonstrate that the inability to strip radioligand at 37 °C was indeed due to its internalization, we performed KSCN-stripping studies after 4 °C binding, a temperature at which internalization does not occur. As expected, even after 180 min of binding with cells stably expressing either of the six receptor constructs with 50 pM 125I-BH-CCK-8 at 4 °C, 85–94% of the radioligand could still be stripped by KSCN (data not shown).
FIG. 3. Internalization of 125I-BH-CCK-8 by NIH/3T3 cells stably expressing WT or mutant CCK receptors.
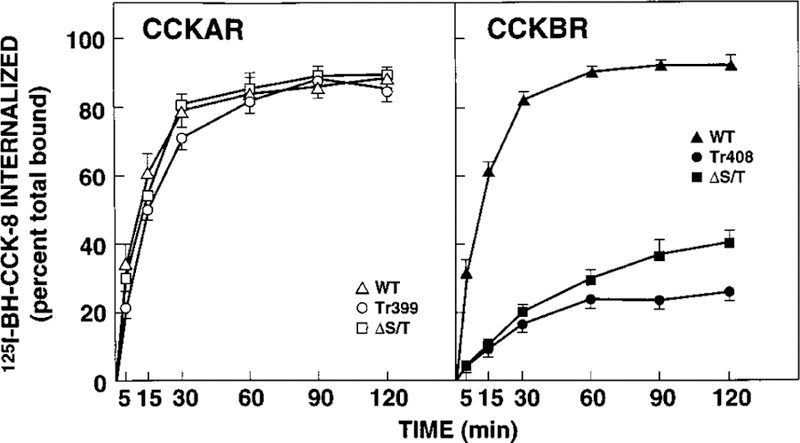
NIH/3T3 cells expressing the indicated WT or mutant CCKAR (left panel) or CCKBR (right panel) were incubated with 125I-BH-CCK-8 (50 pM). At the indicated time points, cells were subjected to a 0.5 M KSCN solution. Internalized radioligand is defined as the 125I-BH-CCK-8 that could not be stripped by KSCN and is expressed as percent of the total saturably bound 125I-BH-CCK-8 in control cells processed in parallel without KSCN exposure. Each value represents the mean ± S.E. of at least three experiments performed in duplicate.
To confirm the effects of the mutations on internalization and to assure that KSCN-resistant 125I-BH-CCK-8 represented internalized receptor, laser scanning confocal microscopy studies were performed. Cells expressing the WT CCKAR or WT CCKBR internalized the majority of the ligand RG-CCK-8 within 15 min away from the cell surface (marked in red with RB-conA) to the interior of the cell, as evidenced by the green intracellular fluorescence (Fig. 4, A and C). Consistent with the radioligand stripping studies, cells transfected with CCKAR Tr399 and CCKAR ΔS/T (Fig. 4B) showed a pattern similar to the WT CCKAR. However, cells expressing CCKBR Tr408 or CCKBR ΔS/T (Fig. 4D) showed colocalization of RB-conA and RG-CCK-8 on the cell surface, which appears as a persistent yellow surface pattern on the overlay of green and red fluorescent images, suggesting that few of the receptors were internalized. Confocal microscopy studies on a population of transiently transfected COS-7 cells with either receptor showed similar results (data not shown), again excluding that differences between WT and mutant receptors were due to clonal variation.
FIG. 4. Internalization of fluorescent Rhodamine Green-conjugated CCK-8 by NIH/3T3 cells stably expressing WT or mutant CCK receptors assessed by confocal laser scanning microscopy.
