

Abstract
Reactivation of Kaposi's sarcoma-associated herpesvirus (KSHV) from latency requires the viral transactivator Rta to contact the host protein Jκ recombination signal-binding protein (RBP-Jκ or CSL). RBP-Jκ normally binds DNA sequence-specifically to determine the transcriptional targets of the Notch-signaling pathway, yet Notch alone cannot reactivate KSHV. We previously showed that Rta stimulates RBP-Jκ DNA binding to the viral genome. On a model viral promoter, this function requires Rta to bind to multiple copies of an Rta DNA motif (called “CANT” or Rta-c) proximal to an RBP-Jκ motif. Here, high-resolution ChIP/deep sequencing from infected primary effusion lymphoma cells revealed that RBP-Jκ binds nearly exclusively to different sets of viral genome sites during latency and reactivation. RBP-Jκ bound DNA frequently, but not exclusively, proximal to Rta bound to single, but not multiple, Rta-c motifs. To discover additional regulators of RBP-Jκ DNA binding, we used bioinformatics to identify cellular DNA-binding protein motifs adjacent to either latent or reactivation-specific RBP-Jκ–binding sites. Many of these cellular factors, including POU class homeobox (POU) proteins, have known Notch or herpesvirus phenotypes. Among a set of Rta- and RBP-Jκ–bound promoters, Rta transactivated only those that also contained POU motifs in conserved positions. On some promoters, POU factors appeared to inhibit RBP-Jκ DNA binding unless Rta bound to a proximal Rta-c motif. Moreover, POU2F1/Oct-1 expression was induced during KSHV reactivation, and POU2F1 knockdown diminished infectious virus production. Our results suggest that Rta and POU proteins broadly regulate DNA binding of RBP-Jκ during KSHV reactivation.
Keywords: Notch pathway, DNA viruses, herpesvirus, viral DNA, DNA-binding protein, chromatin immunoprecipitation (ChiP), tumor virus, KSHV, Oct-1, POU2F1, Rta
Introduction
Kaposi's sarcoma-associated herpesvirus (KSHV),6 the etiologic agent of Kaposi's sarcoma (KS) and primary effusion lymphoma (PEL) (1), is classified as a type I carcinogen by the International Agency for Research in Cancer (IARC) (2). In KS pathogenesis, viral reactivation is required for dissemination from the B-cell reservoir and expression of productive cycle oncogenes (3). Like other herpesviruses, KSHV reactivation proceeds through three stages of sequential gene expression, called immediate early (IE), delayed early (DE), and late (L). Viral DNA replication requires DE gene expression and precedes L stages, and productive reactivation culminates in the release of infectious KSHV virions from the host cell.
Our laboratory and others previously showed that the KSHV Rta transcriptional transactivator is the only viral protein necessary and sufficient for reactivation in cultured PEL cells (4–7). KSHV reactivation requires Rta to form a complex with the cellular protein recombination signal-binding protein (RBP)-Jκ (also known as CSL and CBF-1) to bind and transactivate viral promoters (8–10). RBP-Jκ is best known as the downstream effector of the Notch pathway. Specifically, RBP-Jκ binds to promoters to specify transcriptional targets of activated Notch (NICD) (4, 8, 11–19). The KSHV genome contains 121 putative binding sites that match an RBP-Jκ consensus motif.
Our laboratory has sought to define the molecular mechanisms required for forming transcriptionally productive Rta/RBP-Jκ/DNA complexes necessary for KSHV reactivation. To this end, we have focused on Rta transactivation of the promoter of the essential KSHV DE gene Mta. We showed a new mechanism of RBP-Jκ–dependent transactivation in which Rta stimulates RBP-Jκ DNA binding in infected or uninfected cells (8). Although activated Notch 1 (NICD1) was later shown to stimulate RBP-Jκ DNA binding to cellular DNA (20–23), NICD1 fails to transactivate Mta or reactivate KSHV from latency (8, 24, 25); however, stimulation of RBP-Jκ DNA binding by Rta can restore the ability of NICD1 to transactivate the Mta promoter (8). This dynamic binding of RBP-Jκ to viral DNA suggests that KSHV reactivation does not follow the canonical model for Notch transactivation. Similar to its role in many viral and nonviral cancers (26–32), Notch is constitutively active in KSHV-infected cells and is required for their survival (25, 33–37). KSHV infection thus provides a model system to reveal the mechanisms of Notch pathway target selection by regulation of RBP-Jκ DNA binding.
On the Mta promoter, stimulation of RBP-Jκ DNA binding by Rta requires four molecular interactions as follows: Rta homotetramerization, Rta binding to RBP-Jκ and to repeats of the Rta-c DNA motif (also called a “CANT” motif), and RBP-Jκ binding to a consensus RBP-Jκ motif (8, 15, 16, 38). Rta-c motifs have the consensus sequence ANTGTAACANT(A/T)(A/T)T (the conserved core sequence is underlined), and the motif is repeated seven times near the RBP-Jκ motif in the Mta promoter (Fig. 1). The number of Rta-c motifs is proportional to Rta's promoter–binding affinity, formation of ternary Rta/RBP-Jκ/DNA complexes, and stimulation of RBP-Jκ DNA binding (16).
Figure 1.
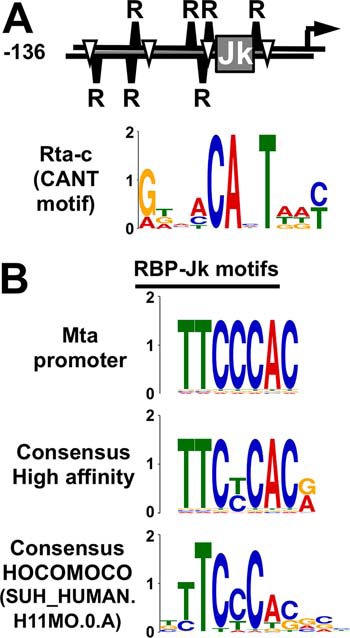
DNA elements required for Rta stimulation of RBP-Jκ DNA binding to the Mta promoter. A, promoter schematic and Rta-c (CANT motif) element logo as in Ref. 16. Jk, RBP-Jκ motif; R, Rta-c (CANT motif); inverted triangle, A/T3 trinucleotide in repeat; arrow, transcription start site. B, sequence logos for RBP-Jκ. Consensus logos as in Ref. 84 and HOCOMOCO as in Refs. 48, 49. Y axes indicate bits for all logos.
Although we used a ChIP/Southern blotting approach to confirm broad enhancement of RBP-Jκ binding to the viral genome during KSHV reactivation, our model had shortcomings (16). We based our model on detailed molecular and biochemical studies of only a single KSHV promoter. In addition, our data did not clearly distinguish the relative contribution of the Rta-c DNA motifs from that of a second overlapping DNA repeat, (A/T)3N17(A/T)3, shown to mediate Rta binding to other viral promoters (39). Our ChIP/Southern blotting also had inherently low resolution, which prohibited us from identifying the architectures of other promoters containing RBP-Jκ–binding sites.
In this study, we identified and characterized KSHV DNA sequences critical for assembling productive Rta/RBP-Jκ complexes in KSHV-infected PEL (B) cells. We employed ChIP/deep sequencing (ChIP/Seq) to classify hundreds of authentic Rta and RBP-Jκ–binding sites on the KSHV genome during latency and reactivation. We demonstrate that DNA binding of RBP-Jκ is dynamic across the viral genome, and very few RBP-Jκ sites are occupied during both latency and reactivation. Stimulation of RBP-Jκ DNA binding to many, but not all promoters, including the Mta promoter, was associated with Rta DNA binding to Rta-c motifs. However, Mta was the only promoter bound by both proteins that contained an Rta-c multimer. To identify additional DNA elements associated preferentially with RBP-Jκ latency or reactivation peaks, plus and minus Rta peaks, we employed discriminative DNA motif discovery (40). Many of the proteins that putatively bind to these motifs have published phenotypes in Notch signaling and/or herpesvirus infection; our data are the first to implicate these proteins as regulators of RBP-Jκ DNA binding. We include a combination of in vitro DNA binding and transactivation assays to evaluate the functional significance of the motifs in candidate target genes, focusing on the bidirectional viral ORF50AS/K-bZIP promoter. Our data point to a crucial role for multiple members of the cellular POU factors in regulating RBP-Jκ DNA binding, and we show that knockdown of one POU factor, Octamer-1, severely debilitates viral reactivation. Our new data set provides a basis for future analyses of regulated target promoter selectivity by RBP-Jκ–dependent transactivators.
Results
Identification of binding sites for Rta and RBP-Jκ across the KSHV genome
We recently tested a series of histone deacetylase inhibitors and showed that valproic acid (VPA) was one of the most efficient chemicals to reactivate KSHV from latency (41). VPA's superior potency was evident by measuring viral gene expression and production of infectious virus. Therefore, we reasoned that VPA would be the ideal stimulus to distinguish latent from reactivation-specific Rta and RBP-Jκ–binding sites on the KSHV genome. Accordingly, we performed a ChIP-Seq experiment on chromatin isolated from infected PEL cells minus/plus VPA treatment. We chose 24 h post-VPA treatment as our reactivation time point; Rta was well-expressed, and a DE protein (ORF59) was beginning to appear (Fig. S1). We observed 104 Rta peaks (Fig. 2 and Table 1), 91 reactivation-specific RBP-Jκ peaks, and 97 latency-specific RBP-Jκ peaks. There were no significant Rta peaks detected during latency, when Rta is not expressed, as expected (data not shown).
Figure 2.

ChIP/Seq of Rta and RBP-Jκ DNA binding on the KSHV genome during latency and reactivation. Visualization of ChIP/Seq peaks by read depth per bp using Integrative Genomics Viewer (IGV) (85). A, genome positions from bp 1 to 68,000. B, genome positions from bp 68,001 to 137,508. Antibodies used are indicated at right, with chromatin from latent (−VPA) or reactivated (+VPA) virus in BC-3 cells. Solid arrows represent open reading frames (ORFs). Numbering above arrows indicate ORF names. Purple arrowheads and two-headed arrows indicate 15 co-localized Rta and RBP-Jκ peaks in reactivation. Asterisks indicate peak positions mapped to PAN promoter (PAN), ORF50AS/K-bZIP promoter (K-bZIP), and Mta promoter (Mta). Orange bars indicate positions of ori-Lyts.
Table 1.
Number of Rta and RBP-Jκ peaks on KSHV genome in latency or reactivation
| Antibody | Treatmenta | No. of peaks |
|||
|---|---|---|---|---|---|
| Total | In promsb | Kineticsc | No. per prom | ||
| Rta | +VPA | 104 | 41 | DE | 1.1 |
| 10 | L | 0.7 | |||
| RBP-Jκ | +VPA | 91 | 39 | DE | 1.1 |
| 4 | L | 0.3 | |||
| −VPA | 97 | 35 | DE | 1.0 | |
| 3 | L | 0.2 | |||
a −VPA corresponds to latency; +VPA corresponds to reactivation.
b Numbers of peaks within promoters are shown.
c Replication kinetics of transcription start sites (as in Ref. 45) corresponding to promoters (DE = delayed early; L = late) are shown.
To identify the peaks that mapped to viral promoters, we compared the peak locations with the KSHV transcription start site (TSS) set (42), and we deduced that most critical transcriptional control elements would lie within 1000 bp upstream of each TSS in the viral genome (one exception is the KSHV ORF50 promoter, which contains control elements located greater than 1000 bp upstream of its TSS (43). Throughout this study, we define ORF50's promoter as the 2.7-kb region lying between the co-regulated ORF45 and ORF50 TSSs (44). RBP-Jκ peaks predominate on DE, rather than L, promoters in both latency and reactivation, but the proportion of RBP-Jκ peaks on promoters increases during reactivation (Table 1; kinetics as in Ref. 45); some IE promoters are further transactivated by Rta with DE kinetics, such as ORF50, so we have scored those promoters as DE promoters. Rta peaks are also over-represented on promoters relative to nonpromoter DNA (data not shown).
The data also show that several known targets of Rta and RBP-Jκ were represented (9, 46, 47). Rta binding was observed on the previously described Rta-direct target, the PAN promoter, during reactivation (Fig. 2, asterisk, and Table S1A, peak 22). Furthermore, RBP-Jκ bound near a consensus RBP-Jκ motif on the PAN promoter during latency and reactivation (Fig. 2 and Table S1B, peak 17, and C, peak 21). These data correlate with our previous report that showed that RBP-Jκ enhanced Rta's transactivation of the PAN promoter (9). We also found that Rta and RBP-Jκ bound to the Mta, Rta, and ORF50AS/K-bZIP promoters during reactivation, and the Rta and RBP-Jκ peaks were close to each other (Fig. 2, asterisks, and Table S1A, peaks 49–54, 57, 63–64, and B, peaks 38–44, 54, 56–57). This finding is consistent with previous publications that showed that Rta transactivation of Mta and K-bZIP requires RBP.
Three of the 12 highest Rta peaks mapped to the PAN, ORF50, and ORF50AS/K-bZIP promoters, and two others mapped to the left and right origins of lytic replication (oriLyts R and L; Fig. 2 and Table S1A). Four of the five highest RBP-Jκ peaks mapped to the ORF50AS/K-bZIP promoter during reactivation (Fig. 2 and Table S1B). The RBP-Jκ peaks at the ORF50/K-bZIP locus and the LANA region both broaden in reactivation relative to latency.
To estimate the resolution of the ChIP/Seq data, we compared the positions of well-established Rta and RBP-Jκ–binding sites in the Mta, PAN, and ORF50AS/K-bZIP promoters to the positions of the nearest peak summits. Those distances ranged from 5 to 61 bp, so we considered peaks to be co-localized if their summits were within 61 bp of each other. Using those criteria, the highest peak in ORF50AS/K-bZIP corresponded to the Rta peak (Fig. 2 and Table S1B). Moreover, the locations of the RBP-Jκ peaks suggested that the association of RBP-Jκ with the viral genome is dynamic (Fig. 2 and Table S1, B and C), with most binding sites used only in latency or reactivation, but not at both times during infection.
We validated the ChIP/Seq results with conventional ChIP/qPCR. In three replicates of ChIP/qPCR, Rta bound to the promoters of ORF4, PAN, K4.1/4.2, and ORF50AS/K-bZIP but not ORF70-K3p (Fig. 3A). RBP-Jκ bound to the ORF70-K3 promoter in latency but not reactivation and to the ORF50AS/K-bZIP promoter in VPA-treated and untreated cells (Fig. 3B). Also similar to the ChIP-Seq, RBP-Jκ failed to bind the promoters from PAN, K5, and K4.1/4.2 in these samples (Fig. 3B). We confirmed that these peaks were not specific to VPA treatment by repeating the ChIP/PCR on a subset of these promoters following TPA-induced reactivation (data not shown).
Figure 3.
