

Abstract
Alcoholic fatty liver disease is often complicated by other pathologic insults, such as viral infection or high-fat diet. Autophagy plays a homeostatic role in the liver but can be compromised by alcohol, high-fat diet, or viral infection, which in turn affects the disease process caused by these etiologies. To understand the full impact of autophagy modulation on alcohol-induced liver injury, several genetic models of autophagy deficiency, which have different levels of functional alterations, were examined after acute binge or chronic-plus-binge treatment. Mice given alcohol with either mode and induced with deficiency in liver-specific Atg7 shortly after the induction of Atg7 deletion had elevated liver injury, indicating the protective role of autophagy. Constitutive hepatic Atg7–deficient mice, in which Atg7 was deleted in embryos, were more susceptible with chronic-plus-binge but not with acute alcohol treatment. Constitutive hepatic Atg5–deficient mice, in which Atg5 was deleted in embryos, were more susceptible with acute alcohol treatment, but liver injury was unexpectedly improved with the chronic-plus-binge regimen. A prolonged autophagy deficiency may complicate the hepatic response to alcohol treatment, likely in part due to endogenous liver injury. The complexity of the relationship between autophagy deficiency and alcohol-induced liver injury can thus be affected by the timing of autophagy dysfunction, the exact autophagy gene being affected, and the alcohol treatment regimen.
The liver plays a crucial role in metabolism in the body.1 When metabolic homeostasis is disturbed in the liver, fatty liver disease (FLD), which is characterized by steatosis, inflammation, and fibrosis, can occur. FLD can be subclassified as being caused by excessive alcohol intake (alcoholic fatty liver disease; AFLD) or by any non–alcohol-related etiology (nonalcoholic fatty liver disease; NAFLD).2
Autophagy is an evolutionarily conserved cellular degradation process that involves the delivery of cytoplasmic cargo (macromolecules or organelles) to the lysosome.3 Three types of autophagy have been defined: macroautophagy, microautophagy, and chaperone-mediated autophagy.4 Macroautophagy is quantitatively the most active form of autophagy and is referred to hereafter as autophagy. In macroautophagy, cytosolic materials are sequestered inside the autophagosome, and transported to, and degraded in, the lysosome.5 Autophagy occurs at the basal level in the liver and can be further enhanced under conditions of stress. It can play an important role in the maintenance of normal liver function and in the pathogenesis of various liver diseases.6, 7 In particular, autophagy could be activated or depressed in such common liver diseases as AFLD,8, 9, 10, 11, 12 NAFLD,13, 14, 15 and hepatitis viral infection.16, 17, 18, 19
The pathologic processes of AFLD are considered to be related to oxidative stress caused by the accumulation of acetaldehyde, increased NADH/NAD+ ratio, or generation of reactive oxidative species.20, 21, 22 During the progress of AFLD, oxidative stress may induce functional and structural mitochondrial changes that may affect oxidative phosphorylation, increase mitochondrial DNA damage, and alter mitochondrial protein profiles.23, 24, 25, 26, 27, 28 Increased reactive oxygen species, together with alcoholic steatosis, lead to lipid peroxidation that can further enhance oxidative damage in AFLD.29 As shown in previous studies, the effects of autophagy can vary at different pathologic stages of AFLD.8, 9, 12, 15, 30 Autophagy is activated in the liver in vivo and in cultured primary hepatocytes after acute alcohol treatment.8, 9, 30 This activation requires alcohol metabolism, and is mediated by reactive oxygen species and several signaling pathways. Acetaldehyde, a major alcohol metabolite and a pro-oxidant, has been implicated in the induction of autophagy.31 Meanwhile, alcohol-induced autophagy can be suppressed by antioxidants, such as N-acetyl cysteine.8, 32 In addition, alcohol treatment alters the mammalian target of rapamycin, 5′ AMP-activated protein kinase, and/or forkhead box O3a signaling pathway,33, 34, 35 which can contribute to changes in the autophagy process. Other autophagy stimulators, including alcohol-induced endoplasmic reticulum stress, proteasome inhibition, and minerals such as zinc, are also implicated in the dynamics of autophagy after alcohol treatment.36, 37, 38
Interestingly, autophagy may be suppressed in chronic alcohol treatment or in other liver diseases. When mice were fed with a Lieber-DeCarli diet for 4 weeks, hepatic autophagy was stimulated at a lower dose of alcohol (accounting for 29% of the caloric need) but was inhibited at a higher alcohol dose (accounting for 36% of the caloric need).15 The suppression of autophagy by long-term alcohol use suggests that the promotion of autophagy can improve the condition. The cause of the autophagy suppression may include decreases in both the amount and the functioning of lysosomes, which were found in long-term alcohol–treated rat livers.10, 11, 39 Indeed, the findings from a recent study suggest that the level of transcription factor EB, which plays a crucial role in lysosomal biogenesis and autophagy, was decreased in the livers of mice given alcohol diet and in patients with alcohol-induced hepatitis.12 Disruption of hepatic transcription factor EB enhanced alcohol-induced liver injury in mice, but the increase in transcription factor EB level led to reduced alcohol-induced liver injury through elevated lysosomal biogenesis and mitochondrial bioenergetics.12
Nonetheless, the assessment of the contribution of autophagy to alcohol-induced liver injury had been mainly conducted through acute pharmacologic manipulation or acute knockout of certain autophagy-related genes. The impact of chronic autophagy deficiency on alcoholic liver injury has not been assessed. This effect may be important as ALDs are often complicated by other disease-causing factors, such as viral infection or high-fat diet, which can compromise autophagy. The present study thus used several genetic models of autophagy deficiency to assess the effects of alcohol in different autophagy functional statuses. The findings suggest that the complicated interaction between alcohol and autophagy in liver injury can be affected by the timing of autophagy dysfunction, the exact autophagy gene being affected, and the alcohol treatment regimen.
Materials and Methods
Mice
Atg5F/F mice (B6.129S-Atg5tm1Myok)40 and Atg7F/F mice41 have been reported in previous studies. Atg5Δhep and Atg7Δhep mice were generated by crossing Atg5F/F or Atg7F/F with Alb:Cre transgenic mice, respectively (The Jackson Laboratory, Bar Harbor, ME). The inducible liver-specific Atg7-deficient mice (Atg7Δhep-ERT2) were generated by crossing Atg7F/F mice with Alb:Cre-ERT2 mice.42 The deletion of Atg7 was induced by the administration of tamoxifen (6 mg/day s.c., for 2 days). Mice were maintained on a 12-hour dark/light cycle with free access to food and water. Both male and female mice were used in the studies. Age- and sex-matched mice were randomly assigned to the treatment or control group.
Animal Models
Acute alcohol binge was conducted as previously described.8 After 6 hours of fasting, mice were given 31.5% (v/v) alcohol by gavage at a total accumulative dosage of 5 g/kg of body weight in four equally divided doses at 20-minute intervals. Atg7Δhep-ERT2 and control Atg7F/F mice were injected with tamoxifen (6 mg/day s.c., for 2 days) 7 days before they were given acute alcohol binge as described in the previous sentence or as indicated in the figure legends. The control mice were given the same volume of water.
The chronic-plus-binge model was conducted as previously described.43 Mice were acclimated with liquid control food for 5 days. The alcohol-treated groups were then given a liquid diet containing 5% (w/v) alcohol for 10 days. Pair-fed groups were given the same volume of control food. In the early morning of day 11, mice in the alcohol-treated groups were given a single dose of 31.5% (v/v) alcohol (5 g/kg of body weight) by gavage. Pair-fed mice were given an isocaloric dose of dextrin maltose by gavage. After 9 hours, the mice were euthanized, and blood and tissue samples were collected.
The protocols of all animal experiments were approved by the Institutional Animal Care and Use Committee of Indiana University (Indianapolis, Indiana).
Antibodies and Chemicals
Antibodies and PCR primers used in this study are listed in Tables 1 and 2, respectively. Tamoxifen (Sigma-Aldrich, St. Louis, MO) was diluted in corn oil. Alcohol (190 proof; Decon Labs, King of Prussia, PA) was diluted in water or added to a formulated liquid diet.43 Bio-Serv Lieber-DeCarli '82 Shake and Pour alcohol liquid diet (catalog number F1258SP; Fisher Scientific, Pittsburgh, PA) was used as the alcohol diet. Bio-Serv Lieber-DeCarli '82 Shake and Pour control liquid diet (catalog number F1259SP; Fisher Scientific) was used as the control diet.
Table 1.
Antibody List
| Antibody name | Company | Catalog number | Host |
|---|---|---|---|
| ACTIN (8H10D10) | Cell Signaling Technology (Beverly, MA) | 3700 | Mouse |
| ADH | Santa Cruz Biotechnology (Dallas, TX) | sc-133207 | Mouse |
| ALDH1/2 | Santa Cruz Biotechnology | sc-166362 | Mouse |
| ATG12 | Cell Signaling Technology | 2011 | Rabbit |
| ATG7 | Cell Signaling Technology | 2631 | Rabbit |
| CK19 | Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA) | TROMA-III | Rat |
| CYP2E1 | Enzo Life Sciences (Lausen, Switzerland) | BML-CR3271 | Rabbit |
| Gapdh | Novus Biologicals (Littleton, CO) | NB 300-221 | Mouse |
| LC3B | Sigma-Aldrich (St. Louis, MO) | L7543 | Rabbit |
| NQO1 | Abcam (Cambridge, MA) | ab34173 | Rabbit |
| SQSTM1/p62 | Abnova (Taipei, Taiwan) | H00008878-M01 | Mouse |
| α-Smooth muscle actin | Thermo Fisher Scientific (Waltham, MA) | PA5-19465 | Rabbit |
Adh, alcohol dehydrogenase; Aldh, aldehyde dehydrogenase; ATG, autophagy-related protein; CK, cytokeratin; CYP, cytochrome P450; Gapdh, glyceraldehyde phosphate dehydrogenase; LC, microtubule-associated protein 1A/1B-light chain; NQO, NAD(P)H quinone dehydrogenase; Sqstm, sequestosome.
Table 2.
Primer List
| Gene/primer name | Sequence (forward) | Sequence (reverse) |
|---|---|---|
| Acaca | 5′-GCCTCTTCCTGACAAACGAG-3′ | 5′-TGACTGCCGAAACATCTCTG-3′ |
| Acadl | 5′-GGTGGAAAACGGAATGAAAGG-3′ | 5′-GGCAATCGGACATCTTCAAAG-3′ |
| Acadm | 5′-TGTTAATCGGTGAAGGAGCAG-3′ | 5′-CTATCCAGGGCATACTTCGTG-3′ |
| Acox1 | 5′-CATATGACCCCAAGACCCAAG-3′ | 5′-CATGTAACCCGTAGCACTCC-3′ |
| Actin | 5′-ACTATTGGCAACGAGCGGTT-3′ | 5′-CAGGATTCCATACCCAAGAAGGA-3′ |
| Adh1 | 5′-TGTTGAGAGCGTTGGAGAAG-3′ | 5′-CGCTTCGGCTACAAAAGTTG-3′ |
| Adh4 | 5′-GTAGACTCTGTCCCAAACCTG-3′ | 5′-AAGGTCAGGATTGTTCGGATG-3′ |
| Adh5 | 5′-AAATCTCCACTCGTCCATTCC-3′ | 5′-CACTCTCCACACTCTTCCATC-3′ |
| Adh7 | 5′-GGTTGTGGAGAGTGTTGGAG-3′ | 5′-CCTGTCAGATCGCTCCTAATG-3′ |
