

Abstract
Human cytochrome P450 (P450) 11B2 catalyzes the formation of aldosterone, the major endogenous human mineralocorticoid. Aldosterone is important for the regulation of electrolyte homeostasis. Mutations and overexpression of P450 11B2 (also known as aldosterone synthase) can lead to hypertension, congestive heart failure, and diabetic nephropathy. The enzyme is therefore a target for drug development to manage these various disorders. P450 11B2 catalyzes aldosterone formation from 11-deoxycorticosterone through three distinct oxidation steps. It is currently unknown to which degree these reactions happen in sequence without the intermediate products dissociating from the enzyme (i.e. processively) or whether these reactions happen solely distributively, in which the intermediate products must first dissociate and then rebind to the enzyme before subsequent oxidation. We present here a comprehensive investigation of processivity in P450 11B2–catalyzed reactions using steady-state, pre-steady–state, pulse-chase, equilibrium-binding titrations, and stopped-flow binding studies. We utilized the data obtained to develop a kinetic model for P450 11B2 and tested this model by enzyme kinetics simulations. We found that although aldosterone is produced processively, the enzyme preferentially utilizes a distributive mechanism that ends with the production of 18-OH corticosterone. This seemingly contradictory observation could be resolved by considering the ability of the intermediate product 18-OH corticosterone to exist as a lactol form, with the equilibrium favoring the ring-closed lactol configuration. In summary, our refined model for P450 11B2 catalysis indicates isomerization of the intermediate to a lactol can explain why P450 11B2 must produce aldosterone through a processive mechanism despite favoring a distributive mechanism.
Keywords: cytochrome P450, steroid, steroidogenesis, enzyme kinetics, kinetics, pre-steady–state kinetics, conformational change, aldosterone synthesis, corticosterone, CYP11B2, deoxycorticosterone, P450 11B2
Introduction
The cytochrome P450 (P450)2 enzymes are a family of enzymes with a wide array of substrates, including compounds both exogenous and endogenous to the human body (1). P450s are typically considered monooxygenases due to their typical reaction catalyzing the addition of a single oxygen atom from molecular oxygen to a substrate as an aliphatic hydroxylation. P450s are also known to catalyze other reactions such as epoxide formation, carbon–carbon bond-cleavage, and subsequent oxidation of alcohols to form carbonyls and carboxylic acids. Among human P450s active on endogenous steroids (P450s 1B1, 7A1, 7B1, 8B1, 11A1, 11B1, 11B2, 17A1, 19A1, 21A2, 27A1, 39A1, 46A1, and 51A1), each of these reaction classes is represented at least once (2, 3). Understanding how this diverse group of enzymes function is essential to human health as alteration to normal human steroid metabolism by mutant variants of the enzymes or by interactions with small molecules can be deleterious (4–10).
P450 11B2 (aldosterone synthase) is expressed in the zona glomerulosa of the adrenal cortex and is the sole enzyme responsible for the production of the major human mineralocorticoid, aldosterone (11). P450 11B2 produces aldosterone from 11-deoxycorticosterone in a three-step catalytic sequence. The first step is an 11β-hydroxylation; the subsequent hydroxylation and oxidation form an aldehyde at the 18-position (Fig. 1) (12). Mineralocorticoids, like aldosterone, are responsible for regulating mineral ion absorption, specifically sodium and potassium. Increased mineralocorticoid levels in the kidneys drive production of sodium channels and sodium/potassium exchange systems that increase blood volume by increasing water absorption. Increased blood volume leads to higher blood pressure (13). Besides renal activity, P450 11B2 has also been associated with heart failure due to its apparent up-regulation in failing hearts (14).
Figure 1.

Primary physiological reactions catalyzed by human P450 11B2. Human P450 11B2 catalyzes subsequent oxidations at the 11- and 18-positions to transform 11-deoxycorticosterone into aldosterone. Human P450 11B1 can perform similar reactions but not the final oxidation to transform the alcohol at the 18-position to an aldehyde.
Decreasing aldosterone levels by inhibiting P450 11B2 is a possible therapeutic approach for decreasing hypertension and increasing overall cardiovascular health. This approach is based on the suggestion that P450 11B2 inhibition may be more successful than mineralocorticoid receptor antagonism for two reasons. First, P450 11B2 inhibition would actually reduce aldosterone levels below chronically elevated levels that may cause unidentified additional side effects. Second, inhibitors of steroidogenic P450 enzymes tend to have fewer side effects on the endocrine system compared with steroidal antagonists (15). This approach is already being explored by laboratories in several pharmaceutical companies (16). Increasing our knowledge of P450 11B2–catalyzed reactions will support development of these and other mineralocorticoid-related therapies.
Our understanding of human P450 11B2 comes from comparisons with similar enzymes as well as direct studies of human P450 11B2. Human P450 11B2 has very-high sequence identity (93%) to human P450 11B1. P450 11B1 is primarily responsible for the synthesis of cortisol from deoxycortisol. This reaction is an 11β-hydroxylation, like the first step of aldosterone production by P450 11B2. Both P450s 11B1 and 11B2 can perform an 11β-hydroxylation reaction on both 11-deoxycorticosterone and 11-deoxycortisol, to varying degrees; however, only P450 11B2 can proceed to produce aldosterone from 11-deoxycorticosterone. A comparison of the amino acid variants between these enzymes does not reveal a clear mechanistic reason for their variance in activity (11, 17–19). Another useful comparison comes from the bovine P450 enzyme. There is only one bovine 11β-hydroxylase isoform. It is responsible for both cortisol and aldosterone production. Mechanistic studies on this system are likely relevant to understanding human P450 11B2 action (20).
Studies with human P450 11B2 from other laboratories have indicated that the enzyme catalyzes its reactions with varied reaction rates and specificities (11, 12, 21). These studies each focused on different aspects of human P450 11B2 reactivity. Hobler et al. (12) identified the ability of P450 11B2 to generate minor steroid products and reported values of kcat (238 min−1) and Km (103 μm), representing total 11-deoxycorticosterone substrate disappearance. Strushkevich et al. (11) described the rates of individual reaction steps and tested the ability of P450 11B2 to utilize other steroids as substrates (e.g. testosterone and progesterone) but did not test substrate specificity or binding. Peng et al. (21) investigated the influence of the stabilizing phospholipid utilized in assays on catalysis. Strushkevich et al. and Martin et al. (11, 22) have both reported crystal structures of human P450 11B2 (Protein Data Bank codes 4DVQ, 4FDH, and 4ZGX).
We build upon the foundational work done in these studies to focus on understanding the kinetic processivity of the P450 11B2 reactions. The subsequent oxidation reactions catalyzed by human P450 11B2 to form aldosterone can occur in a processive or distributive mechanism. A distributive mechanism involves the enzyme releasing the intermediate product after the first reaction and then binding to another intermediate molecule for another reaction. Alternatively, consecutive reactions can occur in a processive manner where multiple reaction events are catalyzed prior to product release. We explored the processivity of human P450 11B2 by more thoroughly investigating the individual reactions under steady-state, pre-steady–state, and pulse–chase conditions. Our assays were designed to mimic the reported preferred conditions, in that we used high Adx/P450 ratios, avoided the use of detergents in assays, and utilized a DLPC phospholipid vesicle system. Our steady-state and pre-steady–state results presented here agree with a number of previous reports and revealed additional insights regarding processivity, rate-limiting steps, and the role of chemical equilibration of a key intermediate with its lactol form.
Results
Steady-state reaction kinetics
Initial investigations were focused on determining steady-state reaction parameters for the three major reactions catalyzed by human P450 11B2. Example data sets are provided in Fig. 2, and averages of hyperbolic fits to data (collected over several days) are provided in Table 1. It was possible to isolate conversion of 11-deoxycorticosterone to corticosterone by working at low enzyme concentrations. The amounts of 18-OH corticosterone and aldosterone produced under these conditions were less than the limit of detection of the assay as defined by background chromatogram signals (2 pmol). Therefore, the reported values of kcat and kcat/Km are correctly defined. Isolated investigation of the final two reactions from each other was not fully possible. When human P450 11B2 was supplied with corticosterone as its substrate, it was not possible to isolate the production of 18-OH corticosterone from the production of aldosterone. The values of kcat and kcat/Km for these reactions were based on hyperbolic fits to the data for the rate of formation of each individual product against initial corticosterone concentration. The reported steady-state parameters are not completely accurate because the values are skewed by reporting for simultaneous reactions. The reported values for kcat and kcat/Km are artificially lowered by this approach in the case of subsequent reactions. The apparent rate of the first reaction (corticosterone → 18-OH corticosterone) appeared to be lower than it actually is because the products that lead to aldosterone were not accounted for in the rate of 18-OH corticosterone production. The apparent rate of the second reaction (18-OH corticosterone → aldosterone) is also artificially changed because by observing conversion from corticosterone to aldosterone we could not account for how much the first reaction might alter the rate of the second reaction, e.g. due to excess substrate.
Figure 2.

Steady-state kinetic experiments of major reactions catalyzed by human P450 11B2. A, example data showing the production of corticosterone; B, 18-OH corticosterone, and C and D, aldosterone when P450 11B2 is incubated with 11-deoxycorticosterone (A), corticosterone (B and C), or 18-OH corticosterone (D) as the initial substrates. A–C were compiled from single time-point data with duplicate points, and both points are shown; the plotted lines indicate nonlinear regression to hyperbolic data. D was compiled from four time points per substrate concentration with each transient fit as a linear regression to produce the single rate (± S.D.) shown in the plot; the plotted line in D is a linear fit to these rates. See Table 1 for kinetic parameters from fits.
Table 1.
Kinetic parameters determined from steady-state kinetic experiments
Results presented are the average values from multiple experiments.
| Reaction | kcat | kcat/Km | Km |
|---|---|---|---|
| min−1 | min−1 μm−1 | μm | |
| 11-Deoxycorticosterone → corticosterone | 33 ± 1 | 13 ± 1 | 2.7 ± 0.3 |
| Corticosterone → 18-OH corticosteronea | 14.9 ± 0.4 | 0.49 ± 0.2 | 31 ± 1 |
| Corticosterone → aldosteronea | 2.84 ± 0.09 | 0.103 ± 0.007 | 28 ± 2 |
| 18-OH corticosterone → aldosteroneb | 6.7 × 10−5 ± 2 × 10−6 |
a The production of 18-OH corticosterone from corticosterone could not be isolated from the production of aldosterone; therefore, the presented values are not true steady-state parameters because the data reports on both reactions simultaneously.
b Saturation of the hyperbolic curve was not achieved due to the limit of substrate solubility. Only kcat/Km was determined; see under “Experimental procedures” for details.
