

Abstract
The synthesis, absolute stereochemical configuration, complete biological characterization, mechanism of action and resistance, and pharmacokinetic properties of (S)-(−)-acidomycin are described. Acidomycin possesses promising antitubercular activity against a series of contemporary drug susceptible and drug-resistant M. tuberculosis strains (MICs = 0.096–6.2 μM), but is inactive against non-tuberculosis mycobacteria, gram-positive and gram-negative pathogens (MICs > 1000 μM). Complementation studies with biotin biosynthetic pathway intermediates and subsequent biochemical studies confirmed acidomycin inhibits biotin synthesis with a Ki of approximately 1 μM through the competitive inhibition of biotin synthase (BioB) and also stimulates unproductive cleavage of S-adenosylmethionine (SAM) to generate the toxic metabolite 5′-deoxyadenosine. Cell studies demonstrate acidomycin selectively accumulates in M. tuberculosis providing a mechanistic basis for the observed antibacterial activity. The development of spontaneous resistance by M. tuberculosis to acidomycin was difficult and only low-level resistance to acidomycin was observed by overexpression of BioB. Collectively, the results provide a foundation to advance acidomycin and highlight BioB as a promising target.
Keywords: Mycobacterium tuberculosis, tuberculosis, biotin biosynthesis, biotin synthase, antimetabolite, acidomycin, accumulation
Graphical Abstract
Tuberculosis (TB) remains the leading cause of bacterial infectious disease mortality from a single agent, with an estimated 1.6 million deaths worldwide in 2016.1 TB is caused by Mycobacterium tuberculosis which belongs to the greater Mycobacterium tuberculosis complex, a group of closely-related human- and animal-adapted strains: M. africanum, M. bovis, M mungi, M. pinnipedii, M. microti, M. caprae, and M. canettii.2–3 The standard treatment of drug-susceptible TB is challenging, and necessitates a 6–9 month dosing regimen of the four first-line antitubercular agents: isoniazid, rifampicin, ethambutol, and pyrazinamide. The emergence of multidrug-resistant (MDR-TB; resistant to isoniazid and rifampicin) and extensively drug-resistant (XDR-TB; MDR-TB resistant to the fluoroquinolones and at least one of the injectable aminoglycosides) further complicates the management of TB.4 Since the approval of rifampicin in 1971, bedaquiline has been the only new TB antibiotic approved by the FDA, illustrating a lack of novel TB antibiotics developed in the last few decades. Motivated to combat drug-resistant TB, researchers have sought to identify and exploit the inhibition of vulnerable pathways for antitubercular drug development.
The output of antibacterial drugs and drug classes has been remarkably low in the past 25 years, stymied by the numerous challenges associated with antibacterial discovery. Effective drug discovery programs applied two different approaches to the development of novel antibiotics: target-oriented screening, and phenotypic or empirical screening.5 Empirical screening was the predominant method of antibiotic drug discovery during the “Golden Age” of antibiotics, where natural products and microbial metabolites were being unearthed at impressive rates with an estimated tens of millions of soil microorganisms screened.6 These screening methods involved the evaluation of growth inhibition of bacterial organisms for natural products, irrespective of whether the mechanism of action was known. The method was successful for many years and engendered the discovery of many most common antibiotics known today, including the focus of this study, the thiazolidinone antitubercular drug acidomycin.
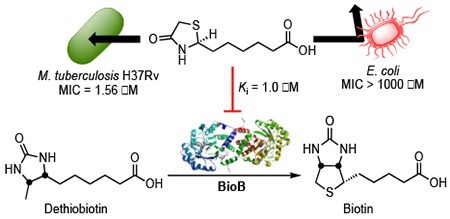
Acidomycin (11, ACM, also known as: actithiazic acid, cinnamonin and mycobacidin) was discovered in 1952 independently by four different research teams,7–10 and was originally isolated from culture filtrates of Streptomyces virginiae,7 Streptomyces acidomyceticus,11 Streptomyces lavendulae,9 and Streptomyces cinnamonensis.12–13 Acidomycin possessed a considerable degree of selective antimicrobial activity against M. tuberculosis in vitro, but was inactive against an M. tuberculosis infection in vivo.7,14 The inhibitory activity of acidomycin depended on the incubation period, amount of inoculum, and concentration of biotin in the medium.7 Concentrations of acidomycin ranging from 0.25 to 10 μg/mL (1.2–46.0 μM) were required to inhibit the growth of mycobacteria in vitro, and conversely acidomycin did not inhibit the growth of certain gram-positive and gram-negative bacteria or fungi at concentrations as high as 1 mg/mL (4.6 mM).7,9 The presence of biotin in growth media as low as 0.064 μg/mL (0.26 μM) was shown to fully antagonize the antibiotic activity of acidomycin and was attributed to the structural similarity between acidomycin and biotin.7,11,13,15–17 Dethiobiotin (DTB, 4) also affected the inhibitory of acidomycin, albeit to a much lesser degree.16 Taken together, these data implicated acidomycin as an inhibitor of the biotin biosynthetic pathway.
The biotin biosynthetic pathway (Figure 1) has been proposed as a promising target for antitubercular drug development. Biotin (vitamin H, vitamin B7, or co-enzyme R) is a required cofactor for the growth of M. tuberculosis, as it is central to cellular metabolism involving the transfer of CO2 during carboxylation, decarboxylation and transcarboxylation reactions. These biotin-dependent enzymes play important roles in many biological pathways such as: membrane lipid biosynthesis, amino acid metabolism, and tricarboxylic acid cycle replenishment.18–22 Biotin biosynthesis involves the conversion of acyl-carrier protein (ACP) conjugate of pimelate (pimeloyl-ACP, 1) to biotin (5, Figure 1) via four highly conserved steps, catalyzed by BioF (7-keto-8-aminopelargonic acid synthase), BioA (7,8-diaminopelargonic acid synthase), BioD (dethiobiotin synthetase), and BioB (biotin synthase). Biotin is then conjugated onto requisite biotin-dependent proteins by BirA (biotin protein ligase). Uniquely, M. tuberculosis relies heavily on this pathway as its genome encodes three putative biotin-dependent acyl-CoA carboxylases (ACCs, 6), which are essential for the synthesis of the extraordinarily complex mycobacterial cell wall, a quintessential hallmark of virulence.23 The biotin biosynthetic pathway has also been shown to be essential for bacterial persistence in murine TB infection models, thus making it a promising target for novel TB therapy.24–25
Figure 1.
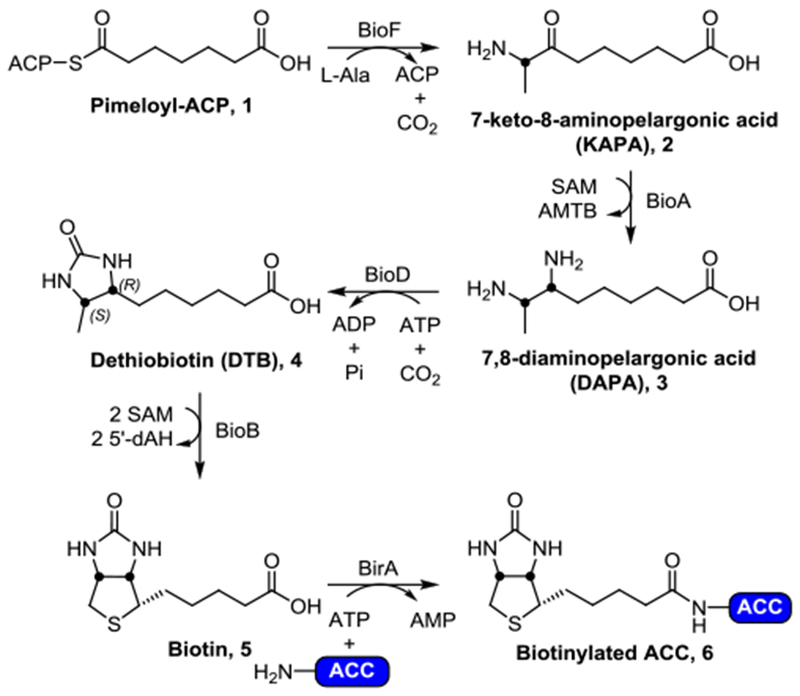
The conserved biotin biosynthetic pathway. Briefly, pimeloyl-ACP (1) is converted to 7-keto-8-aminopelargonic acid (KAPA, 2) by BioF (KAPA synthetase). Transamination by BioA (DAPA synthetase) converts 2 to 7,8-diaminopelargonic acid (DAPA, 3), followed by insertion of a carbonyl by BioD (dethiobiotin synthetase) gives rise to dethiobiotin (DTB, 4). Finally, BioB (biotin synthase) is responsible for the conversion of 4 to biotin (5). The biological fate of biotin (5) is being ligated onto biotin-dependent proteins (such as acyl-CoA carboxylases, ACCs) by BirA (biotin protein ligase, BPL) affording the catalytically active biotinylated holo-ACC (6).
Although innovative screening techniques are important to the development of novel antibiotics, the gap of new drugs discovered compounded with the urgent need for new antibiotics imposed by the looming threat of M(X)DR-TB prompted us to re-investigate acidomycin as a potential candidate for antitubercular drug development. We hypothesize the disinterest in pursuing acidomycin was the lack of in vivo biological activity, unexplored mechanism of action, and the abundance of attractive alternate natural products with potent in vitro and in vivo activity. The goal of this study was to contextualize acidomycin as a potential antitubercular agent in the modern drug discovery landscape as well as to elucidate its mechanism of action. Herein we utilize contemporary synthetic, biochemical, and microbiological techniques to probe the whole-cell activity of acidomycin, to systematically investigate its mechanism of action in terms of both target specificity and selectivity for mycobacteria, and to evaluate its pharmacokinetic properties in vivo.
RESULTS
Synthesis and Stereochemical Determination of Acidomycin.
Acidomycin (ACM, 11) is a structurally simple antibiotic containing a thiazolidinone ring appended to an aliphatic carboxylic acid at the 2-position. The reported syntheses from the groups at Abbott and Pfizer in 1952–1953 involved a low yielding (~40%) condensation of 2-mercaptoacetamide with methyl (9) or ethyl pimelate semialdehyde, the latter of which was prepared in four steps from pimelic acid.14,26–27 We devised an alternate three-step route to 9 from cycloheptanone 7, which was converted to the corresponding eight-membered lactone by Baeyer-Villiger oxidation followed by Fischer transesterification to provide methyl ester 8 (Figure 2A). A mild (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) oxidation afforded methyl pimelate semialdehyde (9) in a 3.5-fold higher overall yield than the route from pimelic acid.26 The key condensation between 2-mercaptoacetamide and methyl pimelate semialdehyde (9) to furnish 4-thiazolidinone (±)-10 was achieved in an improved 79% yield employing freshly prepared 2-mercaptoacetamide.28 Saponification of the methyl ester and recrystallization from water gave racemic (±)-acidomycin [(±)-11], which was readily resolved in gram quantities via preparative chiral HPLC (Chiralpak® IE 5 μm LC column) to afford the natural product (−)-acidomycin and the unnatural antipode (+)-acidomycin in greater than 97.5% optical purities (Figure 2B). Surprisingly, the absolute configuration of (−)-acidomycin had never been established. Thus, the stereochemistry of the levorotatory enantiomer was determined by single crystal X-ray diffraction (XRD) analysis. The low value of the Flack parameter and its uncertainty unambiguously identified the configuration at C-2 of (−)-acidomyin as S (Figure 2C, Table S11).
Figure 2.
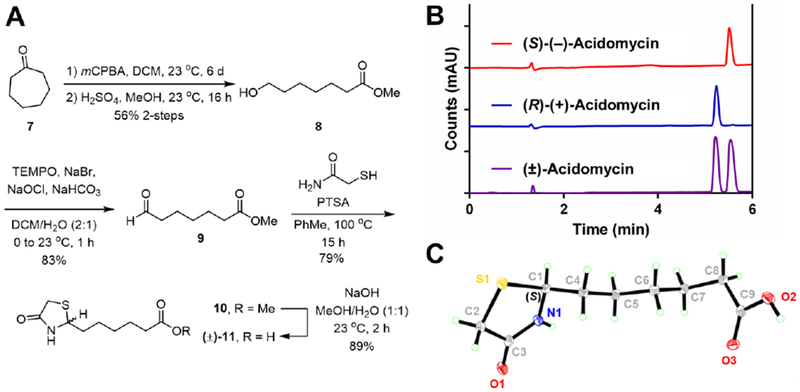
A) Synthetic route to (±)-11 (acidomycin); B) Analytical chiral HPLC trace of (±)-acidomycin (purple), with each resolved enantiomer (S)-(−)-acidomycin (red) and (R)-(+)-acidomycin (blue) using Phenomenex® Cellulose-1 5 μm 250 × 2.0 mm LC column; C) X-ray crystal structure of (−)-acidomycin. See Supporting Information for further details.
Antimicrobial Activity.
