

Abstract
The activated B-cell (ABC-like) subtype of diffuse large B-cell lymphoma (DLBCL) is characterized by the chronic activation of signaling initiated by immunoglobulin-μ (IgM). By analyzing DNA copy profiles of 1,000 DLBCLs, we identified gains of 18q21.2 as the most frequent genetic alteration in ABC-like DLBCL. Using integrative analysis of matched gene expression profiling data we found that the TCF4 (E2–2) transcription factor gene is the target of these alterations. Over-expression of TCF4 led to its occupancy on immunoglobulin and MYC gene enhancers and increased their expression at the transcript and protein level. Inhibition of TCF4 activity with dominant-negative constructs was synthetically lethal to ABC-like DLBCL cell lines harboring TCF4 DNA copy gains, highlighting it as an attractive therapeutic target. Furthermore, the TCF4 gene is one of the top BRD4-regulated genes in DLBCL and a BET proteolysis-targeting chimera (PROTAC) extinguished TCF4, MYC and IgM expression and killed ABC-like DLBCL cells in vitro and in vivo. This highlights a novel genetic mechanism for promoting immunoglobulin signaling in ABC-like DLBCL and provides a functional rationale for the use of BET inhibitors in this disease.
Keywords: DLBCL, TCF4, IgM, BET inhibitor
Summary Sentence:
Targetable DNA copy number gains of the TCF4 gene are the most frequent genetic alteration in ABC-like DLBCL and promote immunoglobulin expression.
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL) is the most common form of lymphoma and is curable in ~60% of patients using a combination chemo-immunotherapy regimen, R-CHOP (1, 2). However, those that are refractory to, or relapse following, first-line therapy have a dismal outcome (3). Chimeric antigen receptor (CAR)-T cells are likely to change the landscape of outcomes in relapsed/refractory patients, but a large number of patients are not eligible for CAR-T therapy and ~50% of those that received CAR-T progress within 12 months (4). Novel rationally-targeted therapeutic strategies are therefore needed for DLBCL.
The clinical heterogeneity of DLBCL is underpinned by molecular heterogeneity, with the major distinction being between the germinal center B-cell (GCB)-like and activated B-cell (ABC)-like ‘cell of origin’ (COO) subtypes that were identified by gene expression profiling (5). The GCB-like subtype shows transcriptional similarities to normal germinal center B-cells, whereas the ABC-like subtype shows transcriptional similarities to CD40-activated B-cells or plasmablasts. Patients with ABC-like DLBCL have significantly worse overall survival compared to patients with GCB-like DLBCL, when treated with the standard-of-care combination chemotherapy (CHOP) plus rituximab (R-CHOP) regimen (6). The ABC-like DLBCL subtype expresses immunoglobulin μ (IgM) (7) in >90% of cases, which forms the B-cell receptor (BCR) signaling complex in association with CD79A and CD79B and drives chronically active BCR signaling. Several genetic alterations have been shown to promote this signaling, including mutations of the CD79A, CD79B, CARD11, and MYD88 genes (8–11). However, these mutations only account for approximately two thirds of ABC-like DLBCL cases(12), suggesting that one or more significant genetic drivers remain to be defined.
A common mechanism for tumorigenesis is the gain or loss of DNA encoding oncogenes or tumor suppressor genes, respectively. These copy number alterations (CNAs) perturb a higher fraction of the cancer genome than somatic nucleotide variants (SNVs) and small insertion/deletions (InDels) and are critically important to cancer etiology (13). Here, we have integrated multiple datasets, including DNA copy number profiles of 1,000 DLBCLs, and identified DNA copy number gain of the E2 transcription factor TCF4 as the most frequent genetic alteration in ABC-like DLBCL. We show that TCF4 is capable of driving IgM expression and is amenable to therapeutic targeting through BET inhibition. These data therefore highlight a novel genetic basis for ABC-like DLBCL with potential implications for future clinical studies.
RESULTS
DNA copy number gains of 18q are the most frequent genetic alteration in the ABC-like subtype of DLBCL.
In order to identify significant CNAs in DLBCL, we interrogated the genomic profiles of 1,000 DLBCLs using the GISTIC2 algorithm (14). These included high-resolution SNP microarrays from 860 previously published cases, in addition to next generation sequencing (NGS)-derived profiles from our own cohort of 140 cases (table S1 and S2). The GISTIC analysis revealed 20 significant DNA copy number gains and 21 significant DNA copy number losses (FDR<0.1; Fig. 1A and table S3). As previously observed, 19.1% of tumors (191/1000) did not bear any of these DNA copy number alterations (15). Using a subset of 448 cases with COO subtype available, we identified 9 CNAs that were significantly more frequent in ABC-like DLBCL and 11 that were significantly more frequent in GCB-DLBCL (Fisher Q-value<0.1; Fig. 1B and fig. S1 and table S4). The most frequent genetic alteration in ABC-like DLBCL was gain of 18q21.2, which was observed in 44% of tumors. In line with the enrichment in ABC-like DLBCL, 18q21.2 gains were associated with significantly reduced overall survival in both CHOP (log-rank P=0.0068) and R-CHOP (log rank P=0.0034) treated patients for which data were available (Fig. 1, C and D). However, this effect was not independent of cell of origin subtype (Fig. S2). Using 199 tumors with matched COO subtype, DNA copy number data and mutation status for 40 genes, we observed that the frequency of 18q21 gains (23.1% of all tumors; 40.7% of ABC-like tumors) were higher than other ABC-like DLBCL-associated somatic mutations including MYD88 mutation (16.6% of all tumors; 33.3% of ABC-like tumors), CD79B mutation (7.5% of all tumors; 18.5% of ABC-like tumors) and other ABC-associated genes (Fig. 1E and fig. S3 and table S5). Because multiple genetic alterations are associated with ABC-like DLBCL, we employed the REVEALER algorithm (16) to identify the set of genetic alterations that best explained ABC-like DLBCL signature. Using a set of 87 CNAs and recurrently mutated genes as the feature set, and MYD88 mutations as the seed feature, REVEALER identified an additional 4 genetic alterations including 18q21.2 gain as those best associating with the ABC-like signature (Fig. 1F). Gains of 18q21 are therefore the most frequent genetic feature of ABC-like DLBCL and are predicted to contribute to this molecular phenotype.
Figure 1: DNA copy number gains of 18q21.2 are the most frequent genetic alteration in ABC-like DLBCL.
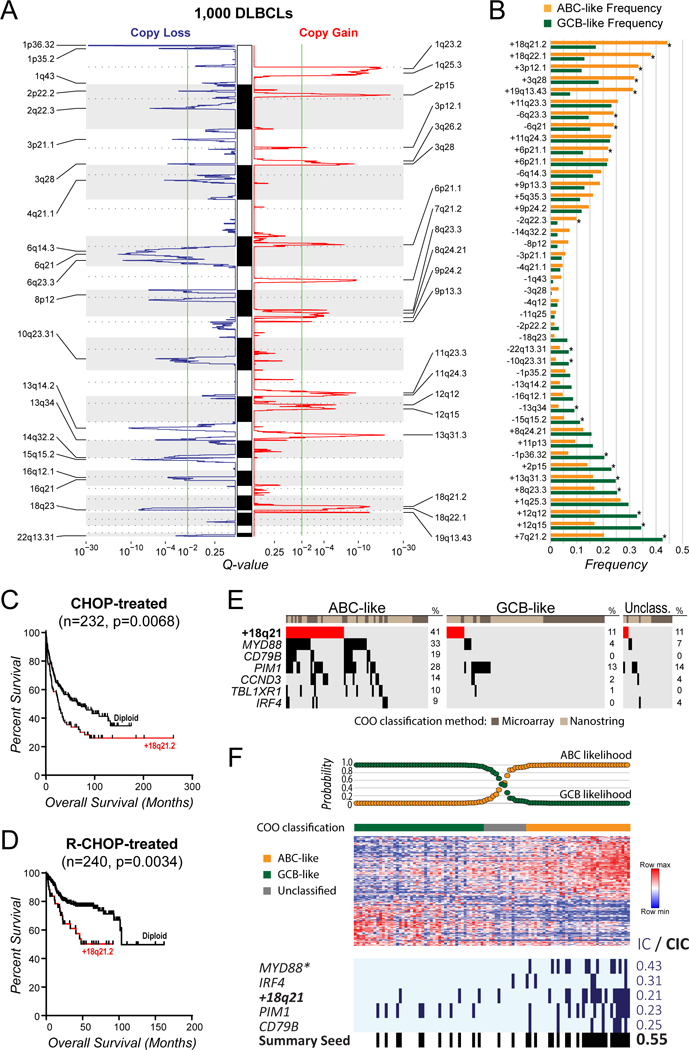
(A) GISTIC analysis of DNA copy number profiles form 1,000 DLBCL tumors identified 21 peaks of DNA copy loss (blue, left) and 20 peaks of DNA copy gain (red, right). The green line indicates the significance threshold of q-value = 0.1. (B) The GISTIC peaks from (A) are shown with reference to their frequency in ABC-like (orange) compared to GCB-like (green) cell of origin subtypes (*Q-value<0.1). DNA copy gains of 18q21.2 were the most frequent alteration in ABC-like DLBCL cases. (C-D) A Kaplan-Meier plot of overall survival for patients treated with CHOP combination chemotherapy (C) or CHOP plus Rituximab (D) shows that the presence of 18q21.2 gain is associated with poor outcome. (E) The frequency of 18q21 gains is shown relative to other somatic mutations that are significantly associated with the ABC-like DLBCL subtype. This shows that gains of 18q21 are the most frequent genetic alteration in ABC-like DLBCL. (F) REVEALER analysis was performed to identify the set functionally-complementary genetic features that likely contribute to the ABC-like DLBCL molecular phenotype. Mutations of MYD88 were used as the seed feature. Mutations of IRF4, PIM1 and CD79B, and DNA copy gains of 18q21 were selected as additional features that likely also contribute to the phenotype (*Seed feature; IC, information coefficient; CIC, conditional information coefficient).
