

Abstract
Phosphonates, often used as isosteric replacements for phosphates, can provide important interactions with an enzyme. Due to their high charge at physiological pH, however, permeation into cells can be a challenge. Protecting phosphonates as prodrugs has shown promise in drug delivery. Thus, a variety of structures and cleavage/activation mechanisms exist, enabling release of the active compound. This review describes the structural diversity of these pro-moieties, relevant cleavage mechanisms and recent advances in the design of phosphonate prodrugs.
Keywords: : drug discovery, medicinal chemistry, phosphonate, prodrug activation, prodrugs
Prodrugs have become increasingly common as replacements for drug candidates that have encountered obstacles during the development process. Since 2008, over 10% of new chemical entities approved by the US FDA have been prodrugs; more impressively, between 2014 and 2017, 17% of new chemical entities with FDA-approval have been prodrugs [1]. The strategy, despite being used for more than a century, was coined by Adrien Albert in 1958, and is defined as an inactive molecule undergoing some biological transformation in order to release the active metabolite [2]. In essence, use of a prodrug strategy enables a problematic molecule to overcome a biological obstacle, such as poor bioavailability, low absorption, instability, poor specificity, formulation difficulties or other adverse effects or toxicity concerns [3–5]. As drug development programs are fast approaching US$3 billion per FDA-approved drug, the consideration of a prodrug strategy early in development may be more efficient than attempting to save a problematic candidate later in the pipeline [6].
A key point of the prodrug strategy is that traditionally problematic functional groups are masked. One such example is the phosphonate: phosphonates often provide an opportunity for unique interactions with a target, but are characterized by a high negative charge and subsequent poor bioavailability [7]. The phosphonate group is often isosteric to a phosphate by replacing one ester oxygen of the phosphate with a carbon atom. This modification can lead to improved metabolic stability, as enzymes typically associated with cleaving an oxygen-phosphorus bond may not be as efficient at cleaving a carbon–phosphorus bond [8,9]. However, due to the charge of phosphonates at physiological pH, diffusion across biological membranes remains difficult (Figure 1) [10]. To remedy this, a variety of protecting groups exist for phosphonates [11]. Each strategy, however, must strike a balance between enabling sufficient absorption and cleavage of the moiety without generation of toxic byproducts.
Figure 1. . Permeation of a phosphonate prodrug and subsequent deprotection to release the active compound.

Phosphonate prodrugs can be classified according to substituents they include, most commonly esters and amides, and the substitution pattern they carry. Prodrugs of phosphonates may be mono- or di-substituted, and, if di-substituted, they can be symmetrical or asymmetrical (Table 1). When asymmetrically di-substituted, a new chiral center is introduced in the molecule at the phosphorus atom, possibly giving rise to slower cleavage of the protecting groups surrounding the phosphonate. In order to determine the optimal pattern of substitution, the reason for using the prodrug must be considered, as does the mechanism of how the protecting group(s) will be cleaved (Figure 2).
Table 1. . Phosphonate prodrugs and nomenclature.
| Symmetrical diesters | ||
|---|---|---|
| R’ = | ||
| Alkyl | -(CH2)nCH3 | |
| Aryl | -C6H5 | |
| Benzyl | -CH2C6H5 | |
| Acyloxyalkyl (POM) | -CH2OC(O)C(CH3)3 | |
| Alkoxycarbonyloxyalkyl (POC) | -CH2OC(O)OCH(CH3)2 | |
| S-Acylthioalkyl (SATE) | -CH2CH2SC(O)R | |
| Asymmetrical diesters | ||
| CycloSal |  |
|
| HepDirect | ||
| Monoesters | ||
| Internal monoester | 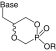 |
|
| Lipid conjugates | 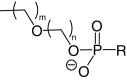 |
|
| Disulfide lipid conjugates | 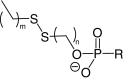 |
|
| Symmetrical bisamidates | ||
| Amino acid esters | 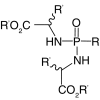 |
|
| Mixed amidate/ester | ||
| ProTides | 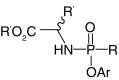 |
|
Figure 2. . Routes of activation of phosphonate prodrugs.

The expanding role of prodrugs in contemporary drug design and development.
Adapted with permission from [1] © Springer Nature (2018).
Phosphonate ester prodrugs
Phosphonate esters are a common prodrug strategy employed, with many examples having been prepared and evaluated for biological activity. A recent comprehensive review by Wiemer and Wiemer [9] thoroughly describes both modern and historical examples of varying prodrug approaches. While their review includes phosphates, this review will specifically focus on phosphonates, including recent examples.
Symmetrical diesters
In order to attain a neutral charge at biological pH, diester prodrugs may be the most obvious choice. Symmetrical esters induce no stereocenter. Thus, both esters are cleaved and biologically activated by the same enzyme or group of enzymes. While simple dialkyl esters of phosphonates have been synthesized, they have been reported as overly stable in mammalian systems [12–14]. In contrast, dibenzyl esters are more readily converted to the active drug, but with rates highly dependent upon the substituent pattern of the aryl ring, as unsubstituted benzyl esters cleave at an undesirably slow rate [13,15]. Because aryl esters are more easily hydrolyzed, they function more readily as prodrugs, with the ability to modify rates of hydrolysis by varying the substituent pattern [14].
More complex examples of phosphonate diesters have been evaluated and may be more advantageous prodrugs. Acyloxyalkyl esters are a general first prodrug for many new phosphonate containing compounds, since their first use to protect fosfomycin in 1969 (Figure 3) [7,16]. The success of this strategy is due in part to the improved stability of acyloxyalkyl phosphonates over their phosphate counterparts [7,17–19]. One such acyloxyalkyl prodrug is the pivaloyloxymethyl (POM) moiety. POM prodrugs are cleaved enzymatically to generate a hydroxymethyl intermediate, followed by spontaneous loss of formaldehyde to release the monoester or active compound (Figure 3) [17]. Nucleotide Adefovir dipivoxil, the di-POM prodrug of 9-[2-(phosphonomethoxy)ethyl]adenine (PMEA), was FDA-approved in 2002 and has been used to treat Hepatitis B through inhibition of reverse transcriptase [20]. The di-POM moiety was compared with a range of other prodrug esters, but was ultimately selected as it displayed the highest oral bioavailability in rats, more than twofold higher than free acid PMEA [21]. More recently, POM prodrugs were shown to increase cell permeability of anti-infective and antitumor agents, demonstrating the continued use of the POM moiety in phosphonate drug design [22–28]. Activities of POM prodrugs of fosmidomycin analogs increased activity in whole cell tuberculosis assays over 50-fold [22], while they also increased activity in whole cell Plasmodium falciparum assays over tenfold [23,24]. Further, concentrations of POM-protected bisphosphonates required to stimulate Vγ2Vδ2 T-cells were up to 1100-fold lower than that of their parent acids [25–28]. Toxic byproducts from the POM moiety, however, raise concern. Pivalic acid lowers carnitine levels and is toxic to small mammals at high concentrations [29,30], while formaldehyde is a known mutagen [31]. However, formaldehyde released through in vivo activation of a prodrug represents a small fraction of daily exposure to formaldehyde when compared with dietary sources and metabolic processes [32]. For these reasons, alternate protecting groups have recently received more attention.
Figure 3. . Activation and examples of acyloxyalkyl prodrugs.

Structurally related to the acyloxyalkyl prodrugs are carbonate esters, most commonly isopropyloxycarbonyloxymethyl (POC) protected phosphonates (Figure 4). Because the structures are related, the metabolic cleavage of POC groups is similar to that of other acyloxyalkyl prodrugs. After enzymatic cleavage of isopropanol, an unstable carboxylate intermediate spontaneously decomposes to lose CO2 and formaldehyde, releasing the mono-ester phosphonate and subsequently, the free phosphonate after a second enzymatic event (Figure 4) [33]. Tenofovir disoproxil fumarate (TDF), the di-POC prodrug of 9-[2-(phosphonomethoxy)propyl]adenine (PMPA), is a second nucleotide reverse transcriptase inhibitor. Alone, it is used to treat chronic Hepatitis B. In combination with other antiretrovirals, TDF is used to treat HIV/AIDS [34]. Use of the disoproxil fumarate prodrug over the free phosphonate results in increased oral absorption and tissue permeability of the drug [35]. TDF may also be used in combination as pre-exposure prophylaxis to prevent acquiring HIV and has undergone multiple clinical trials to be used in vaginal rings for the same purpose [36,37]. Similarly, addition of the POC moiety to an inhibitor of glutamate carboxypeptidase II resulted in a product that is orally bioavailable, with a >20-fold increase in total exposure when compared with an equimolar dose of its parent compound [38]. The enzyme is believed to play a key role in some neurological disorders [38].
Figure 4. . Activation and an example of alkoxycarbonyloxyalkyl prodrug.