Each of the acidomycin enantiomers and racemic acidomycin were initially evaluated against wild type M. tuberculosis H37Rv in biotin-free 7H9 medium to determine the minimum inhibitory concentration (MIC) that resulted in complete growth inhibition. (S)-(−)-Acidomycin had a MIC of 0.6 μM while the unnatural enantiomer (R)-(+)-acidomycin was thirteen-fold less active with an MIC of 7.7 μM and racemic (±)-acidomycin displayed an MIC of 1.6 μM, about 2.5-fold higher than the natural product. (±)-Acidomycin was bacteriostatic and only modestly impacted M. tuberculosis viability (Figure S1). Pre-cultures grown in the presence or absence of biotin did not affect the sensitivity of M. tuberculosis to acidomycin indicating biotin carryover from the initial inoculum was minimal. Further microbiological evaluation against a panel of eight drug sensitive M. tuberculosis clinical isolates, hypervirulent M. tuberculosis HN878, M. tuberculosis Erdman as well as a panel of 15 phenotypically characterized MDR and XDR M. tuberculosis strains demonstrated (±)-acidomycin maintained excellent activity with MICs ranging from 0.096 μM to 6.2 μM for these 25 contemporary M. tuberculosis strains (Table 1, see Table S1 for more details). (±)-Acidomycin was active against other members of the M. tuberculosis complex including M. africanum and M. bovis BCG with MICs of 0.2 and 1.3 μM, respectively. Given the alarming rise of non-tuberculosis mycobacteria (NTM) such as M. abscessus and members of the M. avium complex, which together account for greater than 90% of the total NTM pulmonary diseases in immunocompromised individuals, we also evaluated (±)-acidomycin against these NTMs.29 However, all NTM strains examined were intrinsically resistant to acidomycin. The fast-growing non-pathogenic M. smegmatis was slightly less susceptible than M. tuberculosis H37Rv to (±)-acidomycin requiring 10 μM to achieve ≥90% growth inhibition. As initially reported,7,9 we confirmed acidomycin was highly selective for mycobacteria as (±)-acidomycin was inactive toward several representative gram-negative (Acinetobacter baumannii, Escherichia coli, Pseudomonas aeruginosa) and gram-positive pathogens (Staphylococcus aureus, Enterococcus faecalis) cultured in M9 minimal medium. Importantly, (±)-acidomycin showed no cytotoxicity at 1 mM against HepG2 human liver or African green monkey kidney (Vero) cells providing a large therapeutic index (CC50/MIC99) of >600 based on the MIC against M. tuberculosis H37Rv and more than 10,000 against the most sensitive DS and XDR M. tuberculosis strains.
Table 1.
Biological activity and selectivity of (±)-acidomycin.
| Strain/Cell Line (#strains)a | Classificationb | MIC or EC (μM)c |
|---|---|---|
| M. tuberculosis H37Rv | WT | 1.56 |
| M. tuberculosis (8) | DS | 0.096-6.2 |
| M. tuberculosis HN878 | DS | 0.60 |
| M. tuberculosis Erdman | DS | 1.56 |
| M. tuberculosis (7) | MDR | 0.60-6.2 |
| M. tuberculosis (6) | MDR+ | 0.20-4.8 |
| M. tuberculosis (2) | XDR | 0.096-1.2 |
| M. africanum | MTBC | 0.20 |
| M. bovis BCG | MTBC | 1.30 |
| M. abscessus (4) | NTM | >100 |
| M. avium complex (3) | NTM | >100 |
| M. smegmatis mc2155 | non-pathogenic | 10d |
| E. faecalis | gram positive | >1000d |
| S. aureus | gram positive | >1000d |
| A. baumannii | gram negative | >1000d |
| P. aeruginosa | gram negative | >1000d |
| E. coli ATCC 25922 | gram negative | >1000d |
| HepG2 | mammalian | >1000e |
| Vero | mammalian | >1000e |
Bacterial species and strain or mammalian cell line. The number of strains is indicated in parentheses. Further details of the strain including a complete description of the phenotypic resistance is provided in the Supporting Information.
Classification: WT = wild-type; DS = drug-sensitive Mtb clinical isolates; MDR = multidrug-resistant M. tuberculosis; MDR+ = multidrug resistant M. tuberculosis that are additionally resistant to ethambutol, pyrazinamide, kanamycin, streptomycin, ofloxacin, moxifloxacin, and/or levofloxacin (see Supporting Information for details); XDR = extensively drug resistant M. tuberculosis; MTBC = M. tuberculosis complex; NTM = non-tuberculosis mycobacteria.
MIC = minimum inhibitory concentrations that resulted in complete growth inhibition.
MIC90 = minimum inhibitory concentrations that resulted in ≥90% growth inhibition.
EC = effective concentration that resulted in greater than 50% inhibition of cell viability.
All MIC experiments were performed in triplicate for each concentration and repeated independently at least two times. Mammalian cell viability studies were performed in duplicate for each concentration and repeated independently three times.
Mechanism of Action Studies.
Shortly following the discovery of acidomycin, several groups hypothesized it was a biotin antimetabolite based on the structural similarity to biotin and subsequently demonstrated biotin fully antagonized its antitubercular activity.7,11,13,30 Later investigations by Eisenberg and Hsiung using resting cells of an E. coli mutant, which overexpressed the entire biotin pathway, showed acidomycin inhibited the conversion of DTB to biotin; although these results were never extended to M. tuberculosis.31 To pinpoint the biochemical step targeted by acidomycin in M. tuberculosis, we thus performed complementation studies with biotin pathway intermediates to determine if growth inhibition by acidomycin could be rescued by upstream exogenous biotin precursors. The activity of (±)-acidomycin against M. tuberculosis H37Rv was evaluated in glycerol-alanine-salts (GAS) minimal medium supplemented with either 1 μM of 7-keto-8-aminopelargonic acid (KAPA), DTB or biotin (note: 7,8-diaminopelargonic acid [DAPA] did not complement a ΔbioA M. tuberculosis biotin auxotroph presumably due to poor permeability of this highly polar diamino-carboxylic acid metabolite and hence was not used in our studies).32 Biotin completely rescued growth inhibition by (±)-acidomycin while DTB shifted the MIC approximately two-fold and KAPA had no effect (Figure 3A, Table S2). Illustrating the remarkable selectivity, we confirmed supplementation with each of the biotin pathway intermediates had no impact on the antitubercular activity of rifampicin (Figure 3B). These data are consistent with previous studies,31 and suggest (±)-acidomycin inhibits the conversion of DTB to biotin in M. tuberculosis, catalyzed by biotin synthase (BioB). To further corroborate BioB as the primary target of (±)-acidomycin we analyzed an M. tuberculosis strain carrying two copies of the bioB gene, which is expected to increase BioB expression. In accordance with the conclusion that BioB is the primary target of ACM in M. tuberculosis, the merodiploid shows an increase in the MIC of (±)-acidomycin but not rifampicin (Figure 3CD)
Figure 3.
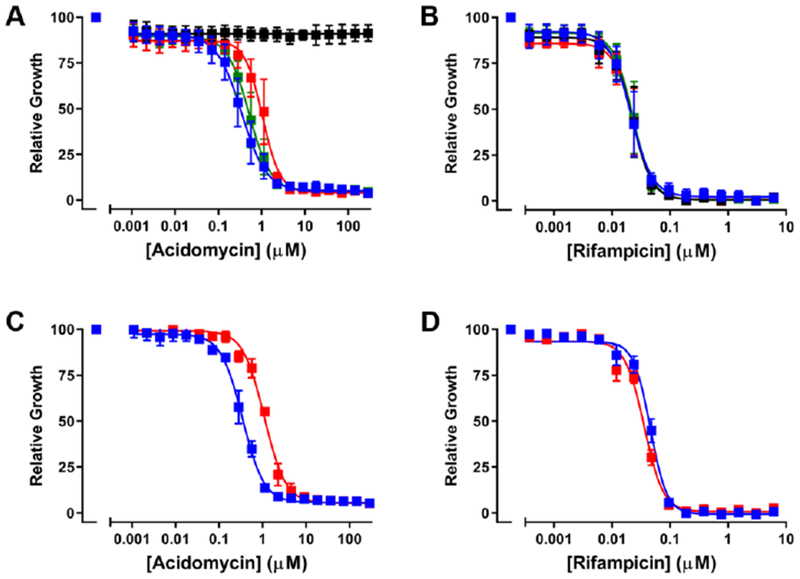
Impact of extrabacterial biotin and overexpression of BioB on susceptibility of M. tuberculosis H37Rv to (±)-acidomycin. Panels A and B show MIC curves for (±)-acidomycin (A) and rifampicin (B) in GAST medium supplemented with 1 μM biotin pathway intermediates (black, biotin; red, DTB; green, KAPA) or DMSO only (blue). Panels C and D show MIC curves for M. tuberculosis H37Rv (blue squares) and the BioB merodiploid strain (red squares) against (±)-acidomycin (C) and rifampicin (D) grown in GAST medium. Normalized growth was calculated at OD580 at the indicated concentration divided by the OD580 with drug (DMSO). Data are averages (± SD) of triplicate cultures and are representative of two independent experiments.
To biochemically validate these findings, we next overexpressed and purified recombinant M. tuberculosis BioB (MtBioB) and attempted to reconstitute activity in an anaerobic chamber (< 5 ppm O2) using E. coli flavodoxin (FLD) and flavodoxin reductase (FLDR) as described.33 Rather than use the initially disclosed high performance liquid chromatography (HPLC)-based assay to monitor MtBioB activity, we developed a more sensitive and specific liquid chromatography-mass spectrometry (LC-MS) assay employing biotin-d4 as an internal standard. However, we never observed product formation under any assay condition examined. As a control, we also overexpressed and purified E. coli BioB (EcBioB) and assayed it under identical conditions (see methods). EcBioB was selected because it has been subject to detailed steady-state and pre-steady-state kinetic analyses as well as structurally characterized with bound substrates SAM and DTB.34–48 Unlike MtBioB, EcBioB displayed time-dependent formation of biotin with kinetic parameters nearly identical to the reported values.37 Both BioB homologues were purified as stable homodimers and exhibited a characteristic brown color from the [2Fe-2S]2+ cluster, thus we hypothesized the inability of MtBioB to turnover substrate was due to failure of the E. coli FLD to promote electron transfer. Unfortunately, the M. tuberculosis genome does not encode for any flavodoxins; consequently, further studies will be required to identify the putative mycobacterial enzyme(s) responsible for single electron transfer to MtBioB. In lieu of a functional assay for MtBioB, we elected to use EcBioB to evaluate inhibition by acidomycin since our homology model of MtBioB (see discussion) indicates both active sites are highly conserved.
EcBioB is marked by slow turnover (kcat ≈ 0.2 min−1) and product inhibition (e.g. 5′-deoxyadenosine and methionine).37 As a result inhibition studies with acidomycin were conducted at a relatively high enzyme concentration (5 μM dimer, Figure 4) under single-turnover conditions at fixed saturating concentrations of DTB (10 μM) and SAM (100 μM).49 Under these conditions (S)-(−)-acidomycin inhibited 50% of EcBioB activity at 13.3 ± 1.8 μM while and (R)-(+)-acidomycin showed less than 30% inhibition at 100 μM. These results are consistent with the observed relative whole-cell antitubercular activities of each enantiomer (Figure 4A). Since BioB is a radical SAM enzyme, we hypothesized acidomycin could potentially act as a mechanism-based inhibitor forming a reactive intermediate that subsequently inactivates the enzyme. To exclude this possibility, we confirmed acidomycin did not exhibit time-dependent inhibition by pre-incubating BioB with all substrates and cofactors for various times (5–30 minutes) followed by initiating the reaction with DTB. To determine the modality of inhibition as well as inhibition constant (Ki) for (±)-acidomycin, initial velocity was measured as a function of DTB concentration (5–30 μM) at several concentrations of (±)-acidomycin ranging from 5 to 30 μM that provided the saturation curves (Figure 4B) and the corresponding double-reciprocal (Figure 4D) which shows (±)-acidomycin is a competitive inhibitor with respect to DTB. While this assay allowed us to operate under initial velocity conditions, the data cannot be interpreted according to Michaelis-Menten kinetics, which assumes substrate concentration is much larger than enzyme concentration ([S] >> [E]), and the concentration of the enzyme-substrate complex does not change over the course of the assay. Instead, we conducted a simultaneous non-linear regression, modeled according to Segel’s treatment of multi-site enzymes, taking into account tight-binding behavior giving a calculated Ki of 2.0 ± 1.3 μM for (±)-acidomycin or approximately 1 μM for the active S-enantiomer of acidomycin (Figure 4C).50
Figure 4.