The TCF4 (E2–2) transcription factor is a novel oncogene in ABC-like DLBCL
Gains of 18q have been previously attributed to the BCL2 oncogene (15, 17). However, our analysis of this large cohort provided the resolution to identify two significant peaks of DNA copy gain on chromosome 18; 18q21.2 (16 genes, Q=4.8×10−14) and 18q22.1 (70 genes, Q=1.1×10−7; table S3). We further integrated GEP data from 249 tumors to identify the likely targets of these lesions by testing for the increase in expression of genes within the most significant peaks of DNA copy gain. This highlighted TCF4 and BCL2 as likely targets of the 18q21.2 and 18q22.1 gains, respectively (Fig. 2A and table S6). Notably, most 18q CNAs incorporated both of these genes (Fig 2, A to B), with only 7.3% or 1.0% of ABC-like DLBCLs having excisional CNAs targeting TCF4 or BCL2 alone, respectively (Fig. 2B). Using paired gene expression profiling data on 249 tumors, we observed that DNA copy number gains were associated with significantly higher TCF4 transcript abundance when comparing within COO subtypes (P<0.001). However, the expression of TCF4 in ABC-like DLBCL tumors was higher than that in GCB-like tumors, such that ABC-like tumors with diploid 18q21.2 had equivalent TCF4 expression to GCB-like tumors with 18q21.2 gain (Fig. 2C). In line with this, ABC-like DLBCL cell lines expressed TCF4 protein at a higher level than GCB-like DLBCL cell lines irrespective of DNA copy number, and these levels were significantly increased by DNA copy gain (Fig. 2, D and E). Therefore, TCF4 is more highly expressed in ABC-like DLBCL compared to GCB-like DLBCL, and is further increased by DNA copy gain. However, this observation should be prospectively validated by immunohistochemistry in patient tumors when appropriate antibodies are available.
Figure 2: The TCF4 gene is the most significant target of 18q DNA copy number gains.
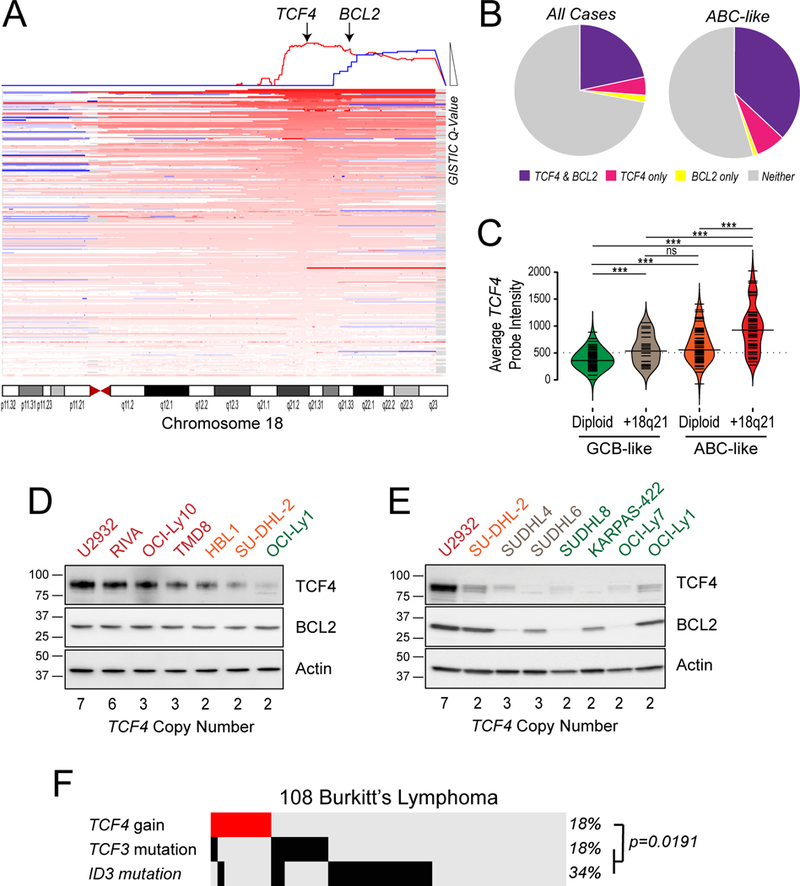
(A) A schematic of 18q DNA copy number gains is shown, with each line representing a single tumor and deeper shades of red indicating higher DNA copy number. The GISTIC q-value is shown at the top of the diagram and the two significant peaks are highlighted with arrows. The most statistically significant peak harbors the TCF4 gene, while the less significant peak harbors the BCL2 gene. However, it can be seen that in many cases the DNA copy number gains span both the TCF4 and BCL2 regions. (B) The frequency of tumors with DNA copy number gains that include both the TCF4 and BCL2 genes (purple), the TCF4 gene and not the BCL2 gene (pink) or the BCL2 gene and not the TCF4 gene (yellow) are shown for all tumors (left) and for the ABC-like only (right). This shows that the majority of 18q DNA copy gains include both TCF4 and BCL2, but TCF4 is more frequently gained independently of BCL2 than vice versa. (C) The gene expression level from microarrays are shown for GCB-like DLBCL with diploid 18q (green), GCB-like DLBCL with 18q copy gain (beige), ABC-like DLBCL tumors with diploid for 18q (orange), and ABC-like DLBCL tumors with 18q DNA copy number gains (red). The expression of TCF4 is significantly higher in ABC-like DLBCL compared to GCB-like DLBCL in diploid cases and further significantly increased by DNA copy number gain. P-values are from students T-test, p>0.05 is denoted as ns, ***p<0.001. (D) The protein level of TCF4 and BCL2 are shown in ABC-like DLBCL cell lines, ordered according to increasing DNA copy number of the TCF4 locus. There is a visible relationship between TCF4 protein abundance and DNA copy number that is less clear for BCL2. (E) The protein level of TCF4 and BCL2 are shown in GCB-like DLBCL cell lines, with the ABC-like cell lines with the highest (U2932) and lowest (SUDHL2) TCF4 expression shown as reference. All of the GCB-like DLBCL cell lines show lower expression of TCF4, and there is no clear relationship with 18q DNA copy number. (F) The frequency of TCF4 DNA copy gains, TCF3 mutation and ID3 mutation are shown for a cohort of 108 Burkitt lymphoma tumors. Gains of the TCF4 locus are present at the same frequency of TCF3 mutations and are significantly mutually exclusive from TCF3 and ID3 mutations.
The TCF4 gene encodes an E2 family transcription factor, E2–2. Mutations of another E2 transcription factor, TCF3, and its negative regulators ID2 and ID3 are frequent in Burkitt’s lymphoma (BL) and promote immunoglobulin signaling (18, 19). We therefore interrogated the mutation status of these genes and TCF4 copy gains in our cohort of 140 DLBCLs and a prior cohort of 108 BLs that were sequenced and analyzed with the same approach (20). We did not observe recurrent mutations of TCF4 or ID2 in this BL cohort, and mutations of TCF3 and ID3 were infrequent in DLBCL. However, gains of TCF4 were present at the same frequency as TCF3 mutations in BL (18%). Furthermore, TCF4 gains were significantly mutually-exclusive from TCF3 and ID3 mutations (Fisher P=0.019; Fig. 2F), suggesting that TCF4 gains may serve a similar function as TCF3/ID3 mutations in promoting immunoglobulin signaling. We speculate based upon gene expression data that the preference for mutating TCF3 and ID3 in BL is due to their higher expression in BL compared to DLBCL, which may be related to the germinal center B-cell specific expression of TCF3 (Fig. S4). In contrast, TCF4 is variably expressed in both BL and DLBCL, and does not decline in expression in normal B-cells following germinal center exit (Fig. S4). These data therefore show that the TCF4 gene is highly expressed in ABC-like DLBCL, with expression further promoted by frequent 18q21.2 DNA copy gains, and implicates TCF4 in immunoglobulin signaling.
