

Abstract
Tryptophanyl-tRNA synthetase (WRS) is a cytosolic aminoacyl-tRNA synthetase essential for protein synthesis. WRS is also one of a growing number of intracellular proteins that are attributed distinct noncanonical “moonlighting” functions in the extracellular milieu. Moonlighting aminoacyl-tRNA synthetases regulate processes such as inflammation, but how these multifunctional enzymes are themselves regulated remains unclear. Here, we demonstrate that WRS is secreted from human macrophages, fibroblasts, and endothelial cells in response to the proinflammatory cytokine interferon γ (IFNγ). WRS signaled primarily through Toll-like receptor 2 (TLR2) in macrophages, leading to phosphorylation of the p65 subunit of NF-κB with associated loss of NF-κB inhibitor α (IκB-α) protein. This signaling initiated secretion of tumor necrosis factor α (TNFα) and CXCL8 (IL8) from macrophages. We also demonstrated that WRS is a potent monocyte chemoattractant. Of note, WRS increased matrix metalloproteinase (MMP) activity in the conditioned medium of macrophages in a TNFα-dependent manner. Using purified recombinant proteins and LC-MS/MS to identify proteolytic cleavage sites, we demonstrated that multiple MMPs, but primarily macrophage MMP7 and neutrophil MMP8, cleave secreted WRS at several sites. Loss of the WHEP domain following cleavage at Met48 generated a WRS proteoform that also results from alternative splicing, designated Δ1–47 WRS. The MMP-cleaved WRS lacked TLR signaling and proinflammatory activities. Thus, our results suggest that moonlighting WRS promotes IFNγ proinflammatory activities, and these responses can be dampened by MMPs.
Keywords: multifunctional protein, aminoacyl tRNA synthetase, proteolysis, matrix metalloproteinase (MMP), interferon, inflammation, toll-like receptor (TLR), monocyte, macrophage, innate immunity
Introduction
Aminoacyl-tRNA synthetases perform the crucial task of ligating amino acids to their cognate tRNA molecules during protein translation. Unexpectedly, many aminoacyl-tRNA synthetases are multifunctional proteins (1, 2) that perform additional cellular functions both within (3, 4) and outside the cell (5, 6). Despite the lack of traditional signal peptides, several tRNA synthetases are secreted by nonclassical means (7–14) and perform extracellular “moonlighting” roles, for example regulation of inflammation (6, 15–17) and angiogenesis (18).
The moonlighting functions of the aminoacyl-tRNA synthetases are primarily mediated by additional domains in higher eukaryotic tRNA synthetases (6, 19). Eukaryotic tryptophanyl-tRNA synthetase (WRS)6 (53 kDa; 471 residues) has N-terminal extensions known as the WHEP domain, named after the eukaryotic tRNA synthetase proteins that possess the domain, or vertebrate-specific extension (residues 82–154), which are not required for aminoacylation and indeed are absent from prokaryotic homologs (20, 21). WRS can be alternatively spliced to a proteoform lacking the N-terminal 47 residues, Δ1–47 WRS (22, 23).
Interferon (IFN) γ is a proinflammatory cytokine that, in human umbilical vein endothelial cells, up-regulates the expression of cytosolic WRS and Δ1–47 WRS (9, 22–27) as well as their secretion (9). WRS is also rapidly secreted from immune cells in response to both bacterial (13) and viral infections (14), suggesting a role in inflammation and immunity. Functional differences between WRS and alternatively spliced Δ1–47 WRS remain unclear. In vitro, neutrophil elastase generates stable WRS products, Δ1–70 WRS and Δ1–93 WRS (20), and WRS is also cleaved by plasmin (9). Unlike full-length WRS, elastase-truncated forms of WRS gain angiostatic properties through inhibition of endothelial cell migration and proliferation (20, 28), to promote angiogenesis (29). The effects of cleavage on inflammatory functions of WRS are unknown.
Macrophages adopt a spectrum of cellular phenotypes depending on their mode of activation (30, 31), ranging from the most polarized proinflammatory (M1) macrophages to anti-inflammatory (M2) macrophages that contribute to the resolution of inflammation and promote healing and extracellular matrix reformation. Differentiation to M1 macrophages is induced by IFNγ as well as tumor necrosis factor α (TNFα) (32–34). IFNγ-activated macrophages express several matrix metalloproteinases (MMPs), including MMP7 that has long been associated with macrophage function and is often stated to be destructive in inflammation (35, 36). M2 macrophage polarization is induced by interleukin 4 (IL4) (32, 33).
MMPs not only degrade extracellular matrix proteins but also process virtually all chemokines and a multitude of other signaling factors (1, 34, 37–41). This has led to a shift in interest to their signaling roles, especially in dampening inflammation that defines MMPs as antitargets in many pathologies (42). We have previously identified WRS as a candidate MMP substrate using degradomics (40, 43, 44), i.e. proteomics techniques for the analysis of proteolysis, with quantification enabled by isotope-coded affinity tags (ICAT) (43), isobaric tags for relative and absolute quantification (iTRAQ) (44), or terminal amine isotopic labeling of substrates (TAILS) that identifies the cleavage sites themselves (38, 40). Here, we report the effects of IFNγ on WRS expression and secretion from macrophages as well as proteolytic processing of WRS by MMPs. We describe how cleavage of the N terminus of WRS to the Δ1–47 WRS proteoform by MMP7 and MMP8 abrogates its proinflammatory functions, aligning with other well-documented anti-inflammatory activities of MMPs.
Results
Secretion of WRS is induced by IFNγ
To investigate potential inflammatory roles of WRS, we profiled the secretion of WRS in response to cytokine stimulation of macrophages. THP1 monocytes were differentiated to a macrophage-like phenotype (THP1 M0) using phorbol 12-myristate 13-acetate (PMA). After treatment with IFNγ (20 ng/ml), TNFα (40 ng/ml), or IL4 (40 ng/ml), we collected cytosolic, membrane, and conditioned medium fractions. Immunoblot analysis using antibodies recognizing the N or C terminus of WRS (αN-WRS and αC-WRS, respectively) showed that only the proinflammatory IFNγ increased cytosolic WRS levels (N = 3; Fig. 1A). IFNγ also increased WRS in the cell membrane and conditioned medium fractions, indicating that WRS translocated to the plasma membrane and was secreted to the medium upon IFNγ stimulation and M1 macrophage polarization (Fig. 1A). Several lower-molecular-weight proteoforms of WRS were identified in the cytosol and conditioned medium following IFNγ stimulation. Predominant detection by the αC-WRS antibody revealed that these were N-terminally truncated. The effect on WRS was IFNγ-selective as neither IL4, TNFα (Fig. 1A), IFNα, nor IFNβ (N = 3; Fig. S1A) had any effect on WRS protein levels or secretion. The absence of tubulin in the conditioned media supported secretion of WRS rather than cell lysis, and the absence of the plasma membrane protein Na+/K+-ATPase in the cytosolic fractions confirmed the fidelity of cell fractionation.
Figure 1.

IFNγ stimulates WRS expression and secretion from human cells. A–E, immunoblots of PMA-differentiated THP1-derived macrophages (THP1 M0) (A and C), human peripheral blood mononuclear cell–derived macrophages (primary M0) (B), HUVECs (D), and skin fibroblasts (BJ) (E). Cells were treated for 24 h or for times shown with IFNγ (20 ng/ml), IL4 (40 ng/ml), or TNFα (40 ng/ml) as indicated. Antibodies specific to the N terminus or C terminus of WRS (αN-WRS and αC-WRS, respectively) identified WRS and proteoforms (arrows). Controls and standards were as follows: recombinant human WRS and Δ1–47 WRS (∼58 and 48 kDa, respectively); α-tubulin, loading control; Na+/K+-ATPase, membrane protein loading control; and polarization markers indoleamine 2,3-dioxygenase (IDO), IFNγ stimulation to M1 macrophage polarization, and TGM2, IL4 stimulation to M2 macrophage polarization. Representative immunoblots after 10% SDS-PAGE are shown from N = 3 independent experiments, N = 2 for fibroblasts. Original uncut immunoblots can be seen in Fig. S6, A–F. Ctrl, control.
We isolated human monocytes from the peripheral blood mononuclear cells (PBMCs) of three healthy human subjects. Monocytes were differentiated into primary M0 macrophages using monocyte colony-stimulating factor and then cultured with IFNγ (20 ng/ml) or IL4 (40 ng/ml) to induce polarization to proinflammatory M1- or M2-type macrophages, respectively. Polarization was confirmed using the M1 marker indoleamine 2,3-dioxygenase and M2 marker transglutaminase 2 (Fig. 1B). Immunoblot analysis of cell lysates showed that, as with the THP1-derived macrophages, full-length WRS and additional smaller WRS proteoforms were markedly increased only in response to IFNγ. This included a form migrating like Δ1–47 WRS that was only detected by the αC-WRS antibody. Again, the absence of tubulin in the conditioned culture medium confirmed that there was little, if any, nonspecific release of cytoplasmic proteins by cell lysis.
A time-dependent increase in WRS expression and secretion up to 48 h occurred in response to 20 ng/ml IFNγ in THP1 M0 cells, endothelial cells (N = 3; Fig. 1, C and D), and BJ human skin fibroblasts (N = 2; Fig. 1E), again with no change in tubulin protein. WRS secretion depended on IFNγ concentration, with 10 ng/ml IFNγ inducing maximal WRS secretion (N = 3; Fig. S1B). The secreted proteoforms detected by each antibody were consistent in size among cell types (αN-WRS, ∼41, 39, and 32 kDa; αC-WRS, ∼48 and 36 kDa), and these matched the electrophoretic migration of intracellular forms.
WRS stimulates proinflammatory monocyte and macrophage activities
Phosphorylation of the p65 subunit of NF-κB leads to up-regulated transcription of multiple cytokines and chemokines, including TNFα, in macrophages and a variety of other cells (45). WRS (100 nm) treatment of THP1 M0 cells induced phosphorylation of p65 coincident with a decrease in the level of NF-κB pathway inhibitor IκB-α protein (N = 3; Fig. 2, A–C). Both responses were maximal at the later time point of 60 min, indicating that NF-κB was activated by an indirect pathway.
Figure 2.

