

Abstract
With a wide range of applications, from energy and environmental engineering, such as in gas separations and water purification, to biomedical engineering and packaging, glassy polymeric materials remain in the core of novel membrane and state-of the art barrier technologies. This review focuses on molecular simulation methodologies implemented for the study of sorption and diffusion of small molecules in dense glassy polymeric systems. Basic concepts are introduced and systematic methods for the generation of realistic polymer configurations are briefly presented. Challenges related to the long length and time scale phenomena that govern the permeation process in the glassy polymer matrix are described and molecular simulation approaches developed to address the multiscale problem at hand are discussed.
Keywords: polymers, diffusion, transition state theory, sorption, permeability, penetrant, separations, kinetic Monte Carlo, coarse-graining, multiscale modeling
1. Introduction
Molecular modeling and simulation play an increasingly important role in the design of products and processes for addressing the grand challenges faced by contemporary society in domains such as health, energy, food, clean water, and the protection of the environment. This is attested by the dramatic increase in the fraction of research papers in science and engineering that use molecular modeling [1]; by the enhanced role of molecular modeling activities in industry [2]; and by economic analyses of the impact of molecular modeling [3]. Very visible and well-funded national initiatives in the area of materials include modeling and simulation as one of their top priorities (see, for example, Materials Genome Initiative in the United States [4]) or are entirely focused on molecular and multiscale modeling (see, for example, OCTA Project in Japan [5] and European Materials Modeling Council in the European Union [6]).
Molecular simulations of sorption and diffusion in membrane materials have been performed since the late 1980s. This is understandable, given that membrane separations constitute a very dynamic and rapidly growing field of modern technology. Early molecular dynamics simulations were instrumental in elucidating atomic-level mechanisms, e.g., of elementary jumps of gaseous penetrants executed between clusters of accessible volume in a glassy polymer. In the 1990s the basic methodology for predicting sorption isotherms and diffusion coefficients from atomistic simulations was established and validated on well-defined model systems. This methodology is exposed in several past reviews of permeation phenomena in amorphous polymers [7,8,9]. Contemporary simulation efforts have expanded to address permeation phenomena in more sophisticated materials of complex chemical constitution, nanostructured and hybrid organic-inorganic systems. As the application of conventional molecular simulation algorithms is often too computationally demanding for such systems, there is increasing interest in multiscale modeling methods based on systematic coarse-graining of the molecular representation. For some categories of new membrane materials whose synthesis has taken off only recently, such as Metal Organic Frameworks, one sees simulation work going hand-in-hand with, and even guiding, experimental efforts. High throughput computational screening for the identification of new nanoporous material structures that best satisfy the requirements of specific gas separation, sequestration, or storage applications has emerged [10]. Machine learning techniques are starting to be used both in the development of more refined quantum mechanics-based classical force fields (or coarse grained ones based on the atomistic representation) for conducting simulations and in mining the copious data generated by molecular simulations for materials design.
In this article we present a brief review of molecular modeling and multiscale simulations (Figure 1) of sorption and diffusion of small molecules in glassy polymers, a broad category of materials which continue being a workhorse of selective membrane separation technology. The treatment is concise and certainly not intended to be all-encompassing. Emphasis is laid on work conducted in the last couple of decades. An extensive reference list is provided, to which the interested reader can resort for details.
Figure 1.

Molecular simulation methods at multiple length- and time scales. Hierarchical multiscale simulations utilize information extracted from the previous scale as input for conducting molecular simulations at longer length and time scales.
The structure of the article is as follows: in Section 2 we present an introduction to basic concepts and discuss equilibrium and non-equilibrium molecular dynamics and Monte Carlo techniques for simulating polymers and sorption and diffusion of small molecules therein. Methodological aspects of the simulations are addressed very briefly—they have been presented extensively in earlier reviews. Emphasis is laid on multiscale methods that incorporate infrequent event analysis of elementary diffusive jumps and on coarse-graining/reverse mapping for the generation of realistic model configurations in which to study permeation phenomena. Both these aspects are necessary for predicting the separation performance of dense glassy polymer matrices, especially if their chemical constitution is complex. Section 3 discusses coarse-grained approaches to the simulation of sorption and diffusion phenomena in polymers per se. The approaches discussed have been designed to address long length and time scale phenomena by cutting down on the computational cost relative to full atomistic analyses; they include methodologies that incorporate mesoscopic kinetic Monte Carlo techniques that utilize information from atomistic simulations (Figure 1) and coarse-grained molecular dynamics simulations accompanied by appropriate time mapping. Some mechanistic aspects of sorption and transport revealed by recent simulation work are presented in Section 4. Emphasis in this Section is laid on the characteristics of the penetrant’s hopping diffusional mechanism, on swelling and plasticization of glassy polymer matrices by compressed CO2 and on mixed gas sorption and diffusion phenomena in polymer glasses. Finally, Section 5 briefly pinpoints challenges that still have to be met, especially in conjunction with new polymer-based membrane materials, and reflects on the future outlook of molecular simulations of sorption and diffusion phenomena in dense amorphous polymeric membranes.
2. Background and Methodology
During the membrane separation process the penetrant molecules are transferred from an upstream fluid phase to the polymer membrane where they dissolve on the membrane surface (sorption phase) and then diffuse through the polymer matrix, exiting eventually at the downstream fluid phase (desorption). The permeation process through dense polymer membranes is described by the widely known solution-diffusion mechanism, considering a thermodynamic process (sorption) and a kinetic process (mass transport). Mass transport phenomena are governed by random molecular motion and permeation is driven by the presence of chemical potential gradients. In practical formulations the gradient of concentration is used instead of the gradient of chemical potential. In a dense polymeric membrane, permeation of a single component is often described locally by the empirical Fick’s laws [11]:
| (1) |
| (2) |
where is the concentration of the diffusing component, is the diffusion coefficient, and is the time. The concentration profile inside the membrane is considered to reach a steady state after a certain time. Assuming that is concentration-independent, which is typically the case at low concentrations, Equation (1) can be integrated to [12]:
| (3) |
where is the membrane thickness and and are the upstream and downstream permeant concentration, respectively. Equation (3) can be also expressed in terms of fluid partial vapor pressures or fugacities, and on the two sides of the membrane:
| (4) |
in which denotes the permeability coefficient. At low concentrations in the Henry regime and considering a constant diffusion coefficient over a range of pressures, the permeability of a membrane to a specific kind of penetrant molecules can be expressed as the product of solubility coefficient, and diffusivity, of the penetrant in the membrane:
| (5) |
where the solubility is considered as the ratio of concentration at a specific pressure to the pressure.
The ideal permselectivity of a membrane material for two penetrant components (i and j) is defined as the ratio of permeabilities for each pair of species in pure:
| (6) |
The ideal permselectivity can be estimated as the product of diffusivity selectivity and solubility selectivity under the assumption that each penetrant component is not affected by the other:
| (7) |
A high performance separation membrane [13] is expected to exhibit high selectivity characteristics combined, simultaneously, with satisfactory, or even, ideally, high permeability to one of the penetrant components. The selectivity performance of membrane materials for gas separations is limited by an upper bound. This empirical limit originates from the fact than an increase in permeability of the more permeable component is accompanied by a decrease in the separation ability. If is the permeability of the more permeable gas, the upper bound [13,14] is determined as a line in the log-log plot of the relationship , where is the slope of the line, above which no data exist. Data near or on the upper bound correspond almost exclusively to glassy polymers and this is attributed primarily to the higher solubility coefficients in glasses compared to rubbery polymers due to the increase of free volume below the glass transition temperature (Tg), and in some cases partially to a better diffusion selectivity of glassy polymers in comparison to the rubbery ones. In Figure 2, data on selectivity for O2/N2 separation are plotted as a function of O2 permeability for rubbery polymers and the upper bounds for both rubbery and glassy polymers [15,16] are shown that exhibit about an order of magnitude difference in favour of the separation performance of glassy polymers.
Figure 2.

Permselectivity performance of glassy polymers in comparison to rubbery polymers for O2/N2 separation. Adapted with permission from [15].
Molecular simulation methods have been developed and are employed to unravel the microscopic mechanisms that are responsible for sorption and transport phenomena in glassy polymeric membranes and to predict their macroscopic permeability and selectivity properties. A compact summary of some representative molecular simulation methods and their applicability is provided in Table 1.
Table 1.
Compact summary of representative computational methods and their applicability for the prediction of solubility and diffusivity in glassy polymers.
| Method | Application | Advantages | Disadvantages | Refs. |
|---|---|---|---|---|
| Molecular Dynamics | Numerical integration the system’s classical equations of motion | Calculation of thermodynamic, dynamic and transport properties | Not able to correctly sample the dynamics of systems that are characterized by a broad range of time scales or rare events | [17,18,19] |
| Monte Carlo | Stochastic method—generation of a Markov chain sequence of configurations | Efficient in sampling long-chain macromolecular systems when coupled with appropriately designed moves ‡ | Does not account for the system’s time evolution—cannot be used to study the system’s dynamics | [18,20,21] ‡[22,23,24,25,26,27,28] |
| Transition State Theory | Infrequent events | Determination of rate constants and penetrant jump pathways | Multidimensional TST that accounts for polymer cooperative motion is computationally intensive | [8,9,29,30,31,32,33,34] |
| Transition Path Sampling | Infrequent events | Determination of realistic pathways at finite temperatures | Dependence on the limited initial transition pathways extracted by MD simulations | [35] |
| Kinetic Monte Carlo | Mesoscopic simulation of a Poisson process | Calculation of penetrant diffusivity by solving numerically the master equations | Requires information on sorption sites network, rate constants and sorption probabilities determined from the atomistic scale | [8,33,34,36,37,38] |
| Coarse-grained MD | Simulation of dynamics at a mesoscopic scale | Simulation of longer time- and length- scales |
|
[39,40,41,42] |
| Widom Test Particle Insertion Method | Sorption | Low concentration sorption of small molecules | Inefficient for dense systems/large solutes | [7,43,44,45] |
| Iterative Widom Schemes | Sorption | Determination of sorption isotherms | Inefficient for dense systems/large solutes | [46,47] |
| Thermodynamic Integration | Sorption | Enhanced efficiency for dense systems | Requires conduction of a series of simulations | [48,49] |
2.1. Molecular Dynamics
Molecular dynamics (MD) [17,18,19,21] is based on the solution of the classical equations of motion of a system in a microscopic representation via numerical integration. During an equilibrium MD simulation the evolution of the multiparticle model representing the system is followed over time, and thermodynamic and dynamical properties are computed as averages over its trajectory. MD is widely used for equilibration and property prediction in computational materials science using atomistic or coarse-grained, fully flexible or constrained model systems and a number of statistical ensembles. MD is demanding in terms of computational time and a lot of effort has been expended in the direction of reducing these demands by implementing for example domain decomposition methods for the parallelization of MD algorithms and by developing multiple time step integration schemes [50,51] that allow the use of longer time steps for the interactions that change at a slower rate. The phase-space points visited along a long MD trajectory constitute a representative sample of the equilibrium ensemble dictated by the macroscopic constraints (e.g., constant number of particles, volume, and energy) under which the trajectory was generated, allowing the estimation of thermodynamic properties as ensemble averages. Despite progress in algorithmic developments, the extremely broad range of time scales that characterize the various modes of motion in macromolecular systems, especially in the glassy state (from tens of femtoseconds for covalent bond vibrations to years for physical aging), renders MD an insufficient method for properly equilibrating and correctly sampling the dynamics of these systems or the transport of small molecules therein.
The non-equilibrium MD (NEMD) methodology has been employed in an effort to accelerate penetrant diffusion. Here external forces are applied that retain the simulated system away from equilibrium and enable the determination of the induced flux that develops in response to the imposed forces. For small external forces, the transport properties under steady-state conditions can be directly extracted from the ratio of the flux to the imposed force, as the system is weakly perturbed, remaining in the linear response regime [52,53]. NEMD has been applied for the study of transport phenomena in amorphous polymers. Müller-Plathe et al. [54] were the first to employ NEMD for the study of the transport properties of gases in polyisobutylene. No substantial computational gain compared to equilibrium MD simulations was detected in their work within the linear response regime. Van der Vegt et al. [55] implemented NEMD for the simulation of penetrants at low concentrations using a harmonic external potential and determining the linear regime by a single simulation. NEMD has also been used for the investigation of the permeation mechanism in microporous polymers under the limitation of very small fluxes observed for small external forces [56]. Recently, Anderson and co-workers [57] employed NEMD to examine gas transport in the interfacial region of three polymeric systems (polyethylene, poly (4-methyl-2-pentyne) and polydimethylsiloxane), incorporating in the prediction of penetrant permeability and diffusivity the effect of the chain oscillation amplitude and of the accessible cavity fraction in the polymer matrices.
2.2. Monte Carlo
Monte Carlo (MC) simulation [18,21] of a microscopic model aims at generating a large number of configurations sampling in an asymptotic manner the probability density of a statistical mechanical ensemble under specific macroscopic conditions. During an MC simulation, each state is generated depending only on the one that precedes it, by implementing an attempted elementary move that may be accepted or rejected [20] based on the energetics and according to specific selection criteria; the latter are designed to follow the principle of microscopic reversibility (or detailed balance) in the generation of the Markov chain sequence of configurations. The attempted moves often involve a very small subset of degrees of freedom, implementing translations and rotations, insertions, deletions, or swaps of molecules, or volume fluctuations of the entire model system. MC is a stochastic technique that does not account for the system’s time evolution and therefore cannot be used for the study of dynamics. This stochastic character of MC, though, coupled with appropriately designed moves, enables the equilibration and sampling of long-chain macromolecular systems orders of magnitude more efficiently than with MD, opening the way to addressing long length scale phenomena.
For the simulation of polymeric systems sophisticated moves [24,27,58] have been introduced, including configurational bias (CBMC) [59,60,61], concerted rotation (CONROT) [58,62], reptation and various connectivity altering moves such as end-bridging (EBMC) [22,23] and double-bridging (DB) [26]. During an end-bridging move, a chain is divided in two pieces via the excision of a trimer segment located in the chain’s inner part as the chain is “attacked” by the end of another chain that lies in close proximity. One piece of the initial chain is then connected to the attacking end by reconstructing the trimer segment that was originally excised. In similar spirit, a double-bridging move is implemented by excising two trimer segments from two individual neighboring chains that are again reconstructed to bridge the remaining chain parts, forming two new chains of the same length as the original ones, but in a completely different conformation. In Figure 3, end-bridging and double bridging MC moves are illustrated for the case of chains with directionality in their macromolecular architecture, in which only the moves depicted with the blue arrows are allowed to be implemented for the chemical structure to be preserved. Using these moves it is possible to equilibrate at all length scales polymer melts with molar mass distributions comparable to those encountered in membrane applications. Equilibrated melt configurations obtained in this way constitute excellent starting points for generating glassy amorphous polymer configurations through cooling. MC methods are often implemented for the study of sorption in membrane materials (see Section 2.3).
Figure 3.

Illustration of the (a) end-bridging and (b) double-bridging MC moves designed to be implemented for polymeric chains with directionality in their chemical structure. In each case, the moves shown by blue arrows are possible. Intramolecular double-bridging MC moves cannot be performed in the case of directional chains.
2.3. Methods for the Molecular Simulation of Penetrant Sorption
The methods that are used within the context of penetrant sorption mainly involve the implementation of Grand Canonical Monte Carlo (GCMC) and Gibbs Ensemble Monte Carlo (GEMC) [63]. In GCMC the chemical potential, the volume and the temperature are kept constant, while the number of particles changes during the simulation, fluctuating around an equilibrium value. During the GCMC simulation the polymer system is considered to be in contact with a penetrant reservoir at specified temperature and chemical potential, with which it exchanges energy and particles. The trial moves in this case are translations, insertions, and deletions of particles.
GEMC enables the study of phase equilibria in one simulation by simultaneously incorporating two individual simulation boxes between which there is no interface. The attempted moves in this case involve particle translation and rotation, volume change (usually in a manner such that the total volume is preserved) and random particle swaps between the two simulation boxes implemented to simulate phase equilibrium of the two phases under the same temperature, pressure and chemical potential conditions. In systems containing a nonvolatile condensed (e.g., polymer) component at equilibrium with a pure or mixed gas phase, the solubilities of the gaseous permeants can be calculated using MC in a hybrid isothermal-isobaric and grand canonical ensemble [9] that does not require explicit simulation of the gas phase. This is termed the N1f2...fnPT ensemble and keeps fixed the number of molecules of the condensed component, N1, the fugacities of the gas components, f2,...,fn, as well as the system temperature and the pressure. Note that only n of the quantities f2,...,fn,P,T are independent.
In some cases the acceptance probability of the attempted insertions and deletions in the above MC schemes is very low, such as in the case of low-accessible volume polymer matrices, very strong penetrant/polymer interactions, or bulky penetrant molecules. Semigrand canonical ensemble MC [22,64,65] can be applied in this type of binary or multicomponent mixtures, during which identity exchange trial moves among the various species is attempted. Considering, for example, a binary system consisting of species A and B, MC moves are attempted that involve conversion of one particle of species A to species B or vice versa. The change in free energy (difference of chemical potentials) brought about by such a change in the chemical identity is taken into account in the acceptance of the move.
At low pressures, the Henry’s law constants can be calculated using the Widom insertion test particle method [66]. The method involves insertion of a ghost penetrant molecule in the polymer matrix at random positions and orientations and calculation of the interaction energy of the inserted molecule with the matrix. The latter is used subsequently for the determination of the excess chemical potential of the test molecule. The solubility of the molecule in the polymer matrix can be calculated as:
| (8) |
with STP corresponding to a temperature of 273.15 K and a pressure of 101.325 kPa and the solubility obtained in units of . A large number of test particle insertions are implemented in well equilibrated configurations of the polymer systems obtained from MD or MC simulations. The method has been widely applied for the estimation of low concentration sorption of small molecules in polymer systems [7,43,44,45].
Sorption isotherms can also be extracted utilizing iterative schemes that involve MD simulations of the polymer-penetrant system at constant composition, calculation of the excess chemical potential and gas fugacity and carrying out new MD simulations at a corrected pressure until convergence is reached [46,47].
More elaborate computational techniques are often necessary in cases that involve sorption of large penetrants, high pressures or strong penetrant-polymer interactions, especially at low temperatures. These methods include configurational bias insertions [67], which are used for the bond-by-bond insertion of multi-atom solutes for which Widom test particle insertion is not successful; thermodynamic integration [48,49] during which a small solute is included in a dense system gradually using progressively increasing coupling parameters for the solute-matrix interactions and conducting a series of simulations; expanded ensemble techniques [68,69] that also involve a stepwise insertion or deletion of the solute and the implementation of free energy perturbation within a single simulation; excluded-volume map sampling [70,71,72,73] and grid search methods [74,75] for efficient sampling in dense systems by blocking regions in which solute insertions are not feasible; the test particle deletion method [76,77] which constitutes an inverse of the Widom method for the calculation of the chemical potential of particles and multisite solutes; extended ensemble MD [78] during which a dynamic coupling parameter is used for the solute-polymer interactions that enhances efficient sampling of the system’s phase space; the scission-fusion MC method [67] for the determination of solubility of oligomer solutes in polydisperse polymeric systems of the same chemical constitution as the solute; grand canonical MD [79] that is implemented for vapor-liquid equilibria and sorption in rigid polymer matrices by conducting two individual simulations in different ensembles without the requirement of particle exchanges between the phases; and fast-growth thermodynamic integration [80] where the chemical potential is calculated efficiently from a number of independent thermodynamic integrations for the estimation of sorption of large molecules in dense matrices.
