

Abstract
Iron–sulfur clusters are protein cofactors with an ancient evolutionary origin. These clusters are best known for their roles in redox proteins such as ferredoxins, but some iron–sulfur clusters have nonredox roles in the active sites of enzymes. Such clusters are often prone to oxidative degradation, making the enzymes difficult to characterize. Here we report a structural and functional characterization of dihydroxyacid dehydratase (DHAD) from Mycobacterium tuberculosis (Mtb), an essential enzyme in the biosynthesis of branched-chain amino acids. Conducting this analysis under fully anaerobic conditions, we solved the DHAD crystal structure, at 1.88 Å resolution, revealing a 2Fe–2S cluster in which one iron ligand is a potentially exchangeable water molecule or hydroxide. UV and EPR spectroscopy both suggested that the substrate binds directly to the cluster or very close to it. Kinetic analysis implicated two ionizable groups in the catalytic mechanism, which we postulate to be Ser-491 and the iron-bound water/hydroxide. Site-directed mutagenesis showed that Ser-491 is essential for activity, and substrate docking indicated that this residue is perfectly placed for proton abstraction. We found that a bound Mg2+ ion 6.5 Å from the 2Fe–2S cluster plays a key role in substrate binding. We also identified a putative entry channel that enables access to the cluster and show that Mtb-DHAD is inhibited by a recently discovered herbicide, aspterric acid, that, given the essentiality of DHAD for Mtb survival, is a potential lead compound for the design of novel anti-TB drugs.
Keywords: biosynthesis, iron–sulfur protein, crystal structure, protein structure, Mycobacterium tuberculosis, protein stability, dihydroxyacid dehydratase
Introduction
Branched-chain amino acid biosynthesis in eubacteria, fungi, and plants is a highly conserved sequence of reactions catalyzed by only eight enzymes, leading to the biosynthesis of the three branched-chain amino acids: l-isoleucine, l-valine, and l-leucine. Six of the eight enzymes in the pathway have been extensively characterized, both mechanistically and structurally (reviewed in Ref. 1). The intermediates, final amino acid products, and enzymes catalyzing these transformations are shown in Fig. 1. The exceptions are the ilvD-encoded dihydroxyacid dehydratase involved in the l-isoleucine and l-valine pathways and the leuC/D-encoded isopropylmalate isomerase in the l-leucine pathway. Both enzymes are essential for bacterial viability (1, 2), and the dehydratase has no known mammalian orthologs. Both enzymes also contain iron–sulfur clusters that are essential for activity but that render them unstable and difficult to work with under normal aerobic conditions.
Figure 1.

Branched-chain amino acid biosynthesis and the role of DHAD (IlvD).
We focus here on the ilvD-encoded dihydroxyacid dehydratase (EC 4.2.1.9; DHAD),3 which catalyzes the synthesis of 2-keto-2-methylvalerate and 2-ketoisovalerate, the penultimate precursor to l-isoleucine and l-valine, respectively (Fig. 1). Importantly, 2-ketoisovalerate is also a precursor of pantothenate, an essential component of coenzyme A (CoA). DHAD orthologs have been studied from bacteria, fungi, and plants (3–7), and to date all are homodimeric iron–sulfur cluster–containing enzymes with monomer molecular masses between 60 and 70 kDa. The spinach enzyme has been shown, largely from spectroscopic analysis, to contain a [2Fe–2S]+2 cluster (6), whereas the Escherichia coli enzyme has been interpreted as containing a [4Fe–4S]+2 cluster (7). In both cases, changes in the EPR spectra upon addition of substrate suggest that the substrate binds directly to the iron–sulfur cluster in a manner similar to that shown for the [4Fe–4S]+2 cluster in the hydrolase aconitase (8, 9). Bacterial DHADs seem to be particularly unstable; the E. coli enzyme becomes inactivated in aerobic conditions with a t½ of only 23 min and even in anaerobic conditions slowly loses activity (7). Unlike aconitase, E. coli DHAD (Ec-DHAD) is not reactivated by the addition of Fe2+ and thiols under reducing conditions, implying that the cluster is degraded completely. The cluster also reacts rapidly with NO to generate a nitrosyl form of the cluster that is irreversibly inactivated (10).
The mechanism proposed for Ec-DHAD (7) relies on the iron–sulfur cluster, assumed to be [4Fe–4S]+2, acting as a Lewis acid, with the C3 hydroxyl group of the substrate binding as the anion to an available coordination site on one iron atom of the cluster. A general base abstracts the C2 proton and eliminates the cluster-bound hydroxyl group to generate the enol. The enol tautomerizes with stereospecific C3 protonation to generate the keto acid product, suggesting that the tautomerization occurs in the active site before release.
Direct structural information on DHAD enzymes has been elusive, largely because of the experimental challenges associated with the instability of the iron–sulfur cluster. Recently, however, crystal structures have been determined for two sugar dehydratases that are members of the wider DHAD family: an l-arabinonate dehydratase from Rhizobium leguminosarum (Rl-ArDHT) at 2.4 Å resolution (11) and a d-xylonate dehydratase from Caulobacter crescentus (Cc-XyDHT) at 2.7 Å resolution (12). These two enzymes contain [2Fe–2S]+2 clusters but appear to be relatively stable to air. The structure of a plant DHAD from Arabidopsis thaliana (At-DHAD) has also been reported in the context of a gene-directed search for novel herbicides (13). This, too, revealed a 2Fe–2S cluster at the active site.
Herein, we focus on the DHAD enzyme from Mycobacterium tuberculosis (Mtb-DHAD). We sought to clarify the nature and role of its iron–sulfur cluster given the apparent use of a 4Fe–4S cluster in Ec-DHAD. The latter, as the only eubacterial DHAD to be investigated in depth, suggested that eubacterial DHADs might contain 4Fe–4S clusters like that of aconitase (8), in contrast to 2Fe–2S plant DHADs and the sugar dehydratases. We also sought to characterize the Mtb enzyme for its importance as an anti-TB drug target. This latter goal is of particular relevance given the recent discovery of a natural product, aspterric acid, that inhibits At-DHAD (13) and could possibly provide a novel lead compound against TB. Inhibition of DHAD activity would impair the synthesis of all three essential branched-chain amino acids as well as CoA (Fig. 1).
Mtb-DHAD is encoded by the ORF Rv0189c, which has no mammalian counterpart and has been shown to be essential for growth by high-density transposon mutagenesis (2). Evidence of its essentiality in Mtb also comes from the observation that, like its E. coli counterpart, Mtb-DHAD is sensitive to oxygen and NO, and the growth of M. tuberculosis treated with NO can be restored only upon supplementation with the three branched-chain amino acids (5). Down-regulation of the Rv0189c ORF with antisense RNA has also been shown to markedly impair Mtb growth in the lungs of infected mice (5). Although a full gene knockout has yet to be reported, these findings suggest Mtb-DHAD to be an outstanding candidate as an antimycobacterial drug target.
Here we have overcome the instability of Mtb-DHAD by carrying out all key steps in an anaerobic environment. We have established its kinetic properties, determined its three-dimensional structure at 1.88 Å resolution, shown it contains a 2Fe–2S cluster, and defined the nature and environment of this cluster, pointing to its likely direct role in the catalytic mechanism. We also show that aspterric acid is indeed a potent inhibitor of the Mtb-DHAD enzyme and thus a potential new lead compound for the development of anti-TB drugs.