Compared with acyloxyalkyl prodrugs, such as POM and POC, S-acylthioethyl (SATE) ester prodrugs are relatively new, as they were first reported in 1993 (Figure 5) [39]. They have also been used as prodrugs of phosphonate compounds. They take advantage of a similar cleavage mechanism to that of the POM and POC moieties, engaging esterases found in the blood and other tissues. After cleaving the terminal thioester to the thiol, the intermediate then decomposes to lose ethylene sulfide and liberate the mono-ester phosphonate [39]. The process repeats to release the active drug (Figure 5). An early example of the SATE moiety was its application to PMEA [40]. The authors found that the bis(tert-butyl-SATE)-PMEA analog retained similar activity to the bis(POM) compound in cells, but saw a half-life increase of over 50-fold in human serum, suggesting the bis(SATE) prodrug may have better in vivo efficacy [40]. Since that time, the SATE moiety has seen use in attempts to improve cell permeation of phosphonoformate and nucleosides [41,42]. Depending on the type of ester used in SATE prodrugs, toxicity concerns may arise. One such concern is the release of a thiol intermediate. However, it may be inferred through negative Ames and Comet assays of SATE phosphates that the released byproducts are not toxic [43]. As the bis(tert-butyl-SATE)-PMEA releases pivalic acid, the aforementioned concerns of carnitine depletion should be noted. Other esters, however, may generate less toxic byproducts and avoid the formaldehyde release seen in both POM and POC moieties.
Figure 5. . Activation and examples of S-acylthioethyl prodrugs.

Asymmetrical diesters
If the two esters of the phosphonate contain different substituents, an asymmetrical diester is formed. These phosphonate ester prodrugs have been employed with favorable results. In these compounds, the phosphorus atom becomes a center of chirality, potentially complicating synthetic routes. One strategy for making an asymmetrical ester is by initially masking the phosphonate using a salicyl alcohol (Figure 6). This strategy was first employed to improve cellular penetration of acylic nucleosides of phosphates, but later was used as a protection of PMEA [44]. Interestingly, the cycloSal analogs rely on aqueous hydrolysis as opposed to enzymatic cleavage [44], potentially enabling protected PMEA to permeate out of a cell before it is cleaved and reaches its target. The hydrolysis is pH dependent, and some analogs cleave more rapidly than others. The substitution pattern of the salicyl alcohol can influence the hydrolysis rate, allowing for fine-tuning of the prodrug’s properties (Figure 6). Despite having less toxic byproducts, the cycloSal prodrugs of PMEA displayed lower activities than those of bis(POM)-PMEA, but a twofold increase over the PMEA phosphonic acid. While the phosphorus atom is a center of chirality, the cycloSal-PMEA enantiomers were not tested separately for activity, but as a racemic mixture, leaving room for future work to synthesize and test the enantiomers. It has been shown in other systems that different phosphorus enantiomers with cycloSal moieties demonstrate varying levels of activity, with reported three to 80-fold differences between enantiomers in cell-based assays [45].
Figure 6. . Activation and an example of a cycloSal prodrug.

A variation of the cycloSal prodrug concept utilizes DNA bases rather than a salicyl alcohol [46]. This modification results in prodrugs known as cyclic nucleoside phosphonates (CNPs, Figure 7). Cidofovir (((S)-1-(3-hydroxy-2-phosphonomethoxypropyl) cytosine, (S)-HPMPC), is a potent inhibitor of various dsDNA viruses and has been US FDA approved for the treatment of cytomegalovirus in patients with AIDS, though nephrotoxic [46]. Cyclic cidofovir (cHPMPC) is less cytotoxic when administered to rats, rabbits and monkeys orally [47], but, like unprotected cidofovir, lacks sufficient oral bioavailability. Where the CNPs struggle, however, is cell penetration due to the single free hydroxyl remaining negatively charged at physiological pH. Prodrugs of cHPMPC and other CNPs, such as a cyclic (S)-9-[3-hydroxy-(2-phosphonomethoxy)propyl]adenosine (cHPMPA), mask the hydroxyl group using a variety of protection strategies (Figure 7). These compounds demonstrate remarkable increases in activity and bioavailability over their parent phosphonates against a range of viral infections [48–52]. Peptidomimetics have shown promise, as they are more readily bioavailable over a single amino acid protecting group, up to eight-times higher than their parent phosphonate when administered to rats orally [48–50]. Other protection strategies have included POM groups, alkoxyalkyl lipid-like chains and alkyl esters, with activity depending on the length of chains for these features [51,52].
Figure 7. . Cyclic cidofovir and prodrugs thereof.

Another approach to developing asymmetric phosphonate ester prodrugs is the HepDirect strategy(Figure 8) [53]. By protecting the phosphonate with a chiral diol, the phosphorus atom is now also a center of chirality. However, unlike cycloSal prodrugs requiring aqueous cleavage or the aforementioned diesters that may be cleaved prior to cell penetration, HepDirect prodrugs are designed to be activated within hepatocytes in order to reach their intended target [53]. To achieve this goal, Erion et al. set out to synthesize molecules that would be activated by enzymes expressed in liver tissue, survive aqueous solution, blood and nonhepatic tissues, and release no toxic byproducts [53]. After oxidation of the benzylic position via a cytochrome P450 isozyme (CYP3A4), the resulting hemiketal spontaneously opens and liberates the active phosphonate and an enone. While a Michael acceptor may prove problematic, Erion et al. suggest that glutathione present in cells is conjugated to the enone, rather than releasing a potential toxic byproduct [53]. As with other diesters, the HepDirect strategy has been applied to PMEA and other phosphonates. Pradefovir, the HepDirect version of PMEA, was synthesized and subsequently found to demonstrate a 12-fold improvement in levels of PMEA in the liver/kidney over POM prodrug adefovir dipivoxil [54]. This is important to note, because adefovir dipivoxil is only approved for treatment at a suboptimal dose due to its renal toxicity [20]. Pradefovir, though, was put on hold for further clinical testing after increased tumor incidence was observed in animal studies [55], but has recently moved forward with safety studies in healthy human subjects [56]. The HepDirect prodrug strategy has also been employed for a thyroid receptor agonist in order to lower cholesterol by increasing oral bioavailability in rats 40-fold [57,58] and as a means to overcome inhibition of CYP3A4 in a molecule targeting glucose production seen with the use of other pro-moieties [59].
Figure 8. . Activation and examples of HepDirect prodrugs.

Monoesters
As shown by several examples above, smaller lipophilic diesters mask both negative charges in a phosphonate, and are effective prodrugs. More recently, monoesters, masking only one anion of the phosphonate, have been reported [60]. These compounds, bearing long hydrocarbon chain, lipid-like substituents, have also shown to work well as prodrugs (Figure 9). Although first designed for use in nucleotide monophosphates, the strategy has also been applied to acyclic nucleoside phosphonates (ANPs). Hostetler et al. have synthesized several lipid-like nucleotide and nucleoside phosphonates in an attempt to increase efficacy against HIV [60]. Specifically, some of these lipid esters were significantly more active against their respective targets relative to the free phosphonate. In some cases, the activity improved by multiple log units. A hexadecyloxypropyl (HDP) prodrug of (S)-9-[3-hydroxy-(2-phosphonomethoxy)propyl]adenosine (HDP-(S)-HPMPA) displayed >10,000-fold increase in activity against HIV-1 [60], The same compound showed >5000-fold improvement in activity against the orf virus [61]. The HDP prodrug of cidofovir (HDP-CDV) showed >2000-fold improvement in activity against Epstein-Barr virus (Figure 9) [62]. These gains in activity are attributed to the increased intracellular concentrations of the drugs, thanks to the HDP lipid moiety.
Figure 9. . Examples of lipid conjugate prodrugs.