2.4. Molecular Simulation Methods for the Study of Infrequent Events
The wide range of time scales that are involved in the relaxation of various modes of motion in polymeric systems renders their simulation a very complicated task. At temperatures near or below the glass transition temperature (Tg), in particular, the polymer matrix becomes almost static, prohibiting the redistribution of accessible volume. This has profound effects on the diffusion of a small penetrant molecule in a selective glassy polymer. The penetrant spends most of its time executing fast local motions while trapped within pockets of accessible volume, or “sorption sites,” within the polymer matrix. Penetrant jumps between sorption sites are infrequent events [8] and cannot be properly sampled by performing even extensively long MD simulations. Transitions from one cavity to another occur rarely and randomly, and generally involve not only translational motion of the penetrant, but also local rearrangements in the polymer matrix that instantaneously open up passages between neighboring pockets of accessible volume.
Transition state theory of infrequent events (TST) [81,82,83] provides appropriate means for the study of systems that are characterized by time scale separation phenomena and crossing of energy barriers that are much higher than the thermal energy, . The dynamical evolution of such systems consists of long periods of vibrational motion within a potential energy basin; transitions between different basins (states) are infrequent, corresponding to average times of many vibrational periods. First order rate constants [84] enable the quantification of the average waiting times (which may be very broadly distributed) that elapse between specific transitions occurring and can be calculated invoking TST. The probabilities of occupancy of two states A and B that are separated by a high energy barrier evolve according to a master Equation:
| (9) |
| (10) |
where and are the probabilities of occupancy of states A and B, respectively, and the rate constant for a transition from state A to state B.
Implementing TST for the study of a penetrant jump in a polymer matrix involves the determination of transition states between sorption states (Figure 4) as well as of the pathways followed by the system while passing from one state to another [8,9]. In a glassy polymer matrix there is a large number of regions in which a penetrant molecule may reside and there is at least one transition state between any two states. TST enables the calculation of rate constants along individual pathways based on the probability of the system to be on the dividing surface between two states compared to the probability of its initial state, neglecting the existence of any re-crossing events. From a mathematical point of view, a transition state is a first-order saddle point of the potential energy in the polymer-penetrant multidimensional configuration space, i.e., a point where the potential energy gradient equals zero and there is one negative eigenvalue of the Hessian matrix of second derivatives. The jump pathway can be considered as the lowest energy path (around which actual crossing trajectories would fluctuate) that connects two neighboring local minima passing through the transition state and is usually determined by calculating the Intrinsic Reaction Coordinate (IRC) [85]. The general TST expression for the calculation of the associated rate constant is given by:
| (11) |
with and denoting the Gibbs energy of the system lying on the dividing surface and in the initial state, respectively. Using a harmonic approximation for the potential energy and considering the quantum mechanical vibrational partition function, the rate constant can be written as [32]:
| (12) |
where , and are the vibrational frequencies and potential energy at the initial state and at the transition state, respectively. If re-crossing events are important in the process, then a dynamical correction factor to has to be calculated and taken into account [82,86,87].
Figure 4.

Trajectory, in 3N dimensional coordinate space q, that passes from one basin to another, crossing a (3N-1) dimensional “dividing surface” separating the two basins. Solid lines correspond to a (3N-1)-dimensional hypersurface of constant energy , is the coordinate along the negative curvature of (reaction coordinate) and is a (3N-1)-dimensional hypersurface normal to the reaction coordinate, constituting the dividing surface between the states A and B.
Gusev et al. [29] were the first to systematically apply TST for the study of diffusivity of spherical probes in rigid minimum energy glassy polymeric matrices generated using molecular mechanics. Identifying the invocation of a static polymer matrix as a crucial drawback of their study, Gusev and Suter [30] implemented the TST method allowing independent harmonic vibrations of the polymer atoms around their equilibrium positions. The elastic motion of the polymer matrix incorporated in their work involved the harmonic motion of polymer atoms at very short times, thus not taking into account any structural relaxation. The amplitude of the elastic motion was determined by an adjustable parameter that directly affected the extracted rate constants. Within the Gusev-Suter approach, TST calculations were conducted in the three-dimensional space of the spherical penetrant. Their method was computationally efficient and in cases that involved very small penetrants able to extract satisfactory results [7,30,88,89,90].
Greenfield and Theodorou [31,32] developed a multidimensional TST methodology in generalized coordinates, in which polymer degrees of freedom are taken into account for the calculation of transition states and diffusion pathways considering a spherical penetrant. The polymer atoms are in this case allowed to move, enabling the realization of local-chain motions that take place during a penetrant passage from one cavity to another. The inclusion of polymer degrees of freedom in the pathway determination becomes very important as the size of the penetrant increases compared to the typical size of clusters of accessible volume in the glassy polymer matrix. Greenfield used a geometric analysis approach [91] for the determination of the accessible volume and its distribution in the polymer matrix to obtain an initial estimate of sorption sites. Narrow-necking regions connecting regions of accessible volume for small spherical probes serve as the starting points for the calculation of the transition states (Figure 5). The reaction path from one state to the other was calculated using a subset of degrees of freedom that were relevant to the specific transition along the pathway. The method was further extended [33,34,92] and applied for the study of polymers with complex chemical constitution in Cartesian coordinates and in the polymer-penetrant multidimensional space using a fully flexible representation for a CO2 penetrant.
Figure 5.
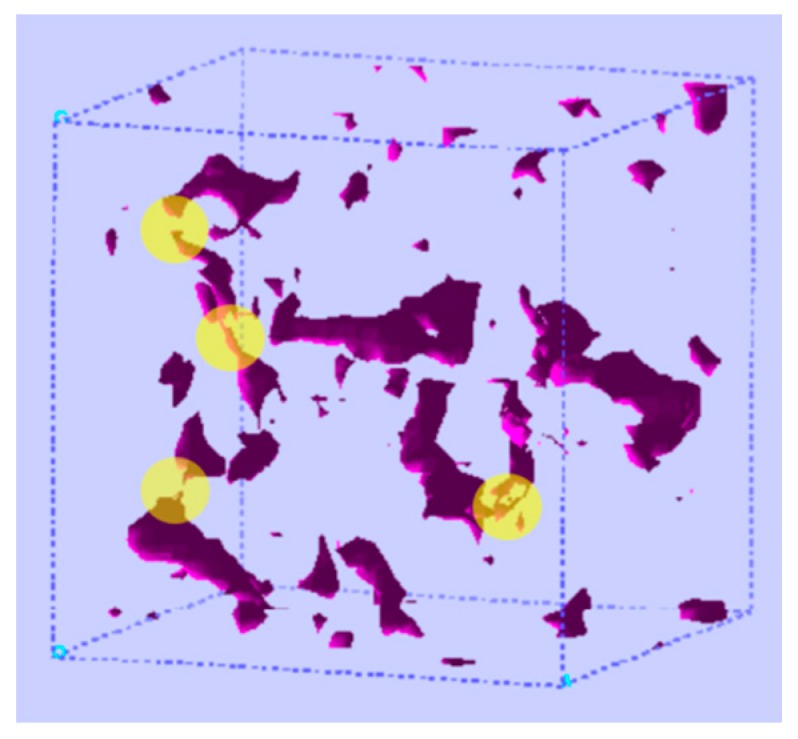
Indicative narrow-necking regions in the accessible volume obtained from geometric analysis, which serve as initial estimates for the transition state search calculations [33]. The simulated polymer is a poly(amide imide) in a cubic box of edge length 28.09 Å with periodic boundary conditions. Atoms are not shown. The dark regions are clusters of accessible volume, as determined with a spherical probe of radius 1.1 Å. Necking regions are identified by probing the same configuration with a smaller probe radius. Some of the necking regions are indicatively highlighted in yellow.
Rare events can be also studied by implementing transition path sampling methods (TPS) [93,94]. The TPS methodology is used for the calculation of rate constants by sampling dynamical reactive trajectories. Trial trajectories are generated based on a pre-determined transition path, a point of which is randomly selected and the momenta corresponding to it are perturbed. The acceptance probability of the generated trial trajectories can be tuned by the amplitude of the momenta perturbation, with the acceptance increasing for small perturbations.
A number of TPS schemes have been developed to address acceptance probability issues [93,95,96,97,98]. TPS generally enables the determination of realistic pathways at finite temperatures, but is computationally intensive. It has been implemented for the study of a wide range of infrequent events, such as nucleation [99,100,101,102], protein folding and conformational changes [103,104,105,106], glassy dynamics [107], reactions [108,109,110] and penetrant diffusion in non-amorphous materials [111,112,113]. The use of TPS methods for the study of penetrant diffusion in polymeric systems is scarce and involves the study of diffusion of water in a glassy hydrophilic polymer [35]. TPS implementation in this case directly depends on the limited initial transition pathways extracted by MD simulations for the systems under study.
2.5. Interactions and Generation of Realistic Structures
Accurate predictions of permeability properties in glassy polymeric systems largely rely on the generation of configurations that are able to resemble the real polymer matrix as well as on the reliable representation of the molecular system under study and the mathematical description of its interactions. A particle-based model may be fully atomistic (AA—all atom); united atom (UA), in which hydrogen atoms are considered to form a single interaction site together with the heavy atom to which they are connected; or coarse-grained (CG), consisting of interacting moieties that are derived by lumping together several atoms. The potential energy function in atomistic models is often derived empirically by fitting the structure, volumetric, thermal, and phase equilibrium properties of small molecule analogues, but also from ab initio electronic structure calculations. It usually consists of a bonded and a nonbonded term, that correspond, respectively, to interactions between atoms that are chemically bonded and to interactions between atoms that are not connected with chemical bonds and may belong to the same or to different molecules. Bonded interactions include contributions to the energy from deviations of chemical bond lengths and bond angles from their equilibrium values and also contributions related to dihedral angles and improper torsions. The non-bonded part includes an electrostatic term, related to the interactions between partial atomic charges (and in general between permanent multipoles), and a van der Waals term. The latter comprises pairwise interactions based on dispersion (attractive) forces due to correlations between electron clouds in different atoms; repulsive (excluded volume) interactions resulting from the overlap of electron clouds of individual atoms at very small distances; and, in some instances, polarization contributions that are also attractive and stem from an induced change in the charges of a moiety owing to the field that it experiences from its neighborhood. The force field parameters for bonded interactions, partial charges, and polarization contributions are often determined by quantum mechanical (QM) calculations, while parameters for the dispersion and excluded volume contributions are typically based on empirical methods or experimental data.
In the general case, the local segment packing that is involved in the penetrant sorption and transport mechanisms, is interconnected with the longer length scale characteristics of the polymeric system [114]. Creation of in silico realistic structures of dense amorphous polymers is a hard and complicated task [34,115]. Amorphous glasses, in particular, are far from equilibrium and are trapped in local energy minima that directly depend on their formation history. Incorporating this fact in molecular simulation strategies for the generation of well-defined and realistic configurations is a great scientific challenge that has still not been adequately addressed.
Several methodologies have been attempted for the generation and equilibration of initial structures of polymers in the melt state [28,114,116,117,118,119,120,121]. Atomistic initial configurations of the macromolecular systems can be rigorously obtained via building the chain in the simulation box bond-by-bond using Flory’s rotational isomeric state model [122], while avoiding excluded volume overlaps based on a modified continuous model for local interactions. These initial configurations are then subjected to energy minimization (molecular mechanics) that can be applied in a stagewise manner [123], removing any remaining atomic overlaps. Molecular mechanics is generally fast but not able to result in an efficient statistical mechanical sampling; nevertheless, it provides a convenient initial configuration for the subsequent implementation of systematic simulation methods towards the generation of realistic glassy polymeric structures. The atomistic structures from molecular mechanics can for example be subjected to equilibration in the melt state using the powerful connectivity altering MC moves [22,23,24,25,26,27,28] or MD or hybrid MC-MD methods. Τhe extracted equilibrated configurations in the melt state have to be subsequently cooled below Tg [124]. The efficiency of the above scheme is very good for polymer systems of rather simple macromolecular architectures and cannot be easily utilized for polymeric matrices with rigid backbones, strong electrostatic and polar interactions, or bulky side groups.
In the case of polymers with complex chemical constitution, systematic hierarchical approaches are required for the generation of realistic structures which are the starting point for studying the permeability of probe molecules in their bulk. These methods may involve the mapping from the atomistic to a coarse-grained level, equilibration at the coarse-grained level and reverse mapping back to the atomistic representation. The atomistic detail is generally necessary for the study of small-molecule sorption and diffusion phenomena in polymeric matrices. Coarse-graining strategies [115,125,126,127,128,129,130,131] involve the substitution of groups of atoms by single interaction sites, thereby reducing the number of system degrees of freedom, while at the same time maintaining the ones that are important for the description of the mechanisms/processes under study. The CG model invoked in each case is thus intimately related to the macromolecular architecture and the scientific problem at hand. Therefore, the mapping to the CG representation is largely empirical, as there is no unique way to coarse-grain. In order to perform simulations at the CG level, an effective potential for the CG model has to be developed based on the corresponding CG degrees of freedom, that is able to reproduce the key characteristics that need to be retained in the CG simulations. The majority of the CG force fields are developed and parameterized by fitting either in a top-down manner, targeting the reproduction of macroscopic properties [132,133,134,135], or aiming at the reproduction of microscopic properties of the molecular system based on atomistic simulations and experimental findings, following a bottom up approach [136] or using a combination of both strategies [137,138]. Fitting schemes for the parameterization of CG effective potentials include inverse Boltzmann and iterative Boltzmann inversion (IBI) [139], force matching [140,141], inverse Monte Carlo [142] and relative entropy [143,144] methods. In addition to those, interaction potentials between CG moieties can be also determined directly from atomistic interactions of groups of atoms that are mapped onto the CG moieties [145,146], which may also be anisotropic [129]. Hybrid particle-field methods have been also proposed and implemented for the generation of polymer atomic structures [147].
An ideal CG effective potential would enable the reproduction of target properties and would be also transferable to other thermodynamic state points [137,138,148,149,150,151]. The CG model should also allow, in a straightforward manner, reverse mapping [152,153,154,155,156,157,158,159,160,161] back to representative atomistic configurations (usually on the basis of geometric criteria) of the well equilibrated structures at the CG level. Special measures must be taken for macromolecular structures with complex chemical constitution to avoid potential overlaps or concatenations [162]. The final atomistic configurations from reverse mapping are then subjected to a final relaxation of the system’s local interactions. Figure 6 illustrates a hierarchical backmapping procedure used for the equilibration of polystyrene models at three different representations [161]. The multi-resolution method proposed in this study enables the generation of well-equilibrated high molecular weight polymer configurations that are initially equilibrated at the largest length scale corresponding, in this case, to a soft-blob based representation (Figure 6c). The detailed description of the system is introduced stagewise, first migrating from the soft-blob based to a moderately CG model (Figure 6b) and equilibrating the system locally at this level and subsequently re-inserting the microscopic detail (Figure 6a) and equilibrating again at this stage. The hierarchical backmapping using a sequence of models aims at the efficient generation of realistic long chain polymer melts in a computationally feasible manner, preserving the long-wavelength characteristics of the macromolecular systems under study.
Figure 6.

Depiction of a hierarchical backmapping corresponding to (a) a united atom representation, (b) a moderate CG model consisting of beads of types A and B, and (c) a blob-based representation of soft spheres by lumping together a number of beads of representation (b). The backmapping procedure is described in (d) for the three resolutions. Reproduced with permission from [161].
The amorphous polymer configurations to be used for the prediction of solubility and diffusivity properties have to be validated. Validation of amorphous polymer configurations can be realized via direct comparison with experimental measurements over a range of thermodynamic conditions (temperatures and pressures). Such comparison between predicted and measured values can be conducted for a number of properties, e.g., thermodynamic (density, cohesive energy, solubility parameters), structural (pair distribution functions and structure factors from X-ray and neutron scattering), and dynamical (local dynamics of the chain segments and glass transition). Among the structural properties, of particular relevance to permeability is the accessible volume and how it is distributed and connected; predictions can be compared against Positron Annihilation Lifetime Spectroscopy (PALS). It is generally advantageous to generate a large number of configurations, thereby obtaining a sample of representative “trapped” glassy states, and also to verify the stability of the final glassy configurations [9] prior to using them in subsequent simulations.
3. Coarse-Graining and Multiscale Approaches in Sorption and Diffusivity Prediction
The broad spectra of length- and time-scales present in macromolecular systems necessitate the implementation of elaborate schemes for the prediction of their properties. Apart from the coarse graining methods related to the generation of realistic initial structures discussed in Section 2.5, hierarchical schemes at multiple length and time-scales have been developed and implemented for the study per se of the permeability of small molecules in polymeric systems.
Transition state theory approaches (see Section 2.4) provide a unique means for calculation of the wide range of interstate rate constants characterizing the elementary jumps executed by a penetrant in an amorphous glassy polymer matrix. By coarse graining to a macrostate level, each penetrant jump can be categorized as an intramacrostate or intermacrostate transition, where a “macrostate” is defined as a collection of basins constructed around neighboring potential energy minima separated by energy barriers that are low compared to the thermal energy . At this level of coarse graining, the diffusivity can be calculated considering a Poisson process of successive uncorrelated penetrant jumps between macrostates. For a transition between two macrostates I and J, the time evolution of the probability that the system is in a macrostate I is calculated as follows:
| (13) |
where:
| (14) |
and and is the rate constant for the transition from macrostate I to macrostate J. A mesoscopic Kinetic Monte Carlo (KMC) simulation [8,163,164,165] is implemented for the calculation of the penetrant diffusion coefficients on the extracted (periodic) networks of macrostates and based on the calculated macrostate-to-macrostate rate constants and sorption probabilities [33,34,92]. This multiscale methodology that combines the TST calculated rate constants and sorption site network with mesoscopic simulations at the macrostate coarse-grained level has been very efficient in the prediction of penetrant diffusivity and in the elucidation of the characteristics of the penetrant’s diffusional motion. Potential artifacts that may arise from the periodic replication of the formed network can be avoided by generating large irregular networks of macrostates using a reverse Monte Carlo (RMC) approach [36]. Analytical approaches for obtaining the diffusivity from penetrant positions representative of each macrostate and from the rate constants between macrostates, taking advantage of the periodic boundary conditions that characterize the model system in each direction, have also been developed [166].