The conversion of 18-OH corticosterone to aldosterone under steady-state conditions was also investigated. When started from 18-OH corticosterone added in solution, this reaction was very slow. Our result contrasts with reports from other laboratories that this reaction does not occur at all (11, 12, 21). The enzyme concentration, substrate concentration, and reaction time all had to be increased significantly for this reaction to be observed (Fig. S1). Saturation of the steady-state rate was not achievable due to the solubility limit of 18-OH corticosterone in aqueous buffer. Thus, only an estimate of the value of kcat/Km is reported in Table 1 based on the slope of the limited number of points presented in Fig. 2D.
Pre-steady–state reaction kinetics
To further examine the kinetics of the P450 11B2 reactions, experiments were also performed under pre-steady–state conditions. Results of these experiments are shown in Fig. 3. These experiments used a much larger concentration of enzyme with an equimolar concentration of starting substrate. Studies done under similar conditions are often termed “single-turnover conditions,” but this term is not strictly accurate in this case because multiple reactions are occurring on the same initial steroid molecule, and the reaction rates are at similar timescales to the product release rates.
Figure 3.

Pre-steady–state kinetic transients of major reactions catalyzed by human P450 11B2. The results shown are from experiments done at a higher P450 concentration (2 μm) than the steady-state experiments, with data points taken at much faster timescales to limit the effects of the product release step on the observed kinetics. A, initial substrate of 2 μm 11-deoxycorticosterone. The black diamonds in A represent 11-deoxycorticosterone. B, initial substrate of 2 μm corticosterone. A and B, red circles represent corticosterone; blue squares represent 18-OH corticosterone; and green triangles represent aldosterone. The lines connect points to help visualize the trends. The limit of detection in this experiment was ∼100 fmol (i.e. 10 nm) when scaled to the given y axis.
The experiments were performed until the initial substrate was below the detection limit (100 fmol). When 11-deoxycorticosterone was the initial substrate, this time period was short (5 s). When corticosterone was the initial substrate, the time period was longer (∼90 s). This behavior is consistent with the changes in kcat/Km reported in Table 1. Also consistent between the steady-state and pre-steady–state data sets is the observation that 18-OH corticosterone was not rapidly converted to aldosterone after 18-OH corticosterone was used as the primary substrate (>45 s). The concentrations of both aldosterone and 18-OH corticosterone were fairly constant for the rest of the observed time. The pre-steady–state data sets differed from one another in the ratio of 18-OH corticosterone to aldosterone near the end of the observation window. At the latest time point of the data set using 11-deoxycorticosterone as substrate (15 s), the corticosterone concentration was ∼0.20 μm, and the ratio of 18-OH corticosterone to aldosterone was 3.4. In the experiment with corticosterone as the starting substrate, at the point with the same concentration of corticosterone remaining (35 s), the ratio of 18-OH corticosterone to aldosterone was 9.8. This difference in final product ratio indicates that the enzyme reactivity was affected by the identity of the initial substrate.
Pulse–chase assays
The difference in reactivity depending on the initial substrate used, seen in both steady-state and pre-steady–state assays, could be considered to support a processive mechanism for aldosterone formation. There was also little to no lag phase in the production of aldosterone observed in pre-steady–state experiments. This result could be further evidence for a processive mechanism for aldosterone production.
To directly probe the processivity of these reactions, pulse–chase assays were performed. In the pulse–chase assays, the enzyme was first incubated with a radioactive substrate for a brief pulse period (1.5 min), and then a chase volume of nonradioactive reaction intermediate was added in large excess, and the reaction was allowed to proceed to obtain an amount of final product that could be observed (e.g. 8.5 min post-chase or 10 min total assay). The 1.5-min pulse period was chosen so that ≤20% of the final product concentration was produced prior to the addition of the chase. In the case of a completely distributive reaction, the excess chase of intermediate will displace the radioactive substrate bound to the enzyme and decrease the final amount of radioactive final product produced. Under our conditions, a completely dissociative reaction would yield only a 20% radioactive final product yield with the chase added, compared with no chase added. In the case of a completely processive reaction, the excess intermediate would be unable to displace the radioactive intermediate from the enzyme, and the amount of radioactive final product should be similar to that obtained with no chase being added. The fraction of radioactive final product yield with and without chase intermediate therefore reports on the degree of processivity of a reaction. Our laboratory has successfully used this method previously with both processive (23, 24) and distributive (25) human P450-catalyzed reactions.
The results of pulse–chase experiments performed with 11-deoxycorticosterone as the initial substrate are shown in Fig. 4. Separate experiments were done using corticosterone or 18-OH corticosterone as the chase compound to investigate the processivity of 18-OH corticosterone production and aldosterone production, respectively. In all of the experiments, the addition of the chase compound decreased the yield of radioactive 18-OH corticosterone and aldosterone (Fig. 4, A and C); this effect was more dramatic when corticosterone was the chase sample, causing the production of these compounds to effectively stop. At the last time point measured for these experiments, the radioactive product yield with chase added relative to no chase added was low. For the assays where corticosterone was the chase compound, the production of radioactive 18-OH corticosterone was 32 ± 1% of the no-chase value, whereas the production of aldosterone was 40 ± 4% (Fig. 4, B and D). Both of these values indicate reaction mechanisms that are only slightly processive. For assays in which 18-OH corticosterone was the chase compound, the production of radioactive aldosterone compared with no chase was 48 ± 5%. This value also indicated only a slightly processive mechanism and is consistent with what was observed when corticosterone was the chase compound.
Figure 4.

Pulse–chase assays with 11-deoxy-[3H]corticosterone as the initial substrate. In this pulse–chase study, 215 nm P450 11B2 was mixed with 10 μm 11-deoxy-[1,2-3H]corticosterone. This mixture reacted (pulsed) for 1.5 min before 750 μm unlabeled corticosterone, 750 μm unlabeled 18-OH corticosterone, or ethanol vehicle was added as a chase. A, production of radioactive 18-OH corticosterone (blue squares) and aldosterone (green triangles) with a corticosterone (Cort) chase (solid symbols, solid line) and without chase (open symbols, dotted line) as duplicate points and the means of the duplicates connected with lines. B, percentage of the radioactive 18-OH corticosterone and aldosterone produced with the chase compared to without the chase. Standard deviations of duplicate experiments are shown with a line above each bar. C and D, experiments were as A and B except that the chase steroid was 18-OH corticosterone, and therefore only aldosterone production is reported. A totally processive mechanism would yield a percent production of 100%.
The results of pulse–chase experiments performed with corticosterone as the initial substrate are shown in Fig. 5. Similar to what was observed in the experiments with 11-deoxycorticosterone as the initial substrate, adding the 18-OH corticosterone chase decreased the yield of radioactive aldosterone. At the last time point measured, the production of radioactive aldosterone compared with no chase was 25 ± 2%. This value indicates a significantly distributive mechanism. This value was significantly lower than what was observed for the same chase compound, starting with 11-deoxycorticosterone as the substrate. Thus, the degree to which a steroid is produced in a processive mechanism may change depending on the initial substrate utilized by the enzyme.
Figure 5.

Pulse–chase assays with [3H]corticosterone as the initial substrate. P450 11B2 (430 nm) was mixed with 20 μm [1,2,6,7-3H]corticosterone. This mixture reacted (pulsed) for 1.5 min before 750 μm unlabeled 18-OH corticosterone or ethanol vehicle was added as a chase. A, production of radioactive aldosterone with chase (solid triangles, solid line) and without chase (open triangles, dotted line) shown as duplicate points, and the means of the duplicates are connected with lines. B, percentage of the radioactive aldosterone produced with the chase compared to without the chase. Standard deviation of duplicate experiments is shown. A totally processive mechanism would yield a percent production of 100%.
Equilibrium-binding titrations
To assess the relative binding of the substrates and products, spectral binding titrations were performed (Fig. 6). The binding of 11-deoxycorticosterone or corticosterone to P450 11B2 induced a type I spectral shift (26). 11-Deoxycorticosterone bound tightly with a Kd of 16 ± 4 nm. The assay with 11-deoxycorticosterone was repeated with a lower concentration of P450 (0.1 μm) and longer pathlength cell (10 cm) to increase the accuracy of the measurement with such a tight-binding substrate. The remaining titrations were performed in a 1-cm pathlength cell with 1 μm P450. Corticosterone bound to P450 11B2 considerably more weakly, with a Kd of 2.1 ± 0.3 μm when 2 μm Adx was included in the solution (like the other titrations). The Kd value increased to 10 ± 2 μm without Adx present, indicating that the presence of Adx affects substrate binding. Furthermore, when the titration was performed without Adx and with 0.5% (w/v) sodium cholate present (12 mm), as has been reported previously (12), the Kd value increased to 150 ± 60 μm indicating that using cholate as a stabilizing detergent disrupts the ability of substrate to bind (see Fig. S2). This result is likely due to the very general similarity of the chemical structure of cholate with the steroidal ligands.
Figure 6.

Equilibrium titrations of substrates and products binding to P450 11B2. A, titration of 11-deoxycorticosterone into 0.1 μm P450 11B2 in a 10-cm pathlength cell. B, titration of corticosterone into 1 μm P450 11B2 in a 1-cm cell. C, titration of corticosterone into 1 μm P450 11B2 premixed with 150 μm 18-OH corticosterone in a 1-cm cell. D, titration of corticosterone into 1 μm P450 11B2 premixed with 200 μm aldosterone in a 1-cm cell. The solid lines are fits to a quadratic binding equation. The insets in each panel show the difference spectra between a sample with titrant added and a sample with no titrant added at each point indicated in the main panels.
18-OH corticosterone and aldosterone did not produce noticeable shifts in the heme Soret band, which has been reported previously (12). To assess the binding potential of these compounds, titrations with these compounds were performed as competition experiments. Specifically, P450 11B2 was preincubated with a relatively high concentration of one of these steroids, 150 μm 18-OH corticosterone or 200 μm aldosterone. Corticosterone was then titrated into the solution, and a spectral shift was observed as the corticosterone displaced the preincubated steroid. Quadratic fits to the resulting difference in absorbance versus corticosterone titrant concentration yielded apparent Kd values of 12 ± 1 μm for 18-OH corticosterone and 7.9 ± 0.8 μm for aldosterone. When we compensated for the presence of the competing ligand and the known Kd value of corticosterone from earlier experiments (see under “Experimental procedures” for more details), the corrected Kd for 18-OH corticosterone was calculated to be 32 ± 6 μm and the Kd for aldosterone was 70 ± 20 μm. In both of these cases, the resulting corticosterone-binding absorbance difference spectra did not have the same appearance as the corticosterone-only titration results (compare insets of Fig. 6, B to C and D). Whereas the corticosterone-only difference spectra had roughly symmetric positive and negative peaks as more corticosterone was added, the difference spectra from the competition experiments show a positive peak similar in intensity to the corticosterone-only experiment but a negative peak with less than half the magnitude of the positive peak.