| Akr1d1 | 5′-CTCATTGGGCTTGGAACCTA-3′ | 5′-CATTGATGGGACATGCTCTG-3′ |
| Aldh16a1 | 5′-GAGGTTCGAGATGGAGATGTG-3′ | 5′-AAAGAGAAAGGAGTCGGCAG-3′ |
| Aldh18a1 | 5′-CAGACATCGTGGAGGGAAAG-3′ | 5′-CTGTTCAGGCTCTAAGGTAGC-3′ |
| Aldh1a1 | 5′-ATCACTGTGTCATCTGCTCTG-3′ | 5′-CCCAGTTCTCTTCCATTTCCAG-3′ |
| Aldh1a2 | 5′-ATGGATGCGTCTGAAAGAGG-3′ | 5′-TGACTCCCTGCAAATCGATG-3′ |
| Aldh1a3 | 5′-AACAAGATAGCCTTCACCGG-3′ | 5′-CCAAGTCCAAGTCAGCATCT-3′ |
| Aldh1b1 | 5′-GGAGTCTTATGTCTTGGATCTGG-3′ | 5′-TGTCGGGTGAAGCAGAAATG-3′ |
| Aldh1l1 | 5′-CATCCAGACCTTCCGATACTTC-3′ | 5′-ACAATACCACAGACCCCAAC-3′ |
| Aldh1l2 | 5′-CATTGACAGCCCAAAGCATG-3′ | 5′-CCCAGAAAACAGAAAACCCAG-3′ |
| Aldh2 | 5′-TGCAGGAGAATGTGTATGACG-3′ | 5′-CGATTTGATGTAGCCGAGGATC-3′ |
| Aldh3a1 | 5′-GGCGTGGTCCTTGTCATAG-3′ | 5′-AGGGATAAGTGTTGAAAGCAGG-3′ |
| Aldh3a2 | 5′-AGCCCTTGTTACATTGACAGAG-3′ | 5′-TTCATGTACTTTCCCCAGGC-3′ |
| Aldh3b1 | 5′-CCTTCGGTCTGGTGCTTATC-3′ | 5′-ACCTCAGCCAGTATCTTTTCAG-3′ |
| Aldh3b2 | 5′-GAATCAGATGTTGGAACGCAC-3′ | 5′-GGAGAAGGTGTCAAAGGAGAAC-3′ |
| Aldh4a1 | 5′-CGATAAGTCTACTGGGTCTGTG-3′ | 5′-GGGCTTATGAGTCTCCTTGATG-3′ |
| Aldh5a1 | 5′-CCAAGATCATAACAGCCGAGAG-3′ | 5′-TCGCTTATCTTTGGCAGAGG-3′ |
| Aldh6a1 | 5′-AAGGGAAGACTCTTGCTGATG-3′ | 5′-GAGGCAGACGGTAGGAATAAAG-3′ |
| Aldh7a1 | 5′-AGATATTCCTGCCCCAAAACG-3′ | 5′-CTGAACCTCGCCTATTCCTTC-3′ |
| Aldh8a1 | 5′-GTATGCATTACACCGTTCGC-3′ | 5′-AAGTCATCTCACTGGGCTTG-3′ |
| Aldh9a1 | 5′-CTGGAATACTATGCAGGGCTG-3′ | 5′-GCGATCTGGAAGGGATAGTTC-3′ |
| Apob | 5′-ATTCGAGCACAGATGACCAG-3′ | 5′-GTACCTTTCACCATCAGACTCC-3′ |
| Apoe | 5′-CAATTGCGAAGATGAAGGCTC-3′ | 5′-TAATCCCAGAAGCGGTTCAG-3′ |
| Bsep | 5′-CTGCCAAGGATGCTAATGCA-3′ | 5′-CGATGGCTACCCTTTGCTTCT-3′ |
| Cat | 5′-CTCGTTCAGGATGTGGTTTTC-3′ | 5′-CTTTCCCTTGGAGTATCTGGTG-3′ |
| Ccl2 | 5′-GTCCCTGTCATGCTTCTGG-3′ | 5′-GCTCTCCAGCCTACTCATTG-3′ |
| Ccl3 | 5′-TTCTCTGTACCATGACACTCTGC-3′ | 5′-CGTGGAATCTTCCGGCTGTAG-3′ |
| Ccr2 | 5′-GCTCTACATTCACTCCTTCCAC-3′ | 5′-ACCACTGTCTTTGAGGCTTG-3′ |
| Cd36 | 5′-GCGACATGATTAATGGCACAG-3′ | 5′-GATCCGAACACAGCGTAGATAG-3′ |
| Col1a1 | 5′-ACGGCTGCACGAGTCACAC-3′ | 5′-GGCAGGCGGGAGGTCTT-3′ |
| Ctgf | 5′-GGGCCTCTTCTGCGATTTC-3′ | 5′-ATCCAGGCAAGTGCATTGGTA-3′ |
| Cyp27a1 | 5′-GCCTCACCTATGGGATCTTCA-3′ | 5′-TCAAAGCCTGACGCAGATG-3′ |
| Cyp2e1 | 5′-TCACTGGACATCAACTGCC-3′ | 5′-TGGTCTCTGTTCCTGCAAAG-3′ |
| Cyp3a11 | 5′-CAGAAGCACCGAGTGGATTT-3′ | 5′-GACTGGGCTGTGATCTCCAT-3′ |
| Cyp7a1 | 5′-AACAACCTGCCAGTACTAGATAGC-3′ | 5′-GTGTAGAGTGAAGTCCTCCTTAGC-3′ |
| Cyp7b1 | 5′-CAGCTATGTTCTGGGCAATG-3′ | 5′-TCGGATGATGCTGGAGTATG-3′ |
| Cyp8b1 | 5′-AGTACACATGGACCCCGACATC-3′ | 5′-GGGTGCCATCCGGGTTGAG-3′ |
| F4/80 | 5′-TGCATCTAGCAATGGACAGC-3′ | 5′-GCCTTCTGGATCCATTTGAA-3′ |
| Fabp1 | 5′-TCTCCGGCAAGTACCAATTG-3′ | 5′-TTGATGTCCTTCCCTTTCTGG-3′ |
| Fasn | 5′-CCCTTGATGAAGAGGGATCA-3′ | 5′-ACTCCACAGGTGGGAACAAG-3′ |
| Fgfr4 | 5′-CTGCCAGAGGAAGACCTCAC-3′ | 5′-GTAGTGGCCACGGATGACTT-3′ |
| Fxr (Nr1h4) | 5′-GGCCTCTGGGTACCACTACA-3′ | 5′-TGTACACGGCGTTCTTGGTA-3′ |
| Gfap | 5′-GAAAACCGCATCACCATTCC-3′ | 5′-CTTAATGACCTCACCATCCCG-3′ |
| Gstm1 | 5′-ACTTGATTGATGGGGCTCAC-3′ | 5′-TCTCCAAAATGTCCACACGA-3′ |
| Hsd3b7 | 5′-CCATCCACAAAGTCAACGTG-3′ | 5′-CTCCATTGACCTTCCTTCCA-3′ |
| Ifng | 5′-GATGCATTCATGAGTATTGCCAAGT-3′ | 5′-GTGGACCACTCGGATGAGCTC-3′ |
| Il6 | 5′-AGTTGCCTTCTTGGGACTGA-3′ | 5′-TCCACGATTTCCCAGAGAAC-3′ |
| Lcn2 | 5′-CTACAATGTCACCTCCATCCTG-3′ | 5′-ACCTGTGCATATTTCCCAGAG-3′ |
| Lipe (Hsl) | 5′-CTGAGATTGAGGTGCTGTCG-3′ | 5′-CAAGGGAGGTGAGATGGTAAC-3′ |
| Lpl | 5′-AACAAGGTCAGAGCCAAGAG-3′ | 5′-CCATCCTCAGTCCCAGAAAAG-3′ |
| Ly6g | 5′-CACCTGAGACTTCCTGCAAC-3′ | 5′-CTTCTATCTCCAGAGCAACGC-3′ |
| Mrp2 (Abcc2) | 5′-GCACTGTAGGCTCTGGGAAG-3′ | 5′-TGCTGAGGGACGTAGGCTAT-3′ |
| Mrp3 | 5′-GGACTTCCAGTGCTCAGAGG-3′ | 5′-AGCTGTGGCCTCGTCTAAAA-3′ |
| Mrp4 | 5′-TGTTTGATGCACACCAGGAT-3′ | 5′-GACAAACATGGCACAGATGG-3′ |
| Nqo1 | 5′-GCACTGATCGTACTGGCTCA-3′ | 5′-CATGGCATAGAGGTCCGACT-3′ |
| Osta | 5′-GTCTCAAGTGATGAACTGCCA-3′ | 5′-TTGAGTGCTGAGTCCAGGTC-3′ |
| Ostb | 5′-GTATTTTCGTGCAGAAGATGCG-3′ | 5′-TTTCTGTTTGCCAGGATGCTC-3′ |
| Pnpla2 (Atgl) | 5′-ATATCCCACTTTAGCTCCAAGG-3′ | 5′-CAAGTTGTCTGAAATGCCGC-3′ |
| Pparg | 5′-ATGCCAGTACTGCCGTTTTC-3′ | 5′-GGCCTTGACCTTGTTCATGT-3′ |
| Saa2 | 5′-CAGGATGAAGCTACTCACCAG-3′ | 5′-CTTCATGTCAGTGTAGGCTCG-3′ |
| Scd1 | 5′-TTCTTACACGACCACCACCA-3′ | 5′-CCGAAGAGGCAGGTGTAGAG-3′ |
| Slc10a1 (Ntcp) | 5′-CACCATGGAGTTCAGCAAGA-3′ | 5′-CCAGAAGGAAAGCACTGAGG-3′ |
| Slco1a1 (Oatp1) | 5′-ATCCAGTGTGTGGGGACAAT-3′ | 5′-GCAGCTGCAATTTTGAAACA-3′ |
| Tgfb1 | 5′-CACCGGAGAGCCCTGGATA-3′ | 5′-TGTACAGCTGCCGCACACA-3′ |
| Timp1 | 5′-ATTCAAGGCTGTGGGAAATG-3′ | 5′-CTCAGAGTACGCCAGGGAAC-3′ |
| Tlr4 | 5′-TTCAGAACTTCAGTGGCTGG-3′ | 5′-TGTTAGTCCAGAGAAACTTCCTG-3′ |
Fxr, farnesoid X receptor; Hsl, hormone-sensitive lipase; Mrp, multidrug-resistance protein.
Serum Biochemistry Analysis
Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), triglycerides (TG), and total cholesterol (TCHO) were measured using kits from Pointe Scientific (Canton, MI) according to the manufacturer's protocol. Serum total bile acids (TBA) were measured using a TBA assay kit from Diazyme Laboratories, Inc. (Poway, CA).
Hepatic Lipid Content Analysis
A piece of liver tissue was weighed and incubated in 1 mL of chloroform-methanol mix (2:1, v/v) with shaking for 1 hour at room temperature to extract lipids. After the addition of 200 μL of water, samples were vortexed and centrifuged at 3000 × g for 5 minutes. The lower lipid phase was collected and dried at room temperature in a chemical hood. The lipid pellet was resuspended in 60 μL of tert-butanol and 40 μL of a Triton X-114 methanol (2:1, v/v) mix. TG and TCHO contents were measured using respective kits from Pointe Scientific. The lipid contents hence were normalized with the tissue weight.
Hepatic TBA Content Analysis
Liver tissue samples (100 mg) were homogenized in 1 mL of 90% alcohol and incubated at 55°C overnight. The lysates were centrifuged at 9600 × g for 10 minutes. The supernatants were collected and measured for TBA concentration using a TBA assay kit from Diazyme Laboratories, Inc.
Immunoblotting Analysis
The liver samples were homogenized in the radio immunoprecipitation assay buffer containing a protease cocktail. Supernatant was collected after centrifugation at 13,800 × g for 12 minutes. Protein concentration was determined using a Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA). The proteins were separated on SDS-PAGE. Proteins were transferred onto polyvinylidene fluoride membranes, which were then blocked with 5% bovine serum albumin or 5% skim milk for 1 hour at room temperature. Membranes were incubated overnight with the appropriate primary antibody and then washed with Tris-buffered saline and 0.1% Tween 20 before being incubated with the horseradish peroxidase–coupled secondary antibodies at room temperature. Protein bands were detected using an enhanced chemiluminescence kit (Thermo Fisher Scientific/Pierce). The images were taken digitally with the ChemiDoc Imaging System (Bio-Rad, Hercules, CA). Densitometry was measured using the companion software, Image Lab software version 6.0.1 (Bio-Rad), and the values were normalized to that of β-actin or glyceraldehyde phosphate dehydrogenase, which were then converted to fold changes of the control.
RNA Isolation and Real-Time Quantitative PCR Analysis
Total RNA was prepared from liver samples using GeneJET RNA Purification Kit (Thermo Fisher Scientific, Grand Island, NY) according to the manufacturer's protocols. cDNA was synthesized using oligo dT primers and an M-MLV Reverse Transcriptase System (Life Technologies, Carlsbad, CA). Real-time quantitative PCR analysis was performed on an Applied Biosystems 7500 Real-Time PCR System (Life Technologies) using SYBR Green master mixes (Life Technologies). All real-time quantitative PCR results were normalized to the level of β-actin, and the gene expression was calculated using the 2−ΔΔCτ method.
Histologic Study
Liver samples were harvested, rinsed with phosphate-buffered saline, and fixed in 10% formalin overnight. Samples were further fixed in 70% alcohol and processed as paraffin-embedded blocks. The paraffin-embedded tissues were sectioned and stained with hematoxylin and eosin, anti-F4/80, or Masson's trichrome C. Photomicrographs were taken using an Eclipse E200 light microscope (Nikon Instruments, Melville, NY) equipped with a SPOT RT Slider color digital camera (Diagnostic Instruments, Inc., Sterling Heights, MI). Area with positive F4/80 or Masson's trichrome C staining were quantified using ImageJ software version 1.51 (NIH, Bethesda, MD; http://imagej.nih.gov/ij); at least four random fields of each section from each mouse liver were used for quantification.
Immunofluorescence Microscopy
Paraffin sections were subjected to antigen retrieval treatment using the citrate buffer (0.01 mol/L, pH 6.0) after deparaffinization. Slides were blocked with 5% goat serum in phosphate-buffered saline containing 0.1% Triton X (PBS-Tx) for 1 hour and then incubated with primary antibodies diluted in 1% bovine serum albumin/PBS-Tx overnight at 4°C. Sections were washed with phosphate-buffered saline, followed by incubation with fluorochrome-conjugated secondary antibodies. Hoechst 33342 was used for nucleus staining. Images were obtained using as Eclipse TE 200 epi-immunofluorescence microscope (Nikon) and the companion NIS-Elements software version AR3.2 (Nikon). Quantification was performed using ImageJ software, at least four random fields of each section from each mouse liver were analyzed.
Alcohol Clearance Analysis
Mice were given a single dose of 31.5% (v/v) alcohol by gavage (5 g/kg of body weight). Blood was sampled from the tail vein before treatment and at 1, 3, and 9 hours after gavage. Alcohol concentration in the plasma was measured using an alcohol colorimetric/fluorometric assay kit (BioVision Inc., Milpitas, CA).
Statistical Analysis
Statistical analysis was performed using SPSS software version 17.0 (SPSS, Inc., Chicago, IL) or Excel 2016 (Microsoft Corp., Redmond, WA). The t-test was used to determine the significance of differences between two groups. The significance of differences between more than two treatment groups was determined using one-way analysis of variance followed by the Duncan post-hoc test. Data are expressed as means ± SEM. Results were considered statistically significant at P < 0.05.
Results
Effects of Genetic Deletion of a Key Autophagy Gene on Liver Injury Induced by Acute Alcohol Binge
To evaluate the effects of hepatic autophagy deficiency on ALDs, one single binge-dose of alcohol was administered to the Atg5Δhep mice and the control mice (Figure 1A). Hepatic Atg5 deficiency was associated with significant hepatomegaly and liver injury (Figure 1B). Serum levels of ALT, AST, and ALP, and hepatic TG, but not hepatic TCHO, were further elevated in Atg5Δhep mice after alcohol treatment (Figure 1B), suggesting that acute alcohol treatment exacerbated the liver pathology in these mice. A protective function of autophagy against hepatic steatosis induced by acute alcohol administration has been shown in several studies.8, 15, 30, 31
Figure 1.