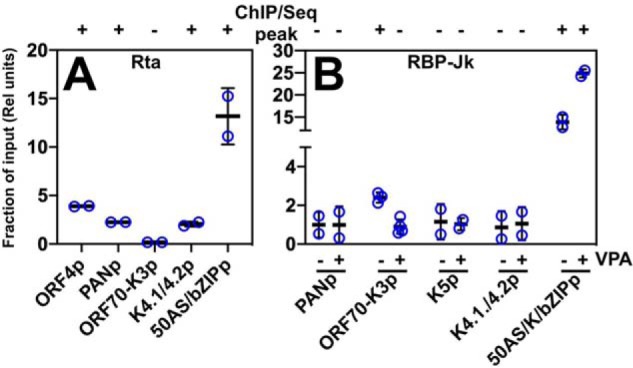
qPCR confirmation of selected ChIP/Seq peaks. Cross-linked chromatin was prepared from triplicate samples of untreated and VPA-treated BC-3 cells at 24 h post-VPA addition. A, Rta antibody was used to precipitate chromatin from VPA-treated cells. B, RBP-Jκ antibody was used to precipitate chromatin from untreated (−VPA) and VPA-treated (+VPA) cells. Chromatin precipitated with anti-Rta or anti-RBP-Jκ was quantitated by qPCR using primers corresponding to each of the promoters indicated below the graphs, normalized to qPCR from chromatin precipitated by control rabbit IgG, and then expressed as a proportion of qPCR from input chromatin. Thick lines indicate means of values; thin lines indicate standard errors; + or − above each of the bars indicates whether the region amplified corresponded to the summit of a ChIP/Seq peak in Fig. 1.
Characteristics of Rta+/RBP-Jκ− peak locations
Our data show that most DNA-bound Rta and RBP-Jκ proteins are not co-localized on the viral genome during reactivation (Fig. 2 and Table S1D). Applying the 61-bp maximum resolution of our ChIP/Seq peaks showed that only 16 Rta and RBP-Jκ peaks co-localized to the same DNA sites in reactivation (Table S1D). An additional set of five promoters bound both Rta and RBP-Jκ, but the proteins were not co-localized.
Many of the Rta peaks that did not co-localize with RBP-Jκ peaks (Rta+/RBP-Jκ−) corresponded to four broad loci of the genome: ORF70 to K4.2, ORF18 to 21, ORF58 to 62, and K12 to K13 (Fig. 2). Interestingly, many Rta+/RBP-Jκ− DE promoters contained Rta peaks in close proximity to consensus RBP-Jκ motifs despite the absence of RBP-Jκ peaks (data not shown).
RBP-Jκ binds dynamically to many sites in the KSHV genome during latency and reactivation
The number of RBP-Jκ peaks that increased or decreased during reactivation was nearly identical (Fig. 4A, left). Only eight peaks remained unchanged following the reactivation switch (black region, Fig. 4A, left). When we consider only the peaks found in promoters, RBP-Jκ peaks that increased in reactivation were as follows: (a) over-represented in both DE and L promoters (Fig. 4A, center and right) relative to the overall number of peaks (Fig. 4A, left), and (b) the average height of RBP-Jκ peaks was greater in DE versus L promoters (data not shown). Interestingly, the RBP-Jκ peaks whose heights did not change during reactivation were only found in DE promoters (Fig. 4A, center).
Figure 4.
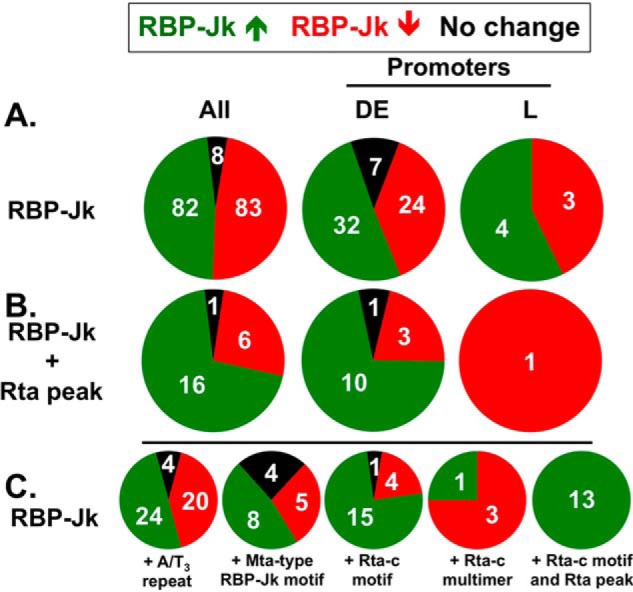
Fraction of RBP-Jκ peaks mapped to DE or L promoters, co-located with Rta peaks and with Mta promoter motifs during latency and reactivation. Green segments represent RBP-Jκ peaks that increase in reactivation; red segments represent RBP-Jκ peaks that decrease in reactivation, and black segments are peaks that are unchanged in reactivation. Numbers of peaks in each category are indicated. A, proportions of all RBP-Jκ peaks in each category and those in DE or L promoters. B, same as A, but showing only the subset of RBP-Jκ peaks with co-localized Rta peaks. C, same as A, but showing subsets of RBP-Jκ peaks associated with each motif and/or Rta peak, as indicated.
Rta binds preferably to KSHV DE promoters at which RBP-Jκ DNA binding is enhanced during reactivation
When we analyzed the subset of RBP-Jκ peaks that corresponded with Rta peaks, Rta DNA-binding preferences were infection-stage–specific. Sixteen of 23 Rta peaks co-localized with increased RBP-Jκ DNA binding overall, and 10 of 14 mapped to viral DE promoters (Fig. 4B, center). Only one Rta peak corresponded to an RBP-Jκ peak on a late promoter and that RBP-Jκ peak was present only in latency (e.g. red) but not reactivation. Rta's preference for binding DE promoters and the high correspondence of Rta and RBP-Jκ DNA binding on DE promoters (Fig. 4B, center) are consistent with the time point (DE; 24 h post-VPA) at which we performed the ChIP.
Rta-c motifs and Rta peaks are positively associated with enhanced RBP-Jκ DNA binding during reactivation
The cis-acting elements that were required for Rta to stimulate RBP-Jκ DNA binding to the Mta promoter were the RBP-Jκ motif and Rta-c DNA multimers (Fig. 1). The Mta promoter also contains an A/T3 DNA repeat that contributes to Rta transactivation of other viral promoters (39), but it has an unknown effect on RBP-Jκ DNA binding (Fig. 1). To determine the potential influence of these cis-elements on regulated RBP-Jκ binding, we determined their co-localization with RBP-Jκ peaks in viral DE promoters.
Among these DNA elements, the A/T3 repeats were found most often near RBP-Jκ peaks. The A/T3 repeats were slightly over-represented with reactivation-specific RBP-Jκ peaks (Fig. 4C, left). Next, high affinity, Mta-like RBP-Jκ motifs localized to 17 of the RBP-Jκ peaks and were over-represented in DE promoters (Fig. 4C). Finally, the Rta-c DNA motif had context-specific associations with RBP-Jκ peaks. Rta-c motifs co-localized with 20 RBP-Jκ peaks and were dramatically over-represented in reactivation-specific peaks (Fig. 4C, center). Thirteen of those RBP-Jκ peaks also co-localized with Rta peaks and were exclusively associated with reactivation but not latency (Fig. 4C, right).
Therefore, all Rta peak/Rta-c motif combinations (Rta+/Rta-c) were associated with reactivation-specific RBP-Jκ peaks, and only three co-localized Rta and RBP-Jκ peaks (Rta+/RBP-Jκ+) did not correspond to Rta-c elements. Interestingly, only four multimers of Rta-c motifs co-localized with RBP-Jκ peaks (Fig. 4C); the only DE reactivation-specific RBP-Jκ peak corresponded with the Rta-c multimers that we previously described on the Mta promoter (see Fig. 1) (16). The Rta-c multimers associated with latent RBP-Jκ peaks were exclusively dimers, and Rta-c multimers with greater than two repeats were over-represented in promoters regardless of co-localization to an RBP-Jκ peak (data not shown).
We also compared the peak heights associated with these motif combinations. The median heights of both RBP-Jκ and Rta peaks were greater when co-localized with the other peak in promoters (Fig. S2, A and B) or in the whole genome (data not shown). RBP-Jκ peaks with co-localized Rta peaks in promoters had a higher median height in the presence of Rta-c motifs (Fig. S2C).
Overall, the close correspondence between stimulated RBP-Jκ DNA binding, Rta DNA binding, and co-localized Rta-c DNA motifs (Fig. 4C) supports the conclusion that the general mechanism we established for Rta stimulation of RBP-Jκ DNA binding to the Mta promoter (Fig. 1) extends to additional viral genes. The exception to that mechanism is that the Rta-c motifs are usually monomers and not multimers. However, 70% of reactivation-specific RBP-Jκ peaks did not co-localize with Rta peaks, suggesting that the model for the Mta promoter is not universally applicable to the whole viral genome.
Classification of RBP-Jκ peaks into five groups
We postulated that a subset of the reactivation-specific RBP-Jκ peaks was regulated by mechanisms distinct from those operating on the Mta promoter. To identify additional DNA elements associated with the regulation of RBP-Jκ DNA binding, we first classified each RBP-Jκ peak into one of five groups based on its presence or absence in latency or reactivation (Table 2 and Table S2).
Table 2.
Classification of RBP-Jκ peaks in latency and reactivation of KSHV
| Class | RBP-Jκ peaka |
No. of peaks |
||||
|---|---|---|---|---|---|---|
| −VPA | +VPA | Total | In promsb | Kineticsc | Examplesd | |
| 1 | + | + | 8 | 7 | DE | ORF8 |
| 0 | L | |||||
| 2 | − | + | 76 | 28 | DE | Mta, ORF50AS/K-bZIP, ORF50 |
| 4 | L | K8.1, K12 | ||||
| 3 | + | ++ | 6 | 4 | DE | ORF50AS/K-bZIP |
| 0 | L | |||||
| 4 | ++ | + | 1 | 0 | DE | |
| 0 | L | |||||
| 5 | + | − | 82 | 24 | DE | ORF50AS/K-bZIP, ORF50 |
| 3 | L | K8.1 | ||||
a Relative magnitudes of RBP-Jκ peak heights (see Table S2) are shown.
b Number of peaks within promoters is shown.
c Replication kinetics of transcription start sites (as in Ref. 45) corresponding to promoters (DE = delayed early and L = late)) are shown.
d Examples of ORFs whose promoters contain a peak of the indicated class corresponding to b are shown.
In this system, Class 1 peaks correspond to sites at which RBP-Jκ is bound equivalently during latency and reactivation (Table 2). The behavior of RBP-Jκ at these Class 1 sites resembles its role in the canonical model of Notch signaling, in which RBP-Jκ remains constitutively bound to DNA. The small fraction of Class 1 RBP-Jκ peaks (4.6% (8/173)) agrees with the conclusion that canonical Notch signaling has modest to no impact on regulating KSHV reactivation.
RBP-Jκ peak Classes 3–5 also represent sites to which RBP-Jκ binds during latency. However, RBP-Jκ binding to those sites is altered during reactivation. In Class 3, RBP-Jκ binding increases during reactivation (Tables 2 and Table S2) and represents about 3.5% (6/173) of peaks. Only one peak is found in Class 4, in which RBP-Jκ binding decreases during reactivation. The Class 4 peak did not map to suspected or known promoters. Class 5 peaks, composing the largest group, represent sites to which RBP-Jκ binding is eliminated during reactivation and encompasses 47.4% of peaks (Table 2).
The second largest peak group was Class 2, in which RBP-Jκ was bound to the genome only during reactivation (Table 2). Because 43.9% of RBP-Jκ peaks are in Class 2, the data suggest that enhanced RBP-Jκ DNA binding is a central mechanism for transactivating KSHV genes during reactivation. Most of the known RBP-Jκ–dependent transcriptional targets of Rta were classified in this group, including Mta/ORF57, ORF50AS/K-bZIP, and ORF50/Rta itself. Additionally, the RBP-Jκ–independent PAN promoter also contained a Class 2 RBP-Jκ peak. Some promoters contained multiple RBP-Jκ peaks from different classes, including ORF50AS/K-bZIP. Although we observed the widespread increase of RBP-Jκ binding to the reactivating KSHV genome in our earlier publication (16), the ChIP/Seq approach in this study facilitates higher resolution determination of sequences associated with RBP-Jκ and Rta DNA binding.
Identification of DNA motifs associated with regulated RBP-Jκ DNA binding
The RBP-Jκ peak classes with the most dramatic potential to regulate reactivation are Classes 2 and 5, because they are exclusive to reactivation or latency, respectively. We reasoned that the Class 2 or 5 peaks in DE promoters should be associated with DNA motifs that bind to proteins that regulate RBP-Jκ DNA binding. In our approach, we were interested in motifs that are preferentially found adjacent to either Class 2 or 5 RBP-Jκ peaks. We used a ratio–of–ratios method for this analysis that compared the expected versus observed association of candidate motifs to Class 2 or 5 RBP-Jκ peaks. The expected association is the actual ratio of Class 2 to Class 5 peaks, which is 1.2 (28 Class 2 peaks/24 Class 5 peaks, or 28 divided by 24). A motif whose observed association with Class 2 peaks exceeded its observed association with Class 5 peaks by greater than 1.2-fold is a candidate to bind a protein that stimulated RBP-Jκ DNA binding during reactivation.
Because the data in Fig. 4 strongly supported a role for both Rta DNA binding and association of an Rta-c motif with reactivation-specific RBP-Jκ DNA binding, we could test the validity of the ratio–of–ratios using the observed association of Rta-c motifs and Rta peaks with Classes 2 and 5 RBP-Jκ peaks. First, for Rta-c motifs, the observed ratio of association with Class 2 or 5 motifs is 3.5, because 14 Rta-c motifs are adjacent to Class 2 peaks and four Rta-c motifs are adjacent to Class 5 peaks (14/4 = 3.5). Because 3.5 exceeds the expected ratio of 1.2, we can state that Rta-c is over-represented at reactivation-specific RBP-Jκ sites and confirms that it participates in stimulating RBP-Jκ DNA binding (Table 3A, right-most column, 3.5/1.2 = 2.9). This conclusion agrees with the data in Fig. 4C and our earlier publications (8, 16). Similarly, the observed Class 2/Class 5 ratio of Rta peaks is 2.7, which also exceeds the observed ratio and agrees with our earlier publication. These examples support the validity of employing the ratio–of–ratios approach to identify other regulators of RBP-Jκ DNA binding.
