

Abstract
Aim:
Limitations in available therapies for trypanosomiases indicate the need for improved medicines. Cysteine proteases cruzain and rhodesain are validated targets for treatment of Chagas disease and human African trypanosomiasis. Previous studies reported a benzimidazole series as potent cruzain inhibitors.
Results & methodology:
Considering the high similarity between these proteases, we evaluated 40 benzimidazoles against rhodesain. We describe their structure-activity relationships (SAR), revealing trends similar to those observed for cruzain and features that lead to enzyme selectivity. This series comprises noncovalent competitive inhibitors (best Ki = 0.21 μM against rhodesain) and micromolar activity against Trypanosoma brucei brucei. A cheminformatics analysis confirms scaffold novelty, and the inhibitors described have favorable predicted physicochemical properties.
Conclusion:
Our results support this series as a starting point for new human African trypanosomiasis medicines.
Keywords: : benzimidazole inhibitors, cruzain, parasite cysteine proteases, rhodesain, Trypanosoma
Sleeping sickness or human African trypanosomiasis (HAT) is caused by the flagellate protozoa, Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense, which are transmitted by the bite of the tsetse fly. The disease is endemic in sub-Saharan Africa where there are approximately 20,000 people infected and 65 million people at risk [1]. Parasite replication takes place initially in the hemolymphatic system but subsequent penetration of the CNS causes a variety of progressive neurological derangements which, if left untreated, are eventually fatal [2].
There are only five medicines available for HAT chemotherapy, some of which are very toxic. The treatment depends on infection stage, and for the first stage pentamidine and suramin are employed. Second stage options include melarsoprol, eflornithine, or a combination of nifurtimox and eflornithine. Among those, nifurtimox–eflornithine has shown success in clinical trials for infections involving T. b. gambiense, for which it is the first-line treatment. However, it has not been studied against T. b. rhodesiense [2]. Thus, the first-line treatment for T. b. rhodesiense and second-line treatment for T. b. gambiense infections is melarsoprol, which is an arsenic compound with an associated 5% drug-fatality rate [2]. Recent studies indicate that new treatment options may be soon available. Specifically, fexinidazole cured T. b. gambiense infections in Phase III clinical trials [3] and the oxaborate, SCYX-7158, is currently awaiting Phase III trials [4]. Nevertheless, the current treatment scenario is still characterized by major shortcomings associated with available chemotherapies, including poor efficacy, significant host toxicity and parasite resistance [5–7]. Accordingly, there is an urgent need to develop safer and effective new drugs.
Among the recent efforts to develop new drugs (recently reviewed by Singh-Grewal et al. [8] and Cullen et al. [9]), several have focused on papain-like cysteine proteases [10–12], which perform critical functions in cell invasion, immune evasion, virulence, parasite growth and differentiation [13,14]. Bloodstream T. brucei parasites express two principal cysteine proteases, the cathepsin L-like, rhodesain (TbCatL), and a cathepsin B-like, TbCatB [15–17]. Rhodesain is the more abundant, facilitates parasite traversal of the blood–brain barrier [18,19] and is essential for parasite survival [20]. Its ortholog in Trypanosoma cruzi, cruzain, is expressed in all developmental stages and is one of the most exploited molecular targets for new therapies to treat Chagas disease [21–26].
Potent inhibitors of both rhodesain and cruzain have been characterized, comprising peptidic [27–32] and nonpeptidic compounds [33–43]. However, especially in the case of rhodesain, the literature is dominated by covalent inhibitors. On the other hand, new classes of noncovalent inhibitors have been described for cruzain, such as the benzimidazole derivative, (N-[2-(1H-benzimidazol-2-yl)ethyl]-2-(2-bromophenoxy)acetamide; compound 1) which competitively inhibits cruzain at nanomolar concentrations (IC50 = 800 nM) [40,44]. Several of these displayed nanomolar or low micromolar inhibition values against both cruzain and T. cruzi in vitro [40], and later quantitative structure–activity relationship studies revealed important features for cruzain inhibition [45].
The structural similarity between cruzain and rhodesain may facilitate the development of inhibitors mutually active against both Chagas disease and HAT, and inhibitor classes with activity against both enzymes have been described [34,46–49]. Here, we characterize this benzimidazole series as potent inhibitors of rhodesain with trypanocidal activity.
Experimental
Compounds
The synthesis and characterization of all compounds evaluated in this work were previously described by our group [40]. Their characterization included 1H and proton-decoupled 13C NMR and high-resolution mass spectrometry, performed using the ESI technique. The purity (≥95%) of all compounds was determined by 1H NMR.
Expression & purification of rhodesain
A DNA plasmid pPICZ_A (Invitrogen, MA, USA) encoding rhodesain without the C-terminal extension (rhodesain ΔC) and containing a N295A mutation to prevent glycosylation, was expressed in Pichia pastoris as previously reported [15]. Transformed colonies grew at 30°C under zeocin (100 μg/ml; Invitrogen) selection on yeast extract-peptone-dextrose agar and expanded in 10 ml liquid medium under the same conditions. The cells were harvested, added to 400 ml of yeast nitrogen base (Sigma-Aldrich Corporation, MO, USA) medium without amino acids but containing 1% methanol, and cultured at 28°C for 72 h under shaking at 250 r.p.m. Throughout this period, enzymatic activity was monitored via hydrolysis of the fluorogenic peptidyl substrate, Z-Phe-Arg-aminomethyl coumarin (Z-FR-AMC; as described below). A signal peptide encoded within the pPICZ_A plasmid leads to protein secretion. Therefore, the enzyme was purified from culture media. Pichia culture supernatants containing rhodesain activity were concentrated with Vivaflow 200 MWCO 3 kDa (Sartorius, Goettingen, Germany) to a volume of approximately 50 ml. To purify the protein, the concentrated supernatant was desalted (HiPrep 26/10 Desalting Column, GE Healthcare, IL, USA) in 0.1 M sodium acetate, pH 5.5 and the pH of the obtained solution was adjusted to 4.5 for overnight incubation at 37°C in the presence of 2 mM β-mercaptoethanol. The desalting procedure was repeated after incubation, and the concentrated rhodesain solution was stored at -80°C.
