

Abstract
X-linked Alport syndrome (XLAS) is a congenital renal disease caused by mutations in COL4A5. In XLAS cases suspected of being caused by aberrant splicing, transcript analysis needs to be conducted to determine splicing patterns and assess the pathogenicity. However, such analysis is not always available. We conducted a functional splicing assay using a hybrid minigene for seven COL4A5 intronic mutations: one was identified by us and six were found in the Human Gene Mutation Database. The minigene assay revealed exon skipping in four variants, exon skipping and a 10-bp insertion in one variant, and no change in one variant, which appeared not to be pathogenic. For one variant, our assay did not work. The results of all three cases for which transcript data were available were consistent with our assay results. Our findings may help to increase the accuracy of genetic test results and clarify the mechanisms causing aberrant splicing.
Subject terms: Interstitial disease, Alport syndrome
Introduction
COL4A5 (NM: 000495.4) is the causative gene of X-linked Alport syndrome (XLAS). XLAS can cause end-stage renal disease accompanied by sensorineural hearing loss and ocular abnormalities in affected patients1. It has been reported that 13.7% or 14.9% of pathogenic COL4A5 mutations are splice site mutations2,3. However, in these previous studies, no intronic variants leading to aberrant COL4A5 splicing outside of the consensus sequence (AG-GT) were identified. Many unclassified pathogenic intronic or exonic mutations result in splicing abnormalities4–6. We previously reported that, of 41 families with COL4A5 aberrant splicing, only 32 (78%) had splice site variants, while the others had deep intronic or exonic variants5. Clinically diagnosed XLAS cases lacking COL4A5 exonic or consensus splice site variants can be caused by intronic or synonymous exonic splicing abnormalities.
Recently, we conducted a genetic analysis of inherited kidney diseases. However, it is sometimes difficult to distinguish intronic variants leading to splicing errors from harmless polymorphisms. The most reliable method to identify splicing aberrations is by in vivo assay. This method is based on the analysis of mRNA derived from the affected tissue. However, this sample type is not always available. Instead, we analyse mRNA isolated from peripheral blood leukocytes, which is unstable and easily destroyed during transportation. To prevent RNA destruction, we use the Paxgene Blood RNA Kit (Qiagen Inc., Chatsworth, CA) or RNAlater RNA Stabilization Reagent (Qiagen Inc.). However, even using these tools, it is sometimes difficult to extract mRNA of sufficient quantity and quality.
Several in silico approaches have been developed to assess the influence of sequence variants on splicing7, but the information acquired using these approaches remains incomplete8,9. They can provide information about changes in the ability of splicing-related proteins to bind to each splicing related element, but do not accurately predict splicing patterns. Recently, the in vitro splicing assay with hybrid minigene has emerged as a relatively fast approach to identify splicing aberrations in inherited kidney disease10–13. Three reports have shown that the hybrid minigene system, using HEK293 or HEK293T cells derived from human embolic kidney cells, was effective for assessing COL4A5 variant splicing patterns5,14,15. Currently, the best approach to identify splicing mutations with a role in disease is to use a combination of these in vivo, in silico and in vitro analyses7.
Many of the components contributing to accurate intron splicing are already known. These include the splicing regulator, consensus sequence, branch point, exonic/intronic splicing enhancer/silencer sequence, and polypyrimidine tract (Fig. 1)16. Therefore, we focused on previously reported COL4A5 intronic mutations separated from exons. We obtained the sequences of all reported disease-causing COL4A5 mutations (at intronic positions 10 to 40 bp upstream of the subsequent exon) from The Human Gene Mutation Database (HGMD) and added one from our case3,17–21. Using a splicing reporter minigene system, we introduced the different mutations and investigated splicing pattern changes. Finally, we speculate on the mechanisms of the splicing pattern changes using evidence from in silico analyses.
Figure 1.
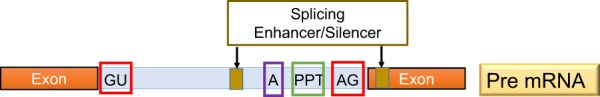
Schema for splicing regulation. Exons, intron, splice consensus sequences (GU or AG), U2 branch point (A), polypyrimidine tract (PPT), splicing enhancer/silencer.
