

Abstract
LonA proteases and ClpB chaperones are key components of the protein quality control system in bacterial cells. LonA proteases form a unique family of ATPases associated with diverse cellular activities (AAA+) proteins due to the presence of an unusual N‐terminal region comprised of two domains: a β‐structured N domain and an α‐helical domain, including the coiled‐coil fragment, which is referred to as HI(CC). The arrangement of helices in the HI(CC) domain is reminiscent of the structure of the H1 domain of the first AAA+ module of ClpB chaperones. It has been hypothesized that LonA proteases with a single AAA+ module may also contain a part of another AAA+ module, the full version of which is present in ClpB. Here, we established and tested the structural basis of this hypothesis using the known crystal structures of various fragments of LonA proteases and ClpB chaperones, as well as the newly determined structure of the Escherichia coli LonA fragment (235–584). The similarities and differences in the corresponding domains of LonA proteases and ClpB chaperones were examined in structural terms. The results of our analysis, complemented by the finding of a singular match in the location of the most conserved axial pore‐1 loop between the LonA NB domain and the NB2 domain of ClpB, support our hypothesis that there is a structural and functional relationship between two coiled–coil fragments and implies a similar mechanism of engagement of the pore‐1 loops in the AAA+ modules of LonAs and ClpBs.
Keywords: AAA+ proteins, ATPase module, ClpB chaperones, coiled‐coil fragments, inserted α‐helical domain, Lon protease
Abbreviations
- AAA+
ATPases associated with diverse cellular activities
- PDB
Protein Data Bank
- PQC
protein quality control
The family of Lon proteases, together with several other energy‐dependent proteases, molecular chaperones, as well as regulatory molecules, forms a system of protein quality control (PQC). These proteins play a key role in the maintenance of cellular proteome in all natural kingdoms 1, 2, 3, 4, 5, 6. Chaperones ensure correct folding of the polypeptide products of biosynthesis and participate in the formation of protein assemblies, as well as in preventing aggregation and refolding of modified cellular proteins. ATP‐dependent proteases degrade damaged or abnormal proteins and control the level of regulatory proteins at every stage of the cell cycle.
Most chaperones and all proteases of the PQC system are heat‐shock proteins (Hsp). The PQC proteases are bifunctional oligomeric enzymes. Their proteolytic domains belong to different classes of peptide hydrolases, whereas the ATPase parts are related to the single Hsp100 superfamily denoted AAA+ proteins (i.e., ATPases associated with diverse cellular activities) 5, 7, 8, 9, 10, 11, 12. Chaperones‐disaggregases Hsp100 (represented by the ClpB family in bacteria or Hsp104 in eukaryotes) also are part of the same superfamily 7, 8, 9.
The domain organization of AAA+ proteins of the PQC system is shown in Fig. 1. The ATPase component of any AAA+ protein is a two‐domain AAA+ module comprising a large nucleotide‐binding (NB or α/β) domain as well as a small α‐helical (H or α) domain. These modules contain a variety of conserved elements consisting of peptide fragments or, in some instances, single amino acid residues 8, 9, 11, 13. Furthermore, all AAA+ proteins include nonhomologous ‘extra’ domains (either located in the protein N‐terminal region or inserted within their NB domains (I domains); see Fig. 1).
Figure 1.
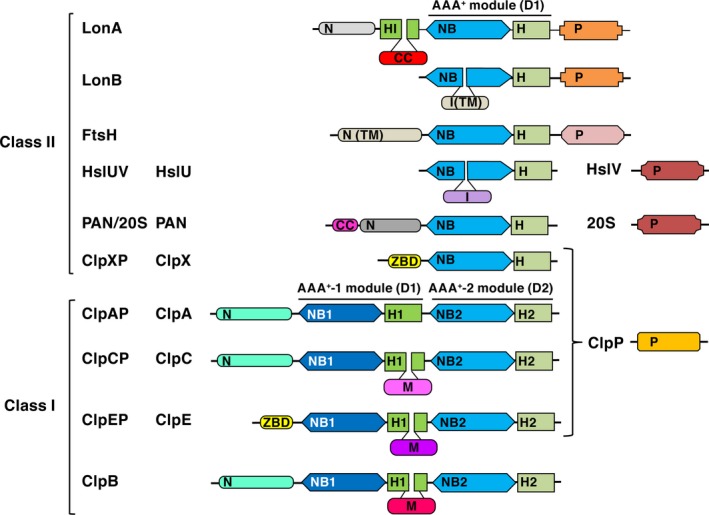
Domain organization in ATP‐dependent proteases and ClpB chaperones of the PQC system. N and ZBD—N‐terminal domains; NB, NB1, and NB2—nucleotide‐binding domains; H, H1, H2, and HI—α‐helical domains; I—inserted domains; TM—transmembrane domains; M—middle CC domains of different sizes, P—proteolytic domains/subunits [clans in MEROPS: SJ (Lon), MA (FtsH), PB (HslV and 20S); SK (ClpP)]; CC‐coiled‐coil regions. Pairs of domains (NB‐H), (NB1‐H1), and (NB2‐H2) form the corresponding AAA+ modules.
The AAA+ proteins are divided into two classes depending on the number of AAA+ modules. The ATPase components of most AAA+ proteases and proteolytic complexes (such as proteasomes) that contain only one such module (Lons, FtsH, HslUV, PAN/20S, ClpXP) belong to class II of AAA+ proteins, whereas components ClpA, ClpC, and ClpE of other proteases, as well as chaperones‐disaggregases ClpB, have two AAA+ modules (D1 and D2) and are assigned to class I (Fig. 1). The H domains of the D1 modules of ClpB, ClpC, and ClpE incorporate inserted coiled‐coil (CC) fragments of different sizes (52 to more than 110 residues). Proteolytic components of the ATP‐dependent proteases can be either domain within a single polypeptide chain of homooligomeric enzymes (Lon and FtsH), or individual subunits of heterooligomeric enzymes (HslUV, ClpXP, ClpAP, and others).
The common pool of Lon proteases comprises a number of different groups, among which the most representative and studied are the subfamilies LonA and LonB 8, 14. Subfamily A includes mainly bacterial and eukaryotic enzymes, whereas most enzymes of the archaeal origin belong to subfamily B. Although proteolytic (P) domains of both, LonA and LonB subfamilies, are serine–lysine hydrolases (clan SJ, family S16 in the MEROPS classification) 15, 16, the environment of the catalytic serine and lysine residues in the P domains of LonA and LonB is different 14. Furthermore, ‘extra’ domains of LonA and LonB proteases are positioned differently. An extra domain in LonA is located at the N terminus of the protein chain, whereas in LonB it is found inside the AAA+ module (Fig. 1). It is not yet clear how these differences are reflected in the structures of the full‐length enzymes, as to date no crystal structure of any full‐length Lon protease has been determined. However, crystal structures of a number of fragments consisting of individual domains of LonA and LonB proteases or their combinations are known.
The most prominent feature of LonA proteases, which distinguishes them from proteases of the LonB subfamily, as well as from other AAA+ proteins, is a very extended N‐terminal region with 300 to over 400 amino acids. It has been suggested that the N‐terminal region represents a combination of two domains—the actual N‐terminal (N) domain, followed by an inserted domain which is located between the N domain and the AAA+ module 17. This inserted domain, referred to as HI(CC) (helical inserted with CC‐fragment 18, Fig. 1), is formed exclusively by α‐helices and is predicted to contain a CC fragment 14, 19 (see also https://embnet.vital-it.ch/software/COILS_form.html). It was suggested that the HI(CC) domain resembles the H1 domain of ClpB chaperone, which also contains a fragment of similar size with CC conformation (namely M domain, Fig. 1) 17, 18.
The role of the N‐terminal region in supporting ATP‐dependent proteolysis and in maintaining the structure of the active enzyme has not yet been fully clarified. There is evidence indicating that this region might play an important role in oligomerization of LonA proteases and could be involved in the binding of protein substrates 20, 21, 22, 23, 24. Furthermore, it was shown that truncations or deletions in the N‐terminal regions of different LonA proteases often disrupt enzymatic activities 20, 21, 22, 25, 26, 27, 28, 29, 30.
With the availability of extensive structural data for the N domain 31, 32, but only incomplete data for the HI(CC) domain 32, 33, 34, we aim here to examine the architecture of the N‐terminal region of LonA proteases, in particular the structure of their HI(CC) domains, focusing on the LonA protease from Escherichia coli (EcLon). The interdomain interactions within monomers and oligomers of EcLon are also analyzed. As part of this study, we determined the structure of the fragment EcLon (235–584) that comprises a part of the HI(CC) domain and the AAA+ module, since no such structure of EcLon has been available to date.