Table 3.
| A. Motifs over-represented at RBP-Jκ reactivation peaks at early promoters | Motif IDa |
||||||
|---|---|---|---|---|---|---|---|
| BCL11A | MNT | MAF B | TCF12 | EN2 | KLF12 | Rta-c | |
| Jk 2/Jk 5b | 6.9 | 5.8 | 4.6 | 3.8 | 3.8 | 3.5 | 2.9 |
| (9.4e-5) | (0.004) | (0.014) | (0.0007) | (0.03) | (0.003) | (0.03) | |
| Jk 2 + Rta/Jk 2c | 1.4 | -4.0 | 1.9 | 0 | 2.8 | 1.7 | 2.7 |
| (0.11) | (0.027) | (0.03) | (0.0004) | (0.0006) | (0.03) | (0.03) | |
| No. of promotersd | 10 | 7 | 7 | 12 | 8 | 14 | 8 |
| B. Motifs over-represented at RBP-Jκ reactivation/Rta co-peaks at early promoters | Motif IDa |
||||||
|---|---|---|---|---|---|---|---|
| POU4F1 | EN2 | Rta-c | POU3F4 | MZF1 | HMGA1/SOX5 | ZNF740/ZIC1 | |
| Jk 2 + Rta/Jk 2c | 3.6 | 2.8 | 2.7 | 2.7 | 2.4 | 2.4 | 2.2 |
| (NA) | (.00006) | (0.03) | (0.02) | (0.05) | (0.02) | (0.005) | |
| Jk 2/Jk 5b | −4.8 | 3.8 | 2.9 | −2.1 | −1.1 | 1.3 | 2.3 |
| (0.06) | (0.03) | (0.03) | (0.12) | (0.42) | (0.35) | (0.067) | |
| No. of promotersd | 5 | 8 | 8 | 9 | 14 | 7 | 10 |
| C. Motifs over-represented at RBP-Jκ latency peaks in early promoters | Motif IDa |
|||||
|---|---|---|---|---|---|---|
| POU3F3 | POU6F1/2 | GRHL1 | POU4F1 | ZEB1/NR1I3 | POU5F1 | |
| Jk 2/Jk 5b | −14.4 | −6.0 | −5.2 | −4.8 | −3.6 | −3.6 |
| (0.0004) | (0.03) | (0.002) | (0.06) | (0.05) | (0.05) | |
| Jk 2 + Rta/Jk 2c | 0 | 0 | 1.2 | 3.6 | 1.8 | 0 |
| (NA) | (NA) | (0.43) | (NA) | (0.25) | (0.18) | |
| No. of promotersd | 8 | 6 | 12 | 5 | 6 | 6 |
| D. Localization of POU Motifs at RBP-Jκ and Rta peaks | Motif IDa |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| POU3F3 | POU6F1/2 | POU4F1 | POU5F1 | POU5F1B | POU3F2 | POU3F4 | Oct-v | POU4F2 | POU2F3 | |
| Jk 2/Jk 5b | −14.4 | −6.0 | −4.8 | −3.6 | −2.4 | −2.4 | −2.1 | −1.5 | 1.0 | 1.9 |
| (0.0004) | (0.03) | (0.06) | (0.05) | (0.24) | (0.24) | (0.12) | (0.29) | (0.46) | (0.14) | |
| Jk 2 + Rta/Jk 2c | 0 | 0 | 3.6 | 0 | 0 | 0 | 2.7 | 2.0 | 2.1 | 1.6 |
| (NA) | (NA) | (NA) | (0.18) | (0.08) | (NA) | (0.02) | (0.04) | (0.06) | (NA) | |
| No. of promotersd | 8 | 6 | 5 | 6 | 4 | 3 | 9 | 10 | 7 | 7 |
a Best Match by TOMTOM Search of Homo sapiens Comprehensive Model Collection (HOCOMOCO) (48, 49). BCL11A = B-Cell CLL/Lymphoma 11A; EN2 = Engrailed Homeobox 2/HME4; GRHL1 = Grainyhead-like Transcription Factor 1; HMGA1/SOX5 = High Mobility Group AT-Hook 1/SRY-Box 5; KLF12 = Kruppel-like Factor 12; MAF B = MAF BZIP Transcription Factor B; MNT = MAX Network Transcriptional Repressor; MZF1 = Myeloid Zinc Finger 1; NR1I3 = Nuclear Receptor Subfamily 1 Group I Member 3; POU2F3 = POU Class 2 Homeobox 3; POU3F2 = POU Class 3 Homeobox 2; POU3F3 = POU Class 3 Homeobox 3; POU3F4 = POU Class 3 Homeobox 4; POU4F1 = POU Class 4 Homeobox 1; POU4F2 = POU Class 4 Homeobox 2; POU5F1 = POU Class 5 Homeobox 1; POU5F1B = POU Class 5 Homeobox 1B; POU6F1/2 = POU Class 6 Homeobox ½; TCF12 = Transcription Factor 12/HTF4; ZEB1 = Zinc Finger E-Box Binding Homeobox 1; ZNF740/ZIC1 = Zinc Finger Protein 740/Zic Family Member 1.
b Number of motifs <61 bp to RBP-Jκ Class 2 (“Jk2”) peak was divided by number of motifs <61 bp to RBP-Jκ Class 5 peak (“Jk5”) in early promoters, expressed as fold relative to actual ratio of Jk2/Jk5 peaks, which was 1.2. Number in parentheses is Z-test p value compared with the observed ratio of peaks.
c Number of motifs <61 bp to RBP-Jκ Class 2 peak/Rta peak (“Jk2 + Rta”) was divided by number of motifs <61 bp to RBP-Jκ Class 2 peak (“Jk2”) in early promoters, expressed as fold relative to actual ratio of Jk2/Jk5 peaks, which was 0.28. Number in parentheses is Z-test p value compared with the observed ratio of peaks; “NA” = not applicable.
d The number of promoters with co-localized Class 2 or Class 5 RBP-Jκ peak is shown.
We used a similar approach to analyze motif associations with co-localized Rta and RBP-Jκ peaks (Rta+/RBP-Jκ+). We first calculated the expected ratio by dividing the number of DE Class 2 RBP-Jκ peaks that were co-localized with Rta peaks (n = 8), by the total number of Class 2 RBP-Jκ peaks (n = 28). The expected ratio was 0.29, revealing that 29% of Class 2 RBP-Jκ peaks in promoters co-localized with Rta peaks. As predicted, Rta-c motifs were over-represented at RBP-Jκ+/Rta+ co-localized peaks (11/14 = 0.79 observed ratio/0.29 expected = 2.7-fold; Table 3A). Both sets of data therefore supported the mechanism we published by studying the viral Mta promoter: Rta binds to Rta-c motifs to stimulate RBP-Jκ DNA binding (16). These data also validate our use of the ratio–of–ratio approach to accurately identify other DNA motifs associated with regulated RBP-Jκ DNA binding.
DNA-binding motifs for cellular proteins associated with promoters at which RBP-Jκ DNA binding is enhanced during reactivation
To identify candidate DNA motifs for ratio–of–ratios analyses, we first used the program MEME in the discriminative mode (40) to discover the motifs that were associated with the highest RBP-Jκ Class 2 and Class 5 peaks. We then calculated the preferential association of each motif with ChIP/Seq peaks across the viral DE promoters using the ratio–of–ratios method described above. Of the 11 motifs that were most over-represented at Class 2 peaks, a TOMTOM search revealed that six had matches in the HOCOMOCO database (Table 3A and Fig. S3) (48, 49): B-cell CLL/Lymphoma (BCL) 11A; MAX Network Transcriptional Repressor (MNT); MAF BZIP Transcription Factor (MAF) B; Transcription Factor 12 (TCF12/also known as HTF4); Engrailed Homeobox 2 (EN2/also known as HME4); and Kruppel-like Factor (KLF) 12. The fold Jk2/Jk5 ratio for BCL11A was highest, at 6.9, and the element was present in 10 promoters (Table 3A). Five of the motifs were also over-represented at Rta+/RBP-Jκ+ peaks relative to RBP-Jκ Class 2 peaks alone, albeit modestly (Table 3A). TCF12 motifs were never found when Rta peaks were co-localized with RBP-Jκ in reactivation, and MNT motifs were negatively correlated with co-localized Rta and RBP-Jκ peaks.
DNA-binding motifs for cellular proteins most associated with promoters at which RBP-Jκ DNA binding is enhanced in the presence of Rta during reactivation
The DNA motifs that were most highly over-represented at RBP-Jκ Class 2 peaks were generally only modestly enriched if an Rta peak was co-localized (Table 3A, compare top row with middle row). Therefore, we repeated our motif discovery approach to find motifs preferentially over-represented at Rta+ Class 2 RBP-Jκ peaks relative to all Class 2 RBP-Jκ peaks. Of the 11 motifs most over-represented at Rta+/RBP-Jκ+ peaks, six had matches in the HOCOMOCO database (Table 3B and Fig. S4): POU Class 4 Homeobox 1 (POU4F1); Engrailed Homeobox 2 (EN2/also known as HME4); POU Class 3 Homeobox 4 (POU3F4); Myeloid Zinc Finger 1 (MZF1); High Mobility Group AT-Hook 1/SRY-Box 5 (HMGA1/SOX5); and Zinc Finger Protein 740/Zic Family Member 1 (ZNF740/ZIC1). The Rta-c motif was ranked third most over-represented at Rta-positive RBP-Jκ reactivation-specific peaks. Interestingly, two of the motifs are predicted to bind Pit/Oct/Unc (POU) family proteins, and both are also under-represented at RBP-Jκ Class 2 peaks relative to Class 5 peaks (negative folds in Table 3B, middle row).
DNA-binding motifs for cellular proteins associated with promoters at which RBP-Jκ DNA binding is eliminated during reactivation
Of the 10 motifs that were most over-represented at Class 5 peaks (expected ratio of 0.86 (24 Class 5 peaks/28 Class 2 peaks)), a TOMTOM search revealed that six had matches in the HOCOMOCO database (48, 49), and four were POU proteins (Table 3C and Fig. S5): POU Class 3 Homeobox 3 (POU3F3); POU Class 6 Homeobox 1/2 (POU6F1/2); Grainyhead-like Transcription Factor (GRHL11); POU Class 4 Homeobox 1 (POU4F1); Zinc Finger E-Box Binding Homeobox 1 (ZEB1); Nuclear Receptor Subfamily 1 Group I Member 3 (NR1I3); POU Class 5 Homeobox 1 (POU5F1). The POU3F3, POU6F1/2, POU5F1 motifs were never found near an Rta+ RBP-Jκ peak (Table 3C, middle row). The remaining motifs were over-represented at RBP-Jκ Class 2 peaks only with a co-localized Rta peak (Table 3C).
Orphan DNA motifs
Our discovery approach also identified a set of motifs with no matches in HOCOMOCO (Table S3 and Fig. S6). These orphan motifs were over- or under-represented at different categories of RBP-Jκ peaks, and they shared no obvious sequence similarity to each other.
Multiple DNA-binding motifs for the POU family of transcription factors are candidates for positive and negative regulation of RBP-Jκ DNA binding
The motif discovery analyses (Table 3, A–C) showed a remarkable association of DNA-binding motifs for multiple members of the cellular POU transcription factor family with regulated RBP-Jκ DNA binding. Moreover, we previously showed that the cellular protein Oct-1 (family member POU2F1) binds to a noncanonical POU motif in the KSHV K-bZIP promoter to modulate Rta transactivation; we termed this motif “Oct-v” (50). This Oct-v motif is localized to the highest RBP-Jκ Class 2 peak (Fig. 2 and Table S1B) in our ChIP/Seq data. The POU proteins are a large family of DNA-binding transcription factors (51–53). Nine of the POU motifs have similar sequences that contain the conserved, central trinucleotides AT(G/T) and are flanked to one or both sides by A/T-rich sequences (Fig. 5). Remarkably, the inverted consensus sequence for the Rta-c motif can also be aligned to the central ATG of the POU motifs (Fig. 5).
Figure 5.
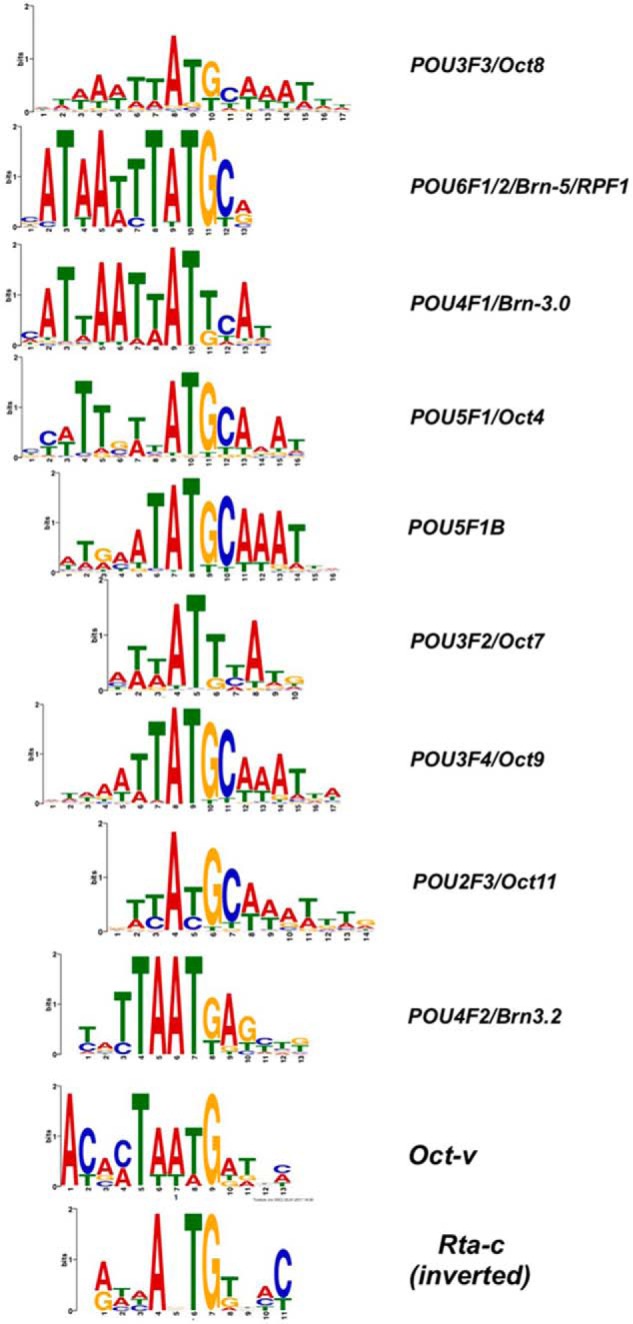
Sequence logos for POU DNA motifs. POU motifs from HOCOMOCO (48, 49), aligned to Oct-v motif (50) and Rta-c (CANT) DNA consensus (16) in inverted orientation.
We determined the potential participation of all of these nine POU proteins in regulating RBP-Jκ DNA binding to the virus by comparing the associations of their DNA motifs (Fig. 5) with RBP-Jκ and Rta peaks in DE promoters. Remarkably, seven of the POU proteins were over-represented at RBP-Jκ latency-specific Class 5 peaks (Table 3D, top row). The Class 2/Class 5 folds ranged from −14.4 for POU3F3 to −2.1 for POU3F4; Oct-v motifs were also over-represented at RBP-Jκ Class 5 peaks, with a fold value of −1.5.
The association of most POU motifs with RBP-Jκ+ peaks was dramatically different when an Rta peak was co-localized (Table 3D, middle row). All of the motifs that were over-represented at RBP-Jκ Class 5 peaks were either completely absent at Rta+/RBP-Jκ+ peaks or over-represented at those dual peaks. This pattern suggested that those POU factors might inhibit RBP-Jκ DNA binding during reactivation, but are counteracted by Rta.