Results
The minigene assay revealed aberrant splicing in five of seven variants. We observed exon skipping in four variants (No. 1, No. 3, No. 5 and No. 7), both exon skipping and a 10-bp insertion in one variant (No. 2), and no change in one variant (No. 6) (Table 1, Fig. 2 and Supplementary Fig. 2). Analysis of minigene construct No. 4 (intron 28-14T>A), containing introns 28–29, was not possible. The inserted sequence produced numerous nonspecific bands, making it difficult to assess the splicing pattern. Among three of the seven variants (No. 2, No. 5 and No. 7), our in vitro assay results were identical to the previously reported patient transcripts (Table 1). In these cases, mRNA samples were extracted from skin fibroblasts (No. 2), hair roots (No. 5) or peripheral blood leukocytes (No. 7). The results of in silico analysis are shown in Table 1. A marked decrease in the original acceptor site score was found for No. 2, No. 4 and No. 5. This suggests that the corresponding changes made it more difficult for the original acceptor site to function in exonic recognition. Novel splicing acceptor sites were created in No. 2, No. 4 and No. 7, suggesting the possibility of insertion or deletion. Indeed, No. 2 leads to a 10-bp insertion in addition to exon 9 skipping (Supplementary Fig. 3). In three variants, reduced polypyrimidine tract scores were observed (No. 3, No. 5 and No. 6). The polypyrimidine tract is preceded by a branch point sequence and is important for downstream exonic recognition. A reduced polypyrimidine tract score may lead to impaired exonic recognition. However, minigene analysis showed no change in splicing pattern in No. 6, even though a reduced polypyrimidine tract score was observed. Of the six cases in which in vitro results were obtained, in four cases (No. 1, No. 2, No. 3 and No. 5) the results could be predicted to some extent by the in silico results, while in No. 6 and No. 7 prediction from the in silico results was difficult (Table 1). At present, this in silico analysis is insufficient to predict the precise splicing pattern. In all cases, no branch point alteration was observed.
Table 1.
mRNA, in vitro (minigene) and in silico assays.
| gDNA mutation | mRNA | In vitro (minigene) | In silico | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| Genetic region cloned | Cloning via | Result | HSF (original ASS) | HSF (novel ASS) | SVM-BPF (PPT score) | ||||
| No. 1 | IVS8-17 T>G | N/A | Intron8–10 | Restriction and ligation | Ex9 skipping | 7.54 → 6.44 | → | Nagel et al.17 | |
| No. 2 | IVS8-12 G>A | Ex9 skipping/ 10 bp ins | Intron8–10 | Restriction and ligation | Ex9 skipping/ 10 bp ins | 7.54 → 2.74 | /→3.4 | → | Wang et al.18 |
| No. 3 | IVS21-20 T>A | N/A | Intron21–22 | Restriction and ligation | Ex22 skipping | 8.09 → 5.99 | ↓ | Bekheirnia et al.3 | |
| No. 4 | IVS28-14 T>A | N/A | Intron28–29 | In-fusion reaction | N/A | 3.87→/ | /→4.68 | → | Weber et al.19 |
| No. 5 | IVS31-10 T>G | Ex 32 skipping | Intron30–33 | In-fusion reaction | Ex32 skipping | 5.68→/ | ↓ | King et al.20 | |
| No. 6 | IVS37-11 C>A | N/A | Intron37–39 | In-fusion reaction | No change | 6.45 → 4.92 | ↓ | Martin et al.21 | |
| No. 7 | IVS26-18 A>G | Ex27 skipping | Intron26–28 | Restriction and ligation | Ex27 skipping | 12.03 → 10.7 | /→6.97 | → | Our case |
IVS: intron, N/A: not available, Ex: exon.
HSF: Human Splicing Finder, ASS: acceptor site score, SVM-BPF: SVM-BPfinder, PPT: polypyrimidine tract.
Figure 2.

Minigene assay transcript analysis. Electrophoresis results and schematic transcript analysis from the minigene constructs. The direct sequence is shown in Supplementary Fig. 2. (a) Wild type (WT) exhibited a single band (full) and No. 1 (case) exhibited a single band (exon 9 skipping), while No. 2 (case) exhibited double bands (exon 9 skipping and 10-bp insertion). (b) WT exhibited a single band (full) and No. 3 (case) exhibited a single band (exon 22 skipping). (c) Both WT and No. 4 (case) exhibited many bands, which were difficult to compare. (d) WT exhibited double bands (full and exon 32 skipping) and No. 5 (case) exhibited a single band (exon 32 skipping). (e) Both WT and No. 6 (case) exhibited single bands (full). (f) WT exhibited a single band (full) and No. 7 (case) exhibited a single band (exon 27 skipping). No cropped pictures are used in this figure.