Materials and methods
The amino acid sequences of the chaperones and proteases analyzed in this study were obtained from the UniProt Knowledgebase (http://www.uniprot.org/) and the MEROPS database (www.merops.sanger.ac.uk) 35, respectively. ExPASy server (https://embnet.vital-it.ch/software/ClustalW.html; https://www.ebi.ac.uk/Tools/msa/clustalo/; https://embnet.vital-it.ch/software/COILS_form.html; http://distill.ucd.ie/porter/) was used to evaluate the degree of conservation in sequences, to identify the CC regions in proteins, and for the subsequent topology analysis.
The fragment of EcLon (235–584) comprising the C‐terminal part of HI(CC) domain and the full‐length AAA+ module was produced by limited proteolysis from purified full‐length EcLonA, as previously described 22. As the final step of purification, concentrated protein was loaded on a Superdex 200 Prep grade 26/600 size exclusion column (GE Healthcare, Waukesha, WI, USA) at a flow rate of 0.5 mL·min−1. The buffer consisted of 20 mm Tris/HCl pH 7.5, 0.2 m NaCl and 1 mm ADP. For crystallization, the protein was concentrated to 8 mg·mL−1 on a Microcon concentrator with 10 kDa MW cutoff (EMD Millipore, Billerica, MA, USA).
Large needle‐shaped crystals were grown by the sitting drop vapor diffusion method with a HydraII crystallization robot (Art Robbins Instruments, Sunnyvale, CA, USA) at 21 °C. The crystals grew in 7–10 days in 30% (w/v) PEG 400, 0.2 m Li2SO4, 0.1 m Na cacodylate pH 6.5 from the Wizard II screen (Beryllium Discovery, Bedford, MA, USA). Crystals were harvested and flash‐frozen in liquid nitrogen.
X‐ray diffraction data were collected at the SER‐CAT 22‐ID beamline of the Advanced Photon Source at Argonne National Laboratory (Argonne, IL, USA) with a MAR225 detector. The best crystal diffracted to ~ 3.0 Å and belonged to space group P61 or its enantiomorph. Diffraction data were processed with HKL2000 36. Since the completeness and quality of data extending beyond 3.5 Å were poor, we only used data up to that resolution limit (Table 1).
Table 1.
Data collection and refinement statistics
| Data collection | |
| Wavelength (Å) | 1.0 |
| Space group | P65 |
| Mol/ASU | 1 |
| X‐ray source | APS‐22‐ID |
| a, b, c (Å) | 88.6, 88.6, 67.7 |
| α, β, γ (°) | 90, 90, 120 |
| d min (Å)a | 3.5 (3.6–3.5) |
| Completeness (%)a | 99.9 (100) |
| Redundancya | 11.1 (11.2) |
| R merge b | 0.12 (0.38) |
| I/σ (I)a | 19.2 (6.4) |
| Refinement | |
| Resolution | 44.3–3.5 |
| Reflections | 3856 |
| Atoms | 2696 |
| Amino acids | 338 |
| R work c/R free d | 0.28/0.32 |
| Validation | |
| Bond angle RMSD (°) | 0.661 |
| Bond length RMSD (Å) | 0.003 |
| Ramachandran favored (%)e | 84.2 |
| Ramachandran allowed (%)e | 12.5 |
| Clashscore | 13.8 |
Indicates statistics for the last resolution shell shown in parentheses.
R merge = Σhkl,j (|Ihkl − <Ihkl>|)/Σhkl,j Ihkl, where <Ihkl> is the average intensity for a set of j symmetry‐related reflections, and Ihkl is the value of the intensity for a single reflection within a set of symmetry‐related reflections.
R work = Σhkl (||F o| − |F c||)/Σhkl|F o| where F o is the observed structure factor amplitude and F c is the calculated structure factor amplitude.
R free = Σhkl,T (||F o| − |F c||)/Σhkl,T|F o|, where a test set, T, is omitted from the refinement.
Calculated with MolProbity 65.
The structure of EcLon (235–584) was solved by molecular replacement using search models derived from the structures of fragments of LonA proteases from Meiothermus taiwanensis [MtLon; Protein Data Bank (PDB) ID 4YPL 34] and Bacillus subtilis (BsLon; PDB ID 3M6A 33). We obtained the best hit with a homology model built from 4YPL with the Phyre2 server 37. Program phaser 38 from the phenix program suite identified a solution in space group P65. The solution was further improved when the search was performed using separately the NB (α/β) and H (α) domains of the AAA+ module. After alternating cycles of refinement with phenix.refine 39 and model building in coot 40, the structure was rebuilt with the help of the crystal structure of E. coli H domain (PDB ID 1QZM 41). The structure was refined to an R work of 28.4% and R free of 32.7% 42 with 86.6% of the residues located in the core region of the Ramachandran plot and 10.7% in the additional allowed region (Table 1). Despite the comparatively low resolution, the electron density is well defined, thus assuring that the overall fold of the structure is correct.
Results and Discussion
Crystal structure of the of EcLon fragment comprising the C‐terminal part of HI(CC) and the AAA+ module
Members of the LonA protease subfamily comprise five domains within a single chain, connected by three linkers:
where domains N and HI(CC) constitute the N‐terminal region, NB and H form the AAA+ module, and P is a serine–lysine peptide hydrolase (Figs 1 and 2A). Structural data are available for only four out of the five domains of LonA proteases from different organisms. They include the functional domains NB, H, P, as well as the N domain from the noncatalytic N‐terminal region.
Figure 2.
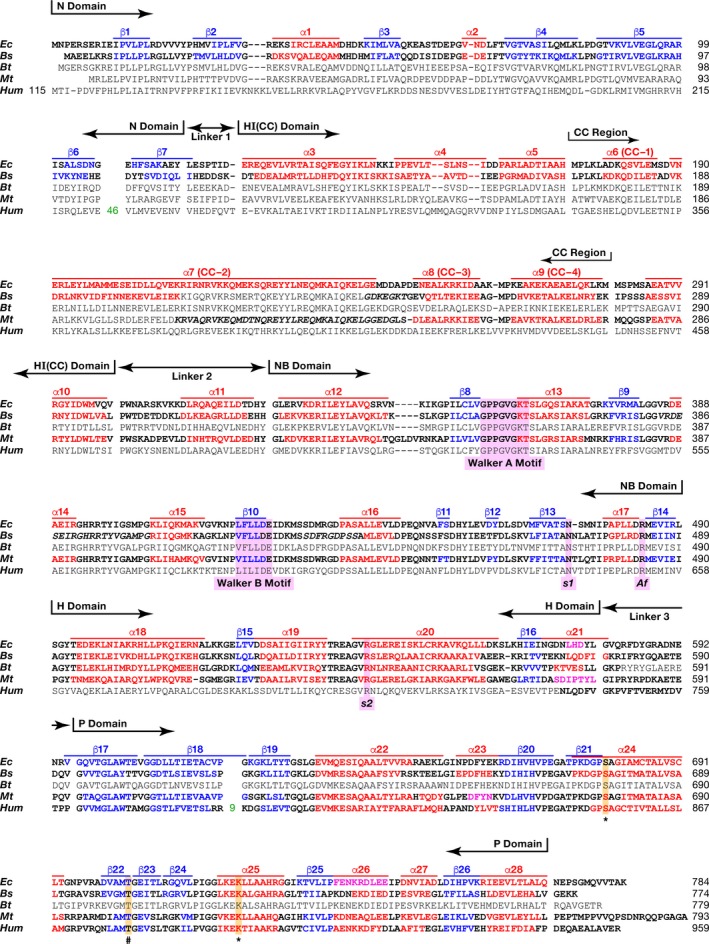
Alignment of the primary and secondary structures of full‐length EcLon, BsLon, BtLon, MtLon, and HumLon. Lon proteases are from Escherichia coli (Ec), Bacillus subtilis (Bs), Brevibacillus thermoruber (Bt), Meiothermus taiwanensis (Mt), and Homo sapiens (Hum) Domain organization of the enzymes and location of the CC region are shown. Fragments of the primary structures were compared using the program http://www.ch.embnet.org/cgi-bin/clustalw_parser; boundaries were determined based on the secondary structures. Experimentally determined secondary structure elements are shown in bold; fragments of sequences not seen in crystal structures are italicized. Red indicates amino acids that form α helices, magenta—3/10 helices, blue—β strands, and black color indicates amino acids that are not included in the secondary structure elements. Consensus sequence elements are highlighted in lavender: Walker motifs A and B, the residues sensor‐1 (s1), sensor‐2 (s2), Arg finger (Af). The catalytic serine (S*) and lysine (K*) residues as well as important for activity threonine (T#) are highlighted in orange.