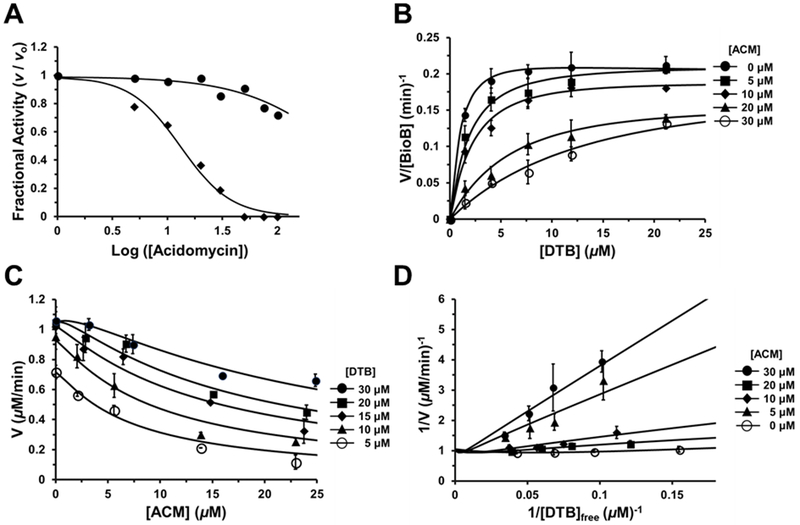
Inhibition of EcBioB by acidomycin. A) Concentration-response plots of the fractional initial velocity of EcBioB as a function of acidomycin concentration. (R)-(+)-acidomycin (solid circles) shows weak inhibition (IC50 of >100 μM) while the calculated IC50 for (S)-(−)-acidomycin (solid diamonds) is 13.3 ± 1.8 μM. B) Saturation curve of initial velocity of EcBioB as a function of DTB concentration at different fixed concentrations of (±)-acidomycin. Each curve was fit to Segel’s model for enzymes with multiple catalytic sites.50 C) Data from panel B replotted and fit to a modified version of the Morrison equation to obtain Ki for (±)-acidomycin (Ki = 2.0 ± 1.3 μM). D) Lineweaver-Burke double reciprocal plot of initial velocity versus free DTB concentration at different concentrations of (±)-acidomycin. The intersection near the y-axis exhibits some curvature because the free ligand and steady-state assumptions are not valid with the implemented assay conditions.
Careful analysis of the reaction byproducts revealed acidomycin significantly enhanced production of 5′-deoxyadenosine (22) by BioB in the absence of DTB compared to the background cleavage rate (Figure S7). We speculate the binding of (S)-(−)-acidomycin to BioB may stimulate unproductive cleavage of SAM to generate 5′-deoxyadenosine, which if continuously produced could accumulate to a sufficient concentration to inhibit BioB and as well as other metabolic processes.
Accumulation Studies.
We hypothesized the selective microbiological activity of acidomycin for mycobacteria, despite potent inhibition of EcBioB, may be caused by differential cellular accumulation. We therefore utilized the detailed method described by Hergenrother employing liquid chromatography tandem mass spectrometry (LC-MS/MS) with slight modification to quantitatively determine antibiotic levels in E. coli and M. tuberculosis.51 Rifampicin, thioridazine and tetracycline were used as control compounds and results are given in Table 2 and Figure 5. The compound accumulation data are expressed in units of nanomoles per 1012 CFU to be consistent with Hergenrother’s data as well as intracellular concentration, which was calculated based on the average volume of cells as detailed in Table 2. The intracellular concentration (IC) divided by the extracellular concentration (EC), which was 50 μM for all compounds, provided the calculated IC/EC ratios. Consistent with previous studies, we observed rifampicin accumulated to low levels in E. coli resulting in an IC/EC ratio of 2.1 while tetracycline displayed high accumulation providing an IC/EC value of 62. The accumulation patterns of tetracycline in M. tuberculosis aligned with the results with E. coli providing an IC/EC ratio of 88. Thioridazine does not accumulate in M. tuberculosis and was thus selected as control for a low accumulator. As expected, the IC/EC value for thioridazine was only 0.7 in M. tuberculosis.52 However, (±)-acidomycin showed a substantial difference reaching an intracellular concentration of 2200 μM in M. tuberculosis and only 80 μM in E. coli resulting in IC/EC ratios of 52 and 1.6, respectively. Thus, (±)-acidomycin accumulated to approximately 30-fold higher levels in M. tuberculosis than in E. coli. We hypothesized that biotin could potentially compete with transport of (±)-acidomycin in M. tuberculosis and thus repeated all studies in the presence of 1 μM biotin; however, exogenous biotin levels had little impact on cellular accumulation of (±)-acidomyin in M. tuberculosis as well as the other antibiotics examined. These data suggest M. tuberculosis is uniquely susceptible to (±)-acidomycin because of selective accumulation that provides an intracellular concentration more than 50-times greater than the extracellular concentration.
Table 2.
Cellular accumulation of antibiotics in E. coli and M. tuberculosis.
| Antibiotic | Organism | Accumulation (nmol/1012 CFUs) |
Intracellular Concentration, −biotin (μM)a | IC/EC Ratiob | ||
|---|---|---|---|---|---|---|
| −biotin | +biotin | |||||
| (±)-Acidomycin | E. coli | 80 ± 20 | 61 ± 2 | 80 ± 20 | 1.6 ± 0.4 | |
| M. tuberculosis | 1300 ± 400 | 1150 ± 40 | 2600 ± 800 | 52 ± 16 | ||
| Tetracycline | E. coli | 3100 ± 120 | 1600 ± 100 | 3100 ± 120 | 62 ± 2 | |
| M. tuberculosis | 2200 ± 200 | 2220 ± 50 | 4400 ± 400 | 88 ± 8 | ||
| Rifampicin | E. coli | 110 ± 60 | 144 ± 4 | 110 ± 60 | 2.14 ± 0.06 | |
| Thioridazine | M. tuberculosis | 18 ± 1 | 5.2 ± 0.4 | 36 ± 2 | 0.72 ± 0.04 | |
Figure 5.
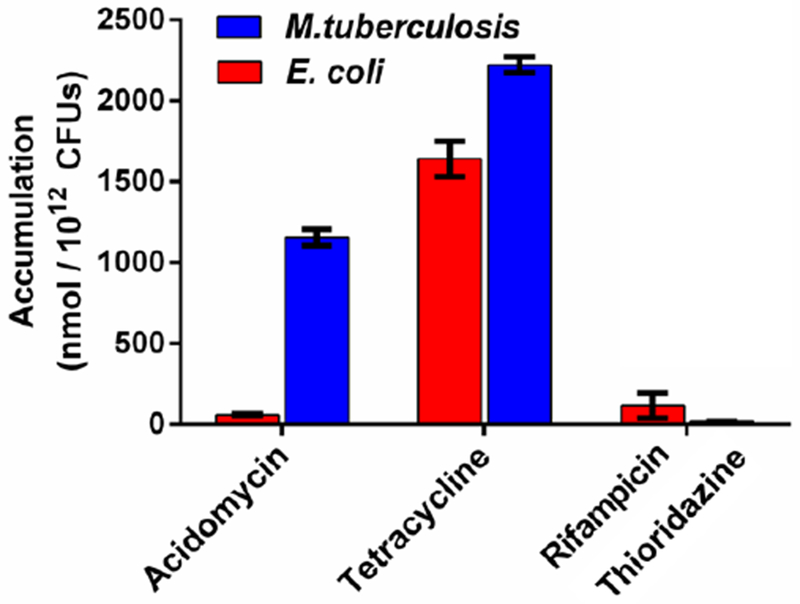
Differential accumulation in M. tuberculosis (blue bars) vs. E. coli (red bars) grown in the absence of biotin. Accumulation levels (represented in nmol / 1012 CFUs) are shown for acidomycin, high accumulating antibiotic (tetracycline) and low accumulating antibiotics (rifampicin and thioridazine).
Resistance Studies.
We were unable to isolate any resistant mutants of (±)-acidomycin by plating at least 1 × 108 colony forming units (CFUs) of M. tuberculosis H37Rv on biotin-free 7H9 agar plates containing (±)-acidomycin at 10-, 25-, and 50-fold its agar MIC (12 μM), indicating the frequency of resistance (FOR) for (±)-acidomycin is less than or equal to 1 × 10−8. To favor isolation of resistant mutants, we next plated M. tuberculosis H37Rv in both the presence and absence of 2 μM DTB and decreased the amount of (±)-acidomycin to be four times its agar MIC. We were unable to isolate any mutants in plates lacking DTB but identified a single mutant with an MIC approximately 16-fold higher than that of WT M. tuberculosis H37Rv in plates containing DTB (Figure S2). Whole-genome sequencing identified a chromosomal duplication spanning from 1750550 to 1807050 which includes the bioAFD- and bioB-containing operons (SRA accession number: PRJNA507782). This suggested that resistance was due to overexpression of proteins in the biotin biosynthetic pathway. Western blot analysis probing for BioA and BioB revealed the mutant to have higher levels of these two enzymes compared to WT H37Rv (Figure S3).
Additional Mechanism of Action Studies.
As a biotin antimetabolite, we hypothesized acidomycin may be recognized by biotin protein ligase (MtBPL), encoded by birA, in M. tuberculosis where it could either act as an inhibitor or potentially act as a substrate. Given the high intracellular concentration achieved by acidomycin, a secondary mechanism of action could be expected to contribute substantially to the observed antitubercular activity; moreover, inhibition of biotin synthase depletes biotin levels, which would serve to potentiate the inhibition of MtBPL by acidomycin. Therefore, we investigated the effect of (S)-(−)-acidomycin on global biotinylated protein levels in M. tuberculosis using a far-western blot assay and discovered acidomycin had very little impact on the degree of protein biotinylation in the time-frame of the assay, even at concentrations that were 10-fold its MIC (Figure 6A, Figure S9). This result contrasted with a known MtBPL inhibitor, 5ʹ-[N-(D-biotinoyl)sulfamoyl]amino-5ʹ-deoxyadenosine (Bio-AMS), which showed a noticeable decrease in protein band intensity as a function of inhibitor concentration.56 Furthermore, we evaluated the binding affinity of (S)-(−)-acidomycin to MtBPL by isothermal titration calorimetry (ITC) in a direct titration experiment. We observed no heat generated when (S)-(−)-acidomycin was titrated into MtBPL, relative to the binding enthalpy generated when a known substrate (biotin) was titrated into MtBPL (Figure 6B, Figure S10). These data indicate (S)-(−)-acidomycin does not bind to MtBPL, and thus does not act as an antimetabolite in protein biotinylation .
Figure 6.
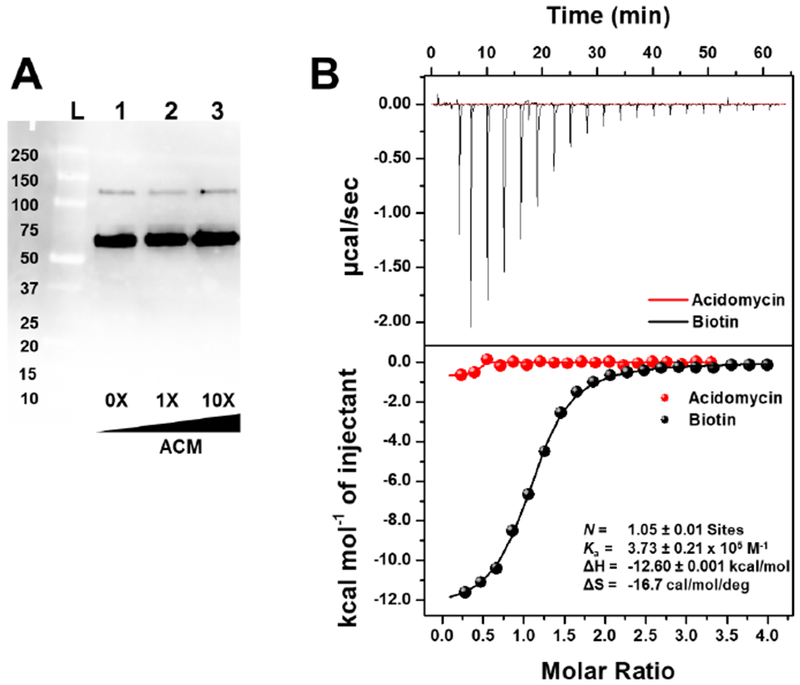
A) Far-western blot of the biotinylated proteome of M. tuberculosis grown in biotin-free 7H9 media, in the presence of acidomycin. Concentration of acidomycin (μM) is depicted at the bottom of the blot. Lane L: Protein ladder; Lanes 1–3: M. tuberculosis grown in the presence of acidomycin at 0× (lane 1), 1× (lane 2) and 10× (lane 3) its MIC. This experiment was performed twice, independently. B) Isothermal titration calorimetry thermogram of (S)-(−)-acidomycin (750 μM, red) and biotin (750 μM, black) titrated into MtBPL (50 μM), with the binding data for biotin shown in black. This experiment was repeated twice, independently.
Pharmacokinetics of Acidomycin.