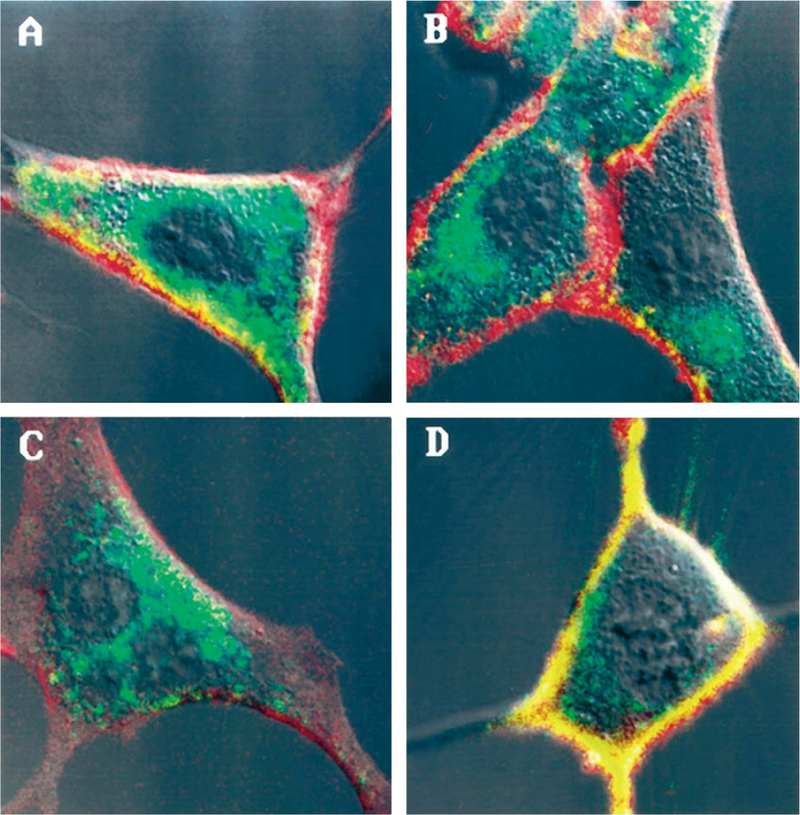
Shown are representative confocal images of NIH/3T3 cells stably expressing the WT CCKAR (A), the CCKAR ΔS/T (B), the WT CCKBR (C), or the CCKBR ΔS/T (D) after a 15-min incubation with Rhodamine Green-conjugated CCK-8. Cell surfaces labeled with Rhodamine B-conjugated concanavalin A are shown in red. Internalized Rhodamine Green-conjugated CCK-8 is shown in green. Colocalization of concanavalin A and Rhodamine Green-conjugated CCK-8 on the cell surface appears as yellow.
For some receptors, intact G protein coupling may be necessary for the internalization process. To determine whether the decreased internalization of the CCKBR Tr408 and the CCKBR ΔS/T was due to an inability to couple to G proteins, we measured the effect of aluminum fluoride on ligand binding. The presence of 30 mM NaF plus 10 µM AlCl3, concentrations previously shown to effectively activate G proteins in intact cells (36, 37) resulting in a decreased ligand affinity of the subsequently uncoupled receptor, decreased the total binding of 125I-BH-CCK-8 for all cells transfected with either of the six receptor constructs to <8% compared with the control with no NaF or AlCl3 in the incubation medium (Fig. 5). Control experiments using 30 mM NaCl plus 10 µM AlCl3 showed a minor effect on total binding (81–99% compared with the control with no additions) (Fig. 5), confirming that the effect of aluminum fluoride was specific and not due to a change in ionic strength or osmolarity.
FIG. 5. Effect of aluminum fluoride on 125I-BH-CCK-8 binding to NIH/3T3 cells stably expressing WT or mutant CCK receptors.