TCF4 regulates IgM and MYC expression in ABC-like DLBCL
To identify potential target genes of TCF4, we performed differential gene expression analysis of primary DLBCL tumors with TCF4 DNA copy gain (n=51) compared to those without (n=59). This was limited to on ABC-like tumors so as to eliminate the confounding effect of genes that differ in expression between COO subtypes. A total of 355 genes (472 probe-sets) and 87 genes (107 probe-sets) were found to be expressed at significantly higher or lower levels in tumors with TCF4 gain, respectively (Q<0.1, fold-change≥1.2; Fig. 3A and table S7). We performed ChIP-seq of ABC-like DLBCL cell lines, SUDHL2 and TMD8, with tetracycline-inducible Myc-DDK-tagged TCF4 in order to define whether these genes were direct transcriptional targets of TCF4 (Fig. 3A). Importantly, TCF4 was expressed at a level comparable to that in the U2932 cell line with TCF4 copy gain (Fig. S5). Using the intersection of significant peaks from both cell lines, we identified TCF4 binding proximal to 180/355 genes with increased expression and 46/87 genes with decreased expression in tumors with TCF4 copy gain (Fig. 3, A and B and table S8). These peaks showed a significant enrichment of motifs containing E-box consensus sequences (CANNTG; Q-value<0.1; Fig. S6), and many of the same regions are also bound by TCF4 in plasmacytoid dendritic cell neoplasm (21) (Fig. S6), providing strong evidence that we detected on-target binding. Among the most significant ChIP-seq peaks were those within the immunoglobulin heavy chain locus (Fig. 3B), in line with the significantly higher expression of IGHM in ABC-like DLBCL tumors with TCF4 copy gain (Fig. 3C). This included peaks immediately upstream and downstream of the IGHM and IGHD genes, respectively, in regions with corresponding H3K27Ac in normal CD20+ B-cells that indicates they are bona fide enhancers (Fig. 3D). We validated TCF4 binding to these two immunoglobulin loci, as well as a downstream enhancer of the MYC gene, using ChIP-qPCR. This confirmed binding to all 3 loci that was significantly above that of a control IgG antibody (P<0.001) in 3 cell lines with tetracycline-inducible expression of TCF4 (SUDHL2, TMD8 and HBL1) and 2 cell lines with high baseline expression of TCF4 (U2932 and RIVA) (P<0.001; Fig. 3E). Immunoglobulin and MYC genes are therefore likely targets of the TCF4 transcription factor in ABC-like DLBCL.
Figure 3: TCF4 regulates IgM expression in ABC-like DLBCL.

(A) Differential gene expression analysis of 110 primary ABC-like DLBCL tumors with or without TCF4 DNA copy number gain identified a large set of genes with increased expression associated with TCF4 gain. This included the direct targets of 18q DNA copy number gains, TCF4 and BCL2, and multiple genes with an important role in the pathophysiology of DLBCL, such as IRF4, MYC and the immunoglobulin heavy chain μ (IGHM), that are upregulated as a secondary effect of TCF4 gain. ChIP-seq of TCF4 from SUDHL2 and TMD8 cell lines showed that the majority of these genes were marked with TCF4 binding in intragenic or distant enhancer elements, suggesting that their up-regulation may be driven by transcriptional activation by TCF4. (B) The significant TCF4 ChIP-seq peaks from SUDHL2 and TMD8 are shown, ordered from strongest (top) to weakest (bottom) signal ratio compared to the input control. Significant peaks were detected for important genes such as MYC and IRF4, but multiple IGHM peaks were amongst those with the highest TCF4 binding. (C) A violin plot shows that primary ABC-like DLBCL tumors with 18q21 gain express significantly higher transcript levels of IGHM compared to ABC-like DLBCL tumors with diploid 18q21. (D) Two of the TCF4 peaks at the immunoglobulin heavy chain locus are shown for TCF4 ChIP (blue) compared to the equivalent input control (grey). Yellow shading indicates the statistically significant peak. For reference, ENCODE data for H3K27 acetylation (H3K27Ac) ChIP-seq in CD20+ B-cells is shown, which support the TCF4 bound regions as bona fide enhancer elements in B-cells. (E) The binding of TCF4 to the two immunoglobulin heavy chain loci and the MYC enhancer locus was confirmed by ChIP-qPCR in two cell lines with high TCF4 DNA copy number and protein expression (U2932 and RIVA) and three cell lines with tetracycline-inducible TCF4 expression (SUDHL2, TMD8, HBL1). The signal was significantly above that of the isotype control IgG antibody for all cell lines and loci (P<0.001). Each bar represents the mean +/− S.E.M. of 3 independent experiments.
Tetracycline-inducible expression of TCF4 in ABC-like DLBCL cell lines led to a marked increase in IGHM at the transcript (Fig. 4A) and protein level (Fig. 4B). Confocal microscopy showed that the TCF4-induced IgM protein accumulated on the cell surface as well as in intracellular compartments (Fig. S7). In comparison, BCL2 expression was not induced by TCF4 over-expression and MYC induction was restricted to the two cell lines (SUDHL2 and HBL1; Fig. S8). To assess the consequence of increased IgM expression on BCR signaling we performed BCR crosslinking with anti-IgM Fab, with or without tetracycline-inducible TCF4 expression, and assessed the activation of the proximal BCR signaling kinases BTK and BLNK. In the absence of BCR crosslinking, TCF4-induced IgM expression had no detectable effect on BTK or BLNK phosphorylation. However, following BCR crosslinking the increase of BTK and BLNK phosphorylation could be significantly potentiated by TCF4 over-expression (P<0.05; Fig. 4C and D). These data therefore show that TCF4-induced IgM expression can promote BCR signaling in ABC-like DLBCL in the presence of BCR stimulation.
Figure 4: Induced expression of TCF4 in ABC-like DLBCL cell lines drives MYC and IgM expression and potentiates BCR signaling.

(A) Tetracycline-induced expression of TCF4 in ABC-like DLBCL cell lines with low TCF4 copy number resulted in a significant increase in IGHM transcript compared to control cells. (B) Tetracycline-induced expression of TCF4 led to a marked increase in IgM protein in ABC-like DLBCL cell lines with low TCF4 copy number. An increase in MYC was also observed in SUDHL2 and HBL1 cell line, but was not significant in TMD8. No change was observed for BCL2. The quantification of triplicate experiments is shown in Figure S5. (C) A representative western blot shows the phosphorylation of downstream kinases from IgM, BTK and BLNK, with or without tetracycline-induced TCF4 and/or BCR stimulation with an αIgM cross-linking antibody. TCF4-induced up-regulation of IgM alone did not significantly increase BTK or BLNK phosphorylation, but it significantly enhanced phosphorylation following BCR stimulation. (D) The quantification of western blots from triplicate experiments combining TCF4 induction and BCR stimulation, as shown in C, demonstrate the significant increase in BCR-induced signaling through BTK and BLNK when TCF4 is over-expressed in each of 3 ABC-like DLBCL cell lines. Each bar represents the mean +/− S.E.M. of 3 independent experiments. Groups were compared by Student’s T-test. P-value>0.05 is denoted at ns. *p<0.05, **p<0.01.
TCF4 is a dependency in ABC-like DLBCLs with 18q gain.
To further validate functional dependencies upon TCF4 in ABC-like DLBCL, we generated two unique TCF4 dominant-negative (dn) constructs, which encode a mutant protein that is capable of sequestering endogenous wild-type TCF4 into DNA-binding incompetent heterodimers. The TCF4ΔBR mutant has a deletion of the basic region (amino acids 567–581) prior to the helix-loop-helix domain, which eliminates DNA binding. The TCF4R582P mutation within the helix-loop-helix domain was previously identified in Pitt-Hopkins Syndrome (22) and shown to completely abrogate the binding of TCF4 to DNA (Fig 5A). These TCF4dn proteins were expressed in DLBCL cell lines using tetracycline-inducible expression construct and used in a competition assay with parental cells. Therein, the expression of either TCF4ΔBR or TCF4R582P was found to cause a time-dependent relative depletion of ABC-like DLBCL cell lines with TCF4 copy gain (U2932, RIVA, OCI-Ly10 and TMD8), but did not have a marked effect on the relative cell growth of ABC-like cell lines with diploid TCF4 (HBL1 and SUDHL2) or GCB-like cell lines (SUDHL4, KARPAS-422, OCI-Ly1; Fig 5B). We performed functional analysis of the effect of TCF4dn at the 48h time-point prior to them inducing toxicity. Using ChIP-qPCR, we confirmed that both constructs significantly inhibited binding of TCF4 to the IgM and MYC enhancer elements in the RIVA and U2932 cell lines (P<0.01; Fig. 5C). This in turn led to a significant reduction in the expression of both IgM and MYC at the protein level, independently of a change in BRD4 expression (P<0.05; Fig. 5, D and E). This trend was also observed using shRNA knock-down of TCF4 (Fig. S9). Therefore, IgM and MYC expression is dependent upon TCF4 function in ABC-like DLBCLs with TCF4 gain, and elimination of this activity is associated with a competitive disadvantage to these cells.