To assess the pharmacokinetic (PK) parameters of acidomycin and compare them to previous pharmacological studies, we administered CD-1 mice a single dose of either natural (S)-(−)-acidomycin or unnatural (R)-(+)-acidomycin (Table 3). The mediocre aqueous solubility of acidomycin (< 4 mM) prevented formulations at higher concentrations without using DMSO, which prompted optimization of the counterion to further improve the solubility. Using the 2-amino-2-methylpropan-1,3-diol (AMP2) salt form of acidomycin, a known counterion used in the formulation of carboxylic acid drugs,57 we were able to improve its aqueous solubility substantially to greater than 50 mM. We therefore used this formulation for the PK experiments. The plasma concentrations of (S)-(−)-acidomycin and (R)-(+)-acidomycin at various time points after a single dose were determined by LC-MS/MS (Figure 7). Each compound was orally (p.o.) dosed at 25 mg/kg in three mice, or intravenously (i.v.) dosed at 5 mg/kg. From these studies, the area under the concentration-time curve (AUC) for each dose and absolute oral bioavailability (F) were determined. Additional parameters that were determined from these experiments were the volume of distribution (Vd), terminal elimination half-life (t1/2) and clearance (CL). Oral administration (p.o.) of (S)-(−)-acidomycin and (R)-(+)-acidomycin gave an oral bioavailability of 8 and 16%, respectively. Following intravenous (i.v.) administration, both (S)-(−)-acidomycin and (R)-(+)-acidomycin exhibited monoexponential elimination with half-lives of 14.4 and 19.2 min, respectively. Consequently, the unnatural (R)-(+)-acidomycin enantiomer possessed roughly the same PK profile, but improved oral bioavailability relative to the natural (S)-(−)-acidomycin.
Table 3.
In vivo Pharmacokinetic Parameters of (R)-(+)-acidomycin and (S)-(−)-acidomycin in CD-1 mice (n = 3).
| Pharmacokinetic indicesa | (R)-(+)-acidomycin | (S)-(−)-acidomycin |
|---|---|---|
| Dose i.v., p.o. (mg/kg) | 5, 25 | 5, 25 |
| AUC0–∞ (p.o., ng·hr/mL) | 1591 ± 718 | 890 ± 106 |
| AUC0–∞ (i.v., ng·hr/mL) | 2055 ± 564 | 2116 ± 115 |
| Vd (i.v. L/kg) | 1.12 ± 0.37 | 0.82 ± 0.05 |
| CL (i.v., mL/kg·hr) | 2433 ± 482 | 2363 ± 126 |
| t1/2 (i.v., min) | 19.2 ± 2.4 | 14.4 ± 0.1 |
| F (%) | 16 ± 7 | 8 ± 1 |
Values are reported as the mean of three replicates.
AUC0–∞, are under the plasma concentration-time curve from time 0 to infinity; Vd, volume of distribution; CL, clearance; t1/2, terminal elimination half-life; F, relative oral bioavailability calculated as follows: F = 100 × [(AUCp.o. × Di.v.)/(AUCi.v. × Dp.o.)] where D is the dose.
Figure 7.
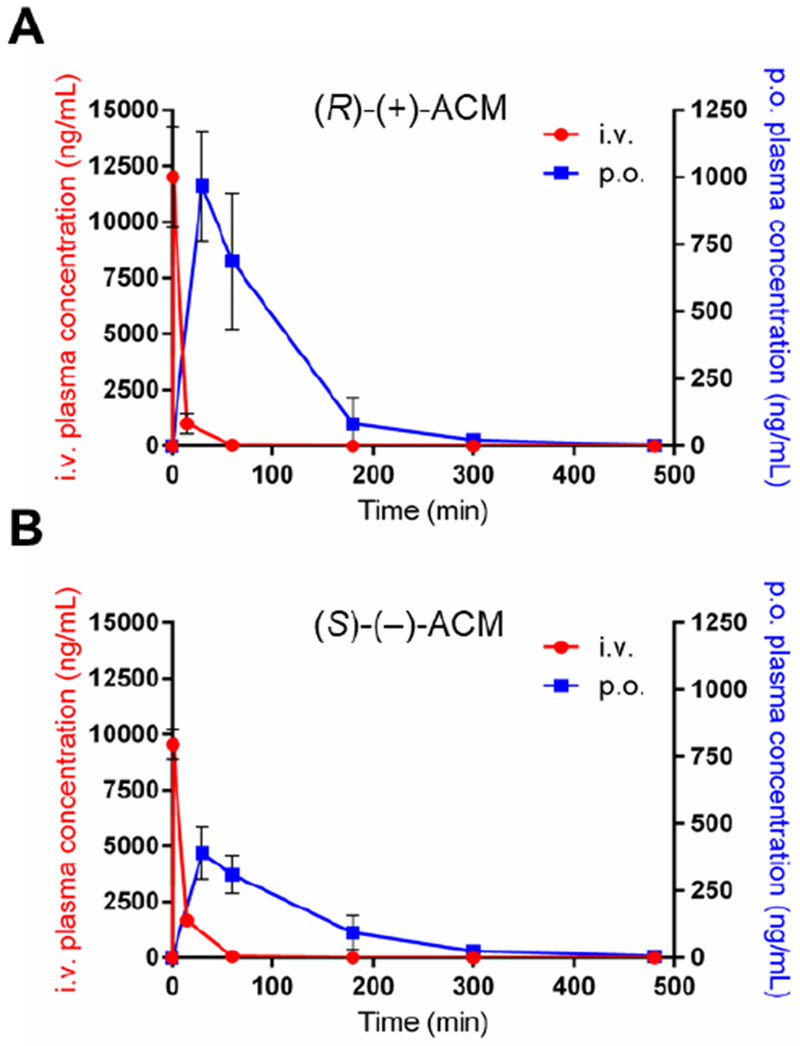
Mean plasma concentration versus time curves after single p.o. (25 mg/kg, blue) and i.v. (5 mg/kg, red) administration of acidomycin to CD-1 mice. Error bars represent standard deviation of the mean (n = 3); A) (R)-(+)-acidomycin; B) (S)-(−)-acidomycin.
DISCUSSION
Since its discovery, acidomycin was recognized as an antimetabolite of the biotin biosynthetic pathway due to the resemblance of the thiazolidinone ring to that of the tetrahydrothiophene/ureido rings of biotin.7–8,13,16,30 Early biochemical characterization and elucidation of the mechanism of acidomycin showed accumulation of biotin pathway intermediates (termed “vitamers”) increased by two- to seventy-fold when acidomycin was added to the culture media of various microorganisms, as determined by a Saccharomyces cerevisiae-based bioassay.58–59 Conversely, the total amount of biotin detected post-assay decreased substantially when given acidomycin, which strongly suggest it directly affects biotin synthesis.58–59 Corroborating these data, we also discovered biotin to rescue activity when M. tuberculosis was grown in the presence of acidomycin, but that other pathway intermediates could not complement growth (Figure 3) suggesting that acidomycin inhibits biotin synthase (BioB), the enzyme responsible for synthesizing biotin (5) from DTB (4).
BioB catalyzes the final step of the biotin biosynthetic pathway (Figures 1 & 8). First characterized in Escherichia coli by Ifuku and coworkers in 1992,60 BioB is part of the radical SAM superfamily and is a homodimeric protein responsible for the formation of the tetrahydrothiophene ring in biotin via insertion of a sulfur atom between the C-6 and C-9 carbons in DTB. Like most radical SAM enzymes, BioB utilizes a reduced [4Fe–4S]+ cluster to cleave SAM (20) into methionine and a high-energy 5′-deoxyadenosyl radical (21, Figure 8A).61–63 This reactive, transient radical is responsible for a single hydrogen atom abstraction from the C-9 carbon and allows for the insertion of sulfur via a newly formed C–S bond originating at a second [2Fe–2S]+ cluster (Figure 8B).44 The tetrahydrothiophene ring is closed from subsequent generation of a second 5′-deoxyadenosyl radical (21), abstracting the hydrogen atom from C-6, and thereby closing the ring (Figure 8B). As a result, BioB requires two equivalents of SAM (20) per complete turnover.44,64 The direct donation of the [2Fe–2S]+ sulfur atom thus makes BioB appear to act as a substrate in vitro, rather than a catalytically functional enzyme, with some studies reporting less than one turnover per protein monomer.65–66 Thus, BioB distinctively possesses a layer of complexity when considering it as a target for drug development that is absent in most enzymatic systems. In the E. coli BioB system, once a successfully completed turnover has occurred, during which the [2Fe–2S]2+ cluster is destroyed, BioB has an increased propensity for oxidative degradation via ATP-dependent proteolysis.40 This begs the question of whether BioB acts as a “true” prototypical enzyme capable of multiple turnovers, or a complex substrate facilitating the conversion of DTB to biotin and dismantling itself in the process.66 However, the extremely tight regulation of biotin production (vis-à-vis BioB degradation) could prove to be promising for antibiotic development targeting BioB, such as acidomycin.
Figure 8.
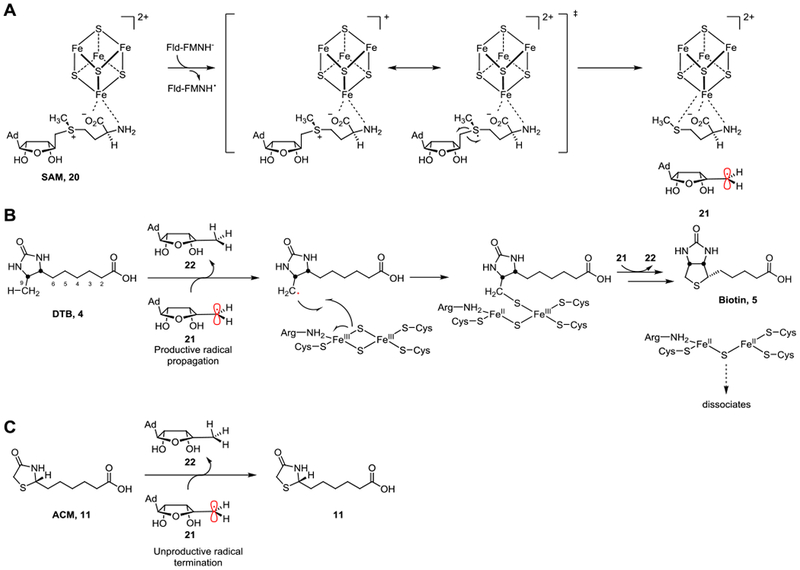
Proposed mechanism of BioB and inhibition by acidomycin (11). A) A single electron from flavodoxin reduces the [4Fe–4S]+ cluster, promoting the homolytic cleavage of SAM (20) into methionine and a high-energy 5′-deoxyadenosyl radical (21). B) Under normal circumstances, the 5′-deoxyadenosyl radical (21) abstracts the C-9 hydrogen atom of the native substrate DTB (4), forming 5′-deoxyadenosine (22) and allows for the insertion of sulfur from the [2Fe–2S]+ cluster (labeled as “productive radical propagation”). Another 5′-deoxyadenosyl radical (21) is responsible for the C-6 hydrogen atom abstraction, completing the thiophane ring in biotin (5). C) When acidomycin (11) is bound to the active site instead, the lack of a C-6 hydrogen atom prevents the highly reactive 5′-deoxyadenosyl radical (21) from H-atom abstraction, leading to the radical to be unproductively quenched as a result (labeled as “unproductive radical termination”).
Our efforts to probe the inhibitory effect of acidomycin on BioB thus led us to the discovery that acidomycin is a competitive inhibitor with respect to DTB (Figure 4D). We resorted to using EcBioB for our inhibition assay as we were unable to develop a functional assay for MtBioB. Although enzymatic activity of MtBioB was obtained previously,33 we were unable to replicate these conditions using the E. coli FLD and FLDR electron transport system. We surmise the electron donation system for BioB is highly tuned to be species-specific and thus the E. coli FLD and FLDR proteins are not functionally active with MtBioB. Genomic mining of M. tuberculosis failed to identify any candidate flavodoxins, which might facilitate the analogous reactions in mycobacteria suggesting a different electron transport mechanism in M. tuberculosis versus E. coli. This is not uncommon, and other organisms are known to utilize ferredoxins in lieu of a flavoprotein for single electron transfer reactions.67 We found five putative ferredoxins in the M. tuberculosis genome that could potentially act as the electron donor: Rv0763c, Rv1177 (potentially FdxC), Rv1786, Rv2007c (potentially FdxA), or Rv3503c (potentially FdxD). Of these, Rv1786 was recently expressed and characterized to couple with flavoprotein reductase A (FprA) and ferredoxin reductase A (FdrA), but its function in vivo is still unknown.68 Investigations into the expression and biochemical evaluation of the MtBioB electron transport partner(s) are ongoing.
To reasonably extrapolate the EcBioB inhibitory data of acidomycin to MtBioB, we therefore constructed a homology model to investigate any differences in the binding site of both EcBioB and MtBioB using the X-ray crystal structure for EcBioB (PDB: 1R30).34 The two sequences have greater than 75% similarity near the binding pocket and some non-conserved residues such as Ser212 (Cys221) and Met246 (Phe253) have backbone interactions with DTB resulting in a nearly identical active site (Figure 9A). The structural similarity between DTB and acidomycin is evident in our model, as it shows that substantial overlap in the active site is possible (Figure 9B). Assuming DTB and acidomycin bind to BioB as illustrated by the model, it is reasonable to extrapolate the EcBioB inhibitory data of acidomycin to MtBioB.
Figure 9.