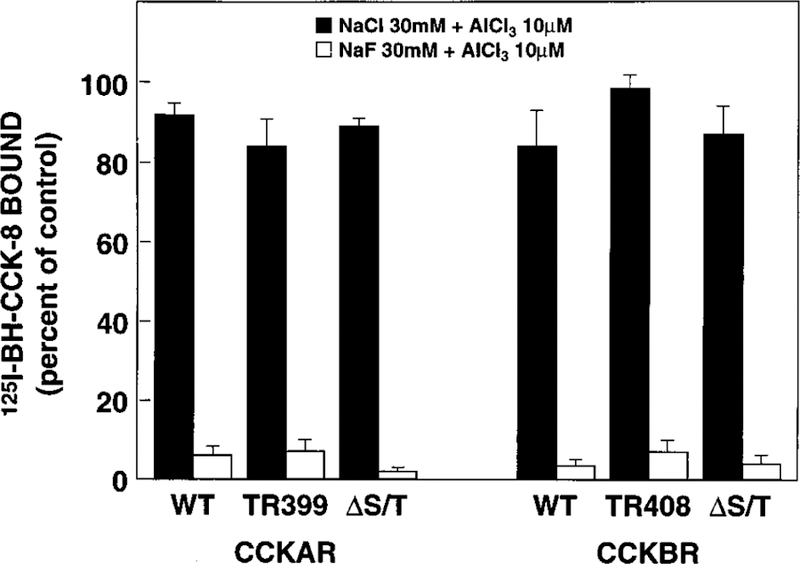
NIH/3T3 cells stably expressing WT or mutant CCKARs (left panel) or CCKBRs (right panel) were incubated with 125I-BH-CCK-8 (50 pM) in the presence of either 30 mM NaCl plus 10 µM AlCl3 (shaded bars) or 30 mM NaF plus 10 µM AlCl3 (open bars). Data are presented as percent of total saturable binding in medium without additions. Each value represents the mean ± S.E. of at least three experiments performed in duplicate.
To exclude that differences in internalization between mutant and WT receptors were due to an altered signal transduction, we determined whether the mutations of the receptors had an effect on the signal transduction cascade by examining 45Ca2+ efflux as a biologic response to CCK-8 stimulation. CCK-8-stimulated 45Ca2+ efflux was maximal at 2 min, in agreement with a previous study (34), and resulted in a 3- to 5-fold increase over basal for all WT and mutant receptors. CCK-8 stimulated 45Ca2+ efflux through WT CCKAR, CCKAR Tr399, and CCKAR ΔS/T in a dose-dependant manner, with nearly identical EC50s (between 1.14 ± 0.06 and 1.5 ± 0.33 nM) (Fig. 6, left panel). Similarly, no significant difference was observed between WT CCKBR, CCKBR Tr408, and CCKBR ΔS/T (EC50s between 0.85 ± 0.1 and 1.65 ± 0.2 nM) (Fig. 6, right panel).
FIG. 6. Ability of CCK-8 to stimulate 45Ca2+ efflux in NIH/3T3 cells stably expressing WT or mutant CCK receptors.
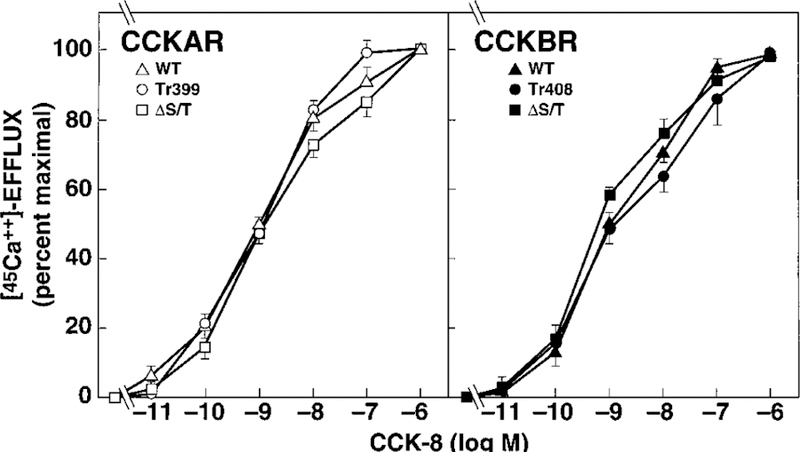
NIH/3T3 cells stably expressing WT or mutant CCKARs (left panel) or CCKBRs (right panel) were stimulated with the indicated concentrations of CCK-8 following a 12–16-h preincubation with 45Ca2+ (8 µCi/ml). Data are presented as percent of maximal stimulation with 1 µM CCK-8. Each value represents the mean ± S.E. of at least three experiments performed in duplicate.