Figure 5: Functional dependency upon TCF4 in ABC-like DLBCL.
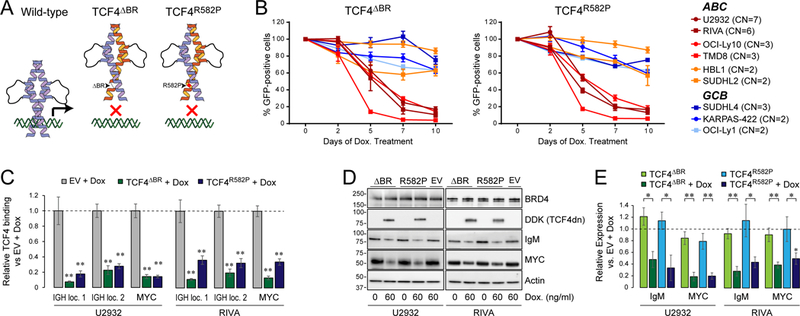
(A) A schematic of the two TCF4 dominant-negative (TCF4dn) constructs. The TCF4ΔBR construct has an in-frame deletion of the basic region prior to the helix-loop-helix domain. The TCF4R582P construct has a single amino acid change within the helix-loop-helix domain. The TCF4dn constructs (orange) are predicted to form heterodimers with wild-type TCF4 (purple) and inhibit DNA binding. (B) A cell competition assay was performed by mixing equal fractions of GFP+ cells possessing the either the TCF4ΔBR or TCF4R582P tetracycline-inducible dominant-negative construct, with the parental cell line. Cells were exposed to doxycycline and the GFP+ fraction measured every 2–3 days for 10 days. The ABC-like DLBCL cell lines with high-level (maroon) or low-level (red) TCF4 DNA copy number gain declined over time with expression of either TCF4dn. This was not observed for ABC-like DLBCL cell lines with diploid TCF4 (orange) or GCB-like DLBCL cell lines selected based upon variable copy number and TCF4 expression. Each point represents the mean +/− S.E.M. of 3 independent experiments. (C) TCF4 ChIP-qPCR was performed for the two immunoglobulin enhancers and the MYC enhancer in the presence or absence of dominant-negative constructs. Both constructs significantly inhibited TCF4 binding at these loci in the U2932 and RIVA cell lines with high TCF4 DNA copy number gain and protein expression. Each bar represented the mean +/− S.E.M. of 3 independent experiments, with statistical significance assessed by Student’s T-test compared to empty vector (EV) control. **p<0.01. (D) A representative western blot shows the expression of the DDK-tagged TCF4dn and the TCF4 target genes, IgM and MYC. Tetracycline-inducible expression of either TCF4dn constructs reduced IgM and MYC expression relative to uninduced cells and doxycycline-treated EV control cells. (E) Quantification of triplicate experiments from D shows that the attenuation of IgM and MYC expression is statistically significant for both of the unique TCF4dn constructs and in both the U2932 and RIVA ABC-like DLBCL cell lines.
TCF4 is critically regulated by BRD4 and can be targeted by BET proteolysis-targeting chimera (PROTAC)
The TCF4 gene is one of the most highly BRD4-loaded genes in DLBCL, including in ABC-like DLBCL cell lines with TCF4 copy gain (Fig. S9). Furthermore, knock-down of BRD4 in U2932 and RIVA markedly reduces TCF4 expression (Fig. S10). We therefore evaluated small molecule BET inhibitors and a BET protein degrader, ARV771, as a potential avenue for reducing TCF4 expression in ABC-like DLBCL cell lines with high-copy number of TCF4. The small molecule BET inhibitors, JQ1 and OTX015, resulted in an up-regulation of BRD4 that was not observed with ARV771 due to its role as a sub-stoichiometric BRD4 degrader (Fig. 6A and Fig. S11). This was associated with a greater efficacy of ARV771 in reducing the BRD4 target genes, MYC and TCF4 (Fig. 6A). In line with prior observations using small molecule inhibitors (23, 24), ARV771 also induced apoptosis of these cell lines (Fig. S12).
Figure 6: BET proteolysis-targeting chimeras (PROTAC) effectively inhibit TCF4.
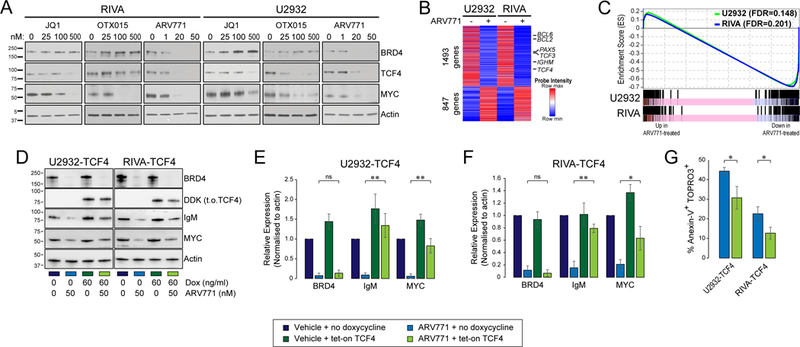
(A) The treatment of ABC-like DLBCL cell lines with high TCF4 DNA copy number using small molecule BET inhibitors, JQ1 and OTX015, leads to an accumulation of BRD4 but can reduce the BRD4-targets TCF4 and MYC. The BET PROTAC, ARV771, effectively degrades BRD4 and leads to a more potent reduction of TCF4 and MYC at 10-fold lower doses than the small molecule inhibitors. (B) The treatment of U2932 and RIVA cell lines with 50nM of ARV771 for 24h led to broad changes in transcript levels. This included the down-regulation of known BRD4 target genes, BCL6 and PAX5, as well as the down-regulation of TCF4 and its target gene, IGHM. (C) Gene set enrichment analyses are overlaid for U2932 (green) and RIVA (blue) for the set of genes that were more highly expressed in primary ABC-like tumors with TCF4 DNA copy number gain compared to those tumors without DNA copy number gain, as shown in Figure 3A. Treatment with ARV771 led to a significant and coordinate down-regulation of this TCF4-associated signature in both the U2932 and RIVA cell lines. (D) The TCF4 target genes IgM and MYC are reduced by ARV771 treatment, but can be partially rescued by enforced expression of DDK-tagged tetratcyline-inducible TCF4 (t.o.TCF4). As expected, the expression of BRD4 was not rescued. (E-F) Quantification of triplicate experiments for U2932 (E) and RIVA (F) shows that IgM and MYC expression are significantly rescued by enforced expression of TCF4. Each bar represents the mean +/− S.E.M. of 3 independent experiments, compared with a Student’s T-test. *p<0.05, **p<0.01. (G) The rescue of TCF4 expression ameliorated, but did not eliminate, the apoptosis induced by ARV771 treatment, as shown by the percentage of Annexin-V+ TOPRO-3+ cells with or without TCF4 rescue by doxycycline-induced expression. Bars represented the mean +/− S.E.M., compared using a Student’s T-test. *p<0.05.
Reductions of TCF4 with ARV771 treatment were accompanied by reduced expression of the TCF4 target genes. This included significant reductions of IgM at the transcript (Q-value<0.1; Fig. 6B) and protein level (P<0.01; Fig. 6, D to F and Table S9) and a coordinate and significant down-regulation of the set of genes that were identified as being increased in association with TCF4 DNA copy number gain in primary tumors (U2932, FDR=0.148; RIVA, FDR=0.201; Fig. 6C). This pattern was not observed in GCB-like DLBCL cell lines (Fig. S13). The MYC gene is a direct target of BRD4 (25), and immunoglobulin genes are also regulated by OCA-B which is reduced by BET inhibition (26). We therefore aimed to determine whether the reduction of MYC and IgM expression, and the induction of apoptosis in by ARV-771 in the cell lines with high DNA copy number of TCF4 were directly related to reduction of TCF4 expression. We investigated this with a rescue experiment, in which TCF4 expression was maintained in RIVA and U2932 cells during ARV771 treatment using the tetracycline-inducible expression construct. The rescue of TCF4 expression significantly reduced the percentage of apoptotic cells following ARV771 treatment in both cell lines (P<0.05), but did not eliminate the cytotoxicity of the compound (Fig. 6G). In addition, enforced TCF4 expression during ARV771 treatment significantly rescued the expression of IgM and MYC (Fig. 6, D to F). Together, these data show that reduction of TCF4 is one of the mechanisms by which BET inhibition reduces IgM and MYC expression and induces apoptosis in ABC-like DLBCL cells with TCF4 DNA copy number gain.