Stopped-flow binding studies
Stopped-flow binding studies were undertaken to study the binding rate of substrates to P450 11B2 (Fig. 7). Only the binding of 11-deoxycorticosterone and corticosterone is reported because, as mentioned above, 18-OH corticosterone and aldosterone do not induce a significant spectral change. Attempts at studying the binding kinetics of 18-OH corticosterone and aldosterone as competition experiments led to inconclusive results due to the significantly weaker binding potential of these steroids. The kon for 11-deoxycorticosterone was calculated (by nonlinear fitting to stopped-flow mixing data) to be 4.20 ± 0.04 μm−1 s−1. The kon for corticosterone was similarly found to be 1.63 ± 0.02 μm−1 s−1. Experimental values for Kd and kon as well as calculated values for koff are summarized in Table 2.
Figure 7.
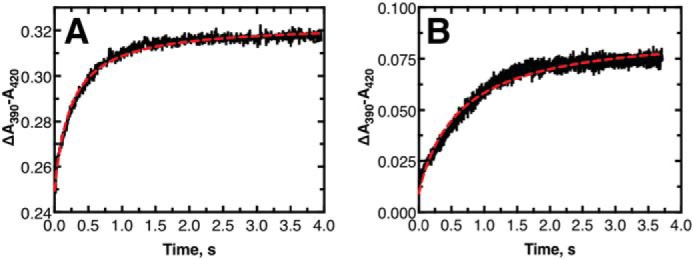
Stopped-flow mixing transients of substrates mixing with P450 11B2. Both panels show the mixing of 1 μm P450 11B2 and 1 μm substrate (final concentrations): A, 11-deoxycorticosterone; B, corticosterone. The red dashed line in each panel is a nonlinear regression fit to the data. See text for more information regarding the fits.
Table 2.
Substrate and product binding parameters to P450 11B2 in the presence of adrenodoxin and lipid
| Ligand | Kd | kon | koff |
|---|---|---|---|
| μm | μm−1 s−1 | s−1 | |
| 11-Deoxycorticosterone | 0.016 ± 0.004a | 4.20 ± 0.04b | 0.07 ± 0.02c |
| Corticosterone | 2.1 ± 0.3a | 1.63 ± 0.02b | 3.4 ± 0.4c |
| 18-OH corticosterone | 32 ± 6d | –e | –e |
| Aldosterone | 70 ± 20d | –e | –e |
a Reported values for Kd come from quadratic fits to the data shown in Fig. 6, A and B.
b Reported values for kon are from nonlinear fits to the data in Fig. 7. See text for details.
c Values calculated from Kd and kon assuming koff = Kd·kon.
d Reported values for Kd come from quadratic fits to the competition titration data in Fig. 6, C and D, which yields an apparent Kd that can be used to calculate the real Kd. See text for details.
e Lack of significant spectral change upon binding prevented determination of these values.
Computational modeling
Initial modeling
Computational modeling studies were performed to assess the qualitative accuracy of the mechanism of human P450 11B2 catalysis shown in Fig. 1. The minimal kinetic mechanism shown in Fig. 8 more thoroughly describes the system by including the required substrate binding (kon) and unbinding steps (koff). The minimal mechanism also assumes that the P450-catalyzed reactions are functionally irreversible in these isolated enzyme experiments. Using the KinTek Explorer software, we tested how well the minimal kinetic mechanism, the steady-state rate constants shown in Table 1 and the binding parameters summarized in Table 2, could reproduce the pre-steady–state experimental data shown in Fig. 3. The result of this initial comparison between simulated (shown as lines) and experimental data (shown as markers) is shown in Fig. 9, A and B. The simulated data fit the experimental data poorly. The steady-state kcat values were too slow to fit the pre-steady–state experimental data. The simulated data also did not reproduce the leveling off of 18-OH corticosterone and aldosterone production. In the case of 11-deoxycorticosterone as the initial substrate (Fig. 9A), the simulated data predicted no aldosterone production during the experimental time frame. In the case of corticosterone as the initial substrate (Fig. 9B), aldosterone is predicted to be produced but does not level off as observed by the experiment. The simulated 18-OH corticosterone trace also indicated the concentration of this product should begin to decrease significantly, which is also not observed by the experiment.
Figure 8.

Initial scheme used for kinetic modeling. This scheme indicates the simplest model possible to represent the major reactions possible between human P450 11B2 and its substrates. It was assumed that the P450-catalyzed reactions are irreversible. Doc = 11-deoxycorticosterone; Cor = corticosterone; 18-OH Cor = 18-OH corticosterone; Aldo = aldosterone.
Figure 9.

Results of kinetic modeling to explain pre-steady–state results. Graphs shown display the same data as in Fig. 3 with the same point shapes and coloring. A, C, and E display the experimental pre-steady–state data starting with 11-deoxycorticosterone as the initial substrate as points. B, D, and F display the experimental pre-steady–state data starting with corticosterone as the initial substrate as points. The lines in each panel represent the expected reaction based on kinetic modeling done with KinTek Explorer. The lines shown in A and B are direct extrapolations of steady-state reaction data and equilibrium titrations. The lines in C and D use the same model as A and B but with optimized fit constants to the catalytic rate constants only. The lines in E and F come from introducing the lactol form of 18-OH corticosterone as a possible state. Kinetic fitting in C and E were performed separately from the fitting in D and F. See text for more information on modeling approach.
We next utilized the nonlinear regression fitting routine in KinTek Explorer to determine whether there was a set of catalytic rate constants (kcat) that could better describe the behavior of the pre-steady–state experiments while still using the simple kinetic mechanism shown in Fig. 8. Only the kcat values were allowed to change for the fitting process to limit degrees of freedom; the values for kon and koff were held constant. Because of the differences in 18-OH corticosterone and aldosterone production observed noted above in the pre-steady–state and pulse–chase experiments (depending on the initial substrate), we performed nonlinear regression analysis on the two pre-steady–state experiments independently. The results of the independent nonlinear regression fitting are shown in Fig. 9, C and D. The simulated data from the nonlinear regression was significantly more similar to the experimental data. Quantitatively, the total X2 value for the 11-deoxycorticosterone as the initial substrate improved from 13,600 to 1090. This improvement came primarily from the 11-deoxycorticosterone- and corticosterone-simulated traces being much closer to the experimental data. However, the 18-OH corticosterone- and aldosterone-simulated traces did not improve much and still did not reproduce the leveling off of formation of 18-OH corticosterone and aldosterone as in the experimental results. The nonlinear regression fitting did not significantly improve the fitting to the data with corticosterone as the initial substrate. The total X2 value was actually poorer, changing from 9640 to 11,400. The simulated 18-OH corticosterone trace does have a stationary behavior, but this is only because all corticosterone is converted 18-OH corticosterone. No aldosterone is produced from this simulated result, which is the fundamental problem with this mechanism. Similar outcomes were achieved with varying the initial estimates used for the kcat values.
Model adaptation
We searched for a meaningful way to adapt the mechanism shown in Fig. 8. It would be simple to add conformational changes to the enzyme, but we had no data to clearly describe or indicate such a feature in P450 11B2 catalysis. Instead, we drew from the chemical properties of the steroids to adjust the model. It has long been known from spectroscopic, chemical degradation, and crystallographic studies that both aldosterone (27, 28) and 18-OH corticosterone (29) can exist in either open forms with aldehyde or alcohol functionality at the 18-position or in closed lactol forms (Fig. 10). These data indicate that both steroids exist primarily in a lactol form when in solution. We confirmed this observation by NMR in 75% CD3OH, 25% D2O (v/v, pH 7) (Fig. S3). The dominant lactol form of aldosterone plays a key role in its physiological role as it makes the steroid a poor substrate for 11β-hydroxysteroid dehydrogenase type 2, which would otherwise inactivate aldosterone from mineralocorticoid receptor activity like it does with glucocorticoids in mineralocorticoid-responsive tissues (30). When 18-OH corticosterone is produced by the enzyme from corticosterone, it must be produced in the open, keto form. However, we expect that when the steroid unbinds from the enzyme it likely rapidly forms the closed, lactol form in solution, in accordance with the previous studies (29). We questioned whether modeling the lactol form of the steroid as an enzyme-inactive version of the steroid could address the issue. The kinetic mechanism was adjusted to include the lactol form 18-OH corticosterone but not aldosterone, in that no form of aldosterone is a substrate for P450 11B2 (Fig. 11). The rate constant for the conversion of the lactol form of 18-OH corticosterone to the open form was set to 1 s−1 based on data for the conversion of fructofuranose to fructose, the closest comparison we found in the literature (31). The rate of lactol formation was then manually adjusted to see whether this could reproduce the leveling off effect seen in the experimental data for 18-OH corticosterone and aldosterone. The lowest value for the rate of lactol formation that reproduced this effect was utilized for subsequent modeling experiments. This value was 2000 s−1, and the value was not allowed to adjust during nonlinear regression to optimize kcat values.
Figure 10.

Open and lactol forms of P450 11B2 products. 18-OH corticosterone can exist in either the keto or hemiketal (lactol) form. Aldosterone can exist in the aldo, hemiacetal (lactol), or acetal (lactol) form. The position of the final oxidation catalyzed by P450 11B2 is the C18 position and is noted in red.
Figure 11.

Final scheme used for kinetic modeling. This scheme adds the lactol forms of 18-OH corticosterone to the scheme shown in Fig. 8. This scheme also defines new kinetic rate constants to describe the lactol ring formation or closing (kclose) and the lactol ring opening to form the carbonyl compound or opening (kopen). Because no form of aldosterone is an active substrate of P450 11B2, the lactol form of aldosterone was not included in the model for simplicity.
Updated modeling with lactol
The results of nonlinear regression to optimize the kcat values with the model, including the nonreactive lactol form (Fig. 11), are shown in Fig. 9, E and F. These results fit the data much better than the previous simulations. The total X2 values were 855 and 4980 for the data sets starting with initial substrates of 11-deoxycorticosterone and corticosterone, respectively. Furthermore, the curvature of all of the simulated transients matches the experimental data well; however, the actual predicted concentrations are higher than the experimental values. The simulated data may be overpredicting the final steroid production levels because we ignored the contributions of minor products previously reported by Hobler et al. (12). Including the contributions of these minor products would likely decrease the fraction of the final steroid that exists as 18-OH corticosterone and aldosterone and would lead to a better match between simulated and experimental data. It is also likely that including various conformational states of the enzyme that might help explain differences in the binding of each substrate and the apparent differences in the rates of subsequent reactions depending on the initial substrate used would also decrease the mismatch between simulated and experimental data; however, we resisted this approach because we have no data to describe these additional states. Regardless of this mismatch, adding the nonreactive lactol form of 18-OH corticosterone to the model, which has definite chemical basis, allows for a much better match between simulated and experimental data.