In an effort to extend the time scale limits of the MD simulations, Neyertz and Brown [37,38] implemented a simplified approach that utilizes MD trajectories of mobile gas penetrants in the polymer matrix to extract information that can be used as input in a kinetic Monte Carlo simulation, referred to as trajectory-extending kinetic Monte Carlo (TEKMC). A sequence of configurations visited during a time interval is chosen for analysis from MD trajectories of total length of a few nanoseconds. Based on the penetrant positions, the primary simulation box is divided into subcells (of the same size and shape) and the probability matrix of jumps between subcells is extracted. The method is of low computational cost, requiring, however, proper adjustments of the parameters related to the sampling interval and subcell grid size [167]. It has been applied for the study of CO2 [38] and other small penetrants [37] in fluorinated glassy polymers, leading to predictions in reasonable agreement with experimental measurements. It has also been used for the investigation of gas diffusion in polymer nanocomposites [168] and ion transport in polymer electrolyte melts [169,170,171,172]. The method can only be applied provided that a percolating network of subcells is sampled and that cases of non-linked channels are taken into account in the error of the extracted diffusivity. In order that MD be able to provide an interconnected network of subcells as required by this method, MD simulations are restricted to medium to high penetrant concentrations, in which plasticization effects are usually present [38].
Coarse-grained molecular dynamics (CG-MD) simulations have been also investigated for the modeling of transport of large penetrants in amorphous polymer systems in an attempt to reach the normal diffusion regime. The CG models in this case should be rather detailed, preserving a minimum of important information on the chemical structure of the macromolecular system under study which directly affects the diffusional behavior of the penetrants in the polymer matrix. The CG-MD simulations are accompanied by atomistic simulations for the validation of the CG model used. Kremer and co-workers [39,40] attempted a combined experimental and simulation methodology of this type for the study of the transport properties of ethylbenzene in mixture with polystyrene (PS) using CG MD with different CG representations for the PS. Lin et al. [41] incorporated the MARTINI CG force field [133,173,174] to perform CG-MD of a fragrance agent, octanal, in a polymer film consisting of four components. In their simulations, they studied the concentration and temperature dependence as well as the effect of the presence of water on the effective time scale of the CG simulations and on the diffusional behavior of octanal in the polymeric film. Three CG models have been employed by Kumar and co-workers to study the penetrant diffusion mechanism in flexible and rigid polymers for various penetrant sizes and in a wide range of temperatures [42]. In CG-MD simulations, the simulated behavior does not correspond to the time scales of the dynamics of the real system. “Time mapping” procedures are attempted in this case based on atomistic simulation in order to link the time scales involved in the CG simulations with the time scales of the real dynamics. The effective time scale in the CG-MD is strongly dependent on the mapping implementation, with coarser schemes corresponding to larger effective time scales in the CG simulations, and the calculated mechanisms are directly affected by the loss of chemical detail in the local environment of the penetrant molecule.
4. Mechanistic Aspects of Sorption and Transport
The permeation of small penetrants in dense glassy polymeric materials is governed by a number of factors that involve the chemical affinity between the molecule and the polymer as well as characteristics of the glassy polymer matrix. Unlike the mechanism involved in many porous membranes, in which the material participates in the permeation process mostly by the pore structure, in dense amorphous polymeric membranes the polymer is actively engaged in the permeation process.
4.1. Sorption
Sorption of solute molecules in a polymer matrix is directly related to both the amount and distribution of free volume in the amorphous phase and to the interactions of the penetrant with the polymer. In the glassy state, the polymeric system is not in thermodynamic equilibrium. Dense polymer glasses are structurally arrested in configurations that depend on the history of their formation (e.g., cooling rate) and have the ability only to fluctuate within a subspace of configurations neighboring the specific local minimum of potential energy in their almost static structure. Structural relaxation in the direction of equilibrium involves the crossing of very high energy barriers that may take place below Tg in time scales of the order of years, corresponding to physical aging of glassy polymers.
In an equilibrium melt state, the pure polymer as well as the polymer plus penetrant system conforms to a Boltzmann distribution in configuration space. At temperatures near or below the glass transition temperature, the dense amorphous polymer system is no longer in thermodynamic equilibrium. Rather, it is trapped in one among many low energy regions surrounding local minima of the energy, within which it fluctuates thermally. The low energy regions (basins) of the configuration space are separated by high energy barriers that require a lot of time to be overcome. The quenched polymer configurations inherently carry information about the formation history of the glass. Upon cooling, the vitrifying system falls out of equilibrium. The rate of structural relaxation (rate of transitions between different basins in configuration space) decelerates precipitously as the temperature is lowered and becomes much slower than the rate of cooling employed. Redistribution in configuration space cannot keep up with the rate of change in temperature. Thus, the distribution in configuration space becomes arrested, unable to follow the demands of equilibrium, which would dictate passage from higher-energy basins into lower energy ones. The system is trapped within disjoint basins in configuration space, the distribution of these basins being much broader than would be dictated by Boltzmann at the prevailing low temperature. Within each basin however, local reconfiguration is possible; locally, the system can be considered as following a Boltzmann distribution within the confines of the basin. At low penetrant concentrations, the solubility can be studied independently in each disjoint, barrier- surrounded basin and then an average can be taken over individual glassy basins.
As a general rule, glassy polymers have a higher sorption capacity than rubbery polymers, with the glassy sorption isotherms exhibiting a concave curvature in contrast to the rubbery ones that are almost linear. During sorption at high concentrations of highly condensable penetrants such as CO2, the potential energy barriers that resulted in disjoint subsets of configurations in the dense amorphous glass are altered, enabling a more frequent barrier crossing. This fact leads to volume swelling phenomena that are observed both in glassy and rubbery polymers, due to a penetrant-induced increase in the fractional free volume and to plasticization of the dense amorphous polymer matrix [175,176,177,178,179,180]. These phenomena directly influence the performance of the material as a separation medium [181], enhancing the permeability and diffusivity as the concentration increases. The presence of the penetrant at high concentrations accelerates the segmental dynamics of the chains, resulting in the case of polymer glasses in the reduction of the glass transition temperature due to polymer softening. Plasticization phenomena are described in terms of a characteristic pressure (known as “plasticization pressure”) which is related to a minimum in the gas permeability (Figure 7). Under those circumstances, sorption isotherms of glassy polymers at high pressures may exhibit a rubber type linear behavior, while following a glassy-type curve at lower pressures. Swelling and plasticization behavior of a polymeric system is affected by temperature [182] and this can be considered as an outcome of the interplay of the opposite trends between sorption capacity and chain mobility as a function temperature.
Figure 7.

Experimental measurement of CO2 permeability, solubility and diffusion coefficient as a function of pressure in a polyimide membrane. The black dashed-line arrow is indicative of the characteristic pressure (“plasticization pressure”) that corresponds to the minimum of permeability. Reproduced with permission from [183].
Sorption and penetrant-induced plasticization and swelling phenomena have been investigated by molecular simulation of bulk polymeric systems [38,183,184,185,186,187,188,189] using either CGMC or implementing iterative multi-stage methods to determine the correct pressure at given penetrant concentration [46,47,190]. Van der Vegt et al. [46,186] have studied sorption of CO2 in a polyethylene-like membrane and have identified below Tg a hole filling mechanism in combination with a finite positive partial molar volume of CO2 being responsible for the gas sorption thermodynamics in the glassy polymer. Spyriouni et al. [47] used well equilibrated polystyrene configurations that were obtained implementing a coarse-graining—reverse-mapping scheme [191] that were loaded up with CO2 in a wide pressure range. They observed a change in the shape of both sorption isotherms and swelling curves with increasing temperature (Figure 8). In this work, a mapping of the accessible volume was conducted to facilitate efficient loading of CO2. The polymer-solute matrices were subjected to NPT MD simulations during which the solute molecules were swapped between accessible volume cavities to achieve an efficient equilibrium repartitioning. The chemical potential calculations were performed using the Direct Particle Deletion method [47]. They also analyzed the polymer segmental relaxation times in the presence of CO2 to obtain estimates of the solvent-induced glass transition pressure at various temperatures below the Tg of pure polystyrene, obtaining good agreement with available experimental measurements. Sorption and dilation effects have been modeled in fluorinated polyimides (6FDA-ODA,6FDA-DPX,6FDA-DAM) [183,192,193], in poly (ether sulfone) (PES) and polysulfone [185] polymers and in copolyimide polymers [188] up to high pressures. Performance and plasticization resistance of an emerging class of polymeric membrane materials called ‘thermally rearranged polymers’ [194,195,196,197,198,199,200,201,202,203,204] that are generated by thermally modifying aromatic polyimide chains, has also been investigated computationally [205,206,207,208,209,210,211] in recent years. These phenomena have been also simulated for high free volume polymers of intrinsic microporosity (PIM) [56,189,212,213].
Figure 8.

Molecular simulation results on (a) isotherms of CO2 sorption and (b) polymer swelling for polystyrene at various temperatures and pressures. Reproduced with permission from [47].
As the penetrant concentrations increase, the volume dilation phenomena are often semi-permanent and, after desorption of the sorbed gas, part of the additional volume that has been created due to swelling during sorption is preserved, a phenomenon that is often referred to as “conditioning” [214]. A hysteretic behavior is thus observed during subsequent sorption cycles, the extent of which depends on the conditions of the experiment and on the amorphous glass thermodynamic history. Plasticization and conditioning effects are in many cases dependent on the thickness of the amorphous polymer membrane [215]. Physical aging generally takes place more rapidly in thin films. [215,216,217] than in the corresponding thicker ones or the ones of bulk glassy matrices and a different permeability trend is observed during the timescale of the measurements [216]. Physical aging of a glassy polymer matrix is characterized by a decrease in the free volume, leading to a densification [218,219,220] that affects sorption and permeability properties [221,222] as well as the plasticization behavior of the polymer matrix.
Molecular simulations have been applied incorporating thin membrane models [57,223,224,225,226,227,228,229,230] and explicit gas feed reservoirs that attempt to approximate experimental conditions using MD or NEMD [57]. Such simulations of interfacial phenomena often result in more intense interfacial effects due to the small thickness of the models and the large surface to volume ratio used in simulations, as modeling of dense polymeric films would require the implementation of a very extended surface area corresponding to system sizes that are currently not computationally feasible [223]. In the course of MD of 6FDA-6FpDA fluorinated polyimide membranes [231] using a CO2 gas reservoir on both sides of the film, a rapid adsorption was initially observed at polymer-gas interfaces that was subsequently followed by a slower uptake. During the sorption process, swelling effects were detected and the additional free volume regions were occupied at short time by the sorbed penetrants. The same models were also used for the study of the effect of structural isomerism in the plasticization of meta-linked and para-linked polyimides, obtaining a plasticization resistance in the meta-linked isomer [228]. In an effort to study the reversibility of polymer membrane dilation, MD simulation has been applied in an explicit film model of 6FDA-6FpDA polyimide with surfaces in contact with a gas reservoir [229]. Two cycles of sorption and post-degassing phases have been applied and metastable states of enhanced solubility after degassing have been identified that depend on the former swelling stages of the material. The two sorption phases were found to differ at short times, as additional void spaces remain after the first cycle of post-degassing relaxation.
Sorption of mixtures of various gases in glassy polymers is common in membrane separations. The mechanism of permeation varies with the concentration of the mixed gas and the competitive character of the sorption among individual species. At low concentrations, each penetrant type may sorb without being affected by the presence of the other components. For an increasing amount of sorbed gases, one species may be more soluble in the polymer matrix leading to plasticization of the membrane, thus modifying the sorption environment of the other components [175,232]. A more complex behavior may arise if strong interactions are present between one of the components and the polymeric membrane material, or among the various penetrants themselves, thus altering the permselectivity performance of the membrane. The mixed gas behavior generally hinges on the chemical identity of the gas pair and its composition along with the pressure conditions and the thickness of the membrane [233]. The actual permselectivity, differs from the ideal one (Equation (6)) [232,234] necessitating the investigation of mixed-gas conditions [235,236] along with the ones of the pure gases for the evaluation of the membrane permselectivity performance.
Mixed-gas simulations [57,223,237] have been recently performed for the study of the actual permeability performance of polymeric membranes. Tanis et al. [237] investigated the permeation properties of pure and mixed gas N2/CH4 in two 6FDA-based fluorinated polyimides and in their block copolymer. For the pure CH4 gas, they extracted a similar methane sorption behavior in the three polyimides, accompanied by a linear-type volume swelling. Small differences were observed in the gas solubility coefficients of each species calculated also for the N2/CH4 mixture in a 2:1 composition, indicating that the permselectivity performance is primarily governed by the kinetics of the gas mixture. MD simulation has been conducted for O2/N2 simultaneous sorption and separation in a 6FDA-6FpDA polyimide glassy polymer film in the presence of gas reservoirs on each side of the film [223].
High initial pressures were applied and very long simulations of 300ns were employed in order for the system to reach equilibrium both in terms of gas concentration and pressure stabilization. The volume swelling effects for the O2/N2 gas mixture were rather limited and the glassy structure was not significantly affected by the air sorption. Figure 9a depicts the average solubility in the film with increasing pressure both including and excluding the probe interactions with the other air molecules, while Figure 9b shows the solubility selectivity with respect to pressure. Both gases were found to occupy the same low energy voids and the calculated solubility selectivity was extracted in good agreement with single gas permeation experimental measurements [238,239,240,241].
Figure 9.

(a) Average (dimensionless) solubility as a function of pressure for O2 and N2 in a 6FDA-6FpDA polyimide glassy polymer film excluding (dashed lines) or including (solid lines) interactions with the other air molecules. (b) Solubility selectivity for O2/N2 separation in the same film. Reprinted with permission from [223].
4.2. Diffusion
Diffusion through a dense amorphous polymer matrix is a very slow process at temperatures near or below Tg. In dilute systems, the diffusivity of the penetrant depends on the penetrant size and its interaction with the polymer and also on the size and connectivity of the unoccupied volume in the polymer matrix. Below Tg, there is nearly no redistribution of the unoccupied space and the penetrant spends most of its time trapped in one cavity; a jump from one cavity to another rarely takes place when local fluctuations of the polymer segments open a channel that closes again directly after the jump. Diffusion in the dense glassy amorphous matrix consists of a sequence of infrequent events described by a hopping mechanism [242,243] and is directly related to the rate constants that govern the jumps between the neighboring cavities as well as the distance and connectivity of the pre-existing cavities in the polymer matrix.
The long time scales of structural decorrelation in amorphous polymers (even above Tg) result in the existence of an anomalous diffusion regime at short times which is characterized by , , with being the penetrant’s mean square displacement and being the time. The penetrant experiences heterogeneities in its local environment and very long time scales are required to enter the Einstein (or Fickian) regime of normal diffusion. Anomalous diffusion can be associated with the wide range of rate constants that govern the penetrant jumps [244,245,246] or with the connectivity of the sorption sites [7,36,43], as well as with the penetrant size [70]. TST-based molecular simulation studies have investigated the diffusion mechanism in glassy polymers, revealing important factors that govern the penetrant’s diffusional behavior such as the range of jump rate constants [32,33], the size, shape and connectivity of the volume clusters that are accessible to the penetrant molecule [33,91], the energetic and entropic contributions to the jump rate constants [9,32,33,247] and the characteristics of the cooperative motion of the polymer chains during the elementary penetrant jumps [32,33,34]. In glassy amorphous solids, a spatially non-uniform dynamics is detected and energy and entropy contributions to the rate constants differ from one region of the polymer matrix to the other. A range of rate constants of the order of 10−3–103 μs−1 were determined for CH4 in glassy atactic polypropylene [32] with a mean jump length of 5 Å. In the TST study of CO2 diffusivity in a complex poly (amide imide) system, [-NH-C6H4-C(CF3)2-C6H4-NH-CO- C6H4(CH3)-N(CO)2C6H3-CO-]n [33] the rate constants appear widely distributed, with the majority of transitions being characterized by rate constants of the order of 10−1–102 μs−1, an average jump length of 5.3 Å and activation energies in the range of 2–11 kcal/mol that fall within range of experimental values of activation energy for diffusion of gases in glassy polymers. The dimensionality of the conducted calculations drastically affects the jump energy barrier, which is significantly decreased by increasing the polymer degrees of freedom in the vicinity of the penetrant included, until an asymptotic behavior in the energy is observed and the energy barrier is no longer influenced by the incorporation of additional polymer degrees of freedom [9,32,33]. Detailed analysis of the CO2 diffusion mechanism reveals the existence of a concerted motion of the polymer segments along the transition path that allows the penetrant to pass from one sorption state to an adjacent one. Along the pathway, changes in the orientation of the CO2 penetrant take place that are important for the realization of the elementary jumps [33,92].
Increasing the penetrant concentration has a diverse effect on penetrant diffusivity, depending on the specific penetrant-polymer system [232]. The diffusion coefficient remains unaffected if low sorbing penetrants are incorporated in the polymer matrix. If penetrant clustering phenomena are present in the system, then a decrease in diffusivity may be observed. For highly condensable penetrants, high concentrations cause volume swelling effects and increase the free volume and segmental mobility [248], a fact that leads to an acceleration in the mobility of the penetrants in the matrix, resulting in higher diffusivities. Even in this case, though, the jump-type hopping mechanism still characterizes the penetrant diffusional motion [38,227,231] with shorter residence times at these conditions.
Interdiffusion phenomena in the presence of more than one penetrant species have a direct effect on the permeability properties of the polymeric system [249]. In some cases, the presence of the more mobile penetrant has the tendency to accelerate the kinetics of the slower one [235,250]. As a result the diffusional behavior of the faster penetrant is subsequently hindered by an increase in the concentration of the slower one. This behavior was also detected in the computational study of Tanis et al. [237], for fluorinated polyimide homopolymers, according to which actual permeabilities cannot be reliably estimated by the ideal ones. The inverse conclusion was drawn from the simulation of air sorption in a 6FDA-6FpDA polyimide glassy polymer film from which it was determined that mixed gas conditions for O2/N2 separation can be extracted based on single gas data [223], a fact that is expected for the case of light gases such as O2 and N2 and in the absence of plasticization and competitive sorption effects [232].
5. New Materials, Challenges and Future Outlook
The need to develop new high-performance task specific functional polymer-based membranes necessitates the investigation and use of hybrid and composite materials [251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271]. Examples of advanced polymer-based separation media include polymer/inorganic membranes incorporating, for example, zeolites, inorganic particles, or nanoparticles in polymer matrices; carbon nanotube (CNT)/polymer composites; glassy polymerized ionic liquids; polymer/ionic liquid composites; and many other multicomponent combinations of materials in polymeric membranes. Several molecular simulation studies in recent years have focused on the investigation of polymer-based composite and mixed matrix membranes [272,273,274,275]. Ionic liquid/ionic polyimide composites [276,277] have been studied using molecular simulations as gas separation media and the effect of the anion structure on gas permeability and selectivity has been studied and is to be confirmed by experimental measurements. Moreover, polymer electrolyte membranes have been studied computationally for use as clean water and desalination membranes [278] and the transport of ions in polymer electrolytes has been simulated [258] also in the presence of nanoparticles [170,171,279,280]. Gas barrier properties in several mixed matrix organic-inorganic membranes have been studied computationally such as in MOF/polymer composites [281,282,283], in zeolite/polymer mixed matrix materials [284], in polyhedral oligomeric silsesquioxane/polymer systems [230,285] and in polymer/nanotube composites [286].