Results
Mtb-DHAD requires anaerobic conditions for full stability
Mtb-DHAD has previously been expressed in E. coli as a thioredoxin fusion protein and shown to be active (5). We have found, however, that anaerobic conditions are required for full characterization. For the present study we could express the N-terminally His6-tagged enzyme in soluble form in both E. coli and Mycobacterium smegmatis. Purification by immobilized metal affinity chromatography (IMAC) and size-exclusion chromatography (SEC) showed evidence of heterogeneity, however. IMAC gave two peaks: one light brown in color (indicating the holo-protein with an iron–sulfur cluster) and the other colorless. SEC of the holo-protein fraction again gave two peaks: one of them in the void volume, colorless and evidently representing aggregated protein, and the other still light brown. Crystallization trials on the latter fraction gave crystals in 3–5 days, but they were colorless and, despite many attempts at improvement, yielded poor diffraction. We concluded that like the E. coli enzyme, Mtb-DHAD is unstable under aerobic conditions, with the iron–sulfur cluster being either lost or degraded.
All subsequent work on Mtb-DHAD, including expression, purification, crystallization, UV-visible spectroscopy, and all functional studies, was therefore conducted in an anaerobic chamber. To ensure complete incorporation of the iron–sulfur cluster and correct folding of the enzyme, we used an E. coli expression system that included the pPH151 plasmid (a kind gift of Dr. Petra Hänzelmann, University of Wurzburg). This plasmid enables overexpression of the entire E. coli suf operon, which orchestrates the efficient synthesis and incorporation of iron–sulfur clusters and has been shown to substantially increase levels of soluble, correctly folded iron–sulfur–containing proteins (14). The expressed Mtb-DHAD was purified by IMAC and SEC. After the IMAC step and before SEC, the protein fractions were supplemented with sodium sulfide and ferric chloride (15) to concentrations of 0.4 and 0.6 mm, respectively, to ensure a homogeneous protein sample fully occupied with its iron–sulfur cluster.
Anaerobically produced Mtb-DHAD is catalytically active
The catalytic activity of Mtb-IlvD was monitored using an assay in which the IlvD-catalyzed reaction was coupled with that of the next enzyme in the isoleucine biosynthesis pathway (IlvE) and with glutamate dehydrogenase. The assay results in absorbance changes at 340 nm caused by the oxidation of NADH. A 10-fold excess of IlvE and glutamate dehydrogenase were used in each assay. All activity measurements and spectral recordings were carried out at 30 °C.
A Michaelis–Menten plot showing the activity of Mtb-DHAD toward its physiological substrate, 2,3-dihydroxy isovalerate, at pH 7.8, is shown in Fig. 2A. Calculated Km and kcat values for (R)2,3-dihydroxy isovalerate were 2025 ± 140 μm and 112 ± 4 min−1, respectively. The enzyme was shown to be specific for the R isomer, with no catalytic activity against the S isomer. The presence of both Mg2+ and carbonate were found to be essential, because no activity could be detected in the absence of either component. We also showed that the coupling enzymes used in the assay, glutamate dehydrogenase and branched-chain aminotransferase (IlvE), do not require Mg2+ or carbonate. To show this, we used 2-ketoisovalerate (the reaction product of DHAD) in the reaction, in the absence of Mg2+ and carbonate, to confirm that neither of these species is required for the activity of the coupling enzymes. We also showed that a serine residue, Ser-491, which projects into the active site, is essential for activity (see “Discussion”). Mutation of this serine to alanine abolishes activity.
Figure 2.

Catalysis by Mtb-DHAD. A, Michaelis–Menten plot showing activity toward (R)2,3-dihydroxy isovalerate, giving calculated Km and kcat values of 2025 ± 140 μm and 112 ± 4 min−1, respectively. B, pH-activity profile, showing a pH optimum of ∼7.8 and implicating two groups with pKa values of 7.35 ± 0.05 and 8.30 ± 0.04 in catalysis. C, inhibition studies with aspterric acid, conducted over substrate concentrations from 0.5 to 5 mm at fixed inhibitor concentrations up to 20 μm. These plots showed competitive inhibition, with a Ki of 10.1 ± 0.4 μm. The kinetic experiments (A) and pH profile (B) were carried out in duplicate, and the standard errors quoted were obtained by global fitting to single data sets as described under “Experimental procedures.” The inhibition experiments (C) were only carried out once, because of the small quantities of aspterric acid available, with the standard error in Ki obtained by global fitting as described.
The pH dependence of the catalytic activity was also determined over the pH range 6.6 to 8.8 to identify potential active site groups involved in the reaction. The pH-activity profile of Mtb-DHAD was bell-shaped (Fig. 2B), with a pH optimum of ∼7.8, suggesting the involvement of two groups exhibiting pKa values of 7.35 ± 0.05 and 8.30 ± 0.04 whose ionization is critical for catalytic activity.
Inhibition studies with aspterric acid (Fig. 2C) were conducted over a range of substrate concentrations (0.5 to 5 mm) using the inhibitor at fixed concentrations up to 20 μm. The data were fitted to a competitive inhibition model, which showed that aspterric acid is a competitive inhibitor of Mtb-DHAD with a Ki of 10.1 ± 0.4 μm.
Crystallization and structure determination of Mtb-DHAD
Mtb-DHAD was crystallized with an N-terminal His6 tag present. All crystallization operations were conducted in the same anaerobic chamber in which the enzyme was produced. Brown hexagonal crystals appeared after 5–10 days, growing out of precipitate, and were flash-cooled in liquid nitrogen prior to data collection. The crystals diffracted to ∼1.8 Å resolution on Beamline 21-ID-D (LS-CAT) at the Advanced Photon Source (Argonne National Laboratory), and the structure was readily determined by single-wavelength anomalous diffraction, using the anomalous scattering from the iron atoms of the intrinsic iron–sulfur cluster. The structure was refined at 1.88 Å resolution to a final R-factor of 0.166 and an Rfree value of 0.206. Full data collection and refinement statistics are in Table 1.
Table 1.
Data collection and refinement statistics for Mtb-DHAD
| Anomalousa,b | Nativea | |
|---|---|---|
| Data processing | ||
| Wavelength (Å) | 1.7389 | 1.0782 |
| Space group | P61 | P61 |
| Cell dimensions | ||
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 |
| a, b, c (Å) | 88.44, 88.44, 483.51 | 88.41, 88.41, 483.31 |
| Resolution (Å)c | 80.58–2.00 (2.11–2.00) | 47.4–1.88 (1.98–1.88) |
| Redundancyc | 19.9 (18.9) | 11.7 (12.1) |
| Completeness (%)c | 100.0 (100.0) | 100.0 (100.0) |
| <I/σI>c | 17.2 (3.0) | 15.3 (2.5) |
| Rmergec | 0.109 (0.813) | 0.100 (0.839) |
| Rmeasurec | 0.115 (0.860) | 0.109 (0.919) |
| CC½c | 0.999 (0.881) | 0.999 (0.79) |
| Wilson B-factor (Å2) | 27.1 | |
| Refinement | ||
| No. reflections | 171,437 (17,067) | |
| Rwork/Rfree | 0.1678/0.2055 | |
| No. atoms | 17,661 | |
| Protein | 16,353 | |
| Fe3+, S2−, Mg2+ ions | 50 | |
| Water | 1258 | |
| B-factors (Å2) | ||
| Protein | 33.6 | |
| Ligand/ion | 41.6 | |
| Water | 37.2 | |
| RMSDs | ||
| Bond lengths (Å) | 0.015 | |
| Bond angles (°) | 1.81 | |
| Ramachandran analysisd (%) | ||
| Favored | 98.05 | |
| Allowed | 1.95 | |
| Outliers | 0.0 |
a Single-wavelength anomalous dispersion data were scaled anomalously.
b These data were collected at LS-CAT Beamline 21-ID-D.
c The highest resolution shells are shown in parentheses.
d Ramachandran analysis was from MolProbity (31).