Enzyme assays
Enzyme kinetics assays were based on the hydrolysis of Z-FR-AMC whereby the fluorescence of free AMC was measured using 340/440 nm excitation/emission filters, following a previously reported protocol [50,51]. The assays were carried out in a Synergy 2 (Biotek®, VT, EUA) microplate reader. Fluorescence was measured at 25°C for 5 min at 12 s intervals, using BioTek’s Gen5™ Reader Control and Data Analysis Software to obtain initial velocity rates. Inhibitory activities were determined based on a control containing 0.5% of DMSO (the same concentration present in assays with inhibitors). The cysteine protease inhibitor, E-64, was utilized as an inhibitor control at a final concentration of 40 nM. The assays were performed in 0.1 M sodium acetate, pH 5.5, containing 0.01% Triton X-100, 1 mM β-mercaptoethanol, 1 nM rhodesain and 2.5 μM Z-FR-AMC substrate.
Given that the compounds evaluated were expected to bind noncovalently to rhodesain, the series was screened without preincubation with the enzyme. Unless explicitly noted, all results were obtained from at least two independent experiments, each in triplicate. Initially, compounds were screened at 100 μM. When the inhibition of rhodesain was >85% at the highest soluble concentration (variable for each compound), the IC50 was determined in at least two independent assays with seven different compound concentrations. IC50 values were calculated using the nonlinear regression method: ‘log (inhibitor) versus response with variable slope – four parameters’ analysis in GraphPad Prism version 6 [52]. Estimated inhibitor constant Ki values were calculated using the Cheng–Prusoff Equation (1)
| (Eq. 1) |
where Ki is the inhibitory constant, [S] is the substrate concentration in the assay ([S] = 2.5 μM for rhodesain; [S] = 5 μM for cruzain) and Km is the substrate concentration at which the enzyme achieves half-maximal velocity catalysis (Km = 1.6 μM for cruzain [40] and Km = 0.5 ± 0.1 μM for rhodesain). The Km for rhodesain was determined by our group, and the value here reported represents the average and standard deviation of nine independent experiments. A representative curve from one experiment is shown in Supplementary Figure 2. The inhibition mechanism of the most potent compound obtained (compound 15) was determined using seven different substrate concentrations (0.3–20.0 μM, in twofold increments) and four inhibitor concentrations (0.0625–1 μM, in twofold increments), based on Michaelis–Menten, Lineweaver–Burk and (Km app/Vmax vs [I]) plots. The Km app/Vmax for each concentration of compound 15 was determined using Michaelis–Menten analysis and the corresponding Ki was obtained by the x-axis intercept of the (Km app/Vmax vs [I]) plot.
To verify that the inhibition measured was not artifactual, the following measures were taken: all compounds were evaluated in the presence of 0.01% Triton X-100, decreasing the chance of compound aggregation; for the most potent compound in the series, a competitive mechanism of inhibition was demonstrated, as described above; fluorescence values at the beginning of the assay were observed and compared with a DMSO control and were not indicative of fluorescence artifacts. Furthermore, in a previous publication, the lead compound 1 in this series has been thoroughly validated as a cruzain competitive inhibitor, including determination of a crystal structure in complex with the enzyme (Protein Data Bank [PDB] code 3KKU) [44]. Compound 1 has also been previously shown to be stable in assay buffer (the same buffer employed here) [44].
Chemical similarity analysis
Papers were retrieved from either PubChem or ScienceDirect using the keywords rhodesain or TBCatL. All studies reporting rhodesain inhibitors were analyzed and compounds demonstrating IC50 or Ki values of at most 100 μM were considered. Chemical structures of reported rhodesain inhibitors were extracted from BindingDB database [53]. Compounds which were reported as detergent-sensitive inhibitors, aggregators and or unspecific inhibitors were discarded. Chemical structures were manually verified against the respective references. Fingerprints were calculated using a hashing algorithm using RDKit [54]. A hashing fingerprint was generated using a daylight-like topological linear algorithm with up to seven-atoms fragments. A 2048 bits long fingerprint was constructed and no bit was discarded regardless of significance. The Tanimoto coefficient of each pair was calculated and similarity matrices were generated. Heat maps were prepared directly from raw similarity matrices data and diagonal elements were discarded to calculate means and statistics. Analyses employed Python scripts.
Whole-organisms screens with T. b. brucei
An established assay [55] was employed that involved bloodstream forms of T. b. brucei (Lister 427) and the DNA binder, SYBR green (Thermo Fisher Cat# S75630, MA, EUA) as the read out. The initial parasite density was 100,000 cells/ml of modified HMI-9 media [56]. The following modifications were employed. Compounds, dissolved in DMSO, were acoustically transferred to Greiner 384 well assay plates (Cat# 781098) using an ATS Gen 4 Plus (Biosero, CA, USA) for a final DMSO concentration of 0.1% in 50 μl. To determine the effective dose at which half the parasites had died (EC50), eight-point concentration-response assays were set up between 50 and 0.25 μM. The reference drug, pentamidine, from Sigma-Aldrich (MO, USA) was kindly provided by Dr Jair Lage de Siqueira-Neto of the Center for Discovery and Innovation in Parasitic Diseases at University of California (UC) San Diego, USA. After 48 h, 12.5 μl lysis buffer (30 mM Tris-HCl, pH 7.5, 7.5 mM EDTA, 0.012% saponin and 0.12% Triton X-100) containing 0.3 μl SYBR Green (10,000× in DMSO) per milliliter was dispensed into each well. After a 60-min incubation, the fluorescence signal was measured in a Perkin Elmer EnVision 2014 plate reader with excitation/emission wavelengths of 485/535 nm. Two experiments were performed, each in triplicate.
Results
Rhodesain inhibitors & comparative SAR against cysteine proteases
A previously characterized series of cruzain inhibitors, composed of benzimidazole derivatives analogous to compound 1 [40] and containing variations in the phenyl and benzimidazole rings, and the linker between them, was evaluated for inhibition of recombinant rhodesain. We performed an initial screen at 100 μM and determined IC50 values for compounds that caused over 85% inhibition at this concentration (or at their highest soluble concentration). Thirteen compounds had IC50 values under 10 μM against rhodesain, allowing the characterization of a new inhibitor series against this enzyme.
Several SAR trends noted with rhodesain were in line with previous reports for cruzain [40]. Specifically, modifications in the phenyl ring of compound 1 were tolerated without significant changes in potency (Table 1). In contrast, attempts to replace the benzimidazole resulted in inactive compounds (Supplementary Table 1), while the addition of substituents in this ring leads to inhibitors with low micromolar IC50 values (Table 2). Finally, modifications in the linker had a significant impact on inhibition potency (Table 3). Significant SAR differences for trypanosomal proteases were also noted, as detailed below.
Table 1. . Inhibition of rhodesain and cruzain by derivatives of compound 1 with substituents on the phenyl ring.