In terms of molecular simulation, for the reliable prediction of properties and the fundamental understanding of the underlying mechanisms in complex and multicomponent materials, it is crucial that the existing hierarchical methods, described in the previous sections, be extended and generalized so that they can be implemented in a straightforward manner. Additionally, unravelling the interfacial interactions [287,288] and characteristics [289,290,291] in polymer-based composite materials is of great importance for an accurate description of their behavior. Development and implementation of efficient, hybrid and adaptive resolution multiscale molecular simulation methods [125,292,293,294,295,296,297,298] may in many cases be necessary for the systematic study of microscopic behavior at the interfaces in composite materials and mixed matrix membranes and for further advancement of the materials-by-design target. In this direction it would be worthwhile to study many technologically and commercially important polymer-based nanocomposite membranes [299,300,301,302,303,304,305] such as Matrimid-based ones [306,307,308], incorporating a number of potential inorganic fillers. Multiscale molecular simulation strategies in the area of next generation multicomponent nanostructured materials are important in identifying how the fillers can be tailored towards an optimum selectivity and permeability behavior, investigating a number of crucial factors such as the effects of the size and the dispersity of the fillers and nanoparticles in the terminal properties as well as the influence of the morphology and surface treatment on the behavior and stability of the multicomponent membrane. Systematic strategies need to be applied for the study of nanocomposite materials [275,309] and for the modeling of complex membranes as for example dense glassy 3D networks, developing in this case efficient cross-linking algorithms [310]. The challenge of modeling materials with increasing complexity and the high demands in terms of system sizes and in the accuracy of predictions is greatly supported by the constant increase in the computational power in the one hand and on the development of novel efficient new algorithms and methods that need to be implemented to enable the in-depth understanding of the composite materials.
In parallel, artificial intelligence and machine learning approaches [311,312,313,314,315,316,317] appear as very promising in the direction of development of improved atomistic interaction potentials utilizing directly the quantum mechanical scale, or of more accurate CG force fields extracted on the basis of the atomistic detail. In the field of force field development in general, the open challenge of optimized transferable interaction potentials is still to be met.
Apart from design of new materials with controlled properties, molecular modeling can also significantly contribute to the design and optimization of novel separation processes. For example, membrane technologies that are based in pervaporation [318,319,320] are used for the dehydration of water-organic mixtures, the separation of organic compounds from water or for separations in organic-organic mixtures. Molecular simulation using NEMD techniques can be utilized to simulate such a process and aid in the optimization of the applied fluxes.
Membrane technology is of great economic, environmental and industrial importance and is directly connected to many societal challenges that need to be faced world-wide in the years to come. Future sustainability relies on the indisputable need for green technologies and security of the water-food-energy nexus around the globe. Efficient membrane materials design is a prerequisite for the development of advanced green cutting-edge separation and barrier processes and the complementary contribution of novel experimental and computational techniques is pivotal in order for the scientific and engineering community to address the critical environmental and sustainability issues.
Acknowledgments
We would like to thank the Guest-Editors, Giulio C. Sarti and Maria Grazia De Angelis, for inviting us to contribute to the “Gas Transport in Glassy Polymers” Special Issue and for their patience with our delay in finishing the manuscript.
Author Contributions
Both authors have contributed in all aspects of this review.
Funding
This review received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Maginn E.J. From discovery to data: What must happen for molecular simulation to become a mainstream chemical engineering tool. AIChE J. 2009;55:1304–1310. doi: 10.1002/aic.11932. [DOI] [Google Scholar]
- 2.Weiß H., Deglmann P., in ‘t Veld P.J., Cetinkaya M., Schreiner E. Multiscale Materials Modeling in an Industrial Environment. Annu. Rev. Chem. Biomol. Eng. 2016;7:65–86. doi: 10.1146/annurev-chembioeng-080615-033615. [DOI] [PubMed] [Google Scholar]
- 3.Goldbeck G. The Economic Impact of Molecular Modelling of Chemicals and Materials. Goldbeck Consulting: 2012. [(accessed on 15 April 2019)]; Available online: https://gerhardgoldbeck.files.wordpress.com/2014/01/the-economic-impact-of-modelling.pdf.
- 4.Materials Genome Initiative USA. [(accessed on 15 April 2019)]; Available online: https://www.mgi.gov.
- 5.OCTA Project Japan. [(accessed on 15 April 2019)]; Available online: http://octa.jp/about-octa/project.
- 6.European Materials Modelling Council European Union. [(accessed on 15 April 2019)]; Available online: https://emmc.info.
- 7.Gusev A.A., Müller-Plathe F., van Gunsteren W.F., Suter U.W. Dynamics of small molecules in bulk polymers. In: Monnerie L., Suter U.W., editors. Atomistic Modeling of Physical Properties. Springer; Berlin/Heidelberg, Germany: 1994. pp. 207–247. [DOI] [Google Scholar]
- 8.Theodorou D.N. Molecular Simulations of Sorption and Diffusion in Amorphous Polymers. In: Neogi P., editor. Diffusion in Polymers. Marcel Dekker Inc.; New York, NY, USA: 1996. p. 67. [Google Scholar]
- 9.Theodorou D.N. Materials Science of Membranes for Gas and Vapor Separation. John Wiley & Sons, Ltd.; Hoboken, NJ, USA: 2006. Principles of Molecular Simulation of Gas Transport in Polymers; pp. 49–94. [Google Scholar]
- 10.Colón Y.J., Fairen-Jimenez D., Wilmer C.E., Snurr R.Q. High-Throughput Screening of Porous Crystalline Materials for Hydrogen Storage Capacity near Room Temperature. J. Phys. Chem. C. 2014;118:5383–5389. doi: 10.1021/jp4122326. [DOI] [Google Scholar]
- 11.Crank J., Park G.S. Diffusion in Polymers. Academic Press; New York, NY, USA: 1968. [Google Scholar]
- 12.Crank J. The Mathematics of Diffusion. Oxford University Press; Oxford, UK: 1985. [Google Scholar]
- 13.Robeson L.M. The upper bound revisited. J. Membr. Sci. 2008;320:390–400. doi: 10.1016/j.memsci.2008.04.030. [DOI] [Google Scholar]
- 14.Robeson L.M. Correlation of separation factor versus permeability for polymeric membranes. J. Membr. Sci. 1991;62:165–185. doi: 10.1016/0376-7388(91)80060-J. [DOI] [Google Scholar]
- 15.Robeson L.M., Liu Q., Freeman B.D., Paul D.R. Comparison of transport properties of rubbery and glassy polymers and the relevance to the upper bound relationship. J. Membr. Sci. 2015;476:421–431. doi: 10.1016/j.memsci.2014.11.058. [DOI] [Google Scholar]
- 16.Robeson L.M., Smith Z.P., Freeman B.D., Paul D.R. Contributions of diffusion and solubility selectivity to the upper bound analysis for glassy gas separation membranes. J. Membr. Sci. 2014;453:71–83. doi: 10.1016/j.memsci.2013.10.066. [DOI] [Google Scholar]
- 17.Haile J.M. Molecular Dynamics Simulation: Elementary Methods. John Wiley & Sons, Inc.; New York, NY, USA: 1992. p. 489. [Google Scholar]
- 18.Frenkel D., Smit B. Understanding Molecular Simulation: From Algorithms to Applications. Academic Press; San Diego, CA, USA: 2002. [Google Scholar]
- 19.Rapaport D.C. The Art of Molecular Dynamics Simulation. Cambridge University Press; New York, NY, USA: 2002. [Google Scholar]
- 20.Metropolis N., Rosenbluth A.W., Rosenbluth M.N., Teller A.H., Teller E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953;21:1087–1092. doi: 10.1063/1.1699114. [DOI] [Google Scholar]
- 21.Allen M.P., Tildesley D.J. Computer Simulation of Liquids. Oxford University Press; New York, NY, USA: 1987. [Google Scholar]
- 22.Pant P.V.K., Theodorou D.N. Variable Connectivity Method for the Atomistic Monte Carlo Simulation of Polydisperse Polymer Melts. Macromolecules. 1995;28:7224–7234. doi: 10.1021/ma00125a027. [DOI] [Google Scholar]
- 23.Mavrantzas V.G., Boone T.D., Zervopoulou E., Theodorou D.N. End-Bridging Monte Carlo: A Fast Algorithm for Atomistic Simulation of Condensed Phases of Long Polymer Chains. Macromolecules. 1999;32:5072–5096. doi: 10.1021/ma981745g. [DOI] [Google Scholar]
- 24.Uhlherr A., Mavrantzas V.G., Doxastakis M., Theodorou D.N. Directed Bridging Methods for Fast Atomistic Monte Carlo Simulations of Bulk Polymers. Macromolecules. 2001;34:8554–8568. doi: 10.1021/ma0102060. [DOI] [Google Scholar]
- 25.Karayiannis N.C., Mavrantzas V.G., Theodorou D.N. A novel monte carlo scheme for the rapid equilibration of atomistic model polymer systems of precisely defined molecular architecture. Phys. Rev. Lett. 2002;88:105503. doi: 10.1103/PhysRevLett.88.105503. [DOI] [PubMed] [Google Scholar]
- 26.Karayiannis N.C., Giannousaki A.E., Mavrantzas V.G., Theodorou D.N. Atomistic Monte Carlo simulation of strictly monodisperse long polyethylene melts through a generalized chain bridging algorithm. J. Chem. Phys. 2002;117:5465–5479. doi: 10.1063/1.1499480. [DOI] [Google Scholar]
- 27.Peristeras L.D., Economou I.G., Theodorou D.N. Structure and Volumetric Properties of Linear and Triarm Star Polyethylenes from Atomistic Monte Carlo Simulation Using New Internal Rearrangement Moves. Macromolecules. 2005;38:386–397. doi: 10.1021/ma048364p. [DOI] [Google Scholar]
- 28.Ramos F.J., Peristeras L., Theodorou D.N. Monte Carlo Simulation of Short Chain Branched Polyolefins in the Molten State. Macromolecules. 2007;40:9640–9650. doi: 10.1021/ma071615k. [DOI] [Google Scholar]
- 29.Gusev A.A., Suter U.W. Dynamics of Small Molecules in Dense Polymers Subject to Thermal Motion. J. Chem. Phys. 1993;99:2228–2234. doi: 10.1063/1.466198. [DOI] [Google Scholar]
- 30.Gusev A.A., Arizzi S., Suter U.W., Moll D.J. Dynamics of light gases in rigid matrices of dense polymers. J. Chem. Phys. 1993;99:2221–2227. doi: 10.1063/1.465283. [DOI] [Google Scholar]
- 31.Greenfield M.L., Theodorou D.N. Coupling of Penetrant and Polymer Motions during Small-Molecule Diffusion in a Glassy Polymer. Mol. Simul. 1997;19:329–361. doi: 10.1080/08927029708024161. [DOI] [Google Scholar]
- 32.Greenfield M.L., Theodorou D.N. Molecular modeling of methane diffusion in glassy atactic polypropylene via multidimensional transition state theory. Macromolecules. 1998;31:7068–7090. doi: 10.1021/ma980750h. [DOI] [Google Scholar]
- 33.Vergadou N. Ph.D. Thesis. University of Athens; Athens, Greece: 2006. Prediction of Gas Permeability of Inflexible Amorphous Polymers via Molecular Simulation. [Google Scholar]
- 34.Theodorou D.N. Hierarchical modelling of polymeric materials. Chem. Eng. Sci. 2007;62:5697–5714. doi: 10.1016/j.ces.2007.04.048. [DOI] [Google Scholar]
- 35.Xi L., Shah M., Trout B.L. Hopping of Water in a Glassy Polymer Studied via Transition Path Sampling and Likelihood Maximization. J. Phys. Chem. B. 2013;117:3634–3647. doi: 10.1021/jp3099973. [DOI] [PubMed] [Google Scholar]
- 36.Greenfield M.L., Theodorou D.N. Coarse-Grained Molecular Simulation of Penetrant Diffusion in a Glassy Polymer Using Reverse and Kinetic Monte Carlo. Macromolecules. 2001;34:8541–8553. doi: 10.1021/ma002157h. [DOI] [Google Scholar]
- 37.Neyertz S., Brown D. A Trajectory-Extending Kinetic Monte Carlo (TEKMC) Method for Estimating Penetrant Diffusion Coefficients in Molecular Dynamics Simulations of Glassy Polymers. Macromolecules. 2010;43:9210–9214. doi: 10.1021/ma1019895. [DOI] [Google Scholar]
- 38.Neyertz S., Brown D., Pandiyan S., van der Vegt N.F. Carbon dioxide diffusion and plasticization in fluorinated polyimides. Macromolecules. 2010;43:7813–7827. doi: 10.1021/ma1010205. [DOI] [Google Scholar]
- 39.Harmandaris V.A., Adhikari N.P., van der Vegt N.F.A., Kremer K., Mann B.A., Voelkel R., Weiss H., Liew C. Ethylbenzene Diffusion in Polystyrene: United Atom Atomistic/Coarse Grained Simulations and Experiments. Macromolecules. 2007;40:7026–7035. doi: 10.1021/ma070201o. [DOI] [Google Scholar]
- 40.Fritz D., Herbers C.R., Kremer K., van der Vegt N.F.A. Hierarchical Modeling of Polymer Permeation. Soft Matter. 2009;5:4556–4563. doi: 10.1039/b911713j. [DOI] [Google Scholar]
- 41.Lin E., You X., Kriegel R.M., Moffitt R.D., Batra R.C. Atomistic to coarse grained simulations of diffusion of small molecules into polymeric matrix. Comput. Mater. Sci. 2017;138:448–461. doi: 10.1016/j.commatsci.2017.07.011. [DOI] [Google Scholar]
- 42.Zhang K., Meng D., Müller-Plathe F., Kumar S.K. Coarse-grained molecular dynamics simulation of activated penetrant transport in glassy polymers. Soft Matter. 2018;14:440–447. doi: 10.1039/C7SM01941F. [DOI] [PubMed] [Google Scholar]
- 43.Müller-Plathe F., Rogers S.C., van Gunsteren W.F. Computational Evidence for Anomalous Diffusion of Small Molecules in Amorphous Polymers. Chem. Phys. Lett. 1992;199:237–243. doi: 10.1016/0009-2614(92)80112-O. [DOI] [Google Scholar]
- 44.Raptis V.E., Economou I.G., Theodorou D.N., Petrou J., Petropoulos J.H. Molecular dynamics simulation of structure and thermodynamic properties of poly(dimethylsilamethylene) and hydrocarbon solubility therein: Toward the development of novel membrane materials for hydrocarbon separation. Macromolecules. 2004;37:1102–1112. doi: 10.1021/ma034332a. [DOI] [Google Scholar]
- 45.Yang Q., Whiting W.I. Molecular-level insight of the differences in the diffusion and solubility of penetrants in polypropylene, poly(propylmethylsiloxane) and poly(4-methyl-2-pentyne) J. Membr. Sci. 2018;549:173–183. doi: 10.1016/j.memsci.2017.12.011. [DOI] [Google Scholar]
- 46.Van der Vegt N.F.A., Briels W.J., Wessling M., Strathmann H. The sorption induced glass transition in amorphous glassy polymers. J. Chem. Phys. 1999;110:11061–11069. doi: 10.1063/1.479042. [DOI] [Google Scholar]
- 47.Spyriouni T., Boulougouris G.C., Theodorou D.N. Prediction of Sorption of CO2 in Glassy Atactic Polystyrene at Elevated Pressures Through a New Computational Scheme. Macromolecules. 2009;42:1759–1769. doi: 10.1021/ma8015294. [DOI] [Google Scholar]
- 48.Knopp B., Suter U.W. Atomistically Modeling the Chemical Potential of Small Molecules in Dense Polymer Microstructures. 2. Water Sorption by Polyamides. Macromolecules. 1997;30:6114–6119. doi: 10.1021/ma970384m. [DOI] [Google Scholar]
- 49.Knopp B., Suter U.W., Gusev A.A. Atomistically Modeling the Chemical Potential of Small Molecules in Dense Polymer Microstructures. 1. Method. Macromolecules. 1997;30:6107–6113. doi: 10.1021/ma970383u. [DOI] [Google Scholar]
- 50.Martyna G.J., Tuckerman M.E., Tobias D.J., Klein M.L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996;87:1117–1157. doi: 10.1080/00268979600100761. [DOI] [Google Scholar]
- 51.Tuckerman M., Berne B.J., Martyna G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992;97:1990–2001. doi: 10.1063/1.463137. [DOI] [Google Scholar]
- 52.McQuarrie D.A. Statistical Mechanics. Harper & Row; New York, NY, USA: 1975. [Google Scholar]
- 53.Hansen J.-P., McDonald I.R. Theory of Simple Liquids. 3rd ed. Academic Press; London, UK: 2006. [Google Scholar]
- 54.Müller-Plathe F., Rogers S.C., van Gunsteren W.F. Gas sorption and transport in polyisobutylene: Equilibrium and nonequilibrium molecular dynamics simulations. J. Chem. Phys. 1993;98:9895–9904. doi: 10.1063/1.464369. [DOI] [Google Scholar]