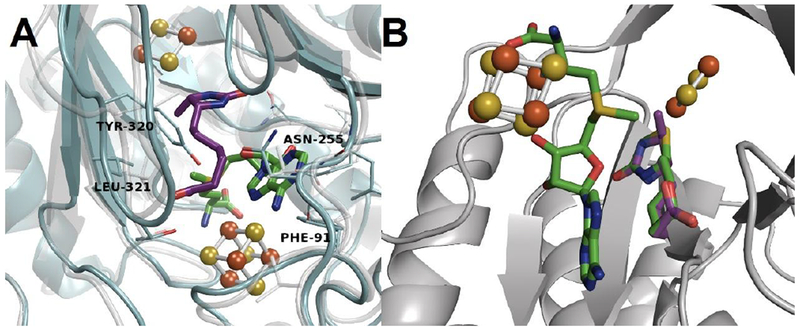
A) Homology model of EcBioB (grey, faded) aligned to MtBioB (blue, solid) depicting DTB (purple) and SAM (green) bound to the active site. All key interactions with residues remain the same between species. B) EcBioB with comparison of acidomycin and DTB (green and purple, respectively) aligned in the active site.
The modest Ki (~1 μM) of acidomycin against EcBioB (and assuming similarity in the inhibitory concentrations between EcBioB and MtBioB), could indicate off-target binding or secondary/indirect effects. In our efforts to explain the difference between the whole-cell activity and enzymatic activity of acidomycin, we discovered an increase in the amount of 5′-deoxyadenosine (22) produced compared to baseline levels in assays containing acidomycin (Figure S7). This unproductive cleavage of SAM does not affect the [2Fe-2S]+ cluster of BioB, which is responsible for the insertion of sulfur into DTB. This implies cleavage of SAM to be catalytic, and thus not obstructed by the Fe-S cluster regeneration requirement that plagues the natural reaction mechanism. Furthermore, the X-ray co-crystal structure of EcBioB and DTB (PDB: 1R30), previously reported by Berkovitch and coworkers, depicts DTB sandwiched between the [4Fe–4S] and [2Fe–2S] clusters with the C-9 only 4.6 Å away from the closest bridging sulfide in the [2Fe–2S] cluster.34 This presumably requires a slight structural change in the conformation of BioB to enable the abstraction of the C-9 H-atom from the SAM-derived 5′-deoxyadenosyl radical (21). With acidomycin bound instead, the reductive homolytic cleavage of SAM (20) could still take place in the active site of BioB in an unproductive manner, whereby the highly reactive 5′-deoxyadenosyl radical (21) is quenched before reacting with acidomycin (Figure 8C). Although it is known that the H-atom abstraction and subsequent sulfide insertion is step-wise, it is still unclear as to whether or not the binding event of SAM and DTB occur in any ordered fashion.42 Therefore, acidomycin could bind to the active site in a competitive manner followed by a “pseudo mechanism-based” inhibition of BioB via depletion of the biologically active SAM. The utilization of the methylthioadenosine/S-adenosylhomocysteine nucleosidase MtnN in our in vitro assays prevented this known substrate inhibition through hydrolysis of the 5′-deoxyadenosine derived from SAM.69 If pools of SAM are depleted in vivo, however, it should come as no surprise that this could be problematic for the survival of the organism, as many essential enzymes – even within the biotin biosynthetic pathway – are SAM-dependent such as BioC and BioA.70 Additionally, M. tuberculosis lacks the ability to scavenge exogenous SAM and intermediates of SAM/methionine biosynthesis, emphasizing a major reliance on intracellular levels of SAM.71 Interestingly, a recent study showed potentiation of para-aminosalacylic acid (PAS), a selective antimetabolite of the folate metabolic pathway, to biotin deprivation by utilizing a biotin auxotrophic strain of M. bovis BCG with a disruption in the bioB gene.72 Methionine is a known potent antagonist of PAS in mycobacteria,73 and thus the connection between PAS, methionine and biotin biosynthesis inhibition could be attributed to SAM as well.74
We hypothesized acidomycin may possess a secondary mechanism of action, wherein it would act as a substrate for MtBPL and become incorporated into the biotin carrier carboxylase protein (BCCP) domain of acyl-CoA carboxylases (ACC) instead of biotin (Figure 1). This modification would not only inactivate biotin-dependent proteins, but also prevent canonical activation, as only non-ligated apo-proteins are recognized by BPLs. Acidomycin ligated to proteins would be unable to facilitate carboxyl-group transfer, as it lacks the proper ureido nitrogen responsible for fixing CO2. The unique bicyclic ring system of biotin is thought to have evolved specifically for carboxylation, as the presence of the sulfur atom reduces unproductive hydrolysis by blocking the approach of water from one side, greatly improving the efficiency of biotinylated enzymes.75–76 The importance of the sulfur in biotin is evident from an evolutionary standpoint as BioB is a highly regulated enzyme with a complex mechanism for sulfur insertion. Therefore, any deviation from this highly specialized ring system (even similar molecules such as DTB) would presumably result in a non-functional enzyme. However, both our whole-cell biotinylated proteome assays (Figure 6A) and direct binding assays (Figure 6B) showed acidomycin does not bind to MtBPL, and thus acidomycin does not appear to possess this intriguing secondary mechanism of action.
Antibiotics with poor cellular accumulation was a major problem faced during many early discovery programs.77–78 As such, the ability to quantify intracellular accumulation of drugs has become a recent trend in the evaluation of antibiotics, new and old.51,79–80 In an effort to reconcile the disparate whole-cell inhibitory activity of acidomycin against E. coli and M. tuberculosis, even though we presented direct inhibition of EcBioB, we chose to look at the levels of intracellular accumulation for these two organisms. Alas, we discovered a thirty-fold increase in intracellular accumulation of acidomycin in M. tuberculosis compared to E. coli, which is a trend we expected, but does not fully explain the >100-fold difference in whole cell activity. These measurements account for drug association within the cells as well as (in the case of M. tuberculosis) embedded in the cell envelope, and these concentrations represent concentrations after a five-minute incubation period. Efflux pumps in E. coli and M. tuberculosis could act on these drugs at different rates, which could also affect the intracellular concentration at a given time. However, solely relying on accumulation to explain the difference in activity oversimplifies the mechanism of selectivity between these two organisms.
A promising result in our studies was the observation of the very low frequency of resistance of acidomycin, consistent with previous studies.7 When M. tuberculosis was grown in the presence of acidomycin and 2 μM DTB, we observed a 16-fold increase in the MIC. The rationale for including DTB was to “force” a mutation of bioB, however when DTB concentrations were reduced to 1 μM, only a two- to three-fold increase in MIC was observed. Grundy and coworkers discovered resistance to acidomycin was not easy acquired, as a meager two-fold increase in inhibitory concentrations were achieved after growth in the presence of acidomycin.7 Sequencing of the mutant identified chromosomal duplication leading to the overexpression of enzymes required for biotin synthesis. The difficulty in obtaining a viable resistant mutant suggests simple point mutations to BioB are unable to confer resistance to acidomycin.
Although acidomycin displayed promising in vitro selective inhibitory activity against mycobacteria, it was shown in this study and in previous studies to be pharmacodynamically inert.11,81–82 The inability of acidomycin to exert an antitubercular effect in vivo prompted researchers to hypothesize that biotin present in tissues antagonized the in vivo activity of acidomycin, when dosed to mice and rats.11,30 Schnappinger and coworkers definitively proved this to not be the case, as their conditionally-regulated bioA knockdown mutant was not only unable to establish an infection in mice, but also chronic infections were cleared when biotin biosynthesis was inhibited, indicating M. tuberculosis requires de novo biotin biosynthesis for the persistence in vivo.24,83 Conditional genetic inactivation of BPL further exemplified that not only de novo biotin biosynthesis but also protein biotinylation is required by M. tuberculosis to sustain an infection.25 Our own PK experiments showed rapid elimination of acidomycin from mice (Table 3). Biotin concentrations in mammalian tissues (including mice) are well under the amount required to antagonize acidomycin, implying the lack of in vivo activity could be explained by rapid elimination. To that end, one study observed approximately 30 % of the intravenous (i.v.) acidomycin dose was recovered from the urine of mice, dogs, cats, and rabbits within 60 minutes, as well as provide evidence that acidomycin is partially inactivated by the abdominal organs.82 Further early in vivo experiments also showed elevated levels of biotin in the urine of rabbits treated with acidomycin.11,15,82 Acidomycin did not significantly change the measured degree of biotin deficiency in egg white-fed rats but rather increased the amount of what was then-termed “avidin uncombinable biotin” and “SC factor,” which was later elucidated to be biotin pathway intermediates.81,84 Combined with our PK data, we can establish the reason for its inactivity to be due to rapid elimination from the host rather than biotin antagonism. Further improvement of these parameters would necessitate the design of prodrugs to increase permeability and extend its terminal half-life.
In addition to the antimicrobial and pharmacological experiments performed, several structurally related acidomycin analogues were synthesized and evaluated for antimicrobial activity against both M. tuberculosis and M. avium.7–8,13,16,30,85–86 The optimal chain length is five methylene units, and removing the carboxylic acid ablates activity.86 Any modifications to the N3 position, as well as oxidizing the sulfur also diminishes activity.85 The primary amide and lower alkyl esters showed improved antitubercular activity, but this could be due to increased cellular permeability.86 The activity of acidomycin derivatives seem to be sensitive to minor changes, indicating a limited scope of SAR. Going forward, it would be prudent to improve the PK properties of acidomycin through prodrug design (via alkyl esters and amides), rather than through analogues of the parent compound.
CONCLUSION
We have explicitly demonstrated the mechanism of acidomycin against M. tuberculosis using both whole-cell and in vitro biochemical assays. Biotin was the only biotin pathway intermediate to antagonize the inhibitory activity of acidomycin against M. tuberculosis. This was further confirmed as (±)-acidomycin displays competitive inhibition against EcBioB, with an inhibitory constant, Ki of 2.0 μM, which is analogous to MtBioB due to the high sequence similarity in the active sites of EcBioB and MtBioB. The purified natural product (S)-(−)-acidomycin shows a 14-fold or greater increase in activity over the non-natural (R)-(+)-acidomycin enantiomer in both whole-cell and in vitro biochemical assays. We observed a 30-fold increase in the amount of accumulated acidomycin in M. tuberculosis over E. coli, owing to its selectivity for M. tuberculosis. The low FOR of acidomycin against M. tuberculosis initially suggested a secondary mechanism of action, but none of our experiments showed evidence of acidomycin acts as a biotin antimetabolite or binds to MtBPL. We also discovered the lack of in vivo activity of (S)-acidomycin in mice was caused by poor PK properties as it is rapidly eliminated with a half-life of 14.4 min. Overall, this study provides the most detailed study of acidomycin’s mechanism of action and selectivity in M. tuberculosis and demonstrates future efforts should focus on improving the pharmacokinetic properties of acidomycin.
METHODS
General materials and methods for synthesis.
Chemicals and solvents were purchased from Acros Organics, Alfa Aesar, Sigma-Aldrich, and TCI America and were used as received. An anhydrous solvent dispensing system using two packed columns of neutral alumina was used for drying MeOH, toluene, THF and CH2Cl2, while two packed columns of molecular sieves were used to dry DMF, and the solvents were dispensed under argon gas (Ar). EtOAc and hexanes were purchased from Fisher Scientific. All reactions were performed under an inert atmosphere of dry argon gas (Ar) in oven-dried (180 °C) glassware. TLC analyses were performed on TLC silica gel 60F254 plates from EMD Chemical Inc. and were visualized with UV light. Optical rotations values were obtained on a polarimeter using a 1 dm cell. Purification by flash chromatography was performed using a medium-pressure flash chromatography system equipped with flash column silica cartridges with the indicated solvent system. Analytical reversed-phase high performance liquid chromatography (HPLC) was performed on a Phenomenex Luna® Omega 3 μm Polar C-18 100 × 2.1 mm LC column operating at 0.300 mL/min with detection at 254 nm and 210 nm employing a linear gradient of 5 to 65 % MeCN + 0.1 % formic acid (solvent B) in water + 0.1 % formic acid (solvent A) for 10 minutes and maintaining 65 % solvent B for an additional 5 min (Method A). Chiral analytical HPLC was performed on a Phenomenex Lux® 5 μm Cellulose-1 100 × 4.6 mm LC column operating at 1.000 mL/min with detection at 230 nm and 210 nm employing a linear gradient of 5 to 40 % MeCN + 0.1 % formic acid (solvent B) in water + 0.1 % formic acid (solvent A) for 5 minutes and maintaining 40 % solvent B for an additional 5 min (Method B). Chiral preparative HPLC was performed on a Chiralpak® IE 5 μm 250 × 2.0 mm LC column operating at 45 g/min with detection at 210 nm employing an isocractic elution of 30 % MeOH (solvent B) in supercritical CO2 (solvent A) for 8 minutes (Method C). 1H and 13C spectra were recorded on a 400 or 500 MHz NMR spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual chloroform (7.27), methanol (3.31) or dimethyl sulfoxide (2.50); carbon chemical shifts are reported in ppm from an internal standard of residual chloroform (77.0), methanol (49.1), or dimethyl sulfoxide (39.5). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublets, dt = doublet of triplets, t = triplet, dq = doublet of quartets, m = multiplet, br = broad, ABqd = AB quartet of doublets, ABq = AB quartet), coupling constant(s), integration. High-resolution mass spectra were obtained on an LTQ Orbitrap Velos instrument (Thermo Scientific, Waltham, MA). All compounds were determined to be >95 % by analytical reverse-phase HPLC (purities for each final compound are given in the experimental section below).