Inflammatory signaling responses are activated by WRS but not Δ1–47 WRS. A–C, representative immunoblots (A) and quantification (B and C) of NF-κB p65, phosphorylated p65 (p-p65), and inhibitor of NF-κB IκB-α protein levels in response to recombinant human WRS (100 nm) incubated with PMA-differentiated THP1-derived macrophages (THP1 M0) for the times shown. α-Tubulin was the loading control (see Fig. S6I for images of the uncut original immunoblots). Relative band densities were plotted as means ± S.D. of N = 3 independent experiments. D and E, TNFα protein levels (determined by ELISA, N = 2 independent experiments) in the conditioned media of THP1 M0 cells treated for 3 h with increasing concentrations of WRS (scatter plots with mean ± S.D. bars shown, n = 4) (D) or WRS versus Δ1–47 WRS (100 nm each), heat-denatured WRS (100 °C WRS), or buffer (scatter plots with means ± S.D., n = 4) (E). F, human cytokine protein array analysis of conditioned media of human peripheral blood mononuclear cell–derived macrophages treated for 3 h with WRS, Δ1–47 WRS (100 nm each), or buffer control. Mean pixel intensities were measured by densitometric analysis of the cytokine protein array shown in Fig. S2 and plotted as -fold change compared with untreated cells (scatter plots with mean ± S.D., n = 4) of N = 2 independent experiments. G and H, transwell chemotaxis migration assay of THP1 monocytes in response to increasing concentrations of WRS (G) or WRS, Δ1–47 WRS, heat-denatured WRS (100 nm each), or buffer over 90 min with CCL7 (MCP-3) (50 nm) as a positive control (H). Migrated cell numbers were plotted as -fold changes compared with buffer (scatter plots with mean ± S.D., n = 3) of N = 2 and N = 3 independent experiments for G and H, respectively. Statistical significance was determined against controls: 0 h for B and C; buffer for D, E, G, and H; and untreated for F using a one-way ANOVA with Dunnett's multiple comparison posttests. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. Error bars represent S.D.
By ELISA, we showed that WRS stimulated TNFα release from THP1 M0 cells, with maximal release at 100 nm WRS (N = 2; Fig. 2D). Neither Δ1–47 WRS nor heat-denatured WRS promoted TNFα release at this concentration (N = 3; Fig. 2E), indicating that both the N terminus and tertiary structure of WRS are important for signaling. We used an antibody array to screen for the secretion of 36 different cytokines and chemokines from human PBMC-derived primary macrophages in response to 100 nm WRS (Fig. S2). The array confirmed that WRS increased TNFα secretion in addition to MIP-1α/β, CXCL8 (IL8), and CXCL1 (N = 2; Fig. 2F). Recombinant Δ1–47 WRS did not stimulate cytokine and chemokine release from PBMCs (Fig. 2F).
Dose-response analyses showed that WRS displayed maximal chemoattractant activity for monocytes in transwell migration assays at 100 nm (N = 2; Fig. 2G) (37). As with chemokine secretion, WRS chemoattractant activity was lost upon removal of the N-terminal 47 residues of WRS and heat denaturation (N = 3; Fig. 2H). Thus, the N-terminal WHEP domain of WRS is essential for its signaling and proinflammatory responses.
WRS signals via Toll-like receptors 2 and 4
Several pattern recognition receptors mediate innate immune signaling, including the Toll-like receptor (TLR) family in monocytes and macrophages (45, 46). We used HEK293 cells transfected with an NF-κB alkaline phosphatase reporter linked to TLR2, TLR4, or TLR9, an intracellular DNA receptor as a negative control (Fig. 3A). We found that TLR2 and TLR4 were significantly stimulated by WRS but not by Δ1–47 WRS (100 nm) (N = 2; Fig. 3A), consistent with the loss of activity of Δ1–47 WRS on TNFα release from THP1-derived M0 macrophages (Fig. 2E). Likewise, boiling of WRS led to loss of activity. Thus, the N-terminal sequence and folding of WRS are critical for TLR2 and TLR4 receptor engagement and signaling.
Figure 3.
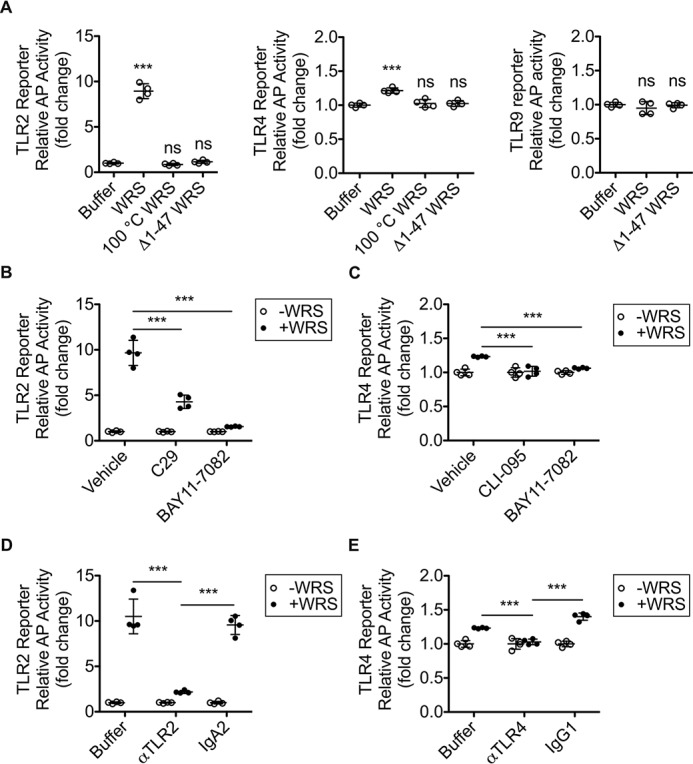
TLR2- and TLR4-mediated signaling responses to WRS. A, HEK293 cells coexpressing TLR2, TLR4, or TLR9 with an NF-κB alkaline phosphatase (AP) reporter system as indicated were treated for 18 h with 100 nm concentrations each of recombinant human WRS, Δ1–47 WRS, heat-denatured WRS (100 °C WRS), or buffer. B and C, reporter cells were pretreated for 1 h with TLR2 inhibitor C29 (100 μm), IκB kinase inhibitor BAY11-7082 (10 μm), TLR4 inhibitor CLI-095 (1 μg/ml), or vehicle (1% (v/v) DMSO) prior to treatment ±WRS (100 nm) for 18 h. D and E, reporter cells were pretreated for 1 h with antibody (5 μg/ml) or buffer prior to treatment ±recombinant human WRS (100 nm) for 18 h. D, TLR2-blocking antibody (αTLR2) and isotype control antibody IgA2. E, TLR4-blocking antibody (αTLR4) or isotype control antibody IgG1.The relative activity of alkaline phosphatase was plotted as follows: A, -fold change for each TLR compared with buffer (scatter plots with mean ± S.D., n = 4) of N = 2 independent experiments; TLR9, N = 1. B–E, -fold change compared with the −WRS control for each treatment (scatter plots with mean ± S.D. bars shown, n = 4) of N = 2 independent experiments. Statistical significance was determined as follows: A, against buffer by a one-way ANOVA with Dunnett's multiple comparison posttests; B–E, between vehicle (or buffer) and inhibitor (or antibody) treatment in the presence of WRS (+WRS) using a two-tailed unpaired Student's t test. ***, p < 0.001; ns, not significant. Error bars represent S.D.
The NF-κB pathway inhibitor BAY11-7082 (10 μm); the IκB kinase inhibitor (47, 48) C29 (100 μm), a specific inhibitor of TLR2 signaling that disrupts the intracellular recruitment of myeloid differentiation primary response gene 88 to TLR2 (49); and CLI-095 (1 μg/ml), a cyclohexane derivative that disrupts the intracellular domain of TLR4 (50, 51) all blocked TLR activation in the WRS-treated reporter cell lines (N = 2; Fig. 3, B and C). This was confirmed by incubating the TLR2 and TLR4 reporter cells with antibodies specific for TLR2 (αTLR2) or TLR4 (αTLR4) (respectively) prior to treatment with 100 nm WRS (N = 2; Fig. 3, D and E). Notably, the alkaline phosphatase readout for TLR2 was ∼10-fold greater than for TLR4, indicating that signaling via TLR2 was the dominant pathway.
The efficacy of the signaling inhibitors in blocking TLR2 and TLR4 signaling at the concentrations used for the TLR reporter cells was investigated in macrophages in response to 100 nm WRS. By ELISA, we measured TNFα release from THP1-derived M0 macrophages incubated in the absence and presence of TLR signaling inhibitors. Each of the inhibitors blocked TNFα release from WRS-treated THP1 M0 cells (N = 2; Fig. 4A), as did the TLR2- and TLR4-blocking antibodies. TNFα release in THP1 M0 cells was reduced 40% by αTLR2 antibody, 25% by αTLR4, and 80% by a combination of these antibodies compared with antibody isotype controls (each at 5 μg/ml) (N = 2; Fig. 4B). Thus, these results and those from the reporter cells demonstrated that WRS triggers TLR2 especially, with a minor contribution by TLR4, to stimulate NF-κB activation and TNFα release in macrophages.
Figure 4.
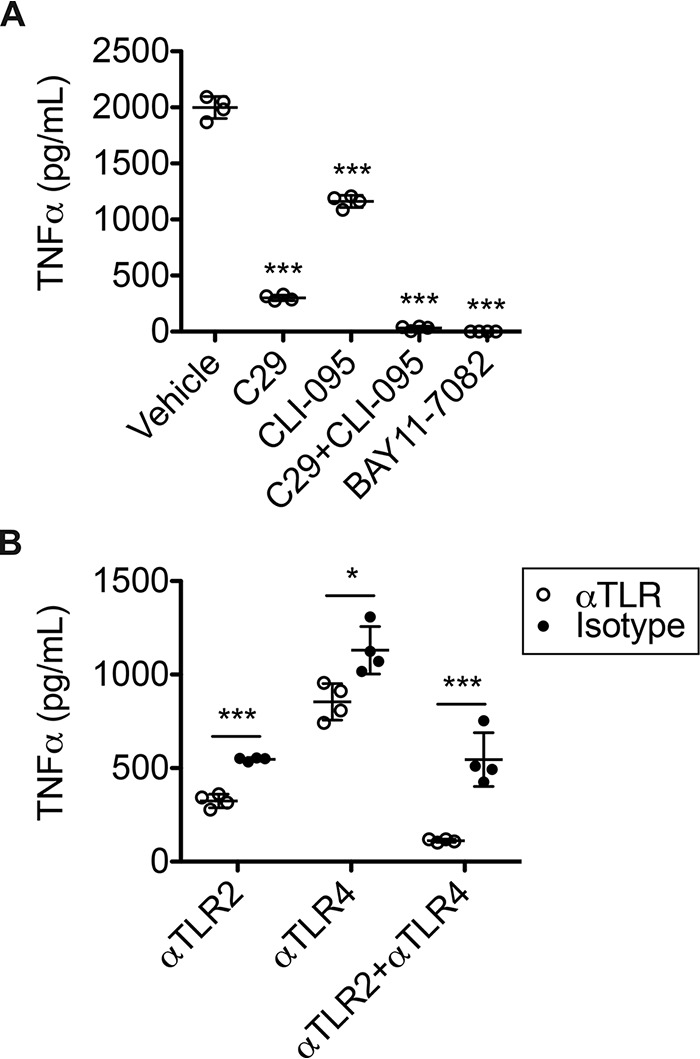
Inhibition of TLR2 and TLR4 signaling reduced WRS-mediated TNFα release from THP1 macrophages. A, ELISA analysis of TNFα released into the conditioned media of PMA-differentiated THP1-derived macrophages in response to treatment with recombinant human WRS (100 nm) in vehicle (1% (v/v) DMSO) for 3 h and after pretreatment for 1 h with TLR2 inhibitor C29 (100 μm), TLR4 inhibitor CLI-095 (1 μg/ml), or IκB kinase inhibitor BAY11-7082 (10 μm). B, TNFα levels in the conditioned media of THP1 M0 cells treated with recombinant human WRS (100 nm) for 3 h after pretreatment for 1 h with TLR2- or TLR4-blocking antibodies (αTLR2 or αTLR4, respectively) or isotype controls (5 μg/ml). Data are presented as scatter plots with mean ± S.D. bars shown, n = 4 of N = 2 independent experiments. Statistical significance was determined against vehicle using a one-way ANOVA with Dunnett's multiple comparison posttests in A and between each isotype control and antibody using a two-tailed unpaired Student's t test in B. *, p < 0.05; ***, p < 0.001; ns, not significant. Error bars represent S.D.