The promising in vitro activity of ARV771 led us to test whether this compound would be efficacious in vivo. In xenografts of the U2932 (Fig. 7, A to E) and RIVA (Fig. 7, F to K) cell lines that express high levels of TCF4, we observed that ARV771 was able to significantly reduce tumor growth. At the end of treatment, tumors were significantly smaller in ARV771-treated mice (P<0.05; Fig 7, B and G) and showed reduced levels of BRD4, TCF4, IgM and MYC expression (Fig. 7, C and H), which demonstrates that the molecule could reach the tumor site at a sufficient concentration to have a functional effect. There were not any signs of toxicity within these mice, and treatment with ARV-771 was associated with a significant prolongation of their survival (P<0.05). Our data provide a clear functional rationale for BET inhibition in ABC-like DLBCL and show that ARV771 is effective at eliminating TCF4 and its target genes and treating ABC-like cell lines DLBCL in vitro and in vivo.
Figure 7: In vivo activity of ARV771 in ABC-like DLBCL.
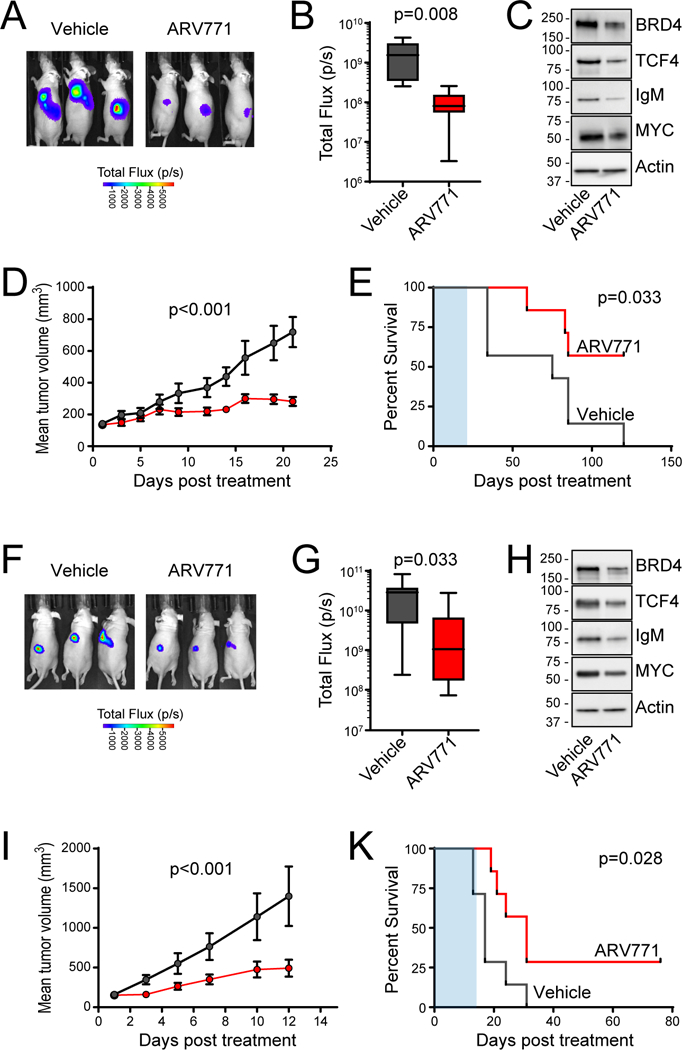
(A-E) Murine xenografts of the U2932 cell line were allowed to become established and then treated with 30mg/kg of ARV771 daily x 5 days per week for 3 weeks. At the end of treatment the luminescence was significantly lower in ARV771-treated mice compared to vehicle control (A, B) and a representative tumor shows on-target reduction of BRD4, TCF4, IgM and MYC expression (C). Treatment significantly inhibited tumor growth (D) and led to significantly prolonged survival in ARV771-treated mice (E) despite the short duration of treatment (E, shaded blue). (F-K) Murine xenografts of the RIVA cell line were allowed to become established and then treated with 30mg/kg of ARV771 daily x 5 days per week for 2 weeks. At the end of treatment the luminescence was significantly lower in ARV771-treated mice compared to vehicle control (F, G) and a representative tumor shows on-target reduction of BRD4, TCF4, IgM and MYC expression (H). Treatment significantly inhibited tumor growth (I) and led to significantly prolonged survival in ARV771-treated mice (K) despite the short duration of treatment (K, shaded blue).
DISCUSSION
The ABC-like subtype is one of two major molecular subtypes of DLBCL that is recognized by the WHO classification (27). These tumors are driven by chronic active BCR signaling that emanates from autoreactive IgM that is localized to the cell surface and intracellular lysosomes (8, 28–30). Mutations in CD79B and MYD88 deregulate this signaling through the reduction of LYN-mediated negative feedback and by activation of the IRAK signaling, respectively (8, 10). However, recent murine studies have shown that MYD88 mutation alone drove a phenotype that was reminiscent of peripheral tolerance, and this was only relieved by the combination of MYD88 and CD79B mutations together, or by increased expression of surface IgM (31). The ABC-like phenotype is therefore the result of cumulative epistatic genetic alterations, rather than a single dominant driver mutation. In further support of this notion, recent genomic studies have defined co-associated sets of genetic alterations that co-segregate with unique genetic subsets of ABC-like DLBCL (32). The “cluster 5” subset of ABC-like DLBCL included frequent MYD88 and CD79B mutations, but the most frequent genetic alteration in this subtype was DNA copy number gain of 18q (32).
We identified the TCF4 (aka E2–2) gene as the most significant target of 18q DNA copy number gains in DLBCL. The TCF4 gene is closely related to TCF3 (aka E2A), with both encoding basic helix-loop-helix (bHLH) transcription factors that form dimers and recognize E-box consensus sequences (CANNTG) (33). Murine conditional knock-out studies showed that Tcf3 and Tcf4 are critical regulators of germinal center B-cell and plasma cell development, in part due to their role in activating immunoglobulin heavy- and light-chain enhancer elements (34, 35). The ID2 and ID3 proteins bind to and inhibit the activity of TCF3 and TCF4 by forming heterodimers that are incapable of binding DNA (33). The TCF3 and ID3 genes are recurrently mutated in another form of B-cell lymphoma, Burkitt lymphoma (BL), with the mutations residing in the interface between TCF3 and ID3 and preventing their interaction (18, 19). We observed that TCF4 DNA copy number gains are also frequent in BL, and that they mutually exclude TCF3 and ID3 mutations, providing further evidence for the importance of TCF3/TCF4 deregulation in this disease. In contrast to BL, TCF3 and ID3 mutations are rare in ABC-like DLBCL, but TCF4 DNA copy number gains are present at more than twice the frequency. The higher mutation rate for ID3 and TCF3 in BL may be due to a higher expression of these genes in BL and an associated greater reliance on TCF3 activity for survival. However, as TCF4 is also variably expressed in these tumors, and ID3 mutations may presumably also reduce the interaction between ID3 and TCF4, the propensity to mutate ID3 at a much higher frequency in BL compared TCF3 may be associated with the ability of these mutations to deregulate both TCF3 and TCF4 activity. We speculate based upon their different expression patterns during B-cell development and phenotypes from single-gene conditional knock-out studies (34, 35) that TCF3 and TCF4 may have some non-redundant roles, with TCF3 being more important in germinal center B-cells and TCF4 playing a more prominent role in post-germinal center B-cells. This may explain the propensity for TCF4 deregulation in ABC-like DLBCL, which align with activated B-cells that are primed for germinal center exit.
In line with the murine studies, we observed a marked up-regulation of IgM transcript expression in primary tumors with TCF4 DNA copy number gain. We identified binding sites for TCF4 in the immunoglobulin heavy chain locus and showed that experimental modulation of TCF4 abundance or activity was sufficient to control IgM expression in ABC-like DLBCL cell lines. Together, this provides strong evidence for a functional role of TCF4 in promoting IgM expression in ABC-like DLBCL. This is particularly important in this disease, because >90% of ABC-like DLBCL cases express IgM and the disease etiology centers on pathogenic signaling downstream of this receptor (7, 8). Notably, TCF4 was more highly expressed in ABC-like DLBCL compared to GCB-like DLBCL generally, even in cases without DNA copy number gain of the locus. This suggests that this axis may be active in all ABC-like DLBCLs, and further enhanced in the ~40% that harbor 18q DNA copy number gains. This is akin to the role of EZH2 in GCB-like DLBCL, which promotes the survival and proliferation of all germinal center B-cells but has enhanced activity in the context of activating somatic mutations (36, 37). We therefore hypothesize that TCF4 may participate in a critical functional axis of immunoglobulin regulation in all ABC-like DLBCL.