Three-dimensional structure
The asymmetric unit of the crystal structure contains one Mtb-DHAD homotetramer, with a 2Fe–2S cluster in each subunit. Each subunit also has a single active site Mg2+ ion, an associated lysine residue with a carbamylated ϵ-amino group, Lys-139, and three cis-peptides, at Pro-328, Pro-461, and Pro-504. The fit of the model to the electron density is generally excellent, although the first 13 residues in each chain are missing (14 in subunit B), and there are several flexible loops, at residues 416–419 and 488–499, that cannot be completely modeled in one or more subunits.
The organization of the Mtb-DHAD structure is shown in Fig. 3. Each polypeptide chain is folded into two domains that can be approximately defined as residues 1–372 and 393–575, connected by a 20-residue linker peptide, residues 373–392, that meanders over the surface (Fig. 3A). The fold is a complex one that has so far been found only for other dehydratase structures. A search for structural homologs using PDBeFold (16) found only the two bacterial sugar dehydratases, l-arabinonate dehydratase (Rl-ArDHT (11), RMSD 1.51 Å over 521 corresponding Cα atom positions, 37% sequence identity) and d-xylonate dehydratase (Cc-XyDHT (12), RMSD 1.70 Å over 495 residues, 34% sequence identity), and the plant dihydroxyacid dehydratase from A. thaliana (At-DHAD (13), RMSD 1.63 Å over 535 residues, 48% sequence identity).
Figure 3.

Three-dimensional structure of Mtb-DHAD. A, the DHAD monomer in rainbow colors from the N terminus (blue) to the C terminus (red). The orange and yellow spheres show the location of the 2Fe–2S cluster, and the blue sphere shows the nearby Mg2+ ion. B, the DHAD dimer (monomers in green and magenta, respectively), showing how the N-terminal subdomain of each monomer sits over the top of the active site of the other monomer (shown by the spheres of the 2Fe–2S cluster and the Mg2+). C, the tetramer seen in the crystal, a dimer of dimers. Each subunit is shown in a different color. D, SEC–multiangle light scattering analysis of Mtb-DHAD. SEC trace of Mtb-DHAD, showing the refractive index (thin lines) and mass average molecular weight (thick lines) data. The left vertical axis refers to the refractive index trace shown for three different protein concentrations (6.0 mg·ml−1 in green, 3.0 mg·ml−1 in blue, and 1.5 mg·ml−1 in purple). The right vertical axis and dotted line show the expected molecular mass of the dimeric form of Mtb-DHAD to be 118 kDa, calculated from the protein sequence.
As in other members of this family, the N-terminal domain is based on a four-stranded parallel β-sheet with helices packed on either face (an αβα sandwich), and a further layer of helices packed against this. The C-terminal domain adopts a mixed eight-stranded β-barrel structure with a few associated α-helices around its periphery. A feature of this structure that seems likely to be functionally important is the presence of a small subdomain, formed by the N-terminal 50 residues, that protrudes from the main body of the N-terminal domain and packs against a neighboring subunit, close to its active site (Fig. 3B). Helix α1 from this subdomain of subunit C approaches to within 8 Å of the 2Fe–2S cluster of subunit A.
The homotetrameric quaternary structure of Mtb-DHAD in the crystal is closely similar to that found for the sugar dehydratases Rl-ArDHT and Cc-XyDHT. It is best regarded as a dimer of dimers (Fig. 3C) in which the two dimers encompass chains AC and BD. The total surface area buried in the AC dimer interface is 6890 Å2 (3445 Å2 per monomer), and in the BD dimer interface it is 6788 Å2 (3394 Å2 per monomer), whereas the tetramer interface is less extensive. The total buried surface between the two dimers of the tetramer is 5060 Å2 (2530 Å2 per dimer). In solution, however, analysis by SEC with multiangle light scattering indicates that Mtb-DHAD is primarily dimeric under the concentrations tested (Fig. 3D), in accord with the dimeric structure found for At-DHAD (13) and other DHAD enzymes, both plant and bacterial.
Active site
Each of the four subunits of Mtb-DHAD contains a 2Fe–2S cluster, which is essential for activity and marks the location of the active site (Fig. 4). The cluster is estimated to be present at full occupancy as the average B-factor for the cluster atoms is 26.5 Å2, closely comparable with the surrounding protein structure. The cluster occupies an internal cavity, which is bounded in part by hydrophobic residues that include Met-100, Ile-180, Phe-184, Tyr-218, and Leu-467, but also contains some 7 or 8 water molecules and the single Mg2+ ion. One iron atom (Fe1) is coordinated tetrahedrally by the thiolate sulfurs of Cys-64, Cys-137, and the two S2− ions of the cluster. In contrast, the other iron atom (Fe2) has only one cysteine ligand (Cys-214) together with the two bridging S2− ions, leaving one tetrahedral coordination position vacant. A protrusion in the electron density at this position (Fig. 3A) clearly indicates, in each of the four subunits, the coordination of a water molecule, or more likely hydroxide, to Fe2 at a distance of 2.1 Å, completing a regular tetrahedral geometry. The iron–sulfur(Cys) bonds are all 2.3 Å in length, the Fe–S2− bonds are 2.2 Å, and the bond angles range from 104 to 117°, with a mean of 109.4° (values taken from subunit A). The iron-iron distance is 2.7 Å.
Figure 4.

Active site of Mtb-DHAD. A, overall view of the active site, showing the 2Fe–2S cluster (iron atoms in orange and sulfur in gold), the Mg2+ ion (blue sphere), and the carbamylated lysine residue (KCX139). Water molecules bound to the Mg2+ and to Fe2 of the cluster are shown as small red spheres. The catalytically essential residue Ser-491 can be seen projecting into the active site cavity. Some water molecules in the cavity are omitted for clarity. B, The electron density for the 2Fe–2S cluster and its bound water molecule, the Mg2+ ion, and the carbamylated lysine is shown, from a bias-removed Fo − Fc electron density map calculated in Phenix (28) and contoured at 3 σ. Model bias was removed by omission of the atoms in question, followed by cycles of refinement prior to calculation of the map. C, omit electron density map for the flexible loop that carries the catalytically essential Ser-491. Shown for molecule A and calculated as for B.
The magnesium ion is ∼6.5 Å from the 2Fe–2S cluster, coordinated octahedrally by carboxylate oxygens of Asp-96, Asp-138, and Glu-465, two water molecules and one oxygen of the carbamylated ϵ-amino group of Lys-139 (Fig. 4). Carbamylated lysine residues have been found in the divalent metal ion sites of a number of other enzymes, notably the amidohydrolase superfamily (17). In Mtb-DHAD the density for this carbamylated lysine is unequivocal in each of the four subunits and is consistent with the essential requirement for both Mg2+ and bicarbonate for the biological activity of the enzyme. The other oxygen of the carbamyl group is hydrogen-bonded to the nearby His-296.