Methyl 7-hydroxyheptanoate (8).
To a solution of 7 (31.3 mL, 0.27 mol, 1.0 equiv) in CH2Cl2 (700 mL) was added 3-chloroperoxybenzoic acid (130 g, 0.40 mol, 1.5 equiv) at 23 °C and stirred for 6 days during which a white precipitate formed. The reaction mixture was then filtered through Celite, and washed thoroughly with saturated aqueous NaHCO3 (5 × 250 mL), saturated aqueous NaCl (250 mL), H2O (250 mL), dried (MgSO4) and concentrated in vacuo to afford crude product as an off-yellow oil which was purified by high vacuum distillation (75–80 °C under 1.0 torr) to give ζ-enantholactone (19 g, 57 %) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.40–1.77 (m, 8H), 2.42 (t, J = 6.3 Hz, 2H), 4.22 (t, J = 5.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 23.6, 25.4, 28.0, 30.5, 31.0, 67.6, 176.4.
To a solution of ζ-enantholactone (20 g, 160 mmol, 1.0 equiv) in MeOH (1000 mL) was added conc. H2SO4 (1 mL) at 23 °C and stirred overnight. After 16 h, the reaction mixture was concentrated in vacuo, and re-dissolved in Et2O (250 mL), washed with H2O (2 × 150 mL), dried (MgSO4) and concentrated in vacuo to yield the title compound (26 g, 99 %) as a colorless oil which was used in the next step without purification: Rf = 0.31 (1:1 EtOAc–hexanes); 1H,13C NMR and MS match previously reported spectra.87
Methyl pimelate semialdehyde (9).
A mixture of 8 (2.0 g, 12.5 mmol, 1.0 equiv.), CH2Cl2 (100 mL), H2O (50 mL), and sodium bromide (1.56 g, 15.0 mmol, 1.2 equiv.) was rapidly stirred in a round-bottom flask (500 mL) and cooled to 0 °C. (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO, 12 mg, 0.024 mmol, 0.6 mol%) was added followed by addition of a mixture of sodium hypochlorite solution (4 – 6 % aqueous, 22 mL, 15.0 mmol, 1.2 equiv.) in NaHCO3 solution (5 % aqueous, 25 mL) at 0 °C. Reaction was monitored by TLC, and at 1 hour showed complete conversion to 9. The reaction was diluted with CH2Cl2 (100 mL), and the CH2Cl2 layer was separated. The aqueous layer was extracted with CH2Cl2 (2 × 100 mL), and combined extracts were washed with 5 % (w/v) aqueous potassium bisulfate (150 mL), 5 % (w/v) aqueous sodium thiosulfate (150 mL), saturated aqueous NaCl (150 mL), dried (MgSO4), and concentrated in vacuo to give an orange oil which was purified by high vacuum fractional distillation (75–85 °C under 0.9 torr) to give the title compound (1.8 g, 91 %) as a colorless oil: Rf = 0.47 (3:7 EtOAc–hexanes); 1H, 13C NMR and MS match previously reported spectra.87
2-Mercaptoacetamide.
To a 30 mL pressure vessel was added a solution of ammonium in methanol (7.0 N, 7.0 mL) and methyl thioglycolate (2.0 mL, 22.3 mmol). The vessel was purged with argon, sealed and stirred for 2 days at 23 °C. The solution was de-gassed with argon, concentrated in vacuo, re-suspended in toluene (10 mL), and concentrated again to afford the title compound (2.03 g, 99%) as an amorphous white powder, which was used directly in the next reaction without purification: 1H NMR (400 MHz, CDCl3) δ 1.95 (t, J = 9.0 Hz, 1H), 3.25 (d, J = 8.6 Hz, 2H), 5.79 (br d, 2H); 13C NMR (125 MHz, CDCl3) δ 29.5, 173.9.
NOTE: Methyl thioglycolate is extremely volatile and both methyl thioglycolate and 2-mercaptoacetamide produces a potent, noisome odor even at extremely low concentrations; much care is necessary when handling. Additionally, as bleach (sodium hypochlorite) is typically used to wash glassware handling thiols, ensure all ammonia is properly and thoroughly removed from the vessel before washing. 2-mercaptoacetamide should be used immediately or stored neat at −20 °C for no more than three days before discarding.
Methyl 6-(4-oxothiazolidin-2-yl)hexanoate (10).
To a solution of 9 (1.0 g, 6.3 mmol, 1.0 equiv) and freshly prepared 2-mercaptoacetamide (2.0 g, 20.7 mmol, 3.1 equiv) in toluene (60 mL) at 23 °C was added p-toluenesulfonic acid monohydrate (60 mg, 0.316 mmol, 5 mol%) and powdered 4Å molecular sieves (0.5 g). The reaction was then heated to 110 °C. After 16 h, the reaction was cooled to 23 °C and filtered through a pad of Celite. The filtrate was concentrated in vacuo, and the residue was dissolved in EtOAc (50 mL), washed with saturated aqueous NaHCO3 (50 mL), saturated aqueous NaCl (50 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash chromatography (1:1–3:1 EtOAc–hexanes, linear gradient) afforded the title compound (1.2 g, 79 %) as an off-white amorphous solid: Rf = 0.29 (7:3 EtOAc–hexanes); HPLC purity: 99.1 %, tR = 9.6 min, k’ = 7.7 (Method A); 1H NMR (400 MHz, CDCl3) δ 1.24–1.52 (m, 4H), 1.63 (dt, J = 14.5, 7.6 Hz, 2H), 1.68–1.77 (m, 1 H), 1.76–1.90 (m, 1H), 2.30 (t, J = 7.2 Hz, 2H), 3.50 (s, 2H), 3.66 (s, 3H), 4.70 (t, J = 6.1 Hz, 1H), 7.37 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 24.6, 25.0 , 28.6, 31.9, 33.8, 38.7, 51.5, 57.3, 174.0, 174.8; HRMS (ESI+) calcd for C10H17NO3SNa+ [M + Na]+ 254.0821, found 252.1559.
6-(4-Oxothiazolidin-2-yl)hexanoic acid/(±)-Acidomycin (11).
To a solution of 10 (0.76 g, 3.29 mmol) in MeOH (10 mL) was added a 10 % (w/v) aqueous NaOH solution (10 mL) at 23 °C. After 2 h, the reaction was concentrated in vacuo, re-dissolved in H2O (2 mL), and cooled to 0 °C. The solution was acidified to pH 1–2 using 6 N HCl during which an off-white precipitate formed. The precipitate was collected by filtration, recrystallized from H2O (2 mL), and dried under high vacuum (P < 0.9 Torr) to afford the title compound (0.63 g, 89 %) as white, needle-like crystals: Mp: 120–124 °C; HPLC purity: 99.9 %, tR = 8.3 min, k’ = 6.5 (Method A); (c 1.00, MeOH); 1H NMR (500 MHz, CD3OD) δ 1.33–1.52 (m, 4H), 1.55–1.76 (m, 3H), 1.84 (ddd, J = 13.8, 9.4, 4.9 Hz, 1H), 2.30 (t, J = 7.5 Hz, 2H), 3.49 (ABqd, JAB = 15.0 Hz, J = 1.8 Hz, 2H), 4.75 (t, J = 5.8 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ 26.02, 26.04, 29.9, 32.9, 34.9, 39.7, 59.2, 177.1, 177.7; HRMS (ESI+) calcd for C9H16NO3S+ [M + H]+ 218.0845, found 218.0668.
(R)-(+)-Acidomycin:
(±)-Acidomycin was resolved by chiral HPLC (method C). Mp: 122–124 °C; HPLC purity: 97.1 %, tR = 5.25 min, k’ = 3.0 (Method B); (c 1.00, MeOH); enantiomeric ratio (er): 97.1:1.0; 1H NMR (500 MHz, CD3OD) δ 1.33–1.52 (m, 4H), 1.55–1.76 (m, 3H), 1.84 (ddd, J = 13.8, 9.4, 4.9 Hz, 1H), 2.30 (t, J = 7.5 Hz, 2H), 3.49 (ABqd, JAB = 15.0 Hz, J = 1.8 Hz, 2H), 4.75 (t, J = 5.8 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ 26.02, 26.04, 29.9, 32.9, 34.9, 39.7, 59.2, 177.1, 177.7; HRMS (ESI+) calcd for C9H16NO3S+ [M + H]+ 218.0845, found 218.0663.
(S)-(−)-Acidomycin:
(±)-Acidomycin was resolved by chiral HPLC (method C). Mp: 122–124 °C; HPLC purity: 99.9 %, tR = 5.55 min, k’ = 3.2 (Method B); (c 1.00, MeOH); enantiomeric ratio (er): 1:97.5; 1H NMR (500 MHz, CD3OD) δ 1.33–1.52 (m, 4H), 1.55–1.76 (m, 3H), 1.84 (ddd, J = 13.8, 9.4, 4.9 Hz, 1H), 2.30 (t, J = 7.5 Hz, 2H), 3.49 (ABqd, JAB = 15.0 Hz, J = 1.8 Hz, 2H), 4.75 (t, J = 5.8 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ 26.02, 26.04, 29.9, 32.9, 34.9, 39.7, 59.2, 177.1, 177.7; HRMS (ESI+) calcd for C9H16NO3S+ [M + H]+ 218.0845, found 218.0678.
Acidomycin AMP2 salt formulation:
To a solution of 2-amino-2-methylpropan-1,3-diol (25 mM, 1.0 equiv) in water was added an equimolar amount of acidomycin (1.0 equiv). The mixture was stirred vigorously for 10 min at 23 °C. The solution was then concentrated overnight via lyophilization to afford the 2-ammonium-2-methyl-propan-1,3-diol salt of acidomycin. The equivalents of AMP2 were determined by NMR: 1H NMR (500 MHz, CD3OD) δ 1.22 (s, 3H, AMP2), 1.33–1.50 (m, 4H), 1.58–1.74 (m, 3H), 1.80–1.89 (m, 1H), 2.17 (t, J = 7.5 Hz, 2H), 3.49 (ovlp ABqd, JAB = 15.0 Hz, J = 1.8 Hz, 2H), 3.56 (ovlp dd, J = 53.4, 11.6 Hz, AMP2), 4.75 (t, J = 6.3 Hz, 1H).
MIC Assays and conditions:
MICs for acidomycin were experimentally determined as previously described using M. tuberculosis H37Rv grown in GAST medium [0.3 g/L Difco Bacto Casitone, 4.0 g/L K2HPO4, 2.0 g/L citric acid, 1.0 g/L L-alanine, 1.2 g/L MgCl2 · 6 H2O, 0.6 g/L K2SO4, 2.0 g/L NH4Cl, 18 mM NaOH, 1% (v/v) glycerol, and 0.05% (v/v) Tyloxapol, pH 6.6] or biotin-free 7H9 medium [0.5 g/L (NH4)2SO4, 0.5 g/L L-glutamic acid, 0.11 g/L sodium citrate tribasic dihydrate, 4.72 g/L Na2HPO4 · 7 H2O, 1 g/L KH2PO4, 40 mg/L ammonium ferric citrate, 0.1 g/L MgSO4 · 7 H2O, 0.66 mg/L CaCl2 · 2 H2O, 1.78 mg/L ZnSO4 · 7 H2O, 1 mg/L CuSO4, and 1 mg/L pyridoxine, 2.5 g/L Bovine Serum Albumin Fraction V protease-free (Roche), 1 g/L dextrose, 0.425 g/L NaCl, 0.2% (v/v) glycerol, and 0.05% (v/v) Tyloxapol, pH 6.6] where noted, with an initial inoculum of approximately 106 CFU/mL in a 384 well plate.56 The plates were incubated at 37 °C under 5% CO2 and OD580 was measured after 11 or 12 days of incubation to monitor growth. MIC90 was defined as the concentration of the compound at which roughly 90 percent growth inhibition was observed compared to the no drug control. Supplementation assays were performed as previously described, supplementing cultures with 7-keto-8-aminopelargonic acid (KAPA), DTB, or biotin at a final concentration of 1 μM.20 M. bovis, M. africanum, M. abscessus, M. avium, and M. smegmatis strains were grown in biotin-free 7H9 medium and MICs were determined as described above, in either 96 or 384 well plates, with incubation times adjusted for strains as needed. All M. tuberculosis DS, MDR, MDR+, XDR, and clinical isolates88 were grown in Dubos-based medium (6.5 g/L Dubos broth base, 0.81 g/L NaCl, 7.5 g/L glucose and 5 g/L BSA fraction V) to an OD650 of 0.2. Cells were diluted 1000-fold in this medium and 50 μL per well dispensed into round-bottom clear sterile polypropylene plates containing either acidomycin or linezolid serially diluted from 50 to 0.049 μM in the same medium at 50 μL per well. Plates were incubated for up to 2 weeks at 37 °C and MIC scored under an inverted enlarging mirror. The ability of biotin to rescue growth was confirmed by performing the MIC under identical conditions with the addition of biotin (final concentration 10 μg/mL) to the media. All experiments were performed in triplicate for each concentration (technical replicates) and repeated independently at least two times (biological replicates).