DISCUSSION
The present study examines the role of the carboxyl terminus of cholecystokinin receptors in regulating ligand-stimulated internalization. CCKAR and CCKBR were mutated by either truncation of the carboxyl terminus or by replacement of serine and threonine residues within the carboxyl terminus with alanines. WT and mutant receptors stably expressed in NIH/3T3 cells displayed similar ligand binding, G protein coupling, and signal transduction, suggesting that the carboxyl termini are not necessary for these processes. Receptor internalization assessed by KSCN stripping of the radioligand and confocal microscopy, was reduced to 26% in the CCKBR truncated in the carboxyl terminus after amino acid residue 408 compared with the WT CCKBR internalization of 92%. The mutation of all 10 serine and threonine residues in the carboxyl terminus of the CCKBR could account for the majority of this effect. In contrast, similar mutations of the CCKAR involving either truncation of the carboxyl terminus after amino acid residue 399 or mutation of all eight serine and threonine residues in the carboxyl terminus to alanines failed to affect internalization.
The carboxyl terminus has been shown to influence internalization in a number of G protein-coupled receptors (5, 8, 10–18). However, these truncations have been shown to have unpredictable effects on internalization. To investigate the role of the carboxyl terminus of CCK receptors for internalization, we truncated the carboxyl terminus including all potential phosphorylation sites for both receptors. Truncation of the CCKAR after amino acid residue 399 failed to affect its internalization, whereas truncation of the CCKBR after amino acid residue 408 profoundly attenuated agonist-induced receptor internalization. This suggests that for the CCKBR, unlike for the CCKAR, structural determinants within the carboxyl terminus are required for internalization. However, a conformational change induced by the truncation of the receptor that alters the accessibility or affinity of the endocytic machinery to another unknown internalization motif cannot be ruled out. A permissive effect of the carboxyl terminus for internalization, like for the CCKBR, has been shown for a number of other GPCRs, such as the angiotensin II (8), the yeast α-pheromone (14), the thyrotropin-releasing hormone (13), and the gastrin-releasing peptide receptors (15). All of these truncations involved the removal of the carboxyl terminus and caused an attenuation of receptor internalization without affecting G protein coupling. On the other hand, an inhibiting influence of the carboxyl terminus on internalization was discovered for the luteinizing hormone receptor and the avian β-adrenergic receptor. Removal of the last 43 amino acid residues of the carboxyl terminus of the luteinizing hormone receptor causes the receptor to internalize faster compared with the full-length receptor (12). Carboxyl-terminal truncation enables the avian β-adrenergic receptor to internalize, whereas the WT receptor is not able to internalize (11). Similar to the CCKAR, truncation of the carboxyl terminus of the Hm1 muscarinic cholinergic receptor, which eliminated all potential phosphorylation sites, did not change receptor internalization. Instead, a serine- and threonine-rich region in the third cytoplasmic loop was found to be critical (20).
A variety of structural determinants within the carboxyl terminus of various receptors have been found to influence internalization. To determine whether potential phosphorylation sites within the carboxyl terminus of the CCKBR are involved in receptor internalization, we mutated all serine and threonine residues to alanines. This resulted in a reduced internalization to nearly the same degree as the deletion of the carboxyl terminus, raising the possibility of a specific role of these residues, perhaps as phosphorylation sites, in the signaling for internalization. The mutation of serine and threonine residues in the carboxyl terminus as potential phosphorylation sites has been shown to attenuate receptor internalization for a number of other GPCRs, such as the human β2-adrenergic (10), the gastrin-releasing peptide (15), and the m3-muscarinic acetylcholine receptors (5). For the human β2-adrenergic receptor, the best characterized receptor for internalization, β-arrestin, acting as an adaptor molecule between the receptor and clathrin-coated pits (23), binds to the receptor with much higher affinity when it is phosphorylated in the carboxyl terminus (22). However, an additional domain, a NPXnY motif conserved in many GPCRs, has been found to be critical for the internalization of the human β2-adrenergic receptor (21), suggesting that the signaling involved in internalization may be more complicated. Furthermore, it is possible that the serine and threonine residues in the carboxyl terminus of the CCKBR do not serve as phosphorylation sites but as a part of a larger internalization consensus motif in the carboxyl terminus as has been found for other GPCRs, such as the yeast α-heromone (14) and the thyrotropin-releasing hormone receptors (13). The slightly but significantly greater extent of internalization of the CCKBR ΔS/T compared with the CCKBR Tr408 (39% versus 26%) may therefore reflect an incomplete disruption of such a motif or that another structural determinant within the carboxyl terminus influences internalization. This could be the putative palmitoylation site that is present in the CCKBR ΔS/T but absent in the CCKBR Tr408 and was found to be important for internalization of the thyrotropin-releasing hormone receptor (13).