Proteins in the BET family, including BRD4, are attractive therapeutic targets in cancer due to their role in the transcriptional activation of oncogenes such as MYC (25, 38). In DLBCL, BRD4 targets include key transcription factors such as BCL6, PAX5 and IRF4 (26). Furthermore, prior studies have shown that BET inhibition is preferentially cytotoxic to ABC-like compared to GCB-like DLBCL cell lines (23, 24) and reduces NFĸB signaling (23). However, the mechanism underlying these observations has not been defined. We have highlighted TCF4 as another prominent target of BRD4 in DLBCL, as has been previously described in plasmacytoid dendritic cell neoplasms (21). Due to the difficulty in directly drugging transcription factors, BET inhibition therefore represents a logical avenue for reducing TCF4 expression in ABC-like DLBCL. However, small molecule inhibitors have also been shown to result in the up-regulation of BRD4 expression (39). We therefore evaluated a novel BET protein PROTAC, ARV771, which combines a BET-targeting warhead from OTX015 with a moiety that recruits the VHL ubiquitin ligase (39). Because the PROTAC is not degraded, this results in the sub-stoichiometric proteolysis of BET proteins, including BRD4. As such, we found that ARV771 was able to inhibit the expression of TCF4 at 10-fold lower concentrations than small molecule BET inhibitors and showed in vivo efficacy against ABC-like cell lines with high TCF4 copy gain. The inhibition of TCF4 expression in these cell lines was accompanied by the coordinate down-regulation of genes that were highly expressed in primary tumors with TCF4 DNA copy gain, such as IgM and MYC. Moreover, the expression of these genes could be rescued by enforced expression of TCF4 during BET inhibition. This was a surprising observation for the MYC gene, which is also a direct BRD4 target (25). Thus, although the effects of BET inhibition are multi-faceted, our data show that a portion of the broad transcriptional changes and toxicity mediated by BET inhibition in ABC-like DLBCLs with 18q DNA copy number gains can be attributed to the downstream effects of reduced TCF4 expression. Together, our data therefore highlight TCF4 DNA copy gains as a functional rationale for BET inhibition in ABC-like DLBCL and show that the BET PROTAC ARV771 has significant activity in this context. However, the over-expression of BCL2 has been described as a resistance mechanism for BET inhibitors (40) and we observed that the majority of 18q DNA copy number gains in DLBCL encompass both the TCF4 and the BCL2 gene. We therefore posit that the promising activity of BET inhibitors in ABC-like DLBCL may be further enhanced by combination with a BCL2 inhibitor such as Venetoclax. In support of this, BET inhibitors have been shown to act synergistically with Venetoclax in myeloid leukemia (40) and in another form of B-cell lymphoma, mantle cell lymphoma (41). Combination of BET and BCL2 inhibition therefore represents an attractive therapeutic avenue for future investigation in ABC-like DLBCL.
There are some caveats to this study that should be addressed in future. We showed TCF4 expression at the transcript level in primary tumor samples, but were unable to perform immunohistochemical staining for TCF4 on these tumors due to the poor performance of available monoclonal antibodies. Prospective validation of TCF4 protein expression and its association with clinical outcome should therefore be performed in a large series of primary DLBCL tumor samples. In addition, we showed a clear functional rationale for targeting TCF4 using BET inhibitors and showed that this was efficacious in in vitro and using cell line xenografts. As xenografts do not predict clinical outcomes, the association between TCF4 DNA copy number status or COO subtype and response to BET inhibitors would be best evaluated as a correlate in future clinical trials. Due to the potential toxicities associated with BET inhibition, it would also be desirable to identify a more direct avenue for specifically targeting TCF4.
In conclusions, we have identified DNA copy number gains of TCF4 as the most frequent genetic alteration in ABC-like DLBCL, and highlighted TCF4 as synthetic dependency in this subset of tumors. Increased expression of TCF4 leads to its occupancy on IgM and MYC enhancer elements and increased expression of these genes at the transcript and protein level. We have shown that BET-targeting PROTACs efficiently reduce the expression of TCF4 and its target genes, induce apoptosis in ABC-like DLBCL cells, and prolong the life of mice bearing ABC-like DLBCL tumors. This study therefore highlights the BRD4-regulated TCF4 and IgM axis as a novel oncogenic pathway and a functional rational for the use of BET inhibitors in ABC-like DLBCL.
MATERIALS AND METHODS
Study Design
The objective of this work is to define the genetic etiology of the ABC-like subtype of DLBCL. We used integrative genomic analysis of a 1,000 primary DLBCL tumors, in vitro analysis of TCF4 function using tetracycline-inducible expression of TCF4 or TCF4 dominant-negative constructs in ABC-like DLBCL cell lines, and in vitro and in vivo testing of BET inhibitors as a therapeutic avenue to directly target TCF4 expression.
DNA copy number data acquisition and processing
Publicly available data for single nucleotide polymorphism microarrays and array comparative genome hybridization platforms with >200,000 markers were downloaded from the gene expression omnibus (15, 17, 42–48) (table S1; www.ncbi.nlm.nih.gov/geo/). Data for all arrays were represented as Log2 copy number change and segmented using the circular binary segmentation (CBS) tool on GenePattern (49). Peaks of significant DNA copy number loss and gain were identified using GISTIC2.0 (14). The thresholds utilized for DNA copy number gain and loss were 0.2 copies over a region encompassing 100 markers. Peaks with a residual Q-value <0.1 were considered significant.
Cell of Origin Subtyping and Integrative Analysis of Gene Expression Profiling Data
Raw cel files for matched Affymetrix U133 Plus 2.0 gene expression microarray data were obtained for 249 previously published DLBCLs (GSE1131816 and GSE3417119) and an additional 98 tumors from UNMC. Data were RMA normalized using the ExpressionFileCreator module of GenePattern (49). The GSE11318 and GSE34171 datasets were batch-corrected and combined using ComBat (50). The additional dataset could not be batch corrected and was analyzed separately only for COO subtype. Cell of origin subtype was determined using a previously described Bayesian classifier (51) and validated using available outcome data (fig. S1 and table S1). For 108 cases, the previously reported Nanostring-derived COO classification was used (44).
The targets of DNA copy number alterations were determined using integrative analysis, as previously described (52). In brief, differential gene expression analysis was performed between tumors (n=249) with or without each lesion, limited to the set of genes within the GISTIC-defined peak (table S6). To identify the signature associated with 18q21 DNA copy number gain, differential gene expression analysis was performed within ABC-like DLBCL cases (n=110). Genes with a fold-change ≥1.2 in the direction of the copy number alteration and a FDR Q-value<0.1 were considered statistically significant. Association between CNAs and COO subtype were determined using a Fisher exact test.
Survival and Cell of Origin Subtype Association
Overall survival data was collated from previous studies for 232 DLBCL patients treated with CHOP combination chemotherapy (15, 17), and 240 DLBCL patients treated with CHOP combination chemotherapy plus the anti-CD20 monoclonal antibody rituximab (R-CHOP) (15, 53) using a log-rank test. Associations between DNA copy number alterations or mutations and COO subtype were determined using a Fishers Exact test, corrected for multiple hypothesis testing using the qvalue R package. The REVEALER tool (16) was implemented in GenePattern, using a set of 98 tumors for which gene expression microarray and targeted NGS data were available (table S1). The seed feature was selected as MYD88 mutation due to its significant association with the ABC-like DLBCL subtype by Fisher analysis (fig. S2). Additional features included all significant DNA copy number alterations (n=24) and recurrently mutated genes (n=63) that were present in 5% or more of the 98 tumors. A total of 10 iterations of feature selection were performed using the ABC probability score as the phenotype and the ABC-like subtype as class.
Targeted next generation sequencing (NGS) and variant calling
Genomic DNA for 140 fresh/frozen DLBCL tumors were obtained from the UNMC lymphoma tissue bank (IRB 161–95-EP) and interrogated by targeted sequencing of a panel of 5.3Mbp hybrid capture panel (Nimblegen) encompassing the full coding regions of 380 genes. Library preparation and data analysis were performed using a previously validated approach (20) that is detailed in the supplementary methods. The average deduplicated sequencing depth was 623X. Variants were called by consensus between VarScan2 and GATK Unified Genotyper, with additional previously described filtering criteria to remove germline variants.