One other notable feature of the active site is the presence of a flexible loop comprising residues 488–499. In subunit A this loop is well-ordered, with well-defined electron density (Fig. 4D), and extends into the active site such that the hydroxyl group of Ser-491, at its tip, is ∼4.5 Å from the iron-bound water/hydroxide ligand. In subunits B, C, and D, however, the loop is much more poorly defined, with some residues that cannot be modeled, and a different conformation in which Ser-491 is ∼4 Å more distant from the cluster and turned away from it. Mutation of Ser-491 to Ala abolishes catalytic activity by Mtb-DHAD, similarly to the equivalent residue in Rl-ArDHT, Ser-480. The loop carrying this serine has the same conformation in Rl-ArDHT as that in subunit A of Mtb-DHAD. We conclude that this configuration corresponds to the active conformation.
UV and EPR spectroscopy suggest substrate binding to the iron–sulfur cluster
As is typical for proteins containing iron–sulfur clusters, the UV spectrum for Mtb-DHAD comprises broad peaks at ∼340 and 420 nm on the shoulder of the 280-nm protein absorption peak. These are not sufficiently well-defined to point to the exact type of cluster. Importantly, however, a change in the spectrum is observed during the time course of the catalytic reaction, following substrate addition (Fig. 5A), providing support for the likelihood that the substrate binds directly to the cluster, either by displacement of the iron-bound hydroxide or by increase in the coordination number. The changes occur over a period of 5–7 min, then stabilize, and are most pronounced in the range 310–320 nm.
Figure 5.

UV and EPR spectroscopy. A, UV-visible spectra of Mtb-DHAD showing the changes that occur during the enzymatic reaction. The spectrum of the enzyme alone is in blue with the spectral changes that follow addition of substrate shown as black lines. The final spectrum is in red. Inset, spectrum of enzyme alone (blue), final spectrum (red, ∼7 min after addition of substrate), and the difference spectrum (red dashes). B, EPR spectrum of Mtb-DHAD as isolated (dashed lines) and following reduction with dithionite (solid line). C, effect of adding (R)2,3-dihydroxy isovalerate substrate. Top line, EPR spectrum of reduced Mtb-DHAD in the absence of added substrate. Bottom line, the EPR spectrum after 7 min of incubation with substrate. The parameters used for the acquisition of EPR spectra are given under “Experimental procedures.”
More definitive evidence comes from EPR spectroscopy. The structure of the cluster consists of three terminal thiolate sulfur ligands, two bridging S2− ions, a single water or hydroxide, and two strongly anti-ferromagnetically coupled iron atoms. The EPR spectrum of the active protein, as isolated, is essentially featureless, consistent with the 2Fe–2S center its oxidized form (Fig. 5B, dashed line) with the anti-ferromagnetically coupled [Fe(III)Fe(III)], forming a diamagnetic electron spin S = 0 state (18). When the protein is reduced with sodium dithionite, a one-electron reduction, the resulting spectrum (Fig. 5B, solid line) is consistent with a [Fe(II)Fe(III)] S = 0.5 species. The EPR spectrum has an average g value of <2.0 and is readily observable at temperatures of >30 K, as expected for the oxidized 2Fe–2S structure (18). Expansion of the spectrum (Fig. 5C, top line) reveals several peaks in the EPR spectrum, perhaps because of slight structural heterogeneity at cluster active sites. Indeed, the EPR spectrum is sensitive to the addition of the substrate (R)2,3-dihydroxy isovalerate, which induces significant changes to peak intensities and positions (Fig. 5C, bottom line). These EPR results suggest that the substrate binds very near, or directly to, the cluster (6, 7).
Substrate docking in the active site
Attempts to cocrystallize Mtb-DHAD with either of its two substrates were unsuccessful. We therefore used the docking program GOLD (19) to predict the positions and orientations of its two substrates: (2R,3R)-dihydroxy-3-methyl valerate (precursor to isoleucine biosynthesis) and (R)2,3-dihydroxy isovalerate (precursor to valine), and the enol intermediates that follow. The location of the active site cavity, bounded by the 2Fe–2S cluster, the Mg2+ site, the catalytically essential Ser-491, and an area of hydrophobic surface, is clear from the foregoing discussion. Each molecule in turn was placed in the active site cavity of molecule A, in which Ser-491 has its presumed active configuration, and each was allowed to find its preferred position and binding mode independently.
The results were clear and convincing. For (2R,3R)-dihydroxy-3-methyl valerate, nine of the ten solutions were essentially identical (Fig. 6A). Five water molecules are displaced by the substrate. The iron-bound water/hydroxide is replaced by the substrate 3-hydroxyl group, and the two waters bound to the Mg2+ ion are replaced by the 2-hydroxyl and one carboxylate oxygen. The Mg2+-bound carboxylate oxygen is also hydrogen-bonded to Thr-291 Oγ, whereas the other carboxylate oxygen receives a hydrogen bond from Thr-291 NH at the N terminus of an α-helix (residues 218–232). The methyl and ethyl groups attached to C3 occupy a highly hydrophobic pocket formed by Met-100, Ile-180, Ile-181, Phe-184, the aromatic ring of Tyr-218, and Leu-467. Importantly, the distance of C2 from Ser-491 Oγ is ∼2.5 Å, and the C2 proton points directly at Oγ. The valine precursor, (R)2,3-dihydroxy isovalerate, binds in exactly the same way, albeit with some slippage in the positions of its two methyl groups, which do not fill the hydrophobic pocket so completely.
Figure 6.

Substrate docking and mechanism. A, stereo view showing the substrate (2R,3R)-dihydroxy-3-methyl valerate (precursor for Ile biosynthesis) docked into the active site of Mtb-DHAD. The substrate is shown with hydrogen atoms in place (small white spheres). The C3–OH is bound to Fe2 of the 2Fe–2S cluster. The Mg2+ ion is shown as a small blue sphere. The proton at C2 is shown pointing directly at Ser-491. B, proposed catalytic mechanism for Mtb-DHAD. The substrate shown here is (2R,3R)-dihydroxy-3-methyl valerate. Hydrogen bonds are shown by thin broken lines, and metal–ligand interactions are shown as bold dotted lines.
Abstraction of the C2 proton by Ser-491 Oγ, acting as an alkoxide base, leads to formation of the enol intermediate with its double bond between C2 and C3 and concomitant loss of the C3 hydroxyl group. Docking the isoleucine-forming intermediate shows its position is largely the same as that of the substrate, maintaining the same interactions of the C2 carboxylate and the C3 ethyl and methyl groups. The change in stereochemistry with double bond formation results, however, in movement of C3 to a position almost equidistant between Ser-491 Oγ on one side and the original position of the iron-bound water on the other. As for the two substrates, the valine-forming intermediate docks almost identically. For these docking experiments with the two intermediates, again there is a high degree of consistency, with nine of ten of the top solutions being essentially identical.
Discussion
Proteins with iron–sulfur centers have crucial roles in all domains of life. Best-known are those that perform redox roles. In recent years, however, increasing interest has focused on iron–sulfur proteins with nonredox activities, including enzymes and transcription factors (20). One of the best-known of these is aconitase, in which a 4Fe–4S cluster is directly involved in catalysis. Such proteins are generally more difficult to characterize, however, because the need for substrate access to the cluster brings with it a susceptibility to cluster degradation or loss induced by reactive oxygen species. It is no coincidence, for example, that the only enzymes from the branched-chain biosynthetic pathway that have so far resisted structural analysis have been the two with intrinsic iron–sulfur clusters.
We have shown here that Mtb-DHAD has a 2Fe–2S cluster with a potentially exchangeable ligand on one of the iron atoms, the water/hydroxide species bound to Fe2. Our EPR and UV spectral results strongly suggest direct substrate binding to the cluster, a conclusion further supported by our ligand-docking experiments. The latter suggest that the iron-bound water is replaced, retaining the tetrahedral geometry, but we cannot exclude the possibility that the water ligand is retained, with the iron adopting a higher coordination number, as occurs for the 4Fe–4S cluster of aconitase (8).