The kinetic modeling results (kcat values and total X2) from the various simulations are summarized in Table 3. The kcat value for the conversion of 11-deoxycorticosterone was not very well resolved but must be faster than the other reactions. The kcat values from the nonlinear regression for the lactol model indicated that the conversion of 18-OH corticosterone to aldosterone is actually faster than the conversion of corticosterone to 18-OH corticosterone. This result was different from what we reported from the steady-state experiments (Table 1) but may be accurate due to our inability to isolate the individual rate constants in the steady-state experiments when there is a significant processive mechanistic component.
Table 3.
Summary of kinetic modeling results
| Simulation | Initial substratea | kcat 11-deoxycorticosterone → corticosterone | kcat corticosterone → 18-OH corticosterone | kcat 18-OH corticosterone → aldosterone | Total Χ2b |
|---|---|---|---|---|---|
| s−1 | s−1 | s−1 | |||
| Initial simulations | 11-DOC | 0.55 | 0.25 | 0.050 | 13,600 |
| Cort | 0.25 | 0.050 | 9600 | ||
| Nonlinear regression fit to Fig. 8 model | 11-DOC | 4000 ± 495,000 | 0.70 ± 0.06 | 0.89 ± 0.15 | 1090 |
| Cort | 0.16 ± 0.02 | 0 (error not determined) | 11,400 | ||
| Nonlinear regression fit to Fig. 11 model | 11-DOC | 8000 ± 1,500,000 | 0.83 ± 0.07 | 13 ± 2 | 855 |
| Cort | 0.021 ± 0.02 | 5.4 ± 0.8 | 4978 |
a 11-DOC = 11-deoxycorticosterone, and Cort = corticosterone.
b X2 = Σi = 0N − 1 ((yi − y(xi))/(σi))2, with the standard deviation of each datum (σi) calculated from a double-exponential analytical fit to the transient for each compound in the data set as suggested by the software manual.
As noted above, the computational modeling results presented here came from independently fitting the experimental data sets. For completeness, the result of simultaneous fitting of the data sets is shown in Fig. S4. Our conclusion is the same with the simultaneous approach, but the results do not fit the data as well, which further indicates other features of the P450 11B2 catalysis are not fully captured by the mechanisms explored here.
Discussion
The P450 11B2-catalyzed three-step synthesis of aldosterone is an important physiological process, and this enzyme is also a target for treatment of hypertension. Several aspects of the mechanism were still unclear following the published studies (11, 12, 20), including the rate-limiting step(s) and the extent of processivity. Searches for pharmacological inhibitors have been done primarily using simple aldosterone end-point assays (in cells) (15, 22, 32–36), but a better understanding of the kinetics of the individual steps of the enzyme could have practical applications as well as yield insight into the more general mechanisms of P450 catalysis.
Consistent among studies of human P450 11B2, including the results presented here, is that the subsequent P450 11B-catalyzed reactions within the overall sequence (Fig. 1) become slower (lower values of kcat) and less specific (lower kcat/Km). Our results indicate slower rate values than that of Hobler et al. (12), but a direct comparison is difficult in that their reported value was generated from all P450 11B2 reactions studied simultaneously, i.e. substrate disappearance. The rate value reported by Strushkevich et al. (11) for the conversion of 11-deoxycorticosterone to corticosterone (37.6 min−1) is similar to our reported value (33 min−1); however, their reported value for the conversion of corticosterone to 18-OH corticosterone (0.7 min−1) is much slower than our estimated value (14.9 min−1). Furthermore, each of the previously published studies reports that P450 11B2 cannot produce aldosterone directly from 18-OH corticosterone (11, 12, 21). Strushkevich et al. (11) reported rates of reaction for several steroids, using only 100 μm concentrations and a single time point. Hobler et al. (12) reported that added 18-OH corticosterone was not converted to aldosterone, but the assay sensitivity was low. We found this reaction is actually possible, albeit significantly less favored than the other reactions (Fig. S1). These differences in actual catalytic values underscore the importance of repeating these basic experiments along with more extensive catalytic assays to better understand how the enzyme functions.
Our initial primary interest was in determining the degree to which P450 11B2 acts processively. As others have noted (11, 12), the steady-state results strongly imply that aldosterone must be produced via a processive mechanism. The production of aldosterone by P450 11B2 is 1000 times more favored (kcat/Km) when corticosterone is the substrate as compared with 18-OH corticosterone. The influence of the initial substrate, and possibly processivity, on aldosterone production is also observed in the pre-steady–state experiments where more aldosterone is produced more rapidly when 11-deoxycorticosterone is the initial substrate as compared with corticosterone. However, the stronger affinity (lower Kd) for 11-deoxycorticosterone for P450 11B2 compared with corticosterone may explain some of the discrepancy in aldosterone production.
In contrast to the evidence from steady-state and pre-steady–state experiments, pulse–chase studies indicate that processivity plays a very limited role in aldosterone production. In all cases, the yield of radioactive aldosterone after a chase of nonradioactive intermediate steroid is significantly reduced. The extent of the reduction in radioactive aldosterone varies by experiment. The less significant the reduction in radioactive aldosterone production, the more processive is the reaction studied. The most processive reaction appears to be when 11-deoxycorticosterone is the initial substrate, and aldosterone is produced (indicated by 18-OH corticosterone as the chase compound). The aldosterone production is less processive when corticosterone is the initial substrate. The production of 18-OH corticosterone from 11-deoxycorticosterone also appears not to be strongly processive.
Steady-state assays strongly suggested processivity in aldosterone production (Fig. 2) but pulse–chase assays did not (Figs. 4 and 5), and we suggest that the evidence for a distributive mechanism can be resolved by considering the nature of the intermediate product 18-OH corticosterone. 18-OH corticosterone, like aldosterone, exists primarily in a lactol structure (Fig. 10) (29). The open keto form of 18-OH corticosterone is the form that must be produced by the enzyme from corticosterone. It is also likely the open form that the enzyme oxidizes to form aldosterone. If the two forms of 18-OH corticosterone have varying reactivity as substrates for P450 11B2, it is possible to resolve the conflict in the kinetic assays.
When 18-OH corticosterone is produced on the enzyme in the open keto form, it can either be rapidly oxidized in a processive mechanism or can unbind from the enzyme, in a distributive manner, and rapidly isomerize to a lactol. The pulse–chase data suggest that a distributive mechanism is favored, so most 18-OH corticosterone will exist in the lactol form. If this form is a significantly less active substrate, then very little aldosterone will be produced. Thus, the great majority of aldosterone is produced through a processive mechanism as supported by the steady-state results. Therefore, the consideration of a less-active lactol form of 18-OH corticosterone is consistent with aldosterone being produced processively while also being consistent with P450 11B2 acting primarily in a distributive manner.
A less active form of 18-OH corticosterone could be defined in various ways. The simplest definition would be that the lactol form of the steroid cannot bind to the enzyme-active site. The result of the competitive corticosterone spectral titration with 18-OH corticosterone present (Fig. 6C) indicates that 18-OH corticosterone binds to P450 11B2 with a Kd of 32 ± 6 μm. This Kd value was calculated by assuming that both forms of 18-OH corticosterone bind to the enzyme. We believe this is a reasonable number because it follows the trend of each successive reaction component being less favored to bind to the enzyme; human P450 19A1 and 17A1 also follow this trend (25, 37). If we instead assume that only the open form of 18-OH corticosterone binds to the enzyme, we can recalculate the Kd value for this new assumption. The NMR data presented in Fig. S3 supports a lower limit for the equilibrium constant between the lactol and open forms of 18-OH corticosterone of 49 (favoring lactol). Using this lower limit, the amount of open form 18-OH corticosterone present during the competitive titration would be ∼3 μm and lead to the actual Kd for 18-OH corticosterone being computed as 0.6 ± 0.1 μm. This is a 3.5-fold stronger binding constant than we measured for corticosterone and therefore seems unlikely. Furthermore, a more realistic equilibrium constant for lactol formation, based on kinetic simulations, is 2000 (favoring the lactol). This more realistic value for the equilibrium constant would mean that the concentration of open form 18-OH corticosterone present during the competitive titration would be ∼0.075 μm and lead to the Kd for 18-OH corticosterone being computed as 0.016 ± 0.002 μm. Whereas this value is very close to what we measured for 11-deoxycorticosterone, it seems unlikely that P450 11B2 would bind 18-OH corticosterone 100 times more strongly than corticosterone. We conclude that the lactol form of 18-OH corticosterone must be able to bind to P450 11B2 to some degree. Attempts to model a nonbinding lactol form using KinTek Explorer were attempted and could match the experimental data reasonably well (data not shown), but this model was rejected for the argument above. We do not have any data that allow us to compare the thermodynamic binding potential of the lactol and open forms. For simplicity, we proceeded with the assumption that each form is equally likely to bind to P450 11B2, but we admit that this could be a caveat in the modeling.
Another way that a less active form of 18-OH corticosterone could be defined is that the lactol form of the steroid can bind to the enzyme but not react with the enzyme to form aldosterone. This is the model we tested with our kinetic modeling work (Fig. 11). The kinetic modeling results clearly indicate that the model without an inactive lactol form (Fig. 8) does not match our pre-steady–state data. However, adding the noncatalytically active lactol form allows the simulated results to match our pre-steady–state data much better. Therefore, we believe incorporating a noncatalytically-active lactol form of the intermediate substrate 18-OH corticosterone into models of P450 11B2 catalysis is essential.
Imai et al. (20) analyzed aspects of the kinetics of deoxycorticosterone oxidation by bovine P450 11B1, which is the only subfamily 11B P450 in that species and has the function of both human P450 11B1 and 11B2. The authors concluded that “… the dissociation of [the] final product, aldosterone, from the enzyme was the slowest step in the synthesis for deoxycorticosterone.” This conclusion was not based on a direct experiment but apparently on modeling of progress curves (Figs. 3 and 4 of Ref. 20), but no experiments were done, and their steady-state results qualitatively resemble our own (Fig. 2). The work in our Fig. 5 was not designed as a test for burst kinetics, which would be expected for a mechanism where a step following aldosterone formation was rate-limiting. A burst experiment could not be done with 18-OH corticosterone for the equilibrium reasons explained here. However, in Fig. 5 the plot of formation of aldosterone from corticosterone (without chase of intermediate) extrapolates to zero product and not to the 0.43 μm value expected (P450 concentration) if a first-step burst were observed due to a rate-limiting step following aldosterone formation.