The confocal microscopy studies visualized the internalization of the fluorescent agonist in cells expressing WT or mutant CCK receptors. Cells transfected with the WT CCKAR or WT CCKBR showed accumulation of the fluorescent ligand in intracellular compartments. For the CCKAR, these results are consistent with the localization of CCKARs expressed in transfected CHO cells by an antiserum directed against the amino terminus of the receptor (38). For the CCKBR, a study on NIH/3T3 cells transfected with the WT CCKBR showed that gastrin was internalized through a clathrin-dependent mechanism accumulating in endosomes and lysosomes (26), a known pathway of internalization for other GPCRs (1). Altogether, these studies indicate that the internalization of the fluorescent ligand in this study reflected the internalization of the receptor. The intracellular pattern of fluorescence for the WT and mutant CCKARs and the WT CCKBR was consistent with the resistance to KSCN stripping of the radioligand. Unlike the WT CCKBR, the confocal pictures of the CCKBR mutants showed that the fluorescent ligand was not translocated to the cell interior, but stayed on the cell surface, suggesting an interruption of the coupling to the endocytic pathway by the truncation or mutation of serine and threonine residues, respectively. This surface pattern was consistent with susceptibility of the radioligand to KSCN stripping.
The relationship between G protein coupling and receptor internalization is still unsettled. Earlier studies on the β-adrenergic and muscarinic receptors suggested a functional relationship between G protein coupling and receptor internalization (39–41). On the other hand, the existence of mutant receptors of the human β2-adrenergic, the α-heromone, and the angiotensin II receptors that are defective in G protein coupling but internalize to the same extent as their respective WT receptors argues that these two processes are unrelated (42–44). A third type of relationship between G protein coupling and internalization is suggested by a recent study on the neurotensin receptor in which a G protein that was not involved in the signal transduction cascade of the receptor was found to affect internalization (18). Regardless, the aluminum fluoride experiments and 45Ca2+ efflux studies showed full G protein coupling and intact signal transduction of all mutant and WT CCKARs and CCKBRs. This excludes the possibility that the internalization defect of the mutants CCKBR Tr408 and CCKBR ΔS/T results from a perturbation of G protein coupling. Finally, these results also indicate that the carboxyl-terminal 45 and 44 amino acids of the CCKAR and CCKBR, respectively, are unnecessary for G protein coupling and signal transduction.
We have shown that the internalization of the CCKBR, unlike that of the CCKAR, depends on structural determinants in the carboxyl-terminal part of the receptor. Therefore, despite a 48% homology in amino acid sequence between the CCKAR and the CCKBR and similar signal transduction pathways, the structural motifs that allow the interaction with the internalization pathway seem to reside in different parts of the receptors. The demonstration that the internalization of CCKBR ΔS/T is impaired almost to the same extent as the internalization of the CCKBR Tr408 would suggest that the serine and threonine residues in the carboxyl terminus are specifically involved in the internalization process. The results of this study will now allow the investigation of the potential relationship between ligand-induced internalization and phosphorylation of the serine and threonine residues in the carboxyl terminus of the CCKBR and the degree to which these processes influence receptor desensitization.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: GPCR, G protein-coupled receptor; CCK, cholecystokinin; CCK-8, CCK octapeptide; CCKAR, cholecystokinin type A receptor; CCKBR, cholecystokinin type B receptor; DMEM, Dulbecco’s modified Eagle’s medium; 125I-BH-CCK-8, 125I-Bolton-Hunter-labeled CCK-8; BSA, bovine serum albumin; WT, wild type; CCKAR Tr399, CCKAR truncated after amino acid residue 399; CCKBR Tr408, CCKBR truncated after amino acid residue 408; PBS, phosphate-buffered saline; RG-CCK-8, rhodamine green-conjugated CCK-8; RB-conA, rhodamine B-conjugated concanavalin A.