Our results thus establish Mtb-DHAD as a member of a novel family of enzymes that utilize a 2Fe–2S cluster as a Lewis acid cofactor in a similar manner to the 4Fe–4S cluster of aconitase. Other enzymes to be characterized from this family in the past year, all with 2Fe–2S clusters, are the two bacterial sugar dehydratases and the plant DHAD from A. thaliana. In the mechanism we put forward (Fig. 6B), adapted from that previously proposed for Ec-DHAD (7), the 2Fe–2S cluster acts as a Lewis acid, polarizing the C3–O3 bond as the C3–OH binds to Fe2. This activates the substrate for β-elimination of a water molecule upon abstraction of a proton from C2.
Proton abstraction from C2 is carried out by the essential Ser-491, providing a parallel with aconitase in which a serine residue, Ser-642, carries out a similar proton abstraction (8). The hydroxyl group of Ser-491 projects into the substrate cavity, positioned (in subunit A) such that Oγ is only ∼2.5 Å from C2 of the docked substrate and the C2 proton points directly at it. Ser-491 is thus identified as one of the two ionizable groups from the kinetic analysis. Proton abstraction by the Ser-491 alkoxide also accounts for the stereospecificity of the enzyme for exclusively the R-isomer at C2. Removal of the proton from C2 is followed by loss of the C3–OH and generation of an enol intermediate. The change in stereochemistry caused by double bond formation in this intermediate is predicted by our docking studies to move C3 away from the 2Fe–2S cluster to a position ∼2.5 Å from the original site of the Fe2-bound water/hydroxide. Our data do not show whether the water/hydroxide on Fe2 is displaced by the substrate C3–OH during this reaction or remains bound to iron with expansion of the coordination number as occurs for aconitase (8). Irrespective of this, proton donation to C3 from an iron-bound water would lead to formation of a product with the correct stereochemistry at C3 and regeneration of a hydroxide on Fe2.
The substrate docking results also explain the essentiality of the Mg2+ ion, which provides a key element of substrate recognition, being chelated by the C2-OH and the carboxylate group of the substrate. Substrate recognition is further enhanced by the hydrophobic pocket that accepts the methyl and ethyl substituents on C3.
Structure–function results on the bacterial l-arabinonate dehydratase Rl-ArDHT (11) confirm a common mechanistic framework. At 2.4 Å resolution for the holo Rl-ArDHT structure, the cluster electron density allowed modeling of an iron-bound water/OH− in only one of the eight subunits (two tetramers) in the asymmetric unit. Substrate docking showed, however, that the C3–OH is directed toward Fe2 of the cluster, and Ser-480, equivalent to Ser-491 in Mtb-DHAD, is in position to extract the C2 proton. The flexible loop that carries Ser-480 also has the same (active) conformation as in subunit A of Mtb-DHAD. The 2Fe–2S cluster and its immediate surrounds, including the Mg2+ ion and the carbamylated lysine are all fully conserved between Mtb-DHAD and Rl-ArDHT, and the docked substrate chelates the Mg2+ ion in the same manner as occurs in Mtb-DHAD. One significant difference is the substitution in Mtb-DHAD of Gly-493, in place of Thr-482 in Rl-ArDHT, which was identified as essential for activity (11). This substitution is a conserved feature of DHADs relative to the sugar DHTs and may have more to do with substrate binding than to catalysis.
The DHAD enzyme from E. coli (7) seems in some respects to be an outlier in this family. Mechanistically it appears very similar, but it is much less stable and is proposed to contain a 4Fe–4S cluster, similar to that present in aconitase (8). Evidence for such a cluster came from detailed EPR, magnetic CD, and resonance Raman studies, but the interpretation was handicapped by the lack of suitable models for an alternative 2Fe–2S species. The nonheme iron and acid-labile sulfur content in Ec-DHAD were consistently measured as 0.9–1.9 and 0.9–1.7 mol, respectively, per protein monomer but were assumed to be lowered by cluster loss given the known instability of the enzyme. Sequence comparisons (Fig. 7) show a pairwise identity of 40% between Mtb-DHAD and Ec-DHAD, which implies very similar protein structures. The E. coli enzyme has several large insertions, but they are on external loops and are unlikely to perturb the internal structure. Interestingly, Ec-DHAD lacks one of the Mtb-DHAD cluster ligands; Cys-64 in Mtb-DHAD is replaced by Gly-49. Ec-DHAD has 12 Cys residues, but based on our Mtb-DHAD structure none apart from Cys-122 and Cys-195 (equivalent to the iron ligands Cys-137 and Cys-214) would be within 10 Å of the cluster. The most likely replacement for Cys-64 may be His-50 at position 65, Mtb-DHAD numbering. His ligands are occasional ligands in 2Fe–2S clusters (21), for example Rieske proteins (22, 23), where they can modulate the redox potential or other properties. Whereas it seems most likely that Ec-DHAD, like Mtb-DHAD, also contains a 2Fe–2S cluster and shares the same mechanism, it is possible that a change in ligation or the larger number of Cys residues in the E. coli enzyme could explain its greater instability. Even the presence of a 4Fe–4S cluster cannot be discounted on sequence grounds alone.
Figure 7.
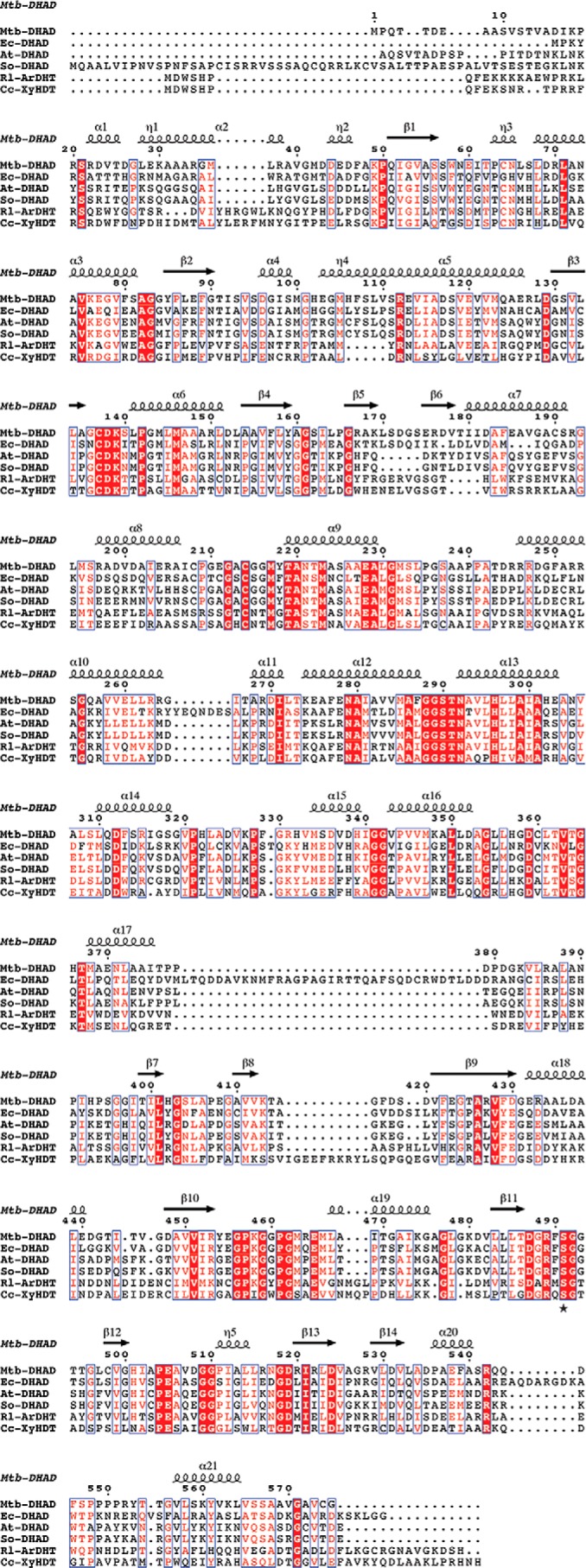
Multiple sequence alignment of dehydratase enzymes of the DHAD family. Sequences are shown for the DHAD enzymes from M. tuberculosis (Mtb-DHAD), E. coli (Ec-DHAD), A. thaliana (At-DHAD), and Spinacia oleracea (So-DHAD) and for two sugar dehydratases l-arabinonate dehydratase from R. leguminosarum (Rl-ArDHT) and d-xylonate dehydratase from C. crescentus (Cc-XyDHT). Numbering and secondary structure elements are given for Mtb-DHAD. Fully conserved residues are highlighted (white letters on a red background).