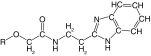 | ||||||
|---|---|---|---|---|---|---|
| Compound | R | Inhibition† (% at 100 μM) | IC50† (μM) | Ki cruzain/Ki rhodesain‡ | ||
| Rhodesain | Cruzain§ | Rhodesain | Cruzain§ | |||
| 1 (lead) | 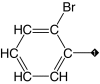 |
93 | 89 | 3.1 ± 0.4 | 0.8 ± 0.1 | 0.4 |
| 2 | 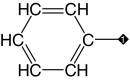 |
99 | 88 | 4.4 ± 0.7 | 10.9 ± 1 | 3.8 |
| 3 | 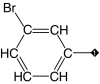 |
96 | 96 | 2.5 ± 1.6 | 0.8 ± 0.5 | 0.5 |
| 4 | 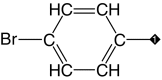 |
97 | 96 | 0.47 ± 0.08 | 2.7 ± 0.4 | 8.7 |
| 5 | 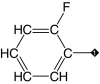 |
95 | 92 | 0.8 ± 0.08 | 5.3 ± 1.1 | 10 |
| 6 | 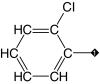 |
77 | 92 | 0.95 | 3.0 ± 0.6 | 4.8 |
| 7 | 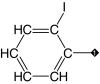 |
83 | 96 | 9.4 ± 2.0 | 1.6 ± 0.2 | 0.3 |
| 8 | 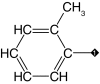 |
83 | 88 | 8.0 ± 0.9 | 4.1 ± 1.1 | 0.8 |
| 9 | 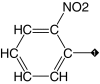 |
83 | 93 | 14.7 ± 9.6 | 13.2 ± 1.2 | 1.4 |
| 10 | 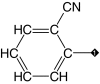 |
84 | 81 | 70.9 ± 0.4 | 65.1 ± 1.2 | 1.4 |
| 11 | 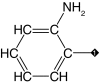 |
55 | 43 | ND | ND | ND |
| 12 | 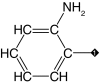 |
20 | 25 | ND | ND | ND |
| 13 | 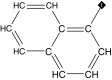 |
85 | 84 | ND | 5.2 ± 0.7 | ND |
| 14 | 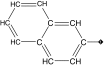 |
71 | 90 | 12.9 ± 2.8 | 3.0 ± 0.5 | 0.4 |
| 15 | 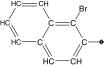 |
88 | 92 | 0.25 ± 0.2 | 0.21 ± 0.04 | 1.3 |
| 16 | 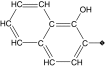 |
89 | 52 | 2.4 ± 0.5 | 8.2 ± 0.9 | 5.2 |
| 17 | 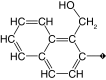 |
100 | 80 | 1.2 ± 0.16 | 12.7 ± 0.8 | 16 |
| 18¶ | 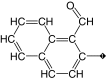 |
100 | 80 | 2.5 ± 1.2 | 13.5 ± 0.6 | 8.2 |
The inhibition percentage at 100 μM of compound, and IC50 against rhodesain and cruzain are represented as the means from at least two independent experiments each performed in triplicate. The IC50 averages are reported with their standard error of the mean, except for compound 6, for which the value was obtained from one curve due to the unavailability of additional material.
Estimated Ki values were calculated using the Cheng-Prusoff equation based on experimental IC50 values.
Data reported in [40].
This compound may form a Schiff base in the presence of an amine. However, this reaction is not expected to occur within our assay conditions, in which this compound is stable.
Ki: Inhibitory constant; ND: Not determined.
Table 2. . Inhibition of rhodesain and cruzain by derivatives containing substituents on the benzimidazole ring.
| Compound | Structure | Inhibition† (% at 100 μM) | IC50† (μM) | Ki cruzain/Ki rhodesain‡ | ||
|---|---|---|---|---|---|---|
| Rhodesain | Cruzain§ | Rhodesain | Cruzain§ | |||
| 19 | 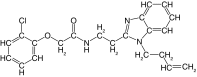 |
92 | 89 | 10.7 ± 6.4 | 23.9 ± 1.2 | 3.4 |
| 20 | 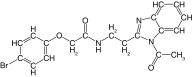 |
89 | 84 | 2.5 | 9.9 ± 0.9 | 6.0 |
| 21 | 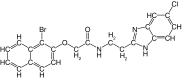 |
74 | 90 | 5.5 ± 0.1 | 0.6 ± 0.2 | 0.2 |
| 22 | 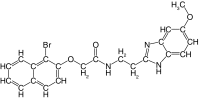 |
69 | 93 | 7.8 ± 0.6 | 0.5 ± 0.2 | 0.1 |
| 23 | 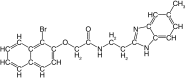 |
72 | 97 | ND | 0.6 ± 0.3 | ND |
The inhibition percentage at 100 μM of compound and IC50 against rhodesain and cruzain are represented as the means from at least two independent experiments each performed in triplicate. The IC50 values are reported with their standard error of the mean, except for compound 20, for which the value was obtained from one curve due to the unavailability of additional material.
Estimated Ki values were calculated using the Cheng–Prusoff equation based on experimental IC50 values.
Data reported in [40].
Ki: Inhibitory constant; ND: Not determined.
Table 3. . Inhibition of rhodesain and cruzain by derivatives that have been modified in the linker between phenyl and benzimidazole rings.
| Compound | Structure | Inhibition† (% at 100 μM) | IC50† (μM) | Ki cruzain/Ki rhodesain‡ | ||
|---|---|---|---|---|---|---|
| Rhodesain | Cruzain§ | Rhodesain | Cruzain§ | |||
| 24 | 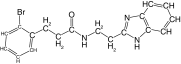 |
86 | 51 | 43.7 ± 11.0 | 77.5 ± 1.4 | 2.7 |
| 25 | 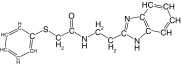 |
84 | 66 | 38.0 ± 1.6 | 38.4 ± 1.2 | 1.5 |
| 26 | 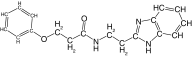 |
58 | 25 | ND | ND | ND |
| 27 | 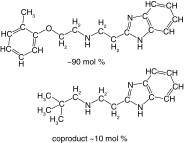 |
8 | 8 | ND | ND | ND |
The inhibition percentage at 100 μM of compound and IC50 against rhodesain and cruzain are represented as the means from at least two independent experiments each performed in triplicate. The IC50 values are reported with their standard error of the mean.
Estimated Ki values were calculated using the Cheng–Prusoff equation based on experimental IC50 values.
Data reported in [40].
Ki: Inhibitory constant; ND: Not determined.