HDP-tenofovir (CMX157) has also been synthesized, originally with the intent of treating HIV and Hepatitis B [63]. It has been tested in combination with each US FDA-approved antiviral used for the treatment of HIV and showed synergistic or additive behavior [64]. Since it was first reported, CMX157 has advanced through a Phase II clinical trial in which it was tested for tolerated dosing in Hepatitis B patients. Because CMX157 is cleaved intracellularly by phospholipase C and/or sphingomyelinase, the nephrotoxicity seen by other tenofovir prodrugs cleaved in plasma may be decreased, and with the aforementioned additive or synergistic behavior, CMX157 may be an intelligent choice to pair with currently approved antivirals to move forward through Phase III studies.
Brincidofovir (CMX001) [65] is an HDP prodrug of cidofovir. CMX001 has excellent activity against a range of viruses including: poxvirus [66–69], cytomegalovirus [70,71] and herpesvirus [62,72], and has been reviewed for its treatment of varying dsDNA viruses [73,74]. Brincidofovir has and is currently undergoing clinical trials in the treatment of adenovirus in pediatric stem cell transplant recipients [75–77]. Krečmerová et al. have synthesized varying alkyloxyalkyl monoester prodrugs that have shown activity against dsDNA viruses, as well, demonstrating potency up to 600-fold higher than that of the parent phosphonate [52]. In attempt to synthesize reduction sensitive lipid prodrugs of tenofovir, monoester disulfide conjugates have been synthesized [78,79]. Multiple linking strategies connecting the phosphonate and disulfide moieties were studied, with a butyl linker improving both the measured activity and therapeutic index of the disulfide prodrug tenfold over TDF when tested against HIV-1, though further studies may be needed to elucidate why the disulfide bond may be advantageous over the alkoxyalkyl lipids.
Phosphonate amidate prodrugs
Phosphonates masked with amidate moieties are a newer concept in the field of phosphonate prodrugs, as the first reported phosphoramidate was reported in 1990 [80]. However, amidates have advantages compared with their ester prodrug cousins, as the prodrug moieties for these compounds are commonly amino acids, generating less problematic byproducts than some of the aforementioned esters. Since 1990, significant advances have been made in phosphor- and phosphon-amidate analogs, and it is important to note the only US FDA-approved phosphonate prodrug since 2007, tenofovir alafenamide, includes an amidate moiety [1,81].
Symmetrical bisamidates
Although more examples of bisamidates have been reported to protect phosphates, this substituent family has found use with respect to phosphonates. Like phosphonate diesters, the simplest amidate prodrugs would be symmetrical in order to avoid introduction of a stereogenic phosphorus. However, because protection by amino acids still allows for a double negative charge, the amino acids are typically esterified to remain neutral. Due to this esterification, activation of the prodrug is a multistep process. An esterase first cleaves a carboxyester. The free carboxylate cyclizes with the phosphonate, liberating the remaining amino acid ester (Figure 10). Water opens the intermediate ring, allowing a phosphoramidase to cleave the final amino acid from the phosphonate [7]. While the cyclic intermediates are unstable, they have recently been characterized for the first time [82].
Figure 10. . Activation and examples of bisamidate prodrugs.

AA: Amino acid.
One bisamidate to advance to clinical trials is CS-917, a bis-alanine ethyl ester-protected phosphonate, which is an inhibitor of fructose 1,6-bisphosphatase [83]. In treating Type 2 diabetes, CS-917 was shown to have a tenfold higher bioavailability than the free phosphonic acid [84]. The compound was ultimately withdrawn when it failed to significantly reduce long-term glucose exposure [85].
In recent years, phosphonodiamidates have received more attention with respect to acyclic nucleosides. They have been used to target Bordetella pertussis adenylate cyclase toxin (ACT) [86]. In targeting ACT, multiple PMEA bisamidates or PMEA analog bisamidates were prepared [86–88]. Despite showing weaker activity than bis(POM)PMEA in cell-based assays, the bisamidates were less cytotoxic and more stable, allowing room for further optimization and developing a better overall profile than bis(POM)PMEA. Bisamidates have also been used to target phosphoribosyltransferases in Mycobacterium tuberculosis and P. falciparum [89,90]. Through synthesizing various bisamidate purine base ANPs, it was found the prodrugs have low cytotoxicity and improved permeability over the free phosphonic acid analogs, furthering their potential as antimalarials [91,92]. Like bisamidates, monoamidates induce no stereocenter at the phosphorus atom through the delocalization of the negative charge. However, monoamidates have received little attention as prodrugs of phosphates and even less with respect to phosphonates, leaving room for exploration in this area.
Mixed amidate/esters
While bisamidates as prodrugs for phosphonates have received considerable attention, mixed amidate/ester prodrugs of phosphonates have received much more (Figure 11). Like asymmetrical diester prodrugs, the mixed substitution pattern introduces a stereogenic center at the phosphorus atom. Also like other phosphonate prodrugs, this strategy was first applied to phosphates to overcome poor cellular uptake, poor conversion to the active component or to release less toxic byproducts [80]. In 2001, the first mixed amidate/ester prodrugs of PMEA and PMPA were reported [93]. It was found the L-alanine methyl ester paired with a phenoxy moiety worked best in combination, while the D-alanine methyl ester resulted in extremely reduced activity. Since this discovery, aryloxy phosphonamidate prodrugs have come to be known as the ‘ProTide’ (pro-nucleotide) [94] technology and have resulted in two US FDA-approved drugs: tenofovir alafenamide for the treatment of HIV (ProTide of PMPA) [81] and sofosbuvir for the treatment of Hepatitis C (Figure 11) [95]. The ProTide technology has been further applied to other areas of interest with favorable results in phosphates, likely serving as precursors to phosphonate analogs [96–105]. The technology has also been extensively reviewed in recent years and is fast becoming a favored approach for nucleotide prodrugs [1,9,106–112].
Figure 11. . Activation and examples of ProTides.

Current & future applications
Whereas phosphate prodrugs have been explored for decades, phosphonates have received increased attention only more recently. Phosphonate prodrug analogs have been prepared to potentially overcome a pharmacokinetic barrier their parent phosphoric or phosphonic acids could not. Innovation of the ProTide technology has expanded the field of phosphonate prodrugs with relation to target and lowered toxicity found with other moieties. What follows is an update on two phosphonate prodrug strategies: as inhibitors of isoprenoid biosynthesis and newer compounds as antiviral agents.
Isoprenoid biosynthesis
Isoprenoids are a class of molecules derived from two five-carbon building blocks, isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) [113]. Thousands of isoprenoids have been identified to date, of which some notable examples are chlorophyll, carotenoids and ubiquinones [114]. Isoprenoids are vital for biological function of an organism; however, humans and certain types of bacteria and protozoa biosynthetically obtain IPP and DMAPP differently. Humans obtain isoprenoids via the mevalonate pathway (MVA) while Gram-negative bacteria and some protozoa utilize the 2C-methyl-D-erythritol 4-phosphate (MEP) pathway.
Because some bacteria obtain isoprenoids in a different manner than humans, the enzymes involved in the MEP pathway serve as attractive targets for inhibition. Two natural product phosphonates, fosmidomycin and its acetyl analog FR900098, have demonstrated inhibition of the MEP pathway. Because both fosmidomycin and FR900098 are free phosphonic acids, prodrugs of each have been synthesized in attempt to increase the oral bioavailability of the active moiety. We and others have performed SAR optimization and synthesized prodrugs of analogs to target M. tuberculosis and P. falciparum [23,115–123]. An α,β-unsaturated bis(POM) analog of fosmidomycin displays potent in vivo activity against P. falciparum (Figure 12) [124]. Other prodrugs have been explored in relation to M. tuberculosis and Gram-negative bacteria with varying results, suggesting the use of prodrugs may be organism dependent [125].
Figure 12. . Inhibitors of the 2C-methyl-D-erythritol 4-phosphate pathway.

While the MEP is an attractive target for infective pathogens, the MVA may be targeted with regards to human diseases. Geranylgeranyl diphosphate synthase and farnesyl diphosphate synthase (FDPS) are attractive targets, as they are essential for growth and metastasis of multiple cancers [126]. Nitrogenous bisphosphonates, structural analogs of natural diphosphates, are inhibitors of FDPS. However, in order to effectively permeate cells, the negative charge of both phosphonate moieties needs to be masked to some degree. Simple alkyl and aryl esters having been tested [127], but the more labile POM prodrugs have yielded better activity over the free nitrogenous or non-nitrogenous bisphosphonate (Figure 13) [128–130]. While the increase in activity varied between compounds and their respective prodrugs, in general, the trend displayed an increase when the charge was masked, displaying the anticancer potential these compounds exhibit. Efforts have continued in optimizing the substituents of the α-carbon, as well. In myeloma cells, bisphosphonates with isoprenoid substituents at the α-carbon and as one of the phosphonate esters have been synthesized as tris(POM) prodrugs, demonstrating submicromolar activity [131]. As the prodrugs had been tested as diastereomeric mixtures, further work in exploring which isomer(s) may be responsible for activity are ongoing. Bisphosphonates have also received attention in the stimulation of Vγ2Vδ2 T cells and inhibiting tumor cell growth [28]. Through inhibition of FDPS, IPP levels will increase and subsequently stimulate Vγ2Vδ2 T cells through the Vγ2Vδ2 T cell receptors. Tetrakis-pivaloyloxymethyl 2-(thiazole-2-ylamino)ethylidene-1,1-bisphosphonate demonstrated remarkable abilities to stimulate the Vγ2Vδ2 T cells from 81- to 1900-fold more than its free acid analog across 21 tumor cell lines, signifying the potential use of bisphosphonate prodrugs as aids in cancer immunotherapy. Further, this compound stimulates Vγ2Vδ2 T cells more effectively compared with zoledronic acid, the most commonly used stimulant in clinical trials [132].
Figure 13. . Anticancer inhibitors of the mevalonate pathway.