The internal location of the 2Fe–2S cluster in Mtb-DHAD implies that there must be an entry portal through which its substrates can enter and products leave. The same portal would presumably account for the susceptibility of the cluster to attack by small molecules that trigger cluster degradation and loss. In the DHAD monomer, the closest access route to the cluster would be via a cleft in the surface between residues 95–99 and 200–207 that exposes the cluster. In the DHAD oligomer, however, this is blocked by the N-terminal subdomain of the other subunit of the dimer; an α-helix residues 31–41 packs into this cleft, inserting a bulky hydrophobic residue (Met-27 for Mtb-DHAD) into van der Waals contact distance of the cluster. This potential entry point could be relevant to cluster assembly and insertion, whereas its blockage in oligomers may explain why the minimal biological unit of DHAD enzymes and related dehydratases appears to be a dimer.
Examination of our Mtb-DHAD structure suggests another potential access channel, however, that is also shared by the sugar dehydratases. Comparisons of the four subunits of the tetramer give some clues. Pairwise superpositions of subunit A on subunits B, C, and D give root-mean-square differences of 1.39, 1.59, and 1.44 Å, respectively, whereas superpositions of B, C, and D on each other are 0.39, 0.42, and 0.57 Å. The differences lie in two main areas: the flexible loop that carries the essential Ser-491 and, in the orientations of two helices, residues 181–190 and residues 468–475, that flank a putative entry channel leading into the active site. In subunit A the Ser-491 loop is well-ordered, the serine is positioned for catalysis, and the two helices are packed closely together such that the entry channel is effectively closed. In subunits B, C, and D, the Ser-491 loop is disordered, Ser-491 is turned away from the substrate cavity, and the two helices have moved apart by ∼4 Å, opening up an entry portal. It is tempting to infer a linkage between these movements, although we see no obvious mechanism. What is most significant is that in the structural analyses of the sugar dehydratase ArDHT both open and closed forms were identified, with the key change being a tilting by ∼7 Å of a helix–loop–helix fragment, residues 166–192. This opens up the same channel as in Mtb-DHAD. There are differences in detail, probably because of sequence changes, but these proteins do appear to share a common substrate entry portal.
Finally, the demonstration that aspterric acid is a competitive inhibitor of Mtb-DHAD, with a Ki of 10 μm, opens up an exciting new avenue for the development of new anti-TB drugs, given the critical role that this enzyme plays in the biosynthesis of not only the branched-chain amino acids but also pantothenate (and thence CoA). Aspterric acid is a tricyclic sesquiterpenoid (24) whose (R)-α-hydroxy, (R)-β-ether oxygen configuration mimics the (2R,3R)-dihydroxy configuration of natural DHAD substrates. If substrates of DHAD bind directly to Fe2 of the 2Fe–2S cluster through their 3-OH group, by analogy aspterric acid is predicted to bind similarly through its β-ether oxygen (13). Aspterric acid is a bulkier molecule than natural DHAD substrates, but it appears to be able to access the active site, and the substrate binding cavity may have sufficient room for some elaboration of this molecule.
Experimental procedures
Protein production
The ORF encoding IlvD (Rv0189c) was amplified from M. tuberculosis H37Rv genomic DNA. The amplified PCR product was then cloned into the E. coli expression vector pProEX. For expression, this construct was transformed into E. coli BL21(DE3) cells containing the pPH151 plasmid overexpressing the suf operon of E. coli (14), a kind gift by Petra Hänzelmann, Institute for Structural Biology, University of Wurzburg, Wurzburg, Germany). Transformants were selected on an LB agar plate containing 50 μg/ml carbenicillin and 34 μg/ml chloramphenicol. A single colony was used to inoculate 20 ml of LB, which, after overnight incubation, was used to start 2 liters of super broth expression media in a 2-liter Pyrex media bottle. After 5 h of growth at 37 °C, cysteine and isopropyl β-d-thiogalactopyranoside were added to final concentrations of 600 and 500 μm, respectively, before transfer of the culture to 22 °C for ∼20 h. The cells were then harvested by centrifugation at 10,000 × g, flash frozen, and stored in liquid nitrogen until purification. All subsequent steps were carried out in an MBraun anaerobic chamber maintained at <0.1 ppm oxygen (MBraun, Stratham, NH).
In a typical purification, ∼30 g of cell paste was resuspended in 30 ml of lysis buffer containing 50 mm HEPES, pH 7.5, 300 mm KCl, 4 mm imidazole, 10 mm 2-mercaptoethanol, 10% glycerol, and 1% Triton X-305. The resuspension was subjected to 50 rounds of sonic disruption (80% output, 3-s pulse on, 12-s pulse off) at 4 °C. The lysate was cleared by centrifugation at 4 °C for 1 h at 15,000 × g, and the supernatant was loaded with an ÄKTA express FPLC system on to a 5-ml fast-flow HisTrapTM column (GE Healthcare Life Sciences) equilibrated in lysis buffer lacking Triton X-305. The column was washed with 10 column volumes of lysis buffer before elution with 5 ml of buffer containing 50 mm HEPES, pH 7.5, 300 mm KCl, 300 mm imidazole, 10 mm 2-mercaptoethanol, and 10% glycerol. The protein fractions were pooled and supplemented with sodium sulfide and ferric chloride to concentrations of 0.4 and 0.6 mm, respectively, to ensure a homogeneous protein sample fully occupied with its iron–sulfur cluster (15). The reconstituted protein was then passed over a HiPrep 16/60 Sephacryl S-200 HR column equilibrated in 20 mm HEPES, pH 7.5, 300 mm KCl, 5 mm DTT, and 10% glycerol. The proteins were concentrated to ∼100 mg/ml with a Vivaspin 20 concentrator (Sartorius). The protein concentration was estimated from its absorbance at 280 nm using the calculated extinction coefficient of 0.243 AU/g·liter.
The WT pProEX-ilvD construct was used as a template to generate the S491A mutant using 5′-phosphorylated primers (GCCGGTGGAACCACCGGCCT and GAACCGGCCGTCGGTCAGCA; Integrated DNA Technologies). After thermal cycling using iProof high-fidelity DNA polymerase (Bio-Rad), the parental DNA was digested with DpnI, and the mutated linear plasmid was circularized using T4 DNA ligase (Roche). The plasmid was transformed into E. coli TOP10 electrocompetent cells for selection, and the mutation was then verified by sequencing. All protein expression and purification steps were performed as for the WT protein.