Our results suggest that the phenyl ring in 1 (IC50 = 3.1 μM) was most amenable to modifications (Table 1). Accordingly, no drastic changes in potency resulted from removal of the bromine (2, IC50 = 4.4 μM), modification of its position to meta or para (3, IC50 = 2.5 μM and 4, IC50 = 0.47 μM, respectively) or replacement by a fluorine, chlorine, iodine, methyl or nitro group (5–9, IC50 values between 0.8 and 14.7 μM). Among these modifications, the greatest impact on potency was a sevenfold improvement for the para-bromine analog. On the other hand, other ortho substituents (nitrile, amino, and formamide, 10–12) led to at least 23-fold lower potencies.
Replacement of the phenyl ring by a naphthyl was also tolerated and led to the most potent compound in the series (15, IC50 = 0.25 μM), which we confirmed as a competitive inhibitor with a Ki = 0.21 μM (Supplementary Figure 3), as reported for cruzain [40]. It is worth noting that replacing the bromine with a hydroxyl (16, IC50 = 2.4 μM), hydroxymethyl (17, IC50 = 1.2 μM) or formyl (18, IC50 = 2.5 μM) had a greater impact on cruzain potency (Table 1). Based on the crystal structure of the complex cruzain-1 and assuming a conserved binding mode within this series, the naphthyl is expected to bind to the S2 subsite (Supplementary Figure 1). An important difference is observed at the bottom of S2: Glu-208 in cruzain is replaced by Ala-208 in rhodesain, resulting in a larger and more open pocket in the latter. This position has been described as a critical determinant for specificity in cruzain and other cysteine proteases [57], and, therefore, the difference observed might explain rhodesain’s better tolerance for derivatives containing larger substituents in the naphthyl ring, as noted for compounds 16–18.
On the other hand, the addition of substituents on the benzimidazole ring had a more significant impact on the inhibition of rhodesain. Whereas the addition of small substituents such as chlorine, methoxy and methyl increased IC50 values for cruzain by approximately threefold, against rhodesain the least impact observed was a 31-fold increase (compare 15 vs 21–23, Tables 1 & 2). The reasons for this are not explained by the crystallographic binding mode of 1, in which the benzimidazole ring is exposed to solvent.
Modification of the linker between the phenyl and benzimidazole rings decreased rhodesain inhibition (Table 3), as previously observed for cruzain [40]. However, it is worth highlighting that replacement of the ether oxygen by a methylene had a much higher impact on the inhibition of cruzain, decreasing potency by 100-fold, whereas with rhodesain a 15-fold decrease was measured (compare 1 vs 24; Tables 1 & 3). Overall, we identified potent rhodesain inhibitors, determined an SAR and identified modifications that resulted in selectivity for rhodesain or cruzain (Figure 1).
Figure 1. . Summary of structure-activity relationships (SAR) for the benzimidazole series against rhodesain and cruzain.

(A) SAR described for rhodesain based on compound 1 structure. (B) Distinctive SAR trends between cruzain and rhodesain.
Chemical similarity analysis
To gain insight into the novelty of inhibitors described in this study, we compared their chemical structures to known rhodesain inhibitors. A library of 513 structures from 46 studies was constructed (Supplementary Table 2). Similarity among compounds here reported and the ones previously published was measured based on pairwise Tanimoto coefficient calculations. Possible Tanimoto coefficient values range from 0 to 1, with higher values indicating more similar structures. The heat map generated from similarity matrices (Figure 2A, see also Supplementary Table 3 as a separate spreadsheet file) shows a clear clustering pattern, indicative of the different structural motifs, which have been explored in the search for rhodesain inhibitors. Furthermore, a relatively low off-cluster similarity was observed, with off-diagonal regions of moderate similarity caused by thoroughly explored motifs (e.g., arylnitriles and thiosemicarbazones). Specifically, the compounds reported here are dissimilar to inhibitors reported in the literature (highest Tanimoto coefficient = 0.395; Figure 2B).
Figure 2. . Chemical similarity among rhodesain inhibitors.

(A) Similarity heat map for rhodesain inhibitors grouped based on reference paper (Supplementary Table 2). A hashing algorithm was used to obtain fingerprints and the Tanimoto coefficient was employed to compare them. (B) Chemical structures of the highest similarity pair for inhibitors described in this study versus a previously reported compound [58].
Evaluation against Trypanosoma brucei in vitro
Next, we evaluated a subset of 23 compounds against bloodstream forms of T. b. brucei (Lister 427). Compounds 1–26 (except 16, 20 and 24), with IC50 values against rhodesain ranging from 0.25 to 70.9 μM or which were inactive against the enzyme, were assayed. Pentamidine, a drug currently employed in HAT treatment, was the positive control and yielded an EC50 value of 0.10 ± 0.02 μM against the parasite. Thirteen compounds inhibited parasite growth, among which ten compounds had EC50 values ≤20 μM, including the two compounds which were most potent against rhodesain (compounds 4 and 15). Ten compounds did not inhibit parasite growth at the concentrations evaluated (up to 50 μM): their EC50 values are indicated as >50 μM (Table 4).
Table 4. . In vitro assay against Trypanosoma brucei (Lister 427).
| Compound | EC50 against T. brucei (μM)‡ | IC50 against rhodesain (μM)§ |
|---|---|---|
| 1 (lead) | 22.3 ± 3.3 | 3.1 ± 0.4 |
| 2 | >50 | 4.4 ± 0.7 |
| 3 | 9.9 ± 1.6 | 2.5 ± 1.6 |
| 4 | 18.8 ± 2.8 | 0.47 ± 0.08 |
| 5 | >50 | 0.8 ± 0.08 |
| 6 | >50 | 0.95 |
| 7 | 18.8 ± 2.2 | 9.4 ± 2.0 |
| 8 | >50 | 8.0 ± 0.9 |
| 9 | >50 | 14.7 ± 9.6 |
| 10 | >50 | 70.9 ± 0.4 |
| 11 | >50 | ND |
| 12 | >50 | ND |
| 13 | 17.7 ± 3.2 | ND |
| 14 | 23.2 ± 4.2 | 12.9 ± 2.8 |
| 15 | 12.2† | 0.25 ± 0.2 |
| 17 | >50 | 1.2 ± 0.2 |
| 18 | 15.1 ± 2.3 | 2.5 ± 1.2 |
| 19 | 12.0† | 10.7 ± 6.4 |
| 21 | 12.2† | 5.5 ± 0.1 |
| 22 | 10.3 ± 0.8 | 7.8 ± 0.6 |
| 23 | 8.6 ± 0.9 | ND |
| 25 | 24.5 ± 5.1 | 38.0 ± 1.6 |
| 26 | >50 | ND |
| Pentamidine | 0.10 ± 0.02 | - |
EC50 values with SEM >25%.