Discussion
This is the first report comprehensively exploring COL4A5 splicing patterns in variants assumed to be pathogenic in intronic regions 10 to 40 bp upstream of exons. Although XLAS is a monogenic disease caused by COL4A5, pathogenic COL4A5 mutations are not detected in all clinically diagnosed XLAS patients. In such cases, intronic mutations or exonic synonymous mutations can cause aberrant splicing4–6. Moreover, 15–60% of pathogenic mutations cause genetic disease through pre-mRNA splicing abnormalities22. Therefore, a renewed focus should be placed on intronic variants that can cause splicing abnormalities.
Recently, splicing has been focused on as a target of treatment. Modifying the splicing pattern is an important goal of some molecular therapies23. Oligonucleotide-based therapies such as Nusinersen or Eteplirsen have been approved by the FDA24,25. Clarification of the involvement of splicing in disease pathogenicity can lead to the development of treatments. For XLAS, we recently reported the phenotype–genotype correlation restricted to cases with COL4A5 splicing abnormalities. These results suggested the possibility of oligonucleotide therapy leading to exon skipping in patients with XLAS5. Taken together, these results show that determining the involvement of splicing in the pathogenicity of inherited kidney diseases is important and may inform treatment approaches.
When nucleotide changes are detected in a gene, it is important to determine whether or not they are pathogenic. This is often difficult, particularly when variants are found in introns. To assess the pathogenicity of intronic variants, it is important to conduct transcript analysis. Tools for in silico analysis, such as the Human Splicing Finder (http://www.umd.be/HSF3/), can be used to predict the effect of transcriptional variants and may aid in predicting the disruption of the original consensus splice sites. We previously reported that, among 41 families with COL4A5 variants inducing aberrant splicing, 19 were completely compatible with the Human Splicing Finder results. Of the remaining families, 17 yielded data that enabled prediction of splicing defects, a precise novel splice site could not be predicted, and the remaining five families yielded insufficient data for the prediction of splicing abnormalities5. In this study, No. 6 showed decreasing polypyrimidine tract scores, but in vitro results showed no difference between this patient and the control. Therefore, the limitation of in silico analysis lies in accurately predicting splicing patterns.
The most accurate method to predict pathogenicity is to conduct transcriptional analysis using the patients’ affected organ tissues. However, obtaining kidney specimens to observe tissue-specific gene expression is always difficult. In addition to renal tissue, peripheral blood lymphocytes, skin biopsies, hair roots and urine-derived cells from patients have also been reported as adjuncts for the diagnosis of AS5,6,14,18,20,26–29. It may be possible to determine the pathogenicity of a mutation by examining the mRNA extracted from each sample. However, this approach is suboptimal as transcripts are not stable within tissues, and abnormal transcripts can be quickly degraded by nonsense-mediated mRNA decay30. Instead, it has been reported that it is effective to examine the expression of type IV collagen α5 chain in the hair root, which can be collected noninvasively, but the sensitivity and specificity have not been fully established14. Therefore, there remains a serious need for another system to assess the pathogenicity of variants of uncertain significance.
Recently, a splicing assay with a hybrid minigene approach has been established as a relatively fast and accurate means to identify splicing aberrations and to study their underlying functional mechanisms10–15,31. This approach has been validated by reports of the same results being obtained from in vivo and in vitro studies10–12,32. In our study, in three of the seven patient transcript analyses, the obtained results were completely consistent with our in vitro assay results. In one case, our minigene system did not work, possibly because of the low original acceptor site score. In addition, the results of No. 5 minigene showed normal and skipping bands in the WT, but only skipping bands in a patient (Fig. 2d). The possibility of identifying abnormal splicing bands in the WT has already been reported in other COL4A5 studies with minigene assay5,15, which can lead to false positive results of aberrant splicing. Therefore, the in vitro results should be evaluated in comparison with controls, bearing in mind variation in splicing patterns among different tissues and differences between in vitro and in vivo conditions.