Genetic deletion of autophagy genes exacerbates liver injury induced by acute alcohol binge. A: Scheme of acute binge treatment. Mice were treated with a single dose of alcohol (ethanol) or the same volume of distilled water (ddH2O) by gavage after 6 hours of fasting. Mice were euthanized 16 hours later. B and C: Liver weight and body weight (L/B) ratio, the serum levels of ALT, AST, ALP, and the hepatic levels of triglycerides (TG) and total cholesterol (TCHO) were measured in liver-specific Atg5-deficient mice 8 to 34 weeks old (B) and Atg7-deficient mice 8 to 16 weeks old (C). D: Scheme of the administration of alcohol to Atg7Δhep-ERT2 mice. Tamoxifen (TMX) was injected on days 1 and 2 to induce the deletion of Atg7. Alcohol or water was then given by oral gavage on day 8. Atg7F/F mice were used as control. E: L/B weight ratio, the serum levels of ALT, AST, ALP, and the hepatic levels of TG and TCHO were measured in induced Atg7Δhep-ERT2 mice 8 to 15 weeks old. Data are expressed as means ± SEM. n = 3 to 5 (E); n = 3 to 6 (C); n = 7 to 14 (B). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
Genetic deletion of Atg7 in hepatic parenchymal cells caused a more severe liver phenotype than that seen in Atg5Δhep mice40 (Figure 1C). In these mice, a single binge dose of alcohol did not further lead to notable increases in serum levels of liver markers or hepatic TG level (Figure 1C). It seemed that a constitute deletion of Atg7 in hepatic parenchymal cells caused too severe an injury so that the relative minor effects of acute alcohol treatment were blunted.
To separate the injury effect caused by constitutive Atg7 deletion and alcohol treatment, Atg7Δhep-ERT2 mice were injected with tamoxifen to induce an acute deletion of Atg7 in the liver before alcohol treatment (Figure 1D). The deficiency of hepatic autophagy in Atg7Δhep-ERT2 mice after tamoxifen injection was confirmed by immunoblotting demonstration of decreases in hepatic microtubule-associated protein 1A/1B–light chain (LC)-3II/LC3I ratio and the accumulation of sequestosome (SQSTM1)-1/p62 protein (Supplemental Figure S1). Obvious hepatomegaly and serum elevation in liver injury markers in the Atg7Δhep-ERT2 mice were not observed at 9 days after induction; however, the serum markers and hepatic TG levels were significantly increased after the acute alcohol treatment in these mice (Figure 1E). The results were consistent with previous results in which autophagy was modified transiently and acutely using chemicals or gene knockout.8, 9, 15 Therefore, an acute deletion of Atg7, while not causing liver injury alone, rendered the hepatocytes more susceptible to alcohol-induced injury.
Effects of Acute Deletion of Atg7 Gene on Alcohol-Induced Liver Injury in a Chronic-Plus-Binge Model
To further examine the impact of hepatic autophagy–deficiency alcohol exposure, the response of these genetically altered mice to a more chronic alcohol exposure was analyzed using a recently established chronic-plus-binge model43 (Figure 2A). Atg7Δhep-ERT2 and Atg7F/F mice were given tamoxifen on days 3 and 4 in a 5-day acclimation period, followed by an alcohol diet for 10 days plus an acute alcohol binge at the end of the feeding period. Autophagy deficiency was confirmed as indicated by reduced levels of Atg7 and LC3II/LC3I ratio, and elevated SQSTM1/p62, in the livers of Atg7Δhep-ERT2 mice (Figure 2, B and C). Consistently, NF erythroid 2–related factor (Nrf)-2 was activated, and its targets, glutathione S-transferase M1 and Nad(p)h quinone dehydrogenase 1, were expressed at much higher levels in the livers of Atg7Δhep-ERT2 mice (Figure 2, B–D). Notably, alcohol treatment did not alter these changes due to Atg7 deletion, although the LC3II/LC3I ratio was increased in the floxed normal mice (Figure 2, B–D).
Figure 2.

Acute deletion of Atg7 enhances alcohol-induced liver injury in a chronic-plus-binge model. A: Scheme of the chronic-plus-binge treatment. Mice were acclimated with liquid diet for 5 days. Atg7F/F and Atg7Δhep-ERT2 mice were injected with tamoxifen (TMX) on days 3 and 4 during acclimation. After acclimation, mice were randomly divided into two groups and given a liquid diet with alcohol (EtOH-fed) or with maltose dextrin (Pair-fed) for 10 days, followed by a single gavage of alcohol (5 g/kg) or isocaloric maltose dextrin. Mice were analyzed 9 hours later. B: Representative Western blot images of protein expression in the livers of Atg7F/F and Atg7Δhep-ERT2 mice after alcohol or pair-fed treatment. Asterisk indicates onspecific bands. C: Densitometry of each protein band was conducted. The density values of Atg7, p62, or Nad(p)h quinone dehydrogenase (Nqo)-1 were normalized to that of β-actin. The density of microtubule-associated protein 1A/1B-light chain (Lc)-3II was normalized to that of Lc3I. D: The mRNA levels of Gstm1 and Nqo1 in the livers of Atg7F/F and Atg7Δhep-ERT2 mice after alcohol or pair-fed treatment. Expression levels were normalized to that of β-actin. E and F: Liver weight and body (L/B) weight ratio, serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and total bile acids (TBA) in Atg7F/F and Atg7Δhep-ERT2 mice after alcohol or pair-fed treatment. G: Hepatic triglycerides (TG) and serum β-hydroxybutyrate (β-OHB) levels in Atg7F/F and Atg7Δhep-ERT2 mice given alcohol or pair-fed treatment. Mice were used at the age of 8 to 12 weeks. Data are expressed as the means ± SEM fold changes over those of pair-fed Atg7F/F mice. n = 4 (A, C, and D); n = 4 to 11 (G). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control. Gstm1, glutathione S-transferase M1; and Nqo, Nad(p)h quinone dehydrogenase 1; Sqstm, sequestosome.
A longer duration of Atg7 deletion in the Atg7Δhep-ERT2 mice resulted in enlarged livers and liver injury in the chronic-plus-binge model (Figure 2E) as compared with the shorter duration of deletion in the binge model (Figure 1E). But as in the binge model, hepatomegaly and liver injury phenotype (Alt and Ast levels), but not ALP or TBA, were further augmented by alcohol treatment (Figure 2F). These results suggest a protective function of Atg7-mediated autophagy in alcohol-induced liver injury.
To determine potential factors that promote injury in autophagy deficiency, the extent of steatosis was examined. Hepatic TG (Figure 2G), but not TCHO (Supplemental Figure S2A), level was altered after alcohol treatment, indicating the presence of steatosis in the livers of both Atg7F/F and Atg7Δhep-ERT2 mice, but the difference was not significant between the two types of mice. Consistently, alcohol-induced elevation of fatty acid oxidation, as measured by the serum level of β-hydroxybutyrate, was not altered in the absence of Atg7 (Figure 2G).
Histologic examination suggested no obvious differences in general structure and fibrosis, but an elevated presence of Kupffer cells in autophagy-deficient livers (Supplemental Figure S2, B–E). Two acute-phase proteins, lipocalin-2 and serum amyloid A2, which are secreted by hepatocytes and other extrahepatic tissues in response to stress or injury, have been recently found to promote alcoholic liver pathology.44, 45 Indeed, the expression levels of both lipocalin-2 and serum amyloid A2 were elevated in Atg7Δhep-ERT2 livers, likely in response to autophagy deficiency–induced stress (Supplemental Figure S3A). After the 10-day chronic-plus-binge alcohol treatment, the expression levels of lipocalin-2 and serum amyloid A1 were elevated in floxed-control mice, but were not higher than the already elevated level in the Atg7-deficient livers (Supplemental Figure S3A). In addition, the mRNA levels of several inflammatory cytokines and inflammatory cell markers were higher in alcohol-treated Atg7Δhep-ERT2 livers than in the same treated floxed mice (Supplemental Figure S3, B and C). But the levels of several of these markers, such as C-C motif chemokine ligand 2 and Toll-like receptor 4, were also elevated in Atg7Δhep-ERT2 livers without alcohol treatment. These findings suggest that the protective effect of autophagy may not be entirely related to modulation of inflammation.
Effects of Constitutive Deletion of Hepatic Atg7 on Alcohol-Induced Liver Injury in a Chronic-Plus-Binge Model
Chronic alcohol treatment caused more injury than did acute binge treatment (Figure 2E versus Figure 1E), which could allow the detection of Atg7-mediated protection even in the constitutive deletion model. Thus, in the Atg7Δhep and the control mice, alcohol diet was administered using the chronic-plus-binge scheme (Figure 3A). As in the inducible Atg7Δhep-ERT2 mice, the reduced Atg7 level and LC3II/LC3I ratio, and the accumulation of SQSTM1 and Nad(p)h quinone dehydrogenase 1 were not significantly altered in the livers of Atg7Δhep mice with an alcohol diet (Figure 3, B and C). Alcohol diet was associated with enhanced hepatomegaly (Figure 3D) and liver injury, based on increased serum levels of Alt and Ast (Figure 3E). There were no significant differences in the serum levels of ALP or TBA with or without alcohol treatment (Figure 3E). In addition, total hepatic TG and TCHO levels were increased in Atg7-deficient liver with alcohol treatment (Figure 3F). These results indicate that constitutive loss of Atg7 may exacerbate injury caused by chronic alcohol treatment as well.
Figure 3.