We had removed lipopolysaccharide (LPS), a potent heat-stable proinflammatory TLR stimulator, from our Escherichia coli–expressed recombinant WRS using Triton X-114 during bacterial cell lysis and using polymyxin B–agarose columns after purification. Furthermore, in all experiments, 10 μg/ml polymyxin B was added to all cultures where WRS was used. To be fully confident that WRS, and not any contaminating LPS, drove the proinflammatory effects seen, the recombinant WRS and Δ1–47 WRS used for all presented experiments was expressed in E. coli ClearColi® BL21 (DE3) cells, which produce a modified LPS lacking both the oligosaccharide chain and two of the six acyl chains required for endotoxin signaling in human cells (52, 53). As the Limulus amoebocyte lysate test for endotoxin is ineffective for proteins prepared from ClearColi (52), we further confirmed that the recombinant WRS proteins were free of LPS by demonstrating that the removal of 10 μg/ml polymyxin B from cell culture experiments did not increase TNFα secretion in response to 100 nm WRS, whereas addition of 100 ng/ml LPS did (N = 3; Fig. S3A). Heat denaturation of WRS also eliminated TNFα release even in the absence of polymyxin B, confirming the absence of heat-stable LPS (N = 3; Fig. S3B).
WRS is cleaved by MMPs
MMPs regulate inflammation by processing bioactive proteins to alter their function (34, 36, 54). MMP cleavage of WRS was explored using nine recombinant MMPs (Fig. 5A), which generated stable cleavage products rather than degrading WRS (Fig. 5, A and B): MMP7, MMP8 and MMP13 processed WRS most efficiently with little intact WRS remaining. MMP2, MMP3, MMP9, and MMP14 also cleaved WRS, whereas MMP1 and MMP10 cleaved WRS poorly. Activity for all MMPs was confirmed using quenched fluorescent peptide cleavage assays (not shown), demonstrating that the lack of activity on WRS by MMP1 and MMP10 was due to specificity differences. The spectrum of activity toward WRS was reflected by kinetic analyses (Fig. 5C), which showed that MMP8 and MMP7 cleaved WRS most efficiently with kcat/Km rate constants of 9.28 × 103 and 7.01 × 103 m−1 s−1, respectively, 10-fold higher than MMP14. Plasmin and neutrophil elastase were included as positive controls.
Figure 5.

SDS-PAGE analysis of WRS cleavage by recombinant MMPs in vitro. A, recombinant human WRS (∼58 kDa) was incubated with recombinant MMPs, neutrophil elastase, or plasmin (1:10 protease:WRS molar ratio) for 18 h at 37 °C and analyzed by 12% SDS-PAGE, N = 2 independent experiments. B, WRS was incubated at the protease:WRS molar ratios shown for 18 h at 37 °C, and the cleavage products were analyzed by 10% SDS-PAGE and Coomassie Brilliant Blue G-250 staining. Original uncut gels are presented in Fig. S7, A and B. Controls (Ctrl), highest concentration of protease without WRS. Cleavage products are indicated by arrows. Vertical line artifacts on the MMP7 and MMP8 images are reflections caused by the scanner. C, kcat/Km values for cleavage of WRS.
Edman N-terminal sequencing of the major MMP7 and MMP8 cleavage products (Fig. S4) revealed that WRS was cleaved at Met48 by both MMPs to generate Δ1–47 WRS (∼48 kDa), homologous to the Δ1–47 WRS alternatively spliced proteoform lacking the WHEP domain, and by MMP8 at Val90 to generate Δ1–90 WRS (∼39 kDa). Both MMPs produced a smaller C-terminal WRS proteoform (Δ1–334 WRS) commencing at Leu335 at the junction of the catalytic and anticodon recognition domains (Fig. S4). Peptide mapping using LC-tandem MS (LC-MS/MS) analysis of SDS-PAGE–resolved MMP-cleaved WRS fragments confirmed both N- and C-terminal truncations of WRS (Fig. S5). Other members of the MMP family generated similar proteoforms of WRS: MMP2, MMP3, MMP7, MMP8, MMP9, MMP13, and MMP14 also produced a ∼48-kDa fragment matching the Δ1–47 WRS alternatively spliced WRS. MMP2, MMP3, MMP8, MMP9, MMP13, and MMP14 generated a ∼43-kDa proteoform, and MMP3, MMP8, MMP9, MMP13, and MMP14 generated a ∼39-kDa band comparable with the Δ1–93 WRS product of neutrophil elastase cleavage (Fig. 5B). Because peptide mapping by shotgun proteomics does not necessarily identify the actual neo N-terminal P1′ residue of the cleavage site, amino-terminal oriented mass spectrometry of substrates (ATOMS), a sensitive targeted MS sequencing method for identifying proteolytically generated N termini (55, 56), was employed. Like Edman degradation, ATOMS identifies N-terminal residues without consideration of their relative abundances in a proteolytically digested mixture, but ATOMS is much more sensitive. ATOMS identified >30 cleavage sites, including those characterized by Edman sequencing, many of which were common to several MMPs and resulted in N- and C-terminal deletions (Fig. 6).
Figure 6.
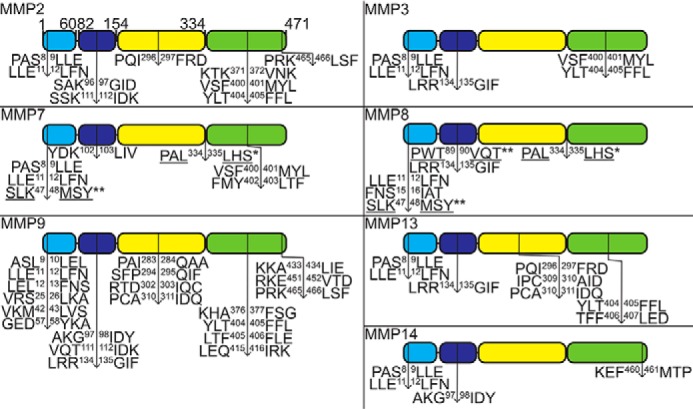
Identification of MMP cleavage sites in WRS. After cleavage of recombinant human WRS by human MMPs, the cleavage sites of all MMPs were identified by ATOMS N-terminal positional proteomics and for MMP7 and MMP8 also by N-terminal sequencing by Edman degradation (WRS cleavage product bands that were sequenced are shown in Fig. S4). Schematic diagrams of WRS with the MMP cleavage sites identified by ATOMS (↓), Edman degradation (*, underlined), or both (**). Only MMP7 and MMP8 cleaved at exactly the same site as the Δ1–47 WRS alternatively spliced form. Light blue, WHEP domain; dark blue, ESE domain; yellow, Rossman fold catalytic domain; green, anticodon recognition domain.
MMP cleavage attenuated the WRS-induced proinflammatory response
We tested how cleavage of WRS by MMP7 and MMP8 affected the WRS-induced proinflammatory responses of monocytes and macrophages (Fig. 7, A and B). Cleavage of WRS by MMP7 or MMP8 decreased chemotaxis of THP1 monocytes by 87 and 76%, respectively, in transwell migration assays (N = 3; Fig. 7, A and C). Similarly, TNFα release from THP1-derived M0 macrophages was reduced by 99 and 84% upon MMP7 or MMP8 cleavage, respectively (100 nm WRS) (N = 3; Fig. 7D). Thus, the removal of the N terminus of WRS by MMP7 or MMP8 abrogated both chemotaxis and TNFα stimulatory responses in THP1 monocytes/macrophages. In other words, proinflammatory TLR signaling by extracellular WRS was inactivated by MMP-mediated removal of the WHEP domain.
Figure 7.

WRS cleavage by MMP7 and MMP8 reduces chemotaxis and TNFα release. A, cleavage of recombinant human WRS (∼58 kDa) by MMP7 and MMP8 (1:10 protease:WRS molar ratio) for 18 h at 37 °C visualized by Coomassie Brilliant Blue G-250–stained 10% SDS-polyacrylamide gels (Fig. S7D shows the original uncut gel). B, schematic diagram of WRS with the MMP7 and MMP8 cleavage sites identified by Edman degradation (↓) (shown in Fig. S4). Light blue, WHEP domain; dark blue, ESE domain; yellow, Rossman fold catalytic domain; green, anticodon recognition domain. C, transwell chemotaxis assay of THP1 monocytes in response to WRS, WRS cleaved by MMP7 or MMP8 (100 nm each), buffer, or MMPs alone (shown in A) over 90 min. Data are presented as -fold change compared with buffer − WRS (scatter plots show means ± S.D., n = 3) of N = 3 independent experiments. D, TNFα released to the conditioned media of PMA-differentiated THP1-derived macrophages treated for 3 h with intact or MMP-cleaved WRS (100 nm each) was measured by ELISA (plotted as scatter plots showing means ± S.D., n = 4) of N = 3 independent experiments. Statistical significance was determined between WRS treated with buffer and WRS treated with each MMP as well as between buffer ± WRS conditions using a two-tailed unpaired Student's t test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. Error bars represent S.D.