Nucleotides & nucleosides
As mentioned previously, concerns for toxicity of metabolic byproducts of a POM or POC prodrug beg for investment into other moieties. Adefovir dipivoxil and TDF are both still in use today, along with multiple groups exploring options to both increase activity and reduce toxicity of their metabolic byproducts by exploring the use of other prodrug moieties. In fact, tenofovir alafenamide, which was approved by the US FDA in 2015 for the treatment of HIV-1, is a testament to the advances of ProTide technology [81,133]. Further clinical trials have been performed with tenofovir alafenamide in attempt to replace TDF, mostly due to the toxicity concerns of the byproducts of long-term use of TDF, specifically in bone and renal tissue [134–137]. In fact, a Phase IIIb study found no inferiority in regards to tenofovir alafenamide replacing TDF in a regimen with rilpivirine and emtricitabine in virally suppressed adults [138]. Tenofovir alafenamide also has shown lower mitochondrial toxicity in T cells when compared with TDF [139]. Because of the toxicity associated with long-term use of TDF, less toxic substitutes are continuously being explored. Mono-protected disulfide lipid conjugates of tenofovir have also shown promise in treating HIV-1 (Figure 14) [78,79]. Other ProTide analogs of TDF and adefovir dipivoxil have also demonstrated better inhibition of HIV and Hepatitis B [99,104]. Despite the demonstration of other prodrugs for tenofovir demonstrating lower toxicity, TDF has still seen further interest in clinical trials and has been tested in vaginal rings as a form of pre-exposure prophylaxis, displaying concentrations higher than needed to inhibit HIV [37]. So, while long-term use of TDF may prove toxic through ingestion, pre-exposure prophylaxis through other means may provide for less systemic side effects.
Figure 14. . Recent examples of nucleoside prodrugs.