DLBCL Cell Lines
The SU-DHL-2 cell line was obtained from ATCC. The RIVA (aka RI-1), HBL1, TMD8, U2932 and OCI-Ly10 cell lines were obtained from DSMZ. The DNA copy number profile of DLBCL cell lines were derived from previously reported SNP6.0 data (52) or targeted next generation sequencing, as described above. Cell of origin subtype was determined according to previous descriptions (8). U2932, RIVA, TMD8, HBL1, SUDHL2, SUDHL4, SUDHL6, SUDHL8 and KARPAS-422 were maintained in RPMI-1640 media with 10% FBS and 1% penicillin/streptomycin. OCI-LY1, OCI-LY7 and OCI-LY10 were maintained in IMDM supplemented with 20% human serum and 1% penicillin/streptomycin. Cell lines were regularly tested for mycoplasma, and identity confirmation by Short Tandem Repeat at core facility of MD Anderson Cancer Center.
Cloning and Tetracycline Inducible Expression of TCF4
The TCF4 gene was cloned into sleeping beauty vector (pSBtet-GP; Addgene 60495) by replacing luciferase gene with TCF4. Tetracycline based TCF4 expressing DLBCL cell lines (HBL1, TMD8 and SU-DHL-2) were generated by co-transfecting transposase expressing vector (pCMV-CATT7-SB100; Addgene 34879) and sleeping beauty vector expressing TCF4 using neon transfection system according to the manufacturer instructions (Neon transfection system; Invitrogen, CA, USA). Transfected cells were selected for stable cell line generation with puromycin (1μg/ml) for one week and maintained in 10% tetracycline negative FBS (Corning; MT35075CV) containing RPMI media. The dose of tetracycline required for physiologically relevant protein expression of TCF4 was determined by a dose titration relative to the expression level in the U2932 cell line (fig. S3). All experiments were performed with 24h of tetracycline treatment at the doses specified in each experiment.
Western blotting
For immunoblotting, cells were lysed in RIPA buffer supplemented with protease inhibitor cocktail and 1mM PMSF. Equal amounts of proteins were resolved using 4–20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to PVDF membranes and probed for following specific primary antibodies: TCF4 (SAB1404449), IgM (ab200541), BCL2 (2872), MYC (13987), BRD4 (13440), and actin (12620). Western blots quantification was performed using ImageJ software, a Java-based image analysis widely used for measurement of density profiles. The intensity of each band was normalized with the intensity of corresponding loading control and calculated as fold changes with respect to control/no-treated samples. All cells were confirmed as being >80% viable by trypan blue exclusion using a Countess FL (Life Technologies) instrument prior to performing analysis of protein expression by western blot.
Quantitative real-time PCR
Total RNA from cell lines were extracted using AllPrep DNA/RNA mini kit (Qiagen; 80204). For each sample, 1μg of RNA was converted into cDNA using Super Script™III first strand synthesis system (Thermo Fisher; 18080051) and quantitative real time PCR analysis was performed using thermal cycler (Applied Biosystems 7500) and SYBR Green/ROX qPCR Master Mix (Applied Biosystem; 4309155). Analysis was performed with the ΔΔCt method and GAPDH was used for normalization. The list of primers used for qPCR have been provided in table S10.
TCF4 ChIP-seq
TMD8-TCF4 or SU-DHL-2-TCF4 cell lines were treated with doxycycline (60ng/ml) for 24 hours to induce TCF4 expression. Five million of cells were fixed with 1% formaldehyde for 10 min, chromatin sheared by sonication, and ChIP performed with anti-TCF4 (21) or control rabbit IgG bound to dynabeads Protein-G. The protein-bound DNA was purified and libraries constructed, with KAPA HyperPrep kits (Roches) and TruSeq indexes. Sequencing reads were aligned to the human genome (hg19) using BWA-Mem (54), realigned around InDels using GATK (55), sorted and deduplicated using Piccard tools. Peaks were called in TCF4 ChIP samples compared to their input control using EaSeq with global thresholding, and annotated according to the transcription start site of the nearest RefSeq gene and filtered. Significantly over-represented DNA sequence motifs (FDR<0.05) were identified in TCF4 ChIP-seq peaks compared to the reference genome (hg19) using CisFinder (56) with the default settings. Motifs with 75% homology were collapsed to motif clusters. For details, refer to supplementary methods.
BRD4 and TCF4 ChIP-qPCR
For ChIP-PCR, chromatin immuno-precipitation was performed with BRD4 antibody (Bethyl, Cat No. A301–985A) or TCF4 antibody (Ceribelli et al) or control rabbit IgG following the protocol as described above for TCF4. Chromatin DNA was also purified from the input samples. The purified DNA was used to perform quantitative PCR using SYBR Green/ROX qPCR Master Mix (Applied Biosystem; 4309155). Percentage of input was quantified from the adjusted input Ct values and further used to determine ΔCt values for BRD4 or TCF4 or IgG ChIP. List of primers used ChIP-PCR have been listed in table S10.
BET Inhibitors and Treatments
BET inhibitors JQ1 and OTX015 were obtained from Selleck Chemicals. BET-PROTAC (ARV771) was provided by Arvinas, Inc. (New Haven, CT). U2932 and RIVA cell lines were treated with indicated concentrations of BET-inhibitors (JQ1, OTX015) or BRD4-PROTAC (ARV771) for 24 hours before immunoblotting. Apoptosis was interrogated by Annexin V (Thermo Fisher; A35122)/To-PRO-3 staining and analyzed using flow cytometry (BD LSRFortessa) and FlowJo software. For gene expression analysis, cell lines (U2932 and RIVA) were un-treated or treated with ARV771 (50ng/ml) for 24 hours, RNA-sequencing libraries constructed using KAPA RNA HyperPrep kit with RiboErase (KK8560), and sequenced on a HiSeq 4000 instrument at the MD Anderson Sequencing and Microarray Core Facility. Fastq files were aligned to the GRCh37 assembly using STAR 2.6.0c, pre-processed with RSEM, quantifyied with rsem-calculate-expression. DESeq2 (v1.18.1) was used to identify differentially expressed genes using a two-variable (Cell line and Treatment) analysis with default settings. Gene set enrichment analysis (57) was performed using GenePattern and a list of all genes from RNA-seq ranked by the fold-change in expression following ARV771 treatment. The gene set consisted of all genes that showed significantly higher expression in ABC-like DLBCL tumors with TCF4 DNA copy number gain compared to those without, as shown in Figure 3.
Murine Xenograft Experiments
All animal studies were performed under a protocol approved by the IACUC at M.D. Anderson Cancer Center, an AAALAC-accredited institution. Five million luciferase-expressing RIVA or U2932 cells (mixed with Matrigel at a volume ratio of 1:1) were s.c. injected in the left flank of male athymic nude mice (nu/nu) (n = 8 per group). Mice were treated with vehicle (10% [1:1 solutol: ethanol] and 90% D5-water, s.c. daily x 5 days per week) or ARV-771 (30 mg/kg, s.c., daily x 5 per week). The RIVA mouse model was treated for two weeks. Due to slower tumor growth, the U2932 mouse model was treated for three weeks. Tumor were monitored by bioluminescent imaging and mice bearing tumors greater than 1500 mm3 were removed from study and humanely euthanized. Tumor size was compared among cohorts by unpaired t-test. The survival of the mice is represented by a Kaplan Meier plot. Differences in survival were calculated by a Mantel-Cox log-rank test. For additional details, refer to supplementary methods.
Statistical Analysis
For the genomic studies we present the significance of tests as a false discovery rate Q-value following correction for multiple hypothesis testing using a Benjamini-Hochberg correction. In addition, the means and standard deviations for each group from differential gene expression profiling analyses are presented in supplementary tables. Categorical variables were tested using a Fisher exact test and continuous variables were tested with a Student’s T-test, each with multiple hypothesis testing correction when appropriate.
Supplementary Material
Figure S1: Validation of cell of origin subtyping
Figure S2: Comparison of outcome in CHOP-treated DLBCL patients stratified by cell-of-origin subtype and 18q21 gain
Figure S3: Recurrently mutated genes enriched in the ABC-like DLBCL subtype
Figure S4: Expression patterns of TCF3 and TCF4 in normal and malignant B-cells
Figure S5: Tetracycline induction of TCF4 expression
Figure S6: ChIP-seq peaks for TCF4
Figure S7: Cellular localization of TCF4-induced IgM
Figure S8: Quantification of TCF4-induced MYC and BCL2 protein expression.
Figure S9: shRNA-mediated knock-down of TCF4
Figure S10: BRD4 regulates TCF4 in DLBCL cell lines
Figure S11: Western blot quantification of triplicate BET inhibitor experiments
Figure S12: ARV771 induces apoptosis in RIVA and U2932 cell lines.