Given the homology between POU motifs and the Rta-c consensus, we also compared the observed ratios of co-occurring Rta-c POU motifs from Table 3D at RBP-Jκ Class 2 and 5 peaks in DE promoters (Table S4). Three of the POU motifs were never found with Rta-c motifs and were eliminated from consideration. POU2F3, P5F1B, and POU3F4 were only found with Rta-c motifs at RBP-Jκ Class 2 peaks, but not Class 5 peaks. Oct-v and Rta-c motifs were also over-represented at Class 2 peaks, at 12.3-fold.
When repeating this analysis but also scoring for co-localized Rta peaks, POU2F3 and Oct-v motifs were the most over-represented (Table S4, middle row). These data suggest that the POU family of proteins has complex roles in regulating RBP-Jκ DNA binding.
Dual Rta-c/Oct-v motifs are also over-represented in viral promoter DNA relative to nonpromoter DNA (Fig. S7A). Within promoters, the Rta-c/Oct-v motifs were vastly over-represented in DE, rather than L promoters, with only one motif localized to the latter promoter set (Fig. S7B). These data suggest a selection for these motifs to co-occur in the fraction of the viral genome that confers Rta regulation on DE gene expression.
Candidate promoters
Guided by the ChIP/Seq peak maps and motif analyses, we investigated whether the binding of Rta and RBP-Jκ on the KSHV genome during reactivation corresponds with RBP-Jκ–dependent Rta transactivation. We selected seven genomic segments for these analyses, named ORF50 distal and proximal, ORF50AS/K-bZIP, ORF56, and Mta distal and proximal (Fig. 6, A and B). Each segment contained at least one consensus RBP-Jκ motif, varying numbers of Rta and RBP-Jκ peaks, and assorted motifs that were identified using MEME, including POU motifs (Tables 3, A–D (40)). Portions of each fragment highlighting RBP-Jκ peaks and motifs are diagrammed in Fig. 6B.
Figure 6.
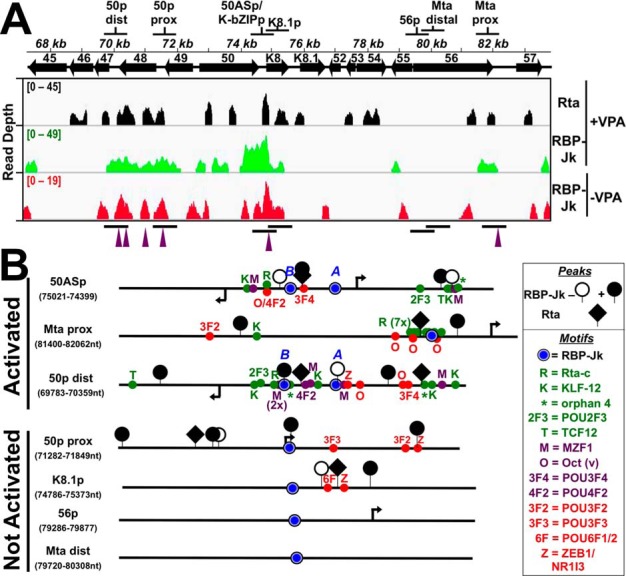
Candidate promoter segments selected from ChIP experiments. A, genomic locations. Top shows the name and location of each segment. Bottom shows the visualization of ChIP/Seq peaks using IGV (85). Antibodies used are indicated at right, and chromatin was from latent (−VPA) or reactivated (+VPA) virus in BC-3 cells. Solid arrows represent open reading frames (ORFs). Numbering above arrows indicates ORF name. Peak heights are given in read depth per genomic position. Purple arrowheads below each panel indicate co-localized Rta and RBP-Jκ peaks in reactivation. B, portions of candidate promoters. Schematics representing portions of candidate promoters containing RBP-Jκ motifs and RBP-Jκ peaks are shown. The names of each promoter and the boundaries of schematics are indicated at left. The boxed legend explains the symbols. Arrows indicate positions of transcription start sites mapped to the viral genome within each candidate promoter. Motif text colors are as follows: red = over-represented at RBP-Jκ latency peaks; green = over-represented at RBP-Jκ reactivation peaks; purple = motif over-represented at coincident RBP-Jk/Rta peaks. Activated and nonactivated refer to results of Rta transactivation shown in Fig. 7.
Rta and RBP-Jκ DNA binding are not sufficient for Rta transactivation
We cloned each candidate promoter (Fig. 6, A and B) into a luciferase reporter vector and tested Rta's ability to activate the selected segments by co-transfecting each reporter plasmid with increasing amounts of Rta expression vector into uninfected (BL-41) B cells. An example of our strategy is shown in Fig. 7A for the ORF50AS promoter–reporter, which demonstrates typical dose-dependent transactivation by Rta. In these experiments, Rta is expressed in a dose-responsive fashion proportional to the amount of Rta expression vector transfected (Fig. 7A, inset). The maximal transactivation of each of the reporter plasmids from similar titration curves is shown in Fig. 7B. The data show that in addition to the ORF50AS promoter, Rta only transactivated the two promoters that also contained Rta- and RBP-Jκ–binding sites (by ChIP analyses), Rta-c motifs, and either Oct-v or POU4F2 motifs (Fig. 7B). Rta failed to transactivate the ORF50-proximal, K8.1, and Mta-distal promoters, which all lacked those motifs regardless of Rta and RBP-Jκ DNA binding
Figure 7.
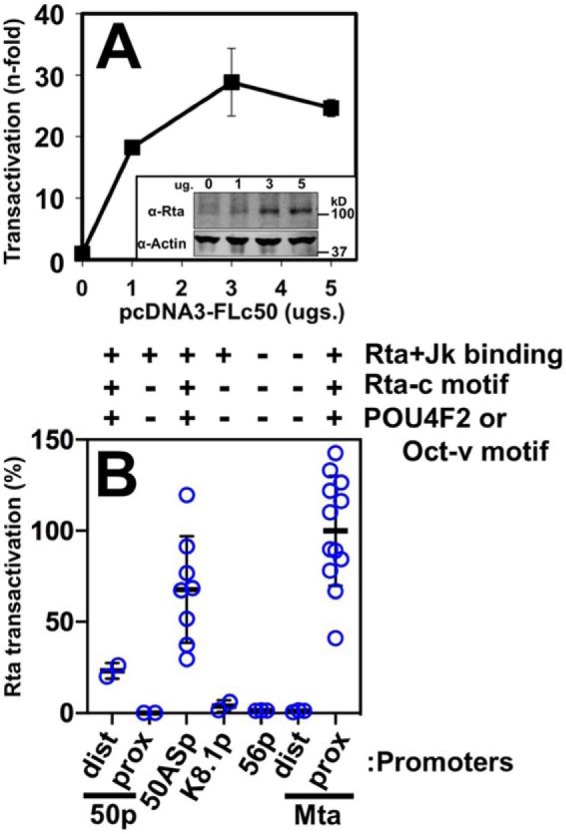
RBP-Jκ DNA binding is not sufficient for Rta transactivation. A, ORF50AS promoter. Reporter plasmid was co-electroporated into BL-41 cells alone or with increasing amounts of Rta expression vector in triplicate. Cells were harvested 48 h post-electroporation, and luciferase was measured. Each luciferase value was normalized to β-gal expression from a second reporter plasmid that was co-electroporated as control. Fold transactivation was calculated by comparison with the promoter reporter transfected alone. Inset, typical results of Western blotting to show expression of ectopic Rta in transactivation experiments. Top panel, Western blotting of total cellular protein probed with anti-Rta serum. Bottom panel, Western blotting from same gel probed with anti-actin. B, other candidate promoters. Each of the indicated reporter plasmids were co-electroporated into BL-41 cells with Rta expression vector following the procedure described in A. The maximal transactivation from each titration curve was normalized to transactivation of the Mta proximal (Mta prox) promoter, which was set at 100%. Thick lines indicate means of values, and thin lines indicate standard errors. Chart at top shows result of Rta + Jk binding from Fig. 2, and the presence or absence of indicated motifs as in Fig. 6. dist prox, distal and proximal.
RBP-Jκ DNA-binding affinity is generally proportional to Rta transactivation
Alignment of the putative RBP-Jκ motifs from the candidate promoters revealed sequence heterogeneity (Fig. 8A). We hypothesized that the observed variation in Rta's magnitudes of transactivation of the candidate promoters (Fig. 7) might be explained by variation in RBP-Jκ DNA-binding affinities to the motifs. We previously used recombinant RBP-Jκ protein in EMSA and footprinting to demonstrate that purified RBP-Jκ protein binds with high affinity (∼15 nm dissociation constant (KD)) to its motif in the RBP-Jκ–proximal promoter (Fig. 8A, top and data not shown) (8, 16, 54). To authenticate the RBP-Jκ ChIP/Seq peaks in this study, we asked whether DNA oligos containing each of the candidate RBP-Jκ motifs could compete for purified RBP-Jκ binding to the 32P-labeled Mta motif in EMSA (Fig. 8A). The control competitors were the WT Mta proximal oligo and an Mta mutant oligo corresponding to a promoter that Rta fails to transactivate (16). To estimate DNA-binding affinities, we titrated each motif to 10-, 30-, and 100-fold molar excesses over Mta proximal WT oligo, respectively.
Figure 8.

Six predicted RBP-Jκ motifs bind to purified RBP-Jκ protein. A, sequences of EMSA oligos chosen from promoters in Fig. 6. Top (+) and bottom (−) strands are indicated. B and C, competition EMSAs. Increasing amounts of the indicated and unlabeled competitor DNAs were preincubated with GST-RBP-Jκ, before addition of 32P-labeled Mta proximal (Mta prox) DNA. Mixtures were electrophoresed and visualized by autoradiography. Vertical line in B indicates cropping of irrelevant lanes in center of image. D, quantitation. Shifted complexes from each gel in B were measured by phosphorimaging. Each complex was normalized to the complexes formed by GST-RBP-Jκ and labeled Mta-proximal DNA alone, in each gel, which was set as 100%. Blue text indicates RBP-Jκ motifs from promoters activated by Rta in Fig. 7. prox dist, proximal distal.
The putative motifs resolved into three groups based on competition for RBP-Jκ binding. The strongest competitors comprised a group containing ORF50AS/K-bZIP motifs A and B, ORF50 distal motifs A and B, and the Mta-proximal motif itself (Fig. 8D); the apparent KD values of these strongest competitors were difficult to distinguish at these probe concentrations. The weakest competitors comprised a group containing the WT K8.1 motif, mutant ORF50AS/K-bZIP motifs A and B, and mutant Mta proximal motifs (Fig. 8, B and D, dotted lines); apparent affinities were ∼45–300 nm. The third group demonstrated an intermediate ability to compete for RBP-Jκ and included the 50p proximal, 56p, and Mta distal motifs (Fig. 8, B and D, open symbols); the apparent affinities of this intermediate group were reduced about 2-fold, but this is likely an underestimate at these probe concentrations.
We conclude that relative magnitudes of Rta transactivation of the candidate WT promoters (Fig. 7) correlate with variations in RBP-Jκ DNA-binding affinities to the WT elements (Fig. 8; motifs from transactivated promoters are indicated by blue traces for easy comparison in Fig. 8D). In addition, the ChIP/Seq data that identified RBP-Jκ–binding motifs in vivo were confirmed by EMSAs for those motifs with the highest RBP-Jκ–binding affinity in vitro (50p distal, ORF50AS/K-bZIP site B, and Mta proximal; compare location of peaks in Fig. 6 with Figs. 7 and 8). The ChIP/Seq and EMSA data did not agree for one strong in vitro RBP-Jκ–binding element (ORF50AS/K-bZIP site A) and two intermediate in vitro binders (ORF56 and Mta distal); because those interactions were not detected by ChIP/Seq, those sites have the potential to bind to RBP-Jκ but fail to do so in vivo and/or reflect limitations of cross-linking efficiency.
Finally, ChIP-Seq showed that RBP-Jκ bound to the K8.1 promoter in both latency and reactivation, but recombinant RBP-Jκ failed to bind the predicted RBP-Jκ motif in vitro. We note the predicted K8.1 motif has the most divergent sequence from the RBP-Jκ consensus of any of the WT motifs tested (Fig. 8A).
Composite Rta-/Oct-v motif represses basal activity but is essential for robust Rta transactivation of the ORF50AS promoter
Despite the correspondence between RBP-Jκ DNA–binding affinity and Rta transactivation (Figs. 7B and 8D), the bioinformatic analyses of the ChIP/Seq data suggested that POU motifs (Tables 3, B–D, and Table S4), rather than the specific sequences of RBP-Jκ motifs (Fig. 4C), were more strongly associated with stimulation of RBP-Jκ DNA binding. To test the significance of a POU motif in Rta transactivation, we focused on the ORF50AS promoter.
ORF50AS is one side of a bidirectional promoter that also controls transcription of the K-bZIP gene in the opposite direction (50, 55). We previously showed that the protein POU2F1/Oct-1 bound to the Oct-v element in the promoter (50). In that publication, when we mutated the Oct-v element, the Octamer-1 binding was reduced, and Rta transactivation of the K-bZIP promoter was debilitated in B cells (50). Closer analysis of the promoter sequence reveals that the element is a composite of an Rta-c and Octamer-v motif, and our mutation affects both parts of the motif (Fig. 9, top). We noted that the Oct-v motif is also homologous to the POU4F2 consensus motif (Fig. 5).
Figure 9.
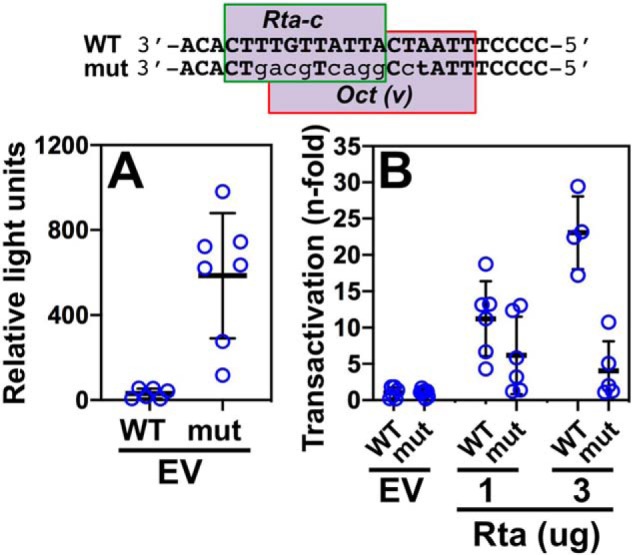
Rta-c/Oct-v motif is necessary for optimal transactivation of the ORF50AS/K-bZIP promoter by Rta. WT and mutant reporters were tested for Rta transactivation using the approach described in the legend to Fig. 8, A and B. The sequence of the Rta-c/Oct-v motif in the ORF50AS/K-bZIP promoter is shown at top. Mutated bps are indicated by lowercase lightface type. A, basal activity of promoter/reporters. B, fold transactivation by Rta. Thick lines indicate means of values, and thin lines indicate standard errors.
To test the requirement for the Oct-v element in Rta transactivation of the ORF50AS promoter (the opposite direction of K-bZIP), we co-transfected the WT and mutant reporter plasmids with increasing amounts of Rta expression vector, or empty vector, in BL-41 cells. With empty vector alone, the basal activity of the promoter increases from ∼30 relative light units (RLU) to ∼585 RLU, suggesting that the mutation eliminates binding of a transcriptional repressor (Fig. 9A) to the promoter.