Though approved by the US FDA for the treatment of Hepatitis B, adefovir dipivoxil suffers from a similar detriment as TDF – the potentially toxic metabolic byproducts. Not only this, but the majority of orally administered adefovir is hydrolyzed to free PMEA in the gastrointestinal tract and is found in the kidneys rather than in circulation, resulting in nephrotoxicity [140]. Various prodrug moieties have been used to mask PMEA in an effort to increase transport to the liver as opposed to hydrolysis while traversing the gastrointestinal tract. A bis(2,2,2-trifluoroethyl)PMEA analog was demonstrated to be at least fourfold more stable in rat plasma in comparison with adefovir dipivoxil (Figure 14), suggesting protecting groups aimed to survive the gastrointestinal tract and deliver to the liver warrant more attention [141].
Recent antiviral advances indicate that phosphonate nucleoside prodrugs show promise outside the realm of HIV/Hepatitis B. Bisamidate L-α-2′-deoxythreosyl prodrugs display a 200-fold increase in potency over their phosphonic acid parent molecule against both HIV-1 and HIV-2 (Figure 14) [142]. Prodrugs of ANPs with purine bases have demonstrated potent inhibition of phosphoribosyltransferases in malaria and tuberculosis [89,90,92,143], with recent advances demonstrating excellent selectivity for the parasitic enzyme over the human homolog [144]. Advances have also been seen with regards to phosphonate prodrugs used in treating DNA viruses, such as Epstein-Barr virus, Herpes virus and cytomegalovirus [52,145]. An N-alkyl tyrosinamide phosphonate ester prodrug has even been shown in vivo to protect against human adenoviruses, which otherwise have no FDA-approved drug specific for this activity (Figure 14) [146]. Further research into replacing phosphate prodrugs with phosphonates is warranted [1].
Future perspective
Over the last decade, prodrugs have accounted for more than 10% of newly approved chemical entities from the US FDA. This demonstrates the need to consider the use of a prodrug prior to clinical evaluation, especially in the case of the traditionally problematic phosphonic acid, in order to overcome the double negative charge at physiological pH. As time has progressed, it has now become almost commonplace to test prodrugs of phosphonates prior to clinical evaluation, due to most prodrugs seeing better activity and availability over their parent acid. This does not come without limits, however. The choice of the pro-moiety is important, both because of where the drug is intended to release and what metabolic byproducts are freed from the protecting group. Despite toxicity concerns from some types of prodrugs, they still see use today, both clinically and at the bench. With the innovative ProTide technology and discovery of other nontoxic or less toxic pro-moieties, it would be wise to delve into testing safer metabolite based pro-moieties on previously potent or clinically approved compounds, as has been seen for TDF. Further, it is important to note the mixed amidate/ester approach has received little attention in phosphonates outside nucleotides and may warrant more focus in this arena, especially due to the success seen with tenofovir alafenamide. With the growing interest in phosphonate analogs of phosphates and concerns for developing safer metabolic byproducts, coupled with the increasing amount of prodrugs in clinical trials and receiving US FDA approval, the potential uses for phosphonate prodrugs will surely remain of interest for years to come.
Executive summary.
Phosphonates, at biological pH, exist as a dianion and have trouble penetrating into cells.
There are many protection strategies and subsequent activation mechanisms that have been developed to deliver phosphonate compounds to their target.
Not all protection strategies work in every situation, so a variety of acyloxyalkyl, amidate and mixed ester/amidate strategies have been developed.
The ProTide technology is a more recent development and has resulted in the only US FDA-approved phosphonate prodrug in the past 10 years.
Footnotes
Financial & competing interests disclosure
We are grateful for support from the NIH (grant number NIAID R01 AI123433) and the George Washington University Department of Chemistry. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Rautio J, Meanwell NA, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 17(8), 559–587 (2018). [DOI] [PubMed] [Google Scholar]; •• Prodrugs exist for a variety of functional groups and this article explains in great detail the importance of prodrugs and their expanding role in drug development.
- 2.Albert A. Chemical aspects of selective toxicity. Nature, 182, 421–422 (1958). [DOI] [PubMed] [Google Scholar]
- 3.Wu K. A new classification of prodrugs: regulatory perspectives. Pharmaceuticals 2, 77–81 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buxton I, Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, Metabolism, and Elimination in Goodman & Gilman’s: The Pharmacological Basis of Therapeutics. Brunton L, Hilal-Dandan R, Knollman B (). McGraw-Hill, NY, USA: (2018). [Google Scholar]
- 5.Goldstein A, Aronow L, Kalman S (). Chapter 13: Drug Development in Principles of Drug Action. John Wiley & Sons, NJ, USA: (1974). [Google Scholar]
- 6.DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J. Health Econ. 47, 20–33 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Hecker SJ, Erion MD. Prodrugs of phosphates and phosphonates. J. Med. Chem. 51(8), 2328–2345 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Engel R. Phosphonates as analogues of natural phosphates. Chem. Rev. 77, 349–367 (1977). [Google Scholar]
- 9.Wiemer AJ, Wiemer DF. Prodrugs of phosphonates and phosphates: crossing the membrane barrier. Top. Curr. Chem., 360, 115–160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Wiemer and Wiemer excellently review not only the history and recent advances within phosphonate and phosphate prodrugs, but also include a thorough description of factors to consider when choosing a pro-moiety and how the active compound would be released.
- 10.Kornberg RD, McNamee MG, McConnell HM. Measurement of transmembrane potentials in phospholipid vesicles. Proc. Natl Acad. Sci. USA, 69(6), 1508–1513 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wuts PGM. (). Protection for the Phosphate Group in Greene's Protective Groups in Organic Synthesis. Wiley, NJ, USA: (2014). [Google Scholar]
- 12.Niemi R, Turhanen P, Vepsalainen J, Taipale H, Jarvinen T. Bisphosphonate prodrugs: synthesis and in vitro evaluation of alkyl and acyloxymethyl esters of etidronic acid as bioreversible prodrugs of etidronate. Eur. J. Pharm. Sci. 11(2), 173–180 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Serafinowska HT, Ashton RJ, Bailey S, Harnden MR, Jackson SM, Sutton D. Synthesis and in vivo evaluation of prodrugs of 9-2-(phosphonomethoxy)ethoxy adenine. J. Med. Chem. 38(8), 1372–1379 (1995). [DOI] [PubMed] [Google Scholar]
- 14.Delombaert S, Erion MD, Tan J. et al. N-phosphonomethyl dipeptides and their phosphonate prodrugs, a new-generation of neutral endopeptidase (NEP, EC-3.4.24.11) inhibitors. J. Med. Chem. 37(4), 498–511 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Dang Q, Liu Y, Rydzewski RM. et al. Bis (para-methoxy)benzyl phosphonate prodrugs with improved stability and enhanced cell penetration. Bioorg. Med. Chem. Lett. 17(12), 3412–3416 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Christensen BG, Albers-Schonberg G, Leanza WJ. Antibiotic (-)-cis-(1,2-Epoxypropyl)phosphonates. DE1805682 Merck, Germany: (1973). [Google Scholar]
- 17.Schultz C. Prodrugs of biologically active phosphate esters. Bioorg. Med. Chem. Lett. 11(6), 885–898 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Farquhar D, Srivastva DN, Kuttesch NJ, Saunders PP. Biologically reversible phosphate-protective groups. J. Pharm. Sci. 72(3), 324–325 (1983). [DOI] [PubMed] [Google Scholar]
- 19.Iyer RP, Phillips LR, Biddle JA. et al. Synthesis of acyloxyanlkyl acylphosphonates as potential prodrugs of the antiviral, trisodium phosphonoformate (foscarnet sodium). Tet. Lett. 30(51), 7141–7144 (1989). [Google Scholar]
- 20.Marcellin P, Chang T, Lim SG. et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. New Eng. J. Med. 348(9), 808–816 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Starrett JE, Tortolani DR, Russell J. et al. Synthesis, oral bioavailability determination, and in-vitro evaluation of prodrugs of the antiviral agent 9-2-(phosphonomethoxy)ethyl adenine (PMEA). J. Med. Chem. 37(12), 1857–1864 (1994). [DOI] [PubMed] [Google Scholar]
- 22.Jackson ER, San Jose G, Brothers RC. et al. The effect of chain length and unsaturation on Mtb Dxr inhibition and antitubercular killing activity of FR900098 analogs. Bioorg. Med. Chem. Lett. 24(2), 649–653 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruecher K, Graewert T, Konzuch S. et al. Prodrugs of reverse fosmidomycin analogues. J. Med. Chem. 58(4), 2025–2035 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Faisca Phillips AM, Nogueira F, Murtinheira F, Barros MT. Synthesis and antimalarial evaluation of prodrugs of novel fosmidomycin analogues. Bioorg. Med. Chem. Lett. 25(10), 2112–2116 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto K, Hayashi K, Murata-Hirai K. et al. Targeting cancer cells with a bisphosphonate prodrug. Chemmedchem 11(24), 2656–2663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kilcollins AM, Li J, Hsiao C-HC, Wiemer AJ. HMBPP analog prodrugs bypass energy-dependent uptake to promote efficient BTN3A1-mediated malignant cell lysis by V gamma 9 V delta 2 T lymphocyte effectors. J. Immunol. 197(2), 419–428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shippy RR, Lin X, Agabiti SS. et al. Phosphinophosphonates and their tris-pivaloyloxymethyl prodrugs reveal a negatively cooperative butyrophilin activation mechanism. J. Med. Chem. 60(6), 2373–2382 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka Y, Iwasaki M, Murata-Hirai K. et al. Anti-tumor activity and immunotherapeutic potential of a bisphosphonate prodrug. Sci. Rep. 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brass EP. Pivalate-generating prodrugs and carnitine homeostasis in man. Pharmacol. Rev. 54(4), 589–598 (2002). [DOI] [PubMed] [Google Scholar]