Enzyme activity
The reaction rates were measured using a coupled assay in which the Mtb-DHAD reaction was coupled to that of the next Mtb enzyme in the biosynthesis of isoleucine, the branched-chain aminotransferase IlvE, and glutamate dehydrogenase. IlvE converts the DHAD reaction product, α-keto isovalerate, into Ile in the presence of Glu while releasing the equivalent amount of α-keto glutarate, which is converted back to Glu in the presence NADH by glutamate dehydrogenase. This reaction could then be monitored spectrophotometrically by following the decrease in absorbance at 340 nm as NADH was consumed. All activity measurements were carried out anaerobically.
A typical reaction mixture in 1 ml contained varying amount of substrate, 150 μm NADH, 10 mm Glu, IlvE, glutamate dehydrogenase, in 50 mm Tris buffer, pH 7.8, containing 2 mm DTT, 10 mm MgCl2, and 25 mm Na2CO3. The reaction was initiated by anaerobically injecting 25 μl of appropriately diluted IlvD into the sealed cuvette, and the reaction was monitored continuously on a Shimadzu UV-2600 spectrophotometer at 30 °C. Enzyme activities were calculated using a molar absorption coefficient of 6220 m−1 cm−1, calculated from the composition of the protein and assuming 100% purity (shown by SDS-PAGE) and minimal contribution to the A280 from the 2Fe-2S cluster. Initial velocity kinetic data were fitted using Sigma Plot 11 to Equation 1,
| (Eq. 1) |
where V is the maximal velocity, A is the substrate concentration, and K is the Km.
The pH dependence of the catalytic rate was measured using 6 mm substrate. Activities were monitored every 0.3 pH unit (or smaller) intervals from pH 6.6 to 8.8. Profiles were generated by plotting the log of kcat versus the pH and fitted using the following equation,
| (Eq. 2) |
where C is the pH-independent plateau value, Ka is the ionization constant for the acidic group, Kb is the ionization constant for the basic group, and H+ is the hydrogen ion concentration.
Aspterric acid was purchased from Santa Cruz Biotechnology and dissolved in 20% DMSO, and its concentration was determined by quantitative NMR using caffeine as standard. Working solutions were further diluted in water such that the final DMSO concentration in inhibition assays was less than 0.05%. The inhibition pattern of DHAD by aspterric acid was determined by measuring the initial velocity at variable concentrations of the substrate at several fixed concentrations of the inhibitor. Equation 3 was used to fit the competitive inhibition.
| (Eq. 3) |
EPR spectroscopy
Continuous wave X-band EPR spectra were obtained on a Bruker ESP 300 equipped with a TE102 mode resonator and an Oxford instruments ESR 10 continuous flow cryostat. Experimental parameters used to acquire the spectra in Fig. 5 were as follows: microwave frequency, 9.49 GHz; microwave power, 1 milliwatt; number of scans, 4; modulation amplitude, 0.8 mT; time constant, 655 ms; and temperature 15 K.
Crystallization and data collection
Diffraction-quality crystals were obtained by sitting-drop vapor diffusion at 20 °C in an anaerobic chamber maintained at <0.1 ppm oxygen (MBraun, Stratham, NH). Drops of 0.4 μl of protein solution (His6-tagged Mtb-DHAD diluted to 20 mg ml−1 in water) were mixed with 0.4 μl of precipitant (0.2 m MgCl2, 20% PEG 3350) and equilibrated against a solution of 0.5 m LiCl. The crystals were removed from the original drop and soaked for 1 min in mother liquor, mounted in nylon loops and flash-cooled in liquid nitrogen, inside the anaerobic chamber, and stored in liquid nitrogen prior to data collection.
Diffraction data were collected at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL) on Beamline 21-ID-D (LS-CAT) and were integrated and scaled using the HKL3000 suite (25). Two data sets were collected: a data set to 2.0 Å resolution was collected at a wavelength of 1.7389 Å to exploit the anomalous signal from the iron atoms of the intrinsic iron–sulfur clusters and a native data set to 1.88 Å resolution, collected at 1.0782 Å, for refinement of the structural model.
Structure determination and refinement
Phases were determined by single-wavelength anomalous diffraction with autoSHARP (26), from the positions of eight iron atoms in the asymmetric unit, giving a figure of merit of 0.61 to 3.0 Å resolution. This enabled an initial polyalanine model to be built with ARP/wARP (27). This initial model was then used to phase the 1.88 Å native data set by molecular replacement, with subsequent rounds of automated model building performed by Phenix AutoBuild (28), interspersed with manual model building and refinement using Coot (29), phenix.refine (28), and Refmac5 (30). The final model comprises four identical polypeptide chains of 575 residues each, with residues missing because of the lack of adequate density being 1–13 from chain A; 1–14, 416–418, and 494–496 from chain B; 1–13 and 416–419 from chain C; and 1–13, 417–418, and 575 from chain D. Validation of the model was performed with MolProbity (31). Full data processing, refinement, and validation details are in Table 1.
Substrate and inhibitor docking
For docking, the crystal structure coordinates were protonated and converted to mol2 format using PyMOL (Schrödinger), and substrate and intermediate molecules were produced using Chem3D (CambridgeSoft) and again saved in mol2 format. Docking of substrate and intermediate molecules was performed using Gold (2019 CSD-Discovery Suite, Cambridge Crystallographic Data Center). Within the GOLD interface, protein coordinates were prepared by removing all water molecules except for the iron-bound OH/water; docking was carried out with and without this water molecule in place. Docking used the CHEMPLP scoring function with default genetic algorithm settings. In each docking run, 10 models were produced without any constraints, and each solution was visually inspected for steric clash and bonding interactions.
Accession codes
The atomic coordinates and structure amplitudes for the structure of Mtb-DHAD have been deposited with the Protein Data Bank with accession code 6ovt.
Author contributions
G. B., T. L. G., and S. S. H. data curation; G. B., T. L. G., C. J. S., and E. N. B. formal analysis; G. B., S. C. A., J. S. B., and E. N. B. supervision; G. B. and T. L. G. validation; G. B., T. L. G., S. S. H., T. L., G. J. G., and C. J. S. investigation; G. B., T. L. G., S. S. H., T. L., G. J. G., S. C. A., C. J. S., J. S. B., and E. N. B. methodology; G. B., T. L. G., S. S. H., G. J. G., C. J. S., J. S. B., and E. N. B. writing-original draft; G. B., T. L. G., S. S. H., G. J. G., S. C. A., J. S. B., and E. N. B. writing-review and editing; S. C. A. and E. N. B. resources; S. C. A., J. S. B., and E. N. B. funding acquisition; C. J. S. software; J. S. B. and E. N. B. conceptualization; E. N. B. project administration.
Acknowledgment
We thank the Advanced Photon Source (Argonne National Laboratory, Argonne, IL) for access to X-ray data collection facilities (Beamline 21-ID-D, LS-CAT).