REFERENCES
- 1.von Zastrow M, and Kobilka BK (1992) J. Biol. Chem 267, 3530–3538 [PubMed] [Google Scholar]
- 2.von Zastrow M, and Kobilka BK (1994) J. Biol. Chem 269, 18448–18452 [PubMed] [Google Scholar]
- 3.Yu SS, Lefkowitz RJ, and Hausdorff WP (1993) J. Biol. Chem 268, 337–341 [PubMed] [Google Scholar]
- 4.Pippig S, Andexinger S, and Lohse MJ (1995) Mol. Pharmacol 47, 666–676 [PubMed] [Google Scholar]
- 5.Yang J, Williams JA, Yule DI, and Logsdon CD (1995) Mol. Pharmacol 48, 477–485 [PubMed] [Google Scholar]
- 6.Holtmann MH, Roettger BF, Pinon DI, and Miller LJ (1996) J. Biol. Chem 271, 23566–23571 [DOI] [PubMed] [Google Scholar]
- 7.Lohse MJ, Benovic JL, Caron MG, and Lefkowitz RJ (1990) J. Biol. Chem 265, 3202–3209 [PubMed] [Google Scholar]
- 8.Thomas WG, Thekkumkara TJ, Motel TJ, and Baker KM (1995) J. Biol. Chem 270, 207–213 [DOI] [PubMed] [Google Scholar]
- 9.Pals-Rylaarsdam R, Xu Y, Witt-Enderby P, Benovic JL, and Hosey MM (1995) J. Biol. Chem 270, 29004–29011 [DOI] [PubMed] [Google Scholar]
- 10.Hausdorff WP, Campbell PT, Ostrowski J, Yu SS, Caron MG, and Lefkowitz RJ (1991) Proc. Natl. Acad. Sci. U. S. A 88, 2979–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hertel C, Nunnally MH, Wong SK-F, Murphy EA, Ross EM, and Perkins JP (1990) J. Biol. Chem 265, 17988–17994 [PubMed] [Google Scholar]
- 12.Rodriguez MC, Xie Y-B, Wang H, Collison K, and Segaloff DL (1992) Mol. Endocrinol 6, 327–336 [DOI] [PubMed] [Google Scholar]
- 13.Nussenzveig DR, Heinflink M, and Gershengorn MC (1993) J. Biol. Chem 268, 2389–2392 [PubMed] [Google Scholar]
- 14.Rohrer J, Benedetti H, Zanolari B, and Riezman H (1993) Mol. Biol. Cell 4, 511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benya RV, Fathi Z, Battey JF, and Jensen RT (1993) J. Biol. Chem 268, 20285–20290 [PubMed] [Google Scholar]
- 16.Huang Z, Chen Y, and Nissenson RA (1995) J. Biol. Chem 270, 151–156 [DOI] [PubMed] [Google Scholar]
- 17.Chabry J, Botto J-M, Nouel D, Beaudet A, Vincent J-P, and Mazella J (1995) J. Biol. Chem 270, 2439–2442 [DOI] [PubMed] [Google Scholar]
- 18.Hermans E, Octave J-N, and Maloteaux J-M (1996) Mol. Pharmacol 49, 365–372 [PubMed] [Google Scholar]
- 19.Arora KK, Sakai A, and Catt KJ (1995) J. Biol. Chem 270, 22820–22826 [DOI] [PubMed] [Google Scholar]
- 20.Lameh J, Philip M, Sharma YK, Moro O, Ramachandran J, and Sadee W (1992) J. Biol. Chem 267, 13406–13412 [PubMed] [Google Scholar]
- 21.Barak LS, Tiberi M, Freedman NJ, Kwatra MM, Lefkowitz RJ, and Caron MG (1994) J. Biol. Chem 269, 2790–2795 [PubMed] [Google Scholar]
- 22.Ferguson SSG, Downey WE, Colapietro A-M, Barak LS, Menard L, and Caron MG (1996) Science 271, 363–366 [DOI] [PubMed] [Google Scholar]