- 30.The National Institute for Occupational Safety and Health Registry of Toxic Effects of Chemical Substances: pivalic acid. (2014). https://www.cdc.gov/niosh-rtecs/TO757E20.html
- 31.The National Institute for Occupational Safety and Health Registry of Toxic Effects of Chemical Substances: formaldehyde. https://www.cdc.gov/niosh-rtecs/LP882F48.html (2014)
- 32.Dhareshwar SS, Stella VJ. Your prodrug releases formaldehyde: should you be concerned? No! J. Pharm. Sci. 97(10), 4184–4193 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Yuan LC, Dahl TC, Oliyai R. Degradation kinetics of oxycarbonyloxymethyl prodrugs of phosphonates in solution. Pharm. Res. 18(2), 234–237 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Martin P, Lau DTY, Nguyen MH. et al. A treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2015 update. Clin. Gastroenterol. Hepatol. 13(12), 2071–2087 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Durand-Gasselin L, Van Rompay KKA, Vela JE. et al. Nucleotide analogue prodrug tenofovir disoproxil enhances lymphoid cell loading following oral administration in monkeys. Mol. Pharm. 6(4), 1145–1151 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehman DA, Baeten JM, McCoy CO. et al. Risk of drug resistance among persons acquiring hiv within a randomized clinical trial of single- or dual-agent preexposure prophylaxis. J. Infect. Dis. 211(8), 1211–1218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keller MJ, Mesquita PM, Marzinke MA. et al. A Phase I randomized placebo-controlled safety and pharmacokinetic trial of a tenofovir disoproxil fumarate vaginal ring. AIDS 30(5), 743–751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majer P, Jancarik A, Krecmerova M. et al. Discovery of Orally Available Prodrugs of the Glutamate Carboxypeptidase II (GCPII) Inhibitor 2-Phosphonomethylpentanedioic Acid (2-PMPA). J. Med. Chem. 59(6), 2810–2819 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Perigaud C, Gosselin G, Lefebvre I. et al. Rational design for cytosolic delivery of nucleoside monophosphate - SATE and DTE as enzyme-labile transient phosphate protecting groups. Bioorg. Med. Chem. Lett. 3(12), 2521–2526 (1993). [Google Scholar]
- 40.Benzaria S, Pelicano H, Johnson R. et al. Synthesis, in vitro antiviral evaluation, and stability studies of bis(S-acyl-2-thioethyl) ester derivatives of 9- 2-(phosphonomethoxy)ethyl adenine (PMEA) as potential PMEA prodrugs with improved oral bioavailability. J. Med. Chem. 39(25), 4958–4965 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Briggs AD, Camplo M, Freeman S, Lundstrom J, Pring BG. S-Acylthioethyl prodrugs of phosphonoformate. Eur. J. Pharm. Sci. 5(4), 199–208 (1997). [Google Scholar]
- 42.Oh CH, Liu LJ, Hong JH. Design and synthesis of dually branched 5'-norcarbocyclic adenosine phosphonodiester analogue as a new anti-HIV prodrug. Nucleosides Nucleotides Nucleic Acids 29(10), 721–733 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Placidi L, De Meo M, Gosselin G. et al. Evaluation of the mutagenic and genotoxic activities of anti-hepatitis B analogs of beta-L-adenosine by the Ames test and the Comet assay. Antivir. Res. 50(2), 139–145 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Meier C, Gorbig U, Muller C, Balzarini J. cycloSal-PMEA and cycloAmb-PMEA: potentially new phosphonate prodrugs based on the cycloSal-pronucleotide approach. J. Med. Chem. 48(25), 8079–8086 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Meier C, Lorey M, De Clercq E, Balzarini J. cycloSal-2′, 3′-dideoxy-2′,3′-didehydrothymidine monophosphate (cycloSal-d4TMP): synthesis and antiviral evaluation of a new d4TMP delivery system. J. Med. Chem. 41(9), 1417–1427 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Lebeau I, Andrei G, Dal Pozzo F. et al. Activities of alkoxvalkyl esters of cidofovir (CDV), cyclic CDV, and (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine against orthopoxviruses in cell monolayers and in organotypic cultures. Antimicrob. Agents Chemother. 50(7), 2525–2529 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bischofberger N, Hitchcock MJM, Chen MS. et al. 1-((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl cytosine, an intracellular prodrug for (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine with improved therapeutic index in vivo. Antimicrob. Agents Chemother. 38(10), 2387–2391 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eriksson U, Peterson LW, Kashemirov BA. et al. Serine peptide phosphoester prodrugs of cyclic cidofovir: synthesis, transport, and antiviral activity. Mol. Pharm. 5(4), 598–609 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peterson LW, Sala-Rabanal M, Krylov IS, Serpi M, Kashemirov BA, McKenna CE. Serine side chain-linked peptidomimetic conjugates of cyclic HPMPC and HPMPA: synthesis and interaction with hPEPT1. Mol. Pharm. 7(6), 2349–2361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zalcharova VM, Serpi M, Krylov IS. et al. Tyrosine-based 1-(S) 3-Hydroxy-2-(phosphonomethoxy)propyl cytosine and -adenine ((S)-HPMPC and (S)-HPMPA) prodrugs: synthesis, stability, antiviral activity, and in vivo transport studies. J. Med. Chem. 54(16), 5680–5693 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keith KA, Wan WB, Ciesla SL, Beadle JR, Hostetler KY, Kern ER. Inhibitory activity of alkoxyalkyl and alkyl esters of cidofovir and cyclic cidofovir against orthopoxvirus replication in vitro. Antimicrob. Agents Chemother. 48(5), 1869–1871 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krecmerova M, Holy A, Andrei G. et al. Synthesis of Ester Prodrugs of 9-(S)- 3-Hydroxy-2-(phosphonomethoxy)propyl -2,6-diaminopurine (HPMPDAP) as anti-poxvirus agents. J. Med. Chem. 53(19), 6825–6837 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Erion MD, Reddy KR, Boyer SH. et al. Design, synthesis, and characterization of a series of cytochrome P-450 3A-activated prodrugs (HepDirect prodrugs) useful for targeting phosph(on)ate-based drugs to the liver. J. Am. Chem. Soc. 126(16), 5154–5163 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Reddy KR, Matelich MC, Ugarkar BG. et al. Pradefovir: a prodrug that targets adefovir to the liver for the treatment of hepatitis B. J. Med. Chem. 51(3), 666–676 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Tillmann HL. Pradefovir, a liver-targeted prodrug of adefovir against HBV infection. Curr. Opin. Investig. Drugs. 8(8), 682–690 (2007). [PubMed] [Google Scholar]
- 56.Ding Y, Zhang H, Li X. et al. Safety, pharmacokinetics and pharmacogenetics of a single ascending dose of pradefovir, a novel liver-targeting, anti-hepatitis B virus drug, in healthy Chinese subjects. Hepatol. Int. 11(4), 390–400 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Erion MD, Cable EE, Ito BR. et al. Targeting thyroid hormone receptor-beta agonists to the liver reduces cholesterol and triglycerides and improves the therapeutic index. Proc. Natl Acad. Sci. USA 104(39), 15490–15495 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boyer SH, Jiang H, Jacintho JD. et al. Synthesis and biological evaluation of a series of liver-selective phosphonic acid thyroid hormone receptor agonists and their prodrugs. J. Med. Chem. 51(22), 7075–7093 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Tsukada T, Tamaki K, Tanaka J. et al. A prodrug approach towards the development of tricyclic-based FBPase inhibitors. Bioorg. Med. Chem. Lett. 20(9), 2938–2941 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Hostetler KY, Aldern KA, Wan WB, Ciesla SL, Beadle JR. Alkoxyakl esters of (S)-9- 3-Hydroxy-2-(phosphonomethoxy)propyl adenine are potent inhibitors of the replication of wild-type and drug-resistant human immunodeficiency virus type 1 in vitro. Antimicrob. Agents Chemother. 50(8), 2857–2859 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dal Pozzo F, Andrei G, Lebeau I. et al. In vitro evaluation of the anti-orf virus activity of alkoxyalkyl esters of CDV, cCDV and (S)-HPMPA. Antivir. Res. 75(1), 52–57 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Williams-Aziz SL, Hartline CB, Harden EA. et al. Comparative activities of lipid esters of cidofovir and cyclic cidofovir against replication of herpesviruses in vitro. Antimicrob. Agents Chemother. 49(9), 3724–3733 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Painter GR, Almond MR, Trost LC. et al. Evaluation of hexadecyloxypropyl-9-R- 2-(phosphonomethoxy)propyl adenine, CMX157, as a potential treatment for human immunodeficiency virus type 1 and hepatitis B virus infections. Antimicrob. Agents Chemother. 51(10), 3505–3509 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lanier ER, Ptak RG, Lampert BM. et al. Development of hexadecyloxypropyl tenofovir (CMX157) for treatment of infection caused by wild-type and nucleoside/nucleotide-resistant HIV. Antimicrob. Agents Chemother. 54(7), 2901–2909 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marty FM, Winston DJ, Rowley SD. et al. CMX001 to prevent cytomegalovirus disease in hematopoietic-cell transplantation. New Eng. J. Med. 369(13), 1227–1236 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Kern ER, Hartline C, Harden E. et al. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob. Agents Chemother. 46(4), 991–995 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. Increased antiviral activity of 1-O-hexadecyloxypropyl- 2-C-14 cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol. Pharm. 63(3), 678–681 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology 318(2), 474–481 (2004). [DOI] [PubMed] [Google Scholar]