The rate of aldosterone release from bovine P450 11B1 was modeled to be 0.03 s−1 by Imai et al. (20). If the association rate were a typical 1–4 × 106 m−1 s−1 (see above), then the Kd would be 0.03 μm for aldosterone, which is extremely low compared with what we estimate for human P450 11B2 using our competitive binding approach (∼70 μm (Fig. 6E)).
This updated understanding of the P450 11B2 kinetic mechanism has ramifications for future studies of the enzyme. First, human P450 11B2 may be difficult to target with drugs while also limiting off-target effects. The major goal of targeting human P450 11B2 would likely be to reduce aldosterone levels; however, because aldosterone is certainly produced in a processive manner, active-site inhibitors of P450 11B2 would also decrease the production of 18-OH corticosterone. This could lead to off-target effects if 18-OH corticosterone is vital to processes other than mineralocorticoid receptor activation. A similar problem has occurred with the development of inhibitors for human P450 17A1 as a therapy for prostate cancer (37). Inhibitors that are specific to P450 11B2 over P450 11B1 would not be expected to decrease corticosterone levels significantly because P450 11B1 is similarly effective at producing this product. Isolating the effects of diminishing 18-OH corticosterone levels would be difficult due to its interconnectedness with aldosterone, and it may only be possible to study these affects in tandem with aldosterone inhibition.
A second ramification of our revised understanding of P450 11B2 is an explanation to the observation among multiple studies that serum levels of 18-OH corticosterone are several times higher than aldosterone (38–40). P450 11B2 mechanistically favors the distributive result of forming 18-OH corticosterone over the processive result of forming aldosterone. Because the equilibrium to the lactol form greatly retards the rate of further oxidation of 18-OH corticosterone by P450 11B2, the steroid is more likely to leave the P450 11B2–containing adrenal glands before it becomes aldosterone. The ratio of 18-OH corticosterone to aldosterone that is observed in the serum varies greatly (38–40), ranging from 1.7 (40) to 23.6 (39); one subject has been reported to be an outlier with a ratio of 104 (39). Our pre-steady–state results do not cover this whole range of results but do indicate a higher ratio when corticosterone is supplied as the substrate as compared with 11-deoxycorticosterone.
A final ramification of our work on human P450 11B2 is the impact on future studies of the enzyme. Although we have been able to explain the role of processivity in P450 11B2 catalysis, several questions still remain. We contend that the open form of 18-OH corticosterone is the form required for the production of aldosterone. Could the active site be natively structured in a way to stabilize the open form or slow its egress from the active site where it presumably forms the lactol? These may be mechanisms to explain differences in P450 11B1 and P450 11B2 aldosterone production. A related question would be whether the active site of P450 11B2 could be intentionally altered to stabilize the open form of 18-OH corticosterone. Such a modification could allow for a catalyst that can more effectively produce aldosterone for commercial or research purposes. Finally, we have seen a difference in the final ratio of 18-OH corticosterone to aldosterone and in the rates of catalytic activity depending on the initial substrate used for studies. How does the initial substrate cause these changes in ratios and rates? We suggest that enzyme conformational changes could play a role. There is no data to suggest this but biophysical studies of protein structure or molecular dynamics simulations may be able to address this hypothesis. Answering these questions and others regarding human P450 11B2 can lead to greater insights into how this enzyme and other P450 enzymes perform sequential reactions. These insights will be important to aid in the better understanding of metabolism of endogenous compounds by P450 enzymes and how to best engage these enzymes for advancing human health.
Experimental procedures
Reagents
Nonradioactive steroids were purchased from MilliporeSigma (Burlington, MA), and radioactive steroids were purchased from American Radiolabeled Chemicals (St. Louis, MO). Solvents, buffer components, and other components used for reactions or protein preparation were obtained from MilliporeSigma or Thermo Fisher Scientific (Hampton, NH) unless otherwise noted.
Recombinant protein
Bovine Adx and AdR were expressed in Escherichia coli DH5α cells (Invitrogen) and purified as described previously (41).
P450 11B2 plasmid
An E. coli codon-optimized sequence (provided in Table S1) was loaded into the pCWOri+ plasmid (42) by GenScript (Piscataway, NJ) using NdeI and HindIII restriction sites. The amino acid sequence used as the basis for the sequence (Table S2) followed the approach for Hobler et al. (12). Briefly, the amino acid sequence is the same as reported as NCBI Reference Sequence NM_000498.3 with modifications to the N terminus to mimic the natural cleavage of the mitochondrial targeting N-terminal peptide and modifications to the N and C termini to facilitate recombinant expression and purification. Specifically, the first 24 N-terminal amino acids were removed, and the following six amino acids were changed from GTRAAR to MATKAAR. A His9 sequence was appended to the C terminus as well.
P450 11B2 expression
E. coli DH5α cells were co-transformed with the P450 11B2 plasmid (ampicillin resistance) and another plasmid containing the E. coli molecular chaperone GroEL/ES (kanamycin resistance) and grown on Difco Luria-Bertani (LB) agar plates containing ampicillin at 50 μg/ml and kanamycin at 20 μg/ml. An individual colony was used to inoculate 100 ml of LB broth containing 100 μg/ml ampicillin and 50 μg/ml kanamycin in a 500-ml Erlenmeyer flask. The solution was incubated overnight at 37 °C and 220 rpm. This overnight culture (5 ml) was then used to inoculate each 2.8-liter Fernbach flask containing 500 ml of Difco Terrific Broth (TB) with 100 μg/ml ampicillin and 50 μg/ml kanamycin. Bulk TB cultures were incubated at 37 °C and 220 rpm. When the bulk culture reached an OD600 = 0.50, expression of P450 11B2 was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (Anatrace Products, Maumee, OH) to a final concentration of 0.5 mm. Expression of GroEL/ES was induced by the addition of solid l-(+)-arabinose to a final concentration of 4 g/liter. Additionally, δ-aminolevulinic acid hydrochloride (Frontier Scientific, Logan, UT) was added to a final concentration of 0.5 mm to induce the expression of heme to support P450 growth. Flasks were incubated at 27.5 °C and 180 rpm for 19 h.
P450 11B2 purification
All purification steps were carried out at 4 °C. The cells from the bulk cultures were pelleted (20 min, 5000 × g), decanted, and then resuspended in 200 ml of Buffer A/liter of bulk culture (Buffer A: 75 mm Tris-HCl (pH 8.0) containing 0.05 mm EDTA and 250 mm sucrose). Chicken egg white lysozyme was added to the resuspended pellets at 75 μg/ml. Resuspended pellets were stirred with lysozyme for 30 min. Solutions were then pelleted (20 min, 5000 × g), decanted, and then resuspended in 17 ml of Buffer B/liter of bulk culture (Buffer B: 50 mm potassium phosphate (pH 7.4) containing 20% glycerol (v/v), 500 mm NaCl, 2% CHAPS detergent (w/v), 1 mm phenylmethanesulfonyl fluoride, and 0.1 mm DTT). The resuspended pellets were sonicated using a 3/8-inch tip on a Branson Digital Sonifier Model 450 (VWR, Radnor, PA) set at 80% power. The solution was kept on ice for the 1-min rounds with 1-min intermissions (5–7 times). The sonicated solution was then centrifuged at 10,000 × g for 25 min. The supernatant was transferred to other tubes and then centrifuged again at 100,000 × g for 1 h. The expressed P450 11B2 was located in the supernatant from the final centrifugation spin.
Approximately 7 ml of Ni-NTA–agarose resin (Qiagen, Hilden, Germany) was prepared in a 2.5-cm diameter open-bed glass column (Bio-Rad). A flow rate of 1 ml/min was maintained with a peristaltic pump. The resin was washed with 10 column volumes of Buffer C (Buffer C: 50 mm potassium phosphate (pH 8.0) containing 20% glycerol (v/v), 500 mm NaCl, 1% sodium cholate (w/v), 1 mm phenylmethanesulfonyl fluoride, and 0.1 mm DTT). The P450 11B2-containing supernatant was then loaded onto the column, washed with Buffer C with 25 mm imidazole added, and then eluted with Buffer C with 200 mm imidazole added. The eluted protein solution was then dialyzed at least twice against 10 volumes of Buffer D for 2 h to reduce the concentrations of potassium phosphate, NaCl, and imidazole (Buffer D: 20 mm potassium phosphate (pH 7.4) containing 20% glycerol (v/v), 1% sodium cholate (w/v), 1 mm phenylmethanesulfonyl fluoride, and 0.1 mm DTT).
Approximately 4 ml of SP-Sepharose Fast Flow resin (GE Healthcare, Uppsala, Sweden) was prepared in a 2.5-cm diameter open-bed glass column. A flow rate of 1 ml/min was maintained with a peristaltic pump. The resin was washed with 10 column volumes of Buffer D. The dialyzed P450 solution was loaded onto the column, washed with Buffer D with 10 mm NaCl added, and then eluted with Buffer D with 250 mm NaCl added. The concentration of potassium phosphate in the elution volume was increased to 500 mm (pH 7.4) to help stabilize the P450. The P450 was then buffer-exchanged into Buffer E and concentrated using Amicon Ultra-15 centrifugal filters with a 10,000-Da molecular mass cutoff (MilliporeSigma) (Buffer E: 500 mm potassium phosphate (pH 7.4) containing 20% glycerol (v/v), 0.1 mm EDTA, and 0.1 mm DTT). It is important to remove the sodium cholate used to stabilize the protein throughout the purification because sodium cholate as low as 0.1% (w/v) can significantly reduce the amount of aldosterone produced (data not shown). We found that P450 11B2 tends to become unstable after elution from the Ni-NTA column. A significant amount of protein (as high as 90%) could precipitate and be removed as a centrifugal pellet. To decrease the loss due to precipitation, the purification should be completed as quickly as possible. The final P450 concentration was determined by measuring the reduced carbon monoxide–binding spectrum using the method of Omura and Sato with the extinction coefficient of ϵ450 nm = 0.091 μm−1 cm−1 (43). The P450 yield ranged from 16 to 55 nmol/liter of bulk culture after the ultracentrifugation spin. After purification and pelleting off the precipitated protein, the overall P450 yield was 1–10 nmol/liter of bulk culture. An example of a gel indicating the purification is shown in Fig. S5. Aliquots of P450 11B2 were stored at −80 °C in small vials.