Crystal structures of LonA fragments that include the ATPase modules from BsLon and MtLon are available 33, 34, but no corresponding structure for EcLon has been previously reported. We have now determined crystal structure of the fragment of EcLon comprising the C‐terminal part of HI(CC) and full length of NB and H domains (residues 235–584) at 3.5 Å resolution (PDB ID 6N2I). Although the N‐terminal residue of the construct is Lys235, the first residue visible in the electron density map is Ala247, located before the α8 helix (Figs 2 and 3A). The α12 helix (aa: 331–341) is considered to be the beginning of the NB domain (Fig. 2) 9.
Figure 3.
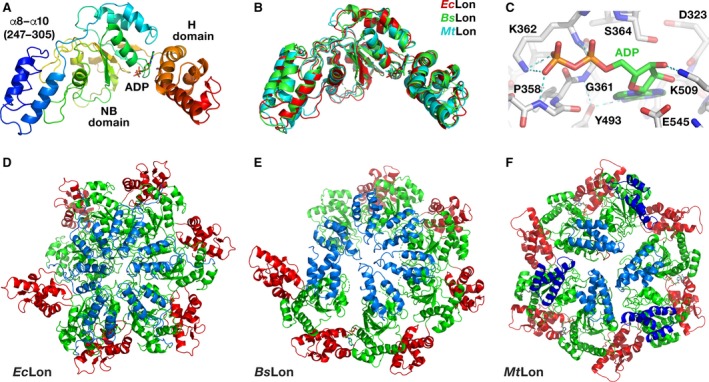
Structures of LonA proteases. (A) Crystal structure of a truncated fragment of EcLon (residues 235–584 in rainbow coloring). Individual domains are marked. The bound ADP molecule is shown in sticks. (B) Superposition of the crystal structures of three similar fragments of EcLon (red), BsLon (green) and MtLon (cyan). (C) Interactions of ADP molecule in the binding pocket of EcLon. (D) Cartoon representation of an open ring hexamer in the structure of EcLon (235–584), with helical arrangement of the monomers around the crystallographic 61 axis passing through the central pore. N‐terminal helices are blue, NB domains are green, and H domains are red. (E) Crystal structure of the open hexamer of BsLon (240–774; PDB ID 3M6A, P domain not shown for clarity). Colors are as in panel D. (F) Crystal structure of the closed hexamer of the fragment (242–793) of MtLon (PDB ID 4YPL, P domain not shown for clarity). Two sets of N‐terminal three‐helix bundles located either at the center or at the periphery of a hexamer are shown in light and dark blue, respectively.
As in all AAA+ proteins, the ATPase module of EcLon is composed of two domains—a large NB (α/β) domain with RecA‐like fold and a small α‐helical (H) domain 8, 9, 11, 43, with an ATP binding site located in the junction between these two domains (Fig. 3A). The overall fold of the AAA+ modules of EcLon, BsLon, and MtLon is very similar (Fig. 3B). The structure of the NB domain of EcLon is characterized by a central β‐sheet comprising five parallel β strands (with strand order 51432), flanked by α helices, while the H domain adapts a four‐helix bundle fold.
A set of conserved consensus fragments of the AAA+ module includes Walker motifs A (residues 356–363) and B (419–424), as well as residues forming sensor‐1 (Asn473), sensor‐2 (Arg542), and the arginine finger Arg484 (Fig. 2) 8, 9, 10, 18. All these fragments are crucial for binding and hydrolyzing ATP. The motif A coordinates the γ‐phosphate of ATP, and the motif B coordinates a magnesium ion and activates a water molecule for nucleophilic attack on the γ‐phosphate 43. Sensor‐1 residue coordinates a nucleophilic water molecule, while sensor‐2 interacts with the α‐phosphate of ATP; the arginine finger provides the interactions between neighboring subunits of the enzyme.
An ADP molecule with well‐defined electron density is found in the nucleotide‐binding site (Fig. 3A,C). Lys362 of the consensus Walker A motif (Fig. 2) is involved in interactions with both phosphates of the ADP. Its NZ atom is hydrogen bonded to the β phosphate, whereas its amide interacts with the α phosphate. Another interaction with the β phosphate is provided by the amide nitrogen of Gly359, while orientation of the base is supported by its interactions with Tyr493, and the sugar oxygen interacts with Lys509 (Fig. 3C).
While the fragment of the HI(CC) domain at the N terminus of our structure has poorer density suggesting mobility, the electron density unambiguously defines the overall fold of the NB and H domains of EcLon, as well as packing of the individual molecules in the oligomer (Fig. 3D). The oligomers in the crystal structures of BsLon and MtLon fragments demonstrate two different kinds of hexameric arrangements—hexamers with open and closed rings, respectively (Fig. 3E,F). In our structure of EcLon, the AAA+ modules form an open helical hexameric ring (Fig. 3D), as was observed in the crystal structure of BsLon (Fig. 3E) 33.
The unusual N‐terminal region of LonA proteases
Structural data are available for two N‐terminal fragments of EcLon, consisting of 116 and 245 amino acid residues, respectively 31, 32, as well as for a fragment of BsLon (1–209) 33. These structures, combined with prediction of the secondary structures for several other LonA proteases (http://www.ch.embnet.org), revealed that the extended N‐terminal region of LonA proteases preceding the AAA+ module is formed by two domains, the predominantly β‐structured N domain, and the α‐helical HI(CC) domain (Figs 1 and 2) 17, 18, 32.
In contrast, the ATPase subunits of ClpAP and ClpCP proteases, as well as of ClpB chaperones, have homologous α‐helical N domains 44, 45, 46, whereas subunits ClpX and ClpE (proteases ClpXP and ClpEP) include specific Zn‐binding N domains (ZBD) 47, 48. Membrane‐binding bacterial FtsH proteases contain periplasmic N domains which have α/β‐fold formed by two α‐helices and five β‐strands 49. All these N domains were shown to be involved in recognition and binding of either target proteins or adapter molecules 44, 45, 46, 47, 48. Thus, two‐domain organization of the N‐terminal region radically distinguishes LonA proteases from the other AAA+ proteases and ClpB chaperones of the PQC system that bear single N‐terminal domains with varying folds (Fig. 1).
The N domains of bacterial LonAs are composed of seven β strands and two α‐helical fragments (Fig. 2; see also Fig. 1A in 31). A large insert that may include up to 100 amino acids is present in most eukaryotic LonA proteases in the C‐terminal hairpin formed by the sixth and seventh β strands (Fig. 2). The predicted secondary structures of various N domains agree with the experimentally determined secondary structures of the N domains of EcLon (PDB ID 2ANE ) and BsLon (PDB ID 3M65), suggesting conservation of the fold. The linker 1 regions between the N domain and the subsequent HI(CC) domain in bacterial and eukaryotic enzymes are of similar size and include 7–8 amino acid residues (Fig. 2).
The second domain in the N‐terminal region of LonA is α‐helical domain, HI(CC), located between the N and NB domains (Fig. 1). HI(CC) domains are similar in size in all LonAs and show a fairly high degree of sequence similarity in groups of bacterial and eukaryotic enzymes (data not shown). A comparative analysis of the primary and the experimental or predicted secondary structures of various LonAs revealed that the HI(CC) domain is formed by eight helices (α3–α10, Fig. 2). According to a prediction (https://embnet.vital-it.ch/software/COILS_form.html), four helices (α6–α9 or CC‐1–CC‐4) represent a CC region, with helix α7 (CC‐2) being unusually long (~ 85 Å, aa V189‐E243 in EcLon; Fig. 2). It should be emphasized that the degrees of similarity of the CC regions exceed those of the full‐sized HI(CC) domains, and the most conserved fragment of the CC regions is the extended helix α7 (CC‐2).