- 55.Van der Vegt N.F.A., Briels W.J., Wessling M., Strathmann H. A Nonequilibrium Simulation Method for Calculating Tracer Diffusion Coefficients of Small Solutes in N-Alkane Liquids and Polymers. J. Chem. Phys. 1998;108:9558–9565. doi: 10.1063/1.476404. [DOI] [Google Scholar]
- 56.Frentrup H., Hart E.K., Colina M.C., Müller A.E. In Silico Determination of Gas Permeabilities by Non-Equilibrium Molecular Dynamics: CO2 and He through PIM-1. Membranes. 2015;5:99–119. doi: 10.3390/membranes5010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anderson L.R., Yang Q., Ediger A.M. Comparing gas transport in three polymers via molecular dynamics simulation. Phys. Chem. Chem. Phys. 2018;20:22123–22133. doi: 10.1039/C8CP02829J. [DOI] [PubMed] [Google Scholar]
- 58.Theodorou D.N. Variable-Connectivity Monte Carlo Algorithms for the Atomistic Simulation of Long-Chain Polymer Systems. In: Nielaba P., Mareschal M., Ciccotti G., editors. Bridging Time Scales: Molecular Simulations for the Next Decade. Springer; Berlin/Heidelberg, Germany: 2002. pp. 67–127. [Google Scholar]
- 59.Rosenbluth M.N., Rosenbluth A.W. Monte Carlo Calculation of the Average Extension of Molecular Chains. J. Chem. Phys. 1955;23:356–359. doi: 10.1063/1.1741967. [DOI] [Google Scholar]
- 60.Siepmann J.I., Frenkel D. Configurational bias Monte Carlo: A new sampling scheme for flexible chains. Mol. Phys. 1992;75:59–70. doi: 10.1080/00268979200100061. [DOI] [Google Scholar]
- 61.De Pablo J.J., Laso M., Suter U.W. Estimation of the chemical potential of chain molecules by simulation. J. Chem. Phys. 1992;96:6157–6162. doi: 10.1063/1.462658. [DOI] [Google Scholar]
- 62.Dodd L.R., Boone T.D., Theodorou D.N. A concerted rotation algorithm for atomistic Monte Carlo simulation of polymer melts and glasses. Mol. Phys. 1993;78:961–996. doi: 10.1080/00268979300100641. [DOI] [Google Scholar]
- 63.Panagiotopoulos A.Z. Direct determination of phase coexistence properties of fluids by Monte Carlo simulation in a new ensemble. Mol. Phys. 1987;61:813–826. doi: 10.1080/00268978700101491. [DOI] [Google Scholar]
- 64.Briano J.G., Glandt E.D. Statistical thermodynamics of polydisperse fluids. J. Chem. Phys. 1984;80:3336–3343. doi: 10.1063/1.447087. [DOI] [Google Scholar]
- 65.Kofke D.A., Glandt E.D. Monte carlo simulation of multicomponent equilibria in a semigrand canonical ensemble. Mol. Phys. 1988;64:1105–1131. doi: 10.1080/00268978800100743. [DOI] [Google Scholar]
- 66.Widom B. Some Topics in the Theory of Fluids. J. Chem. Phys. 1963;39:2808–2812. doi: 10.1063/1.1734110. [DOI] [Google Scholar]
- 67.Zervopoulou E., Mavrantzas V.G., Theodorou D.N. A new Monte Carlo simulation approach for the prediction of sorption equilibria of oligomers in polymer melts: Solubility of long alkanes in linear polyethylene. J. Chem. Phys. 2001;115:2860–2875. doi: 10.1063/1.1383050. [DOI] [Google Scholar]
- 68.Lyubartsev A.P., Martsinovski A.A., Shevkunov S.V., Vorontsov-Velyaminov P.N. New approach to Monte Carlo calculation of the free energy: Method of expanded ensembles. J. Chem. Phys. 1992;96:1776–1783. doi: 10.1063/1.462133. [DOI] [Google Scholar]
- 69.De Pablo J.J., Yan Q., Escobedo F.A. Simulation of Phase Transitions in Fluids. Annu. Rev. Phys. Chem. 1999;50:377–411. doi: 10.1146/annurev.physchem.50.1.377. [DOI] [PubMed] [Google Scholar]
- 70.Cuthbert T.R., Wagner N.J., Paulaitis M.E. Molecular Simulation of Glassy Polystyrene: Size Effects on Gas Solubilities. Macromolecules. 1997;30:3058–3065. doi: 10.1021/ma960776d. [DOI] [Google Scholar]
- 71.Deitrick G.L., Scriven L.E., Davis H.T. Efficient molecular simulation of chemical potentials. J. Chem. Phys. 1989;90:2370–2385. doi: 10.1063/1.455979. [DOI] [Google Scholar]
- 72.Tamai Y., Tanaka H., Nakanishi K. Molecular Simulation of Permeation of Small Penetrants through Membranes. 2. Solubilities. Macromolecules. 1995;28:2544–2554. doi: 10.1021/ma00111a058. [DOI] [Google Scholar]
- 73.Fukuda M. Solubilities of small molecules in polyethylene evaluated by a test-particle-insertion method. J. Chem. Phys. 1999;112:478–486. doi: 10.1063/1.480594. [DOI] [Google Scholar]
- 74.Dömötör G., Hentschke R. Atomistically Modeling the Chemical Potential of Small Molecules in Dense Systems. J. Phys. Chem. B. 2004;108:2413–2417. doi: 10.1021/jp0367427. [DOI] [Google Scholar]
- 75.Dömötör G., Hentschke R. Equilibrium Swelling of an Epoxy-Resin in Contact with Water—A Molecular Dynamics Simulation Study. Macromol. Theory Simul. 2004;13:506–511. doi: 10.1002/mats.200400018. [DOI] [Google Scholar]
- 76.Boulougouris G.C., Economou I.G., Theodorou D.N. On the calculation of the chemical potential using the particle deletion scheme. Mol. Phys. 1999;96:905–913. doi: 10.1080/00268979909483030. [DOI] [Google Scholar]
- 77.Boulougouris G.C., Economou I.G., Theodorou D.N. Calculation of the chemical potential of chain molecules using the staged particle deletion scheme. J. Chem. Phys. 2001;115:8231–8237. doi: 10.1063/1.1405849. [DOI] [Google Scholar]
- 78.Van der Vegt N.F.A., Briels W.J. Efficient sampling of solvent free energies in polymers. J. Chem. Phys. 1998;109:7578–7582. doi: 10.1063/1.477379. [DOI] [Google Scholar]
- 79.Vrabec J., Hasse H. Grand Equilibrium: Vapour-liquid equilibria by a new molecular simulation method. Mol. Phys. 2002;100:3375–3383. doi: 10.1080/00268970210153772. [DOI] [Google Scholar]
- 80.Hess B., Peter C., Ozal T., van der Vegt N.F.A. Fast-Growth Thermodynamic Integration: Calculating Excess Chemical Potentials of Additive Molecules in Polymer Microstructures. Macromolecules. 2008;41:2283–2289. doi: 10.1021/ma702070n. [DOI] [Google Scholar]
- 81.Glasstone S., Laidler K.J., Eyring H. The Theory of Rate Processes: The Kinetics of Chemical Reactions, Viscosity, Diffusion and Electrochemical Phenomena. McGraw-Hill; New York, NY, USA: 1941. [Google Scholar]
- 82.Voter A.F., Doll J.D. Dynamical Corrections to Transition-State Theory for Multistate Systems—Surface Self-Diffusion in the Rare-Event Regime. J. Chem. Phys. 1985;82:80–92. doi: 10.1063/1.448739. [DOI] [Google Scholar]
- 83.Eyring H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935;3:107–115. doi: 10.1063/1.1749604. [DOI] [Google Scholar]
- 84.van Kampen N.G. Stochastic Processes in Physics and Chemistry. 3rd ed. North Holland Publishing Company; Amsterdam, The Netherlands: 1983. [Google Scholar]
- 85.Fukui K. The Path of Chemical Reactions—The IRC Approach. Acc. Chem. Res. 1981;14:363–368. doi: 10.1021/ar00072a001. [DOI] [Google Scholar]
- 86.Kotelyanskii M., Theodorou D.N. Simulation Methods for Polymers. Marcel Dekker Inc; New York, NY, USA: 2004. [Google Scholar]
- 87.Chandler D. Statistical mechanics of isomerization dynamics in liquids and the transition state approximation. J. Chem. Phys. 1978;68:2959–2970. doi: 10.1063/1.436049. [DOI] [Google Scholar]
- 88.Hofmann D., Fritz L., Ulbrich J., Paul D. Molecular simulation of small molecule diffusion and solution in dense amorphous polysiloxanes and polyimides. Comp. Theor. Polymer Sci. 2000;10:419–436. doi: 10.1016/S1089-3156(00)00007-6. [DOI] [Google Scholar]
- 89.Hofmann D., Fritz L., Ulbrich J., Schepers C., Bohning M. Detailed-Atomistic Molecular Modeling of Small Molecule Diffusion and Solution Processes in Polymeric Membrane Materials. Macromol. Theory Simul. 2000;9:293–327. doi: 10.1002/1521-3919(20000701)9:6<293::AID-MATS293>3.0.CO;2-1. [DOI] [Google Scholar]
- 90.Karayiannis N.C., Mavrantzas V.G., Theodorou D.N. Detailed Atomistic Simulation of the Segmental Dynamics and Barrier Properties of Amorphous Poly(ethylene Terephthalate) and Poly(ethylene Isophthalate) Macromolecules. 2004;37:2978–2995. doi: 10.1021/ma0352577. [DOI] [Google Scholar]
- 91.Greenfield M.L., Theodorou D.N. Geometric analysis of diffusion pathways in glassy and melt atactic polypropylene. Macromolecules. 1993;26:5461–5472. doi: 10.1021/ma00072a026. [DOI] [Google Scholar]
- 92.Vergadou N., Theodorou D.N. Molecular simulation of CO2 Diffusion in Glassy Polymers using Multidimensional Transition State Theory. 2019 In preparation. [Google Scholar]
- 93.Bolhuis P.G. Transition path sampling on diffusive barriers. J. Phys. Condens. Matter. 2002;15:S113–S120. doi: 10.1088/0953-8984/15/1/314. [DOI] [Google Scholar]
- 94.Bolhuis P.G., Dellago C., Chandler D. Sampling Ensembles of Deterministic Transition Pathways. Faraday Discuss. 1998;110:421–436. doi: 10.1039/a801266k. [DOI] [Google Scholar]
- 95.Peters B., Trout B.L. Obtaining Reaction Coordinates by Likelihood Maximization. J. Chem. Phys. 2006;125:054108. doi: 10.1063/1.2234477. [DOI] [PubMed] [Google Scholar]
- 96.Peters B., Beckham G.T., Trout B.L. Extensions to the Likelihood Maximization Approach for Finding Reaction Coordinates. J. Chem. Phys. 2007;127:034109. doi: 10.1063/1.2748396. [DOI] [PubMed] [Google Scholar]
- 97.Grünwald M., Dellago C., Geissler P.L. Precision shooting: Sampling long transition pathways. J. Chem. Phys. 2008;129:194101. doi: 10.1063/1.2978000. [DOI] [PubMed] [Google Scholar]
- 98.Mullen R.G., Shea J.-E., Peters B. Easy Transition Path Sampling Methods: Flexible-Length Aimless Shooting and Permutation Shooting. J. Chem. Theory Comput. 2015;11:2421–2428. doi: 10.1021/acs.jctc.5b00032. [DOI] [PubMed] [Google Scholar]
- 99.Pan A.C., Chandler D. Dynamics of nucleation in the ising model. J. Phys. Chem. B. 2004;108:19681. doi: 10.1021/jp0471249. [DOI] [Google Scholar]
- 100.Moroni D., Ten Wolde P.R., Bolhuis P.G. Interplay between structure and size in a critical crystal nucleus. Phys. Rev. Lett. 2005;94:235703. doi: 10.1103/PhysRevLett.94.235703. [DOI] [PubMed] [Google Scholar]
- 101.Lechner W., Dellago C., Bolhuis P.G. Role of the Prestructured Surface Cloud in Crystal Nucleation. Phys. Rev. Lett. 2011;106:085701. doi: 10.1103/PhysRevLett.106.085701. [DOI] [PubMed] [Google Scholar]
- 102.Beckham G.T., Peters B.J. Optimizing Nucleus Size Metrics for Liquid-Solid Nucleation from Transition Paths of Near-Nanosecond Duration. Phys. Chem. Lett. 2011;2:1133–1138. doi: 10.1021/jz2002887. [DOI] [PubMed] [Google Scholar]
- 103.Chong L.T., Saglam A.S., Zuckerman D.M. Path-sampling strategies for simulating rare events in biomolecular systems. Curr. Opin. Struct. Biol. 2017;43:88–94. doi: 10.1016/j.sbi.2016.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Juraszek J., Bolhuis P.G. Rate Constant and Reaction Coordinate of Trp-Cage Folding in Explicit Water. Biophys. J. 2008;95:4246–4257. doi: 10.1529/biophysj.108.136267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang B.W., Jasnow D., Zuckerman D.M. Efficient and verified simulation of a path ensemble for conformational change in a united-residue model of calmodulin. Proc. Natl. Acad. Sci. USA. 2007;104:18043. doi: 10.1073/pnas.0706349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Roy A., Taraphder S. Transition Path Sampling Study of the Conformational Fluctuation of His-64 in Human Carbonic Anhydrase II. J. Phys. Chem. B. 2009;113:12555–12564. doi: 10.1021/jp9010982. [DOI] [PubMed] [Google Scholar]
- 107.Keys A.S., Hedges L.O., Garrahan J.P., Glotzer S.C., Chandler D. Excitations Are Localized and Relaxation Is Hierarchical in Glass-Forming Liquids. Phys. Rev. X. 2011;1:021013. [Google Scholar]
- 108.Pan B., Ricci M.S., Trout B.L. A molecular mechanism of hydrolysis of peptide bonds at neutral pH using a model compound. J. Phys. Chem. B. 2011;115:5958–5970. doi: 10.1021/jp1076802. [DOI] [PubMed] [Google Scholar]
- 109.Quaytman S.L., Schwartz S.D. Reaction coordinate of an enzymatic reaction revealed by transition path sampling. Proc. Natl. Acad. Sci. USA. 2007;104:12253–12258. doi: 10.1073/pnas.0704304104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Basner J.E., Schwartz S.D. How Enzyme Dynamics Helps Catalyze a Reaction in Atomic Detail: A Transition Path Sampling Study. J. Am. Chem. Soc. 2005;127:13822–13831. doi: 10.1021/ja043320h. [DOI] [PubMed] [Google Scholar]
- 111.Vlugt T.J.H., Dellago C., Smit B. Diffusion of Isobutane in Silicalite Studied by Transition Path Sampling. J. Chem. Phys. 2000;113:8791–8799. doi: 10.1063/1.1318771. [DOI] [Google Scholar]
- 112.Peters B., Zimmermann N.E.R., Beckham G.T., Tester J.W., Trout B.L. Path Sampling Calculation of Methane Diffusivity in Natural Gas Hydrates from a Water-Vacancy Assisted Mechanism. J. Am. Chem. Soc. 2008;130:17342–17350. doi: 10.1021/ja802014m. [DOI] [PubMed] [Google Scholar]
- 113.Boulfelfel S.E., Ravikovitch P.I., Sholl D.S. Modeling Diffusion of Linear Hydrocarbons in Silica Zeolite LTA Using Transition Path Sampling. J. Phys. Chem. C. 2015;119:15643–15653. doi: 10.1021/acs.jpcc.5b01633. [DOI] [Google Scholar]
- 114.Auhl R., Everaers R., Grest G.S., Kremer K., Plimpton S.J. Equilibration of long chain polymer melts in computer simulations. J. Chem. Phys. 2003;119:12718–12728. doi: 10.1063/1.1628670. [DOI] [Google Scholar]
- 115.Theodorou D.N. Equilibration and Coarse-Graining Methods for Polymers. In: Ferrario M., Ciccotti G., Binder K., editors. Computer Simulations in Condensed Matter Systems: From Materials to Chemical Biology Volume 2. Springer; Berlin/Heidelberg, Germany: 2006. pp. 419–448. [Google Scholar]
- 116.Brown D., Clarke J.H.R., Okuda M., Yamazaki T. The preparation of polymer melt samples for computer simulation studies. J. Chem. Phys. 1994;100:6011–6018. doi: 10.1063/1.467111. [DOI] [Google Scholar]
- 117.Gao J. An efficient method of generating dense polymer model melts by computer simulation. J. Chem. Phys. 1995;102:1074–1077. doi: 10.1063/1.469455. [DOI] [Google Scholar]
- 118.Mondello M., Grest G.S., Webb E.B., Peczak P. Dynamics of n-alkanes: Comparison to Rouse model. J. Chem. Phys. 1998;109:798–805. doi: 10.1063/1.476619. [DOI] [Google Scholar]
- 119.Kröger M. Efficient hybrid algorithm for the dynamic creation of wormlike chains in solutions, brushes, melts and glasses. Comput. Phys. Commun. 1999;118:278–298. doi: 10.1016/S0010-4655(98)00160-X. [DOI] [Google Scholar]
- 120.Sliozberg Y.R., Kröger M., Chantawansri T.L. Fast equilibration protocol for million atom systems of highly entangled linear polyethylene chains. J. Chem. Phys. 2016;144:154901. doi: 10.1063/1.4946802. [DOI] [PubMed] [Google Scholar]
- 121.Martínez L., Andrade R., Birgin E.G., Martínez J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009;30:2157–2164. doi: 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- 122.Flory P. Statistical Mechanics of Chain Molecules. Interscience Publishers; New York, NY, USA: 1969. [Google Scholar]
- 123.Theodorou D.N., Suter U.W. Detailed Molecular-Structure of a Vinyl Polymer Glass. Macromolecules. 1985;18:1467–1478. doi: 10.1021/ma00149a018. [DOI] [Google Scholar]
- 124.Rigby D., Roe R.J. Molecular dynamics simulation of polymer liquid and glass. I. Glass transition. J. Chem. Phys. 1987;87:7285–7292. doi: 10.1063/1.453321. [DOI] [Google Scholar]
- 125.Gooneie A., Schuschnigg S., Holzer C. A Review of Multiscale Computational Methods in Polymeric Materials. Polymers. 2017;9:16. doi: 10.3390/polym9010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Müller-Plathe F. Coarse-Graining in Polymer Simulation: From the Atomistic to the Mesoscopic Scale and Back. ChemPhysChem. 2002;3:754–769. doi: 10.1002/1439-7641(20020916)3:9<754::AID-CPHC754>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 127.Tschöp W., Kremer K., Batoulis J., Bürger T., Hahn O. Simulation of polymer melts. I. Coarse-graining procedure for polycarbonates. Acta Polym. 1998;49:61–74. doi: 10.1002/(SICI)1521-4044(199802)49:2/3<61::AID-APOL61>3.0.CO;2-V. [DOI] [Google Scholar]
- 128.Karimi-Varzaneh H.A., van der Vegt N.F.A., Müller-Plathe F., Carbone P. How Good Are Coarse-Grained Polymer Models? A Comparison for Atactic Polystyrene. ChemPhysChem. 2012;13:3428–3439. doi: 10.1002/cphc.201200111. [DOI] [PubMed] [Google Scholar]
- 129.Zacharopoulos N., Vergadou N., Theodorou D.N. Coarse graining using pretabulated potentials: Liquid benzene. J. Chem. Phys. 2005;122:244111. doi: 10.1063/1.1948370. [DOI] [PubMed] [Google Scholar]