However, the intact motif is required for robust Rta transactivation of the ORF50AS promoter (Fig. 9B). Mutating the motif severely impaired, but did not completely eliminate, Rta transactivation (Fig. 9B).
Both the affinity of RBP-Jκ DNA binding and proximity to Oct-v motifs are essential for robust RBP-Jκ–dependent Rta transactivation
The DNA that lies between the divergent start sites in the bidirectional ORF50AS/K-bZIP promoter contains two RBP-Jκ motifs, which we have named “Jk A,” and “Jk B” (Fig. 10A). Although purified RBP-Jκ binds to both motifs with high affinities, motif B competes to bind RBP-Jκ more potently than motif A competes to bind RBP-Jκ (Fig. 8D, bottom). Moreover, the RBP-Jκ–binding mutations are more debilitating in motif B than in motif A in EMSA (Fig. 8D, bottom). To test the effects of these mutations on Rta transactivation, we introduced them, alone or together, into the cognate promoter construct, and then tested each reporter for transactivation by Rta using the approach described in Fig. 7. Mutation of either site alone reduced transactivation to different magnitudes. Mutation of site B, alone, but not site A, eliminated Rta transactivation of the ORF50AS promoter (Fig. 10A). The single site B mutant was equally as debilitated as the double mutation in both sites A and B.
Figure 10.

RBP-Jκ element location is important for Rta transactivation of the ORF50AS and K-bZIP promoters. A, transactivation of the WT and mutant ORF50AS and K-bZIP promoters. Each RBP-Jκ–binding site in the promoter was mutated alone, or together, and then tested for Rta transactivation using the approach described in the legend to Fig. 7, A and B. Thick lines indicate means of values, and thin lines indicate standard errors. B, distance of the RBP-Jκ motif to the Rta-c/Oct-v motif determines Rta transactivation magnitude. Each RBP-Jκ–binding site in the promoter was altered as shown in the schematic and then tested for Rta transactivation using the approach described in the legend to Fig. 7, A and B. For both panels, results are shown as percentage of transactivation of WT promoters by Rta divided by empty vector. Promoter schematics follow the design described in the legend to Fig. 6. Thick lines indicate means of values, and thin lines indicate standard errors.
To study the requirement of the individual RBP-Jκ elements by Rta on the K-bZIP promoter, we tested each WT and mutant promoter cloned in the inverse orientation relative to the luciferase reporter gene. Consistent with the results for ORF50AS, mutation of either of the RBP-Jκ sites reduced Rta's ability to transactivate the promoter. Therefore, site B was essential for Rta transactivation in both promoter directions (Fig. 10A). Because site B binds RBP-Jκ with somewhat higher affinity (Fig. 8), these transactivation results agreed with our earlier observations that relative affinities of RBP-Jκ DNA binding are directly proportional to Rta transactivation levels (Figs. 7B and 8).
However, RBP-Jκ motif B is also closer to the Rta-c/Oct-v motif, so we could not eliminate the possibility that the proximity of motif B to Rta-c/Oct-v affected the magnitude of Rta transactivation. Therefore, we exchanged the position of motifs A (CCTGGGAA) and B (CGTGAGAA) in the ORF50AS promoter using site-directed mutagenesis (Fig. 10B). Importantly, the relative binding affinities of RBP-Jκ to the “swapped” motifs were not affected, indicating that the affinities were intrinsic to the sequences of each RBP-Jκ motif themselves, but not the sequence surrounding the motifs (compare 50AS A WT with 50AS site A inv B, and 50AS B WT with 50AS site B inv A, Fig. 8D, bottom). We found that exchanging the position of the two motifs reduced Rta transactivation by 60% (Fig. 10B, compare WT with invAB). Moreover, completely removing motif A in reporter invAB further debilitated Rta transactivation to 10% of WT levels (Fig. 10B, compare WT with invAB and invB). These data suggest that the context of an RBP-Jκ motif is more important for Rta transcriptional activation than the sequence of the RBP-Jκ motif in an otherwise identical promoter.
However, because a single copy of the RBP-Jκ motif A is never sufficient for Rta transactivation even at position B (compare invA in Fig. 10B to mutB in Fig. 10A), these data also reiterate that the sequence of an RBP-Jκ motif, and thus RBP-Jκ DNA-binding affinity, contributes to Rta transactivation.
We also found that a single copy of RBP-Jκ motif B is sufficient at either its cognate position or at position A, but is favored in its cognate site (compare invB in Fig. 10B with mutA in Fig. 10A). Adding a second A or B motif to a promoter containing only one RBP-Jκ motif always enhances Rta transactivation relative to the promoter with a single motif (Fig. 10B).
Overall, these data confirm the crucial role of the Rta-c and Oct motifs in Rta transactivation. Because RBP-Jκ motif B is closer to the Rta-c and Oct-v elements in the native promoter, and moving motif B from its native position to the distal site A reduces transactivation, the data support our conclusion that Rta transactivation and stimulation of RBP-Jκ DNA binding depends upon proximity between particular RBP-Jκ motifs to Rta-c and/or Oct-v elements (Fig. 10).
ORF50AS/K-bZIP, Mta, and ORF50 distal promoters have similar architectures of RBP-Jκ, Rta-c, and POU motifs
Close inspection of the ORF50AS/K-bZIP promoter shows that Jk motif B is positioned 26 bp from the Rta-c motif and 29 bp from the Oct-v motif (Fig. 11A). Comparison of ORF50AS/K-bZIP with the two other promoters that were activated by Rta, Mta proximal and ORF50 distal (Fig. 7), shows that they contain similar motif architectures (Fig. 11, B and C). The Mta promoter RBP-Jκ motif at position 81,981, which is essential for Rta transactivation and stimulation of RBP-Jκ DNA binding, is spaced 25 and 31 bp to Rta-c and Oct-v DNA motifs, respectively (Fig. 11B). The ORF50 distal promoter contains an RBP-Jκ motif (Fig. 11C) spaced 25 bp from a POU4F2 motif, and 39 bp from an Rta-c motif (Fig. 11C). Although the distance separating the Rta-c and RBP-Jκ motifs in the ORF50 distal promoter is greater than those in the ORF50AS/K-bZIP and Mta promoters, we note that the ORF50 distal promoter contains a 2nd sequence with slight divergence from the Rta-c consensus that is spaced 26 bp from RBP-Jκ (light green outline in Fig. 11C). These observations suggest that KSHV has conserved specific architectures of POU and Rta-c motifs to facilitate regulated RBP-Jκ DNA binding and Rta transactivation (Fig. 11D).
Figure 11.

Architecture of RBP-Jκ, POU, and Rta-c motifs are similar in the ORF50AS/K-bZIP (A), Mta (B), and ORF50 (C) distal promoters. Established or predicted cis-regulatory elements are shown by rectangles. The arrows on the thick line in A indicate positions of DE transcriptional start sites, with genomic coordinates listed below (8). Numbers below each RBP-Jκ motif (blue outlined Jk) indicate genomic coordinates and/or positions of motifs relative to transcriptional start sites. Horizontal arrows on DNA sequences indicate bp distances between motifs. D, summary of motif arrangements in Rta-transactivated promoters.
The proximities of the POU, Rta-c, and RBP-Jκ motifs in the three activated promoters were reflected in the relative distances between these motifs and RBP-Jκ peaks across the viral DE promoters (Fig. S8). Distance medians and ranges of Rta-c motifs to RBP-Jκ peaks decreased in the order RBP-Jκ latency > RBP-Jκ reactivation > RBP-Jκ reactivation + Rta (Fig. S8, left). Distance medians and ranges of Oct-v and POU4F2 to RBP-Jκ motifs were not significantly different during latency or reactivation, but they were dramatically shorter for co-localized RBP-Jκ and Rta peaks in reactivation (Fig. S8). Conversely, the medians and ranges of distances between RBP-Jκ peaks and POU3F3 motifs, which were not found in any Rta-transactivated promoters, increased in the reverse order of Rta-c, with the shortest distances to latency-specific peaks (Fig. S8). These data suggest the general conservation of proximities of Rta-c, POU, and RBP-Jκ sites across the viral DE promoters, supporting a critical overall role for these factors in regulating viral reactivation.
Rta binds the composite Rta-c/Octamer motif from the ORF50AS/K-bZIP promoter in a sequence-specific fashion
We used competition EMSAs to compare binding of Rta to the ORF50AS/K-bZIP Rta-c/Oct-v motif to its homologous element in the Mta proximal promoter and to the nonhomologous Rta-responsive element from the PAN promoter. Purified Rta forms two robust complexes with the ORF50AS/K-bZIP probe alone (Fig. S9, lane 2). Preincubation of Rta with DNAs containing the intact Rta-c motif from the Mta promoter (Mta Rta-c WT, m2, and m3) inhibit formation of the Rta/ORF50AS/K-bZIP DNA complexes (Fig. S9). However, the mutant Rta-c/Oct-v motif, Mta proximal m1, is severely impaired in its ability to compete for Rta binding (Fig. S9). Furthermore, the Rta-responsive element from the PAN promoter also competes with the ORF50AS/K-bZIP element to bind Rta. The Rta-binding activity of the PAN element exceeds that of the single Mta Rta-c motif, confirming a previous report (56). We conclude that Rta binds to the single Rta-c/Oct-v motif from the bidirectional ORF50AS/K-bZIP promoter in a fashion similar to the single Rta-c motif from the Mta promoter. Our current data therefore cannot distinguish between the importance of Rta or Oct-1 binding to the Rta-c/Oct-v element in transactivating the ORF50AS/K-bZIP promoter.
Oct-1 is induced during KSHV reactivation
Because our data implicate POU factors, and particularly Oct-1, in regulating KSHV transcription, we evaluated Oct-1 protein expression in infected cells. Western blotting shows that Oct-1 protein is induced by treatment of BC-3 cells with TPA to reactivate KSHV (Fig. 12A). Induction of Oct-1 expression is at least mediated at the level of transcription, as we also detected increased Oct-1 mRNA following TPA-stimulated reactivation in infected B cells using RNA-Seq (data not shown).
Figure 12.
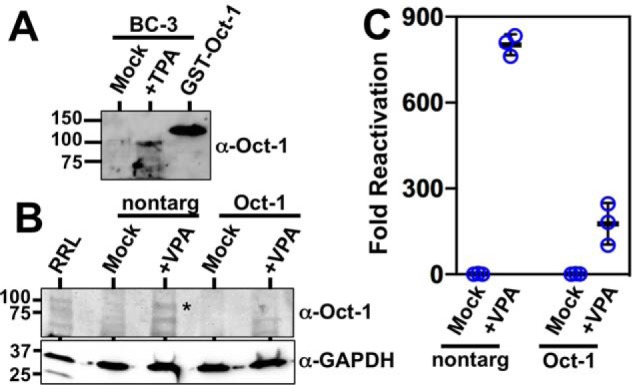
Oct-1 is required for optimal KSHV reactivation. A, Oct-1 is induced during KSHV reactivation in PEL cells. KSHV-infected BC-3 cells were treated with TPA or with vehicle control (mock) and tested for Oct-1 expression by Western blotting. Purified recombinant GST–Oct-1 protein was included as a Western blotting control. Numbers at left indicate positions of protein apparent molecular weight standards. B, Oct-1 siRNA specifically knocks down Oct-1. Whole-cell protein extracts from Vero cells transfected in Fig. 10A were pooled, and equal concentrations were analyzed by SDS-PAGE/Western blotting. RRL = rabbit reticulocyte lysate programmed to express Oct-1 protein. The asterisk indicates migration of Oct-1–specific protein band. Numbers at left indicate positions of protein apparent molecular weight standards. C,. Oct-1 knockdown debilitates KSHV reactivation. KSHV-infected Vero rKSHV.294 cells were transfected with Oct-1–specific siRNA and control siRNA (scrambled), incubated for 48 h, and then treated with 1.0 mm VPA for 6 h. Media were replaced with fresh media and incubated an additional 48 h. Infectious virus was quantitated by SeAP assay. Fold reactivation was calculated by normalizing SeAP activity to that for control cells, which had been incubated with supernatants from Vero rKSHV.294 cells that were transfected with control siRNA without VPA. Thick lines indicate means of values, and thin lines indicate standard errors. Nontarg = transfected with control (nontargeting) siRNAs.
Oct-1 is required for optimal KSHV reactivation
Transactivation by Rta is essential for KSHV reactivation (4). If the proximity of the Rta-c/Oct-v element to the ORF50AS/K-bZIP RBP-Jκ site B is relevant to Rta transactivation, then Oct-1 should also contribute to KSHV reactivation. To evaluate the significance of Oct-1 for KSHV reactivation, we knocked down endogenous Oct-1 by transfecting infected Vero cells with Oct-1–specific siRNAs. In the presence of nontargeting siRNA, VPA treatment of infected Vero cells induced Oct-1 protein expression (Fig. 12B), similar to induction of Oct-1 in infected human PEL cells (Fig. 12A). Transfection of Oct-1–specific siRNAs reduced Oct-1 protein in both untreated and VPA-treated Vero cells (Fig. 12B).
Finally, inhibiting Oct-1 expression decreased VPA-stimulated reactivation from ∼800- to 175-fold (Fig. 12C). Control (nontargeting) siRNA showed no effects on reactivation (Fig. 12C). Therefore, these data confirmed that Oct-1 is required for KSHV reactivation.
Discussion
KSHV reactivation requires the viral lytic switch protein Rta to form ternary complexes with the cellular Notch signaling effector RBP-Jκ and viral DNA (8–10, 16, 19). Previous publications demonstrated that Rta transactivation of many viral promoters requires RBP-Jκ in uninfected cells. Our data identify hundreds of Rta- and RBP-Jκ–binding sites on the KSHV genome during latency and reactivation (Fig. 2 and Table 1), suggesting Rta's potential to transactivate almost all viral promoters during reactivation. The large number of RBP-Jκ and Rta peaks is commensurate with the large number of predicted RBP-Jκ motifs on the KSHV genome. Indeed, we count 121 matches to the RBP-Jκ consensus motifs in the genome, and RBP-Jκ can bind to additional motifs that have a relaxed consensus (17). The abundance of RBP-Jκ viral–binding sites identified in VPA-induced reactivation in this study agrees with the broad stimulation of RBP-Jκ binding to the KSHV genome during TPA-stimulated reactivation that we previously observed at a lower resolution (16). ChIP/PCR of peak specificity after TPA treatment also agrees with our VPA peak identification (data not shown) (8). Thus, Rta's effect on RBP-Jκ DNA binding is not limited by the viral reactivation signal. Several known targets for Rta and RBP-Jκ were identified in our data, including the Mta, K-bZIP, and PAN promoters (Table S1). Whereas previous papers also suggested a large number of potential Rta and RBP-Jκ targets on the viral genome (19, 57–59), the breadth of the genome bound by Rta and RBP-Jκ in our work exceeds those papers and may be due to technical differences.
We devised a strategy to classify each RBP-Jκ peak into one of five categories based on DNA binding during latency and/or reactivation (Table 2). Our classification confirms that RBP-Jκ binding to the KSHV genome is not static, but rather is dramatically dynamic during reactivation. The vast majority of RBP-Jκ peaks was unique to latency or reactivation, but not both states (Tables 2 and Table S2).