Constitutive deletion of hepatic Atg7 enhances alcohol-induced liver injury in a chronic-plus-binge model. A: Scheme of the chronic-plus-binge treatment. Atg7F/F and Atg7Δhep mice were acclimated with liquid diet for 5 days. After acclimation, mice were randomly divided into two groups and given alcohol (EtOH) or control diet as in Figure 2. B: Representative Western blot images of protein expression in the liver from Atg7F/F and Atg7Δhep mice after alcohol or pair-fed treatment. C: The density of Atg7, p62, or Nad(p)h quinone dehydrogenase (Nqo)-1 was normalized to that of glyceraldehyde phosphate dehydrogenase (Gapdh). The density of microtubule-associated protein 1A/1B-light chain (Lc)-3II was normalized to that of Lc3I. D and E: Liver weight and body (L/B) weight ratio, serum levels of alanine aminotransferase (Alt), aspartate aminotransferase (Ast), alkaline phosphatase (ALP), and total bile acids (TBA) in Atg7F/F and Atg7Δhep mice after alcohol or pair-fed treatment. F: Hepatic triglycerides (TG) and total cholesterol (TCHO) levels in Atg7F/F and Atg7Δhep mice given alcohol or pair-fed treatment. Mice were used at the age of 8 to 15 weeks. Data are expressed as the means ± SEM fold changes over those of pair-fed Atg7F/F mice. n = 3 (C), n = 3 to 5 (F). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control. LE, long exposure; SE, short exposure; Sqstm, sequestosome.
Effects of Constitutive Deletion of Hepatic Atg5 on Liver Injury in a Chronic-Plus-Binge Model
A single dose of alcohol-binge treatment was associated with enhanced liver injury in Atg5Δhep mice (Figure 1B). Chronic alcohol treatment in Atg5Δhep mice (Figure 4A) was not associated with significant alterations in the repression of autophagy in these mice (Figure 4, B–D). Alcohol treatment was associated with slightly increased hepatomegaly in younger Atg5Δhep mice (Figure 4E), and with significant decreases in the serum levels of Alt, Ast, and ALP in both younger and older Atg5Δhep mice (Figure 4, F–H). The reduced injury was observed in both male and female mice (Supplemental Figures S4A and S5A). Total hepatic TG level but not TCHO level was still increased in Atg5Δhep and in Atg5F/F mice after alcohol treatment (Figure 4, I–L, and Supplemental Figures S4B and S5B), indicating an expected hepatic response to alcohol. Consistent with a higher level of total hepatic TG in Atg5Δhep mice after alcohol treatment, hematoxylin and eosin staining of the liver sections also showed a more prominent hepatic steatosis in these mice (Figure 5A).
Figure 4.

Constitutive deletion of hepatic Atg5 does not enhance liver injury in a chronic-plus-binge model. A: Scheme of the chronic-plus-binge alcohol treatment. Atg7F/F and Atg7Δhep-ERT2 mice were injected with tamoxifen on days 3 and 4 during acclimation. After acclimation, mice were randomly divided into two groups and given a liquid diet with alcohol (EtOH-fed) or with maltose dextrin (Pair-fed) for 10 days, followed by a single gavage of alcohol (5 g/kg) or isocaloric maltose dextrin. Mice were analyzed 9 hours later. B: Representative Western blot images of protein expression in the liver from Atg5F/F and Atg5Δhep mice after alcohol or pair-fed treatment. Anti-Agt12 antibody was used to detect the Atg12-Atg5 conjugate. C: The density of Atg5-Atg12, p62, or Nad(p)h quinone dehydrogenase (Nqo)-1 was normalized to that of β-actin. The density of microtubule-associated protein 1A/1B-light chain (Lc)-3II was normalized to that of Lc3I. D: mRNA levels of Gstm1 and Nqo1 in livers of Atg5F/F and Atg5Δhep mice after alcohol or pair-fed treatment. Expression levels were normalized to that of β-actin. E–H: Liver weight and body (L/B) weight ratio, serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) in Atg5F/F and Atg5Δhep mice after alcohol or pair-fed treatment. I–L: Hepatic triglycerides (TG) and total cholesterol (TCHO) levels in Atg5F/F and Atg5Δhep mice given alcohol or pair-fed treatment. Data are expressed as the means ± SEM fold changes over pair-fed Atg5F/F mice. n = 4 (C); n = 6 (D); n = 7 to 18 (E–H). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
Figure 5.

Reduced inflammation and fibrosis in constitutive Atg5–deficient livers after chronic-plus-binge alcohol treatment. A–E:Atg7F/F and Atg7Δhep-ERT2 mice (18 to 24 weeks old) were injected with tamoxifen on days 3 and 4 during acclimation. After acclimation, mice were randomly divided into two groups and given a liquid diet with alcohol (EtOH-fed) or with maltose dextrin (Pair-fed) for 10 days, followed by a single gavage of alcohol (5 g/kg) or isocaloric maltose dextrin. Mice were analyzed 9 hours later. Liver sections were subjected to hematoxylin and eosin (H&E) staining (A), F4/80 stain and quantified (B and C), and Masson's trichrome stain and quantified (D and E). F: The hepatic hydroxyproline level was determined. G: Representative Western blot images of α-smooth muscle actin (Sma) protein and densitometric analysis. The density of α-Sma protein was normalized to that of β-actin. H: The mRNA expression of fibrosis-related genes in the livers of Atg5F/F and Atg5Δhep mice given alcohol or control diet. Expression levels of indicated genes were determined by real-time quantitative -PCR and were normalized to that of β-actin. Data are expressed as the means ± SEM fold-changes over those of Atg5F/F mice given the pair-fed diet. n = 4 (G); n = 5 to 6 (F); n = 6 (H). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control. Original magnification, ×200 (A, B, and D). Ctgf, connective tissue growth factor; Gfap, glial fibrillary acidic protein; Tgf, transforming growth factor; Timp1, tissue inhibitor of metallopeptidase 1.
It is well established that elevation of hepatic TG level is an immediate consequence of alcohol stimulation, and that steatosis is also a major pathologic phenomenon in AFLD.46, 47 Generally, alcohol can affect hepatic lipid metabolism by accelerating lipogenesis, decelerating lipid breakdown, and causing defective hepatic lipid export.47 The mRNA expression levels of several lipid metabolism related–genes in the Atg5Δhep mice were altered from those in the Atg5F/F mice before and/or after alcohol treatment (Supplemental Figure S6, A–E). The expression levels of genes involved in lipid secretion, such as apolipoprotein B (Apob) and Apoe, were reduced, whereas the expression levels of genes involved in lipid update, such as cluster of differentiation 36 (Cd36) and lipoprotein lipase (Lpl), were increased before or after alcohol treatment. Although the expression levels of the genes involved in lipogenesis, except stearoyl-Coenzyme (Co)-A desaturase 1 (Scd1), were not elevated, the expression levels of the genes involved in lipolysis, such as adipose triglyceride lipase (Atgl) and hormone-sensitive lipase E (Lipe), and the genes in lipid oxidation, such as acyl-CoA dehydrogenase, long-chain (Acadl), acyl-CoA dehydrogenase medium chain (Acadm), peroxisomal acyl-CoA oxidase 1 (Acox1), and peroxisome proliferator-activated receptor α (Ppara), were repressed. Moreover, the expression levels of the genes involved in lipid oxidation were overall reduced in Atg5Δhep mice (Supplemental Figure S6E); however, the serum level of β-hydroxybutyrate remained elevated in response to alcohol stimulation (Supplemental Figure S6F). These changes may overall favor the accumulation of lipids in the cells and decrease lipid use.
The number of F4/80+-staining macrophages was significantly increased in Atg5Δhep livers, similar to what was observed in Atg7Δhep livers.48 The level of macrophage expansion was noticeably reduced with alcohol treatment (Figure 5, B and C). The expression levels of many inflammation-related genes were not significantly altered in Atg5-deficient livers, except C-C motif chemokine ligand 2, which was increased in Atg5-deficient livers (Supplemental Figure S7). Alcohol treatment was associated with reduce levels of C-C motif chemokine ligand 2 and several other cytokines and inflammatory cell markers in these mice (Supplemental Figure S7, A and B). On the other hand, the expression levels of lipocalin-2 and serum amyloid A2 remained higher (Supplemental Figure S7C). Inflammation plays important roles in the pathogenesis of ALFD.49 In particular, C-C motif chemokine ligand 2 is associated with the severity of, and neutrophil infiltrates in, ALD.50 Thus, the reduction of inflammation in chronic alcohol treatment condition may contribute to the reduced injury level in Atg5Δhep livers.
Interestingly, the significantly elevated fibrosis in Atg5-deficient livers was also decreased by chronic alcohol treatment, as shown by Masson's trichrome staining (Figure 5, D and E), hydroxyproline level (Figure 5F), and the expression of α-smooth muscle actin, at the protein (Figure 5G) and the mRNA levels (Figure 5H). The levels of other fibrosis-related genes, such as connective tissue growth factor (Ctgf), transforming growth factor β1 (Tgfb1), tissue inhibitor of metallopeptidase 1 (Timp1), and glial fibrillary acidic protein (Gfap), were not significantly different between alcohol treatment and control diet (Figure 5H). This improving effect of alcohol is reminiscent of findings from an earlier study showing a potentially protective effect of light alcohol consumption on hepatocellular injury and liver fibrosis in patients with NAFLD.51 It seems that alcohol may have variable effects on hepatic fibrosis in different pathologic conditions, which may partially explain the improvement in liver fibrosis in Atg5-deficient livers after alcohol treatment.
Reduction in Cholestatic Injury in Atg5-Deficient Livers after Alcohol Treatment
Autophagy deficiency in the liver can cause cholestatic injury, which is accompanied by a significant ductular reaction.48, 52 Alcohol treatment was associated with a reduction in this pathologic change in Atg5Δhep livers, as measured by the level of cytokeratin-19–positive ductular cell (Figure 6, A and B). Consistently, the serum level of TBA was also decreased after alcohol treatment in Atg5Δhep mice if both sexes and at both the older (Figure 6C) and younger (Supplemental Figure S8A) ages. Similar chronic alcohol treatment in Atg7Δhep-ERT2 (Figure 2F) or Atg7Δhep (Figure 3E) mice was not associated with reductions in the elevated serum TBA levels in these mice. Unexpectedly, the hepatic TBA load remained high in Atg5Δhep mice even after alcohol treatment, regardless of age (Figure 6, D and E, and Supplemental Figure S8, B and C). Interestingly, Atg5Δhep mice given one dose of alcohol binge also showed a reduced serum bile acid level, in contrast to Atg7Δhep and Atg7Δhep-ERT2 mice given the same acute treatment (Supplemental Figure S8D).
Figure 6.
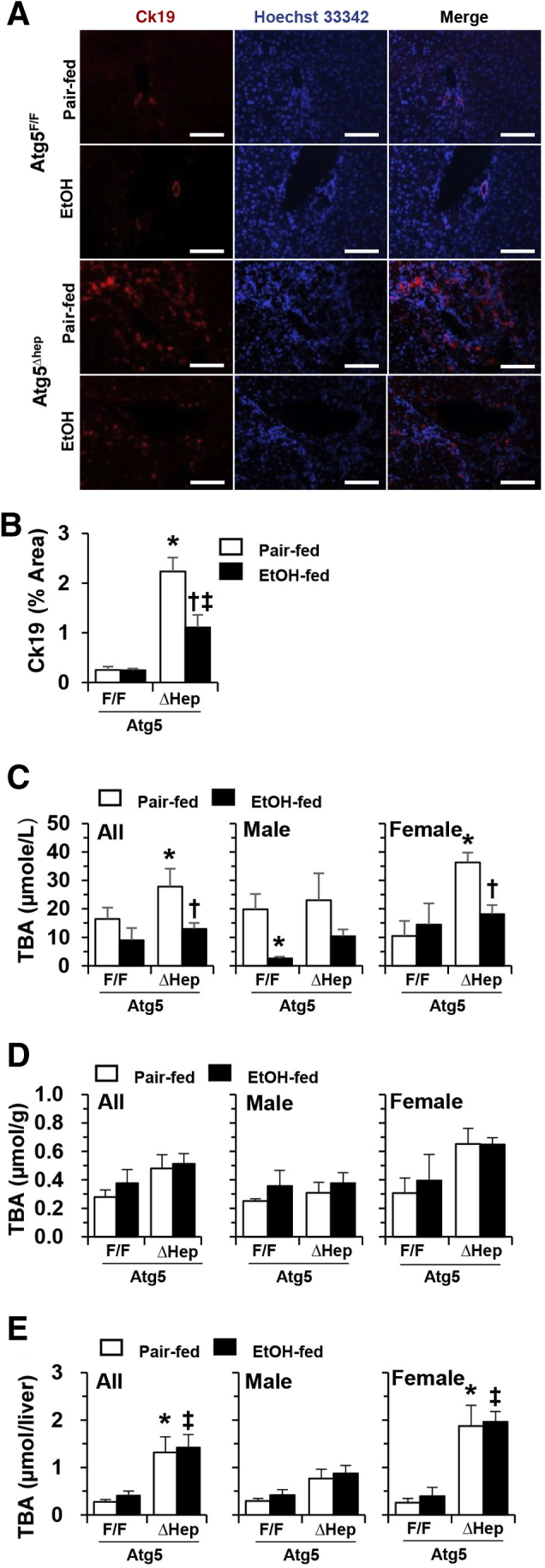
Reduced cholestasis in constitutive Atg5-deficient livers after chronic-plus-binge alcohol treatment. Atg7F/F and Atg7Δhep-ERT2 mice (18 to 24 weeks old) were injected with tamoxifen on days 3 and 4 during acclimation. After acclimation, mice were randomly divided into two groups and given a liquid diet with alcohol (EtOH-fed) or with maltose dextrin (Pair-fed) for 10 days, followed by a single gavage of alcohol (5 g/kg) or isocaloric maltose dextrin. Mice were analyzed 9 hours later. A: Liver sections were immunostained with anti–cytokeratin (Ck)-19. B: Ck19-positive areas were quantified. C: Serum levels of total bile acids (TBA). D and E: The levels of TBA in each gram of liver (D) or in the whole liver (E) were determined. Data are expressed as means ± SEM. n = 3 (D and E); n = 4 to 5 (B); n = 4 to 7 (A, female); n = 6 to 12 (A, male). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control. Scale bar = 0.1 mm.
The mRNA levels of genes related to bile acid metabolism were analyzed to determine any potential effects of alcohol. The expression levels of many of these genes were decreased in Atg5-deficient livers, as previously reported52 (Supplemental Figure S9). However, overall, the mRNA levels of genes related to bile acid synthesis were not further significantly altered in Atg5-deficient liver after alcohol treatment (Supplemental Figure S9, A and B). In addition, the expression of genes related to farnesoid X receptor (FXR) signaling remained low in Atg5-deficient livers (Supplemental Figure S9C). The impact of autophagy deficiency on the expression of the bile acid transporters, most of which were not further significantly affected by the alcohol (Supplemental Figure S9, D–F), has been reported before.52 Exceptions were the expression levels of organic solute transporter β in Atg5Δhep liver (Supplemental Figure S9E) and organic anion transporting polypeptide 1 (OATP1) in Atg5F/F liver (Supplemental Figure S9F), which were significantly decreased after alcohol treatment. Gene-expression results indicated that the influence of alcohol on bile acid metabolism in Atg5-deficient livers would likely be limited.
Hepatic Atg5 Deficiency and the Expression of Genes Related to Alcohol Metabolism and the Plasma Alcohol Clearance Rate in Mice
Acetaldehyde along with other alcohol metabolites are considered to be involved in ALDs.53 Since alcohol-induced liver injury is affected by alcohol metabolism, it was tested whether alcohol metabolism was affected in Atg5-deficient livers. The expression levels of genes related to alcohol metabolism were analyzed. Notably, the mRNA levels of several alcohol dehydrogenase (Adh)-related genes, including Adh classes I to IV (Adh1, -4, -5, and -7, respectively), were decreased in Atg5-deficient livers (Figure 7A). The expression levels of cytochrome P450 (Cyp) family 2 subfamily E member 1 and catalase were also decreased in Atg5-deficient livers (Figure 7A). Interestingly, the mRNA levels of Adh1, Adh4, Adh5, Cyp2e1, and Cat were also decreased in the livers of Atg5F/F mice after alcohol treatment.
Figure 7.