TNFα is known to induce the expression of MMPs (57). Because we showed that WRS induced TNFα secretion from macrophages, we tested whether WRS promoted MMP expression via TNFα. Infliximab (100 ng/ml), a mAb inhibitor of TNFα, was added to THP1-derived M0 macrophages treated with 100 nm WRS or Δ1–47 WRS or 40 ng/ml TNFα as a positive control. Using a quenched fluorescent peptide cleavage assay, we assayed for MMP activity in the conditioned medium. WRS treatment increased MMP activity with similar potency to TNFα (N = 3; Fig. 8), but Δ1–47 WRS had no effect, indicating that the increase in MMPs depended on the N-terminal WHEP domain of WRS. Furthermore, the inclusion of infliximab with WRS abrogated the increase in MMP activity. Therefore, WRS mediates increased MMP protein secretion and activity through the downstream mediator TNFα.
Figure 8.
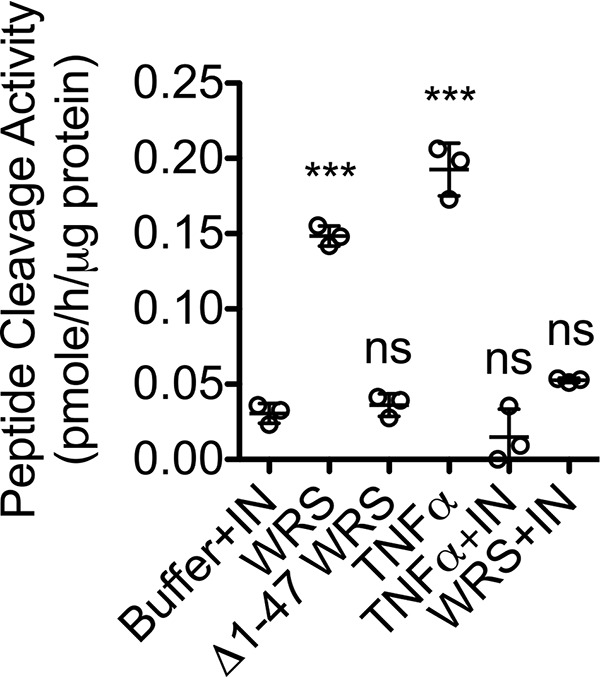
WRS increases MMP activity in macrophage-conditioned media by a TNFα-dependent pathway. PMA-differentiated THP1-derived macrophages were treated for 24 h with recombinant human WRS (100 nm), Δ1–47 WRS (100 nm) as an analog of the MMP7 and MMP8 cleavage product, TNFα (40 ng/ml), or buffer with or without a mAb inhibitor of TNFα (infliximab (IN)) (100 ng/ml). MMPs in 20-fold–concentrated conditioned media were activated with 1 mm APMA, and cleavage of the quenched fluorescence peptide substrate Mca-Pro-Leu-Gly↓Leu-Dpa-Ala-Arg-NH2 was measured (scatter plots show means ± S.D., n = 3) in N = 3 independent experiments. Statistical significance was determined against buffer + infliximab using a one-way ANOVA with Dunnett's multiple comparison posttests. ***, p < 0.001; ns, not significant. Error bars represent S.D.
Discussion
Our study reveals that WRS, one of 37 nuclearly and mitochondrially encoded tRNA synthetases (58), is specifically secreted from human macrophages and other cell types in response to IFNγ, a cytokine critical to both innate and adaptive immunity that is produced during the initiation of immune responses (59, 60). The mechanism of WRS secretion remains uncertain. In endothelial cells stimulated with IFNγ, cytosolic WRS interacts with two known exocytosis-regulating proteins, cytoplasmic annexin A2 and S100A10 (9), to control WRS secretion. In proteomics studies of macrophage secretomes, WRS was identified in extracellular vesicles and exosomes from human primary macrophages (61, 62), PMA-differentiated THP1 cells (63), and exosomes from the murine macrophage cell line Ana-1 (64). It is likely that local concentrations of WRS, like MMPs, could be high because the influx of neutrophils and macrophages can be considerable. 100 nm WRS is ∼5 μg/ml, a concentration that is likely attainable in inflammation. Indeed, concentrations of other innate immune regulatory proteins, such as pentraxin 3 (itself an MMP substrate (65)) and C-reactive protein, increase in concentration 100–1,000-fold during inflammation, up to 5,000 μg/ml for C-reactive protein in plasma (66, 67).
We found several extracellular proinflammatory functions for WRS, including induction of monocyte chemotaxis and stimulation of the release of proinflammatory TNFα and several other chemokines from macrophages, most notably the potent neutrophil chemokine IL8. We also determined that WRS activated TLR2 and, to a lesser extent, TLR4 followed by phosphorylation of NF-κB subunit p65 and loss of the NF-κB inhibitor IκB-α. This is consistent with two recent reports showing that WRS engages the TLR4–myeloid differentiation factor 2 complex to stimulate an innate immune response in murine bone marrow–derived macrophages (13, 14). These authors found that the first 154 residues of WRS were necessary to form the TLR4 complex (13). Our data refined this observation where we showed that recombinant Δ1–47 WRS, corresponding to the WRS splice form that was up-regulated in the cytosol by IFNγ and the major cleavage product of WRS generated by both MMP7 and MMP8, did not activate TLR2 or TLR4 and was inactive in all signaling and inflammatory pathways that we investigated.
In our study, the maximal NF-κB response occurred at 1 h, which is considerably later than is typical for an immediate direct response (68), suggesting a signaling relay, which we showed included TNFα. Indeed, TLR2- and TLR4-dependent release of TNFα into the medium led to increased MMP expression and activity. We showed that MMPs, particularly MMP7 and MMP8 that are secreted from macrophages and neutrophils, respectively, cleaved in the N-terminal domain of WRS to generate Δ1–47 WRS as one of their products. This major cleaved form of WRS did not activate TLR2 or TLR4. In line with these observations, MMP cleavage of WRS reduced TNFα release and monocyte chemotaxis, revealing a negative feedback loop mediated by MMPs that suppresses the proinflammatory actions of WRS (Fig. 9). Thus, MMPs in this context dampen inflammatory responses.
Figure 9.
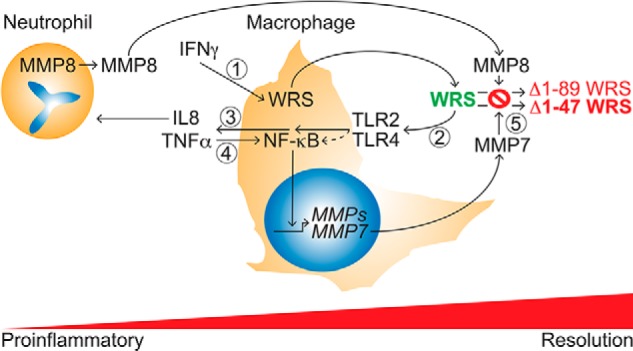
Model of the WRS–MMP temporal relationship and feedback loops regulating IFNγ responses in inflammation. 1, IFNγ activation of M1 macrophage polarization induces secretion of WRS. 2 and 3, WRS activation of TLR2 (and to a lesser extent TLR4) signaling induces monocyte/macrophage chemotaxis (2) and cytokine/chemokine release, notably of TNFα and the neutrophil chemoattractant IL8 (3). 4, TNFα activates NF-κB and up-regulates MMP expression, notably of MMP7, in macrophages. 5, with time, MMP cleavage of WRS, particularly by macrophage MMP7 and neutrophil-specific MMP8, inactivates proinflammatory activities of WRS and so dampens these inflammatory pathways.
The distinct activities of full-length WRS and truncated proteoforms suggested that MMPs modulate IFNγ-induced inflammatory processes through proteolysis of WRS. It is interesting that both MMPs and serine proteases (neutrophil elastase (20) and plasmin (9)) from different cellular and tissue sources generate similar lower-molecular-weight forms through extracellular proteolysis as alternative splicing does within cells, namely the N-terminal truncation of WRS to Δ1–47 WRS. Redundancy of proteases cleaving WRS would afford different cell types and tissues the ability to temporally control the resolution of innate immune responses, further indicating the importance of WRS in host defense. As initially proposed by Jin (17) and because both alternatively spliced Δ1–47 WRS and neutrophil elastase–generated WRS proteoforms exhibit antiangiogenic activity (20), we hypothesize that increased secretion of WRS and accumulation of MMP-generated N-terminally truncated antiangiogenic WRS proteoforms could pause angiogenesis at a site of injury to allow proper resolution of inflammation prior to angiogenesis. Furthermore, neutrophils, which quickly infiltrate a site of tissue injury along IL8 chemoattractant gradients and play an important role in wound healing, secrete neutrophil elastase and MMP8, which potently truncate the N terminus of WRS, terminating its proinflammatory function while promoting angiogenesis (20). Therefore, cleavage of WRS by immune cell MMPs to generate Δ1–47 WRS expands the potential functions of MMPs in regulating angiogenesis and wound healing.
In conclusion, our study shows that, in human cells, MMPs exert post-translational control over moonlighting WRS to down-regulate inflammation. Our degradomics analyses previously indicated that MMPs cleave WRS in secretomes of human MDA-MB-231 cells expressing MMP14 (43) and murine MMP2−/− fibroblasts (44), and we have now elucidated a new MMP–WRS–TNFα axis for regulating inflammation. MMPs process a variety of substrates to regulate inflammation (36, 54), and our data here show that WRS is another such substrate. It would be interesting to examine WRS processing in inflammatory disease, for example both WRS and MMPs are increased in the blood of sepsis patients (13, 69), so it is possible that processing of WRS by MMPs during sepsis has a role in regulating systemic inflammation. To establish the significance of WRS proteolysis by MMPs in the immune response, noncleavable mutant WRS animal models would be valuable because knocking out MMPs would likely be ineffective due to proteolytic redundancy. Overall, our data reveal that WRS is an MMP substrate that is entwined in inflammation due to its moonlighting functions, eclipsing mere matrix-modeling roles for MMPs.
Experimental procedures
WRS expression and purification
Human full-length WRS and Δ1–47 WRS with a C-terminal His tag in pET28a were expressed in ClearColi BL21 (DE3) (Lucigen) in Lennox broth under kanamycin (20 μg/ml) selection. Cell pellets were resuspended in ice-cold column buffer (20 mm KH2PO4, 500 mm NaCl, 10 mm imidazole, 2 mm β-mercaptoethanol, pH 7.8) supplemented with protease inhibitors (1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), 1 mm EDTA, 10 μm E-64, 10 μm leupeptin, 1 μm pepstatin A). Clarified cell lysates were treated with Triton X-114 as described previously (70). Treated lysates were loaded onto a nickel-nitrilotriacetic acid Perfect Pro (Qiagen) column and washed with column buffer at pH 7.8, pH 6, and then pH 5.2 before elution using a 25–250 mm imidazole gradient. Fractions containing WRS were pooled; dialyzed against PBS, 15% glycerol; passed through a polymyxin B–agarose column (Millipore-Sigma); and stored at −80 °C. Protein concentrations were determined by bicinchoninic acid assay (Thermo Fisher Scientific).