Based on our previous work that focused on regulation of the KSHV Mta promoter (8, 16), we hypothesized that specific, conserved elements on the viral genome would define the dynamic relationship between DNA binding of Rta and RBP-Jκ. In that work, we showed that Rta stimulates RBP-Jκ DNA binding to the Mta promoter through interactions with Rta-c (CANT) and (A/T)3 repeats in close proximity to, or straddling, an RBP-Jκ motif (Fig. 1) (16). Among those elements of our working model, the ChIP/Seq data show that coincident Rta DNA binding and a nearby Rta-c DNA motif are both independently associated with stimulation of RBP-Jκ DNA binding to promoters across the viral genome during reactivation (Fig. 4 and Tables 3, A and B). Moreover, all 13 instances of Rta bound to an Rta-c promoter motif are found at reactivation-specific RBP-Jκ peaks (Fig. 4C). Importantly, these data support the conclusion that the mechanism we established for Rta stimulation of RBP-Jκ DNA binding to the Mta promoter (16) extends to additional viral genes.
While this work confirms the significant roles of the Rta and the Rta-c motif in regulating RBP-Jκ DNA binding, it reveals important exceptions to our earlier publication (16). On the Mta promoter, we identified seven Rta-c motifs arranged in tandem and in palindromes; the number of Rta-c motifs was proportional to Rta DNA-binding affinity, formation of ternary complexes with DNA and RBP-Jκ, and stimulation of RBP-Jκ DNA binding (16). This work shows that the multiple copies of Rta-c in the Mta promoter are the exception in the KSHV genome, and none of the other reactivation-specific RBP-Jκ peaks coincide with Rta-c multimers (Fig. 4C). The ChIP/Seq data also suggest that the (A/T)3 repeats and the Mta-specific RBP-Jκ motif sequence are not major influences on regulating RBP-Jκ DNA binding to viral DNA (Fig. 4C). Most significantly, 20 of the 33 reactivation-specific RBP-Jκ peaks in DE promoters lacked the Rta peak/Rta-c motif combination (Table S1D). We propose three scenarios for those RBP-Jκ Class 2 peaks at which Rta was not found: 1) Rta does not remain uniformly associated to all RBP-Jκ/DNA sites; 2) Rta epitope is hidden at some of the Rta/RBP-Jκ/DNA sites; or 3) RBP-Jκ DNA binding can be stimulated independently of Rta. Scenario 1 is supported by our observation that many Rta-negative, reactivation-specific RBP-Jκ peaks have corresponding Rta-c or PAN-like DNA motifs (data not shown). Scenario 3 implies that additional unknown DNA-binding proteins stimulate RBP-Jκ DNA binding.
To reveal those additional candidate regulators of RBP-Jκ DNA binding, we used a ratio–of–ratios calculation to identify DNA motifs that were over-represented at RBP-Jκ peaks in either reactivation (Class 2) or latency (Class 5) (Tables 3, A and C). We reasoned that motifs over-represented in latency (Class 5) would bind proteins that repressed RBP-Jκ DNA binding during reactivation and that motifs over-represented in reactivation would bind proteins that stimulated RBP-Jκ DNA binding (Class 2). This motif discovery approach was validated by our observation that Rta-c motifs were over-represented at Class 2 peaks with and without corresponding Rta peaks (2.7- and 2.9-fold, respectively (Table 3A)).
Despite our initial expectation that Class 2 motif–binding proteins would be stimulators of RBP-Jκ DNA binding, we found that the published transcriptional functions of many of the cellular motif-binding proteins contradict their grouping into Class 2 or 5. For example, we identified the Oct-v motif as a Class 5 negative regulator of RBP-Jκ DNA binding during reactivation (Fig. 3, C and D). However, we previously showed that the Oct-v motif binds to a supporter of Rta transactivation, the cellular protein POU2F1/Oct-1 (50). Comparing co-localization of Rta and RBP-Jκ DNA binding in the ChIP data instead revealed a de-repression/stimulation mechanism for POU proteins and Rta to regulate RBP-Jκ DNA binding. In the absence of Rta, the Oct-v motif is over-represented at latency-specific RBP-Jκ peaks (Class 5), but in the presence of an Rta peak, the Oct-v motif is over-represented at reactivation-specific RBP-Jκ peaks (Class 2; Table 3D). These data are consistent with a mechanism in which the Oct-v motif binds a protein that eliminates RBP-Jκ DNA binding during reactivation unless Rta is present at the promoter. In reactivation, Rta induces Oct-1 expression, which binds Oct-v and cooperates with Rta to eliminate the repressive Oct-v binder and to sustain and stimulate RBP-Jκ DNA binding (Fig. 13).
Figure 13.
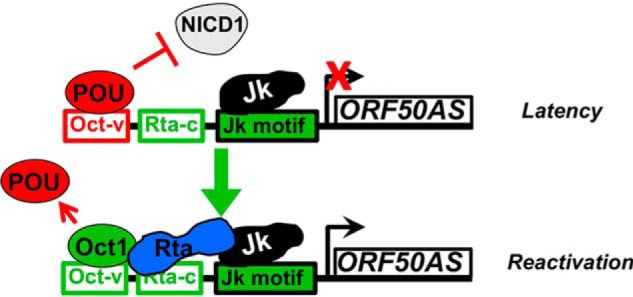
Model for regulation of RBP-Jκ DNA binding on the ORF50AS promoter. POU proteins inhibit NICD1/RBP-Jκ interaction in latency. Rta/Oct-1 complex removed POU and sustains and stimulates Jk DNA binding in reactivation.
Our classification of the Oct-v/POU4F2 site in the ORF50AS/K-bZIP promoter as a Class 5 motif alone but a Class 2 motif in the presence of Rta also agreed with the promoter reporter experiments: mutation of the Oct-v motif dramatically increased the basal activity of the promoter but also debilitated Rta transactivation (Fig. 9). Furthermore, we hypothesize that an Oct-v–binding protein excludes constitutively-active Notch1 from the ORF50AS/K-bZIP promoter during latency and in uninfected B cells; in this scenario, mutation of Oct-v prohibits binding of the POU protein, thus permitting Notch access to RBP-Jκ to increase the apparent basal activity of the promoter (Fig. 13).
Further support for the POU proteins as major regulators of RBP-Jκ DNA binding and KSHV reactivation comes from analyses of Rta-c and POU motif combinations. Dual Rta-c/Oct-v motifs are over-represented at RBP-Jκ Class 2 peaks by 12.3-fold, and the motif combination is only found at one Class 5 peak (Table S4 and data not shown). Moreover, the highest Class 2 RBP-Jκ peak localizes to the Rta-c/Oct-v motif combo in the ORF50AS/K-bZIP promoter (peak 53, Table S1B). Additional POU motifs are only found at RBP-Jκ Class 2 peaks when co-localized with an Rta-c motif (POU2F3, POU5F1B, and POU3F4; Table S4) or with DNA-bound Rta (POU4F1, POU4F2, POU2F3, and POU 3F4; Table 3D).
We predict that the latent Oct-v–binding protein is in the POU family because POU proteins bind to DNA motifs that share sequence homology with Oct-1–binding sites (49, 52, 53). Indeed, many of the highest-scoring Class 5 motifs are predicted to bind to various members of the POU transcription factor family (Table 3C and Fig. S5). A well-established mechanism for transcriptional control in the POU family occurs when different family members or alternatively-spliced forms compete for the same binding site (53, 60); in this regard, although we did not detect over-representation of the POU2F2 (Oct-2) motif near the RBP-Jκ peaks, Oct-2 has previously been shown to inhibit auto-activation of Rta by preventing binding of Oct-1 and Rta to the Rta promoter (61). However, we think it is unlikely that Oct-1 regulates the reactivation switch independently because its expression is undetectable during latency and is only induced during reactivation, and ectopic Oct-1 siRNAs did not reactivate latent virus (Fig. 12). To our knowledge, our viral model is the first to propose that POU DNA binding can regulate RBP-Jκ DNA binding.
Overall, our data suggest that a broad set of POU factors have the potential to positively and negatively regulate RBP-Jκ DNA binding to the viral genome. Indeed, Oct-1 is not the only protein from KSHV-infected B-cell extracts that binds to the Oct-v motif (50). Further work is required to identify the spectrum of Oct-v–binding proteins and distinguish their DNA-binding specificities from those of Rta (Fig. S9).
It is likely that regulation of RBP-Jκ DNA binding by POU proteins has broader biologic significance. Indeed, many publications describe POU protein associations with Notch and other herpesviruses. POU5F1/Oct-4 is one of the four transcription factors involved in pluripotency of embryonic stem (ES) cells (62) and binds to RBP-Jκ in ES protein extracts (63). POU3F2/Brn2 enhances expression of Notch pathway proteins to promote transformation in melanoma tumor models (64). The Drosophila homolog of Oct-1, dOct-1/nubbin, represses Notch target genes in that organism (65). Oct-1's classic and seminal function among herpesviruses is to specify transactivation for the herpes simplex virus virion protein (VP)-16 as part of a quaternary complex with host cell factor and IE promoters (66, 67). Like KSHV Rta, Epstein-Barr virus Rta also binds to Oct-1 to transactivate viral promoters (50, 68). Many of the other motifs that scored highest for Class 2 or Class 5 peaks with matches in the HOCOMOCO database are also putative binding sites for cellular proteins with known functions in Notch signal transduction, γ-herpesvirus infection, or both.
The significance of POU DNA motifs to regulating RBP-Jκ DNA binding is further supported by our Rta transactivation studies. Our work shows that stimulation of RBP-Jκ DNA binding to a promoter is not sufficient for Rta transactivation (compare Fig. 6 with Fig. 7), but instead it depends on both the inherent affinity of RBP-Jκ DNA binding and proximity to Oct-v/POU4F2 and Rta-c motifs (Figs. 10 and 11). In this regard, five of the seven promoters we tested for transactivation contained RBP-Jκ class 2 peaks, yet Rta only activated the three DNAs that contained both Rta-c and Oct-v/POU4F2 motifs (Fig. 7). Those three promoters also contained the three RBP-Jκ motifs with the highest affinity for purified RBP-Jκ (Fig. 8). Whereas the correspondence between RBP-Jκ affinity and transactivation also applied to the two RBP-Jκ motifs in the ORF50AS/K-bZIP promoter, the higher-affinity motif B functioned poorly as a target for Rta transactivation when moved to the context of motif A (Fig. 10). Indeed, only the RBP-Jκ motif proximal to the Rta-c/Oct-v site corresponded to an RBP-Jκ Class 2 peak (Figs. S2 and S6) and supported robust Rta transactivation (Fig. 10). In the three transactivated promoters, the distances between the Rta-c, Oct-v/POU4F2, and the nearest RBP-Jκ motifs were also similar (Fig. 11). Conservation of that motif architecture suggests that Rta transactivation may involve a direct interaction between Oct-v/POU4F2 and RBP-Jκ. We recognize, however, that our data do not eliminate the explanation that DNA fragments that fail to respond to Rta simply are intrinsically incompetent to initiate transcription.
We can draw additional mechanistic predictions by comparing the association of other DNA motifs with Class 2 or Class 5 RBP-Jκ peaks and Rta peaks. For example, the Class 2 TCF12 motif was never found at RBP-Jκ/Rta coincident peaks (Table 3A), implicating that factor as an Rta-independent regulator of RBP-Jκ DNA binding and suggesting that its positive effect on RBP-Jκ may be mutually exclusive of Rta. Overall, we did not identify a uniform motif by which every RBP-Jκ Class 2 peak was defined. However, pairwise analyses of motifs revealed a few combinations that were always associated with Class 2 and never Class 5 peaks, including Rta-c plus TCF12 (data not shown).
We acknowledge that our bioinformatic analyses of the ChIP/Seq data are limited by our definition of viral promoters. We defined promoters as DNA sequences lying within 1000 bp upstream of known TSSs. However, that promoter definition is based upon many publications that established the general span of DNA sufficient for Rta to transactivate heterologous reporter genes outside the context of the virus. In our previous studies, we used that promoter size to define the DNA elements critical for Rta to stimulate RBP-Jκ binding to an Mta reporter plasmid (4, 5, 8, 16, 54). Hence, we chose that promoter definition for the ChIP/Seq to test our hypothesis that those Mta DNA elements would also be critical for stimulating binding of RBP-Jκ more broadly to other promoters in the viral genome. However, it is likely that many of the RBP-Jκ sites that we deemed nonpromoter-associated have greater significance in Rta transactivation than considered herein. TSS identification in the virus is probably not complete, and, like the Rta promoter (43), other KSHV promoters are probably larger than 1000 bp. Moreover, we chose a single time point during reactivation in an attempt to maximize the number of Rta target sites for our analyses; it is possible that the number of targets would be different earlier in reactivation.
The great number of RBP-Jκ and Rta peaks contrast dramatically with the small number of direct transcriptional targets of Rta that we previously identified at a kinetically similar time after reactivation (69). In that study, we expressed a conditionally active allele of Rta that transactivated only eight viral genes when we extinguished ongoing protein translation. That study also suggested that new cellular gene expression is crucial for full viral reactivation, which was confirmed in a more recent publication (59). The proteins with binding sites that are over-represented at RBP-Jκ Class 2 peaks in this study are prime candidates whose expressions are required to support Rta transactivation and progression of reactivation. Indeed, we have seen that Oct-1 protein is induced during KSHV reactivation in response to two different chemicals in two different cell types (Fig. 12, B and C); our preliminary data suggest that Oct-1 is also induced downstream of TLR7 in PEL cells (data not shown). Furthermore, we published that stimulation of RBP-Jκ DNA binding by Rta could convert the Mta promoter from Notch-nonresponsive to Notch-responsive (8). Because Notch is constitutively active in KSHV-infected cells (25, 33–36), stimulation of RBP-Jκ DNA binding by Rta might make promoter-bound RBP-Jκ newly available to Notch, resulting in reprogramming of the Notch-responsive cellular and viral transcriptomes. In this scenario, we predict that NICD1's inability to reactivate KSHV is the result of its inability to stimulate Oct-1 expression and/or de-repress RBP-Jκ DNA binding by a Class 5 POU protein. For other proteins implicated by our motif discovery that have Notch-related phenotypes, our work supports the notion that they impact Notch by regulating RBP-Jκ DNA binding.
In summary, our results demonstrate that RBP-Jκ DNA binding is dynamic to the KSHV genome during latency and reactivation, in a promoter-specific fashion. These data support previous reports on RBP-Jκ DNA binding from our laboratory and others in which the canonical view of RBP-Jκ isoform constitutively bound to viral or cellular DNA is not always sufficient for Notch target specification. Our approaches demonstrate the strength of the KSHV model system for revealing mechanisms that regulate RBP-Jκ–dependent promoter specification and their biological consequences. Our work implicates the POU protein family as regulators of RBP-Jκ DNA binding and Notch-signaling activity.
Experimental procedures
BCBL-1, BC-3, and BL-41 B lymphoma cell lines were maintained as described previously (70). Vero rKSHV.294 and -.293 MSR Tet-Off cells were propagated as described previously (24). Viral reactivation was induced with 1 mm valproic acid or 20 ng/ml TPA.