In silico analysis assisted the interpretation of in vivo or in vitro analysis results, but was insufficient to predict the splicing pattern by itself. Three intronic variants analyzed by COL4A5 minigene assay have been reported so far5,14,15. Among them, the coexistence of exon skipping and a 43-bp deletion was detected in one case. It was possible to predict exon skipping by in silico analysis from reduced branch point scores in that case; however, the 43-bp deletion could not be predicted. Furthermore, although exon skipping occurred in the remaining two cases, their splice site scores decreased only slightly, supporting the difficulty of predicting exon skipping by in silico analysis itself (Supplementary Table 1). However, although it is difficult to predict the splicing pattern, clarifying the mechanisms of aberrant splicing by in silico analysis allows us to understand how to correct the splicing pattern to weaken or ameliorate the pathogenicity. Combining in vivo and/or in vitro and in silico analyses can thus be a powerful tool for assessing pathogenicity and for the development of appropriate therapeutic approaches.
In conclusion, our splicing assay with hybrid minigene makes it possible to assess whether the mutation in question causes aberrant splicing. In addition, in silico tools can predict the aberrant splicing mechanisms. Our system is useful to increase the accuracy of genetic tests through determining the pathogenicity of intronic mutations and may help inform treatment discovery strategies.
Methods
In vitro splicing assay
To create hybrid minigene constructs, we used the H492 vector that we developed previously, which is based on the pcDNA 3.0 mammalian expression vector and contains a multicloning site (Invitrogen, Carlsbad, CA, USA) (Supplementary Fig. 1)10. We cloned DNA fragments containing a couple of exons and introns around the target variant in the COL4A5 gene using classical restriction and ligation methods or In-fusion cloning methods, as shown in Table 1. As for No. 1–3 and No. 7, cloning was performed by classical restriction and ligation methods using NheI and BamHI. As for No. 4–6, we used infusion cloning methods with the HD Cloning Kit (Takara Bio Inc., Tokyo, Japan), in accordance with the manufacturer’s instructions. As for No. 7, the patients’ gDNA was available, so gDNA of the patients and WT was cloned. As for No. 1–6, because the patients’ gDNA was not available (just the sequence was reported), we initiated cloning with WT gDNA and then introduced mutations by site-directed mutagenesis using PrimeSTAR mutagenesis basal kit (Takara Bio Inc.), in accordance with the manufacturer’s instructions. The primers used are shown in Supplementary Table 1.
The hybrid minigenes were confirmed by sequencing and transfected into HEK293T and HeLa cells using Lipofectamine® 2000 (Thermo Fisher Scientific, Waltham, MA, CA). Total RNA was extracted from cells after 24 h using the Rneasy® Plus Mini Kit (QIAGEN, GmbH, Hilden, Germany). Total RNA (1 µg) was reverse-transcribed using RNA to cDNA EcoDry Premix (Double Primed) (Takara Bio Inc). PCR was performed using a forward primer corresponding to a segment upstream of exon A (YH307: 5′-ATTACTCGCTCAGAAGCTGTGTTGC-3′) and a reverse primer complementary to a segment downstream of exon B (YH308: 5′-CTGCCAGTTGCTAAGTGAGAGACTT-3′). PCR products were analysed by electrophoresis on an 1.5% agarose gel using φX174-Hae III digest marker and direct sequencing.
In silico splicing assay
We predicted splicing domain strength in each variant, using Human Splicing Finder (http://www.umd.be/HSF3/) and SVM-BP Finder (http://regulatorygenomics.upf.edu/Software/SVM_BP/)33,34. As for potential splice sites, scores obtained using MaxEnt Scan matrix are shown.
Compliance with ethical standards
All procedures were reviewed and approved by the Institutional Review Board of Kobe University School of Medicine.
Supplementary information
Acknowledgements
We thank Rebecca Porter, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript. This study was supported by Grants-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (subject ID: 18K15712 to Tomoko Horinouchi, 19K08726 to Kandai Nozu, 16K19642 to Tomohiko Yamamura and 17H04189 to Kazumoto Iijima), and by AMED under Grant Number JP19ek0109231h0003 to Kandai Nozu and Kazumoto Iijima. Kazumoto Iijima has received grant support from Daiichi Sankyo Co., Ltd., and consulting fees from Takeda Pharmaceutical Company, Ono Pharmaceutical Co., Ltd., Boehringer Ingelheim, Astellas Pharma Inc. and Kyowa Hakko Kirin Co., Ltd. Kandai Nozu has received lecture fees from Novartis Pharmaceuticals Corporation and consulting fees from Kyowa Hakko Kirin Co., Ltd. Kazumoto Iijima and Kandai Nozu have filed a patent application on the development of antisense nucleotides for exon skipping therapy in Alport syndrome.