Cell culture
The human skin fibroblast cell line BJ (ATCC, catalog number CRL-2522) was cultured in Dulbecco's modified Eagle's medium (DMEM), 4.5 g/liter glucose, 10% (v/v) Cosmic calf serum (CCS; Hyclone), 50 μg/ml streptomycin, and 50 units/ml penicillin. Human primary umbilical vein endothelial cells (HUVECs) (Lonza, catalog number C2519A) were cultured in vascular cell basal medium plus Endothelial Cell Growth kit-VEGF (both from ATCC).
Blood was donated by healthy volunteers using a University of British Columbia Clinical Research Ethics Board–approved protocol (number H06-00047). Informed consent was obtained from all donors, and research was conducted in accordance with the Declaration of Helsinki. PBMCs were prepared from buffy coats obtained using Ficoll-PacqueTM PLUS (GE Healthcare). Monocytes were isolated from PBMCs using an EasySepTM Human Monocyte Isolation kit (Stemcell) following the manufacturer's instructions. Monocytes (2 × 105/ml) were differentiated into primary macrophages (M0) by culturing in 50 ng/ml monocyte colony-stimulating factor in ImmunoCultTM-SF macrophage medium (both Stemcell) for 4 days. For cytokine/chemokine array analysis, PBMC-derived M0 cells were serum-starved and treated for 3 h in serum-free Roswell Park Memorial Institute (RPMI) growth medium, 10 μg/ml polymyxin B (Millipore-Sigma) with 100 nm WRS, Δ1–47 WRS, or PBS. Clarified conditioned medium was assayed using a Human Cytokine Array® (R&D Systems, catalog number ARY005B) according to the manufacturer's instructions. Membranes were blocked with Odyssey Blocking Buffer (LI-COR Biosciences) for 20 min and detected with Alexa Fluor® 680-streptavidin (Thermo Fisher Scientific). Imaging was conducted with a LI-COR Odyssey IR imager (LI-COR Biosciences). Spots were quantified by densitometry using NIH ImageJ software.
THP1 cells (human monocytic cell line; ATCC, catalog number TIB-202) were cultured in RPMI growth medium (RPMI 1640 medium, 4.5 g/liter glucose, 10% (v/v) CCS, 50 μg/ml streptomycin, 50 units/ml penicillin). THP1 M0 macrophages were differentiated from THP1 monocytes (1 × 106 cells/ml) using 100 ng/ml PMA as described previously (34). Experiments were performed in serum-free growth medium containing 10 μg/ml polymyxin B on PMA-differentiated THP1 M0 cells synchronized by serum starvation. For NF-κB pathway analyses, cells were treated with 100 nm WRS for up to 3 h. PBS-washed cells were lysed with 50 mm Tris-HCl, 150 mm NaCl, 10 mm EDTA, 0.2% (w/v) Zwittergent, pH 8, supplemented with a protease and phosphatase inhibitor mixture (Biotool). Cleared lysates were analyzed by immunoblotting (see below). For TNFα release, THP1 M0 cells were treated for 3 h with 100 nm WRS, heat-denatured WRS (boiled for 5 min), Δ1–47 WRS, E. coli 0111:B4 LPS (Millipore-Sigma), or PBS. TNFα in conditioned media was quantified using a human TNFα ELISA kit (Duoset® ELISA, R&D Systems). For TLR inhibitor experiments, 5 μg/ml TLR2-blocking antibody (InvivoGen, catalog number maba2-htlr2), IgA2 (InvivoGen, catalog number maba2-ctrl) isotype control antibody, TLR4-blocking antibody (InvivoGen, catalog number mabg-htlr4), IgG1 isotype control (InvivoGen, catalog number mabg1-crtlm), 1% (v/v) DMSO vehicle, 100 μm C29 (MedChemExpress), 1 μg/ml CLI-095 (InvivoGen), 10 μm BAY11-7082 (Selleckchem), or combinations thereof were added to THP1 M0 cells 1 h prior to treatment with 100 nm WRS for 3 h.
For chemotaxis, 100 nm WRS, heat-denatured WRS, Δ1–47 WRS, 50 nm CCL7, or PBS in chemotaxis buffer (RPMI 1640 medium, 20 mm HEPES, 0.1% BSA, 10 μg/ml polymyxin B) was dispensed into the lower reservoir of chemotaxis chambers (Neuroprobe). A 5-μm membrane was placed over the wells. Serum-starved THP1 monocytes were resuspended (1 × 106 cells/ml) in chemotaxis buffer, and cells (2 × 105 cells) were added to the top reservoir. After 90 min at 37 °C, cells were collected from the bottom chamber and quantified using CyQUANT® (Thermo Fisher Scientific). CCL7 was synthesized as described previously (71).
HEK-BlueTM (human embryonic kidney 293) cells coexpressing the NF-κB reporter system and TLR2, TLR4, TLR9, or receptor-null counterparts (Null1 and Null2) were cultured in DMEM with selective antibiotics. Cells and reagents were from InvivoGen, and assays were carried out according to their instructions. For TLR reporter assays, 96-well plates were seeded with TLR2 and Null1, TLR4 and Null2, or TLR9 and Null1 HEK-Blue cells and incubated with 100 nm WRS, Δ1–47 WRS, heat-denatured WRS, or PBS with 10 μg/ml polymyxin B for 18 h. Conditioned media were assayed for NF-κB activation in QUANTI-BlueTM detection medium. For TLR inhibitor experiments, cells were incubated with antibodies and inhibitors for 1 h prior to treatment with 100 nm WRS for 18 h as for THP1 M0 cells above.
WRS secretion assays
Serum-free media containing various concentrations of human IFNα (Cedarlane), IFNβ, IFNγ, IL4 (all Peprotech), TNFα (Millipore-Sigma), or combinations thereof were added to PBMC-derived M0 cells, PMA-differentiated THP1 M0 cells, HUVECs, and fibroblasts at 80% confluence. Clarified conditioned media were harvested at times shown, and protease inhibitor mixture (Biotool) and 1 mm EDTA were added. PBS-washed cells were lysed with Zwittergent buffer containing protease inhibitor mixture as above. Membrane and cytosolic fractions were isolated from THP1 M0 cells using a Mem-PERTM Plus membrane protein extraction kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
To precipitate proteins, 15% (v/v) TCA was added to conditioned media samples and centrifuged, and the protein pellet was washed with 100% acetone. Pellets were air-dried, resolubilized by boiling for 5 min in 4× SDS-PAGE loading buffer (0.5 m Tris, 8 m urea, 8% (w/v) SDS, pH 6.8), and diluted 4-fold. Protein concentrations were determined by A280 nm.
Immunoblotting
Samples were diluted in SDS-PAGE loading buffer with DTT and denatured by boiling before separation using 10% SDS-PAGE and transfer to PVDF membrane (Immobilon-FL, Millipore-Sigma). After blocking with Odyssey Blocking Buffer for 20 min, primary antibodies (at concentrations recommended by the manufacturers) were incubated overnight at 4 °C. Primary antibodies were as follows: affinity-purified polyclonal antibodies against WRS (Bethyl Laboratories, catalog numbers A304-274A (N-terminal) and A304-275A (C-terminal)); monoclonal antibodies indoleamine 2,3-dioxygenase (IDO) (catalog number 86630), transglutaminase 2 (TGM2) (catalog number 3557), total NF-κB p65 (catalog number 4764), phospho-NF-κB p65 (Ser536; catalog number 3033), and total IκB-α (catalog number 4814) (all from Cell Signaling Technology); α-tubulin (Santa Cruz Biotechnology, catalog number sc-53646); and Na+/K+-ATPase (Abcam, catalog number ab76020). Secondary antibodies goat anti-mouse IgG conjugated to IRDye 800CW (LI-COR Biosciences) and goat anti-rabbit IgG conjugated to Alexa Fluor 680 (Thermo Fisher Scientific) were applied for 1 h at 25 °C. Imaging was conducted with an Odyssey IR imager. Where indicated, bands were quantified by densitometry using NIH ImageJ software.
MMP expression and purification
Human MMP7 cDNA was subcloned into pGW1GH, both generously provided by J. M. Clements (British Biotech Pharmaceuticals, Oxford, UK). MMP7-pGW1GH was electroporated into CHO-K1 cells (ATCC, catalog number CCL-61) grown in DMEM, 4.5 g/liter glucose, 10% (v/v) CCS, 50 μg/ml streptomycin, 50 units/ml penicillin. Stable clones were selected with 25 μg/ml mycophenolic acid, cultured to 80% confluence in flasks, transferred to 1,700-cm2 roller bottles (Corning), and grown with rotation in serum-free medium (1:1 (v/v) CHO-SFM (Thermo Fisher Scientific):DMEM). One-tenth volume of buffer (500 mm MES, 1 m NaCl, 50 mm CaCl2, pH 6) was added to clarified conditioned medium before loading onto a green Sepharose column (Millipore-Sigma). After washing, bound protein was eluted using a 1–2 m NaCl gradient. The eluate, dialyzed against 20 mm Tris, 5 mm CaCl2, pH 7.4, was loaded onto a Q-Sepharose (Millipore-Sigma) column. After washing, bound protein was eluted with 20 mm Tris, 200 mm NaCl, 5 mm CaCl2, pH 7.4. Fractions containing MMP7 were assayed for activity as described below and stored at −80 °C.
C-terminally FLAG-tagged murine MMP10 in pGW1GH was expressed and purified from CHO-K1 cells. Conditioned medium was loaded onto a green Sepharose column and eluted as described for MMP7. Eluate, dialyzed against 50 mm Tris, 150 mm NaCl, pH 7.4, was loaded onto an αFLAG-agarose column (Millipore-Sigma) and washed with dialysis buffer, and MMP10 was eluted with 100 mm glycine, pH 3.5, into tubes containing 50 μl of 1 m Tris, pH 8. Fractions containing MMP10 were exchanged into 50 mm HEPES, 150 mm NaCl, 5 mm CaCl2, pH 7.2, using Ultra-4 centrifugal filter units (Amicon). Activity was assayed as described below, and aliquots were stored at −80 °C.