Figure S13: Effect of ARV-771 on IgM expression in GCB-like DLBCL cell lines.
Table S1: Genomic and clinical data from DLBCL tumors included in this study
Table S2: NGS statistics
Table S3: Genes in GISTIC Peaks
Table S4: COO association of GISTIC peaks
Table S5: COO association of recurrently mutated genes
Table S6: Intregrative analysis of DNA copy number alterations
Table S7: Differential gene expression analysis of ABC-like tumors with or without TCF4 copy gain
Table S8: ChIP-seq peaks for TCF4 signature genes
Table S9: Differentially expressed genes following ARV-771 treatment
Table S10: Primer sequences
ACKNOWLEDGEMENTS
Arvinas Inc. provided ARV771 for the studies. pSBtet-GP was a gift from E. Kowarz. pCMV(CAT)T7-SB100 was a gift from Z. Izsvak. pHIV-Luc-ZsGreen was a gift from B. Welm. Funding: This research was supported by the Nebraska Department of Health and Human Services (LB506 2016–16 to M.R.G.), the Schweitzer Family Fund (to J.W., R.E.D., and M.R.G.), RO1 CA210250 (to K.B.), R01 CA201380 (to M.R.G.), the Fred & Pamela Buffet Cancer Center Support Grant (P30 CA036727), and the MD Anderson Cancer Center NCI CORE Grant (P30 CA016672).
Footnotes
COMPETING INTERESTS
The authors have no competing interests to declare.
AVAILABILITY OF DATA
The data produced in this study are available in the gene-expression omnibus (www.ncbi.nlm.nih.gov/geo/), accession number GSE119241. The SNP and gene expression microarray accessions for the previously published data are listed in table S1. Raw next generation sequencing data will be provided upon reasonable request to the corresponding author.
SUPPLEMENTARY MATERIALS
Supplementary Materials and Methods
REFERENCES
- 1.Coiffier B et al. , CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. The New England journal of medicine 346, 235–242 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Armitage JO, How I treat patients with diffuse large B-cell lymphoma. Blood 110, 29–36 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Crump M et al. , Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 130, 1800–1808 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neelapu SS et al. , Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. The New England journal of medicine 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alizadeh AA et al. , Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Lenz G et al. , Stromal gene signatures in large-B-cell lymphomas. The New England journal of medicine 359, 2313–2323 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruminy P et al. , The isotype of the BCR as a surrogate for the GCB and ABC molecular subtypes in diffuse large B-cell lymphoma. Leukemia 25, 681–688 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Davis RE et al. , Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenz G et al. , Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Ngo VN et al. , Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morin RD et al. , Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476, 298–303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roschewski M, Staudt LM, Wilson WH, Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nature reviews. Clinical oncology 11, 12–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peifer M et al. , Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature genetics 44, 1104–1110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mermel CH et al. , GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology 12, R41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monti S et al. , Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer cell 22, 359–372 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim JW et al. , Characterizing genomic alterations in cancer by complementary functional associations. Nature biotechnology 34, 539–546 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lenz G et al. , Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proceedings of the National Academy of Sciences of the United States of America 105, 13520–13525 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmitz R et al. , Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490, 116–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richter J et al. , Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nature genetics 44, 1316–1320 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Bouska A et al. , Adult High Grade B-cell Lymphoma with Burkitt Lymphoma Signature: Genomic features and Potential Therapeutic Targets. Blood, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ceribelli M et al. , A Druggable TCF4- and BRD4-Dependent Transcriptional Network Sustains Malignancy in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancer cell 30, 764–778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sepp M, Pruunsild P, Timmusk T, Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet 21, 2873–2888 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Ceribelli M et al. , Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proceedings of the National Academy of Sciences of the United States of America 111, 11365–11370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boi M et al. , The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clinical cancer research : an official journal of the American Association for Cancer Research 21, 1628–1638 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Delmore JE et al. , BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chapuy B et al. , Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer cell 24, 777–790 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swerdlow SH et al. , The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127, 2375–2390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Havranek O et al. , Tonic B-cell receptor signaling in diffuse large B-cell lymphoma. Blood 130, 995–1006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young RM et al. , Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proceedings of the National Academy of Sciences of the United States of America 112, 13447–13454 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phelan JD et al. , A multiprotein supercomplex controlling oncogenic signalling in lymphoma. Nature 560, 387–391 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang JQ et al. , Synergistic cooperation and crosstalk between MYD88L265P and mutations that dysregulate CD79B and surface IgM. The Journal of experimental medicine 214, 2759–2776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chapuy B et al. , Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nature medicine, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Engel I, Murre C, The function of E- and Id proteins in lymphocyte development. Nature reviews. Immunology 1, 193–199 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Wohner M et al. , Molecular functions of the transcription factors E2A and E2–2 in controlling germinal center B cell and plasma cell development. The Journal of experimental medicine 213, 1201–1221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gloury R et al. , Dynamic changes in Id3 and E-protein activity orchestrate germinal center and plasma cell development. The Journal of experimental medicine 213, 1095–1111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beguelin W et al. , EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer cell 23, 677–692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Green MR, Chromatin modifying gene mutations in follicular lymphoma. Blood 131, 595–604 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stathis A, Bertoni F, BET Proteins as Targets for Anticancer Treatment. Cancer discovery 8, 24–36 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Lu J et al. , Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & biology 22, 755–763 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bui MH et al. , Preclinical characterization of BET family bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer research, (2017). [DOI] [PubMed] [Google Scholar]
- 41.Sun B et al. , BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia, (2017). [DOI] [PubMed] [Google Scholar]
- 42.Pasqualucci L et al. , Analysis of the coding genome of diffuse large B-cell lymphoma. Nature genetics 43, 830–837 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chigrinova E et al. , Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood 122, 2673–2682 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Karube K et al. , Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets. Leukemia 32, 675–684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato M et al. , Frequent inactivation of A20 in B-cell lymphomas. Nature 459, 712–716 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Compagno M et al. , Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 459, 717–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Green MR et al. , Integrative genomic profiling reveals conserved genetic mechanisms for tumorigenesis in common entities of non-Hodgkin’s lymphoma. Genes, chromosomes & cancer 50, 313–326 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Bodker JS et al. , Performance comparison of Affymetrix SNP6.0 and cytogenetic 2.7M whole-genome microarrays in complex cancer samples. Cytogenet Genome Res 139, 80–87 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Reich M et al. , GenePattern 2.0. Nature genetics 38, 500–501 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Johnson WE, Li C, Rabinovic A, Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Wright G et al. , A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America 100, 9991–9996 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Green MR et al. , Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116, 3268–3277 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scandurra M et al. , Genomic lesions associated with a different clinical outcome in diffuse large B-Cell lymphoma treated with R-CHOP-21. British journal of haematology 151, 221–231 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Li H, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 1303.3997, (2013). [Google Scholar]
- 55.McKenna A et al. , The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharov AA, Dudekula DB, Ko MS, CisView: a browser and database of cis-regulatory modules predicted in the mouse genome. DNA research : an international journal for rapid publication of reports on genes and genomes 13, 123–134 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Subramanian A et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Validation of cell of origin subtyping
Figure S2: Comparison of outcome in CHOP-treated DLBCL patients stratified by cell-of-origin subtype and 18q21 gain
Figure S3: Recurrently mutated genes enriched in the ABC-like DLBCL subtype
Figure S4: Expression patterns of TCF3 and TCF4 in normal and malignant B-cells
Figure S5: Tetracycline induction of TCF4 expression
Figure S6: ChIP-seq peaks for TCF4
Figure S7: Cellular localization of TCF4-induced IgM
Figure S8: Quantification of TCF4-induced MYC and BCL2 protein expression.
Figure S9: shRNA-mediated knock-down of TCF4
Figure S10: BRD4 regulates TCF4 in DLBCL cell lines
Figure S11: Western blot quantification of triplicate BET inhibitor experiments
Figure S12: ARV771 induces apoptosis in RIVA and U2932 cell lines.
Figure S13: Effect of ARV-771 on IgM expression in GCB-like DLBCL cell lines.
Table S1: Genomic and clinical data from DLBCL tumors included in this study
Table S2: NGS statistics
Table S3: Genes in GISTIC Peaks
Table S4: COO association of GISTIC peaks
Table S5: COO association of recurrently mutated genes
Table S6: Intregrative analysis of DNA copy number alterations
Table S7: Differential gene expression analysis of ABC-like tumors with or without TCF4 copy gain
Table S8: ChIP-seq peaks for TCF4 signature genes
Table S9: Differentially expressed genes following ARV-771 treatment
Table S10: Primer sequences