Constitutive deletion of hepatic Atg5 alters the expression of genes related to alcohol metabolism and serum alcohol clearance. Atg7F/F and Atg7Δhep-ERT2 mice (18 to 24 weeks old) were injected with tamoxifen on days 3 and 4 during acclimation. After acclimation, mice were randomly divided into two groups and given a liquid diet with alcohol (EtOH-fed) or with maltose dextrin (Pair-fed) for 10 days, followed by a single gavage of alcohol (5 g/kg) or isocaloric maltose dextrin. Mice were analyzed 9 hours later. A and B: The mRNA levels of genes related to the oxidation of alcohol (A) or acetaldehyde (B) in the liver. C: The protein levels of key alcohol-metabolizing enzymes in the liver. β-Actin [for cytochrome P450 (Cyp)-2e1] and glyceraldehyde phosphate dehydrogenase (Gapdh) [for aldehyde dehydrogenase (Aldh)-1/2 and alcohol dehydrogenase (Adh)] were used as loading control. D: Densitometry was conducted, and the density levels were normalized to that of loading control. E:Atg5F/F and Atg5Δhep mice at the age of 13 to 20 weeks were given a single dose of alcohol binge (5 g/kg). Plasma alcohol concentrations were then measured at 1, 3, and 9 hours later. Data are expressed as the means ± SEM fold changes over Atg5F/F mice given pair-fed treatment. n = 3 to 5 (E); n = 4 (D); n = 6 (A and B). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control; §§§P < 0.001. Atg, autophagy-related protein; Cat, catalase.
The mRNA levels of several aldehyde dehydrogenases (Aldh) in the liver were analyzed. Significant decreases in the mRNA expression levels of Aldh2, -4a1, -5a1, -7a1, -8a1, and -9a1 were observed in the Atg5-deficient livers (Figure 7B). Although alcohol did not seem to cause further changes in the expression levels of most of these genes, it was associated with elevated Aldh1a3 gene expression in Atg5-deficient livers, and elevated Aldh18a1 gene expression in Atg5F/F livers (Figure 7B). The expression levels of several other Aldh family members, including Aldh1a1, -1a2, -1b1, -1l1, -1l2, -3a2, -3b1, and -6a1, were not significantly changed in the livers of Atg5-deficient mice or with alcohol treatment (Supplemental Figure S10).
The protein levels of Adh, Cyp2e1, and Aldh1/2 were further analyzed. Protein stability of CYP2E1 is crucial to its induction by alcohol.54 The protein levels of Cyp2e1 and Adh were decreased in older (18 to 24 weeks) (Figure 7, C and D) and younger (8 to 11 weeks) (Supplemental Figure S11) Atg5-deficient livers. However, consistent with findings from a previous study,54 the protein level of Cyp2e1 was significantly increased after alcohol treatment in the livers of both Atg5Δhep and Atg5F/F mice (Figure 7, C and D, and Supplemental Figure S9). No consistent changes in Adh or Aldh1/2 in either older (18 to 24 weeks) or younger (8 to 1 week) mice were observed (Figure 7, C and D, and Supplemental Figure S9). The gene-expression and protein-expression data indicate that Atg5 deficiency in the liver alters the expression of genes related to alcohol metabolism; however, alcohol itself did not seem to have affected the mRNA levels of most of these genes. These changes could result in a lower metabolism of alcohol and therefore lower levels of metabolites of alcohol that are known to be involved in its toxicity.
The lower metabolism of alcohol might cause a higher plasma alcohol level in the Atg5-deficient mice. To examine this possibility, Atg5Δhep and Atg5F/F mice were given a single dose of alcohol by gavage, and the plasma levels of alcohol were measured at different time points (Figure 7E). The plasma alcohol in nontreated mice was undetectable. The plasma levels of alcohol at 1 hour after gavage were comparable between Atg5Δhep and Atg5F/F mice, suggesting that there was no difference in the intestinal absorption of alcohol (Figure 7E). Interestingly, plasma levels of alcohol were slightly lower at 3 hours and dramatically lower at 9 hours in Atg5Δhep mice than those in Atg5F/F mice (Figure 7E). These results indicate an increased plasma alcohol clearance in Atg5Δhep mice despite reductions in the expression of genes in the alcohol-metabolism pathway. Other mechanisms may therefore account for the alcohol elimination in the autophagy-deficient mice.
Discussion
Effects of How Autophagy Is Inhibited and How Alcohol Is Administered on Alcohol-Induced Liver Injury
This is the first study in which the role of autophagy in alcohol-induced liver injury was assessed through a genetic approach to disrupt the formation of autophagosomes using either inducible or constitutive knockout of two different autophagy genes, Atg7 and Atg5. These models differ in the duration of autophagy deficiency, the specific gene being affected, and the severity of endogenous liver injury. This study thus allows an extensive analysis of the interaction of alcohol administration and autophagy function in the context of acute and chronic liver injury.
Previous studies have examined the role of autophagy by using siRNA-mediated knockdown of Atg7, or by interference of the lysosomal functions with chloroquine or transcription factor EB.8, 9, 12, 15, 31 In these studies, the protective role of autophagy in alcohol-induced liver injury had been shown in the models of an acute binge, a chronic 4-week Liber-DeCarli feeding, and a chronic-plus-binge feeding. The protective effects of autophagy in the gastric-infusion model had also been observed with the use of chloroquine (J.Z. and X.-M.Y., unpublished data). In the infusion model, alcohol (up to 37.1% of calorie uptake) was given in either an 8% low-fat or 40% high-fat base over a period of 28 days.55 The administration of chloroquine led to increased hepatic TG and blood ALT.
Here we have confirmed the protective effect of autophagy in the inducible Atg7-deficient livers (Atg7Δhep-ERT2) using both the acute binge model and a new chronic-plus-binge model. In both models, autophagy deficiency via Atg7 deletion was induced only shortly before alcohol treatment. The inducible approach minimized the liver injury caused by autophagy deficiency alone, so that the impact of autophagy deficiency on alcohol-induced injury could be directly examined. However, mice harboring constitutive deletion of Atg7 (Atg7Δhep) had different responses to alcohol treatment in the acute binge and chronic-plus-binge models. Liver injury was noticeably enhanced in Atg7Δhep mice after the chronic-plus-binge treatment, but not after acute binge treatment. The difference in the liver injury after alcohol treatments between the two Atg7-deletion models may have been due to the difference in the durations of gene deletion, which would have resulted in varying levels of autophagy dysfunction and liver injury. It is possible that a higher level of detrimental SQSTM1/p62 accumulation and NRF2 activation in the Atg7Δhep mice (Figure 3, B and C, and Figure 2, B and C) may have accounted for this resistance.
In mice with constitutive Atg5 deletion in the liver (Atg5Δhep), liver injury was noticeably enhanced after a single dose of binge treatment, but paradoxically improved after the chronic-plus-binge treatment. In the latter, the improved presentation included lower blood levels of liver enzymes and TBA and reduced hepatic inflammation, ductular reaction, and fibrosis. It is possible that the level of NRF2 activation could have been in part involved in the difference between the Atg5Δhep and Atg7Δhep mice. Thus the levels of SQSTM1/p62 and Nad(p)h quinone dehydrogenase 1 were higher in the Atg7Δhep livers than in the Atg5Δhep livers (Figure 3, B and C, and Figure 4, B and C). The extent of Nrf2 activation may determine the severity of endogenous injury,41, 48, 56 which in turn may affect the response to alcohol treatment.
These results demonstrate that the outcomes of the interaction of alcohol and hepatic autophagy function could depend on how autophagy function is disabled (constitutively or acutely), which autophagy gene is deleted (Atg7 or Atg5), and how alcohol is administered. However, published and current results clearly demonstrate that acute inactivation of autophagy or lysosome function, via a genetic, chemical (siRNA), or pharmacologic approach, promotes alcohol-induced liver injury in a variety of alcohol-administration models with different dosages and durations. But the more complex findings of alcohol-induced phenotypes in chronically autophagy-deficient mice perhaps more realistically reflect clinical scenarios in which autophagy functions could be compromised through a more chronic process by alcohol and other coexisting factors such as viral infection16, 17, 18, 19 or a high-fat diet.13, 14, 15
Variable Liver Pathology in Atg5Δhep and Atg7Δhep Mice and Their Differential Responses to Alcohol Given through Different Regimens
Embryonic deletion of Atg5 or Atg7 in the parenchymal cells under the albumin promotor leads to severe liver pathology.48, 56 Both Atg7 and Atg5 are crucial for LC3 to conjugate to phosphatidylalcoholamine on the autophagosomal membrane, which is required for autophagosome formation. However, the hepatic phenotypes of Atg5Δhep and Atg7Δhep mice are interestingly different in the degree and progression of the injury. Overall, Atg5Δhep mice seemed to present a less severe phenotype of liver injury (Figure 1, B and C). Similar observations could be found in earlier sudies.48, 56 In addition, Atg5-deficient mice seem to undergo a spontaneous improvement when they became older (4 months and beyond) (Figure 4, F–H).56 Also, the improvement was mainly contributed by the male mice (Supplemental Figure S4A), not the female mice (Supplemental Figure S5A). The mechanisms for the more severe phenotype of Atg7Δhep mice are not clear, but may be related to the more crucial function of Atg7 being the only E1-like molecule involved in both Atg5-Atg12 conjugation and LC3-phosphatidylalcoholamine conjugation.5 Alternatively, the possibility that Atg7 might have additional functions cannot be ruled out.
The mild liver injury associated with Atg5 deficiency could have allowed the detection of the added alcohol-induced insults in the acute binge model (Figure 1B). On the other hand, Atg7Δhep mice might have had a blunted response to an acute alcohol binge and failed to show an increased injury. Since the elevation of hepatic TG level is an immediate consequence of alcohol stimulation and steatosis is a major pathologic phenomenon in AFLD,46, 47 the change in the hepatic total TG level was examined in Atg7Δhep and Atg5Δhep mice. Indeed, TG level was not elevated in Atg7Δhep mice (Figure 1C) but was elevated in Atg5Δhep (Figure 1B) mice receiving an acute binge treatment. This observation supports that Atg7Δhep mice had a blunted response to acute alcohol stimulation. In the chronic-plus-binge regimen, Atg7Δhep mice responded to the prolonged alcohol stimulation since the total TG level was elevated (Figure 3E), and consequently an elevated injury was observed (Figure 3D).
The response of Atg5Δhep mice to the chronic-plus-binge treatment was unexpected. The liver injury phenotype was paradoxically reduced in these mice despite that they seemed to respond to alcohol as evidenced by elevated hepatic TG level (Figure 4, I and J). The improvement in liver injury despite an elevation in hepatic TG was evident in both sexes and regardless of age (Supplemental Figures S4 and S5). An earlier study had also shown that Atg5Δhep mice were paradoxically resistant to acetaminophen-induced liver injury.57 A common feature of alcohol- and acetaminophen-induced liver injury is the induction of oxidative stress. Thus it is possible that Atg5Δhep mice had pathologic changes that rendered them resistant to oxidative stress.
The deletion of either Atg5 or Atg7 in the liver causes SQSTM1/p62 accumulation in the liver, which in turn activates NRF2.40, 41, 56, 58 The latter may create a somewhat reduced environment to resist damage caused by oxidative stress.57 However, the elevated NRF2 activation in autophagy deficiency is likely correlated with the pathology, as shown by the co-deletion of Nrf2 and the ensuing blockage of most of the liver phenotypes in Atg7Δhep and Atg5Δhep mice.41, 48, 56 Thus the improved hepatic presentation of Atg5Δhep mice given chronic-plus-binge alcohol treatment may have been due to other factors.
The effect of alcohol in mice with chronic liver injury due to autophagy deficiency is thus complicated by the baseline liver changes in the autophagy-deficient condition. Caution must be exercised in reaching conclusions on the potential mechanisms that are involved. Notably, clinically ALD is often complicated by other conditions such as viral infection16, 17, 18, 19 and NAFLD,13, 14, 15 both of which could compromise autophagy function. Long-term alcohol use can also suppress autophagy by itself.9, 10, 11, 12, 15 The findings from this study demonstrate that the effects of alcohol can be quite diverse in livers with varying degrees of autophagy deficiency.
Potential Multiple Factors in Improvement of Liver Injury in Atg5Δhep Mice by Chronic Alcohol Treatment
Other factors to consider include the potential role of inflammation, which is known to play important roles in the pathogenesis of ALFD.49, 50 The reduced injury in Atg5Δhep livers by the chronic alcohol treatment may have been related to reduced inflammation. In addition, changes related to the metabolism of bile acids and alcohol itself may deserve attention as well.
As previously published,52, 56 Atg5Δhep and Atg7Δhep mice develop cholestatic injury, which is accompanied by an increased level of cytokeratin-19–positive ductular cells (ductular reaction), altered mRNA levels of genes in the bile acid–metabolism pathways, and an increase in circulating TBA level. Interestingly, several studies have shown a relationship between alcohol administration and bile acid metabolism.59, 60, 61 Indeed, serum levels of TBA were decreased by alcohol treatment in both Atg5F/F and Atg5Δhep mice. However, the improving effect in Atg5Δhep mice by alcohol may not have been related to intrahepatic bile acid metabolism since the hepatic bile acid level in Atg5Δhep remained high (Figure 6, D and E) and the expression of genes that are related to bile acid metabolism did not change significantly (Supplemental Figure S9). Nonetheless, this reduction in serum TBA may still be beneficial, as suggested by the decrease in ductular reaction (Figure 6A) and serum ALT/AST (Figure 4, F–G). How chronic alcohol treatment and Atg5 deficiency lead to improved cholestatic outcome is an interesting topic for future studies.
Alcohol is generally metabolized in hepatic parenchymal cells, which express the highest levels of alcohol-oxidizing enzymes, including ADHs, CYP2E1, and catalase.46 The first step of alcohol breakdown in the liver by these enzymes generates toxic metabolites, including acetaldehyde and other potentially damaging molecules.53 Interestingly, the mRNA levels of the Adh, Cyp2e1, and Cat genes were decreased in Atg5-deficient livers. Reduced levels of these alcohol-oxidizing enzymes may reduce the generation of toxic metabolites, and thus less or no alcohol-mediated toxicity. However, there was no consistent difference in the expression of these genes between Atg5Δhep livers and Atg7Δhep livers (data not shown), which may thus not account for the difference in how these mice responded to alcohol.
The toxic acetaldehyde can be enzymatically removed by ALDH. Aldh2 is one of the most important Aldh family of genes for alcohol metabolism. In other studies in mice, the pharmacologic activation of either Aldh2 or global Aldh2 overexpression seemed to ameliorate chronic alcohol-induced hepatic steatosis and improved ALD,62, 63 and Aldh2 deficiency paradoxically resulted in improved alcoholic fatty livers, although worse liver inflammation and fibrosis.64 In the present study, the expression levels of several Aldh genes, including Aldh2, were decreased in Atg5-deficient livers, although the protein levels remained unchanged. The significance of these changes is not known, but the overall findings would suggest that alcohol metabolism might have been altered in Atg5Δhep livers, which may have contributed to the observed phenotypes.
A lower expression of these enzymes may result in a slower blood alcohol clearance, as in Adh1 and Adh4 knockout mice.65 Surprisingly, a faster clearance rate of plasma alcohol in Atg5Δhep mice was observed. It is notable that alcohol could be cleared from circulation by other mechanisms independent of these enzymes, such as through breath. The potential involvement of these independent mechanisms needs to be investigated in the future.
In summary, the findings from this study demonstrate a protective role of autophagy in alcohol-induced liver injury, which was best illustrated in models in which an autophagy gene Atg7 was acutely deleted. In mice with constitutive deletion of Atg7 or Atg5, the effects of autophagy deficiency on alcohol-induced liver phenotype become complicated, in large part due to the coexistence of liver pathology caused by autophagy deficiency. Collectively, the findings from this study indicate the complexity of the relationship between autophagy status and alcohol-induced liver injury. This relationship may be dictated by the timing of the autophagy inhibition and alcohol exposure, the regimen of alcohol administration, and the particular autophagy-related gene being inactivated. These conditions may draw some similarities to the clinical ALDs, which are often complicated by additional confounding factors, such as viral infection16, 17, 18, 19 and NAFLD,13, 14, 15 in which autophagy function can be compromised. This study may provide a model to understand the complex interactions of alcohol with autophagy deficiency at various degrees caused by other underlying conditions.
Footnotes
Supported in part by NIH grants R21AA-021450 (X-M.Y) and R01AA-021751 (X-M.Y).
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.ajpath.2019.05.011.
Supplemental Data
Supplemental Figure S1.