We expressed and purified recombinant human MMP1, MMP2, MMP3, MMP8, MMP9, MMP13, and soluble MMP14 (lacking the transmembrane domain) as described previously (72). MMP activity was validated by quenched fluorescence synthetic peptide substrate cleavage assays using Mca-Pro-Leu-Gly↓Leu-Dpa-Ala-Arg-NH2 (R&D systems) as described previously (72, 73).
To assess MMP activity in conditioned medium, THP1 M0 cells were cultured in phenol red–free, serum-free growth medium for 24 h with 10 μg/ml polymyxin B plus 100 nm WRS, TNFα 40 μg/ml, or PBS ± 100 μg/ml infliximab (Novus Biologicals, catalog number NBP2-52655). Clarified conditioned medium was concentrated 20× using Ultra-4 centrifugal filter units, and 40 μg of protein was assayed for MMP activity using the quenched fluorescence peptide cleavage activity as described above.
WRS cleavage assays
Pro-MMPs were activated for 20 min at 25 °C with 1 mm p-aminophenylmercuric acetate (APMA) in HEPES buffer (50 mm HEPES, 150 mm NaCl, 5 mm CaCl2, pH 7.2). APMA was removed by dialysis against HEPES buffer at 4 °C for 1 h. WRS cleavage assays were performed at protease:WRS molar ratios described in the figures. Human serine proteases neutrophil elastase (Millipore-Sigma) and plasmin (Biovision) were reconstituted in 50 mm HEPES, 150 mm NaCl, 5 mm CaCl2, pH 7.2. For kinetic analyses, electrophoretic bands of WRS cleavage fragments were quantified by densitometry as described previously (74) using NIH ImageJ software, and the results were fitted to the following equation.
| (Eq. 1) |
LC-MS/MS analysis of MMP cleavage of WRS
WRS (1 μg) was digested at 37 °C ±MMP for 18 h at molar ratios selected from prior digests. Reactions were diluted into sample buffer (0.5 m Tris, 8% (w/v) SDS, pH 6.8, 20% (v/v) β-mercaptoethanol), boiled for 5 min, and resolved by 10% SDS-PAGE. Gels were stained with Coomassie Brilliant Blue G-250. Cleaved WRS bands were excised, destained, lyophilized, and rehydrated in 15 μl of MS-grade trypsin (12 ng/μl in 50 mm ammonium bicarbonate) for 45 min at 4 °C. Excess buffer was removed, and the gel plugs were resuspended in 15 μl of ammonium bicarbonate for 18 h at 37 °C. Centrifuged plugs were discarded, and supernatants were desalted using StageTips (75). Samples were run on an Easy nLC-1000 (Thermo Fisher Scientific) online-coupled to a UHR Q-TOF Impact II mass spectrometer (Bruker Daltonics) with a CaptiveSpray nanoBooster ionization interface. Peptides (1 μg) were loaded on to a 75-μm × 400-mm analytical column containing ReproSil-Pur C18 1.8-μm stationary phase resin (packed in house; Dr. Maisch GmbH), and the column temperature was maintained at 50 °C. Samples were automatically loaded onto the analytical column at 800 bars using buffer A (0.1% formic acid) and 8-μl injection flush volume. Peptides were eluted using a 125-min gradient established with the nLC at 200 nl/min from 2 to 24% buffer B (99.9% acetonitrile, 0.1% formic acid) over 90 min, then increased to 30% over a 10-min period, further increased to 95% buffer B over 5 min, and finally held at 95% for 15 min. Alternatively, a 60-min gradient with 20-min separation was utilized with similar washing parameters. Peptides were ionized by electrospray ionization (2.2 kV), and MS analysis was performed in positive ion polarity with precursor ions detected from 150 to 1750 m/z. Spectra were acquired using a Top17 data-dependent method with precursor intensity–adjusted MS/MS summation time (Compass oTOF control 1.9, Bruker Daltonics).
For database searching, Bruker .d files were converted to .mgf using DataAnalysis v4.3 (Bruker Daltonics) and searched against the reverse concatenated Homo sapiens UniProt database (downloaded January 10, 2014; 69,085 sequences) with the Mascot search algorithm (Matrix Science) using the following parameters: 25-ppm MS1 tolerance; 0.08-Da MS2 tolerance; semitryptic enzyme specificity; and variable modifications oxidation (Met), propionamide (Cys), N-terminal ammonia loss, and N-terminal cyclization at glutamine and glutamic acid. Result files were imported into Scaffold v4.8.7 (Proteome Software) and searched with X!Tandem (76). Search results were filtered for 1% false discovery rate at the peptide and protein level. The N-terminal residue of the most N-terminal semitryptic peptide detected and C-terminal residue of the most C-terminal semitryptic peptide detected were defined as the N and C termini of the excised band, respectively. The MS proteomics data have been deposited with the ProteomeXchange Consortium (77) via the PRIDE partner repository (78) with the data set identifier PXD013217.
Determination of WRS cleavage sites
WRS cleavage sites were determined by Edman degradation (performed by Tufts University Core Facility) as described previously (55, 56) and by positional MS using ATOMS (55, 56). Briefly, WRS (100 μg) was incubated ±MMP (at molar ratios selected from prior digests) for 18 h at 37 °C. Digests were denatured with 4 m guanidine HCl. Cysteines were reduced with 5 mm DTT for 1 h at 37 °C and alkylated with 15 mm iodoacetamide for 15 min at room temperature in the dark. Excess iodoacetamide was quenched with 15 mm DTT for 30 min at 37 °C. Lysine and N termini were labeled with 40 mm heavy (13CD2O) (+MMP) or light (CH2O) (control) formaldehyde with 20 mm sodium cyanoborohydride for 18 h at 37 °C. Excess formaldehyde was quenched with 50 mm ammonium bicarbonate for 2 h at 37 °C. Samples were mixed, split in half, and digested for 16 h at 37 °C with either MS-grade trypsin (1 μg/ml; Thermo Fisher Scientific) or Glu-C (Staphylococcus aureus protease V8; 1 μg/ml; Worthington). Samples were desalted and analyzed by LC-MS/MS as described above.
Data were analyzed using MaxQuant software v1.6.0.1 (79) and searched against a custom protein database including WRS, all proteases used, and 247 protein contaminants frequently observed in MS experiments. Enzyme specificity was set as semispecific free N terminus. Quantitation of peptides was performed using the MS1 signal from heavy (13C2D4; 34.063 Da) and light (C2H4; 28.031 Da) dimethylated peptides. Carbamidomethylation on cysteine was set as a fixed modification, and methionine oxidation and asparagine deamidation were set as variable modifications. Delta score–based false discovery rates of 1 and 5% were set for peptide and protein identification, respectively. Peptide spectrum matches corresponding to N-terminal dimethylated peptides from WRS were manually validated. P1′ residues of the cleavage site were identified from peptides dimethylated at the N terminus that can only be labeled after a cleavage event, with a heavy to light ratio >2, and identified (i) in both trypsin and Glu-C analyses, (ii) in either trypsin and Glu-C analyses based on three or more peptide spectrum matches, or (iii) by Edman degradation. Cleavage sites beginning at the P1′ position with a charged or a hydrophilic residue with the exception of glutamine and cysteine were excluded due to known substrate preferences for MMPs (72). The data have been deposited as above under the data set identifier PXD013367.
Statistics
All statistical tests were performed using GraphPad Prism version 5.0b software.
Author contributions
P. G. J., N. S., and Y. M. data curation; P. G. J., N. S., Y. M., and P. A. B. formal analysis; P. G. J. investigation; P. G. J. methodology; P. G. J. writing-original draft; P. G. J., N. S., Y. M., P. A. B., N. H. K., S. K., C. M. O., and G. S. B. writing-review and editing; N. H. K., S. K., and C. M. O. resources; C. M. O. and G. S. B. conceptualization; C. M. O. and G. S. B. supervision; C. M. O. and G. S. B. funding acquisition; C. M. O. and G. S. B. project administration.
Supplementary Material
Acknowledgment
We thank Peter M. Grin for critical reading of the manuscript and Dr. R. Kappelhoff for laboratory management.
This work was supported by Canadian Institutes of Health Research (CIHR) Grant MOP-111055 (to C. M. O. and G. S. B.), CIHR Foundation Grant FDN-148408 (to C. M. O.), Michael Smith Foundation for Health Research (MSFHR) Grant IN-NPG-00105 to establish the British Columbia Proteomics Network (to C. M. O.), and Canada Foundation for Innovation Grant 31059 (to C. M. O.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S8.
The mass spectrometric raw data and spectral libraries associated with this manuscript are available from ProteomeXchange with the accession numbers PXD013217 and PXD013367.
- WRS
- tryptophanyl-tRNA synthetase
- APMA
- p-aminophenylmercuric acetate
- ATOMS
- amino-terminal oriented mass spectrometry of substrates
- CCS
- Cosmic calf serum
- DMEM
- Dulbecco's modified Eagle's medium
- ESE
- eukaryotic specific extension
- IFN
- interferon
- IL
- interleukin
- LPS
- lipopolysaccharide
- MMP
- matrix metalloproteinase
- PBMC
- peripheral blood mononuclear cell
- PMA
- phorbol 12-myristate 13-acetate
- RPMI
- Roswell Park Memorial Institute
- TLR
- Toll-like receptor
- TNF
- tumor necrosis factor
- IκB-α
- NF-κB inhibitor α
- HUVEC
- human umbilical vein endothelial cell
- CHO
- Chinese hamster ovary
- HEK
- human embryonic kidney
- Mca
- 7-methoxycoumarin-4-yl)acetyl
- Dpa
- N-3-(2,4-dinitrophenyl)-l-2,3-diaminopropionyl
- TGM2
- transglutaminase 2
- ANOVA
- analysis of variance.