The packing of the individual helices in the HI(CC) domain is still unknown, as no structure containing an intact HI(CC) domain has been determined. However, the helical nature of the HI(CC) domain is confirmed by combining data from the independently obtained crystal structures of EcLon (1–245) 32 that includes the N domain and the first five α helices of the HI(CC) domain (Fig. 4A), and the structure of BsLon (240–774) 33 that comprises the three C‐terminal helices of the HI(CC) domain, AAA+ module, and P domain (Fig. 4B). Considering the extensive sequence similarity between EcLon and BsLon that exceeds 86%, it might be expected that a hybrid model of a full‐length bacterial LonA could be assembled from these two structures that contain four overlapping residues (Fig. 2). However, in practice such modeling cannot be done due to the large discrepancy in the size and packing of some individual helices comprising the HI(CC) fragment in the structures of differently truncated molecules of EcLon and BsLon. A simple structural comparison of the N‐terminal fragments of EcLon and BsLon illustrates that phenomenon very clearly (Fig. 5). The sizes of the first helices of HI(CC) domains in these two structures, as well as the topological arrangement of the following helical fragments, are different. Although the variation in the size of the helices and their packing within the HI(CC) fragments could be a result of differences in crystallization or truncation (Fig. 5), it emphasizes the extreme flexibility of this region, substantiating the difficulties encountered in attempts to grow crystals of the full‐length enzyme.
Figure 4.
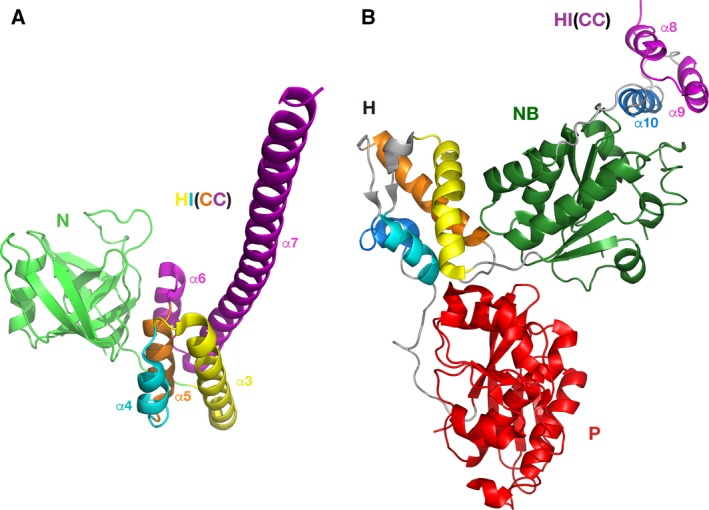
Crystal structures of the fragments EcLon (1–245) and BsLon (240–774). (A) N domain of EcLon (PDB ID 3LJC; residues 1–116) is shown in green; the first five helices of the HI(CC) domain are yellow (α3, residues 124–145), blue (α4, residues 149–159), orange (α5, residues 162–172), and magenta (α6 and α7, residues 181–185 and 189–243), respectively. (B) The last three helices α8–α10 of the HI(CC) domain of BsLon (PDB ID 3M6A, three N‐terminal helices of the construct) are shown in magenta (α8, α9) and dark blue (α10); the NB domain is green; helices in the H domain are shown with the same color scheme as the first three helices and the last helix of HI(CC) domain, respectively; the P domain is red.
Figure 5.
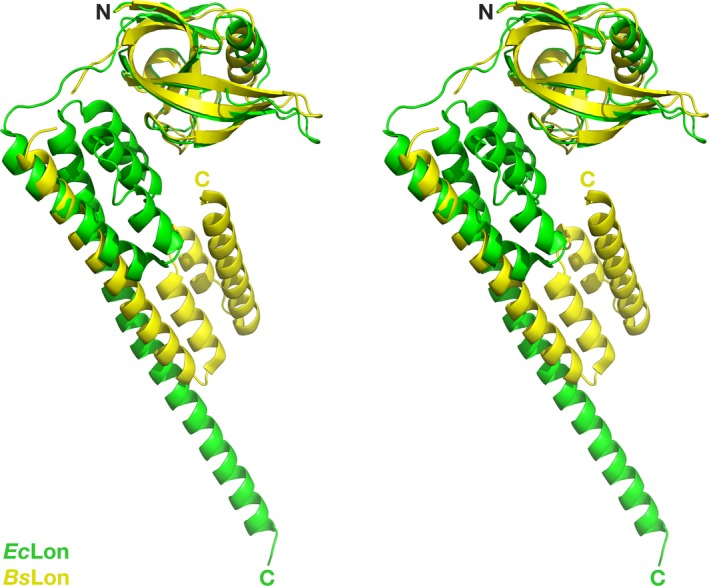
Superposition of the structures of the N‐terminal fragments of EcLon and BsLon. The structures of EcLon (1–245; PDB ID 3LJC, green) and BsLon (1–209; PDB ID 3M65, yellow) are superimposed by aligning Cα atoms in their N domains.
These conclusions are supported by the recently determined structures of two fragments of MtLon comprising different C‐terminal parts of the HI(CC) domain (PDB IDs 4YPL and 4YPN) 34, as well as by the structure of EcLon (235–584) presented here. Although the starting residues in the two fragments of MtLon are different (residue 207 in 4YPN and residue 242 in 4YPL), the segment comprising residues 207–242 in 4YPN is not visible. Similarly, residues 235–246 are not seen in the structure of EcLon (235–584). These results confirm the extensive flexibility of this region in the truncated constructs of LonAs.
The HI(CC) domain resembles very closely the H1 domain of first AAA+ module of ClpB chaperone with an inserted coiled‐coil M domain, with more than 30% sequence similarity between them. One of the helices in the M domain is composed of 58 residues, similar to the long α7 helix in the EcLon HI(CC) domain. This resemblance prompted a conjecture that the HI(CC) domain might correspond to an α‐helical domain of a hypothetical AAA+ module that has lost its own NB domain, and is embedded between the N domain and the existing single AAA+ module of LonA 18. This conjecture is supported by the prior observation that organization of the sole ATPase modules in the class II AAA+ proteins (ClpX and HslU) is similar to the organization of the D2 modules in the class I AAA+ proteins 13, 50, 51, 52, 53.
Structural comparison of the HI(CC) domain of LonA and the H1(M) fragment of ClpB
Whereas a resemblance of the HI(CC) domain of LonA to the H1(M) fragment of ClpB has been previously suggested 18, no structural basis for such a topological similarity has been established. A comparison of the primary and secondary structures of the HI(CC) domains of EcLon, BsLon, and MtLon on the one hand, and the H1(M) fragments of E. coli and Thermus thermophilus ClpB chaperones (EcClpB and TtClpB) 46 on the other hand (Fig. 6), reveals noticeable similarity. Based on the sequence alignment, it was suggested that eight helices of the HI(CC) domain [five helices (α3–α7) seen in the EcLon (1–245) and three N‐terminal helices (α8–α10) from the structures of EcLon (235–584), BsLon (240–774), and MtLon (242–793)], might correspond to the C1–C4 helices of the H1 domain of the chaperone, combined with the helices L1–L4 of the inserted M domain (Fig. 6). The fragment (302–324) of EcLon that includes helix α11 and the equivalent fragments of BsLon and MtLon correspond to the linker region connecting the H1 and NB2 domains in EcClpB and TtClpB.
Figure 6.
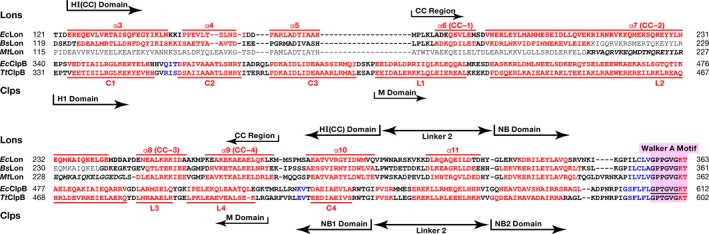
Alignment of the primary and secondary structures of the HI(CC) domains of EcLon, BsLon, and MtLon with the H1(M) fragments of EcClpB and TtClpB. The secondary structure elements are designated and highlighted as in Fig. 2. Helices α3–α10 form the Lon HI(CC) domain, helices C1–C4 form the ClpB H1 domain, and helices L1–L4 form the ClpB M domain. Helices in Clp structures are labeled as in original report 46.
The long α7 (CC‐2) helix, consisting of 55 residues, is a distinctive feature of EcLon (1–245) fragment (Fig. 4A, magenta). A helix of similar length (L2) is the key element of the propeller‐like M domain of ClpB chaperone (magenta in Fig. 7A) that has CC conformation and is inserted in the chaperone H1 domain (Figs 1 and 6) between its α helices C3 and C4 46. The degree of similarity of these helices in EcLon and EcClpB (Fig. 6) is > 45%. An important addition to the similarities listed above is the conservation of consensus elements of AAA+ proteins—positively charged ‘sensor‐2’ residues at the beginning of the third helix (Arg164 in EcLon, Arg162 in BsLon, and Lys388 in EcClpB), as well as the Walker A motifs located in the following nucleotide‐binding domains (Fig. 6).