Author Contributions
T.H. and T.Y. designed the study concept and wrote the manuscript. Ka.N. interpreted the data and wrote the manuscript. S.M., C.N., N.S., N.M., S.I., Y.A. and R.R. established and conducted molecular analysis and interpreted the data. M.M. established the minigene assay. Ko.N., Y.S., H.N., H.T. and K.I. critically reviewed the manuscript. All authors read and approved the final version of the manuscript.
Data Availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-48990-9.
References
- 1.Kashtan CE. Alport syndrome and thin glomerular basement membrane disease. Journal of the American Society of Nephrology: JASN. 1998;9:1736–1750. doi: 10.1681/ASN.V991736. [DOI] [PubMed] [Google Scholar]
- 2.Jais JP, et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. Journal of the American Society of Nephrology: JASN. 2000;11:649–657. doi: 10.1681/ASN.V114649. [DOI] [PubMed] [Google Scholar]
- 3.Bekheirnia MR, et al. Genotype-phenotype correlation in X-linked Alport syndrome. Journal of the American Society of Nephrology: JASN. 2010;21:876–883. doi: 10.1681/asn.2009070784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu XJ, et al. X-linked Alport syndrome associated with a synonymous p.Gly292Gly mutation alters the splicing donor site of the type IV collagen alpha chain 5 gene. Clinical and experimental nephrology. 2016;20:699–702. doi: 10.1007/s10157-015-1197-9. [DOI] [PubMed] [Google Scholar]
- 5.Horinouchi T, et al. Detection of Splicing Abnormalities and Genotype-Phenotype Correlation in X-linked Alport Syndrome. Journal of the American Society of Nephrology: JASN. 2018;29:2244–2254. doi: 10.1681/asn.2018030228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nozu K, et al. X-linked Alport syndrome caused by splicing mutations in COL4A5. Clinical journal of the American Society of Nephrology: CJASN. 2014;9:1958–1964. doi: 10.2215/cjn.04140414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baralle D, Lucassen A, Buratti E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. EMBO reports. 2009;10:810–816. doi: 10.1038/embor.2009.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartmann L, Theiss S, Niederacher D, Schaal H. Diagnostics of pathogenic splicing mutations: does bioinformatics cover all bases? Frontiers in bioscience: a journal and virtual library. 2008;13:3252–3272. doi: 10.2741/2924. [DOI] [PubMed] [Google Scholar]
- 9.Houdayer C, et al. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Human mutation. 2008;29:975–982. doi: 10.1002/humu.20765. [DOI] [PubMed] [Google Scholar]
- 10.Nozu K, et al. In vivo and in vitro splicing assay of SLC12A1 in an antenatal salt-losing tubulopathy patient with an intronic mutation. Human genetics. 2009;126:533–538. doi: 10.1007/s00439-009-0697-7. [DOI] [PubMed] [Google Scholar]
- 11.Nakanishi Keita, Nozu Kandai, Hiramoto Ryugo, Minamikawa Shogo, Yamamura Tomohiko, Fujimura Junya, Horinouchi Tomoko, Ninchoji Takeshi, Kaito Hiroshi, Morisada Naoya, Ishimori Shingo, Nakanishi Koichi, Morioka Ichiro, Awano Hiroyuki, Matsuo Masafumi, Iijima Kazumoto. A comparison of splicing assays to detect an intronic variant of the OCRL gene in Lowe syndrome. European Journal of Medical Genetics. 2017;60(12):631–634. doi: 10.1016/j.ejmg.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Yamamura T, et al. An in vitro splicing assay reveals the pathogenicity of a novel intronic variant in ATP6V0A4 for autosomal recessive distal renal tubular acidosis. BMC nephrology. 2017;18:353. doi: 10.1186/s12882-017-0774-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamamura T, et al. Functional splicing analysis in an infantile case of atypical hemolytic uremic syndrome caused by digenic mutations in C3 and MCP genes. Journal of human genetics. 2018;63:755–759. doi: 10.1038/s10038-018-0436-9. [DOI] [PubMed] [Google Scholar]