References
- 1. Butler G. S., and Overall C. M. (2009) Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 8, 935–948 10.1038/nrd2945 [DOI] [PubMed] [Google Scholar]
- 2. Jobin P. G., Butler G. S., and Overall C. M. (2017) New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim. Biophys. Acta Mol. Cell Res. 1864, 2043–2055 10.1016/j.bbamcr.2017.05.013 [DOI] [PubMed] [Google Scholar]
- 3. Han J. M., Jeong S. J., Park M. C., Kim G., Kwon N. H., Kim H. K., Ha S. H., Ryu S. H., and Kim S. (2012) Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 149, 410–424 10.1016/j.cell.2012.02.044 [DOI] [PubMed] [Google Scholar]
- 4. Ko Y. G., Kim E. Y., Kim T., Park H., Park H. S., Choi E. J., and Kim S. (2001) Glutamine-dependent antiapoptotic interaction of human glutaminyl-tRNA synthetase with apoptosis signal-regulating kinase 1. J. Biol. Chem. 276, 6030–6036 10.1074/jbc.M006189200 [DOI] [PubMed] [Google Scholar]
- 5. Jeffery C. J. (1999) Moonlighting proteins. Trends Biochem. Sci. 24, 8–11 10.1016/S0968-0004(98)01335-8 [DOI] [PubMed] [Google Scholar]
- 6. Guo M., Yang X.-L., and Schimmel P. (2010) New functions of aminoacyl-tRNA synthetases beyond translation. Nat. Rev. Mol. Cell Biol. 11, 668–674 10.1038/nrm2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wakasugi K., and Schimmel P. (1999) Two distinct cytokines released from a human aminoacyl-tRNA synthetase. Science 284, 147–151 10.1126/science.284.5411.147 [DOI] [PubMed] [Google Scholar]
- 8. Park S. G., Kim H. J., Min Y. H., Choi E.-C., Shin Y. K., Park B.-J., Lee S. W., and Kim S. (2005) Human lysyl-tRNA synthetase is secreted to trigger proinflammatory response. Proc. Natl. Acad. Sci. U.S.A. 102, 6356–6361 10.1073/pnas.0500226102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kapoor M., Zhou Q., Otero F., Myers C. A., Bates A., Belani R., Liu J., Luo J. K., Tzima E., Zhang D. E., Yang X. L., and Schimmel P. (2008) Evidence for annexin II-S100A10 complex and plasmin in mobilization of cytokine activity of human TrpRS. J. Biol. Chem. 283, 2070–2077 10.1074/jbc.M706028200 [DOI] [PubMed] [Google Scholar]
- 10. Park M. C., Kang T., Jin D., Han J. M., Kim S. B., Park Y. J., Cho K., Park Y. W., Guo M., He W., Yang X.-L., Schimmel P., and Kim S. (2012) Secreted human glycyl-tRNA synthetase implicated in defense against ERK-activated tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 109, E640–E647 10.1073/pnas.1200194109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Williams T. F., Mirando A. C., Wilkinson B., Francklyn C. S., and Lounsbury K. M. (2013) Secreted threonyl-tRNA synthetase stimulates endothelial cell migration and angiogenesis. Sci. Rep. 3, 1317 10.1038/srep01317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Casas-Tintó S., Lolo F. N., and Moreno E. (2015) Active JNK-dependent secretion of Drosophila tyrosyl-tRNA synthetase by loser cells recruits haemocytes during cell competition. Nat. Commun. 6, 10022 10.1038/ncomms10022 [DOI] [PubMed] [Google Scholar]
- 13. Ahn Y. H., Park S., Choi J. J., Park B.-K., Rhee K. H., Kang E., Ahn S., Lee C.-H., Lee J. S., Inn K.-S., Cho M.-L., Park S.-H., Park K., Park H. J., Lee J.-H., et al. (2016) Secreted tryptophanyl-tRNA synthetase as a primary defence system against infection. Nat. Microbiol. 2, 16191 10.1038/nmicrobiol.2016.191 [DOI] [PubMed] [Google Scholar]
- 14. Lee H.-C., Lee E.-S., Uddin M. B., Kim T.-H., Kim J.-H., Chathuranga K., Chathuranga W. A. G., Jin M., Kim S., Kim C.-J., and Lee J.-S. (2019) Released tryptophanyl-tRNA synthetase stimulates innate immune responses against viral infection. J. Virol. 93, e01291–18 10.1128/JVI.01291-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo M., and Schimmel P. (2013) Essential nontranslational functions of tRNA synthetases. Nat. Chem. Biol. 9, 145–153 10.1038/nchembio.1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim S., You S., and Hwang D. (2011) Aminoacyl-tRNA synthetases and tumorigenesis: more than housekeeping. Nat. Rev. Cancer 11, 708–718 10.1038/nrc3124 [DOI] [PubMed] [Google Scholar]
- 17. Jin M. (2019) Unique roles of tryptophanyl-tRNA synthetase in immune control and its therapeutic implications. Exp. Mol. Med. 51, 1 10.1038/s12276-018-0196-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tzima E., Reader J. S., Irani-Tehrani M., Ewalt K. L., Schwartz M. A., and Schimmel P. (2005) VE-cadherin links tRNA synthetase cytokine to anti-angiogenic function. J. Biol. Chem. 280, 2405–2408 10.1074/jbc.C400431200 [DOI] [PubMed] [Google Scholar]
- 19. Lo W.-S., Gardiner E., Xu Z., Lau C.-F., Wang F., Zhou J. J., Mendlein J. D., Nangle L. A., Chiang K. P., Yang X.-L., Au K.-F., Wong W. H., Guo M., Zhang M., and Schimmel P. (2014) Human tRNA synthetase catalytic nulls with diverse functions. Science 345, 328–332 10.1126/science.1252943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wakasugi K., Slike B. M., Hood J., Otani A., Ewalt K. L., Friedlander M., Cheresh D. A., and Schimmel P. (2002) A human aminoacyl-tRNA synthetase as a regulator of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 99, 173–177 10.1073/pnas.012602099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu X., Zhou H., Zhou Q., Hong F., Vo M.-N., Niu W., Wang Z., Xiong X., Nakamura K., Wakasugi K., Schimmel P., and Yang X.-L. (2018) An alternative conformation of human TrpRS suggests a role of zinc in activating non-enzymatic function. RNA Biol. 15, 649–658 10.1080/15476286.2017.1377868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tolstrup A. B., Bejder A., Fleckner J., and Justesen J. (1995) Transcriptional regulation of the interferon-γ-inducible tryptophanyl-tRNA synthetase includes alternative splicing. J. Biol. Chem. 270, 397–403 10.1074/jbc.270.1.397 [DOI] [PubMed] [Google Scholar]
- 23. Turpaev K. T., Zakhariev V. M., Sokolova I. V., Narovlyansky A. N., Amchenkova A. M., Justesen J., and Frolova L. Y. (1996) Alternative processing of the tryptophanyl-tRNA synthetase mRNA from interferon-treated human cells. Eur. J. Biochem. 240, 732–737 10.1111/j.1432-1033.1996.0732h.x [DOI] [PubMed] [Google Scholar]
- 24. Rubin B. Y., Anderson S. L., Xing L., Powell R. J., and Tate W. P. (1991) Interferon induces tryptophanyl-tRNA synthetase expression in human fibroblasts. J. Biol. Chem. 266, 24245–24248 [PubMed] [Google Scholar]
- 25. Fleckner J., Rasmussen H. H., and Justesen J. (1991) Human interferon γ potently induces the synthesis of a 55-kDa protein (γ2) highly homologous to rabbit peptide chain release factor and bovine tryptophanyl-tRNA synthetase. Proc. Natl. Acad. Sci. U.S.A. 88, 11520–11524 10.1073/pnas.88.24.11520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reano A., Richard M. H., Denoroy L., Viac J., Benedetto J. P., and Schmitt D. (1993) γ interferon potently induces tryptophanyl-tRNA synthetase expression in human keratinocytes. J. Invest. Dermatol. 100, 775–779 10.1111/1523-1747.ep12476463 [DOI] [PubMed] [Google Scholar]
- 27. Fleckner J., Martensen P. M., Tolstrup A. B., Kjeldgaard N. O., and Justesen J. (1995) Differential regulation of the human, interferon inducible tryptophanyl-tRNA synthetase by various cytokines in cell lines. Cytokine 7, 70–77 10.1006/cyto.1995.1009 [DOI] [PubMed] [Google Scholar]
- 28. Zeng R., Chen Y. C., Zeng Z., Liu W. Q., Jiang X. F., Liu R., Qiang O., and Li X. (2011) Effect of mini-tyrosyl-tRNA synthetase/mini-tryptophanyl-tRNA synthetase on ischemic angiogenesis in rats: proliferation and migration of endothelial cells. Heart Vessels 26, 69–80 10.1007/s00380-010-0032-1 [DOI] [PubMed] [Google Scholar]
- 29. Otani A., Slike B. M., Dorrell M. I., Hood J., Kinder K., Ewalt K. L., Cheresh D., Schimmel P., and Friedlander M. (2002) A fragment of human TrpRS as a potent antagonist of ocular angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 99, 178–183 10.1073/pnas.012601899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mantovani A., Sica A., Sozzani S., Allavena P., Vecchi A., and Locati M. (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25, 677–686 10.1016/j.it.2004.09.015 [DOI] [PubMed] [Google Scholar]
- 31. Gordon S. (2003) Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35 10.1038/nri978 [DOI] [PubMed] [Google Scholar]
- 32. Martinez F. O., and Gordon S. (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray P. J., Allen J. E., Biswas S. K., Fisher E. A., Gilroy D. W., Goerdt S., Gordon S., Hamilton J. A., Ivashkiv L. B., Lawrence T., Locati M., Mantovani A., Martinez F. O., Mege J. L., Mosser D. M., et al. (2014) Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dufour A., Bellac C. L., Eckhard U., Solis N., Klein T., Kappelhoff R., Fortelny N., Jobin P., Rozmus J., Mark J., Pavlidis P., Dive V., Barbour S. J., and Overall C. M. (2018) C-terminal truncation of IFN-γ inhibits proinflammatory macrophage responses and is deficient in autoimmune disease. Nat. Commun. 9, 2416 10.1038/s41467-018-04717-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wilson C. L., Ouellette A. J., Satchell D. P., Ayabe T., López-Boado Y. S., Stratman J. L., Hultgren S. J., Matrisian L. M., and Parks W. C. (1999) Regulation of intestinal α-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 286, 113–117 10.1126/science.286.5437.113 [DOI] [PubMed] [Google Scholar]
- 36. Parks W. C., Wilson C. L., and López-Boado Y. S. (2004) Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 4, 617–629 10.1038/nri1418 [DOI] [PubMed] [Google Scholar]
- 37. McQuibban G. A., Gong J. H., Tam E. M., McCulloch C. A., Clark-Lewis I., and Overall C. M. (2000) Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 289, 1202–1206 10.1126/science.289.5482.1202 [DOI] [PubMed] [Google Scholar]
- 38. auf dem Keller U., Prudova A., Eckhard U., Fingleton B., and Overall C. M. (2013) Systems-level analysis of proteolytic events in increased vascular permeability and complement activation in skin inflammation. Sci. Signal. 6, rs2 10.1126/scisignal.2003512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rodríguez D., Morrison C. J., and Overall C. M. (2010) Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 1803, 39–54 10.1016/j.bbamcr.2009.09.015 [DOI] [PubMed] [Google Scholar]