Figure 7.
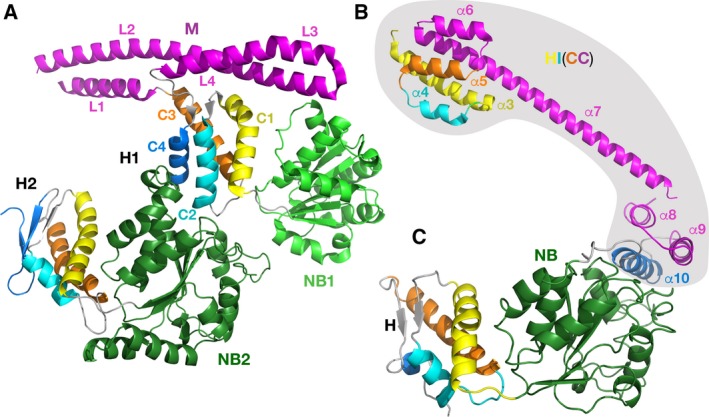
Crystal structures of fragments of TtClpB chaperone and EcLon. (A) TtClpB chaperone (150–854; PDB ID 1QVR); (B) EcLon (124–245; PDB ID 3LJC); (C) EcLon (235–584; PDB ID 6N2I). The nucleotide‐binding domains are colored in light (NB1, TtClpB) and dark green (NB2, TtClpB and NB, EcLon). Corresponding helices in the H1 and H2 domains of TtClpB, as well as in HI(CC) and H domains of EcLon, are colored identically.
We postulate that the first three helices (α3–α5), combined with the eighth helix (α10) of the HI(CC) domain, form a putative α‐helical domain in the N‐terminal part of the LonA proteases (α‐helical inserted domain, HI domain). Such a domain would topologically resemble the H1 domain of the first AAA+ module of ClpB (D1), since it includes the inserted CC region formed by four helices (α6–α9 or CC‐1–CC‐4), equivalent to the arrangement of the helices L1–L4 in the M domain of ClpB. In order to clarify this proposition in structural terms, we marked with identical colors the corresponding helices in the structure of the fragment (150–854) of TtClpB (PDB ID 1QVR, Fig. 7A) and in the structure of the fragments of EcLon (124–245) and EcLon (235–584; Fig. 7B,C). The fragments of EcLon (124–245) and EcLon (247–299), highlighted by a gray background in Fig. 7B,C, comprise the structure of the HI(CC) domain.
As mentioned earlier, the mutual arrangements of the helices in HI(CC) in respect to the other domains in the partial structures of EcLon and BsLon might be affected by truncations introduced into the constructs that have been used for crystallization. In the absence of a LonA structure with the intact HI(CC) domain, the existing structural fragments containing HI(CC) helices cannot be properly compared between themselves and to ClpB. Therefore, in order to test this hypothesis using the available structural data, we analyzed the resemblance between the two proteins on the level of both the secondary and tertiary structures, comparing either the individual helices, or the whole domains.
We started by superimposing selected helices comprising the HI(CC) domain of EcLon with their counterparts in the H1 domain of the first AAA+ module of ClpB 54 (Fig. 8). As can be seen in Fig. 8A, the first three helices of the LonA HI(CC) domain (α3–α5) could be superimposed quite well onto the corresponding helices of the H1 domain of ClpB (C1–C3). The short helix α6, preceding the long helix α7 in the structure of the N‐terminal fragment of EcLon (yellow in Fig. 8A), is oriented differently than the corresponding first helix L1 of ClpB M domain (shown in a different shade of yellow), which leads to a dramatic difference in the orientation of the long helices α7 and L2 (shown in two different shades of green).
Figure 8.
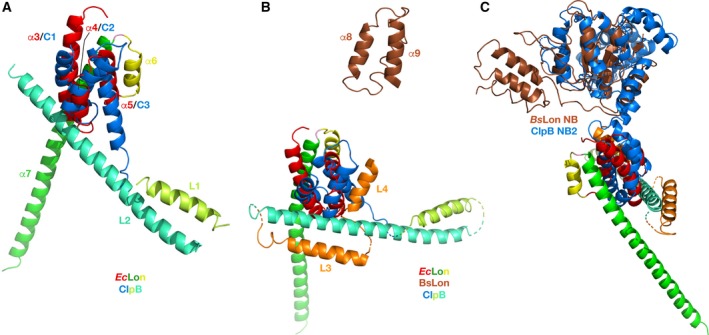
Structural comparison of the HI(CC) domain of LonAs and H1 domain of EcClpB. (A) The first three helices of the EcLon HI(CC) domain (α3–α5, red, PDB ID 3LJC) are superimposed on the first three helices of the H1 domain of EcClpB (blue, PDB ID 4D2U). Two following helices in both proteins are colored in different shades of yellow and green, correspondingly. (B) Topological assignment of the remaining three helices of the HI(CC) domain. Helices α8–α10 of the EcLon HI(CC) domain (brown, PDB ID 6N2I) are placed within the frame of Fig. 8A by superimposing the NB domain of EcLon and NB2 domain of EcClpB. Corresponding helices in ClpB are shown in orange. (C) Flexible linker region between the N‐terminal three‐helix bundle and NB domain in EcLon (brown). The NB2 domain of EcClpB, superimposed with the latter, is shown in blue.
Furthermore, helices α8 (CC‐3), α9 (CC‐4), and α10 at the N terminus of EcLon (235–584) correspond to helices L3, L4, and C4 of ClpB (Figs 6 and 8B, shown in brown and orange, respectively). In the hexamers of EcLon (235–584) and BsLon (240–774), these helices are oriented toward the central pore (shown in blue in Fig. 3D,E). The authors attribute the open ring arrangement of the monomers in the hexamer of BsLon to the observed position of this helical bundle in the crystal structure that ‘is incompatible’ with formation of a closed ring 33. The helices α8–α10 in EcLon are connected to the NB domain via a long, extended linker (Fig. 8C, colors as above), which would permit flexibility in the relative positioning of these helical fragments in truncated molecules of EcLon and BsLon. That conclusion is also supported by variations in the relative position of the N‐terminal three‐helix bundle in six monomers, as well as by increased temperature factors for this area. Therefore, the observed localization of the N‐terminal helices of EcLon and BsLon may not reflect their position in the structure of a Lon molecule in which the HI(CC) domain would be intact.
Two crystal structures of the closed ring hexamers of the fragments (242–793) and (207–492) of MtLon (PDB ID 4YPL, 4YPN) with a similar three‐helix bundle at the N terminus of the molecule also indicate that formation of a closed hexameric ring does not tolerate the presence of all three‐helix bundles in the central pore, but requires to move three bundles out of six to the periphery of the ring (Fig. 3F).
Similarities and differences in the structures of the NB domains in AAA+ modules of LonA proteases and Clp chaperones
Structural data on the NB domains of LonA proteases include our structure of EcLon presented here (Fig. 3A), as well as two high‐resolution crystal structures of BsLon (PDB ID 3M6A, Fig. 4B) 33 and MtLon (PDB ID 4YPN) 34. The overall fold of the LonA NB domain is typical of many other AAA+ proteins, in particular of the ATPase components of ClpXP (PDB ID 3HTE) 55 and ClpAP proteases (PDB ID 1KSF) 44, as well as both NB domains of ClpB chaperones (PDB IDs 1QVR, 1JBK) 46, 56.
Similarly to other AAA+ ATPases, LonA proteases are expected to function as hexamers with a central pore that is lined with axial loops, required for protein unfolding and translocation. Three different pore loops, called ‘GYVG’ (or pore‐1), pore‐2 and ‘RKH’, are usually found in almost all AAA+ unfoldases 55, 57.
The homologs of GYVG (pore‐1) loops are found in all AAA+ proteases and chaperones of the ClpB/Hsp104 family 55, 57, 58, 59, 60. As currently established 57, 61, GYVG loops are involved during all stages of molecular machinery work. They interact with polypeptide substrates and pull them into the pore in a concerted way with nucleotide‐dependent loop movements unfolding those substrates that cannot enter the pore otherwise. A comparison of the fragments containing GYVG loops in the structures of AAA+ modules of chaperones ClpA and ClpB revealed a distinct difference in the location of the functional motif within the individual loops in their NB1 and NB2 domains. Structure‐based sequence alignment of the NB domains of LonA proteases with the NB1 and NB2 domains of ClpA and ClpB chaperones (Fig. 9) indicates variability in the placement of GYVG motif in these proteins. A structural superposition of the corresponding fragments, comprising GYVG signature motives in these proteins (Fig. 10), provides a good illustration of this feature.