Expression of autophagy-related protein in Atg7Δhep-ERT2 livers after acute ethanol (EtOH) treatment. A: Representative Western blot images of p62 and microtubule-associated protein 1A/1B-light chain (LC)-3. β-Actin was used as loading control (CTRL). B: The density of p62 was normalized to β-actin, and the density of Lc3BII was normalized to Lc3BI. Data are expressed as means ± SEM fold changes of those of Atg7F/F without ethanol. n = 3 (all groups, A and B). ∗P < 0.05 versus nontreated F/F control; ‡P < 0.05 versus treated F/F control. Sqstm, sequestosome.
Supplemental Figure S2.

Pathologic changes in Atg7Δhep-ERT2 mice after alcohol treatment in a chronic-plus-binge model. Mice were acclimated with liquid diet for 5 days. Atg7F/F and Atg7Δhep-ERT2 mice were injected with tamoxifen on days 3 and 4 during acclimation. After acclimation, mice were randomly divided into two groups and given a liquid diet with alcohol (EtOH-fed) or with maltose dextrin (Pair-fed) for 10 days, followed by a single gavage of alcohol (5 g/kg) or isocaloric maltose dextrin. Mice were analyzed 9 hours later. A: Hepatic total cholesterol (TCHO) levels as measured in whole liver or normalized by the liver weight. B–E: Mouse livers were examined histologically by hematoxylin and eosin (H&E) staining (B), immunostaining for F4/80 and quantification (C and D), and trichrome staining (E). Data are expressed as means ± SEM. n = 4–11 (A); n = 4 (B–E). ∗P < 0.05 versus nontreated F/F control; ‡P < 0.05 versus treated F/F control.
Supplemental Figure S3.