‡ EC50 values are reported as the means from two independent experiments each performed in triplicate, with their SEM.
Values extracted from Tables 1–3, and copied here to facilitate comparison with data against the parasite.
ND: Not determined; SEM: Standard error of the mean.
All of the compounds with EC50 values <50 μM against the parasite were rhodesain inhibitors, and inhibited by at least 70% the enzyme’s activity at 100 μM. Also, compounds with low activity against rhodesain, as observed for 11, 12 and 26, were inactive against the parasite. Some potent enzyme inhibitors (such as compounds 5, 6 and 17) had no trypanocidal activity. This might be due to their physicochemical properties. The overall agreement between inhibition of the enzyme and reduction of parasite growth is coherent with inhibition of rhodesain as a possible mechanism underlying the antiparasite activity.
Prediction of pharmacokinetic properties
Finally, to evaluate overall drug likeness of the series described, we estimated several relevant physicochemical and pharmacokinetic descriptors, using the Qikprop v4.9 (Schrödinger) software [59]. Among 43 calculated properties for these compounds, we highlight a subset in Table 5. This analysis revealed the high drug likeness of these molecules, including compliance with the Lipinski’s rule of five properties [60], and most of their estimated properties (polarizability, cell permeability, logP, water solubility, number of predicted metabolites) within the range observed for 95% of known drugs. Among the highlighted properties, the only one for which predicted values are consistently outside the range normally observed for drugs is QPlogHERG inhibition. It is worth noting that none of the compounds is predicted to have poor permeability against Caco-2 cells (QPPCaco) and most (15/21) are predicted to have great permeability with values >500 nm/s for this parameter. Blood–brain barrier permeability, which is an essential requirement for any new HAT drug to treat second-stage infection, was also in the expected range.
Table 5. . Computed physico-chemical descriptors to assess the ‘drug likeness’ of the inhibitor series.
| Comp | MW† | HBD‡ | HBA§ | QPpolrz¶ | QPlogPo/w# | QPlogS†† | QPlogHERG‡‡ | QPPCaco§§ | QPlogBB¶¶ | met## | ROF††† |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 374.2 | 2 | 4.8 | 36.1 | 3.2 | -4.2 | -5.1 | 707.5 | -0.6 | 3 | 0 |
| 2 | 295.3 | 2 | 4.8 | 34.5 | 2.7 | -3.6 | -5.4 | 657.7 | -0.8 | 3 | 0 |
| 3 | 374.2 | 2 | 4.8 | 36.2 | 3.2 | -4.4 | -5.3 | 657.5 | -0.7 | 3 | 0 |
| 4 | 374.2 | 2 | 4.8 | 36.2 | 3.2 | -4.4 | -5.3 | 657.5 | -0.7 | 3 | 0 |
| 5 | 313.3 | 2 | 4.8 | 34.9 | 2.9 | -3.9 | -5.2 | 679.3 | -0.7 | 3 | 0 |
| 6 | 329.8 | 2 | 4.8 | 35.8 | 3.1 | -4.1 | -5.1 | 700.4 | -0.6 | 3 | 0 |
| 7 | 421.2 | 2 | 4.8 | 36.4 | 3.3 | -4.3 | -5.1 | 705.5 | -0.6 | 3 | 0 |
| 8 | 309.4 | 2 | 4.8 | 36.5 | 3.0 | -4.3 | -5.2 | 863.9 | -0.8 | 4 | 0 |
| 9 | 340.3 | 2 | 5.8 | 36.1 | 2.1 | -3.5 | -5.1 | 126.9 | -1.6 | 4 | 0 |
| 10 | 320.4 | 2 | 6.3 | 36.1 | 2.0 | -4.4 | -5.2 | 184.3 | -1.5 | 3 | 0 |
| 11 | 310.4 | 3.5 | 5.8 | 34.7 | 1.8 | -3.4 | -5.2 | 222.5 | -1.5 | 6 | 0 |
| 12 | 352.4 | 3 | 7.3 | 38.9 | 2.2 | -3.5 | -5.1 | 378.1 | -1.1 | 4 | 0 |
| 13 | 345.4 | 2 | 4.8 | 40.6 | 3.6 | -5.0 | -5.9 | 839.2 | -0.8 | 3 | 0 |
| 14 | 345.4 | 2 | 4.8 | 40.7 | 3.6 | -4.8 | -6.0 | 657.9 | -0.9 | 3 | 0 |
| 15 | 424.3 | 2 | 4.8 | 42.1 | 4.1 | -5.2 | -5.7 | 710.1 | -0.7 | 3 | 0 |
| 17 | 375.4 | 3 | 6.5 | 41.8 | 2.8 | -4.6 | -5.8 | 298.2 | -1.5 | 4 | 0 |
| 18 | 373.4 | 2 | 6.8 | 41.9 | 2.8 | -4.3 | -5.8 | 213.0 | -1.5 | 3 | 0 |
| 19 | 383.9 | 1 | 4.8 | 42.4 | 4.7 | -5.3 | -5.4 | 1074.6 | -0.6 | 4 | 0 |
| 21 | 458.7 | 2 | 4.8 | 43.4 | 4.6 | -6.0 | -5.6 | 710.3 | -0.5 | 3 | 0 |
| 22 | 454.3 | 2 | 5.5 | 43.9 | 4.2 | -5.5 | -5.6 | 703.8 | -0.8 | 4 | 0 |
| 23 | 438.3 | 2 | 4.8 | 43.9 | 4.4 | -5.8 | -5.6 | 704.2 | -0.7 | 4 | 0 |
| 25 | 311.4 | 2 | 4.5 | 35.8 | 3.1 | -4.0 | -5.3 | 690.7 | -0.7 | 2 | 0 |
| 26 | 295.3 | 2 | 4.8 | 34.6 | 2.7 | -3.6 | -5.3 | 657.7 | -0.8 | 3 | 0 |
| 27 | 295.4 | 2 | 3.8 | 35.8 | 3.8 | -4.0 | -7.3 | 819.0 | 0.0 | 4 | 0 |
Desirable range values for the descriptors according to the program QikProp (based on the ranges observed for 95% of known drugs):
Compound MW: 130–725 g/mol.
Average number of HBD: 0.0–6.0.
Average number of HBA: 2.0–20.0.
QPpolrz: 13.0–70.0 Å3.
QPlogPo/w: -2.0–6.5.
QPlogS: -6.5–0.5 mol.dm-3.
QPlogHERG concern values are below -5.
QPPCaco recommended values are <25 poor and >500 great.
QPlogBB for orally delivered drugs: -3.0–1.2.
met: 1–8.