Figure 9.

Structure‐based sequence alignment of the NB domains of EcLon, BsLon, and MtLon with the NB1 and NB2 domains of EcClpA and TtClpB chaperones. The secondary structure elements are designated and highlighted as in Fig. 2. Axial pore loops are highlighted in green.
Figure 10.
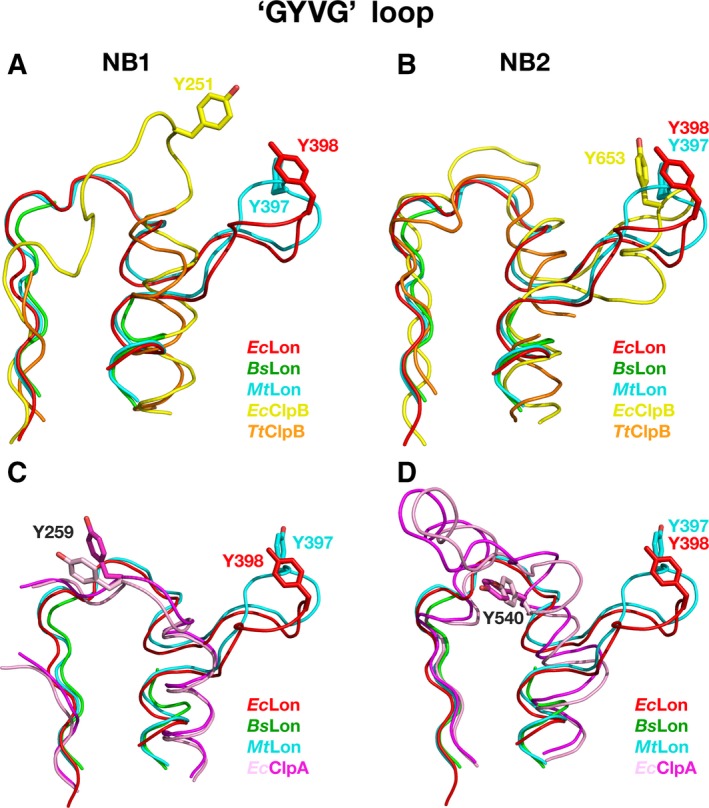
A comparison of the fragments containing axial GYVG (pore‐1) loops in the crystal structures of NB domains of LonAs and NB1–2 domains of ClpA and ClpB chaperones. GYVG comprising fragments from EcLon (PDB ID 6N2I, residues 378–409, Tyr398 shown in sticks, red), BsLon (PDB ID 3M6A, residues 376–408 with a break between residues 382 and 403, green) and MtLon (PDB ID 4YPL, residues 377–409, Tyr397 shown in sticks, blue) are superimposed: (A) with corresponding fragments from the NB1 domains of EcClpB (PDB ID 5OFO, residues 237–267, Tyr251 shown in sticks, yellow) and TtClpB (PDB ID 1QVR, residues 230–259 with a break between 234 and 246, orange); (B) with corresponding fragments from the NB2 domains of EcClpB (PDB ID 5OFO, residues 628–668, Tyr653 shown in sticks, yellow) and TtClpB (PDB ID 1QVR, residues 620–656 with a break between 636 and 651, orange); (C) with corresponding fragments from the NB1 domains of EcClpA (PDB ID 1KSF, residues 246–273 with a break between 251 and 255, Tyr259 shown in sticks, pink) and EcClpA (PDB ID 1R6B, residues 246–273 with a break between 251 and 255, Tyr259 shown in sticks, magenta); (D) with corresponding fragments from the NB2 domains of EcClpA (PDB ID 1KSF, residues 517–552, Tyr540 shown in sticks, pink) and EcClpA (PDB ID 1R6B, residues 517–552, Tyr540 shown in sticks, magenta).
A pairwise comparison of the fragments containing the GYVG loops in the structures of NB domains of EcLon, BsLon, and MtLon with those in the NB1 and NB2 domains of ClpA and ClpB chaperones reveals a striking similarity in the location of GYVG motif with a conserved tyrosine residue within the helical fragment of these loops between the LonA NB and ClpB NB2 domains (Figs 9 and 10B). It must be stressed that although the pore‐1 loop is partially disordered in the NB1 domain of TtClpB, GYVG location is unambiguously different in both, EcClpB and TtClpB, from the one described above (Figs 9 and 10A). It is reminiscent of the position of the GYVG loop in EcClpA NB2 domain, although the placement of the conserved tyrosine is different among them (Figs 9 and 10D). It should be stressed, however, that the conformation of the GYVG loop in the EcClpA NB2 domain might be affected by the presence of a Mg ion bound in the vicinity of this loop in the crystals 62. On the other hand, location of the conserved tyrosine residues in the NB1 domains of ClpA and ClpB is similar (Figs 9 and 10A,C).
Our analysis indicates that there is a correlation between the presence of the CC fragment, embedded in the α‐helical domain that precedes either the single or second AAA+ module, and the location of GYVG motif in the pore‐1 loop of the following NB domain. Other proteins with a single AAA+ module (ClpX, HslU, FtsH) that do not contain a CC insertion have their GYVG signature located on the top of the loop, as shown in Fig. 10A.
Location of two other axial loops (pore‐2 and RKH) is highly conserved in the structures of LonA proteases and of both chaperones, ClpA and ClpB (Fig. 11A,B). Pore‐2 loops are also pointing into the central pore and are expected to interact with the substrate, as pore‐1 loops do, but at a different stage of substrate translocation 55, 59, 63. In LonA proteases, they are more similar in length to their counterparts in the NB1 domains of both ClpA and ClpB (Figs 9 and 11A).
Figure 11.
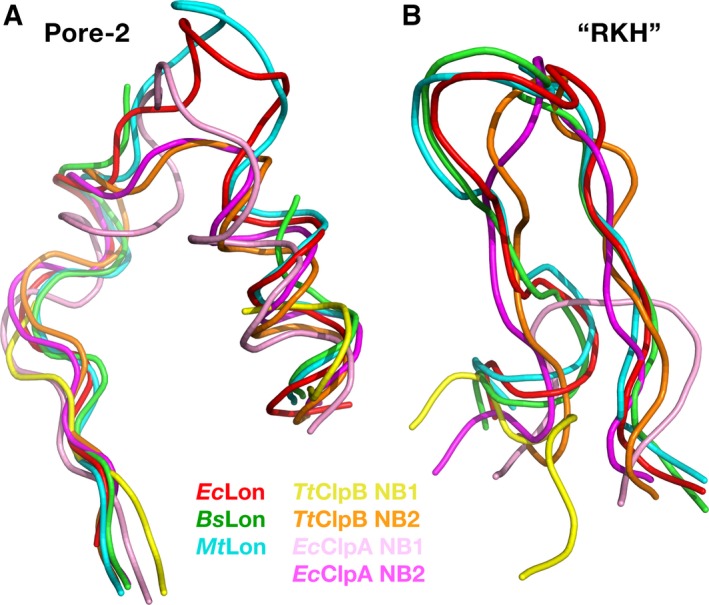
A comparison of the fragments containing axial loops pore‐2 and RKH in the crystal structures of the NB domains of LonAs and NB1–2 domains of ClpA and ClpB chaperones. (A) Pore‐2 comprising fragments from EcLon (PDB ID 6N2I, residues 419–445, red), BsLon (PDB ID 3M6A, residues 416–442 with a break between 427 and 437, green), and MtLon (PDB ID 4YPL, residues 417–443, blue) are superimposed onto corresponding fragments from TtClpB (PDB ID 1QVR) of both the NB1 domain (residues 264–295 with a break between 271 and 291, yellow), and the NB2 domain (residues 662–684, orange), as well as with the corresponding fragments from EcClpA (PDB ID 1R6B) of both the NB1 (residues 280–307, pink), and NB2 domains (residues 559–581, magenta); (B) RKH comprising fragments from EcLon (PDB ID 6N2I, residues 447–465, red), BsLon (residues 445–463, green) and MtLon (residues 446–464, blue) are superimposed onto the corresponding fragments from TtClpB of both the NB1 (residues 296–302, yellow) and NB2 domains (residues 686–700, orange), as well as with the corresponding fragments from EcClpA of both the NB1 (residues 308–316, pink), and NB2 domains (residues 582–597, magenta).