BL-41 cells were electroporated and assayed for luciferase and β-gal (control) as in Refs. 8, 15, 16, 38, 50, with the following modifications: 3.3 × 106 cells per electroporation sample, with 27 μg of total DNA, in 300 μl of serum-free RPMI 1640 medium using an exponential decay pulse, 230 volts, and 975 microfarad capacitance.
All promoters were cloned into the luciferase reporter vector pGL3-Basic (Promega) by PCR amplification. Amplicons with approximate lengths of 500 bp were selected by centering RBP-Jκ motifs having associated RBP-Jκ ChIP/Seq peaks on the viral genome. PCR primers introduced SmaI restriction sites to both ends to permit cloning into pGL3-Basic (Promega). Candidate promoters and their end points in the viral genome are as follows: 50p distal (69,783–70,359); 50p proximal (71,282–71,849); 50ASp (74,399–75,021); 56p (79,286–79,877); Mta distal (79,720–80,308); and Mta proximal (81,668–82,168). pFL57-GL3 (full-length Mta promoter), p57Δ5-GL3 (Mta promoter from −106 to +1), and K-bZIP WT and mutant (m 1 + 2) Rta-c (CANT) motif reporters were described previously (54, 71).
Mutations of RBP-Jκ motifs on the ORF50AS/K-bZIP promoters were produced by PCR site-directed mutagenesis using Pfu Ultra High-Fidelity DNA polymerase (Agilent; sequences available upon request), as described previously (72). All plasmid DNA sequences were confirmed by restriction digestion and sequencing (Macrogen).
pcDNA3-FLc50 expresses Rta/ORF50 protein from cDNA in animal cells (4, 5, 54). pGST-RBP-Jκ expresses full-length RBP-Jκ fused to the GSH S-transferase (GST) tag. pMalc2X-FL Rta and pMalc2X-RtaΔSTAD plasmid express full-length Rta and RtaΔSTAD fused to the maltose-binding protein (MBP) (8). pGST–Oct-1 expresses full-length Oct-1 fused to GST (50). GST–RBP-Jκ and GST–Oct-1 were expressed in Escherichia coli and purified in NETN+ buffer as described (54). MBP–FL-Rta and MBP–RtaΔSTAD were expressed and purified from E. coli BL21 codon plus RIL/DE3 (Agilent) as described (8).
For ChIP/Seq, 7 × 106 BC-3 cells (1 × 106 cells per sample) were treated with 1 mm VPA or left untreated. After 24 h, cells were washed once with 1× phosphate-buffered saline (PBS) and cross-linked by treatment with 1% formaldehyde at room temperature for 10 min. The reaction was stopped by adding 2 m glycine to a final concentration of 0.125 m. Cells were washed twice with ice-cold 1× PBS and centrifuged at 2000 rpm for 10 min. Chromatin was sheared by sonication (BioRuptorTM; Diagenode) in 2 ml of Buffer III (1% SDS, 10 mm EDTA, 50 mm Tris-HCl) for eight 8-min cycles, 30 s on/off, each, to an average size of 200 bp (73) in 15-ml polystyrene tubes. Cell debris was removed by micro-centrifugation at full speed for 10 min at 4 °C.
The chromatin supernatants were divided into 300-μl aliquots (1 × 106 cells) and then increased to final volumes of 2 ml/sample in ChIP pre-clearing buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm Tris-HCl, 167 mm NaCl) (74). Samples were pre-cleared with 60 μl of protein A–agarose/salmon sperm DNA beads (Millipore) for 1 h at 4 °C with nutation and then incubated with 4 μg of each antibody overnight at 4 °C with nutation to immunoprecipitate the protein/DNA complexes. Test antibodies were our well-characterized anti-Rta rabbit serum (from rabbit D3861) (4, 5) and rabbit anti-RBP-Jκ (Millipore, AB5790). Negative control antibody was rabbit total IgG (Sigma, 15006). Immunocomplexes were precipitated with 60 μl of protein A–agarose/salmon sperm DNA beads (Millipore) for 1 h at 4 °C with nutation, then washed with 1 ml of ChIP pre-clearing buffer three times at 4 °C and once at room temperature for 30 min, with nutation. Beads were resuspended in 100 μl of 1× TE, and RNA was degraded by incubating with 1.6 μl of RNase A (50 μg/ml) for 30 min at 37 °C. Proteins were degraded by adding 5 μl of 10% SDS and 1.4 μl of proteinase K (20 mg/ml) for 2 h at 55 °C, then overnight at 65 °C, to completely reverse protein–DNA cross-links. DNA was purified by sequential phenol-chloroform/chloroform extraction and precipitated with 2 volumes of 100% ethanol, 1/10th volume of 3 m sodium acetate (pH 5.2), and 1 μl of glycogen (10 mg/ml) at −20 °C overnight. After centrifugation (full speed at 4 °C for 15 min), pellets were washed once with 2 volumes of 70% ethanol, centrifuged again (full speed at 4 °C for 15 min), and air-dried. DNA was resuspended in 50 μl of double-distilled H2O.
Fragment sizes and quantities of enriched ChIP samples from three independent experiments were confirmed using the high-sensitivity DNA assay (2100 Bioanalyzer, Agilent Technologies). DNA libraries were constructed using the 5500 SOLiDTM fragment library core kit (Applied Biosystems) by the Molecular Resource Facility at Rutgers-New Jersey Medical School, sequenced on ABI 5500 system, and adapter sequences were removed using CLC genomics workbench. Raw sequence files and base quality files were combined and groomed specifying ASCII quality encoding using Galaxy (75). Read quality was determined using the tool FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).7 Reads were quality-trimmed using the tool TrimSequences, then mapped to the KSHV genome using Bowtie 2 with a single-end strategy on the Galaxy server (GenBankTM U75698; usegalaxy.org (75–79)). Mapping was evaluated using the tool Flagstat, and unmapped and duplicate reads were removed using the tool Filter BAM (80). The average read density of the +VPA input sample was ∼227. Peaks were called using MACS2; treatment files were mapped reads from ChIPs performed with anti-Rta or anti-RBP-Jκ antibodies and control files from ChIPs performed with total IgG; scaling of files was enabled. Peak detection used the highest p value that correctly called the Rta peak on the PAN promoter and the RBP-Jκ peak on the Mta promoter (81). Transcriptional start site locations were carried over from Ref. 42 to the U75698 genome manually; the file is available upon request. RBP-Jκ Classes 1, 3, and 4 peaks were determined by subtracting the fold enrichment of the peak in the absence of VPA from the fold enrichment plus VPA, and then compared with the standard deviation (S.D.) of the fold enrichments of the peaks in the absence of VPA. A difference of <1 S.D. was considered unchanged (Class 1); a difference of >1 S.D. was considered class 3 if positive and class 4 if negative. DNA motifs were identified using the FIMO and MEME algorithms (40, 82)
To confirm DNA enrichment from VPA or TPA ChIPs, real-time PCR (qPCR) was performed using 2× SYBR Green® select master mix (Applied Biosystems) (primer sequences available on request). The cycling parameters were 5 min at 95 °C, 40 cycles of 15 s at 95 °C, and 1 min at 60 °C.
Competition EMSA was performed as described previously (8, 16, 50, 54). The 32P-containing complexes were quantitated by phosphorimaging (Typhoon 9410; GE Healthcare).
For siRNA-mediated knockdowns of Oct-1, Vero-rKSHV.294 cells were seeded at a density of 2 × 105 cells/well in a 6-well plate and grown for 24 h. siRNA oligonucleotides targeting the Oct-1 gene (sc-36119) and a nontargeting control (sc-37007) were from Santa Cruz Biotechnology. The Oct-1 siRNA was chosen by analysis of the Vero cell genome and consultation with the manufacturer (83). Thirty picomoles of siRNA oligonucleotides and 9 μl of Lipofectamine RNAiMax reagent (Invitrogen) were diluted separately in Opti-MEM medium (Invitrogen), mixed together, and incubated for 5 min at room temperature. The siRNA/lipid complex was added to the cells and incubated at 37 °C for 48 h before VPA treatment. Production of infectious virus was quantitated 48 h later by transferring cell supernatants to 293 reporter cells as described previously (41).
Western blotting was performed as described previously (4), subsequent to PAGE in 8% Tris-glycine or 4–12% Bis-Tris glycine (ThermoFisher Scientific) gels. Blots were washed with PBS, 0.1% Tween 20 (PBST); primary antibody dilutions were 1:200 for Oct-1 (Santa Cruz Biotechnology), 1:1000 for Rta, 1:1000 for actin (Cytoskeleton), and 1:1000 for glyceraldehyde-6-phosphate dehydrogenase (Biolegend). Immunocomplexes were visualized by autoradiography or by myECLTM imager (Thermo Fisher Scientific).
Author contributions
O. G.-L., J. D., C. G., H. M., K. E. D., D. P., and D. M. L. conceptualization; O. G.-L., J. D., C. G., H. M., B. A. V.-O., H. J. S., K. E. D., P. D., D. P., and D. M. L. data curation; O. G.-L., J. D., C. G., H. M., H. J. S., K. E. D., D. P., and D. M. L. formal analysis; O. G.-L., J. D., C. G., B. A. V.-O., H. J. S., K. E. D., D. P., and D. M. L. investigation; O. G.-L., J. D., C. G., H. J. S., K. E. D., P. D., D. P., and D. M. L. methodology; O. G.-L. and D. M. L. writing-original draft; O. G.-L., J. D., C. G., H. M., B. A. V.-O., H. J. S., K. E. D., P. D., D. P., and D. M. L. writing-review and editing; P. D. software; D. P. and D. M. L. visualization; D. M. L. resources; D. M. L. supervision; D. M. L. funding acquisition; D. M. L. validation; D. M. L. project administration.
Supplementary Material
Acknowledgments
We thank Carolina Arias for providing TSS data and Kentaro Hanada and Jennifer Hillman-Jackson for helpful discussions. We gratefully acknowledge Robert Donnelly and the Molecular Resource Facility at Rutgers-New Jersey Medical School for technical support in the ChIP/Seq experiments.
This work was supported by National Institutes of Health Grants AI 078138 and AI 117127. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9 and Tables S1–S4.
The GEO accession number for the ChIP/Seq data is GSE122752.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- KSHV
- Kaposi's sarcoma-associated herpesvirus
- KS
- Kaposi's sarcoma
- PEL
- primary effusion lymphoma
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- IGV
- Integrative Genomics Viewer
- qPCR
- quantitative PCR
- Seq
- sequencing
- oligo
- oligonucleotide
- TPA
- 12-O-tetradecanoyl phorbol acetate
- VPA
- valproic acid
- RBP
- recombination signal-binding protein
- IE
- immediate early
- DE
- delayed early
- L
- late
- TSS
- transcription start site
- RLU
- relative light units
- ES
- embryonic stem
- GST
- GSH S-transferase
- MBP
- maltose-binding protein
- siRNA
- small interfering RNA
- AS
- antisense.