Number of violations of Lipinski’s ROF [60]. The rules are: mol_MW <500. QPlogPo/w <5. HBD ≤5. HBA ≤10. Compounds that satisfy at least three of these rules are considered drug like. Predicted properties, which fall within the range observed for 95% of known drugs are shown in green cells, while those outside this range (or not classified as ‘great’ in the case of QPPCaco) are shown in red cells.
HBA: Hydrogen bond acceptor; HBD: Hydrogen bond donor; met: Number of likely metabolic reactions; QPlogBBQ: Predicted brain/blood partition coefficient; PlogHERG: Predicted IC50 value for blockage of HERG K+ channel; QPlogPo/w: Predicted octanol/water partition coefficient; QPlogS: Predicted aqueous solubility; QPPCaco: Predicted apparent Caco-2 cell permeability in nanometer per second; QPpolrz: Predicted polarizability; ROS: Rule of five.
Discussion
Motivated by the high structural similarity between cruzain and rhodesain, and by the need to develop new drugs for treatment of HAT, a previously characterized series of cruzain inhibitors [40] was evaluated in enzymatic assays against rhodesain. This resulted in an important contribution to HAT drug discovery that comprised the characterization of a new compound class of competitive, noncovalent and submicromolar rhodesain inhibitors. Other classes of inhibitors, such as nitriles [49], thiosemicarbazones [34,50], vinyl sulfones [46,61] and nitroalkenes [47] have been shown to inhibit both cruzain and rhodesain. However, to the best of our knowledge, we report the first example of a noncovalent competitive series active against both enzymes.
We comprehensively characterized the SAR for this series through the evaluation of over 40 analogs containing various structural modifications. These assays also allowed a comparison of the SAR between cruzain and rhodesain, which highlighted modifications that lead to selectivity for one or other enzyme. Considering the potential impact of developing dual inhibitors as potential leads for both Chagas disease and HAT, such contributions are important to guide the design of new compound series. To better understand the observed SAR and selectivity trends, our group has also performed molecular modeling studies, including molecular docking, molecular dynamics simulations and free energy calculations through thermodynamic integration. These results have been recently published [62].
Our compounds were active against T. b. brucei parasites at mid-low micromolar concentrations. Even though it would be desirable to obtain compounds more potent against the parasite, as described in some studies [32,35], the potency range observed here is similar to the one reported in other recent publications [30,63] and characterizes a new classes of molecules active against T. brucei. Our data are coherent with a mechanism based on rhodesain inhibition, but these results are not sufficient to prove the trypanocidal mechanism of this series. It is also possible that off-target effects are involved. In a previous study, these compounds were evaluated against fibroblast cells infected by T. cruzi trypomastigotes, and several of them have also shown activity against T. cruzi (EC50 values between 1.6 and 46.1 μM) [40]. Together with those data, our present findings validate benzimidazoles as a source of trypanocidal compounds that potently inhibit the requisite cysteine proteases. In particular, we highlight the enzyme inhibition and parasiticidal activities of compounds 4 and 15. Both also have drug-like properties and good predicted physicochemical properties. They were previously shown to be noncytotoxic against human fibroblasts and safe in acute toxicity assays in mice [40].
Conclusion
Benzimidazoles previously characterized as inhibitors of the T. cruzi enzyme, cruzain, have been shown to also inhibit the T. b. brucei enzyme, rhodesain, in some cases with nanomolar potency. Together with their drug likeness, chemical novelty and activity against T. b. brucei, our results support future optimization of this series to develop a novel HAT chemotherapy. Our data also provide relevant information on the comparative SAR for this series against both trypanosomal cysteine proteases.
Future perspective
In future studies with this series, it will be highly desirable to improve potency against T. brucei and to obtain further evidence of target engagement, confirming the mechanism of trypanomicidal activity. In parallel, it will be also important to experimentally determine and optimize physicochemical properties, as well as to verify efficacy in animal models of the disease. After optimization of these properties, this series may progress through the drug-discovery pipeline, potentially generating a novel drug to treat trypanosomiasis.
Summary points.
Rhodesain is a validated target for human African trypanosomiasis (HAT), for which new drugs are urgently needed.
Benzimidazoles were characterized as a novel scaffold of rhodesain inhibitors.
Potent competitive rhodesain inhibitors were identified (Ki as low as 0.21 μM).
This series contains drug-like compounds, with favorable physicochemical properties predicted.
Chemoinformatic analysis revealed the novelty of this series when compared with known rhodesain inhibitors.
Comparative structure–activity relationships for rhodesain and cruzain are reported.
Several compounds were active against T. b. brucei in vitro.
This benzimidazole series constitutes a starting point for the development of a new human African trypanosomiasis therapy.
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/full/10.4155/fmc-2018-0523
Acknowledgments
The authors acknowledge the Center of Flow Cytometry and Fluorimetry at the Biochemistry and Immunology Department (UFMG) for allowing access to the Synergy 2 (Biotek®) fluorimeter. They are thankful to JC Borges and PR Dores-Silva, from Instituto de Química de São Carlos (USP-São Carlos, Brazil) for help in characterizing the purity and oligomeric state of rhodesain.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Financial & competing interests disclosure
The authors are grateful to the Brazilian agencies CAPES, FAPEMIG and CNPq for funding this research. GAN Pereira was a postdoctoral scholar supported by the grants Special Visitant Researcher (PVE A118/2013) and Edital 51/2013 Biologia Computacional (CAPES). KR Liedl was a Special Visitant Researcher (PVE A118/2013) scholar, and LH Santos received a CAPES Programa de Doutorado Sanduíche no Exterior (PDSE 99999.010357/2014-09) scholarship. RS Ferreira holds a CNPq researcher fellowship (Bolsa de Produtividade em Pesquisa) and has received the L'Oréal-UNESCO-ABC ‘For Women in Science Award, Category Chemistry, Brazil, 2017’ and L'Oréal-UNESCO ‘International Rising Talent Award 2018’. Whole organism screens with Trypanosoma brucei (CR Caffrey and SC Wang) were supported by a grant from the US National Institute of Allergy and Infectious Diseases of the NIH under the award R21AI133394. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Büscher P, Cecchi G, Jamonneau V, Priotto G. Human African trypanosomiasis. Lancet 390(10110), 2397–2409 (2017). [DOI] [PubMed] [Google Scholar]
- 2.WHO. Human African trypanosomiasis. (sleeping sickness). https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness)
- 3.Maxmen A. Sleeping sickness can now be cured with pills. Nature 550(7677), 441–441 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Steketee PC, Vincent IM, Achcar F. et al. Benzoxaborole treatment perturbs S-adenosyl-L-methionine metabolism in Trypanosoma brucei. PLoS Negl. Trop. Dis. 12(5), e0006450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bern C. Antitrypanosomal therapy for chronic Chagas’ disease. N. Engl. J. Med. 364(26), 2527–2534 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Steverding D. The development of drugs for treatment of sleeping sickness: a historical review. Parasit. Vectors 3(1), 15 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coura JR, Viñas PA. Chagas disease: a new worldwide challenge. Nature 465(n7301_supp), S6–S7 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Singh Grewal A, Pandita D, Bhardwaj S, Lather V. Recent updates on development of drug molecules for human African trypanosomiasis. Curr. Top. Med. Chem. 16(20), 2245–2265 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Cullen D, Mocerino M. A brief review of drug discovery research for human African trypanosomiasis. Curr. Med. Chem. 24(7), 701–717 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Mayorga K, Byler KG, Ramirez-Hernandez AI, Terrazas-Alvares DE. Cruzain inhibitors: efforts made, current leads and a structural outlook of new hits. Drug Discov. Today 20(7), 890–898 (2015). [DOI] [PubMed] [Google Scholar]
- 11.da Silva EB, do Nascimento Pereira GA, Ferreira RS. Trypanosomal cysteine peptidases: target validation and drug design strategies. : Comprehensive Analysis of Parasite Biology: From Metabolism to Drug Discovery, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 121–145 (2016). [Google Scholar]
- 12.Rocha DA, Silva EB, Fortes IS, Lopes MS, Ferreira RS, Andrade SF. Synthesis and structure–activity relationship studies of cruzain and rhodesain inhibitors. Eur. J. Med. Chem. 157, 1426–1459 (2018). [DOI] [PubMed] [Google Scholar]; • Recent review describing synthetic routes and structure-activity relationships for varied chemotypes of cruzain and rhodesain inhibitors.