- 69.Quenelle DC, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Kern ER. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob. Agents Chemother. 48(2), 404–412 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kern ER, Collins DJ, Wan WB, Beadle JR, Hostetler KY, Quenelle DC. Oral treatment of murine cytomegalovirus infections with ether lipid esters of cidofovir. Antimicrob. Agents Chemother. 48(9), 3516–3522 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wan WB, Beadle JR, Hartline C. et al. Comparison of the antiviral activities of alkoxyalkyl and alkyl esters of cidofovir against human and murine cytomegalovirus replication in vitro. Antimicrob. Agents Chemother. 49(2), 656–662 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beadle JR, Hartline C, Aldern KA. et al. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob. Agents Chemother. 46(8), 2381–2386 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hostetler KY. Synthesis and early development of hexadecyloxypropyl-cidofovir: an oral antipoxvirus nucleoside phosphonate. Viruses 2(10), 2213–2225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Florescu DF, Keck MA. Development of CMX001 (Brincidofovir) for the treatment of serious diseases or conditions caused by dsDNA viruses. Expert Rev. Anti Infect. Ther. 12(10), 1171–1178 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Grimley MS, Chemaly RF, Englund JA. et al. Brincidofovir for asymptomatic adenovirus viremia in pediatric and adult allogeneic hematopoietic cell transplant recipients: a randomized placebo-controlled Phase II trial. Biol. Blood Marrow Transplant. 23(3), 512–521 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Hiwarkar P, Amrolia P, Sivaprakasam P. et al. Brincidofovir is highly efficacious in controlling adenoviremia in pediatric recipients of hematopoietic cell transplant. Blood 129(14), 2033–2037 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Chimerix. The AdAPT trial; adenovirus after allogeneic pediatric transplantation. (2017). https://clinicaltrials.gov/ct2/show/NCT03339401
- 78.Giesler KE, Marengo J, Liotta DC. Reduction sensitive lipid conjugates of tenofovir: synthesis, stability, and antiviral activity. J. Med. Chem. 59(15), 7097–7110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giesler KE, Liotta DC. Next-generation reduction sensitive lipid conjugates of tenofovir: antiviral activity and mechanism of release. J. Med. Chem. 59(22), 10244–10252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Devine KG, McGuigan C, Oconnor TJ, Nicholls SR, Kinchington D. Novel phosphate derivatives of zidovudine as anti-HIV compounds. AIDS 4(4), 371–373 (1990). [PubMed] [Google Scholar]
- 81.Ray AS, Fordyce MW, Hitchcock MJM. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus. Antivir. Res. 125, 63–70 (2016). [DOI] [PubMed] [Google Scholar]
- 82.Procházková E, Navrátil R, Janeba Z, Roithová J, Baszczyňski O. Reactive cyclic intermediates in the ProTide prodrugs activation: trapping the elusive pentavalent phosphorane. Org. Biomol. Chem. 17(2), 315–320 (2019). [DOI] [PubMed] [Google Scholar]
- 83.Erion MD, van Poelje PD, Dang Q. et al. MB06322 (CS-917): a potent and selective inhibitor of fructose 1,6-bisphosphatase for controlling gluconeogenesis in Type 2 diabetes. Proc. Natl Acad. Sci. USA 102(22), 7970–7975 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dang Q, Kasibhatla SR, Jiang T. et al. Discovery of phosphonic diamide prodrugs and their use for the oral delivery of a series of fructose 1,6-bisphosphatase inhibitors. J. Med. Chem. 51(14), 4331–4339 (2008). [DOI] [PubMed] [Google Scholar]
- 85.van Poelje PD, Potter SC, Erion MD. Fructose-1, 6-bisphosphatase inhibitors for reducing excessive endogenous glucose production in Type 2 diabetes. Handb. Exp. Pharmacol. 203, 279–301 (2011). [DOI] [PubMed] [Google Scholar]
- 86.Cesnek M, Jansa P, Smidkova M. et al. Bisamidate prodrugs of 2-substituted 9- 2-(phosphonomethoxy)ethyl adenine (PMEA, adefovir) as selective inhibitors of adenylate cyclase toxin from bordetella pertussis. Chemmedchem 10(8), 1351–1364 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Brehova P, Smidkova M, Skacel J. et al. Design and synthesis of fluorescent acyclic nucleoside phosphonates as potent inhibitors of bacterial adenylate cyclases. Chemmedchem 11(22), 2534–2546 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smidkova M, Dvorakova A, Tloust'ova E, Cesnek M, Janeba Z, Mertlikova-Kaiserova H. Amidate prodrugs of 9-[2-(phosphonomethoxy)ethyl]adenine as inhibitors of adenylate cyclase toxin from Bordetella pertussis. Antimicrob. Agents Chemother. 58(2), 664–671 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eng WS, Hockova D, Spacek P. et al. First crystal structures of mycobacterium tuberculosis 6-oxopurine phosphoribosyltransferase: complexes with GMP and pyrophosphate and with acyclic nucleoside phosphonates whose prodrugs have antituberculosis activity. J. Med. Chem. 58(11), 4822–4838 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Hockova D, Janeba Z, Naesens L. et al. Antimalarial activity of prodrugs of N-branched acyclic nucleoside phosphonate inhibitors of 6-oxopurine phosphoribosyltransferases. Bioorg. Med. Chem. 23(17), 5502–5510 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Spacek P, Keough DT, Chavchich M. et al. Synthesis and evaluation of symmetric acyclic nucleoside bisphosphonates as inhibitors of the Plasmodium falciparum, Plasmodium vivax and human 6-oxopurine phosphoribosyltransferases and the antimalarial activity of their prodrugs. Bioorg. Med. Chem. 25, 4008–4030 (2017). [DOI] [PubMed] [Google Scholar]
- 92.Spacek P, Keough DT, Chavchich M. et al. Synthesis and evaluation of asymmetric acyclic nucleoside bisphosphonates as inhibitors of plasmodium falciparum and human hypoxanthine-guanine-(xanthine) phosphoribosyltransferase. J. Med. Chem. 60(17), 7539–7554 (2017). [DOI] [PubMed] [Google Scholar]
- 93.Ballatore C, McGuigan C, De Clercq E, Balzarini J. Synthesis and evaluation of novel amidate prodrugs of PMEA and PMPA. Bioorg. Med. Chem. Lett. 11(8), 1053–1056 (2001). [DOI] [PubMed] [Google Scholar]
- 94.Mehellou Y, Balzarini J, McGuigan C. Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. Chemmedchem 4(11), 1779–1791 (2009). [DOI] [PubMed] [Google Scholar]
- 95.Sofia MJ, Bao D, Chang W. et al. Discovery of a beta-D-2′-Deoxy-2′-alpha-fluoro-2′-beta-C-methyluridine nucleotide prodrug (psi-7977) for the treatment of hepatitis c virus. J. Med. Chem. 53(19), 7202–7218 (2010). [DOI] [PubMed] [Google Scholar]
- 96.Mackman RL, Ray AS, Hui HC. et al. Discovery of GS-9131: design, synthesis and optimization of amidate prodrugs of the novel nucleoside phosphonate HIV reverse transcriptase (RT) inhibitor GS-9148. Bioorg. Med. Chem. 18(10), 3606–3617 (2010). [DOI] [PubMed] [Google Scholar]
- 97.McGuigan C, Murziani P, Slusarczyk M. et al. Phosphoramidate ProTides of the anticancer agent FUDR successfully deliver the preformed bioactive monophosphate in cells and confer advantage over the parent nucleoside. J. Med. Chem. 54(20), 7247–7258 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Quintiliani M, Balzarini J, McGuigan C. Design, synthesis, and biological evaluation of C1-phosphonamidate analogues of 2-deoxy-D-ribose-l-phosphate. Tetrahedron 69(43), 9111–9119 (2013). [Google Scholar]
- 99.Pertusati F, Hinsinger K, Flynn AS. et al. PMPA and PMEA prodrugs for the treatment of HIV infections and human papillomavirus (HPV) associated neoplasia and cancer. Eur. J. Med. Chem. 78, 259–268 (2014). [DOI] [PubMed] [Google Scholar]
- 100.McGuigan C, Derudas M, Gonczy B. et al. ProTides of N-(3-(5-(2′-deoxyuridine))prop-2-ynyl)octanamide as potential anti-tubercular and anti-viral agents. Bioorg. Med. Chem. 22(9), 2816–2824 (2014). [DOI] [PubMed] [Google Scholar]
- 101.Zano SP, Pate C, Frank M, Rock CO, Jackowski S. Correction of a genetic deficiency in pantothenate kinase 1 using phosphopantothenate replacement therapy. Mol. Gen. Metab. 116(4), 281–288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu C, Dumbre SG, Pannecouque C. et al. Amidate prodrugs of deoxythreosyl nucleoside phosphonates as dual inhibitors of HIV and HBV replication. J. Med. Chem. 59(20), 9513–9531 (2016). [DOI] [PubMed] [Google Scholar]
- 103.James E, Pertusati F, Brancale A, McGuigan C. Kinase-independent phosphoramidate S1P(1) receptor agonist benzyl ether derivatives. Bioorg. Med. Chem. Lett. 27(6), 1371–1378 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Luo M, Groaz E, Andrei G. et al. Expanding the antiviral spectrum of 3-fluoro-2-(phosphonomethoxy)propyl acyclic nucleoside phosphonates: diamyl aspartate amidate prodrugs. J. Med. Chem. 60(14), 6220–6238 (2017). [DOI] [PubMed] [Google Scholar]
- 105.Osgerby L, Lai Y-C, Thornton PJ. et al. Kinetin riboside and its ProTides activate the parkinson's disease associated PTEN-induced putative kinase 1 (PINK1) independent of mitochondrial depolarization. J. Med. Chem. 60(8), 3518–3524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pertusati F, Serpi M, McGuigan C. Medicinal chemistry of nucleoside phosphonate prodrugs for antiviral therapy. Antivir. Chem. Chemother. 22(5), 181–203 (2012). [DOI] [PubMed] [Google Scholar]