Catalytic assays
Steady-state conditions
The steady-state assays exemplified in Fig. 2, A–C, were performed using a reconstituted enzyme system with a final reaction volume of 1.0 ml. The reaction mixture contained 0.7 nm P450 11B2 (with 11-deoxycorticosterone as substrate) or 200 nm P450 11B2 (with corticosterone as substrate), 0.5 μm AdR, 1.0 μm Adx, 50 μm DLPC (added as lipid vesicles after sonication of a 1 mg/ml aqueous stock, Enzo Life Sciences, Farmingdale, NY), and 50 mm potassium phosphate buffer (pH 7.4). Substrate concentrations in assays ranged from 0.5 to 150 μm. Substrates were added from stock solutions in 95% ethanol so that the final concentration of ethanol was <1% (v/v). Duplicate samples were preincubated at 37 °C for 5 min (shaking water bath) prior to the addition of an NADPH-generating system to initiate the reaction (10 mm glucose 6-phosphate, 0.5 mm NADP+, and 2 μg/ml yeast glucose-6-phosphate dehydrogenase) (44). Reactions were quenched after a 7.5-min incubation at 37 °C by the addition of 4 ml of CH2Cl2, mixing with a vortex device, and chilling on ice. Samples were then centrifuged at 2000 × g for 2 min to separate the aqueous and organic layers. A 3.8-ml aliquot of the organic layer from each sample was transferred to a new vessel. The organic layers were dried under a nitrogen stream. Dried samples were resuspended in 150 μl of a 9:1 mixture of UHPLC mobile phases A:B described below (v/v) and then transferred to vials for UHPLC injection.
The chromatography system used was a Waters (Milford, MA) Acquity UPLC system with a photodiode array detector for absorbance measurements. Aliquots of the samples (20 μl) were injected on an Acquity BEH C18 UPLC octadecylsilane column (2.1 × 100 mm, 1.7 μm). Mobile phase A was 95% H2O, 5% CH3CN, and 0.1% HCO2H (v/v). Mobile phase B was 99% CH3CN, 1% H2O, and 0.1% HCO2H (v/v). The mobile phase linear gradient ran at 0.35 ml/min as follows: 0 min, 0% B; 7.5 min, 62.5% B; 8.0 min, 62.5% B; 8.25 min, 0% B; 10 min, 0% B (all v/v). The column temperature was maintained at 35 °C. The steroid components were identified by absorbance at 245 nm, compared with commercial standards. An example chromatogram is shown in Fig. S6. Integration of chromatogram peak areas was performed using the Waters MassLynx software. Peak areas were transformed to moles using a set of external standards of various concentrations made of corticosterone based on the assumption that the extinction coefficient of the steroids would change only negligibly between steroids because the A-ring chromophore is unaltered by P450 11B2. Data were then analyzed by nonlinear regression of hyperbolic fits in Prism software (GraphPad, San Diego). Hyperbolic fits were done to solve for kcat and kcat/Km (ksp) directly instead of Km (see Equation 1). For a discussion of the advantages of this approach, see Ref. 45.
| (Eq. 1) |
The steady-state assay shown in Fig. 2D was performed similar to the above steady-state assays. Samples of various concentrations of 18-OH corticosterone (180, 350, or 700 μm) were made up in total volumes of 450 μl. The samples contained 2.15 μm P450 11B2, 1.5 μm AdR, 7.5 μm Adx, 50 μm DLPC, 0.1 mg/ml catalase (to reduce H2O2 buildup in the longer assays), and 50 mm potassium phosphate buffer (pH 7.4). Reactions were initiated by addition of the same NADPH-generating system as above and then incubated at 37 °C in a shaking water bath. Aliquots (100 μl) from these 450-μl samples were removed at 0, 10, 20, and 30 min after reaction initiation, quenched by the addition of 100 μl of 1 m HCl, mixed with a vortex device, and placed on ice. Samples were centrifuged at 15,000 × g for 10 min to pellet acid-precipitated protein. A measured aliquot (125 μl) of the supernatant of each sample was transferred to a new vial and analyzed as described above for the other steady-state assays. The four time points from each concentration of initial substrate were fit by linear regression to produce a value for the rate of aldosterone production at each concentration. These rates were then plotted against initial substrate concentration (Fig. 2D) and fit again by linear regression to estimate the value of kcat/Km because the rate of the reaction did not saturate. The 18-OH corticosterone used as substrate (MilliporeSigma) contained some contaminant that gave a background signal at the same retention time as aldosterone. This problem was compensated for by preparing a sample the same way as above but with no NADPH-generating system. The background integration of the no NADPH sample was subtracted from the other sample prior to any data fitting.
Pre-steady–state conditions
Pre-steady–state experiments were performed with a KinTek RQF-3 quench-flow instrument (KinTek, Snow Shoe, PA). The instrument rapidly mixes two small volumes of different solutions and holds them in a reservoir for input amount of time before rapidly mixing them with a reaction quenching solution. Prior to and during mixing, solutions were kept at 37 °C. Mixing solution 1 contained 4 μm P450 11B2, 12 μm AdR, 60 μm Adx, 100 μm DLPC, 50 mm potassium phosphate buffer (pH 7.4), and either 4 μm (20 Ci/mmol) 11-deoxy-[1,2-3H]corticosterone or [1,2,6,7-3H]corticosterone. Mixing solution 2 contained 10 mm NADPH in the same buffer. These two solutions were mixed in equal volumes (19 μl each); therefore, the reaction concentrations were half that of those listed above. The reaction mixture was quenched with ∼160 μl (5× dilution) of 1 m HCl and collected in an Eppendorf tube. Reaction times of 0–120 s were collected from the same mixing solutions on the same day. Fresh mixing solutions were made each day data were collected. Replicate experiments were consistent from day to day. Quenched reaction solutions were centrifuged at 25,000 × g for 5 min to pellet acid-precipitated protein. The supernatant was transferred to vials for analysis by HPLC-UV in-line liquid scintillation counting. Samples (50-μl injections) were resolved on a Beckman Ultrasphere-C18 octadecylsilane column (4.6 × 250 mm, 5 μm) with the same mobile phases described for steady-state reactions. The mobile phase linear gradient ran at 1.0 ml/min as follows: 0 min, 20% B; 2 min, 20% B; 23.25 min, 50% B; 23.5 min, 20% B; 31.5 min, 20% B (all v/v). The column temperature was maintained at 25 °C. After UV detection, the fluid was mixed with Liquiscint scintillation fluid (National Diagnostic, Atlanta, GA) flowing at a rate of 3 ml/min. The mixture was measured with an INSUS Systems β-RAM detector, and the resulting radiochromatograms were analyzed with LabLogic Systems Laura software (Tampa, FL). Product retention times were compared with commercial standards. Integrated radioactivity peak areas were converted to concentration based on the fraction of total radioactive signal.
Pulse–chase assays
The pulse–chase assays were done under steady-state conditions. P450 11B2 (215 nm for 11-deoxycorticosterone as substrate or 430 nm for corticosterone as substrate), 1 μm AdR, 2 μm Adx, 50 μm DLPC, radioactive substrate (10 μm, 2 Ci/mmol 11-deoxy-[1,2-3H]corticosterone, or 20 μm, 2 Ci/mmol [1,2,6,7-3H]corticosterone), and 50 mm potassium phosphate buffer (pH 7.4) were incubated for 5 min at 37 °C in a shaking water bath (final concentrations listed following addition of NADPH). An NADPH-generating system was added to initiate the reaction with final concentrations of 10 mm glucose 6-phosphate, 0.5 mm NADP+, and 2 μg/ml yeast glucose-6-phosphate dehydrogenase. The reactions were allowed to incubate at 37 °C in a shaking water bath for 1.5 min before the addition of chase intermediate sample, either 750 μm (unlabeled) corticosterone, 750 μm (unlabeled) 18-OH corticosterone, or ethanol vehicle. Aliquots (100 μl) of the reaction mixtures were taken at 0, 2, 4, 6, 8, and 10 min after the addition of the NADPH-generating system. Aliquots were immediately quenched by the addition of 100 μl of 1 m HCl, mixing with a vortex device, and storage on ice. Aliquots were centrifuged for 10,000 × g for 10 min to pellet acid-precipitated protein. A measured aliquot (125 μl) of the sample supernatant was transferred to a new vial for HPLC analysis. Radiochromatographic measurement and analysis were done in the same way as described for pre-steady–state assays. Duplicate samples were used.
Binding studies
Equilibrium-binding titrations
Spectral binding titrations were performed in two different ways. For titrations with 11-deoxycorticosterone as the titrant, a 10-cm cell (Starna Cells, Atascadero, CA, catalogue no. 34Q-100, 25 ml) was used with an OLIS-Cary 14 spectrophotometer (On-Line Instrument Systems, Bogart, GA). A 25-ml solution of 0.09 μm P450 11B2 with 2 μm Adx, 50 μm DLPC, and 50 mm potassium phosphate buffer (pH 7.4) was prepared and used to generate the reference absorbance spectrum, scanning from 350 to 500 nm. Small volumes of 11-deoxycorticosterone stock solutions in 95% ethanol (0.50–4 mm) were then titrated into the 10-cm cell, final ethanol concentrations <0.1% (v/v). The cell was then rocked gently to mix the solution prior to the absorbance spectrum being taken. The reference spectrum was subtracted from each titrant point spectrum to generate a difference spectrum. The difference between the maximum (390 nm) and minimum (420 nm) of the difference spectra was calculated and plotted against total titrant concentration.