- 14.Malone AF, Funk SD, Alhamad T, Miner JH. Functional assessment of a novel COL4A5 splice region variant and immunostaining of plucked hair follicles as an alternative method of diagnosis in X-linked Alport syndrome. Pediatric nephrology (Berlin, Germany) 2017;32:997–1003. doi: 10.1007/s00467-016-3565-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chiereghin C, et al. Alport syndrome cold cases: Missing mutations identified by exome sequencing and functional analysis. PloS one. 2017;12:e0178630. doi: 10.1371/journal.pone.0178630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic acids research. 2006;34:3494–3510. doi: 10.1093/nar/gkl498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagel M, Nagorka S, Gross O. Novel COL4A5, COL4A4, and COL4A3 mutations in Alport syndrome. Human mutation. 2005;26:60. doi: 10.1002/humu.9349. [DOI] [PubMed] [Google Scholar]
- 18.Wang F, Wang Y, Ding J, Yang J. Detection of mutations in the COL4A5 gene by analyzing cDNA of skin fibroblasts. Kidney international. 2005;67:1268–1274. doi: 10.1111/j.1523-1755.2005.00204.x. [DOI] [PubMed] [Google Scholar]
- 19.Weber S, et al. Identification of 47 novel mutations in patients with Alport syndrome and thin basement membrane nephropathy. Pediatric nephrology (Berlin, Germany) 2016;31:941–955. doi: 10.1007/s00467-015-3302-4. [DOI] [PubMed] [Google Scholar]
- 20.King K, Flinter FA, Nihalani V, Green PM. Unusual deep intronic mutations in the COL4A5 gene cause X linked Alport syndrome. Human genetics. 2002;111:548–554. doi: 10.1007/s00439-002-0830-3. [DOI] [PubMed] [Google Scholar]
- 21.Martin, P. et al. Spectrum of COL4A5 mutations in Finnish Alport syndrome patients. Human mutation15, 579, 10.1002/1098-1004(200006)15:6<579::Aid-humu13>3.0.Co;2-k (2000). [DOI] [PubMed]
- 22.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nature reviews. Genetics. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 23.Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. The Journal of clinical investigation. 2003;112:481–486. doi: 10.1172/jci19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Traynor K. Eteplirsen approved for Duchenne muscular dystrophy. American journal of health-system pharmacy: AJHP: official journal of the American Society of Health-System Pharmacists. 2016;73:1719. doi: 10.2146/news160063. [DOI] [PubMed] [Google Scholar]
- 25.Ottesen EW. ISS-N1 makes the First FDA-approved Drug for Spinal Muscular Atrophy. Translational neuroscience. 2017;8:1–6. doi: 10.1515/tnsci-2017-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hashimura Y, et al. Milder clinical aspects of X-linked Alport syndrome in men positive for the collagen IV alpha5 chain. Kidney international. 2014;85:1208–1213. doi: 10.1038/ki.2013.479. [DOI] [PubMed] [Google Scholar]
- 27.Nakanishi K, et al. Immunohistochemical study of alpha 1-5 chains of type IV collagen in hereditary nephritis. Kidney international. 1994;46:1413–1421. doi: 10.1038/ki.1994.413. [DOI] [PubMed] [Google Scholar]
- 28.Tazón-Vega Bárbara, Ars Elisabet, Burset Moisès, Santín Sheila, Ruíz Patricia, Fernández-Llama Patricia, Ballarín José, Torra Roser. Genetic Testing for X-Linked Alport Syndrome by Direct Sequencing of COL4A5 cDNA From Hair Root RNA Samples. American Journal of Kidney Diseases. 2007;50(2):257.e1-257.e14. doi: 10.1053/j.ajkd.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 29.Daga S, et al. Urine-derived podocytes-lineage cells: A promising tool for precision medicine in Alport Syndrome. Human mutation. 2018;39:302–314. doi: 10.1002/humu.23364. [DOI] [PubMed] [Google Scholar]
- 30.Doma MK, Parker R. RNA quality control in eukaryotes. Cell. 2007;131:660–668. doi: 10.1016/j.cell.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 31.Tournier I, et al. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Human mutation. 2008;29:1412–1424. doi: 10.1002/humu.20796. [DOI] [PubMed] [Google Scholar]
- 32.van der Klift HM, et al. Splicing analysis for exonic and intronic mismatch repair gene variants associated with Lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Molecular genetics & genomic medicine. 2015;3:327–345. doi: 10.1002/mgg3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desmet FO, et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids research. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corvelo A, Hallegger M, Smith CW, Eyras E. Genome-wide association between branch point properties and alternative splicing. PLoS computational biology. 2010;6:e1001016. doi: 10.1371/journal.pcbi.1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.