- 107.Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 114(18), 9154–9218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thornton PJ, Kadri H, Miccoli A, Mehellou Y. Nucleoside phosphate and phosphonate prodrug clinical candidates. J. Med. Chem. 59(23), 10400–10410 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Mehellou Y. The ProTides boom. Chemmedchem 11(11), 1114–1116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sinokrot H, Smerat T, Najjar A, Karaman R. Advanced prodrug strategies in nucleoside and non-nucleoside antiviral agents: a review of the recent five years. Molecules, 22(10), (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mehellou Y, Rattan HS, Balzarini J. The ProTide prodrug technology: from the concept to the clinic. J. Med. Chem. 61(6), 2211–2226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Offers a fascinating look into the history of how ProTides came to be and how they have advanced since their inception.
- 112.Slusarczyk M, Serpi M, Pertusati F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir. Chem. Chemother. 26, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Not only does the paper offer a brief introduction into the realm of antiviral nucleosides, but synthetic strategies to employ the ProTide method are discussed thoroughly.
- 113.Wang X, Dowd CS. The methylerythritol phosphate pathway: promising drug targets in the fight against tuberculosis. ACS Infect. Dis. 4(3), 278–290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gershenzon J, Dudareva N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 3(7), 408–414 (2007). [DOI] [PubMed] [Google Scholar]
- 115.Reichenberg A, Wiesner J, Weidemeyer C. et al. Diaryl ester prodrugs of FR900098 with improved in vivo antimalarial activity. Bioorg. Med. Chem. Lett. 11(6), 833–835 (2001). [DOI] [PubMed] [Google Scholar]
- 116.Ortmann R, Wiesner J, Reichenberg A. et al. Acyloxyalkyl ester prodrugs of FR900098 with improved in vivo anti-malarial activity. Bioorg. Med. Chem. Lett. 13(13), 2163–2166 (2003). [DOI] [PubMed] [Google Scholar]
- 117.Ortmann R, Wiesner J, Reichenberg A. et al. Alkoxycarbonyloxyethyl ester prodrugs of FR900098 with improved in vivo antimalarial activity. Archiv. Der. Pharmazie. 338(7), 305–314 (2005). [DOI] [PubMed] [Google Scholar]
- 118.Kurz T, Schlueter K, Kaula U, Bergmann B, Walter RD, Geffken D. Synthesis and antimalarial activity of chain substituted pivaloyloxymethyl ester analogues of Fosmidomycin and FR900098. Bioorg. Med. Chem. 14(15), 5121–5135 (2006). [DOI] [PubMed] [Google Scholar]
- 119.Kurz T, Behrendt C, Kaula U, Bergmann B, Walter RD. alpha-phenylethyl substituted bis(pivaloyloxymethyl) ester analogues of fosmidomycin and FR900098. Aus. J. Chem. 60(3), 154–158 (2007). [DOI] [PubMed] [Google Scholar]
- 120.Ponaire S, Zingle C, Tritsch D, Grosdemange-Billiard C, Rohmer M. Growth inhibition of Mycobacterium smegmatis by prodrugs of deoxyxylulose phosphate reducto-isomerase inhibitors, promising anti-mycobacterial agents. Eur. J. Med. Chem. 51, 277–285 (2012). [DOI] [PubMed] [Google Scholar]
- 121.Jose GS, Jackson ER, Uh E. et al. Design of potential bisubstrate inhibitors against Mycobacterium tuberculosis (Mtb) 1-deoxy-D-xylulose 5-phosphate reductoisomerase (Dxr)-evidence of a novel binding mode. Medchemcomm 4(7), 1099–1104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.San Jose G, Jackson ER, Haymond A. et al. Structure-activity relationships of the MEPicides: N-acyl and O-linked analogs of fr900098 as inhibitors of Dxr from mycobacterium tuberculosis and yersinia pestis. ACS Infect. Dis. 2(12), 923–935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Edwards RL, Brothers RC, Wang X. et al. MEPicides: potent antimalarial prodrugs targeting isoprenoid biosynthesis. Sci.Rep. 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang X, Edwards RL, Bal H. et al. MEPicides: alpha,beta-unsaturated fosmidomycin analogues as DXR inhibitors against malaria. J. Med. Chem. 61(19), 8847–8858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Uh E, Jackson ER, Jose GS. et al. Antibacterial and antitubercular activity of fosmidomycin, FR900098, and their lipophilic analogs. Bioorg. Med. Chem. Lett. 21(23), 6973–6976 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wiemer AJ, Wiemer DF, Hohl RJ. Geranylgeranyl diphosphate synthase: an emerging therapeutic target. Clin. Pharmacol. Ther. 90(6), 804–812 (2011). [DOI] [PubMed] [Google Scholar]
- 127.Monteil M, Migianu-Griffoni E, Sainte-Catherine O, Di Benedetto M, Lecouvey M. Bisphosphonate prodrugs: synthesis and biological evaluation in HuH7 hepatocarcinoma cells. Eur. J. Med. Chem. 77, 56–64 (2014). [DOI] [PubMed] [Google Scholar]
- 128.Troutman JM, Chehade KAH, Kiegiel K, Andres DA, Spielmann HP. Synthesis of acyloxymethyl ester prodrugs of the transferable protein farnesyl transferase substrate farnesyl methylenediphosphonate. Bioorg. Med. Chem. Lett. 14(19), 4979–4982 (2004). [DOI] [PubMed] [Google Scholar]
- 129.Zhang Y, Leon A, Song Y. et al. Activity of nitrogen-containing and non-nitrogen-containing bisphosphonates on tumor cell lines. J. Med. Chem. 49(19), 5804–5814 (2006). [DOI] [PubMed] [Google Scholar]
- 130.Wiemer AJ, Yu JS, Shull LW. et al. Pivaloyloxymethyl-modified isoprenoid bisphosphonates display enhanced inhibition of cellular geranylgeranylation. Bioorg. Med. Chem. 16(7), 3652–3660 (2008). [DOI] [PubMed] [Google Scholar]
- 131.Foust BJ, Allen C, Holstein SA, Wiemer DF. A new motif for inhibitors of geranylgeranyl diphosphate synthase. Bioorg. Med. Chem. 24(16), 3734–3741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tanaka Y, Murata-Hirai K, Iwasaki M. et al. Expansion of human T cells for adoptive immunotherapy using a bisphosphonate prodrug. Cancer Sci. 109(3), 587–599 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.U.S. Food and Drug Administration approves Gilead's single tablet regimen genvoya (elvitegravir, cobicistat, emtricitabine and tenofovir alafenamide) for treatment of HIV-1 infection (2015). http://www.gilead.com/news/press-releases/2015/11/us-food-and-drug-administration-approves-gileads-single-tablet-regimen-genvoya-elvitegravir-cobicistat-emtricitabine-and-tenofovir-alafenamide-for-treatment-of-hiv1-infection
- 134.Agarwal K, Fung SK, Nguyen TT. et al. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J. Hepatol. 62(3), 533–540 (2015). [DOI] [PubMed] [Google Scholar]
- 135.Chan HLY, Fung S, Seto WK. et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg-positive chronic hepatitis B virus infection: a randomised, double-blind, Phase III, non-inferiority trial. Lancet Gastroenterol. Hepatol. 1(3), 185–195 (2016). [DOI] [PubMed] [Google Scholar]
- 136.Gallant JE, Daar ES, Raffi F. et al. Efficacy and safety of tenofovir alafenamide versus tenofovir disoproxil fumarate given as fixed-dose combinations containing emtricitabine as backbones for treatment of HIV-1 infection in virologically suppressed adults: a randomised, double-blind, active-controlled Phase III trial. Lancet HIV 3(4), E158–E165 (2016). [DOI] [PubMed] [Google Scholar]
- 137.Sax PE, Wohl D, Yin MT. et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, Phase III, non-inferiority trials. Lancet 385(9987), 2606–2615 (2015). [DOI] [PubMed] [Google Scholar]
- 138.Orkin C, DeJesus E, Ramgopal M. et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide coformulated with rilpivirine and emtricitabine in virally suppressed adults with HIV-1 infection: a randomised, double-blind, multicentre, phase 3b, non-inferiority study. Lancet HIV 4(5), E195–E204 (2017). [DOI] [PubMed] [Google Scholar]
- 139.Stray KM, Park Y, Babusis D. et al. Tenofovir alafenamide (TAF) does not deplete mitochondrial DNA in human T-cell lines at intracellular concentrations exceeding clinically relevant drug exposures. Antivir. Res., 140, 116–120 (2017). [DOI] [PubMed] [Google Scholar]
- 140.Naesens L, Balzarini J, Declercq E. Pharmacokinetics in mice of the antiretrovirals agent 9-(2-phosphonylmethoxyethyl)adenine. Drug Metab. Dispos. 20(5), 747–752 (1992). [PubMed] [Google Scholar]
- 141.Liao S, Fan S-Y, Liu Q. et al. In vitro evaluation of 9-(2-phosphonylmethoxyethyl)adenine ester analogues, a series of anti-HBV structures with improved plasma stability and liver release. Arch. Pharmacal Res. 37(11), 1416–1425 (2014). [DOI] [PubMed] [Google Scholar]
- 142.Liu C, Dumbre SG, Pannecouque C, Korba B, De Jonghe S, Herdewijn P. Synthesis and antiviral evaluation of base-modified deoxythreosyl nucleoside phosphonates. Org. Biomolec. Chem. 15(26), 5513–5528 (2017). [DOI] [PubMed] [Google Scholar]
- 143.Kaiser MM, Hockova D, Wang T-H. et al. Synthesis and evaluation of novel acyclic nucleoside phosphonates as inhibitors of plasmodium falciparum and human 6-oxopurine phosphoribosyltransferases. Chemmedchem 10(10), 1707–1723 (2015). [DOI] [PubMed] [Google Scholar]
- 144.Kaiser MM, Baszczynski O, Hockova D. et al. Acyclic nucleoside phosphonates containing 9-deazahypoxanthine and a five-membered heterocycle as selective inhibitors of plasmodial 6-oxopurine phosphoribosyltransferases. Chemmedchem 12(14), 1133–1141 (2017). [DOI] [PubMed] [Google Scholar]
- 145.Coen N, Duraffour S, Naesens L. et al. Evaluation of novel acyclic nucleoside phosphonates against human and animal gammaherpesviruses revealed an altered metabolism of cyclic prodrugs upon epstein-barr virus reactivation in P3HR-1 cells. J. Virol. 87(22), 12422–12432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Toth K, Spencer JF, Ying B. et al. USC-087 protects Syrian hamsters against lethal challenge with human species C adenoviruses. Antivir. Res. 153, 1–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]