RKH loops are located at the upper entry to the central pore and are expected to interact with the substrate at early stages of its approach to the AAA+ molecular machine, stabilizing the contacts during initial complex formation 55, 64. RKH loops of LonA proteases are much more similar in length to their structural equivalents in the NB2 domains of ClpA and ClpB chaperones, than to the shorter ones in their NB1 domains (Figs 9 and 11B).
Conclusions
The principal aim of this study was to provide structural support for the hypothesis that LonA proteases, which bear a single classical AAA+ module, may also contain a part of the second AAA+ module, which is present in full in the Clp enzymes of class I AAA+ proteins. A comparative analysis of the corresponding domains in the available structures of LonA proteases and ClpB chaperones revealed similarities on all four levels of structural organization of these proteins (from primary to quaternary). There is a strong indication that the architecture of the unique HI(CC) domain of LonA proteases resembles the structure of the H domain of the D1 AAA+ module of ClpB chaperones. The number of helices, their topology, and the presence of the long helix in the HI(CC) domain, the structural equivalent of which in the M domain of D1 is engaged in regulation of the necessary dynamic rearrangement of the subunits in a hexamer of ClpB 54, are similar in both proteins. However, discrepancies in the sizes of the corresponding helices would imply that their packing within the individual domains might be different.
The other argument in favor of this hypothesis comes from studies evaluating functional characteristics of an E. coli LonA mutant with deleted HI(CC) domain (residues 124–304) 29. The ATPase activity of this mutant was shown to decrease by more than an order of magnitude, and its proteolytic activity was almost totally lost. At the same time, the enzyme retained partial ability to hydrolyze peptide substrates (about 30%), although, unlike intact EcLon, binding of nucleotides and their complexes with magnesium ions did not affect the efficiency of peptide hydrolysis. It was concluded that the inserted HI(CC) domain is needed for formation of a functionally active enzyme, and the absence of this domain alters coupling between ATP hydrolysis and substrate proteolysis, as well as it affects interactions between LonA and a protein substrate.
This idea is also supported by comparison of the functionally important axial loops in the NB domain of the single AAA+ module of LonA with the corresponding ones in the NB1 and NB2 domains of two AAA+ modules of ClpA and ClpB chaperones, respectively. Significant results of that analysis are provided for the most conserved axial pore‐1 loops (GYVG) that are engaged in the interactions with substrate during all stages of protein function. A comparison of the fragment comprising this loop in LonA with the corresponding fragments of both the NB1 and NB2 domains of ClpA and ClpB chaperones unambiguously reveals a singular match in the location of the GYVG loop between the LonA NB domain and the NB2 domain of ClpB. This finding suggests that the mechanism of engagement of pore‐1 loops in these AAA+ modules of LonA and ClpB might be indeed similar. Our observations, combined with the data derived from the structure of ClpB with bound substrate 61 that designate the AAA+‐2 module as being the main motor of ClpB, give a new perspective to a functional meaning of the resemblance between the sole ATPase module of class II and the AAA+‐2 module of class I of the AAA+ proteins, and also correlate with the matching topology between the HI(CC) domain of LonAs and H1 domain of ClpBs.
In the absence of a structure of full‐length LonA protease, the structural elements that carry the functional or regulatory properties for these molecular machines cannot be accurately matched up between different partial structures. However, available structural data strongly support the hypothesis of their revised structural and functional relationship, thus suggesting that LonA proteases, exhibiting the structural features of both classes of AAA+ proteins, may represent a novel subclass of AAA+ proteins, distinctive from the established class I and class II.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
TVR and AG conceived this study and, together with AW, supervised its conduct. IB and ML performed the crystallographic experiments. AGA and AMK performed sequence alignments and generated the illustrations. All authors analyzed and discussed the data and contributed to writing the manuscript.
Acknowledgements
This work was supported in part with Federal funds from the National Cancer Institute, NIH, under contract HHSN261200800001E, by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by the Russian Foundation for Basic Research (project no. 19‐04‐00646). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government.
References
- 1. Gottesman S, Wickner S and Maurizi MR (1997) Protein quality control: triage by chaperones and proteases. Genes Dev 11, 815–823. [DOI] [PubMed] [Google Scholar]
- 2. Bukau B, Weissman J and Horwich A (2006) Molecular chaperones and protein quality control. Cell 125, 443–451. [DOI] [PubMed] [Google Scholar]
- 3. Saibil HR (2008) Chaperone machines in action. Curr Opin Struct Biol 18, 35–42. [DOI] [PubMed] [Google Scholar]
- 4. Buchberger A, Bukau B and Sommer T (2010) Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 40, 238–252. [DOI] [PubMed] [Google Scholar]
- 5. Gur E, Ottofueling R and Dougan DA (2013) Machines of destruction ‐ AAA+ proteases and the adaptors that control them. Subcell Biochem 66, 3–33. [DOI] [PubMed] [Google Scholar]
- 6. Balchin D, Hayer‐Hartl M and Hartl FU (2016) In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- 7. Neuwald AF, Aravind L, Spouge JL and Koonin EV (1999) AAA+: A class of chaperone‐like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9, 27–43. [PubMed] [Google Scholar]
- 8. Lupas AN and Martin J (2002) AAA proteins. Curr Opin Struct Biol 12, 746–753. [DOI] [PubMed] [Google Scholar]
- 9. Iyer LM, Leipe DD, Koonin EV and Aravind L (2004) Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol 146, 11–31. [DOI] [PubMed] [Google Scholar]
- 10. Striebel F, Kress W and Weber‐Ban E (2009) Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol 19, 209–217. [DOI] [PubMed] [Google Scholar]
- 11. Sauer RT and Baker TA (2011) AAA+ proteases: ATP‐fueled machines of protein destruction. Annu Rev Biochem 80, 587–612. [DOI] [PubMed] [Google Scholar]
- 12. Bittner LM, Arends J and Narberhaus F (2016) Mini review: ATP‐dependent proteases in bacteria. Biopolymers 105, 505–517. [DOI] [PubMed] [Google Scholar]
- 13. Wendler P, Ciniawsky S, Kock M and Kube S (2012) Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta 1823, 2–14. [DOI] [PubMed] [Google Scholar]
- 14. Rotanova TV, Melnikov EE, Khalatova AG, Makhovskaya OV, Botos I, Wlodawer A and Gustchina A (2004) Classification of ATP‐dependent proteases Lon and comparison of the active sites of their proteolytic domains. Eur J Biochem 271, 4865–4871. [DOI] [PubMed] [Google Scholar]
- 15. Rotanova TV, Melnikov EE and Tsirulnikov KB (2003) Catalytic dyad Ser‐Lys at the active site of Escherichia coli ATP‐dependent Lon‐proteinase. Russ J Bioorgan Chem 29, 85–87. [DOI] [PubMed] [Google Scholar]
- 16. Botos I, Melnikov EE, Cherry S, Tropea JE, Khalatova AG, Rasulova F, Dauter Z, Maurizi MR, Rotanova TV, Wlodawer A et al. (2004) The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser‐Lys dyad in the active site. J Biol Chem 279, 8140–8148. [DOI] [PubMed] [Google Scholar]
- 17. Rotanova TV and Melnikov EE (2010) A novel view on the architecture of the non‐catalytic N‐terminal region of ATP‐dependent LonA proteases. Biochemistry (Mosc) 4, 404–408. [Google Scholar]
- 18. Rotanova TV, Dergousova NI and Morozkin AD (2013) Unique structural organization of ATP‐dependent LonA proteases. Russ J Bioorgan Chem 39, 268–282. [DOI] [PubMed] [Google Scholar]
- 19. Ebel W, Skinner MM, Dierksen KP, Scott JM and Trempy JE (1999) A conserved domain in Escherichia coli Lon protease is involved in substrate discriminator activity. J Bacteriol 181, 2236–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roudiak SG and Shrader TE (1998) Functional role of the N‐terminal region of the Lon protease from Mycobacterium smegmatis . Biochemistry 37, 11255–11263. [DOI] [PubMed] [Google Scholar]
- 21. Vasilyeva OV, Martynova NI, Potapenko NA and Ovchinnikova TV (2004) Isolation and characterization of fragments of ATP‐dependent protease Lon from Escherichia coli obtained by limited proteolysis. Russ J Bioorgan Chem 30, 306–314. [PubMed] [Google Scholar]