- 23.Goodman OB, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, and Benovic JL (1996) Nature 383, 447–450 [DOI] [PubMed] [Google Scholar]
- 24.Wank SA (1995) Am. J. Physiol 269, G628–G646 [DOI] [PubMed] [Google Scholar]
- 25.Roettger BF, Rentsch RU, Hadac EM, Hellen EH, Burghardt TP, and Miller LJ (1995) J. Cell Biol 130, 579–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarasova NI, Wank SA, Hudson EA, Romanov VI, Czerwinski G, Resau JH, and Michejda CJ (1997) Cell Tissue Res 287, 325–333 [DOI] [PubMed] [Google Scholar]
- 27.Abdelmoumene S, and Gardner JD (1980) Am. J. Physiol 239, G272–G279 [DOI] [PubMed] [Google Scholar]
- 28.Cherner JA, Naik L, and Singh G (1989) Am. J. Physiol 256, G837–G845 [DOI] [PubMed] [Google Scholar]
- 29.Boden PR, and Hill RG (1988) Neuropeptides 12, 95–103 [DOI] [PubMed] [Google Scholar]
- 30.Ozcelebi F, and Miller LJ (1995) J. Biol. Chem 270, 3435–3441 [DOI] [PubMed] [Google Scholar]
- 31.Gates LK, Ulrich CD, and Miller LJ (1993) Am. J. Physiol 264, G840–G847 [DOI] [PubMed] [Google Scholar]
- 32.Takebe Y, Seiki M, Fujisawa J-I, Hoy P, Yokota K, Arai K-I, Yoshida M, and Arai N (1988) Mol. Cell. Biol 8, 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Lean A, Munson PJ, and Rodbard D (1978) Am. J. Physiol 235, E97–E102 [DOI] [PubMed] [Google Scholar]
- 34.Mendoza SA, Schneider JA, Lopez-Rivas A, Sinnett-Smith JW, and Rozengurt E (1986) J. Cell Biol 102, 2223–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Civan MM, Sinnett-Smith J, Bouzyk M, and Rozengurt E (1993) Am. J. Physiol 265, C1658–C1662 [DOI] [PubMed] [Google Scholar]
- 36.Bigay J, Deterre P, Pfister C, and Chabre M (1987) EMBO J 6, 2907–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marc S, Leiber D, and Harbon S (1988) Biochem. J 255, 705–713 [PMC free article] [PubMed] [Google Scholar]
- 38.Roettger BF, Rentsch RU, Pinon D, Holicky E, Hadac E, Larkin JM, and Miller LJ (1995) J. Cell Biol 128, 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strader CD, Sigal IS, Blake AD, Cheung AH, Register RB, Rands E, Zemcik BA, Candelore MR, and Dixon RAF (1987) Cell 49, 855–863 [DOI] [PubMed] [Google Scholar]
- 40.Cheung AH, Sigal IS, Dixon RAF, and Strader CD (1989) Mol. Pharmacol 34, 132–138 [PubMed] [Google Scholar]
- 41.Thompson AK, Mostafapour SP, Denlinger LC, Bleasdale JE, and Fisher SK (1991) J. Biol. Chem 266, 23856–23862 [PubMed] [Google Scholar]
- 42.Cheung AH, Dixon RAF, Hill WS, Sigal IS, and Strader CD (1990) Mol. Pharmacol 37, 775–779 [PubMed] [Google Scholar]
- 43.Hunyady L, Baukal AJ, Balla T, and Catt KJ (1994) J. Biol. Chem 269, 24798–24804 [PubMed] [Google Scholar]
- 44.Zanolari B, Raths S, Singer-Kruger B, and Riezman H (1992) Cell 71, 755–763 [DOI] [PubMed] [Google Scholar]