Expression of acute-phase proteins and cytokines in Atg7Δhep-ERT2 mice after alcohol treatment in a chronic-plus-binge model. Mice were acclimated with liquid diet for 5 days. Atg7F/F and Atg7Δhep-ERT2 mice were injected with tamoxifen on days 3 and 4 during acclimation. After acclimation, mice were randomly divided into two groups and given a liquid diet with alcohol (EtOH-fed) or with maltose dextrin (Pair-fed) for 10 days, followed by a single gavage of alcohol (5 g/kg) or isocaloric maltose dextrin. Mice were analyzed 9 hours later. Expression levels of indicated genes were determined by real-time quantitative PCR and were normalized to that of β-actin. A: Acute-phase proteins: lipocalin (Lcn)-2 and serum amyloid A (Saa)-2A2. B: Cytokines: C-C motif chemokine ligand (Ccl)-2/monocyte chemotactic protein (Mcp)-1, Ccl3/Mip1α, interferon (Ifn)-γ, tumor necrosis factor (Tnf)-α, and Il-6. C: Inflammatory cell markers: C-C motif chemokine receptor (Ccr)-2, Toll-like receptor (Tlr)-4, F4/80 [alias, adhesion G protein-coupled receptor (Adgr)-E1], and lymphocyte antigen 6 complex locus G6D (Ly6g). Data are expressed as mean ± SEM fold changes over Atg7F/F mice given pair-fed treatment. n = 3 or 4. ∗P < 0.05 versus nontreated F/F control; ‡P < 0.05 versus treated F/F control.
Supplemental Figure S4.

Constitutive deletion of hepatic Atg5 in male mice reduces ethanol-induced liver injury in a chronic-plus-binge model. Younger (8 to 11 weeks) and older (18 to 24 weeks) male Atg5F/F and Atg5Δhep mice were exposed to chronic-plus-binge ethanol (EtOH) treatment or control treatment. Liver weight and body (L/B) weight ratio, and serum levels of alanine aminotransferase (Alt), aspartate aminotransferase (Ast), and alkaline phosphatase (ALP) (A), and total hepatic levels of triglycerides (TG) and cholesterol (TCHO) (B) were measured. Data are expressed as means ± SEM. n = 4 to 6 (8 to 11 weeks); n = 4 to 12 (18 to 24 weeks). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
Supplemental Figure S5.

Constitutive deletion of hepatic Atg5 in female mice reduces ethanol-induced liver injury in a chronic-plus-binge model. Younger (8 to 11 weeks) and older (18 to 24 weeks) female Atg5F/F and Atg5Δhep mice were exposed to chronic-plus-binge ethanol (EtOH) treatment or control treatment. A and B: Liver weight and body (L/B) weight ratio, and serum levels of alanine aminotransferase (Alt), aspartate aminotransferase (Ast), and alkaline phosphatase (ALP) (A), and total hepatic levels of triglycerides (TG) and cholesterol (TCHO) (B) were measured. Data are expressed as means ± SEM. n = 3 to 5 (8 to 11 weeks); n = 4 to 7 (18 to 24 weeks). ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
Supplemental Figure S6.

Changes in mRNA levels of lipid metabolism–related genes in older constitutive Atg5-deficient livers after chronic-plus-binge ethanol (EtOH) treatment. Atg5F/F and Atg5Δhep mice at the age of 18 to 24 weeks were exposed to chronic-plus-binge ethanol treatment or control treatment. A–E: Expression levels of indicated genes were determined by real-time quantitative PCR and were normalized to that of β-actin. A: Genes related to lipid secretion: Apob (apolipoprotein B) and Apoe. B: Genes related to lipid uptake: Cd36 (cluster of differentiation 36), Fabp1 (fatty acid binding protein 1), and Lpl (lipoprotein lipase). C: Genes related to lipogenic: Fasn (fatty acid synthase), Scd1 [stearoyl coenzyme (Co)-A desaturase 1], and Acca (acetyl-CoA carboxylase α). D: Genes related to lipolysis: Atgl (adipose triglyceride lipase) and Lipe (lipase E, hormone-sensitive type). E: Genes related to lipid oxidation: Acadl (long-chain acyl-CoA dehydrogenase), Acadm (medium-chain Acad), Acox1 (acyl-CoA oxidase 1), and Ppara (peroxisome proliferator activated receptor-α). F: Serum level of β-hydroxybutyrate (β-OHB) was determined. Data are expressed as the means ± SEM fold changes over Atg5F/F mice given pair-fed treatment. n = 6. ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
Supplemental Figure S7.

Changes in mRNA levels of inflammation-related genes in older constitutive Atg5-deficient livers after chronic-plus-binge ethanol (EtOH) treatment. Atg5F/F and Atg5Δhep mice at the age of 18 to 24 weeks were exposed to chronic-plus-binge ethanol treatment or control treatment. Expression levels of indicated genes were determined by real-time quantitative PCR and were normalized to that of β-actin. A: Cytokines: C-C motif chemokine ligand (Ccl)-2, Ccl3, interferon (Ifn)-γ, tumor necrosis factor (Tnf)-α, and Il-6. B: Inflammatory cell markers: C-C motif chemokine receptor (Ccr)-2, Toll-like receptor (Tlr)-4, F4/80 [alias, adhesion G protein–coupled receptor (Adgr)-E1], and lymphocyte antigen 6 complex locus G6D (Ly6g). C: Acute-phase proteins: lipocalin (Lcn)-1 and serum amyloid A (Saa)-2. Data are expressed as the means ± SEM fold changes over Atg5F/F mice given pair-fed treatment. n = 6. ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
Supplemental Figure S8.

Serum and hepatic bile acid levels in younger constitutive Atg5-deficient mice after chronic-plus-binge ethanol (EtOH) treatment. Atg5F/F and Atg5Δhep mice at the age of 8 to 11 weeks were exposed to chronic-plus-binge ethanol treatment or control treatment. A: Serum total bile acids (TBA) levels. B: Hepatic TBA content per gram of liver. C: Hepatic TBA content per liver. D: Serum TBA levels in mice given a single dose of acute binge alcohol treatment as in Figure 1, A and D. n = 3 female (A); n = 3 to 4 male (A); n = 3 to 6 Atg7-ERT2 (D); n = 4 to 6 Atg7 (D); n = 5 Atg5 (D). Data are expressed as means ± SEM. ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
Supplemental Figure S9.

Changes in mRNA levels of bile acid metabolism–related genes in older constitutive Atg5-deficient livers after chronic-plus-binge ethanol (EtOH) treatment. Atg5F/F and Atg5Δhep mice at the age of 18 to 24 weeks were exposed to chronic-plus-binge ethanol treatment or control treatment. Expression levels of indicated genes were determined by real-time quantitative PCR and were normalized to that of β-actin. A: Genes related to the classic bile acid synthesis pathway: Cyp7a1 (cytochrome P450 family 7 subfamily A member 1), Hsd3b7 (hydroxy-δ-5-steroid dehydrogenase, 3 β- and steroid δ-isomerase 7), Cyp8b1 (Cyp family 8 subfamily B member 1), and Akr1d1 (aldo-keto reductase family 1-member D1). B: Genes related to the alternative bile acid synthesis pathway: Cyp27a1 (Cyp family 27 subfamily A member 1) and Cyp7b1 (Cyp family 7 subfamily B member 1). C: Other genes related to bile acid metabolism: Fxr (farnesoid X receptor), Cyp3a11 (Cyp family 3 subfamily A member 11), and Fgfr4 (fibroblast growth factor receptor 4). D: Genes of apical transporters: Bsep (bile salt export pump) and Mrp2 (multidrug-resistance protein 2). E: Genes of basolateral transporters: Mrp3, Mrp4, Osta (organic solute transporter-α), and Ostb. F: Genes of basolateral transporters related to enterohepatic recirculation: Oatp1 (organic anion transporting polypeptide), Ntcp (Na+-taurocholate co-transporting protein). Data are expressed as the means ± SEM fold changes over Atg5F/F mice given pair-fed treatment. n = 6. ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
Supplemental Figure S10.

Constitutive deletion of hepatic Atg5 alters the expression of ethanol metabolism–related genes in the liver. Atg5F/F and Atg5Δhep mice at the age of 18 to 24 weeks were exposed to chronic-plus-binge ethanol treatment (EtOH) or control treatment. Expression levels of indicated genes were determined by real-time quantitative PCR and were normalized to that of β-actin. Statistical significance was not found when groups were compared to nontreated F/F control, nontreated Δhep control, or treated F/F control. Data are expressed as the means ± SEM fold changes over Atg5F/F mice given pair-fed treatment. n = 6. Aldh, aldehyde dehydrogenase.
Supplemental Figure S11.