- 130.Deichmann G., Dallavalle M., Rosenberger D., van der Vegt N.F.A. Phase Equilibria Modeling with Systematically Coarse-Grained Models—A Comparative Study on State Point Transferability. J. Phys. Chem. B. 2019;123:504–515. doi: 10.1021/acs.jpcb.8b07320. [DOI] [PubMed] [Google Scholar]
- 131.Sun Q., Faller R. Systematic Coarse-Graining of a Polymer Blend: Polyisoprene and Polystyrene. J. Chem. Theory Comput. 2006;2:607–615. doi: 10.1021/ct600065v. [DOI] [PubMed] [Google Scholar]
- 132.Mognetti B.M., Virnau P., Yelash L., Paul W., Binder K., Müller M., MacDowell L.G. Coarse-grained models for fluids and their mixtures: Comparison of Monte Carlo studies of their phase behavior with perturbation theory and experiment. J. Chem. Phys. 2009;130:044101. doi: 10.1063/1.3050353. [DOI] [PubMed] [Google Scholar]
- 133.Marrink S.J., Risselada H.J., Yefimov S., Tieleman D.P., de Vries A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 134.Maerzke K.A., Siepmann J.I. Transferable Potentials for Phase Equilibria−Coarse-Grain Description for Linear Alkanes. J. Phys. Chem. B. 2011;115:3452–3465. doi: 10.1021/jp1063935. [DOI] [PubMed] [Google Scholar]
- 135.Allison J.R., Riniker S., van Gunsteren W.F. Coarse-grained models for the solvents dimethyl sulfoxide, chloroform, and methanol. J. Chem. Phys. 2012;136:054505. doi: 10.1063/1.3681140. [DOI] [PubMed] [Google Scholar]
- 136.Brini E., Algaer E.A., Ganguly P., Li C., Rodríguez-Ropero F., van der Vegt N.F.A. Systematic coarse-graining methods for soft matter simulations—A review. Soft Matter. 2013;9:2108–2119. doi: 10.1039/C2SM27201F. [DOI] [Google Scholar]
- 137.Huang H., Wu L., Xiong H., Sun H. A Transferrable Coarse-Grained Force Field for Simulations of Polyethers and Polyether Blends. Macromolecules. 2019;52:249–261. doi: 10.1021/acs.macromol.8b01802. [DOI] [Google Scholar]
- 138.Kuo A.-T., Okazaki S., Shinoda W. Transferable coarse-grained model for perfluorosulfonic acid polymer membranes. J. Chem. Phys. 2017;147:094904. doi: 10.1063/1.4986287. [DOI] [PubMed] [Google Scholar]
- 139.Reith D., Pütz M., Müller-Plathe F. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem. 2003;24:1624–1636. doi: 10.1002/jcc.10307. [DOI] [PubMed] [Google Scholar]
- 140.Ercolessi F., Adams J.B. Interatomic Potentials from First-Principles Calculations: The Force-Matching Method. Europhys. Lett. (EPL) 1994;26:583–588. doi: 10.1209/0295-5075/26/8/005. [DOI] [Google Scholar]
- 141.Izvekov S., Voth G.A. Modeling real dynamics in the coarse-grained representation of condensed phase systems. J. Chem. Phys. 2006;125:151101. doi: 10.1063/1.2360580. [DOI] [PubMed] [Google Scholar]
- 142.Lyubartsev A.P., Laaksonen A. Calculation of effective interaction potentials from radial distribution functions: A reverse Monte Carlo approach. Phys. Rev. E. 1995;52:3730–3737. doi: 10.1103/PhysRevE.52.3730. [DOI] [PubMed] [Google Scholar]
- 143.Chaimovich A., Shell M.S. Coarse-graining errors and numerical optimization using a relative entropy framework. J. Chem. Phys. 2011;134:094112. doi: 10.1063/1.3557038. [DOI] [PubMed] [Google Scholar]
- 144.Shell M.S. The relative entropy is fundamental to multiscale and inverse thermodynamic problems. J. Chem. Phys. 2008;129:144108. doi: 10.1063/1.2992060. [DOI] [PubMed] [Google Scholar]
- 145.Villa A., Peter C., van der Vegt N.F.A. Self-assembling dipeptides: Conformational sampling in solvent-free coarse-grained simulation. Phys. Chem. Chem. Phys. 2009;11:2077–2086. doi: 10.1039/b818144f. [DOI] [PubMed] [Google Scholar]
- 146.Li C., Shen J., Peter C., van der Vegt N.F.A. A Chemically Accurate Implicit-Solvent Coarse-Grained Model for Polystyrenesulfonate Solutions. Macromolecules. 2012;45:2551–2561. doi: 10.1021/ma202490h. [DOI] [Google Scholar]
- 147.De Nicola A., Kawakatsu T., Milano G. Generation of Well-Relaxed All-Atom Models of Large Molecular Weight Polymer Melts: A Hybrid Particle-Continuum Approach Based on Particle-Field Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014;10:5651–5667. doi: 10.1021/ct500492h. [DOI] [PubMed] [Google Scholar]
- 148.Rosenberger D., van der Vegt N.F.A. Addressing the temperature transferability of structure based coarse graining models. Phys. Chem. Chem. Phys. 2018;20:6617–6628. doi: 10.1039/C7CP08246K. [DOI] [PubMed] [Google Scholar]
- 149.Potter T.D., Tasche J., Wilson M.R. Assessing the transferability of common top-down and bottom-up coarse-grained molecular models for molecular mixtures. Phys. Chem. Chem. Phys. 2019;21:1912–1927. doi: 10.1039/C8CP05889J. [DOI] [PubMed] [Google Scholar]
- 150.Dunn N.J.H., Foley T.T., Noid W.G. Van der Waals Perspective on Coarse-Graining: Progress toward Solving Representability and Transferability Problems. Acc. Chem. Res. 2016;49:2832–2840. doi: 10.1021/acs.accounts.6b00498. [DOI] [PubMed] [Google Scholar]
- 151.Guenza M.G., Dinpajooh M., McCarty J., Lyubimov I.Y. Accuracy, Transferability, and Efficiency of Coarse-Grained Models of Molecular Liquids. J. Phys. Chem. B. 2018;122:10257–10278. doi: 10.1021/acs.jpcb.8b06687. [DOI] [PubMed] [Google Scholar]
- 152.Tschöp W., Kremer K., Hahn O., Batoulis J., Bürger T. Simulation of polymer melts. II. From coarse-grained models back to atomistic description. Acta Polym. 1998;49:75–79. doi: 10.1002/(SICI)1521-4044(199802)49:2/3<75::AID-APOL75>3.0.CO;2-5. [DOI] [Google Scholar]
- 153.Hess B., León S., van der Vegt N., Kremer K. Long time atomistic polymer trajectories from coarse grained simulations: Bisphenol-A polycarbonate. Soft Matter. 2006;2:409–414. doi: 10.1039/B602076C. [DOI] [PubMed] [Google Scholar]
- 154.Wu C. Multiscale simulations of the structure and dynamics of stereoregular poly(methyl methacrylate)s. J. Mol. Modeling. 2014;20 doi: 10.1007/s00894-014-2377-3. [DOI] [PubMed] [Google Scholar]
- 155.Ghanbari A., Böhm M.C., Müller-Plathe F. A Simple Reverse Mapping Procedure for Coarse-Grained Polymer Models with Rigid Side Groups. Macromolecules. 2011;44:5520–5526. doi: 10.1021/ma2005958. [DOI] [Google Scholar]
- 156.Krajniak J., Pandiyan S., Nies E., Samaey G. Generic Adaptive Resolution Method for Reverse Mapping of Polymers from Coarse-Grained to Atomistic Descriptions. J. Chem. Theory Comput. 2016;12:5549–5562. doi: 10.1021/acs.jctc.6b00595. [DOI] [PubMed] [Google Scholar]
- 157.Krajniak J., Zhang Z., Pandiyan S., Nies E., Samaey G. Reverse mapping method for complex polymer systems. J. Comput. Chem. 2018;39:648–664. doi: 10.1002/jcc.25129. [DOI] [PubMed] [Google Scholar]
- 158.Rzepiela A.J., Schäfer L.V., Goga N., Risselada H.J., De Vries A.H., Marrink S.J. Reconstruction of atomistic details from coarse-grained structures. J. Comput. Chem. 2010;31:1333–1343. doi: 10.1002/jcc.21415. [DOI] [PubMed] [Google Scholar]
- 159.Wassenaar T.A., Pluhackova K., Böckmann R.A., Marrink S.J., Tieleman D.P. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014;10:676–690. doi: 10.1021/ct400617g. [DOI] [PubMed] [Google Scholar]
- 160.Ohkuma T., Kremer K., Daoulas K. Equilibrating high-molecular-weight symmetric and miscible polymer blends with hierarchical back-mapping. J. Phys. Condens. Matter. 2018;30:174001. doi: 10.1088/1361-648X/aab684. [DOI] [PubMed] [Google Scholar]
- 161.Zhang G., Chazirakis A., Harmandaris V.A., Stuehn T., Daoulas K.C., Kremer K. Hierarchical modelling of polystyrene melts: From soft blobs to atomistic resolution. Soft Matter. 2019;15:289–302. doi: 10.1039/C8SM01830H. [DOI] [PubMed] [Google Scholar]
- 162.Santangelo G., Di Matteo A., Müller-Plathe F., Milano G. From Mesoscale Back to Atomistic Models: A Fast Reverse-Mapping Procedure for Vinyl Polymer Chains. J. Phys. Chem. B. 2007;111:2765–2773. doi: 10.1021/jp066212l. [DOI] [PubMed] [Google Scholar]
- 163.Bortz A., Kalos M., Lebowitz J. A new algorithm for Monte Carlo simulation of Ising spin systems. J. Comput. Phys. 1975;17:10–18. doi: 10.1016/0021-9991(75)90060-1. [DOI] [Google Scholar]
- 164.Young W.M., Elcock E.W. Monte Carlo studies of vacancy migration in binary ordered alloys: I. Proc. Phys. Soc. Lond. 1966;89:735. doi: 10.1088/0370-1328/89/3/329. [DOI] [Google Scholar]
- 165.Fichthorn K.A., Weinberg W.H. Theoretical foundations of dynamical Monte Carlo simulations. J. Chem. Phys. 1991;95:1090–1096. doi: 10.1063/1.461138. [DOI] [Google Scholar]
- 166.Kolokathis P.D., Theodorou D.N. On solving the master equation in spatially periodic systems. J. Chem. Phys. 2012;137:034112. doi: 10.1063/1.4733291. [DOI] [PubMed] [Google Scholar]
- 167.Tien W.-J., Chiu C.-C. Generic parameters of trajectory-extending kinetic Monte Carlo for calculating diffusion coefficients. AIP Adv. 2018;8:065311. doi: 10.1063/1.5035553. [DOI] [Google Scholar]
- 168.Hanson B., Pryamitsyn V., Ganesan V. Computer Simulations of Gas Diffusion in Polystyrene–C60 Fullerene Nanocomposites Using Trajectory Extending Kinetic Monte Carlo Method. J. Phys. Chem. B. 2012;116:95–103. doi: 10.1021/jp209294t. [DOI] [PubMed] [Google Scholar]
- 169.Mogurampelly S., Borodin O., Ganesan V. Computer Simulations of Ion Transport in Polymer Electrolyte Membranes. Annu. Rev. Chem. Biomol. Eng. 2016;7:349–371. doi: 10.1146/annurev-chembioeng-080615-034655. [DOI] [PubMed] [Google Scholar]
- 170.Hanson B., Pryamitsyn V., Ganesan V. Mechanisms Underlying Ionic Mobilities in Nanocomposite Polymer Electrolytes. ACS Macro Lett. 2013;2:1001–1005. doi: 10.1021/mz400234m. [DOI] [PubMed] [Google Scholar]
- 171.Mogurampelly S., Ganesan V. Effect of Nanoparticles on Ion Transport in Polymer Electrolytes. Macromolecules. 2015;48:2773–2786. doi: 10.1021/ma502578s. [DOI] [Google Scholar]
- 172.Mogurampelly S., Ganesan V. Structure and mechanisms underlying ion transport in ternary polymer electrolytes containing ionic liquids. J. Chem. Phys. 2017;146:074902. doi: 10.1063/1.4976131. [DOI] [PubMed] [Google Scholar]
- 173.Marrink S.J., de Vries A.H., Mark A.E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B. 2004;108:750–760. doi: 10.1021/jp036508g. [DOI] [Google Scholar]
- 174.Lee H., de Vries A.H., Marrink S.-J., Pastor R.W. A Coarse-Grained Model for Polyethylene Oxide and Polyethylene Glycol: Conformation and Hydrodynamics. J. Phys. Chem. B. 2009;113:13186–13194. doi: 10.1021/jp9058966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bos A., Pünt I.G.M., Wessling M., Strathmann H. CO2-induced plasticization phenomena in glassy polymers. J. Membr. Sci. 1999;155:67–78. doi: 10.1016/S0376-7388(98)00299-3. [DOI] [Google Scholar]
- 176.Wessling M., Huisman I., Boomgaard T.v.d., Smolders C.A. Dilation kinetics of glassy, aromatic polyimides induced by carbon dioxide sorption. J. Polym. Sci. Part B: Polym. Phys. 1995;33:1371–1384. doi: 10.1002/polb.1995.090330907. [DOI] [Google Scholar]
- 177.Coleman M.R., Koros W.J. Conditioning of Fluorine Containing Polyimides. 1. Effect of Exposure to High Pressure Carbon Dioxide on Permeability. Macromolecules. 1997;30:6899–6905. doi: 10.1021/ma961323b. [DOI] [Google Scholar]
- 178.Coleman M.R., Koros W.J. Conditioning of Fluorine-Containing Polyimides. 2. Effect of Conditioning Protocol at 8 Volume Dilation on Gas-Transport Properties. Macromolecules. 1999;32:3106–3113. doi: 10.1021/ma981376o. [DOI] [Google Scholar]
- 179.Ismail A.F., Lorna W. Penetrant-induced plasticization phenomenon in glassy polymers for gas separation membrane. Sep. Purif. Technol. 2002;27:173–194. doi: 10.1016/S1383-5866(01)00211-8. [DOI] [Google Scholar]
- 180.Visser T., Wessling M. When Do Sorption-Induced Relaxations in Glassy Polymers Set In? Macromolecules. 2007;40:4992–5000. doi: 10.1021/ma070202g. [DOI] [Google Scholar]
- 181.Reijerkerk S.R., Nijmeijer K., Ribeiro C.P., Freeman B.D., Wessling M. On the effects of plasticization in CO2/light gas separation using polymeric solubility selective membranes. J. Membr. Sci. 2011;367:33–44. doi: 10.1016/j.memsci.2010.10.035. [DOI] [Google Scholar]
- 182.Duthie X., Kentish S., Powell C., Nagai K., Qiao G., Stevens G. Operating temperature effects on the plasticization of polyimide gas separation membranes. J. Membr. Sci. 2007;294:40–49. doi: 10.1016/j.memsci.2007.02.004. [DOI] [Google Scholar]
- 183.Zhang L., Xiao Y., Chung T.S., Jiang J. Mechanistic understanding of CO2-induced plasticization of a polyimide membrane: A combination of experiment and simulation study. Polymer. 2010;51:4439–4447. doi: 10.1016/j.polymer.2010.07.032. [DOI] [Google Scholar]
- 184.Hölck O., Böhning M., Heuchel M., Siegert M.R., Hofmann D. Gas sorption isotherms in swelling glassy polymers—Detailed atomistic simulations. J. Membr. Sci. 2013;428:523–532. doi: 10.1016/j.memsci.2012.10.023. [DOI] [Google Scholar]
- 185.Heuchel M., Boehning M., Hoelck O., Siegert M.R., Hofmann D. Atomistic Packing Models for Experimentally Investigated Swelling States Induced by CO2 in Glassy Polysulfone and Poly(ether sulfone) J. Polym. Sci. Part B: Polym. Phys. 2006;44:1874–1897. doi: 10.1002/polb.20844. [DOI] [Google Scholar]
- 186.Van der Vegt N.F.A. Ph.D. Thesis. University of Twente; Enschede, The Netherlands: 1998. Molecular dynamics simulations of sorption and diffusion in rubbery and glassy polymers. [Google Scholar]
- 187.Lock S.S.M., Lau K.K., Shariff A.M., Yeong Y.F., Bustam M.A., Jusoh N., Ahmad F. An atomistic simulation towards elucidation of operating temperature effect in CO2 swelling of polysulfone polymeric membranes. J. Nat. Gas Sci. Eng. 2018;57:135–154. doi: 10.1016/j.jngse.2018.07.002. [DOI] [Google Scholar]
- 188.Balçık M., Ahunbay M.G. Prediction of CO2-induced plasticization pressure in polyimides via atomistic simulations. J. Membr. Sci. 2018;547:146–155. doi: 10.1016/j.memsci.2017.10.038. [DOI] [Google Scholar]
- 189.Kupgan G., Demidov A.G., Colina C.M. Plasticization behavior in polymers of intrinsic microporosity (PIM-1): A simulation study from combined Monte Carlo and molecular dynamics. J. Membr. Sci. 2018;565:95–103. doi: 10.1016/j.memsci.2018.08.004. [DOI] [Google Scholar]
- 190.Pandiyan S., Brown D., Neyertz S., van der Vegt N.F.A. Carbon Dioxide Solubility in Three Fluorinated Polyimides Studied by Molecular Dynamics Simulations. Macromolecules. 2010;43:2605–2621. doi: 10.1021/ma902507d. [DOI] [Google Scholar]
- 191.Spyriouni T., Tzoumanekas C., Theodorou D., Müller-Plathe F., Milano G. Coarse-Grained and Reverse-Mapped United-Atom Simulations of Long-Chain Atactic Polystyrene Melts: Structure, Thermodynamic Properties, Chain Conformation, and Entanglements. Macromolecules. 2007;40:3876–3885. doi: 10.1021/ma0700983. [DOI] [Google Scholar]
- 192.Velioğlu S., Ahunbay M.G., Tantekin-Ersolmaz S.B. Investigation of CO2-induced plasticization in fluorinated polyimide membranes via molecular simulation. J. Membr. Sci. 2012;417-418:217–227. doi: 10.1016/j.memsci.2012.06.043. [DOI] [Google Scholar]
- 193.Velioğlu S., Ahunbay M.G., Tantekin-Ersolmaz S.B. Propylene/propane plasticization in polyimide membranes. J. Membr. Sci. 2016;501:179–190. doi: 10.1016/j.memsci.2015.11.034. [DOI] [Google Scholar]
- 194.Tullos G., Mathias L. Unexpected thermal conversion of hydroxy-containing polyimides to polybenzoxazoles. Polymer. 1999;40:3463–3468. doi: 10.1016/S0032-3861(98)00555-2. [DOI] [Google Scholar]
- 195.Liu Q., Borjigin H., Paul D.R., Riffle J.S., McGrath J.E., Freeman B.D. Gas permeation properties of thermally rearranged (TR) isomers and their aromatic polyimide precursors. J. Membr. Sci. 2016;518:88–99. doi: 10.1016/j.memsci.2016.06.026. [DOI] [Google Scholar]
- 196.Park H.B., Jung C.H., Lee Y.M., Hill A.J., Pas S.J., Mudie S.T., Van Wagner E., Freeman B.D., Cookson D.J. Polymers with Cavities Tuned for Fast Selective Transport of Small Molecules and Ions. Science. 2007;318:254–258. doi: 10.1126/science.1146744. [DOI] [PubMed] [Google Scholar]