The MICs of acidomycin against E. faecalis (V583), S. aureus (USA300), A. baumannii (AB5075), P. aeruginosa (PA14) and E. coli (ATCC: 25922) were determined by broth microdilution method as previously described.89 E. faecalis, A. baumannii, P. aeruginosa and E. coli were grown in LB medium overnight and sub-cultured into M9 minimal medium supplemented with 0.2% (v/v) glucose; S. aureus sub-cultured into M9 medium supplemented with: 0.2% (v/v) glucose, 0.5% (v/v) Casamino acids, 2 μg/mL pantothenate, 2 μg/mL nicotinamide, and 2 μg/mL thiamine. MIC was defined as the concentration of the compound which completely prevented visible grown after incubation at 37 °C for 24 h. All experiments were performed in triplicate for each concentration (technical replicates) and repeated independently at least two times (biological replicates).
Cell Cytotoxicity Assay:
Human hepatocellular carcinoma cells (HepG2, ATCC: HB-8065) and African green monkey kidney cells (Vero, ATCC: CCL-81) were cultivated in Dulbecco’s minimum essential medium (MEM) supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C and 5% CO2 in a humidified incubator. HepG2 and Vero cells were used to seed white opaque 96-well plates at a density of 20,000 cells/well in a total volume of 100 μL. After incubation for 24 h, the medium was carefully removed from adherent cells and acidomycin (formulated as the 2-ammonium-2-methylpropan-1,3-diol [AMP2] salt) was added to each well in yielding final concentrations of 1 mM, 0.5 mM and 0.1 mM, and subsequently allowed to incubate for a further 72 h. Control wells contained either no compound (negative control) or 1 % Triton X-100 (positive control). All experiments were performed in duplicate for each concentration (technical replicates) and repeated independently three times (biological replicates). Following the 72 h incubation, MEM containing acidomycin was aspirated and 50 μL of fresh MEM was added with 50 μL of the Celltiter-Glo® reagent (Promega). Cells were lysed via shaking at 350 rpm for 2 min on an orbital shaker, and then incubated at 23 °C for 10 min. The luminescent signal was measured in a Synergy H1 hybrid multimode microplate reader (BioTek).
Acidomycin Frequency of Resistance (FOR):
The MIC of acidomycin (2-ammonium-2-methylpropan-1,3-diol [AMP2] salt) on agar plates was determined by spotting approximately 106 bacteria onto biotin-free 7H9 agar [same as broth recipe above without BSA, dextrose, and NaCl, but including 15 g/L agar, 10% (v/v) OADC, 0.5% (v/v) glycerol] plates and varying drug concentrations. The lowest drug concentration where no growth was observed upon visual inspection after incubation at 37 °C for three weeks was used as the agar MIC. To isolate resistant mutants, approximately 108 bacteria were plated onto biotin-free 7H9 agar plates containing acidomycin at a concentration of 4×, 10×, 25×, or 50× the agar MIC (the experiment with 4× was performed both with and without 2 μM DTB supplementation in the agar). After incubation at 37 °C for 3 weeks, colonies were counted. Plates without colonies were incubated for a further 6 weeks and a final count was done at the end of 9 weeks of incubation. The initial number of bacteria was determined by plating dilutions of the original culture. FOR was calculated as the number of CFUs divided by the total number of bacteria plated for each concentration of acidomycin.
Chromosomal DNA Isolation:
This method was adapted from a previously-described protocol.91 Briefly, mid-exponential growth bacteria were suspended to a final OD580 of 0.1–0.2 in 30 mL fresh 7H9 [4.7 g/L Middlebrook 7H9 Broth Base, 2.5 g/L Bovine Serum Albumin Fraction V protease-free (Roche), 1 g/L dextrose, 0.425 g/L NaCl, 0.2% (v/v) glycerol, and 0.05% (v/v) Tyloxapol] in a vented T175 flask, and were incubated for 7 d at 37 °C and 5% CO2 in a humid incubator. Bacteria in 20 mL of this culture were pelleted by centrifugation. The pellet was re-suspended in 1 mL TE Buffer [10 mM Tris, 1 mM EDTA, pH 8.0]. 50 μL of 10 mg/mL lysozyme (Sigma) prepared in TE Buffer and 0.25 μL of 100 mg/mL RNaseA (Qiagen) was added to the suspension, which was then briefly vortexed. The suspension was distributed equally between two 1.5-mL Eppendorf tubes and was incubated at 37 °C overnight. 5 μL of 20 mg/mL Proteinase K (Invitrogen) and 45 μL of 10% (w/v) SDS solution was added to each tube followed by a brief vortex. The tubes were incubated at 65 °C for 1 h. 50 μL of 3 M NaCl and 40 μL of 10% (w/v) CTAB (hexadecyltrimethyl ammonium bromide) was added to each tube, followed by a brief vortex. The tubes were further incubated at 65 °C for 1 h. The contents of the two tubes derived from the same suspended pellet were re-combined in a single 2-mL Eppendorf tube. 700 μL of ice-cold chloroform was added to the tube which was then shaken to mix the contents well. The layers were separated by centrifugation in a microcentrifuge at 13,000 rpm and 4 °C for 5 min. The upper aqueous layer was transferred to a new 2-mL tube. 1 mL of phenol–chloroform solution (from phenol:chloroform:isoamyl alcohol 25:24:1, saturated with 10 mM Tris, pH 8.0, 1 mM EDTA [Sigma]) was added to the tube which was then shaken to mix the contents well. The layers were separated by centrifugation in a microcentrifuge at 13,000 rpm and 4 °C for 5 min. The upper aqueous layer was transferred to a new 2-mL tube and was mixed with 1 mL of chloroform. The layers were separated by centrifugation in a microcentrifuge at 13,000 rpm and 4 °C for 5 min. The upper aqueous layer was transferred to a new 1.5-mL tube. 420 μL of isopropanol was added to the tube and the solution was mixed by inversion to precipitate out the DNA. 10 μL of 3 M sodium acetate was added to the tube with mixing by inversion, and the DNA was pelleted by centrifugation in a microcentrifuge at 13,000 rpm and 4 °C for 30 min. The supernatant was removed from the pellet, and the pellet was then washed with 500 μL of 75% (v/v) ethanol. The DNA was re-pelleted by centrifugation in a microcentrifuge at 13,000 rpm and 4 °C for 5 min. The supernatant was removed and the pellet was allowed to air dry. The DNA was re-suspended in 50 μL of TE Buffer.
Whole genome sequencing.
Between 150 and 200 ng of genomic DNA was sheared acoustically and HiSeq sequencing libraries were prepared using the KAPA Hyper Prep Kit (Roche). PCR amplification of the libraries was carried out for 10 cycles. 5–10×106 50-bp paired-end reads were obtained for each sample on an Illumina HiSeq 2500 using the TruSeq SBS Kit v3 (Illumina). Post-run demultiplexing and adapter removal were performed and fastq files were inspected using fastqc. Trimmed fastq files were then aligned to the reference genome (M. tuberculosis H37RvCO; NZ_CM001515.1) using bwa mem. Bam files were sorted and merged using samtools. Read groups were added and bam files de-duplicated using Picard tools and GATK best-practices were followed for SNP detection. The circular binary segmentation algorithm was used to identify genomic regions with abnormal copy number.
Immunoblot analysis of WT H37Rv and Acidomycin-resistant mutant whole-cell lysates:
Mid-exponential growth bacteria were suspended to a final OD580 of 0.01 in 50 mL fresh biotin-free 7H9 in vented T175 flasks and were incubated for 11 or 12 d at 37 °C and 5% CO2 in a humid incubator. Bacteria were pelleted by centrifugation and pellets were washed three times with 10 mL of 10% glycerol. After the final wash, pellets were re-suspended in 500 μL PBS containing cOmplete EDTA-free Protease Inhibitor Cocktail (Roche) and transferred to 2-mL O-ringed, screw-capped tubes containing about 300 μL of 0.1-mm diameter zirconia/silica beads (BioSpec). The pellets were mechanically lysed three times with a BeadBug Microtube Homogenizer (Southern Labware) for 30 s at the highest speed setting, with at least a 2-min rest at ambient temperature between cycles. 50 μL of 20% SDS solution was added to each lysate, followed by a quick vortex and incubation at 65 °C for 1 h. Tubes were vortexed again briefly and beads/insoluble debris were pelleted by centrifugation in a microcentrifuge at 13,000 rpm for 5 min. The supernatants were transferred to fresh 2-mL O-ringed, screw-capped tubes and were centrifuged once more at 13,000 rpm for 5 min to pellet any remaining beads and/or insoluble debris. The lysates were sterilized by centrifugation at 13,000 rpm for 5 min through 0.22-μm cellulose acetate Spin-X Centrifuge Tube Filters (Corning). Total lysate protein concentrations were measured by Pierce BCA Protein Assay Kit (Thermo) according to manufacturer instructions. Quantified lysates were diluted as appropriate to 0.4 μg/μL with 4× loading buffer (240 mM Tris·HCl pH 6.8, 40% glycerol, 8% SDS, 0.04% bromophenol blue, and 5% β-mercaptoethanol) and 2% SDS in PBS. Prepared samples were heated at 100 °C for 10 min and 8 μg per sample was separated via SDS-PAGE at 150 V using 4–15% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad) according to manufacturer instructions. Proteins were transferred to a nitrocellulose blotting membrane via iBlot 2 Dry Blotting System (Invitrogen) according to manufacturer instructions, using the instrument’s P0 preset transfer protocol. Membranes were first incubated with Odyssey Blocking Buffer (LI-COR) for 30 min at ambient temperature. Membranes were then probed with rabbit-generated antisera against BioA or BioB at a dilution of 1:1000, and NadE (loading control) at a dilution of 1:500 in Odyssey Blocking Buffer with 0.1% tyloxapol, for 1 h at ambient temperature. Primary antibodies were washed away with three incubations at ambient temperature in PBS with 0.1% tyloxapol (5 min each). The membranes were then probed with IRDye 800CW Donkey anti-Rabbit IgG (H + L) (LI-COR), prepared according to manufacturer instructions, at a dilution of 1:12,500 in Odyssey Blocking Buffer with 0.1% tyloxapol, for 1 h at ambient temperature. The secondary antibody was washed away with three incubations at ambient temperature in PBS with 0.1% tyloxapol (5 min each). The membranes were dried at ambient temperature on paper towels and were scanned with an Odyssey Infrared Imaging System (LI-COR).
Construction of an M. tuberculosis strain carrying two copies of bioB.
The M. tuberculosis bioB gene was PCR amplified from genomic DNA using primers clo-SD1-bioB-attB2 (5′-GGGGACAGCTTTCTTGTACAAAGTGGCTAAGGAGGTATCTCCATGACGCAAGCGGAACGCGACCGA-3’) and clo-bioB-attB3 (5′-GGGGACAACTTTGTATAATAAAGTTGTTACAGGCTGGCGTTGAGTGCCT-3′) and was cloned into a pDE43-MCZ destination vector with hsp60 promoter of mycobacteria. The cloning was performed as described previously.91 The resulting plasmid pGMCZ-phsp60-SD1-BioBtb was integrated into the attachment site of the phage L5 by transformation of M. tuberculosis H37Rv and selection with zeocin.
In vitro BioB inhibition assay.
General methods:
E. coli biotin synthase (EcBioB),37 M. tuberculosis biotin synthase (MtBioB),33 flavodoxin (FLD),42 ferredoxin(flavodoxin):NADP+ oxidoreductase (FNR)42 and methylthioadenosine nucleosidase (MtnN)90 were expressed and purified as previously reported. S-adenosyl-L-methionine (SAM) was synthesized enzymatically as previously described.37 Nicotinamide adenine dinucleotide phosphate (NADPH) stock solution concentration was determined spectrophotometrically using ε340 = 2600 M−1 cm−1 at pH 7. All other reagents were purchased from commercial sources and used without further purification.
Enzyme assay:
All assays were performed as previously described to obtain baseline activity for EcBioB.37 Briefly, 45 min prior to the start of the assay, all materials were transferred into an anaerobic chamber (< 1 ppm O2). Each reaction was carried out in a final volume of 200 μL, and final concentrations of materials are indicated in parentheses. EcBioB (5 μM dimer) was added to Tris-HCl pH 8.0 (50 mM), KCl (100 mM), and DTT (5 mM). Na2S (80 μM) and (NH4)2Fe(SO4)2 (80 μM) were added and incubated for 10 min at 23 ⁰C to reconstitute the [4Fe-4S]2+ cluster. FLD (15 μM), FNR (5 μM), NADPH (1 mM), MtnN (70 nM), dethiobiotin (5–30 μM) and acidomycin (0–30 μM) were added and incubated for 10 min at 37 ⁰C. The reaction was initiated by the addition of SAM (100 μM), and reactions were incubated for 6 min at 37 ⁰C (unless otherwise stated). The reaction was then quenched with 5 M sodium acetate pH 4 (20 μL) and incubated at 0 ⁰C for 15 min to precipitate proteins. The precipitate was removed by centrifugation (18,000 × g for 10 min), and the supernatant was collected and analyzed by LC-MS.