For titrations with corticosterone as the titrant, 1-cm semi-microvolume disposable cuvettes (Bio-Rad, 1.5 ml) were used with an Aminco DW2/OLIS spectrophotometer (On-Line Instrument Systems). A 2-ml solution of 1 μm P450 11B2 with 15 μm Adx, 50 μm DLPC, and 50 mm potassium phosphate buffer (pH 7.4) was prepared and split between two cuvettes, one reference and one sample. The difference between these cuvettes was recorded as the difference absorbance spectrum from 350 to 500 nm. Small volumes of corticosterone stock solutions in 95% ethanol (1.0–20 mm) were then titrated into the 1-cm cell, final ethanol concentration of <1.1% (v/v). The cell was mixed with a cuvette plumper prior to the difference spectrum being recorded. The difference between the maximum (395 nm) and minimum (430 nm) of the difference spectra was calculated and plotted against total titrant concentration. Competition titrations were done to determine the binding constant for 18-OH corticosterone and aldosterone. The competition titrations were performed as described for the corticosterone titrations except either 150 μm 18-OH corticosterone or 200 μm aldosterone was added to the P450 solution before the titration and allowed to mix for at least 5 min. All plots of absorbance difference against total titrant concentration were fit via nonlinear regression to a quadratic binding equation using GraphPad Prism software (GraphPad, San Diego). Equation 2 is presented below, where y is the spectral difference; y0 is background signal; A is a spectral coefficient; E is the total enzyme concentration; Kapp is the apparent Kd, and x is the total titrant concentration.
| (Eq. 2) |
Competition experiments were further analyzed by comparison with the corticosterone-only titration result (Kd, cort) to transform the apparent Kd (Kapp) from the quadratic fit of the competition experiment into the actual Kd (Kreal) at a given concentration of the competing ligand ([A], 18-OH corticosterone, or aldosterone). The form of Equation 3 below assumes direct competition for the two ligands and no allosteric binding (46).
| (Eq. 3) |
Stopped-flow binding studies
Substrate-binding rates to P450 11B2 were determined for 11-deoxycorticosterone and corticosterone using an OLIS RSM-1000 stopped-flow instrument. This instrument gives full absorbance spectra (300–535 nm) with a temporal resolution of 1 ms after rapid mixing of two solutions by a stopped-flow apparatus. Slit widths were 1.24 mm; the pathlength was 20 mm; and 600 line/500 nm gratings were used (at 23 °C). The total recorded time after mixing was 4 s with a dead time of ∼2 ms. Solution 1 in both experiments contained 2 μm P450, 30 μm Adx, 100 μm DLPC, and 50 mm potassium phosphate buffer (pH 7.4). Solution 2 was 2 μm 11-deoxycorticosterone or corticosterone in 50 mm potassium phosphate buffer (pH 7.4). The two solutions were mixed in equal volumes, so that the final concentration during the reaction was half what is listed above. The absorbance difference between 390 and 420 nm was found at each time point and exported to generate mixing transients. These transients were then fit by nonlinear regression in GraphPad Prism to avoid the heteroskedasticity that occurs with linear fits (47). The fitting Equation 4 assumes that the enzyme and substrate are initially at the same equation and is shown below. y is the observed spectroscopic signal; A is a spectroscopic scaling constant; C0 is the initial concentration of enzyme or substrate, which must be the same; k is the binding rate constant; x is the time since mixing the two solutions, and y0 is the background spectroscopic signal.
| (Eq. 4) |
Computational modeling
Computational modeling of the human P450 11B2 catalytic system was performed using KinTek Explorer software (version 8.0, KinTek Corp., Snow Shoe, PA) (48, 49). The data shown in Fig. 3 were used as input data. Because the measured rates of binding for 11-deoxycorticosterone and corticosterone were 1.6–4.2 μm−1 s−1, a consistent approximation for the binding rate of all the steroids of 1 μm−1 s−1 was used and set so as to be unchangeable during global fitting. The value of the unbinding rate for each steroid was determined by the formula koff = Kd·kon. The Kd values used for these calculations are taken from Table 2. Therefore, because kon was set to 1 μm−1 s−1, the value of koff is equal to the value of Kd except with different units. These koff values were also set to be unchangeable during fitting. The values of kcat from Table 1 were used as initial estimates for the reaction rate constants. Analysis of the data proceeded in three steps. Step 1 was to analyze how well a simple model for P450 11B2 catalysis (Fig. 8) and the initial predictions for kcat led to a simulation that matched the experimental data. Step 2 was to have the KinTek Explorer software to attempt to minimize the differences in the simulated and experimental data by adjusting the three values for kcat. When step 2 failed to give acceptable results, an adjusted kinetic scheme that included 18-OH corticosterone lactol formation was developed (Fig. 10). The rate of the lactol form opening to form the aldehyde form of the steroid was set to 1 s−1 based on experimental results for fructofuranose opening to fructose presented elsewhere and adjusted to pH 7.4 (31). The lactol formation rate was adjusted manually until the lowest possible value was that forced the simulated production of 18-OH corticosterone and aldosterone to level off like the experimental data. This rate of formation was 2000 s−1. This is significantly higher than the reported values for fructose (7.4 or 41 s−1 for the α and β anomers, respectively), but it is hard to predict how the more rigid steroid structure contributes to the rate of lactol formation compared with the flexible fructose structure (31). Therefore, we decided this value could be reasonable and consistent with the strong preference for lactol formation. Both the lactol formation and lactol opening rates were set as unchangeable for step 3. Step 3 was to use this adjusted model and allow the KinTek Explorer software to again try and minimize differences by adjusting the kcat values. In rate optimization of steps 2 and 3, the data sets starting with 11-deoxycorticosterone and corticosterone were treated independently. This choice was made because we had already found different behavior depending on the starting substrate, and we wanted to emphasize the contribution of the lactol to the simulations.
Author contributions
M. J. R. data curation; M. J. R. formal analysis; M. J. R. validation; M. J. R. investigation; M. J. R. methodology; M. J. R. and F. P. G. writing-original draft; M. J. R. and F. P. G. writing-review and editing; F. P. G. conceptualization; F. P. G. supervision; F. P. G. funding acquisition; F. P. G. project administration.
Supplementary Material
Acknowledgments
We thank C. J. Wilkey and T. T. N. Phan for preparation of AdR, A. Bambach for assistance developing the expression of P450 11B2, and K. Trisler for assistance in preparation of the manuscript.
This work was supported by National Institutes of Health Grants R01 GM118122 (to F. P. G.) and T32 ES007028 (to F. P. G. for support of M. J. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Tables S1 and S2.
- P450
- cytochrome P450
- Adx
- adrenodoxin
- AdR
- NADPH–Adx reductase
- DLPC
- 1,2-dilauroyl-sn-glycero-3-phosphocholine
- OH
- hydroxy
- Ni-NTA
- nickel-nitrilotriacetic acid.
References
- 1. Rendic S., and Guengerich F. P. (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem. Res. Toxicol. 28, 38–42 10.1021/tx500444e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guengerich F. P. (2017) Intersection of the roles of cytochrome P450 enzymes with xenobiotic and endogenous substrates: relevance to toxicity and drug interactions. Chem. Res. Toxicol. 30, 2–12 10.1021/acs.chemrestox.6b00226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guengerich F. P. (2015) Human cytochrome P450 enzymes, in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano P. R., ed), 4th Ed., pp. 523–785, Springer, New York [Google Scholar]
- 4. Katsumata N., Ohtake M., Hojo T., Ogawa E., Hara T., Sato N., and Tanaka T. (2002) Compound heterozygous mutations in the cholesterol side-chain cleavage enzyme gene (CYP11A) cause congenital adrenal insufficiency in humans. J. Clin. Endocrinol. Metab. 87, 3808–3813 10.1210/jcem.87.8.8763 [DOI] [PubMed] [Google Scholar]
- 5. Kim C. J., Lin L., Huang N., Quigley C. A., AvRuskin T. W., Achermann J. C., and Miller W. L. (2008) Severe combined adrenal and gonadal deficiency caused by novel mutations in the cholesterol side chain cleavage enzyme, P450scc. J. Clin. Endocrinol. Metab. 93, 696–702 10.1210/jc.2007-2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parajes S., Loidi L., Reisch N., Dhir V., Rose I. T., Hampel R., Quinkler M., Conway G. S., Castro-Feijóo L., Araujo-Vilar D., Pombo M., Dominguez F., Williams E. L., Cole T. R., Kirk J. M., et al. (2010) Functional consequences of seven novel mutations in the CYP11B1 gene: four mutations associated with nonclassic and three mutations causing classic 11β-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 95, 779–788 10.1210/jc.2009-0651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pascoe L., Curnow K. M., Slutsker L., Rösler A., and White P. C. (1992) Mutations in the human CYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency. Proc. Natl. Acad. Sci. U.S.A. 89, 4996–5000 10.1073/pnas.89.11.4996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Portrat-Doyen S., Tourniaire J., Richard O., Mulatero P., Aupetit-Faisant B., Curnow K. M., Pascoe L., and Morel Y. (1998) Isolated aldosterone synthase deficiency caused by simultaneous E198D and V386A mutations in the CYP11B2 gene. J. Clin. Endocrinol. Metab. 83, 4156–4161 10.1210/jcem.83.11.5258 [DOI] [PubMed] [Google Scholar]
- 9. Auchus R. J. (2017) Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J. Steroid Biochem. Mol. Biol. 165, 71–78 10.1016/j.jsbmb.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simpson E. R. (2000) Genetic mutations resulting in loss of aromatase activity in humans and mice. J. Soc. Gynecol. Investig. 7, S18–S21 10.1177/1071557600007001S07 [DOI] [PubMed] [Google Scholar]
- 11. Strushkevich N., Gilep A. A., Shen L., Arrowsmith C. H., Edwards A. M., Usanov S. A., and Park H. W. (2013) Structural insights into aldosterone synthase substrate specificity and targeted inhibition. Mol. Endocrinol. 27, 315–324 10.1210/me.2012-1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hobler A., Kagawa N., Hutter M. C., Hartmann M. F., Wudy S. A., Hannemann F., and Bernhardt R. (2012) Human aldosterone synthase: recombinant expression in E. coli and purification enables a detailed biochemical analysis of the protein on the molecular level. J. Steroid Biochem. Mol. Biol. 132, 57–65 10.1016/j.jsbmb.2012.03.002 [DOI] [PubMed] [Google Scholar]