- 22. Melnikov EE, Andrianova AG, Morozkin AD, Stepnov AA, Makhovskaya OV, Botos I, Gustchina A, Wlodawer A and Rotanova TV (2008) Limited proteolysis of E. coli ATP‐dependent protease Lon ‐ a unified view of the subunit architecture and characterization of isolated enzyme fragments. Acta Biochim Pol 55, 281–296. [PMC free article] [PubMed] [Google Scholar]
- 23. Vieux EF, Wohlever ML, Chen JZ, Sauer RT and Baker TA (2013) Distinct quaternary structures of the AAA+ Lon protease control substrate degradation. Proc Natl Acad Sci USA 110, E2002–E2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wohlever ML, Baker TA and Sauer RT (2014) Roles of the N domain of the AAA+ Lon protease in substrate recognition, allosteric regulation and chaperone activity. Mol Microbiol 91, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee AY, Hsu CH and Wu SH (2004) Functional domains of Brevibacillus thermoruber Lon protease for oligomerization and DNA binding: role of N‐terminal and sensor and substrate discrimination domains. J Biol Chem 279, 34903–34912. [DOI] [PubMed] [Google Scholar]
- 26. Chir JL, Liao JH, Lin YC and Wu SH (2009) The N‐terminal sequence after residue 247 plays an important role in structure and function of Lon protease from Brevibacillus thermoruber WR‐249. Biochem Biophys Res Commun 382, 762–765. [DOI] [PubMed] [Google Scholar]
- 27. Cheng I, Mikita N, Fishovitz J, Frase H, Wintrode P and Lee I (2012) Identification of a region in the N‐terminus of Escherichia coli Lon that affects ATPase, substrate translocation and proteolytic activity. J Mol Biol 418, 208–225. [DOI] [PubMed] [Google Scholar]
- 28. Kudzhaev AM, Dubovtseva ES, Serova OV, Adrianova AG and Rotanova TV (2016) Influence of the (1–106) fragment of Escherichia coli Lon protease on the enzyme function and DNA binding. Russ J Bioorgan Chem 42, 381–388. [Google Scholar]
- 29. Kudzhaev AM, Andrianova AG, Dubovtseva ES, Serova OV and Rotanova TV (2017) Role of the inserted α‐helical domain in E. coli ATP‐dependent Lon protease function. Acta Naturae 9, 75–81. [PMC free article] [PubMed] [Google Scholar]
- 30. Kudzhaev A, Dubovtseva E, Serova O, Andrianova A and Rotanova T (2018) Effect of the deletion of the (173–280) fragment of the inserted α‐helical domain on the functional properties of ATP‐dependent Lon protease from E. coli . Russ J Bioorgan Chem 44, 518–527. [Google Scholar]
- 31. Li M, Rasulova F, Melnikov EE, Rotanova TV, Gustchina A, Maurizi MR and Wlodawer A (2005) Crystal structure of the N‐terminal domain of E. coli Lon protease. Protein Sci 14, 2895–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li M, Gustchina A, Rasulova FS, Melnikov EE, Maurizi MR, Rotanova TV, Dauter Z and Wlodawer A (2010) Structure of the N‐terminal fragment of E. coli Lon protease. Acta Crystallogr D66, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duman RE and Lowe J (2010) Crystal structures of Bacillus subtilis Lon protease. J Mol Biol 401, 653–670. [DOI] [PubMed] [Google Scholar]
- 34. Lin CC, Su SC, Su MY, Liang PH, Feng CC, Wu SH and Chang CI (2016) Structural insights into the allosteric operation of the Lon AAA+ protease. Structure 24, 667–675. [DOI] [PubMed] [Google Scholar]
- 35. Rawlings ND, Barrett AJ and Finn R (2016) Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 44, D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Otwinowski Z and Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 37. Kelley LA, Mezulis S, Yates CM, Wass MN and Sternberg MJ (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC and Read RJ (2007) Phaser crystallographic software. J Appl Cryst 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al. (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emsley P and Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 41. Botos I, Melnikov EE, Cherry S, Khalatova AG, Rasulova FS, Tropea JE, Maurizi MR, Rotanova TV, Gustchina A and Wlodawer A (2004) Crystal structure of the AAA+ α domain of E. coli Lon protease at 1.9 Å resolution. J Struct Biol 146, 113–122. [DOI] [PubMed] [Google Scholar]
- 42. Brunger AT (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. [DOI] [PubMed] [Google Scholar]
- 43. Ye J, Osborne AR, Groll M and Rapoport TA (2004) RecA‐like motor ATPases–lessons from structures. Biochim Biophys Acta 1659, 1–18. [DOI] [PubMed] [Google Scholar]
- 44. Guo F, Maurizi MR, Esser L and Xia D (2002) Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J Biol Chem 277, 46743–46752. [DOI] [PubMed] [Google Scholar]
- 45. Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J and Shi Y (2011) Structure and mechanism of the hexameric MecA‐ClpC molecular machine. Nature 471, 331–335. [DOI] [PubMed] [Google Scholar]
- 46. Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M and Tsai FT (2003) The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 115, 229–240. [DOI] [PubMed] [Google Scholar]
- 47. Donaldson LW, Wojtyra U and Houry WA (2003) Solution structure of the dimeric zinc binding domain of the chaperone ClpX. J Biol Chem 278, 48991–48996. [DOI] [PubMed] [Google Scholar]
- 48. Derre I, Rapoport G, Devine K, Rose M and Msadek T (1999) ClpE, a novel type of HSP100 ATPase, is part of the CtsR heat shock regulon of Bacillus subtilis . Mol Microbiol 32, 581–593. [DOI] [PubMed] [Google Scholar]
- 49. Scharfenberg F, Serek‐Heuberger J, Coles M, Hartmann MD, Habeck M, Martin J, Lupas AN and Alva V (2015) Structure and evolution of N domains in AAA metalloproteases. J Mol Biol 427, 910–923. [DOI] [PubMed] [Google Scholar]
- 50. Gottesman S, Squires C, Pichersky E, Carrington M, Hobbs M, Mattick JS, Dalrymple B, Kuramitsu H, Shiroza T, Foster T et al. (1990) Conservation of the regulatory subunit for the Clp ATP‐dependent protease in prokaryotes and eukaryotes. Proc Natl Acad Sci USA 87, 3513–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schirmer EC, Glover JR, Singer MA and Lindquist S (1996) HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem Sci 21, 289–296. [PubMed] [Google Scholar]
- 52. Mogk A, Haslberger T, Tessarz P and Bukau B (2008) Common and specific mechanisms of AAA+ proteins involved in protein quality control. Biochem Soc Trans 36, 120–125. [DOI] [PubMed] [Google Scholar]
- 53. Zolkiewski M (2006) A camel passes through the eye of a needle: protein unfolding activity of Clp ATPases. Mol Microbiol 61, 1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carroni M, Kummer E, Oguchi Y, Wendler P, Clare DK, Sinning I, Kopp J, Mogk A, Bukau B and Saibil HR (2014) Head‐to‐tail interactions of the coiled‐coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. Elife 3, e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Glynn SE, Martin A, Nager AR, Baker TA and Sauer RT (2009) Structures of asymmetric ClpX hexamers reveal nucleotide‐dependent motions in a AAA+ protein‐unfolding machine. Cell 139, 744–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li J and Sha B (2002) Crystal structure of E. coli Hsp100 ClpB nucleotide‐binding domain 1 (NBD1) and mechanistic studies on ClpB ATPase activity. J Mol Biol 318, 1127–1137. [DOI] [PubMed] [Google Scholar]
- 57. Martin A, Baker TA and Sauer RT (2008) Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor‐dependent recognition of ssrA‐tagged substrates. Mol Cell 29, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Siddiqui SM, Sauer RT and Baker TA (2004) Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates. Genes Dev 18, 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Iosefson O, Olivares AO, Baker TA and Sauer RT (2015) Dissection of axial‐pore loop function during unfolding and translocation by a AAA+ proteolytic machine. Cell Rep 12, 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iosefson O, Nager AR, Baker TA and Sauer RT (2015) Coordinated gripping of substrate by subunits of a AAA+ proteolytic machine. Nat Chem Biol 11, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Deville C, Carroni M, Franke KB, Topf M, Bukau B, Mogk A and Saibil HR (2017) Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci Adv 3, e1701726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lum R, Tkach JM, Vierling E and Glover JR (2004) Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem 279, 29139–29146. [DOI] [PubMed] [Google Scholar]
- 63. Martin A, Baker TA and Sauer RT (2007) Distinct static and dynamic interactions control ATPase‐peptidase communication in a AAA+ protease. Mol Cell 27, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Farrell CM, Baker TA and Sauer RT (2007) Altered specificity of a AAA+ protease. Mol Cell 25, 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS and Richardson DC (2010) MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]