- 40. Bellac C. L., Dufour A., Krisinger M. J., Loonchanta A., Starr A. E., Auf dem Keller U., Lange P. F., Goebeler V., Kappelhoff R., Butler G. S., Burtnick L. D., Conway E. M., Roberts C. R., and Overall C. M. (2014) Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. 9, 618–632 10.1016/j.celrep.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 41. Dean R. A., Cox J. H., Bellac C. L., Doucet A., Starr A. E., and Overall C. M. (2008) Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 112, 3455–3464 10.1182/blood-2007-12-129080 [DOI] [PubMed] [Google Scholar]
- 42. Dufour A., and Overall C. M. (2013) Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol. Sci. 34, 233–242 10.1016/j.tips.2013.02.004 [DOI] [PubMed] [Google Scholar]
- 43. Tam E. M., Morrison C. J., Wu Y. I., Stack M. S., and Overall C. M. (2004) Membrane protease proteomics: isotope-coded affinity tag MS identification of undescribed MT1-matrix metalloproteinase substrates. Proc. Natl. Acad. Sci. U.S.A. 101, 6917–6922 10.1073/pnas.0305862101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dean R. A., and Overall C. M. (2007) Proteomics discovery of metalloproteinase substrates in the cellular context by iTRAQTM labeling reveals a diverse MMP-2 substrate degradome. Mol. Cell. Proteomics 6, 611–623 10.1074/mcp.M600341-MCP200 [DOI] [PubMed] [Google Scholar]
- 45. Kawai T., and Akira S. (2007) Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469 10.1016/j.molmed.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 46. Henning L. N., Azad A. K., Parsa K. V., Crowther J. E., Tridandapani S., and Schlesinger L. S. (2008) Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J. Immunol. 180, 7847–7858 10.4049/jimmunol.180.12.7847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pierce J. W., Schoenleber R., Jesmok G., Best J., Moore S. A., Collins T., and Gerritsen M. E. (1997) Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 272, 21096–21103 10.1074/jbc.272.34.21096 [DOI] [PubMed] [Google Scholar]
- 48. Melisi D., and Chiao P. J. (2007) NF-κB as a target for cancer therapy. Expert Opin. Ther. Targets 11, 133–144 10.1517/14728222.11.2.133 [DOI] [PubMed] [Google Scholar]
- 49. Mistry P., Laird M. H., Schwarz R. S., Greene S., Dyson T., Snyder G. A., Xiao T. S., Chauhan J., Fletcher S., Toshchakov V. Y., MacKerell A. D. Jr., and Vogel S. N. (2015) Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc. Natl. Acad. Sci. U.S.A. 112, 5455–5460 10.1073/pnas.1422576112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ii M., Matsunaga N., Hazeki K., Nakamura K., Takashima K., Seya T., Hazeki O., Kitazaki T., and Iizawa Y. (2006) A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol. Pharmacol. 69, 1288–1295 10.1124/mol.105.019695 [DOI] [PubMed] [Google Scholar]
- 51. Kawamoto T., Ii M., Kitazaki T., Iizawa Y., and Kimura H. (2008) TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur. J. Pharmacol. 584, 40–48 10.1016/j.ejphar.2008.01.026 [DOI] [PubMed] [Google Scholar]
- 52. Mamat U., Wilke K., Bramhill D., Schromm A. B., Lindner B., Kohl T. A., Corchero J. L., Villaverde A., Schaffer L., Head S. R., Souvignier C., Meredith T. C., and Woodard R. W. (2015) Detoxifying Escherichia coli for endotoxin-free production of recombinant proteins. Microb. Cell Fact. 14, 57 10.1186/s12934-015-0241-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Planesse C., Nativel B., Iwema T., Gasque P., Robert-Da Silva C., and Viranaïcken W. (2015) Recombinant human HSP60 produced in ClearColi BL21(DE3) does not activate the NFκB pathway. Cytokine 73, 190–195 10.1016/j.cyto.2015.01.028 [DOI] [PubMed] [Google Scholar]
- 54. Butler G. S., and Overall C. M. (2013) Matrix metalloproteinase processing of signaling molecules to regulate inflammation. Periodontol. 2000 63, 123–148 10.1111/prd.12035 [DOI] [PubMed] [Google Scholar]
- 55. Doucet A., and Overall C. M. (2011) Broad coverage identification of multiple proteolytic cleavage site sequences in complex high molecular weight proteins using quantitative proteomics as a complement to Edman sequencing. Mol. Cell. Proteomics 10, M110.003533 10.1074/mcp.M110.003533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Doucet A., and Overall C. M. (2011) Amino-terminal oriented mass spectrometry of substrates (ATOMS): N-terminal sequencing of proteins and proteolytic cleavage sites by quantitative mass spectrometry. Methods Enzymol. 501, 275–293 10.1016/B978-0-12-385950-1.00013-4 [DOI] [PubMed] [Google Scholar]
- 57. Vincenti M. P. (2001) The matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) genes. Transcriptional and posttranscriptional regulation, signal transduction and cell-type-specific expression. Methods Mol. Biol. 151, 121–148 [DOI] [PubMed] [Google Scholar]
- 58. Antonellis A., and Green E. D. (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genomics Hum. Genet. 9, 87–107 10.1146/annurev.genom.9.081307.164204 [DOI] [PubMed] [Google Scholar]
- 59. Schroder K., Hertzog P. J., Ravasi T., and Hume D. A. (2004) Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189 10.1189/jlb.0603252 [DOI] [PubMed] [Google Scholar]
- 60. Hu X., and Ivashkiv L. B. (2009) Review cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity 31, 539–550 10.1016/j.immuni.2009.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cypryk W., Ohman T., Eskelinen E.-L., Matikainen S., and Nyman T. A. (2014) Quantitative proteomics of extracellular vesicles released from human monocyte-derived macrophages upon β-glucan stimulation. J. Proteome Res. 13, 2468–2477 10.1021/pr4012552 [DOI] [PubMed] [Google Scholar]
- 62. Lorey M. B., Rossi K., Eklund K. K., Nyman T. A., and Matikainen S. (2017) Global characterization of protein secretion from human macrophages following non-canonical caspase-4/5 inflammasome activation. Mol. Cell. Proteomics 16, S187–S199 10.1074/mcp.M116.064840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yao Z., Jia X., Megger D. A., Chen J., Liu Y., Li J., Sitek B., and Yuan Z. (2019) Label-free proteomic analysis of exosomes secreted from THP-1-derived macrophages treated with IFN-α identifies antiviral proteins enriched in exosomes. J. Proteome Res. 18, 855–864 10.1021/acs.jproteome.8b00514 [DOI] [PubMed] [Google Scholar]
- 64. Zhu Y., Chen X., Pan Q., Wang Y., Su S., Jiang C., Li Y., Xu N., Wu L., Lou X., and Liu S. (2015) A comprehensive proteomics analysis reveals a secretory path- and status-dependent signature of exosomes released from tumor-associated macrophages. J. Proteome Res. 14, 4319–4331 10.1021/acs.jproteome.5b00770 [DOI] [PubMed] [Google Scholar]
- 65. Butler G. S., Dean R. A., Tam E. M., and Overall C. M. (2008) Pharmacoproteomics of a metalloproteinase hydroxamate inhibitor in breast cancer cells: dynamics of membrane type 1 matrix metalloproteinase-mediated membrane protein shedding. Mol. Cell. Biol. 28, 4896–4914 10.1128/MCB.01775-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Daigo K., Inforzato A., Barajon I., Garlanda C., Bottazzi B., Meri S., and Mantovani A. (2016) Pentraxins in the activation and regulation of innate immunity. Immunol. Rev. 274, 202–217 10.1111/imr.12476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sproston N. R., and Ashworth J. J. (2018) Role of C-reactive protein at sites of inflammation and infection. Front. Immunol. 9, 754 10.3389/fimmu.2018.00754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Klein T., Fung S.-Y., Renner F., Blank M. A., Dufour A., Kang S., Bolger-Munro M., Scurll J. M., Priatel J. J., Schweigler P., Melkko S., Gold M. R., Viner R. I., Régnier C. H., Turvey S. E., et al. (2015) The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-κB signalling. Nat. Commun. 6, 8777 10.1038/ncomms9777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yazdan-Ashoori P., Liaw P., Toltl L., Webb B., Kilmer G., Carter D. E., and Fraser D. D. (2011) Elevated plasma matrix metalloproteinases and their tissue inhibitors in patients with severe sepsis. J. Crit. Care 26, 556–565 10.1016/j.jcrc.2011.01.008 [DOI] [PubMed] [Google Scholar]
- 70. Aida Y., and Pabst M. J. (1990) Removal of endotoxin from protein solutions by phase separation using Triton X-114. J. Immunol. Methods 132, 191–195 10.1016/0022-1759(90)90029-U [DOI] [PubMed] [Google Scholar]
- 71. McQuibban G. A., Gong J.-H., Wong J. P., Wallace J. L., Clark-Lewis I., and Overall C. M. (2002) Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 100, 1160–1167 [PubMed] [Google Scholar]
- 72. Eckhard U., Huesgen P. F., Schilling O., Bellac C. L., Butler G. S., Cox J. H., Dufour A., Goebeler V., Kappelhoff R., Keller U. A. D., Klein T., Lange P. F., Marino G., Morrison C. J., Prudova A., et al. (2016) Active site specificity profiling of the matrix metalloproteinase family: proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses. Matrix Biol. 49, 37–60 10.1016/j.matbio.2015.09.003 [DOI] [PubMed] [Google Scholar]
- 73. Knight C. G., Willenbrock F., and Murphy G. (1992) A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 296, 263–266 10.1016/0014-5793(92)80300-6 [DOI] [PubMed] [Google Scholar]
- 74. Starr A. E., and Overall C. M. (2009) Chapter 13. Characterizing proteolytic processing of chemokines by mass spectrometry, biochemistry, neo-epitope antibodies and functional assays. Methods Enzymol. 461, 281–307 10.1016/S0076-6879(09)05413-5 [DOI] [PubMed] [Google Scholar]
- 75. Rappsilber J., Mann M., and Ishihama Y. (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 10.1038/nprot.2007.261 [DOI] [PubMed] [Google Scholar]
- 76. Craig R., and Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 10.1093/bioinformatics/bth092 [DOI] [PubMed] [Google Scholar]
- 77. Deutsch E. W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D. S., Bernal-Llinares M., Okuda S., Kawano S., Moritz R. L., Carver J. J., Wang M., Ishihama Y., Bandeira N., et al. (2017) The ProteomeXchange Consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 45, D1100–D1106 10.1093/nar/gkw936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 10.1093/nar/gky1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.