References
- 1. Carbone A., Gaidano G., Gloghini A., Larocca L. M., Capello D., Canzonieri V., Antinori A., Tirelli U., Falini B., and Dalla-Favera R. (1998) Differential expression of BCL-6, CD138/syndecan-1, and Epstein-Barr virus-encoded latent membrane protein-1 identifies distinct histogenetic subsets of acquired immunodeficiency syndrome-related non-Hodgkin's lymphomas. Blood 91, 747–755 [PubMed] [Google Scholar]
- 2. Gramolelli S., and Schulz T. F. (2015) The role of Kaposi sarcoma-associated herpesvirus in the pathogenesis of Kaposi sarcoma. J. Pathol. 235, 368–380 10.1002/path.4441 [DOI] [PubMed] [Google Scholar]
- 3. Mesri E. A., Cesarman E., and Boshoff C. (2010) Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer 10, 707–719 10.1038/nrc2888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lukac D. M., Kirshner J. R., and Ganem D. (1999) Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73, 9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lukac D. M., Renne R., Kirshner J. R., and Ganem D. (1998) Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252, 304–312 10.1006/viro.1998.9486 [DOI] [PubMed] [Google Scholar]
- 6. Sun R., Lin S. F., Gradoville L., Yuan Y., Zhu F., and Miller G. (1998) A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U.S.A. 95, 10866–10871 10.1073/pnas.95.18.10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu Y., AuCoin D. P., Huete A. R., Cei S. A., Hanson L. J., and Pari G. S. (2005) A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79, 3479–3487 10.1128/JVI.79.6.3479-3487.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carroll K. D., Bu W., Palmeri D., Spadavecchia S., Lynch S. J., Marras S. A., Tyagi S., and Lukac D. M. (2006) Kaposi's sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jκ/CSL to activate the Notch pathway. J. Virol. 80, 9697–9709 10.1128/JVI.00746-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liang Y., Chang J., Lynch S. J., Lukac D. M., and Ganem D. (2002) The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jκ, the target of the Notch signaling pathway. Genes Dev. 16, 1977–1989 10.1101/gad.996502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liang Y., and Ganem D. (2003) Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 8490–8495 10.1073/pnas.1432843100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Song M. J., Deng H., and Sun R. (2003) Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 77, 9451–9462 10.1128/JVI.77.17.9451-9462.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song M. J., Li X., Brown H. J., and Sun R. (2002) Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 76, 5000–5013 10.1128/JVI.76.10.5000-5013.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y., Tang Q., Maul G. G., and Yuan Y. (2006) Kaposi's sarcoma-associated herpesvirus ori-Lyt–dependent DNA replication: dual role of replication and transcription activator. J. Virol. 80, 12171–12186 10.1128/JVI.00990-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang P. J., Shedd D., Gradoville L., Cho M. S., Chen L. W., Chang J., and Miller G. (2002) Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 76, 3168–3178 10.1128/JVI.76.7.3168-3178.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guito J., and Lukac D. M. (2012) KSHV Rta promoter specification and viral reactivation. Front. Microbiol. 3, 30 10.3389/fmicb.2012.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palmeri D., Carroll K. D., Gonzalez-Lopez O., and Lukac D. M. (2011) Kaposi's sarcoma-associated herpesvirus Rta tetramers make high-affinity interactions with repetitive DNA elements in the Mta promoter to stimulate DNA binding of RBP-Jκ/CSL. J. Virol. 85, 11901–11915 10.1128/JVI.05479-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Persson L. M., and Wilson A. C. (2010) Wide-scale use of Notch signaling factor CSL/RBP-Jκ in RTA-mediated activation of Kaposi's sarcoma-associated herpesvirus lytic genes. J. Virol. 84, 1334–1347 10.1128/JVI.01301-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scholz B. A., Harth-Hertle M. L., Malterer G., Haas J., Ellwart J., Schulz T. F., and Kempkes B. (2013) Abortive lytic reactivation of KSHV in CBF1/CSL deficient human B cell lines. PLoS Pathog. 9, e1003336 10.1371/journal.ppat.1003336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang L., Zhu C., Guo Y., Wei F., Lu J., Qin J., Banerjee S., Wang J., Shang H., Verma S. C., Yuan Z., Robertson E. S., and Cai Q. (2014) Inhibition of KAP1 enhances hypoxia-induced Kaposi's sarcoma-associated herpesvirus reactivation through RBP-Jκ. J. Virol. 88, 6873–6884 10.1128/JVI.00283-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Castel D., Mourikis P., Bartels S. J., Brinkman A. B., Tajbakhsh S., and Stunnenberg H. G. (2013) Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 27, 1059–1071 10.1101/gad.211912.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang H., Zang C., Taing L., Arnett K. L., Wong Y. J., Pear W. S., Blacklow S. C., Liu X. S., and Aster J. C. (2014) NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc. Natl. Acad. Sci. U.S.A. 111, 705–710 10.1073/pnas.1315023111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krejcí A., and Bray S. (2007) Notch activation stimulates transient and selective binding of Su(H)/CSL to target enhancers. Genes Dev. 21, 1322–1327 10.1101/gad.424607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H., Zou J., Zhao B., Johannsen E., Ashworth T., Wong H., Pear W. S., Schug J., Blacklow S. C., Arnett K. L., Bernstein B. E., Kieff E., and Aster J. C. (2011) Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. U.S.A. 108, 14908–14913 10.1073/pnas.1109023108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeCotiis J. L., Ortiz N. C., Vega B. A., and Lukac D. M. (2017) An easily transfectable cell line that produces an infectious reporter virus for routine and robust quantitation of Kaposi's sarcoma-associated herpesvirus reactivation. J. Virol. Methods 247, 99–106 10.1016/j.jviromet.2017.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang H., Dittmer D. P., Shin Y. C., Chul S. Y., Hong Y., and Jung J. U. (2005) Role of Notch signal transduction in Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 79, 14371–14382 10.1128/JVI.79.22.14371-14382.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Giancotti F. G. (2013) Mechanisms governing metastatic dormancy and reactivation. Cell 155, 750–764 10.1016/j.cell.2013.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Penton A. L., Leonard L. D., and Spinner N. B. (2012) Notch signaling in human development and disease. Semin. Cell Dev. Biol. 23, 450–457 10.1016/j.semcdb.2012.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kamath B. M., Spinner N. B., and Rosenblum N. D. (2013) Renal involvement and the role of Notch signalling in Alagille syndrome. Nat. Rev. Nephrol. 9, 409–418 10.1038/nrneph.2013.102 [DOI] [PubMed] [Google Scholar]
- 29. Aquila G., Pannella M., Morelli M. B., Caliceti C., Fortini C., Rizzo P., and Ferrari R. (2013) The role of Notch pathway in cardiovascular diseases. Glob. Cardiol. Sci. Pract. 2013, 364–371 10.5339/gscp.2013.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aster J. C. (2014) In brief: Notch signalling in health and disease. J. Pathol. 232, 1–3 10.1002/path.4291 [DOI] [PubMed] [Google Scholar]
- 31. Ntziachristos P., Lim J. S., Sage J., and Aifantis I. (2014) From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell 25, 318–334 10.1016/j.ccr.2014.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Louvi A., and Artavanis-Tsakonas S. (2012) Notch and disease: a growing field. Semin. Cell Dev. Biol. 23, 473–480 10.1016/j.semcdb.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Curry C. L., Reed L. L., Golde T. E., Miele L., Nickoloff B. J., and Foreman K. E. (2005) γ-Secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi's sarcoma tumor cells. Oncogene 24, 6333–6344 10.1038/sj.onc.1208783 [DOI] [PubMed] [Google Scholar]
- 34. Gasperini P., Espigol-Frigole G., McCormick P. J., Salvucci O., Maric D., Uldrick T. S., Polizzotto M. N., Yarchoan R., and Tosato G. (2012) Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through Notch-dependent signaling. Cancer Res. 72, 1157–1169 10.1158/0008-5472.CAN-11-3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lan K., Choudhuri T., Murakami M., Kuppers D. A., and Robertson E. S. (2006) Intracellular activated Notch1 is critical for proliferation of Kaposi's sarcoma-associated herpesvirus-associated B-lymphoma cell lines in vitro. J. Virol. 80, 6411–6419 10.1128/JVI.00239-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lan K., Verma S. C., Murakami M., Bajaj B., Kaul R., and Robertson E. S. (2007) Kaposi's sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein. Proc. Natl. Acad. Sci. U.S.A. 104, 16287–16292 10.1073/pnas.0703508104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lan K., Murakami M., Bajaj B., Kaul R., He Z., Gan R., Feldman M., and Robertson E. S. (2009) Inhibition of KSHV-infected primary effusion lymphomas in NOD/SCID mice by γ-secretase inhibitor. Cancer Biol. Ther. 8, 2136–2143 10.4161/cbt.8.22.9743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bu W., Carroll K. D., Palmeri D., and Lukac D. M. (2007) The Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 ORF50/Rta Lytic switch protein functions as a tetramer. J. Virol. 81, 5788–5806 10.1128/JVI.00140-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liao W., Tang Y., Kuo Y. L., Liu B. Y., Xu C. J., and Giam C. Z. (2003) Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 transcriptional activator Rta is an oligomeric DNA-binding protein that interacts with tandem arrays of phased A/T-trinucleotide motifs. J. Virol. 77, 9399–9411 10.1128/JVI.77.17.9399-9411.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bailey T. L., and Elkan C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 [PubMed] [Google Scholar]
- 41. Shin H. J., DeCotiis J., Giron M., Palmeri D., and Lukac D. M. (2014) Histone deacetylase classes I and II regulate Kaposi's sarcoma-associated herpesvirus reactivation. J. Virol. 88, 1281–1292 10.1128/JVI.02665-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arias C., Weisburd B., Stern-Ginossar N., Mercier A., Madrid A. S., Bellare P., Holdorf M., Weissman J. S., and Ganem D. (2014) KSHV 2.0: a comprehensive annotation of the Kaposi's sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 10, e1003847 10.1371/journal.ppat.1003847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu J., Verma S. C., Cai Q., Saha A., Dzeng R. K., and Robertson E. S. (2012) The RBP-Jκ binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog. 8, e1002479 10.1371/journal.ppat.1002479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhu F. X., Cusano T., and Yuan Y. (1999) Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73, 5556–5567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu M., Suen J., Frias C., Pfeiffer R., Tsai M. H., Chuang E., and Zeichner S. L. (2004) Dissection of the Kaposi's sarcoma-associated herpesvirus gene expression program by using the viral DNA replication inhibitor cidofovir. J. Virol. 78, 13637–13652 10.1128/JVI.78.24.13637-13652.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Song M. J., Brown H. J., Wu T.-T., and Sun R. (2001) Transcription activation of polyadenylated nuclear RNA by Rta in human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus. J. Virol. 75, 3129–3140 10.1128/JVI.75.7.3129-3140.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang Y., and Yuan Y. (2007) Essential role of RBP-Jκ in activation of the K8 delayed-early promoter of Kaposi's sarcoma-associated herpesvirus by ORF50/RTA. Virology 359, 19–27 10.1016/j.virol.2006.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gupta S., Stamatoyannopoulos J. A., Bailey T. L., and Noble W. S. (2007) Quantifying similarity between motifs. Genome Biol. 8, R24 10.1186/gb-2007-8-2-r24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kulakovskiy I. V., Medvedeva Y. A., Schaefer U., Kasianov A. S., Vorontsov I. E., Bajic V. B., and Makeev V. J. (2013) HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 41, D195–D202 10.1093/nar/gks1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carroll K. D., Khadim F., Spadavecchia S., Palmeri D., and Lukac D. M. (2007) Direct interactions of KSHV/HHV-8 ORF50/Rta protein with the cellular protein Octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 81, 8451–8467 10.1128/JVI.00265-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kang J., Shakya A., and Tantin D. (2009) Stem cells, stress, metabolism and cancer: a drama in two Octs. Trends Biochem. Sci. 34, 491–499 10.1016/j.tibs.2009.06.003 [DOI] [PubMed] [Google Scholar]
- 52. Tantin D. (2013) Oct transcription factors in development and stem cells: insights and mechanisms. Development 140, 2857–2866 10.1242/dev.095927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao F. Q. (2013) Octamer-binding transcription factors: genomics and functions. Front. Biosci. 18, 1051–1071 10.2741/4162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lukac D. M., Garibyan L., Kirshner J. R., Palmeri D., and Ganem D. (2001) DNA binding by the Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 75, 6786–6799 10.1128/JVI.75.15.6786-6799.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu Y., and Ganem D. (2010) Making sense of antisense: seemingly noncoding RNAs antisense to the master regulator of Kaposi's sarcoma-associated herpesvirus lytic replication do not regulate that transcript but serve as mRNAs encoding small peptides. J. Virol. 84, 5465–5475 10.1128/JVI.02705-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song M. J., Hwang S., Wong W., Round J., Martinez-Guzman D., Turpaz Y., Liang J., Wong B., Johnson R. C., Carey M., and Sun R. (2004) The DNA architectural protein HMGB1 facilitates RTA-mediated viral gene expression in γ-2 herpesviruses. J. Virol. 78, 12940–12950 10.1128/JVI.78.23.12940-12950.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen J., Ye F., Xie J., Kuhne K., and Gao S. J. (2009) Genome-wide identification of binding sites for Kaposi's sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 386, 290–302 10.1016/j.virol.2009.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ellison T. J., Izumiya Y., Izumiya C., Luciw P. A., and Kung H. J. (2009) A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi's sarcoma-associated herpesvirus. Virology 387, 76–88 10.1016/j.virol.2009.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Papp B., Motlagh N., Smindak R. J., Jin Jang S., Sharma A., Alonso J. D., and Toth Z. (2019) Genome-wide identification of direct RTA targets reveals key host factors for Kaposi's sarcoma-associated herpesvirus lytic reactivation. J. Virol. 93, e01978–18 10.1128/JVI.01978-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pankratova E. V., Stepchenko A. G., Krylova I. D., Portseva T. N., and Georgieva S. G. (2018) The regulatory interplay between Oct-1 isoforms contributes to hematopoiesis and the isoforms imbalance correlates with a malignant transformation of B cells. Oncotarget 9, 29892–29905 10.18632/oncotarget.25648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Di Bartolo D. L., Hyjek E., Keller S., Guasparri I., Deng H., Sun R., Chadburn A., Knowles D. M., and Cesarman E. (2009) Role of defective Oct-2 and OCA-B expression in immunoglobulin production and Kaposi's sarcoma-associated herpesvirus lytic reactivation in primary effusion lymphoma. J. Virol. 83, 4308–4315 10.1128/JVI.02196-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Okita K., Ichisaka T., and Yamanaka S. (2007) Generation of germline-competent induced pluripotent stem cells. Nature 448, 313–317 10.1038/nature05934 [DOI] [PubMed] [Google Scholar]
- 63. van den Berg D. L., Snoek T., Mullin N. P., Yates A., Bezstarosti K., Demmers J., Chambers I., and Poot R. A. (2010) An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 6, 369–381 10.1016/j.stem.2010.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Thurber A. E., Douglas G., Sturm E. C., Zabierowski S. E., Smit D. J., Ramakrishnan S. N., Hacker E., Leonard J. H., Herlyn M., and Sturm R. A. (2011) Inverse expression states of the BRN2 and MITF transcription factors in melanoma spheres and tumour xenografts regulate the NOTCH pathway. Oncogene 30, 3036–3048 10.1038/onc.2011.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Neumann C. J., and Cohen S. M. (1998) Boundary formation in Drosophila wing: Notch activity attenuated by the POU protein Nubbin. Science 281, 409–413 10.1126/science.281.5375.409 [DOI] [PubMed] [Google Scholar]
- 66. Roizman B., and Whitley R. J. (2013) An inquiry into the molecular basis of HSV latency and reactivation. Annu. Rev. Microbiol. 67, 355–374 10.1146/annurev-micro-092412-155654 [DOI] [PubMed] [Google Scholar]
- 67. Wysocka J., and Herr W. (2003) The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem. Sci. 28, 294–304 10.1016/S0968-0004(03)00088-4 [DOI] [PubMed] [Google Scholar]
- 68. Robinson A. R., Kwek S. S., Hagemeier S. R., Wille C. K., and Kenney S. C. (2011) Cellular transcription factor Oct-1 interacts with the Epstein-Barr virus BRLF1 protein to promote disruption of viral latency. J. Virol. 85, 8940–8953 10.1128/JVI.00569-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bu W., Palmeri D., Krishnan R., Marin R., Aris V. M., Soteropoulos P., and Lukac D. M. (2008) Identification of direct transcriptional targets of the KSHV Rta lytic switch protein by conditional nuclear localization. J. Virol. 82, 10709–10723 10.1128/JVI.01012-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guito J., Gavina A., Palmeri D., and Lukac D. M. (2014) The cellular peptidyl-prolyl cis/trans isomerase Pin1 regulates reactivation of Kaposi's sarcoma-associated herpesvirus from latency. J. Virol. 88, 547–558 10.1128/JVI.02877-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Spadavecchia S., Gonzalez-Lopez O., Carroll K. D., Palmeri D., and Lukac D. M. (2010) Convergence of KSHV reactivation with EBV latency and cellular growth mediated by the Notch signal transduction pathway. J. Virol. 84, 10488–10500 10.1128/JVI.00894-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zheng L., Baumann U., and Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Günther T., and Grundhoff A. (2010) The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 6, e1000935 10.1371/journal.ppat.1000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Carey M. F., Peterson C. L., and Smale S. T. (2009) Chromatin immunoprecipitation (ChIP). Cold Spring Harb. Protoc. 2009, pdb prot5279 10.1101/pdb.prot5279 [DOI] [PubMed] [Google Scholar]
- 75. Blankenberg D., Von Kuster G., Coraor N., Ananda G., Lazarus R., Mangan M., Nekrutenko A., and Taylor J. (2010) Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. Chapter 19, Unit 19.10.1-21 10.1002/0471142727.mb1910s89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Goecks J., Nekrutenko A., Taylor J., and Galaxy T. (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11, R86 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Giardine B., Riemer C., Hardison R. C., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J., Miller W., Kent W. J., and Nekrutenko A. (2005) Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455 10.1101/gr.4086505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Russo J. J., Bohenzky R. A., Chien M. C., Chen J., Yan M., Maddalena D., Parry J. P., Peruzzi D., Edelman I. S., Chang Y., and Moore P. S. (1996) Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U.S.A. 93, 14862–14867 10.1073/pnas.93.25.14862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Langmead B., and Salzberg S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., and 1000 Genome Project Data Processing Subgroup. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., and Liu X. S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Grant C. E., Bailey T. L., and Noble W. S. (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Osada N., Kohara A., Yamaji T., Hirayama N., Kasai F., Sekizuka T., Kuroda M., and Hanada K. (2014) The genome landscape of the african green monkey kidney-derived Vero cell line. DNA Res. 21, 673–683 10.1093/dnares/dsu029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nellesen D. T., Lai E. C., and Posakony J. W. (1999) Discrete enhancer elements mediate selective responsiveness of enhancer of split complex genes to common transcriptional activators. Dev. Biol. 213, 33–53 10.1006/dbio.1999.9324 [DOI] [PubMed] [Google Scholar]
- 85. Thorvaldsdóttir H., Robinson J. T., and Mesirov J. P. (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.