- 13. Wilczynski C., Shah L., Emanuele M. A., Emanuele N., and Mazhari A. (2015) Selective hypoaldosteronism: a review. Endocr. Pract. 21, 957–965 10.4158/EP14551.RA [DOI] [PubMed] [Google Scholar]
- 14. Young M. J., Clyne C. D., Cole T. J., and Funder J. W. (2001) Cardiac steroidogenesis in the normal and failing heart. J. Clin. Endocrinol. Metab. 86, 5121–5126 10.1210/jcem.86.11.7925 [DOI] [PubMed] [Google Scholar]
- 15. Ulmschneider S., Müller-Vieira U., Klein C. D., Antes I., Lengauer T., and Hartmann R. W. (2005) Synthesis and evaluation of (pyridylmethylene)tetrahydronaphthalenes/−indanes and structurally modified derivatives: potent and selective inhibitors of aldosterone synthase. J. Med. Chem. 48, 1563–1575 10.1021/jm0492397 [DOI] [PubMed] [Google Scholar]
- 16. Schumacher C. D., Steele R. E., and Brunner H. R. (2013) Aldosterone synthase inhibition for the treatment of hypertension and the derived mechanistic requirements for a new therapeutic strategy. J. Hypertens. 31, 2085–2093 10.1097/HJH.0b013e328363570c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Böttner B., Schrauber H., and Bernhardt R. (1996) Engineering a mineralocorticoid- to a glucocorticoid-synthesizing cytochrome P450. J. Biol. Chem. 271, 8028–8033 10.1074/jbc.271.14.8028 [DOI] [PubMed] [Google Scholar]
- 18. Curnow K. M., Mulatero P., Emeric-Blanchouin N., Aupetit-Faisant B., Corvol P., and Pascoe L. (1997) The amino acid substitutions Ser288Gly and Val320Ala convert the cortisol producing enzyme, CYP11B1, into an aldosterone producing enzyme. Nat. Struct. Biol. 4, 32–35 10.1038/nsb0197-32 [DOI] [PubMed] [Google Scholar]
- 19. Mulatero P., Curnow K. M., Aupetit-Faisant B., Foekling M., Gomez-Sanchez C., Veglio F., Jeunemaitre X., Corvol P., and Pascoe L. (1998) Recombinant CYP11B genes encode enzymes that can catalyze conversion of 11-deoxycortisol to cortisol, 18-hydroxycortisol, and 18-oxocortisol. J. Clin. Endocrinol. Metab. 83, 3996–4001 10.1210/jcem.83.11.5237 [DOI] [PubMed] [Google Scholar]
- 20. Imai T., Yamazaki T., and Kominami S. (1998) Kinetic studies on bovine cytochrome P45011β catalyzing successive reactions from deoxycorticosterone to aldosterone. Biochemistry 37, 8097–8104 10.1021/bi9802768 [DOI] [PubMed] [Google Scholar]
- 21. Peng H. M., Barlow C., and Auchus R. J. (2018) Catalytic modulation of human cytochromes P450 17A1 and P450 11B2 by phospholipid. J. Steroid Biochem. Mol. Biol. 181, 63–72 10.1016/j.jsbmb.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Martin R. E., Aebi J. D., Hornsperger B., Krebs H. J., Kuhn B., Kuglstatter A., Alker A. M., Märki H. P., Müller S., Burger D., Ottaviani G., Riboulet W., Verry P., Tan X., Amrein K., and Mayweg A. V. (2015) Discovery of 4-aryl-5,6,7,8-tetrahydroisoquinolines as potent, selective, and orally active aldosterone synthase (CYP11B2) inhibitors: in vivo evaluation in rodents and cynomolgus monkeys. J. Med. Chem. 58, 8054–8065 10.1021/acs.jmedchem.5b00851 [DOI] [PubMed] [Google Scholar]
- 23. Chowdhury G., Calcutt M. W., and Guengerich F. P. (2010) Oxidation of N-nitrosoalkylamines by human cytochrome P450 2A6: sequential oxidation to aldehydes and carboxylic acids and analysis of reaction steps. J. Biol. Chem. 285, 8031–8044 10.1074/jbc.M109.088039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chowdhury G., Calcutt M. W., Nagy L. D., and Guengerich F. P. (2012) Oxidation of methyl and ethyl nitrosamines by cytochrome P450 2E1 and 2B1. Biochemistry 51, 9995–10007 10.1021/bi301092c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sohl C. D., and Guengerich F. P. (2010) Kinetic analysis of the three-step steroid aromatase reaction of human cytochrome P450 19A1. J. Biol. Chem. 285, 17734–17743 10.1074/jbc.M110.123711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schenkman J. B., Remmer H., and Estabrook R. W. (1967) Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol. Pharmacol. 3, 113–123 [PubMed] [Google Scholar]
- 27. Simpson S. A., Tait J. F., Wettstein A., Neher R., v, Euw J., Schindler O., and Reichstein T. (1954) Aldosteron. Isolierung und eigenschaften. Über bestandteile der nebennierenrinde und verwandte stoffe. 91. Mitteilung. Helv. Chim. Acta 37, 1163–1200 10.1002/hlca.19540370423 [DOI] [Google Scholar]
- 28. Simpson S. A., Tait J. F., Wettstein A., Neher R., Voneuw J., Schindler O., and Reichstein T. (1954) Konstitution des aldosterons, des neuen mineralocorticoids. Experientia 10, 132–133 10.1007/BF02158515 [DOI] [PubMed] [Google Scholar]
- 29. Boudi A., Lemoine P., Viossat B., Tomas A., Fiet J., and Galons H. (1999) A convenient synthesis of 18-hydroxycorticosterone and 18-hydroxy-11-desoxycorticosterone via stereospecific hypoiodination of 20-hydroxysteroids. Tetrahedron 55, 5171–5176 10.1016/S0040-4020(99)00187-8 [DOI] [Google Scholar]
- 30. Ferrari P. (2010) The role of 11β-hydroxysteroid dehydrogenase type 2 in human hypertension. Biochim. Biophys. Acta 1802, 1178–1187 10.1016/j.bbadis.2009.10.017 [DOI] [PubMed] [Google Scholar]
- 31. Goux W. J. (1985) Complex isomerization of ketoses: a carbon-13 NMR study of the base-catalyzed ring-opening and ring-closing rates of d-fructose isomers in aqueous solution. J. Am. Chem. Soc. 107, 4320–4327 10.1021/ja00300a041 [DOI] [Google Scholar]
- 32. Hu Q., Jagusch C., Hille U. E., Haupenthal J., and Hartmann R. W. (2010) Replacement of imidazolyl by pyridyl in biphenylmethylenes results in selective CYP17 and dual CYP17/CYP11B1 inhibitors for the treatment of prostate cancer. J. Med. Chem. 53, 5749–5758 10.1021/jm100317b [DOI] [PubMed] [Google Scholar]
- 33. Pinto-Bazurco Mendieta M. A., Hu Q., Engel M., and Hartmann R. W. (2013) Highly potent and selective nonsteroidal dual inhibitors of CYP17/CYP11B2 for the treatment of prostate cancer to reduce risks of cardiovascular diseases. J. Med. Chem. 56, 6101–6107 10.1021/jm400484p [DOI] [PubMed] [Google Scholar]
- 34. Hoyt S. B., Petrilli W., London C., Liang G. B., Tata J., Hu Q., Yin L., van Koppen C. J., Hartmann R. W., Struthers M., Wisniewski T., Ren N., Bopp C., Sok A., Cai T. Q., et al. (2015) Discovery of triazole CYP11B2 inhibitors with in vivo activity in Rhesus monkeys. ACS Med. Chem. Lett. 6, 861–865 10.1021/acsmedchemlett.5b00048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu Q., Yin L., Ali A., Cooke A. J., Bennett J., Ratcliffe P., Lo M. M., Metzger E., Hoyt S., and Hartmann R. W. (2015) Novel pyridyl substituted 4,5-dihydro-[1,2,4]triazolo[4,3-a]quinolines as potent and selective aldosterone synthase inhibitors with improved in vitro metabolic stability. J. Med. Chem. 58, 2530–2537 10.1021/acs.jmedchem.5b00079 [DOI] [PubMed] [Google Scholar]
- 36. Gobbi S., Hu Q., Zimmer C., Engel M., Belluti F., Rampa A., Hartmann R. W., and Bisi A. (2016) Exploiting the chromone scaffold for the development of inhibitors of corticosteroid biosynthesis. J. Med. Chem. 59, 2468–2477 10.1021/acs.jmedchem.5b01609 [DOI] [PubMed] [Google Scholar]
- 37. Gonzalez E., and Guengerich F. P. (2017) Kinetic processivity of the two-step oxidations of progesterone and pregnenolone to androgens by human cytochrome P450 17A1. J. Biol. Chem. 292, 13168–13185 10.1074/jbc.M117.794917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mulatero P., di Cella S. M., Monticone S., Schiavone D., Manzo M., Mengozzi G., Rabbia F., Terzolo M., Gomez-Sanchez E. P., Gomez-Sanchez C. E., and Veglio F. (2012) 18-Hydroxycorticosterone, 18-hydroxycortisol, and 18-oxocortisol in the diagnosis of primary aldosteronism and its subtypes. J. Clin. Endocrinol. Metab. 97, 881–889 10.1210/jc.2011-2384 [DOI] [PubMed] [Google Scholar]
- 39. Kem D. C., Tang K., Hanson C. S., Brown R. D., Painton R., Weinberger M. H., and Hollifield J. W. (1985) The prediction of anatomical morphology of primary aldosteronism using serum 18-hydroxycorticosterone levels. J. Clin. Endocrinol. Metab. 60, 67–73 10.1210/jcem-60-1-67 [DOI] [PubMed] [Google Scholar]
- 40. Oelkers W., Boelke T., and Bähr V. (1988) Dose-response relationships between plasma adrenocorticotropin (ACTH), cortisol, aldosterone, and 18-hydroxycorticosterone after injection of ACTH-(1–39) or human corticotropin-releasing hormone in man. J. Clin. Endocrinol. Metab. 66, 181–186 10.1210/jcem-66-1-181 [DOI] [PubMed] [Google Scholar]
- 41. Yoshimoto F. K., Jung I. J., Goyal S., Gonzalez E., and Guengerich F. P. (2016) Isotope-labeling studies support the electrophilic compound I iron active species, FeO3+, for the carbon–carbon bond cleavage reaction of the cholesterol side-chain cleavage enzyme, cytochrome P450 11A1. J. Am. Chem. Soc. 138, 12124–12141 10.1021/jacs.6b04437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barnes H. J., Arlotto M. P., and Waterman M. R. (1991) Expression and enzymatic activity of recombinant cytochrome P450 17α-hydroxylase in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 88, 5597–5601 10.1073/pnas.88.13.5597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Omura T., and Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes: I. Evidence for its hemoprotein nature. J. Biol. Chem. 239, 2370–2378 [PubMed] [Google Scholar]
- 44. Guengerich F. P. (2014) Analysis and characterization of enzymes and nucleic acids relevant to toxicology, in Hayes' Principles and Methods of Toxicology (Hayes A. W., and Kruger C. L., eds) 6th Ed., pp. 1905–1964, CRC Press-Taylor & Francis, Boca Raton, FL [Google Scholar]
- 45. Johnson K. A. (2019) New standards for collecting and fitting steady state kinetic data. Beilstein J. Org. Chem. 15, 16–29 10.3762/bjoc.15.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hulme E. C., and Trevethick M. A. (2010) Ligand binding assays at equilibrium: validation and interpretation. Br. J. Pharmacol. 161, 1219–1237 10.1111/j.1476-5381.2009.00604.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perrin C. L. (2017) Linear or nonlinear least-squares analysis of kinetic data? J. Chem. Educ. 94, 669–672 10.1021/acs.jchemed.6b00629 [DOI] [Google Scholar]
- 48. Johnson K. A., Simpson Z. B., and Blom T. (2009) Global Kinetic Explorer: a new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 10.1016/j.ab.2008.12.024 [DOI] [PubMed] [Google Scholar]
- 49. Johnson K. A., Simpson Z. B., and Blom T. (2009) FitSpace Explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 10.1016/j.ab.2008.12.025 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.