Constitutive deletion of hepatic Atg5 alters the protein levels of key ethanol-metabolizing enzymes in the liver. Atg5F/F and Atg5Δhep mice at the age of 8 to 11 weeks were exposed to chronic-plus-binge ethanol treatment or control treatment. A: Representative Western blot images of cytochrome P450 (Cyp)-2e1, aldehyde dehydrogenase (Aldh)-1/2, and alcohol dehydrogenase (Adh). β-Actin (for Cyp2e1) and glyceraldehyde phosphate dehydrogenase (Gapdh) (for Aldh1/2 and Adh) were used as loading control. B: Densitometry was conducted, and the density levels were normalized to that of loading control. Data are expressed as the means ± SEM fold changes over those of Atg5F/F mice given pair-fed treatment. n = 4. ∗P < 0.05 versus nontreated F/F control; †P < 0.05 versus nontreated Δhep control; ‡P < 0.05 versus treated F/F control.
References
- 1.Abdel-Misih S.R., Bloomston M. Liver anatomy. Surg Clin North Am. 2010;90:643–653. doi: 10.1016/j.suc.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddy J.K., Rao M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–G858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- 3.Deter R.L., De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. 1967;33:437–449. doi: 10.1083/jcb.33.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meijer A.J., Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Mizushima N., Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 6.Yin X.M., Ding W.X., Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- 7.Czaja M.J., Ding W.X., Donohue T.M., Jr., Friedman S.L., Kim J.S., Komatsu M., Lemasters J.J., Lemoine A., Lin J.D., Ou J.H., Perlmutter D.H., Randall G., Ray R.B., Tsung A., Yin X.M. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding W.X., Li M., Chen X., Ni H.M., Lin C.W., Gao W., Lu B., Stolz D.B., Clemens D.L., Yin X.M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomes P.G., Trambly C.S., Thiele G.M., Duryee M.J., Fox H.S., Haorah J., Donohue T.M., Jr. Proteasome activity and autophagosome content in liver are reciprocally regulated by ethanol treatment. Biochem Biophys Res Commun. 2012;417:262–267. doi: 10.1016/j.bbrc.2011.11.097. [DOI] [PubMed] [Google Scholar]
- 10.Kharbanda K.K., McVicker D.L., Zetterman R.K., Donohue T.M., Jr. Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochim Biophys Acta. 1996;1291:45–52. doi: 10.1016/0304-4165(96)00043-8. [DOI] [PubMed] [Google Scholar]
- 11.Dolganiuc A., Thomes P.G., Ding W.X., Lemasters J.J., Donohue T.M., Jr. Autophagy in alcohol-induced liver diseases. Alcohol Clin Exp Res. 2012;36:1301–1308. doi: 10.1111/j.1530-0277.2012.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao X., Wang S., Zhao K., Li Y., Williams J.A., Li T., Chavan H., Krishnamurthy P., He X.C., Li L., Ballabio A., Ni H.M., Ding W.X. Impaired TFEB-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology. 2018;155:865–879.e12. doi: 10.1053/j.gastro.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L., Li P., Fu S., Calay E.S., Hotamisligil G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H., Yan S., Khambu B., Ma F., Li Y., Chen X., Martina J.A., Puertollano R., Li Y., Chalasani N., Yin X.M. Dynamic MTORC1-TFEB feedback signaling regulates hepatic autophagy, steatosis and liver injury in long-term nutrient oversupply. Autophagy. 2018;14:1779–1795. doi: 10.1080/15548627.2018.1490850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin C.W., Zhang H., Li M., Xiong X., Chen X., Chen X., Dong X.C., Yin X.M. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–999. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang H., Da L., Mao Y., Li Y., Li D., Xu Z., Li F., Wang Y., Tiollais P., Li T., Zhao M. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology. 2009;49:60–71. doi: 10.1002/hep.22581. [DOI] [PubMed] [Google Scholar]
- 17.Liu B., Fang M., Hu Y., Huang B., Li N., Chang C., Huang R., Xu X., Yang Z., Chen Z., Liu W. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy. 2014;10:416–430. doi: 10.4161/auto.27286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sir D., Chen W.L., Choi J., Wakita T., Yen T.S., Ou J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim S.J., Khan M., Quan J., Till A., Subramani S., Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathogens. 2013;9:e1003722. doi: 10.1371/journal.ppat.1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lumeng L., Crabb D.W. Alcoholic liver disease. Curr Opin Gastroenterol. 2000;16:208–218. doi: 10.1097/00001574-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Lieber C.S. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. doi: 10.1016/j.alcohol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Zakhari S., Li T.K. Determinants of alcohol use and abuse: impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46:2032–2039. doi: 10.1002/hep.22010. [DOI] [PubMed] [Google Scholar]
- 23.Bailey S.M., Cunningham C.C. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 24.Demeilliers C., Maisonneuve C., Grodet A., Mansouri A., Nguyen R., Tinel M., Letteron P., Degott C., Feldmann G., Pessayre D., Fromenty B. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology. 2002;123:1278–1290. doi: 10.1053/gast.2002.35952. [DOI] [PubMed] [Google Scholar]
- 25.Mansouri A., Demeilliers C., Amsellem S., Pessayre D., Fromenty B. Acute ethanol administration oxidatively damages and depletes mitochondrial DNA in mouse liver, brain, heart, and skeletal muscles: protective effects of antioxidants. J Pharmacol Exp Ther. 2001;298:737–743. [PubMed] [Google Scholar]
- 26.Mansouri A., Gaou I., De Kerguenec C., Amsellem S., Haouzi D., Berson A., Moreau A., Feldmann G., Letteron P., Pessayre D., Fromenty B. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology. 1999;117:181–190. doi: 10.1016/s0016-5085(99)70566-4. [DOI] [PubMed] [Google Scholar]
- 27.Cahill A., Cunningham C.C., Adachi M., Ishii H., Bailey S.M., Fromenty B., Davies A. Effects of alcohol and oxidative stress on liver pathology: the role of the mitochondrion. Alcohol Clin Exp Res. 2002;26:907–915. [PMC free article] [PubMed] [Google Scholar]
- 28.Cahill A., Stabley G.J., Wang X., Hoek J.B. Chronic ethanol consumption causes alterations in the structural integrity of mitochondrial DNA in aged rats. Hepatology. 1999;30:881–888. doi: 10.1002/hep.510300434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L., Khambu B., Zhang H., Yin X.M. Autophagy in alcoholic liver disease, self-eating triggered by drinking. Clin Res Hepatol Gastroenterol. 2015;39 Suppl 1:S2–S6. doi: 10.1016/j.clinre.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L., Zhou J., Yan S., Lei G., Lee C.H., Yin X.M. Ethanol-triggered lipophagy requires SQSTM1 in AML12 hepatic cells. Sci Rep. 2017;7:12307. doi: 10.1038/s41598-017-12485-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomes P.G., Ehlers R.A., Trambly C.S., Clemens D.L., Fox H.S., Tuma D.J., Donohue T.M. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy. 2013;9:63–73. doi: 10.4161/auto.22490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu D., Wang X., Zhou R., Yang L., Cederbaum A.I. Alcohol steatosis and cytotoxicity: the role of cytochrome P4502E1 and autophagy. Free Radic Biol Med. 2012;53:1346–1357. doi: 10.1016/j.freeradbiomed.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sid B., Verrax J., Calderon P.B. Role of AMPK activation in oxidative cell damage: implications for alcohol-induced liver disease. Biochem Pharmacol. 2013;86:200–209. doi: 10.1016/j.bcp.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Ding W.X., Li M., Yin X.M. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy. 2011;7:248–249. doi: 10.4161/auto.7.2.14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ni H.M., Du K., You M., Ding W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol. 2013;183:1815–1825. doi: 10.1016/j.ajpath.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding W.X., Yin X.M. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 37.Ding W.X., Ni H.M., Gao W., Yoshimori T., Stolz D.B., Ron D., Yin X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liuzzi J.P., Yoo C. Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol Trace Elem Res. 2013;156:350–356. doi: 10.1007/s12011-013-9816-3. [DOI] [PubMed] [Google Scholar]
- 39.Kharbanda K.K., McVicker D.L., Zetterman R.K., MacDonald R.G., Donohue T.M., Jr. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology. 1997;26:929–934. doi: 10.1002/hep.510260419. [DOI] [PubMed] [Google Scholar]
- 40.Takamura A., Komatsu M., Hara T., Sakamoto A., Kishi C., Waguri S., Eishi Y., Hino O., Tanaka K., Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y.S., Ueno I., Sakamoto A., Tong K.I., Kim M., Nishito Y., Iemura S., Natsume T., Ueno T., Kominami E., Motohashi H., Tanaka K., Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 42.Schuler M., Dierich A., Chambon P., Metzger D. Efficient temporally controlled targeted somatic mutagenesis in hepatocytes of the mouse. Genesis. 2004;39:167–172. doi: 10.1002/gene.20039. [DOI] [PubMed] [Google Scholar]
- 43.Bertola A., Mathews S., Ki S.H., Wang H., Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu M., Yin H., Mitra M.S., Liang X., Ajmo J.M., Nadra K., Chrast R., Finck B.N., You M. Hepatic-specific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology. 2013;58:1953–1963. doi: 10.1002/hep.26589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wieser V., Tymoszuk P., Adolph T.E., Grander C., Grabherr F., Enrich B., Pfister A., Lichtmanegger L., Gerner R., Drach M., Moser P., Zoller H., Weiss G., Moschen A.R., Theurl I., Tilg H. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J Hepatol. 2016;64:872–880. doi: 10.1016/j.jhep.2015.11.037. [DOI] [PubMed] [Google Scholar]
- 46.Osna N.A., Donohue T.M., Kharbanda K.K. Alcoholic liver disease: pathogenesis and current management. Alcohol Res. 2017;38:147–161. [PMC free article] [PubMed] [Google Scholar]
- 47.Sozio M., Crabb D.W. Alcohol and lipid metabolism. Am J Physiol Endocrinol Metab. 2008;295:E10–E16. doi: 10.1152/ajpendo.00011.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khambu B., Huda N., Chen X., Antoine D.J., Li Y., Dai G., Kohler U.A., Zong W.X., Waguri S., Werner S., Oury T.D., Dong Z., Yin X.M. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J Clin Invest. 2018;128:2419–2435. doi: 10.1172/JCI91814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H.J., Gao B., Zakhari S., Nagy L.E. Inflammation in alcoholic liver disease. Annu Rev Nutr. 2012;32:343–368. doi: 10.1146/annurev-nutr-072610-145138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Degre D., Lemmers A., Gustot T., Ouziel R., Trepo E., Demetter P., Verset L., Quertinmont E., Vercruysse V., Le Moine O., Deviere J., Moreno C. Hepatic expression of CCL2 in alcoholic liver disease is associated with disease severity and neutrophil infiltrates. Clin Exp Immunol. 2012;169:302–310. doi: 10.1111/j.1365-2249.2012.04609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamada K., Mizukoshi E., Seike T., Horii R., Kitahara M., Sunagozaka H., Arai K., Yamashita T., Honda M., Kaneko S. Light alcohol consumption has the potential to suppress hepatocellular injury and liver fibrosis in non-alcoholic fatty liver disease. PLoS One. 2018;13:e0191026. doi: 10.1371/journal.pone.0191026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khambu B., Li T., Yan S., Yu C., Chen X., Goheen M., Li Y., Lin J., Cummings O.W., Lee Y.A., Friedman S., Dong Z., Feng G.S., Wu S., Yin X.M. Hepatic autophagy deficiency compromises FXR functionality and causes cholestatic injury. Hepatology. 2019;69:2196–2213. doi: 10.1002/hep.30407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lieber C.S. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health. 2003;27:220–231. [PMC free article] [PubMed] [Google Scholar]
- 54.Roberts B.J., Song B.J., Soh Y., Park S.S., Shoaf S.E. Ethanol induces CYP2E1 by protein stabilization. Role of ubiquitin conjugation in the rapid degradation of CYP2E1. J Biol Chem. 1995;270:29632–29635. doi: 10.1074/jbc.270.50.29632. [DOI] [PubMed] [Google Scholar]
- 55.Ueno A., Lazaro R., Wang P.Y., Higashiyama R., Machida K., Tsukamoto H. Mouse intragastric infusion (iG) model. Nat Protoc. 2012;7:771–781. doi: 10.1038/nprot.2012.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ni H.M., Woolbright B.L., Williams J., Copple B., Cui W., Luyendyk J.P., Jaeschke H., Ding W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61:617–625. doi: 10.1016/j.jhep.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ni H.M., Boggess N., McGill M.R., Lebofsky M., Borude P., Apte U., Jaeschke H., Ding W.X. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci. 2012;127:438–450. doi: 10.1093/toxsci/kfs133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hartmann P., Hochrath K., Horvath A., Chen P., Seebauer C.T., Llorente C., Wang L., Alnouti Y., Fouts D.E., Starkel P., Loomba R., Coulter S., Liddle C., Yu R.T., Ling L., Rossi S.J., DePaoli A.M., Downes M., Evans R.M., Brenner D.A., Schnabl B. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology. 2018;67:2150–2166. doi: 10.1002/hep.29676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu X., Jogasuria A., Wang J., Kim C., Han Y., Shen H., Wu J., You M. MitoNEET deficiency alleviates experimental alcoholic steatohepatitis in mice by stimulating endocrine adiponectin-Fgf15 axis. J Biol Chem. 2016;291:22482–22495. doi: 10.1074/jbc.M116.737015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J., Kim C., Jogasuria A., Han Y., Hu X., Wu J., Shen H., Chrast R., Finck B.N., You M. Myeloid cell-specific lipin-1 deficiency stimulates endocrine adiponectin-FGF15 axis and ameliorates ethanol-induced liver injury in mice. Sci Rep. 2016;6:34117. doi: 10.1038/srep34117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhong W., Zhang W., Li Q., Xie G., Sun Q., Sun X., Tan X., Sun X., Jia W., Zhou Z. Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol- induced hepatic steatosis and cell death in mice. J Hepatol. 2015;62:1375–1381. doi: 10.1016/j.jhep.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo R., Xu X., Babcock S.A., Zhang Y., Ren J. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. J Hepatol. 2015;62:647–656. doi: 10.1016/j.jhep.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Kwon H.J., Won Y.S., Park O., Chang B.X., Duryee M.J., Thiele G.E., Matsumoto A., Singh S., Abdelmegeed M.A., Song B.J., Kawamoto T., Vasiliou V., Thiele G.M., Gao B. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology. 2014;60:146–157. doi: 10.1002/hep.27036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Deltour L., Foglio M.H., Duester G. Metabolic deficiencies in alcohol dehydrogenase Adh1, Adh3, and Adh4 null mutant mice. Overlapping roles of Adh1 and Adh4 in ethanol clearance and metabolism of retinol to retinoic acid. J Biol Chem. 1999;274:16796–16801. doi: 10.1074/jbc.274.24.16796. [DOI] [PubMed] [Google Scholar]