This work was supported by the Tertiary Education Commission of New Zealand (Centre of Research Excellence funding to the Maurice Wilkins Centre); a Health Research Council of New Zealand program grant (to E. N. B.); a Hercus Health Research Fellowship (to G. B.); and National Institutes of Health Grants RO1–060899 (to J. S. B.), R21-AI133329 (to T. L. G. and S. C. A.), P01-GM118303-01 (to J. A. Gerlt and S. C. A.), U54-GM093342 (to J. A. Gerlt and S. C. A.), and U54-GM094662 (to S. C. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 6ovt) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- DHAD
- dihydroxyacid dehydratase
- RMSD
- root-mean-square deviation
- CoA
- coenzyme A
- Ec
- E. coli
- Rl
- R. leguminosarum
- Cc
- C. crescentus
- At
- A. thaliana
- Mtb
- M. tuberculosis
- IMAC
- immobilized metal affinity chromatography
- SEC
- size-exclusion chromatography
- ArDHT
- l-arabinonate dehydratase
- XyDHT
- d-xylonate dehydratase
- LS-CAT
- Life Sciences Collaborative Access Team.
References
- 1. Amorim Franco T. M., and Blanchard J. S. (2017) Bacterial branched-chain amino acid biosynthesis: structure, mechanisms and drugability. Biochemistry 56, 5849–5865 10.1021/acs.biochem.7b00849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sassetti C. M., Boyd D. H., and Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84 10.1046/j.1365-2958.2003.03425.x [DOI] [PubMed] [Google Scholar]
- 3. Myers J. W. (1961) Dihydroxy acid dehydrase; an enzyme involved in the biosynthesis of isoleucine and valine. J. Biol. Chem. 236, 1414–1418 [PubMed] [Google Scholar]
- 4. Armstrong F. B., Muller U. S., Reary J. B., Whitehouse D., and Croute D. H. (1977) Stereoselectivity and stereospecificity of the α,β-dihydroxyacid dehydratase from Salmonella typhimurium. Biochim. Biophys. Acta 498, 282–293 10.1016/0304-4165(77)90266-5 [DOI] [PubMed] [Google Scholar]
- 5. Singh V., Chandra D., Srivastava B. S., and Srivastava R. (2011) Downregulation of Rv0189c, encoding a dihydroxyacid dehydratase, affects growth of Mycobacterium tuberculosis in vitro and in mice. Microbiology 157, 38–46 10.1099/mic.0.042358-0 [DOI] [PubMed] [Google Scholar]
- 6. Flint D. H., and Emptage M. H. (1988) Dihydroxyacid dehydratase from spinach contains a [2Fe–2S] cluster. J. Biol. Chem. 263, 3558–3564 [PubMed] [Google Scholar]
- 7. Flint D. H., Emptage M. H., Finnegan M. G., Fu W., and Johnson M. K. (1993) The role and properties of the iron–sulfur cluster in Escherichia coli dihydroxyacid dehydratase. J. Biol. Chem. 268, 14732–14742 [PubMed] [Google Scholar]
- 8. Beinert H., Kennedy M. C., and Stout C. D. (1996) Aconitase as iron–sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev. 96, 2335–2374 10.1021/cr950040z [DOI] [PubMed] [Google Scholar]
- 9. Lauble H., Kennedy M. C., Beinert H., and Stout C. D. (1992) Crystal structures of aconitase with isocitrate and nitroisocitrate bound. Biochemistry 31, 2735–2748 10.1021/bi00125a014 [DOI] [PubMed] [Google Scholar]
- 10. Duan X., Yang J., Ren B., Tan G., and Ding H. (2009) Reactivity of nitric oxide with the [4Fe–4S] cluster of dihydroxyacid dehydratase from Escherichia coli. Biochem. J. 417, 783–789 10.1042/BJ20081423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rahman M. M., Andberg M., Thangaraj S. K., Parkkinen T., Penttilä M., Jänis J., Koivula A., Rouvinen J., and Hakulinen N. (2017) The crystal structure of a bacterial l-arabinonate dehydratase contains a [2Fe–2S] cluster. ACS Chem. Biol. 12, 1919–1927 10.1021/acschembio.7b00304 [DOI] [PubMed] [Google Scholar]
- 12. Rahman M. M., Andberg M., Koivula A., Rouvinen J., and Hakulinen N. (2018) The crystal structure of d-xylonate dehydratase reveals functional features of enzymes from the Ilv/ED dehydratase family. Sci. Rep. 8, 865 10.1038/s41598-018-19192-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan Y., Liu Q., Zang X., Yuan S., Bat-Erdene U., Nguyen C., Gan J., Zhou J., Jacobsen S. E., and Tang Y. (2018) Resistance-gene-directed discovery of a natural-product herbicide with a new mode of action. Nature 559, 415–418 10.1038/s41586-018-0319-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hänzelmann P., Hernández H. L., Menzel C., García-Serres R., Huynh B. H., Johnson M. K., Mendel R. R., and Schindelin H. (2004) Characterization of MOCS1A, an oxygen-sensitive iron–sulfur protein involved in human molybdenum cofactor biosynthesis. J. Biol. Chem. 279, 34721–34732 10.1074/jbc.M313398200 [DOI] [PubMed] [Google Scholar]
- 15. Booth M. P. S., Challand M. R., Emery D. C., Roach P. L., and Spencer J. (2010) High-level expression and reconstitution of active Cfr, a radical-SAM rRNA methyltransferase that confers resistance to ribosome-acting antibiotics. Prot. Exp. Purif. 74, 204–210 10.1016/j.pep.2010.07.010 [DOI] [PubMed] [Google Scholar]
- 16. Krissinel E., and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 17. Seibert C. M., and Raushel F. M. (2005) Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry 44, 6383–6391 10.1021/bi047326v [DOI] [PubMed] [Google Scholar]
- 18. Orme-Johnson W. H., and Orme-Johnson N. R. (1982) Iron–sulfur proteins: the problem of determining cluster type. In Metals in Biology: Iron–Sulfur Proteins (Spiro T. G., ed) pp. 67–96, Wiley-Interscience, New York [Google Scholar]
- 19. Jones G., Willett P., Glen R. C., Leach A. R., and Taylor R. (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748 10.1006/jmbi.1996.0897 [DOI] [PubMed] [Google Scholar]
- 20. Flint D. H., and Allen R. M. (1996) Iron–sulfur proteins with non-redox functions. Chem. Rev. 96, 2315–2334 10.1021/cr950041r [DOI] [PubMed] [Google Scholar]
- 21. Bak D. W., and Elliott S. J. (2014) Alternative FeS cluster ligands: tuning redox potentials and chemistry. Curr. Opin. Chem. Biol. 19, 50–58 10.1016/j.cbpa.2013.12.015 [DOI] [PubMed] [Google Scholar]
- 22. Rieske J. S., Maclennan D. H., and Coleman R. (1964) Isolation and properties of an iron protein from the (reduced coenzyme Q)-cytochrome c reductase complex of the respiratory chain. Biochem. Biophys. Res. Commun. 15, 338–344 10.1016/0006-291X(64)90171-8 [DOI] [Google Scholar]
- 23. Schneider D., and Schmidt C. L. (2005) Multiple Rieske proteins in prokaryotes: where and why? Biochim. Biophys. Acta 1710, 1–12 10.1016/j.bbabio.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 24. Tsuda Y., Kaneda M., Tada A., Nitta K., Yamamoto Y., and Iitaka Y. (1978) Aspterric acid, a new sesquiterpenoid of the carotene group, a metabolite from Aspergillus terreus IFO-6123. X-ray crystal and molecular structure of its p-bromobenzoate. J. Chem. Soc. Chem. Commun. 160–161 [Google Scholar]
- 25. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 26. Vonrhein C., Blanc E., Roversi P., and Bricogne G. (2007) Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 [DOI] [PubMed] [Google Scholar]
- 27. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 10.1038/nprot.2008.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) Molprobity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]