- 197.Sanders D.F., Smith Z.P., Ribeiro C.P., Guo R., McGrath J.E., Paul D.R., Freeman B.D. Gas permeability, diffusivity, and free volume of thermally rearranged polymers based on 3,3′-dihydroxy-4,4′-diamino-biphenyl (HAB) and 2,2′-bis-(3,4-dicarboxyphenyl) hexafluoropropane dianhydride (6FDA) J. Membr. Sci. 2012;409–410:232–241. doi: 10.1016/j.memsci.2012.03.060. [DOI] [Google Scholar]
- 198.Scholes C.A., Freeman B.D. Thermal rearranged poly(imide-co-ethylene glycol) membranes for gas separation. J. Membr. Sci. 2018;563:676–683. doi: 10.1016/j.memsci.2018.06.027. [DOI] [Google Scholar]
- 199.Scholes C.A., Freeman B.D., Kentish S.E. Water vapor permeability and competitive sorption in thermally rearranged (TR) membranes. J. Membr. Sci. 2014;470:132–137. doi: 10.1016/j.memsci.2014.07.024. [DOI] [Google Scholar]
- 200.Smith Z.P., Hernández G., Gleason K.L., Anand A., Doherty C.M., Konstas K., Alvarez C., Hill A.J., Lozano A.E., Paul D.R., et al. Effect of polymer structure on gas transport properties of selected aromatic polyimides, polyamides and TR polymers. J. Membr. Sci. 2015;493:766–781. doi: 10.1016/j.memsci.2015.06.032. [DOI] [Google Scholar]
- 201.Stevens K.A., Smith Z.P., Gleason K.L., Galizia M., Paul D.R., Freeman B.D. Influence of temperature on gas solubility in thermally rearranged (TR) polymers. J. Membr. Sci. 2017;533:75–83. doi: 10.1016/j.memsci.2017.03.005. [DOI] [Google Scholar]
- 202.Calle M., Doherty C.M., Hill A.J., Lee Y.M. Cross-Linked Thermally Rearranged Poly(benzoxazole-co-imide) Membranes for Gas Separation. Macromolecules. 2013;46:8179–8189. doi: 10.1021/ma4014115. [DOI] [Google Scholar]
- 203.Park H.B., Han S.H., Jung C.H., Lee Y.M., Hill A.J. Thermally rearranged (TR) polymer membranes for CO2 separation. J. Membr. Sci. 2010;359:11–24. doi: 10.1016/j.memsci.2009.09.037. [DOI] [Google Scholar]
- 204.Kim S., Jo H.J., Lee Y.M. Sorption and transport of small gas molecules in thermally rearranged (TR) polybenzoxazole membranes based on 2,2-bis(3-amino-4-hydroxyphenyl)-hexafluoropropane (bisAPAF) and 4,4′-hexafluoroisopropylidene diphthalic anhydride (6FDA) J. Membr. Sci. 2013;441:1–8. doi: 10.1016/j.memsci.2013.03.054. [DOI] [Google Scholar]
- 205.Jiang Y., Willmore F.T., Sanders D., Smith Z.P., Ribeiro C.P., Doherty C.M., Thornton A., Hill A.J., Freeman B.D., Sanchez I.C. Cavity size, sorption and transport characteristics of thermally rearranged (TR) polymers. Polymer. 2011;52:2244–2254. doi: 10.1016/j.polymer.2011.02.035. [DOI] [Google Scholar]
- 206.Park C.H., Tocci E., Lee Y.M., Drioli E. Thermal Treatment Effect on the Structure and Property Change between Hydroxy-Containing Polyimides (HPIs) and Thermally Rearranged Polybenzoxazole (TR-PBO) J. Phys. Chem. B. 2012;116:12864–12877. doi: 10.1021/jp307365y. [DOI] [PubMed] [Google Scholar]
- 207.Chang K.-S., Wu Z.-C., Kim S., Tung K.-L., Lee Y.M., Lin Y.-F., Lai J.-Y. Molecular modeling of poly(benzoxazole-co-imide) membranes: A structure characterization and performance investigation. J. Membr. Sci. 2014;454:1–11. doi: 10.1016/j.memsci.2013.11.047. [DOI] [Google Scholar]
- 208.Park C.H., Tocci E., Kim S., Kumar A., Lee Y.M., Drioli E. A Simulation Study on OH-Containing Polyimide (HPI) and Thermally Rearranged Polybenzoxazoles (TR-PBO): Relationship between Gas Transport Properties and Free Volume Morphology. J. Phys. Chem. B. 2014;118:2746–2757. doi: 10.1021/jp411612g. [DOI] [PubMed] [Google Scholar]
- 209.Rizzuto C., Caravella A., Brunetti A., Park C.H., Lee Y.M., Drioli E., Barbieri G., Tocci E. Sorption and Diffusion of CO2/N2 in gas mixture in thermally-rearranged polymeric membranes: A molecular investigation. J. Membr. Sci. 2017;528:135–146. doi: 10.1016/j.memsci.2017.01.025. [DOI] [Google Scholar]
- 210.Velioğlu S., Ahunbay M.G., Tantekin-Ersolmaz S.B. An atomistic insight on CO2 plasticization resistance of thermally rearranged 6FDA-bisAPAF. J. Membr. Sci. 2018;556:23–33. doi: 10.1016/j.memsci.2018.03.047. [DOI] [Google Scholar]
- 211.Brunetti A., Tocci E., Cersosimo M., Kim J.S., Lee W.H., Seong J.G., Lee Y.M., Drioli E., Barbieri G. Mutual influence of mixed-gas permeation in thermally rearranged poly(benzoxazole-co-imide) polymer membranes. J. Membr. Sci. 2019;580:202–213. doi: 10.1016/j.memsci.2019.01.058. [DOI] [Google Scholar]
- 212.Hart K.E., Colina C.M. Estimating gas permeability and permselectivity of microporous polymers. J. Membr. Sci. 2014;468:259–268. doi: 10.1016/j.memsci.2014.06.017. [DOI] [Google Scholar]
- 213.Hart K.E., Springmeier J.M., McKeown N.B., Colina C.M. Simulated swelling during low-temperature N2 adsorption in polymers of intrinsic microporosity. Phys. Chem. Chem. Phys. 2013;15:20161–20169. doi: 10.1039/C3CP53402B. [DOI] [PubMed] [Google Scholar]
- 214.Merkel T.C., Bondar V.I., Nagai K., Freeman B.D., Pinnau I. Gas sorption, diffusion, and permeation in poly(dimethylsiloxane) J. Polym. Sci. Part B: Polym. Phys. 2000;38:415–434. doi: 10.1002/(SICI)1099-0488(20000201)38:3<415::AID-POLB8>3.0.CO;2-Z. [DOI] [Google Scholar]
- 215.Zhou C., Chung T.-S., Wang R., Liu Y., Goh S.H. The accelerated CO2 plasticization of ultra-thin polyimide films and the effect of surface chemical cross-linking on plasticization and physical aging. J. Membr. Sci. 2003;225:125–134. doi: 10.1016/j.memsci.2003.07.006. [DOI] [Google Scholar]
- 216.Horn N.R., Paul D.R. Carbon dioxide plasticization and conditioning effects in thick vs. thin glassy polymer films. Polymer. 2011;52:1619–1627. doi: 10.1016/j.polymer.2011.02.007. [DOI] [Google Scholar]
- 217.Rowe B.W., Pas S.J., Hill A.J., Suzuki R., Freeman B.D., Paul D.R. A variable energy positron annihilation lifetime spectroscopy study of physical aging in thin glassy polymer films. Polymer. 2009;50:6149–6156. doi: 10.1016/j.polymer.2009.10.045. [DOI] [Google Scholar]
- 218.Kim J.H., Koros W.J., Paul D.R. Physical aging of thin 6FDA-based polyimide membranes containing carboxyl acid groups. Part I. Transport properties. Polymer. 2006;47:3094–3103. doi: 10.1016/j.polymer.2006.02.083. [DOI] [Google Scholar]
- 219.Kim J.H., Koros W.J., Paul D.R. Physical aging of thin 6FDA-based polyimide membranes containing carboxyl acid groups. Part II. Optical properties. Polymer. 2006;47:3104–3111. doi: 10.1016/j.polymer.2006.02.079. [DOI] [Google Scholar]
- 220.Kim J.H., Koros W.J., Paul D.R. Effects of CO2 exposure and physical aging on the gas permeability of thin 6FDA-based polyimide membranes: Part 1. Without crosslinking. J. Membr. Sci. 2006;282:21–31. doi: 10.1016/j.memsci.2006.05.004. [DOI] [Google Scholar]
- 221.Rowe B.W., Freeman B.D., Paul D.R. Physical aging of ultrathin glassy polymer films tracked by gas permeability. Polymer. 2009;50:5565–5575. doi: 10.1016/j.polymer.2009.09.037. [DOI] [Google Scholar]
- 222.Murphy T.M., Freeman B.D., Paul D.R. Physical aging of polystyrene films tracked by gas permeability. Polymer. 2013;54:873–880. doi: 10.1016/j.polymer.2012.12.010. [DOI] [Google Scholar]
- 223.Neyertz S., Brown D. Air Sorption and Separation by Polymer Films at the Molecular Level. Macromolecules. 2018;51:7077–7092. doi: 10.1021/acs.macromol.8b01423. [DOI] [Google Scholar]
- 224.Lock S.S.M., Lau K.K., Shariff A.M., Yeong Y.F., Bustam M.A. Thickness dependent penetrant gas transport properties and separation performance within ultrathin polysulfone membrane: Insights from atomistic molecular simulation. J. Polym. Sci. Part B Polym. Phys. 2018;56:131–158. doi: 10.1002/polb.24523. [DOI] [Google Scholar]
- 225.Neyertz S., Douanne A., Brown D. Effect of Interfacial Structure on Permeation Properties of Glassy Polymers. Macromolecules. 2005;38:10286–10298. doi: 10.1021/ma051463y. [DOI] [Google Scholar]
- 226.Neyertz S., Douanne A., Brown D. A molecular dynamics simulation study of surface effects on gas permeation in free-standing polyimide membranes. J. Membr. Sci. 2006;280:517–529. doi: 10.1016/j.memsci.2006.02.011. [DOI] [Google Scholar]
- 227.Neyertz S., Brown D. Molecular Dynamics Simulations of Oxygen Transport through a Fully Atomistic Polyimide Membrane. Macromolecules. 2008;41:2711–2721. doi: 10.1021/ma7026676. [DOI] [Google Scholar]
- 228.Neyertz S., Brown D. The effect of structural isomerism on carbon dioxide sorption and plasticization at the interface of a glassy polymer membrane. J. Membr. Sci. 2014;460:213–228. doi: 10.1016/j.memsci.2014.03.002. [DOI] [Google Scholar]
- 229.Neyertz S., Brown D. Nanosecond-time-scale reversibility of dilation induced by carbon dioxide sorption in glassy polymer membranes. J. Membr. Sci. 2016;520:385–399. doi: 10.1016/j.memsci.2016.08.003. [DOI] [Google Scholar]
- 230.Gopalan P., Brachet P., Kristiansen A., Männle F., Brown D. Oxygen Transport in Amino-Functionalized Polyhedral Oligomeric Silsesquioxanes (POSS) AU—Neyertz, S. Soft Mater. 2014;12:113–123. doi: 10.1080/1539445X.2013.781037. [DOI] [Google Scholar]
- 231.Neyertz S., Brown D. Molecular Dynamics Study of Carbon Dioxide Sorption and Plasticization at the Interface of a Glassy Polymer Membrane. Macromolecules. 2013;46:2433–2449. doi: 10.1021/ma302073u. [DOI] [Google Scholar]
- 232.Yampol’skii Y.P., Pinnau I., Freeman B.D. Materials Science of Membranes for Gas and Vapor Separation. John Wiley; Hoboken, NJ, USA: 2006. [Google Scholar]
- 233.Xia J., Chung T.-S., Paul D.R. Physical aging and carbon dioxide plasticization of thin polyimide films in mixed gas permeation. J. Membr. Sci. 2014;450:457–468. doi: 10.1016/j.memsci.2013.09.047. [DOI] [Google Scholar]
- 234.O’Brien K.C., Koros W.J., Barbari T.A., Sanders E.S. A new technique for the measurement of multicomponent gas transport through polymeric films. J. Membr. Sci. 1986;29:229–238. doi: 10.1016/S0376-7388(00)81262-4. [DOI] [Google Scholar]
- 235.Genduso G., Litwiller E., Ma X., Zampini S., Pinnau I. Mixed-gas sorption in polymers via a new barometric test system: Sorption and diffusion of CO2-CH4 mixtures in polydimethylsiloxane (PDMS) J. Membr. Sci. 2019;577:195–204. doi: 10.1016/j.memsci.2019.01.046. [DOI] [Google Scholar]
- 236.Fraga S.C., Monteleone M., Lanč M., Esposito E., Fuoco A., Giorno L., Pilnáček K., Friess K., Carta M., McKeown N.B., et al. A novel time lag method for the analysis of mixed gas diffusion in polymeric membranes by on-line mass spectrometry: Method development and validation. J. Membr. Sci. 2018;561:39–58. doi: 10.1016/j.memsci.2018.04.029. [DOI] [Google Scholar]
- 237.Tanis I., Brown D., Neyertz S., Heck R., Mercier R., Vaidya M., Ballaguet J.-P. A comparison of pure and mixed-gas permeation of nitrogen and methane in 6FDA-based polyimides as studied by molecular dynamics simulations. Comput. Mater. Sci. 2018;141:243–253. doi: 10.1016/j.commatsci.2017.09.028. [DOI] [Google Scholar]
- 238.Coleman M.R., Koros W.J. Isomeric polyimides based on fluorinated dianhydrides and diamines for gas separation applications. J. Membr. Sci. 1990;50:285–297. doi: 10.1016/S0376-7388(00)80626-2. [DOI] [Google Scholar]
- 239.Tanaka K., Kita H., Okano M., Okamoto K.-I. Permeability and permselectivity of gases in fluorinated and non-fluorinated polyimides. Polymer. 1992;33:585–592. doi: 10.1016/0032-3861(92)90736-G. [DOI] [Google Scholar]
- 240.Coleman M.R., Koros W.J. The transport properties of polyimide isomers containing hexafluoroisopropylidene in the diamine residue. J. Polym. Sci. Part B: Polym. Phys. 1994;32:1915–1926. doi: 10.1002/polb.1994.090321109. [DOI] [Google Scholar]
- 241.Cornelius C.J., Marand E. Hybrid silica-polyimide composite membranes: Gas transport properties. J. Membr. Sci. 2002;202:97–118. doi: 10.1016/S0376-7388(01)00734-7. [DOI] [Google Scholar]
- 242.Takeuchi H. A jump motion of small molecules in glassy polymers: A molecular dynamics simulation. J. Chem. Phys. 1990;93:2062–2067. doi: 10.1063/1.459083. [DOI] [Google Scholar]
- 243.Müller-Plathe F., Laaksonen L., van Gunsteren W.F. Cooperative Effects in the Transport of Small Molecules Through an Amorphous Polymer Matrix. J. Mol. Graph. 1993;11:118. doi: 10.1016/0263-7855(93)87006-Q. [DOI] [PubMed] [Google Scholar]
- 244.Haus J.W., Kehr K.W. Diffusion in regular and disordered lattices. Phys. Rep. 1987;150:263–406. doi: 10.1016/0370-1573(87)90005-6. [DOI] [Google Scholar]
- 245.Havlin S., Ben-Avraham D. Diffusion in disordered media. Adv. Phys. 1987;36:695–798. doi: 10.1080/00018738700101072. [DOI] [Google Scholar]
- 246.Stauffer D., Aharony A. Introduction to Percolation Theory. Taylor & Francis; London, UK: 1992. [Google Scholar]
- 247.Rallabandi P.S., Thompson A.P., Ford D.M. A Molecular Modeling Study of Entropic and Energetic Selectivities in Air Separation with Glassy Polymers. Macromolecules. 2000;33:3142–3152. doi: 10.1021/ma991599u. [DOI] [Google Scholar]
- 248.Wind J.D., Sirard S.M., Paul D.R., Green P.F., Johnston K.P., Koros W.J. Carbon Dioxide-Induced Plasticization of Polyimide Membranes: Pseudo-Equilibrium Relationships of Diffusion, Sorption, and Swelling. Macromolecules. 2003;36:6433–6441. doi: 10.1021/ma0343582. [DOI] [Google Scholar]
- 249.Pinnau I., Toy L.G. Transport of organic vapors through poly(1-trimethylsilyl-1-propyne) J. Membr. Sci. 1996;116:199–209. doi: 10.1016/0376-7388(96)00041-5. [DOI] [Google Scholar]
- 250.Koros W.J., Chern R.T., Stannett V., Hopfenberg H.B. A model for permeation of mixed gases and vapors in glassy polymers. J. Polym. Sci. Polym. Phys. Ed. 1981;19:1513–1530. doi: 10.1002/pol.1981.180191004. [DOI] [Google Scholar]
- 251.Park H.B., Kamcev J., Robeson L.M., Elimelech M., Freeman B.D. Maximizing the right stuff: The trade-off between membrane permeability and selectivity. Science. 2017;356:eaab0530. doi: 10.1126/science.aab0530. [DOI] [PubMed] [Google Scholar]
- 252.Du Y.-C., Huang L.-J., Wang Y.-X., Yang K., Tang J.-G., Wang Y., Cheng M.-M., Zhang Y., Kipper M.J., Belfiore L.A., et al. Recent developments in graphene-based polymer composite membranes: Preparation, mass transfer mechanism, and applications. J. Appl. Polym. Sci. 2019;136:47761. doi: 10.1002/app.47761. [DOI] [Google Scholar]
- 253.Deng L., Hägg M.-B. Carbon nanotube reinforced PVAm/PVA blend FSC nanocomposite membrane for CO2/CH4 separation. Int. J. Greenh. Gas Control. 2014;26:127–134. doi: 10.1016/j.ijggc.2014.04.018. [DOI] [Google Scholar]
- 254.Ihsanullah Carbon nanotube membranes for water purification: Developments, challenges, and prospects for the future. Sep. Purif. Technol. 2019;209:307–337. doi: 10.1016/j.seppur.2018.07.043. [DOI] [Google Scholar]
- 255.Qu X., Alvarez P.J.J., Li Q. Applications of nanotechnology in water and wastewater treatment. Water Res. 2013;47:3931–3946. doi: 10.1016/j.watres.2012.09.058. [DOI] [PubMed] [Google Scholar]
- 256.Celik E., Park H., Choi H., Choi H. Carbon nanotube blended polyethersulfone membranes for fouling control in water treatment. Water Res. 2011;45:274–282. doi: 10.1016/j.watres.2010.07.060. [DOI] [PubMed] [Google Scholar]
- 257.Gong H., Chuah C.Y., Yang Y., Bae T.-H. High performance composite membranes comprising Zn(pyrz)2(SiF6) nanocrystals for CO2/CH4 separation. J. Ind. Eng. Chem. 2018;60:279–285. doi: 10.1016/j.jiec.2017.11.014. [DOI] [Google Scholar]
- 258.Dettori R., Yang Q., Whiting W.I. Exploration of anion transport in a composite membrane via experimental and theoretical methods. J. Membr. Sci. 2018;563:270–276. doi: 10.1016/j.memsci.2018.05.052. [DOI] [Google Scholar]
- 259.Vinoba M., Bhagiyalakshmi M., Alqaheem Y., Alomair A.A., Pérez A., Rana M.S. Recent progress of fillers in mixed matrix membranes for CO2 separation: A review. Sep. Purif. Technol. 2017;188:431–450. doi: 10.1016/j.seppur.2017.07.051. [DOI] [Google Scholar]
- 260.Favvas E.P., Katsaros F.K., Papageorgiou S.K., Sapalidis A.A., Mitropoulos A.C. A review of the latest development of polyimide based membranes for CO2 separations. React. Funct. Polym. 2017;120:104–130. doi: 10.1016/j.reactfunctpolym.2017.09.002. [DOI] [Google Scholar]