- 13.Sajid M, McKerrow JH. Cysteine proteases of parasitic organisms. Mol. Biochem. Parasitol. 120(1), 1–21 (2002). [DOI] [PubMed] [Google Scholar]
- 14.McKerrow JH, Caffrey C, Kelly B, Loke P, Sajid M. Proteases in parasitic diseases. Annu. Rev. Pathol. 1, 497–536 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Caffrey CR, Hansell E, Lucas KD. et al. Active site mapping, biochemical properties and subcellular localization of rhodesain, the major cysteine protease of Trypanosoma brucei rhodesiense. Mol. Biochem. Parasitol. 118(1), 61–73 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Mackey ZB, O'Brien TC, Greenbaum DC, Blank RB, McKerrow JH. A cathepsin B-like protease is required for host protein degradation in Trypanosoma brucei. J. Biol. Chem. 279(46), 48426–48433 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Kerr ID, Wu P, Marion-Tsukamaki R, Mackey ZB, Brinen LS. Crystal structures of TbCatB and rhodesain, potential chemotherapeutic targets and major cysteine proteases of Trypanosoma brucei. PLoS Negl. Trop. Dis. 4(6), e701 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdulla M-H, O'Brien T, Mackey ZB, Sajid M, Grab DJ, McKerrow JH. RNA interference of Trypanosoma brucei cathepsin B and L affects disease progression in a mouse model. PLoS Negl. Trop. Dis. 2(9), e298 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nikolskaia OV. Blood–brain barrier traversal by African trypanosomes requires calcium signaling induced by parasite cysteine protease. J. Clin. Invest. 116(10), 2739–2747 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steverding D, Sexton DW, Wang X, Gehrke SS, Wagner GK, Caffrey CR. Trypanosoma brucei: chemical evidence that cathepsin L is essential for survival and a relevant drug target. Int. J. Parasitol. 42(5), 481–488 (2012). [DOI] [PubMed] [Google Scholar]; •• An important paper demonstrating that rhodesain is an essential enzyme for Trypanosoma brucei.
- 21.Jose Cazzulo J, Stoka V, Turk V. The major cysteine proteinase of Trypanosoma cruzi: a valid target for chemotherapy of Chagas disease. Curr. Pharm. Des. 7(12), 1143–1156 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Harth G, Andrews N, Mills AA, Engel JC, Smith R, McKerrow JH. Peptide-fluoromethyl ketones arrest intracellular replication and intercellular transmission of Trypanosoma cruzi. Mol. Biochem. Parasitol. 58(1), 17–24 (1993). [DOI] [PubMed] [Google Scholar]
- 23.McKerrow JH, Doyle PS, Engel JC. et al. Two approaches to discovering and developing new drugs for Chagas disease. Mem. Inst. Oswaldo Cruz 104(Suppl.), 263–269 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vieira RP, Santos VC, Ferreira RS. Structure-based approaches targeting parasite cysteine proteases. Curr. Med. Chem. 24, 1 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Ferreira LG, Andricopulo AD. Targeting cysteine proteases in trypanosomatid disease drug discovery. Pharmacol. Ther. 180, 49–61 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Sajid M, Robertson SA, Brinen LS, McKerrow JH. Cruzain. : Advances in Experimental Medicine and Biology, Springer, MA, USA, 100–115 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Kerr ID, Lee JH, Farady CJ. et al. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 284(38), 25697–25703 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choe Y, Brinen LS, Price MS. et al. Development of α-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorg. Med. Chem. 13(6), 2141–2156 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Huang L, Brinen LS, Ellman JA. Crystal structures of reversible ketone-based inhibitors of the cysteine protease cruzain. Bioorg. Med. Chem. 11(1), 21–29 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Previti S, Ettari R, Cosconati S. et al. Development of novel peptide-based Michael acceptors targeting rhodesain and falcipain-2 for the treatment of neglected tropical diseases (NTDs). J. Med. Chem. 60(16), 6911–6923 (2017). [DOI] [PubMed] [Google Scholar]; • Recent paper describing potent rhodesain inhibitors.
- 31.Mallari JP, Guiguemde WA, Guy RK. Antimalarial activity of thiosemicarbazones and purine derived nitriles. Bioorg. Med. Chem. Lett. 19(13), 3546–3549 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giroud M, Kuhn B, Saint-Auret S. et al. 2 H -1,2,3-triazole-based dipeptidyl nitriles: potent, selective, and trypanocidal rhodesain inhibitors by structure-based design. J. Med. Chem. 61(8), 3370–3388 (2018). [DOI] [PubMed] [Google Scholar]; •• Recent paper describing potent rhodesain inhibitors with trypanocidal activity.