- 261.Etxeberria-Benavides M., David O., Johnson T., Łozińska M.M., Orsi A., Wright P.A., Mastel S., Hillenbrand R., Kapteijn F., Gascon J. High performance mixed matrix membranes (MMMs) composed of ZIF-94 filler and 6FDA-DAM polymer. J. Membr. Sci. 2018;550:198–207. doi: 10.1016/j.memsci.2017.12.033. [DOI] [Google Scholar]
- 262.Li W., Samarasinghe S.A.S.C., Bae T.-H. Enhancing CO2/CH4 separation performance and mechanical strength of mixed-matrix membrane via combined use of graphene oxide and ZIF-8. J. Ind. Eng. Chem. 2018;67:156–163. doi: 10.1016/j.jiec.2018.06.026. [DOI] [Google Scholar]
- 263.Barooah M., Mandal B. Synthesis, characterization and CO2 separation performance of novel PVA/PG/ZIF-8 mixed matrix membrane. J. Membr. Sci. 2019;572:198–209. doi: 10.1016/j.memsci.2018.11.001. [DOI] [Google Scholar]
- 264.Li H., Han L., Hou J., Liu J., Zhang Y. Oriented Zeolitic imidazolate framework membranes within polymeric matrices for effective N2/CO2 separation. J. Membr. Sci. 2019;572:82–91. doi: 10.1016/j.memsci.2018.10.086. [DOI] [Google Scholar]
- 265.Ding X., Tan F., Zhao H., Hua M., Wang M., Xin Q., Zhang Y. Enhancing gas permeation and separation performance of polymeric membrane by incorporating hollow polyamide nanoparticles with dense shell. J. Membr. Sci. 2019;570–571:53–60. doi: 10.1016/j.memsci.2018.10.033. [DOI] [Google Scholar]
- 266.Xie K., Fu Q., Qiao G.G., Webley P.A. Recent progress on fabrication methods of polymeric thin film gas separation membranes for CO2 capture. J. Membr. Sci. 2019;572:38–60. doi: 10.1016/j.memsci.2018.10.049. [DOI] [Google Scholar]
- 267.Akther N., Phuntsho S., Chen Y., Ghaffour N., Shon H.K. Recent advances in nanomaterial-modified polyamide thin-film composite membranes for forward osmosis processes. J. Membr. Sci. 2019;584:20–45. doi: 10.1016/j.memsci.2019.04.064. [DOI] [Google Scholar]
- 268.Liu G., Labreche Y., Chernikova V., Shekhah O., Zhang C., Belmabkhout Y., Eddaoudi M., Koros W.J. Zeolite-like MOF nanocrystals incorporated 6FDA-polyimide mixed-matrix membranes for CO2/CH4 separation. J. Membr. Sci. 2018;565:186–193. doi: 10.1016/j.memsci.2018.08.031. [DOI] [Google Scholar]
- 269.Liu Y., Liu G., Zhang C., Qiu W., Yi S., Chernikova V., Chen Z., Belmabkhout Y., Shekhah O., Eddaoudi M., et al. Enhanced CO2/CH4 Separation Performance of a Mixed Matrix Membrane Based on Tailored MOF-Polymer Formulations. Adv. Sci. 2018;5:1800982. doi: 10.1002/advs.201800982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Teodoro R.M., Tomé L.C., Mantione D., Mecerreyes D., Marrucho I.M. Mixing poly(ionic liquid)s and ionic liquids with different cyano anions: Membrane forming ability and CO2/N2 separation properties. J. Membr. Sci. 2018;552:341–348. doi: 10.1016/j.memsci.2018.02.019. [DOI] [Google Scholar]
- 271.Kanehashi S., Kishida M., Kidesaki T., Shindo R., Sato S., Miyakoshi T., Nagai K. CO2 separation properties of a glassy aromatic polyimide composite membranes containing high-content 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ionic liquid. J. Membr. Sci. 2013;430:211–222. doi: 10.1016/j.memsci.2012.12.003. [DOI] [Google Scholar]
- 272.Li J., Liu B., Zhang X., Cao D., Chen G. Hydrogen Bond Networks of Glycol Molecules on ZIF-8 Surfaces as Semipermeable Films for Efficient Carbon Capture. J. Phys. Chem. C. 2017;121:25347–25352. doi: 10.1021/acs.jpcc.7b09068. [DOI] [Google Scholar]
- 273.Gonciaruk A., Althumayri K., Harrison W.J., Budd P.M., Siperstein F.R. PIM-1/graphene composite: A combined experimental and molecular simulation study. Microporous Mesoporous Mater. 2015;209:126–134. doi: 10.1016/j.micromeso.2014.07.007. [DOI] [Google Scholar]
- 274.Kupgan G., Abbott L.J., Hart K.E., Colina C.M. Modeling Amorphous Microporous Polymers for CO2 Capture and Separations. Chem. Rev. 2018;118:5488–5538. doi: 10.1021/acs.chemrev.7b00691. [DOI] [PubMed] [Google Scholar]
- 275.Vogiatzis G.G., Theodorou D.N. Multiscale Molecular Simulations of Polymer-Matrix Nanocomposites. Arch. Comput. Methods Eng. 2018;25:591–645. doi: 10.1007/s11831-016-9207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Abedini A., Crabtree E., Bara J.E., Turner C.H. Molecular Simulation of Ionic Polyimides and Composites with Ionic Liquids as Gas-Separation Membranes. Langmuir. 2017;33:11377–11389. doi: 10.1021/acs.langmuir.7b01977. [DOI] [PubMed] [Google Scholar]
- 277.Abedini A., Crabtree E., Bara J.E., Turner C.H. Molecular analysis of selective gas adsorption within composites of ionic polyimides and ionic liquids as gas separation membranes. Chem. Phys. 2019;516:71–83. doi: 10.1016/j.chemphys.2018.08.039. [DOI] [Google Scholar]
- 278.Aryal D., Ganesan V. Reversal of Salt Concentration Dependencies of Salt and Water Diffusivities in Polymer Electrolyte Membranes. ACS Macro Lett. 2018;7:739–744. doi: 10.1021/acsmacrolett.8b00333. [DOI] [PubMed] [Google Scholar]
- 279.Mogurampelly S., Sethuraman V., Pryamitsyn V., Ganesan V. Influence of nanoparticle-ion and nanoparticle-polymer interactions on ion transport and viscoelastic properties of polymer electrolytes. J. Chem. Phys. 2016;144:154905. doi: 10.1063/1.4946047. [DOI] [PubMed] [Google Scholar]
- 280.Mogurampelly S., Ganesan V. Influence of nanoparticle surface chemistry on ion transport in polymer nanocomposite electrolytes. Solid State Ion. 2016;286:57. doi: 10.1016/j.ssi.2015.12.018. [DOI] [Google Scholar]
- 281.Zhang L., Hu Z., Jiang J. Metal–Organic Framework/Polymer Mixed-Matrix Membranes for H2/CO2 Separation: A Fully Atomistic Simulation Study. J. Phys. Chem. C. 2012;116:19268–19277. doi: 10.1021/jp3067124. [DOI] [Google Scholar]
- 282.Azar A.N.V., Velioglu S., Keskin S. Large-Scale Computational Screening of Metal Organic Framework (MOF) Membranes and MOF-Based Polymer Membranes for H2/N2 Separations. ACS Sustain. Chem. Eng. 2019;7:9525–9536. doi: 10.1021/acssuschemeng.9b01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Yilmaz G., Keskin S. Predicting the Performance of Zeolite Imidazolate Framework/Polymer Mixed Matrix Membranes for CO2, CH4, and H2 Separations Using Molecular Simulations. Ind. Eng. Chem. Res. 2012;51:14218–14228. doi: 10.1021/ie302290a. [DOI] [Google Scholar]
- 284.Dutta R.C., Bhatia S.K. Structure and Gas Transport at the Polymer–Zeolite Interface: Insights from Molecular Dynamics Simulations. ACS Appl. Mater. Interfaces. 2018;10:5992–6005. doi: 10.1021/acsami.7b17470. [DOI] [PubMed] [Google Scholar]
- 285.Zhang Q.G., Liu Q.L., Wu J.Y., Chen Y., Zhu A.M. Structure-related diffusion in poly(methyl methacrylate)/polyhedral oligomeric silsesquioxanes composites: A molecular dynamics simulation study. J. Membr. Sci. 2009;342:105–112. doi: 10.1016/j.memsci.2009.06.030. [DOI] [Google Scholar]
- 286.Wang C., Jagirdar P., Naserifar S., Sahimi M. Molecular Simulation Study of Gas Solubility and Diffusion in a Polymer-Boron Nitride Nanotube Composite. J. Phys. Chem. B. 2016;120:1273–1284. doi: 10.1021/acs.jpcb.5b10493. [DOI] [PubMed] [Google Scholar]
- 287.Bonakala S., Lalitha A., Shin J.E., Moghadam F., Semino R., Park H.B., Maurin G. Understanding of the Graphene Oxide/Metal-Organic Framework Interface at the Atomistic Scale. ACS Appl. Mater. Interfaces. 2018;10:33619–33629. doi: 10.1021/acsami.8b09851. [DOI] [PubMed] [Google Scholar]
- 288.Hwang S., Semino R., Seoane B., Zahan M., Chmelik C., Valiullin R., Bertmer M., Haase J., Kapteijn F., Gascon J., et al. Revealing the Transient Concentration of CO2 in a Mixed-Matrix Membrane by IR Microimaging and Molecular Modeling. Angew. Chem. Int. Ed. 2018;57:5156–5160. doi: 10.1002/anie.201713160. [DOI] [PubMed] [Google Scholar]
- 289.Semino R., Dürholt J.P., Schmid R., Maurin G. Multiscale Modeling of the HKUST-1/Poly(vinyl alcohol) Interface: From an Atomistic to a Coarse Graining Approach. J. Phys. Chem. C. 2017;121:21491–21496. doi: 10.1021/acs.jpcc.7b07090. [DOI] [Google Scholar]
- 290.Lin R., Villacorta Hernandez B., Ge L., Zhu Z. Metal organic framework based mixed matrix membranes: An overview on filler/polymer interfaces. J. Mater. Chem. A. 2018;6:293–312. doi: 10.1039/C7TA07294E. [DOI] [Google Scholar]
- 291.Zhao J., He G., Liu G., Pan F., Wu H., Jin W., Jiang Z. Manipulation of interactions at membrane interfaces for energy and environmental applications. Prog. Polym. Sci. 2018;80:125–152. doi: 10.1016/j.progpolymsci.2017.12.002. [DOI] [Google Scholar]
- 292.Zavadlav J., Bevc S., Praprotnik M. Adaptive resolution simulations of biomolecular systems. Eur. Biophys. J. 2017;46:821–835. doi: 10.1007/s00249-017-1248-0. [DOI] [PubMed] [Google Scholar]
- 293.Kreis K., Tuckerman M.E., Donadio D., Kremer K., Potestio R. From Classical to Quantum and Back: A Hamiltonian Scheme for Adaptive Multiresolution Classical/Path-Integral Simulations. J. Chem. Theory Comput. 2016;12:3030–3039. doi: 10.1021/acs.jctc.6b00242. [DOI] [PubMed] [Google Scholar]
- 294.Kreis K., Potestio R., Kremer K., Fogarty A.C. Adaptive Resolution Simulations with Self-Adjusting High-Resolution Regions. J. Chem. Theory Comput. 2016;12:4067–4081. doi: 10.1021/acs.jctc.6b00440. [DOI] [PubMed] [Google Scholar]
- 295.Kreis K., Kremer K., Potestio R., Tuckerman M.E. From classical to quantum and back: Hamiltonian adaptive resolution path integral, ring polymer, and centroid molecular dynamics. J. Chem. Phys. 2017;147:244104. doi: 10.1063/1.5000701. [DOI] [PubMed] [Google Scholar]
- 296.Praprotnik M., Delle Site L., Kremer K. Adaptive resolution molecular-dynamics simulation: Changing the degrees of freedom on the fly. J. Chem. Phys. 2005;123:224106. doi: 10.1063/1.2132286. [DOI] [PubMed] [Google Scholar]
- 297.Delle Site L., Abrams C.F., Alavi A., Kremer K. Polymers near Metal Surfaces: Selective Adsorption and Global Conformations. Phys. Rev. Lett. 2002;89:156103. doi: 10.1103/PhysRevLett.89.156103. [DOI] [PubMed] [Google Scholar]
- 298.Dong D., Zhang W., Barnett A., Lu J., Van Duin C.A., Molinero V., Bedrov D. Multiscale Modeling of Structure, Transport and Reactivity in Alkaline Fuel Cell Membranes: Combined Coarse-Grained, Atomistic and Reactive Molecular Dynamics Simulations. Polymers. 2018;10:1289. doi: 10.3390/polym10111289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Maghami S., Mehrabani-Zeinabad A., Sadeghi M., Sánchez-Laínez J., Zornoza B., Téllez C., Coronas J. Mathematical modeling of temperature and pressure effects on permeability, diffusivity and solubility in polymeric and mixed matrix membranes. Chem. Eng. Sci. 2019;205:58–73. doi: 10.1016/j.ces.2019.04.037. [DOI] [Google Scholar]
- 300.Castro-Muñoz R., Iglesia Ó.d., Fíla V., Téllez C., Coronas J. Pervaporation-Assisted Esterification Reactions by Means of Mixed Matrix Membranes. Ind. Eng. Chem. Res. 2018;57:15998–16011. doi: 10.1021/acs.iecr.8b01564. [DOI] [Google Scholar]
- 301.Ursino C., Castro-Muñoz R., Drioli E., Gzara L., Albeirutty H.M., Figoli A. Progress of Nanocomposite Membranes for Water Treatment. Membranes. 2018;8:18. doi: 10.3390/membranes8020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Sarango L., Paseta L., Navarro M., Zornoza B., Coronas J. Controlled deposition of MOFs by dip-coating in thin film nanocomposite membranes for organic solvent nanofiltration. J. Ind. Eng. Chem. 2018;59:8–16. doi: 10.1016/j.jiec.2017.09.053. [DOI] [Google Scholar]
- 303.Navarro M., Benito J., Paseta L., Gascón I., Coronas J., Téllez C. Thin-Film Nanocomposite Membrane with the Minimum Amount of MOF by the Langmuir–Schaefer Technique for Nanofiltration. ACS Appl. Mater. Interfaces. 2018;10:1278–1287. doi: 10.1021/acsami.7b17477. [DOI] [PubMed] [Google Scholar]
- 304.Echaide-Górriz C., Navarro M., Téllez C., Coronas J. Simultaneous use of MOFs MIL-101(Cr) and ZIF-11 in thin film nanocomposite membranes for organic solvent nanofiltration. Dalton Trans. 2017;46:6244–6252. doi: 10.1039/C7DT00197E. [DOI] [PubMed] [Google Scholar]
- 305.Sánchez-Laínez J., Zornoza B., Orsi A.F., Łozińska M.M., Dawson D.M., Ashbrook S.E., Francis S.M., Wright P.A., Benoit V., Llewellyn P.L., et al. Synthesis of ZIF-93/11 Hybrid Nanoparticles via Post-Synthetic Modification of ZIF-93 and Their Use for H2/CO2 Separation. Chem. A Eur. J. 2018;24:11211–11219. doi: 10.1002/chem.201802124. [DOI] [PubMed] [Google Scholar]
- 306.Castro-Muñoz R., Martin-Gil V., Ahmad M.Z., Fíla V. Matrimid® 5218 in preparation of membranes for gas separation: Current state-of-the-art. Chem. Eng. Commun. 2018;205:161–196. doi: 10.1080/00986445.2017.1378647. [DOI] [Google Scholar]
- 307.Castro-Muñoz R., Fíla V., Ahmad M.Z. Enhancing the CO2 Separation Performance of Matrimid 5218 Membranes for CO2/CH4 Binary Mixtures. Chem. Eng. Technol. 2019;42:645–654. doi: 10.1002/ceat.201800111. [DOI] [Google Scholar]
- 308.Castro-Muñoz R., Fíla V., Martin-Gil V., Muller C. Enhanced CO2 permeability in Matrimid® 5218 mixed matrix membranes for separating binary CO2/CH4 mixtures. Sep. Purif. Technol. 2019;210:553–562. doi: 10.1016/j.seppur.2018.08.046. [DOI] [Google Scholar]
- 309.Mathioudakis I.G., Vogiatzis G.G., Tzoumanekas C., Theodorou D.N. Molecular modeling and simulation of polymer nanocomposites at multiple length scales. IEEE Trans. Nanotechnol. 2016;15:416–422. doi: 10.1109/TNANO.2016.2538460. [DOI] [Google Scholar]
- 310.Li C., Strachan A. Molecular simulations of crosslinking process of thermosetting polymers. Polymer. 2010;51:6058–6070. doi: 10.1016/j.polymer.2010.10.033. [DOI] [Google Scholar]
- 311.Chmiela S., Sauceda H.E., Poltavsky I., Müller K.-R., Tkatchenko A. sGDML: Constructing accurate and data efficient molecular force fields using machine learning. Comput. Phys. Commun. 2019 doi: 10.1016/j.cpc.2019.02.007. [DOI] [Google Scholar]
- 312.Bejagam K.K., Singh S., An Y., Deshmukh S.A. Machine-Learned Coarse-Grained Models. J. Phys. Chem. Lett. 2018;9:4667–4672. doi: 10.1021/acs.jpclett.8b01416. [DOI] [PubMed] [Google Scholar]
- 313.Chan H., Narayanan B., Cherukara M.J., Sen F.G., Sasikumar K., Gray S.K., Chan M.K.Y., Sankaranarayanan S.K.R.S. Machine Learning Classical Interatomic Potentials for Molecular Dynamics from First-Principles Training Data. J. Phys. Chem. C. 2019;123:6941–6957. doi: 10.1021/acs.jpcc.8b09917. [DOI] [Google Scholar]
- 314.Wang J., Olsson S., Wehmeyer C., Pérez A., Charron N.E., de Fabritiis G., Noé F., Clementi C. Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. ACS Cent. Sci. 2019;5:755–767. doi: 10.1021/acscentsci.8b00913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Zhang L., Han J., Wang H., Car R., Weinan E. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics. Phys. Rev. Lett. 2018;120:143001. doi: 10.1103/PhysRevLett.120.143001. [DOI] [PubMed] [Google Scholar]
- 316.Zhang L., Han J., Wang H., Car R., Weinan E. DeePCG: Constructing coarse-grained models via deep neural networks. J. Chem. Phys. 2018;149:034101. doi: 10.1063/1.5027645. [DOI] [PubMed] [Google Scholar]
- 317.Wang H., Zhang L., Han J., Weinan E. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun. 2018;228:178–184. doi: 10.1016/j.cpc.2018.03.016. [DOI] [Google Scholar]
- 318.Castro-Muñoz R., Galiano F., de la Iglesia Ó., Fíla V., Téllez C., Coronas J., Figoli A. Graphene oxide—Filled polyimide membranes in pervaporative separation of azeotropic methanol–MTBE mixtures. Sep. Purif. Technol. 2019;224:265–272. doi: 10.1016/j.seppur.2019.05.034. [DOI] [Google Scholar]
- 319.Baker R.W., Wijmans J.G., Huang Y. Permeability, permeance and selectivity: A preferred way of reporting pervaporation performance data. J. Membr. Sci. 2010;348:346–352. doi: 10.1016/j.memsci.2009.11.022. [DOI] [Google Scholar]
- 320.Castro-Muñoz R., Buera-González J., de la Iglesia Ó., Galiano F., Fíla V., Malankowska M., Rubio C., Figoli A., Téllez C., Coronas J. Towards the dehydration of ethanol using pervaporation cross-linked poly(vinyl alcohol)/graphene oxide membranes. J. Membr. Sci. 2019;582:423–434. doi: 10.1016/j.memsci.2019.03.076. [DOI] [Google Scholar]