- 33.Du X, Guo C, Hansell E. et al. Synthesis and structure−activity relationship study of potent trypanocidal thio semicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J. Med. Chem. 45(13), 2695–2707 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Greenbaum DC, Mackey Z, Hansell E. et al. Synthesis and structure−activity relationships of parasiticidal thiosemicarbazone cysteine protease inhibitors against Plasmodium falciparum, Trypanosoma brucei, and Trypanosoma cruzi. J. Med. Chem. 47(12), 3212–3219 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Giroud M, Dietzel U, Anselm L. et al. Repurposing a library of human cathepsin L ligands: identification of macrocyclic lactams as potent rhodesain and Trypanosoma brucei inhibitors. J. Med. Chem. 61(8), 3350–3369 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Mallari JP, Shelat AA, Obrien T. et al. Development of potent purine-derived nitrile inhibitors of the trypanosomal protease TbcatB. J. Med. Chem. 51(3), 545–552 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Mott BT, Ferreira RS, Simeonov A. et al. Identification and optimization of inhibitors of trypanosomal cysteine proteases: cruzain, rhodesain, and TbCatB. J. Med. Chem. 53(1), 52–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferreira RS, Simeonov A, Jadhav A. et al. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 53(13), 4891–4905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiggers HJ, Rocha JR, Fernandes WB. et al. Non-peptidic cruzain inhibitors with trypanocidal activity discovered by virtual screening and in vitro assay. PLoS Negl. Trop. Dis. 7(8), e2370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferreira RS, Dessoy MA, Pauli I. et al. Synthesis, biological evaluation, and structure–activity relationships of potent noncovalent and nonpeptidic cruzain inhibitors as anti-Trypanosoma cruzi agents. J. Med. Chem. 57(6), 2380–2392 (2014). [DOI] [PubMed] [Google Scholar]; •• Describes the activity the benzimidazole series studied here against cruzain and Trypanosoma cruzi.
- 41.Fonseca NC, Da Cruz LF, Da Silva Villela F. et al. Synthesis of a sugar-based thiosemicarbazone series and structure–activity relationship versus the parasite cysteine proteases rhodesain, cruzain, and Schistosoma mansoni cathepsin B1. Antimicrob. Agents Chemother. 59(5), 2666–2677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Espíndola JWP, De Oliveira Cardoso MV, De Oliveira Filho GB. et al. Synthesis and structure–activity relationship study of a new series of antiparasitic aryloxyl thiosemicarbazones inhibiting Trypanosoma cruzi cruzain. Eur. J. Med. Chem. 101, 818–835 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Braga SFP, Martins LC, da Silva EB. et al. Synthesis and biological evaluation of potential inhibitors of the cysteine proteases cruzain and rhodesain designed by molecular simplification. Bioorg. Med. Chem. 25(6), 1889–1900 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Ferreira RS, Simeonov A, Jadhav A. et al. Complementarity between a docking and a high-throughput screen in discovering new cruzain inhibitors. J. Med. Chem. 53(13), 4891–4905 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pauli I, Ferreira LG, de Souza ML. et al. Molecular modeling and structure–activity relationships for a series of benzimidazole derivatives as cruzain inhibitors. Future Med. Chem. 9(7), 641–657 (2017). [DOI] [PubMed] [Google Scholar]; • Employs several QSAR techniques to investigate features relevant for inhibition of cruzain by the benzimidazoles here described.
- 46.Jaishankar P, Hansell E, Zhao D-M, Doyle PS, McKerrow JH, Renslo AR. Potency and selectivity of P2/P3-modified inhibitors of cysteine proteases from trypanosomes. Bioorg. Med. Chem. Lett. 18(2), 624–628 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Latorre A, Schirmeister T, Kesselring J. et al. Dipeptidyl nitroalkenes as potent reversible inhibitors of cysteine proteases rhodesain and cruzain. ACS Med. Chem. Lett. 7(12), 1073–1076 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen YT, Lira R, Hansell E, McKerrow JH, Roush WR. Synthesis of macrocyclic trypanosomal cysteine protease inhibitors. Bioorg. Med. Chem. Lett. 18(22), 5860–5863 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mott BT, Ferreira RS, Simeonov A. et al. Identification and optimization of inhibitors of trypanosomal cysteine proteases: cruzain, rhodesain, and TbCatB. J. Med. Chem. 53(1), 52–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fonseca NC, da Cruz LF, da Silva Villela F. et al. Synthesis of a sugar-based thiosemicarbazone series and structure–activity relationship versus the parasite cysteine proteases rhodesain, cruzain, and Schistosoma mansoni cathepsin B1. Antimicrob. Agents Chemother. 59(5), 2666–2677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Braga SFP, Martins LC, da Silva EB. et al. Synthesis and biological evaluation of potential inhibitors of the cysteine proteases cruzain and rhodesain designed by molecular simplification. Bioorg. Med. Chem. 25(6), 1889–1900 (2017). [DOI] [PubMed] [Google Scholar]
- 52.GraphPad Prism, version 6.0 for Windows (2015). https://www.graphpad.com/scientific-software/prism/
- 53.Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44(D1), D1045–D1053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.RDKit: Open-source cheminformatics (2015). https://www.rdkit.org/
- 55.Faria J, Moraes CB, Song R. et al. Drug discovery for human African trypanosomiasis. J. Biomol. Screen. 20(1), 70–81 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 75(6), 985–989 (1989). [PubMed] [Google Scholar]
- 57.Gillmor SA, Craik CS, Fletterick RJ. Structural determinants of specificity in the cysteine protease cruzain. Protein Sci. 6(8), 1603–1611 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watts KR, Ratnam J, Ang K-H. et al. Assessing the trypanocidal potential of natural and semi-synthetic diketopiperazines from two deep water marine-derived fungi. Bioorg. Med. Chem. 18(7), 2566–2574 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Small Molecule Discovery Suite 2016-3: QikProp, version 4.9 Schrödinger, LLC, New York, NY, USA: (2016). https://www.schrodinger.com/qikprop [Google Scholar]
- 60.Lipinski CA. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 44(1), 235–249 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Bryant C, Kerr ID, Debnath M. et al. Novel non-peptidic vinylsulfones targeting the S2 and S3 subsites of parasite cysteine proteases. Bioorg. Med. Chem. Lett. 19(21), 6218–6221 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Santos LH, Waldner BJ, Fuchs JE. et al. Understanding structure–activity relationships for trypanosomal cysteine protease inhibitors by simulations and free energy calculations. J. Chem. Inf. Model. 59(1), 137–148 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Royo S, Schirmeister T, Kaiser M. et al. Antiprotozoal and cysteine proteases inhibitory activity of dipeptidyl enoates. Bioorg. Med. Chem. 26(16), 4624–4634 (2018). [DOI] [PubMed] [Google Scholar]