Instrumentation and quantification:
Samples were analyzed by LC-MS (Agilent 1100 series, Agilent Technologies, Inc., Santa Clara, CA). Reverse-phase LC was performed on an Eclipse XDB C-18 column (50 mm × 4.6 mm, 5.0 μm particle size; Agilent Technologies, Inc.) operating at 0.700 mL/min with UV detection at 254 nm employing a linear gradient of 5 to 30 % MeCN + 0.1 % formic acid (solvent B) in water + 0.1 % formic acid (solvent A) for 9 minutes. The injection volume was 10 μL. The LC system was coupled to an Agilent 6400 triple-quandrupole mass spectrometer (Agilent Technologies, Inc., Santa Clara, CA) and operated in ESI positive mode with the mass range m/z 100–400 at 0.99 scan/sec. Biotin (tR = 6.0 min, m/z 245.1 [M + H]+, m/z 227.1 [M – H2O + H]+, m/z 267.1 [M + Na]+), acidomycin (tR = 7.8 min, m/z 218.1 [M + H]+, m/z 200.0 [M – H2O + H]+, m/z 240.1 [M + Na]+) and caffeine (tR = 4.5 min, m/z 195.1 [M + H]+, internal standard) were quantified by integration of peak area in the extracted ion chromatogram and peak areas were normalized to internal standard peak areas, and the analyte concentrations were determined with an appropriate standard curve.
Intracellular accumulation of antibiotics:
Accumulation assay:
Assay conditions were adapted and slightly modified from previously reported conditions.51 Briefly, E. coli K-12 or M. tuberculosis H37Ra were grown overnight at 37 °C in either: minimal (M9) medium, or M9 medium supplemented with 1 μM biotin for E. coli; biotin-free 7H9 medium (described above), or 7H9 medium for M. tuberculosis. Overnight cultures were then diluted (1:10, 250 mL) into fresh M9, LB (E. coli) or biotin-free 7H9, 7H9 medium (M. tuberculosis) and grown to an optical density (OD600) of ~0.40–0.60. The bacteria were pelleted by centrifugation (3,000 × g for 10 min), resuspended in 40 mL phosphate buffered saline (PBS), and pelleted again as before. The supernatant was discarded, the cells resuspended in PBS (10 mL) and aliquoted into Eppendorf tubes (450 μL each). The colony-forming units (CFUs) were obtained from the remaining stock of resuspended cells in PBS by a calibration curve on LB agar (E. coli) or 7H9 (M. tuberculosis) agar plates. The samples were equilibrated at 37 °C for 5 min, then drug (acidomycin, tetracycline, rifampicin, or thioridazine) was added (50 μL of a 500 μM stock, giving a final concentration of 50 μM) to each sample, which were incubated at 37 °C for 10 min. After incubation, 400 μL of each sample was layered onto 400 μL of silicone oil (9:1 AR-20:Sigma high temperature oil, cooled to −78 °C). Bacteria were pelleted through the oil by centrifugation (13,000 × g for 5 min). Both the supernatant layers were removed, and the pellets were resuspended in H2O (200 μL). The cells were lysed by multiple rounds of freeze-thaw (–78 to 65 °C) cycles. The lysates were diluted (1:1) with 10% (w/v) aqueous TCA (trichloroacetic acid) + 500 nM biotin-d4 (as an internal standard), filtered through 0.2 μm filters, and analyzed by LC-MS/MS. Each compound was run in triplicate and experimental results were verified with two independent experiments.
Instrumentation and quantification:
Samples were analyzed by LC-MS/MS (Shimadzu UFLC XR-AB SCIEX QTRAP 5500). Reverse-phase LC was performed on a Kinetix C-18 column (50 mm × 2.1 mm, 2.6 μm particle size; Phenomenex, Torrance, CA) operating with a flow rate was 0.500 mL/min and column oven at 40 °C. Solvent A was 5% (v/v) MeCN in 10 mM ammonium formate pH 4.5 + 0.1% aqueous formic acid and solvent B was 90% (v/v) MeCN in 10 mM ammonium formate pH 4.5 + 0.1% aqueous formic acid for acidomycin, biotin, rifampicin and tetracycline. Solvent A was water + 0.1% aqueous formic acid and solvent B was MeCN + 0.1% aqueous formic acid for thioridazine. Initial conditions were 5% B from 0 to 0.5 min, after which the %B was increased to 95% from 0.5 to 3 min. The column was washed in 95% B for 2 min, returned to 5% over 0.2 min, and allowed to re-equilibrate for 2.8 min in 5% B to provide a total run time of 8 min. The injection volume was 10 μL. All analytes were analyzed by MS in positive ionization mode by Multiple Reaction Monitoring (MRM), and quantified by integration of peak area in the extracted ion chromatogram. The mass spectrometry settings were optimized by direct infusion of authentic standards for: acidomycin, biotin, rifampicin, tetracycline and thioridazine. The collision energy ranged from +20 to +36 V; the declustering potential was +35 V. The following transitions were observed: acidomycin (tR = 1.9 min, m/z 227.1 [M + H]+ → m/z 200.1 [M – H2O + H]+), biotin (tR = 1.5 min, m/z 245.1 [M + H]+ → m/z 227.1 [M – H2O + H]+), biotin-d4 (tR = 1.5 min, m/z 249.1 [M + H]+ → m/z 231.1 [M – H2O + H]+), rifampicin (tR = 2.6 min, m/z 823.2 [M + H]+ → m/z 791.4 [M – OH – Me + H]+), rifampicin quinone (tR = 2.6 min, m/z 821.2 [M + H]+ → m/z 789.4 [M – OH – Me + H]+), tetracycline (tR = 1.5 min, m/z 445.2 [M + H]+ → m/z 410.1 [M – H2O – OH + H]+), thioridazine (tR = 3.1 min, m/z 371.1 [M + H]+ → m/z 126.1). Analyte and internal standard peak areas were calculated (MultiQuant, version 2.0.2, AB SCIEX). Analyte peak areas were normalized to internal standard peak areas, and the analyte concentrations were determined with an appropriate standard curve.
Biotinylated Proteome Assay.
This experiment was performed according to a previous procedure.56 Briefly, a 30 mL culture of E. coli K12 in either biotin-containing M9 (supplemented with 0.2% glucose and 1 mM biotin) or M9 (supplemented with 0.2% glucose, biotin-free) media were grown at 37 °C until an OD600 ~0.1 was achieved. Each culture was split unto 3 × 10 mL cultures, and were treated with 0, 1, 10, or 100 μM of acidomycin. The cultures were then grown until OD600 ~0.6 was reached, and the cells were harvested via centrifugation (10 min, 5,000 × g) and the cell pellets were washed once with PBS (10 mM sodium phosphate and 150 mM NaCl, pH 7.4) and harvested via centrifugation (10 min, 5,000 × g). The pellets were resuspended in 1 mL PBS + inhibitor cocktail (1 EDTA-free tablet per 10 mL PBS, Roche), and subjected to bead beating (250 μL worth of zirconium beads + 500 μL of cell suspension). The lysate was separated from the cellular debris via centrifugation (30 min, 13,000 × g). For M. tuberculosis lysates: a 30 mL culture of M. tuberculosis H37Ra in either 7H9 or biotin-free 7H9 media were grown at 37 °C until an OD600 ~0.1 was achieved. Each culture was split unto 3 × 10 mL cultures, and were treated with 0, 1, or 10 μM of (S)-(−)-acidomycin or 0, 0.78, or 7.8 μM of 5ʹ-[N-(D-biotinoyl)sulfamoyl]amino-5ʹ-deoxyadenosine (Bio-AMS, synthesized as previously described).56 The cultures were then grown until OD600 ~0.6 was reached (24 h), and the cells were harvested, lysed and lysates were obtained the same way as described above. The total protein concentrations were determined for each sample and diluted with PBS until they were all 4.0 mg/mL. To the samples was then added 5× SDS reducing gel electrophoresis loading buffer, which were subsequently heated for 5 min at 80 °C. A 30 μL aliquot of each sample was separated by denaturing gel electrophoresis (4–20% Tris/Glycine Mini-Protean TGX, Bio-Rad) and was transferred to a nitrocellulose membrane (iBlot System, Invitrogen). The membrane was blocked with Tris-buffered saline (TBS) containing 0.05% Tween-20 (TBST) containing 1.0% BSA for 30 min at 25 °C. The membrane was incubated with an AlexaFluor 647 fluorescent-streptavidin conjugate (Invitrogen) at a dilution of 1:1000 in TBST for 1 h at 25 °C, after which the membrane was washed 3 × 10 mL TBST. Biotinylated proteins within the membrane were visualized by fluorescence scanning using a Typhoon imager (FLA 7000, General Electric). Band intensities were normalized for protein concentration using the ImageQuant software (v 7.0, General Electric) to provide the normalized percentage band intensity values.
Isothermal titration calorimetry (ITC).
All ITC experiments were conducted on an automated microcalorimeter (Malvern Instruments). The experiments were performed at 25 °C in ITC buffer (10 mM Tris pH 7.5, 30 mM NaCl, 200 mM KCl, 2.5 mM MgCl2). MtBPL was exchanged (3 × 10 mL) into ITC buffer using an Amicon Ultra concentrator, and the final filtrate was used to prepare a solution of the analogs from a 10 mM stock in DMSO. Final sample concentrations were 50 μM for MtBPL, 750 μM acidomycin and 750 μM biotin (determined by weighing sample on an ultramicrobalance [Mettler Toledo] accurate to 0.001 mg). In the direct titration experiments, the analogue was injected into a solution of the enzyme. Titrations were carried out with a stirring speed of 750 rpm and 200 s interval between 2 μL injections. The first injection for each sample was excluded from data fitting. Titrations were run past the point of enzyme saturation to correct for heats of dilution. The experimental data were fitted to a theoretical titration curve using the Origin software package (version 7.0) provided with the instrument to afford values of KA (the binding constant of the ligands in the presence of biotin in M−1), n (stoichiometry of binding) and ΔH (the binding enthalpy change in kcal/mol).
Pharmacokinetic studies.
All studies were carried out in accordance with the guide for the fair and ethical use of Laboratory Animals of the National Institutes of Health, with approval from the Institutional Animal Care and Use Committee (IACUC) of the New Jersey Medical School, Rutgers University, Newark under IACUC protocol number 15114. Drug administration: Acidomycin [both (+)- and (−)-] were evaluated in uninfected CD-1 mice to determine pharmacokinetic (PK) parameters. A single dose of (+)- or (−)-acidomycin was administered either intravenously (IV) at 5 mg/kg or orally (PO) at 25 mg/kg to groups of three mice (N = 3). Both the IV and PO formulations were the 2-ammonium-2-methylpropan-1,3-diol (AMP2) salt of (+)- and (−)-acidomycin in water. Blood was collected in K2EDTA-coated tubes pre-dose and at 1 min, 15 min, 1, 3, 5, and 8 h post-dose following IV administration or pre-dose and at 30 min, 15 min, 1, 3, 5, 8, and 24 h post-dose following PO administration. Plasma was obtained by centrifugation for 10 min at 5,000 rpm and stored at −80 °C until analyzed. Concentrations of both (+)- and (−)-acidomycin were determined as described below. The lower limit of quantification was 5 ng/mL. The PK parameters (area under the curve [AUC] from time 0 to time t [AUC0–t] and AUC from time 0 to 24 h [AUC0–24], peak [maximum] plasma concentration [Cmax], and half-life [t1/2]) were calculated from the mean concentrations using Microsoft Excel software (Office 2010; Microsoft Corp., Redmond, WA). AUCs were calculated using the linear trapezoidal rule. Half-life and elimination rate constants were calculated by linear regression using semilogarithmic concentration-versus-time data.
Instrumentation and quantification:
Neat 1 mg/mL DMSO stocks of all tested compounds (described above) were first serially diluted in MeCN:water (1:1), and subsequently serially diluted in drug-free CD-1 mouse plasma (K2EDTA; Bioreclamation IVT, NY) to create standard curves and quality control (QC) spiking solutions. 20 μL of standards, QC samples, control plasma, and study samples were extracted by adding 200 μL of MeCN:MeOH (1:1) protein precipitation solvent containing the internal standard (10 ng/mL verapamil). Extracts were vortexed for 5 min and centrifuged at 4,000 rpm for 5 min. One hundred microliters of supernatant were transferred for high-pressure liquid chromatography tandem mass spectrometry (LC–MS/MS) diluted with 100 μL of deionized water.
Extracts were then analyzed by LC–MS/MS (SCIEX QTRAP 6500+ triple-quadrupole mass spectrometer coupled to a Shimadzu 30ACMP HPLC). Reverse-phase LC was performed on an Agilent Zorbax SB-C8 column (30 mm × 2.1 mm, 3.6 μm particle size). Mobile phase A was 0.1% aqueous formic acid while mobile phase B was 0.1% formic acid in acetonitrile. Initial conditions were 5% to 90% B from 0 to 2 min, after which the %B was maintained at 90% from 2 to 3 min. The column was then returned to 5% B, and allowed to re-equilibrate for 0.5 min to provide a total run time of 3.5 min. The flow rate was 0.7 mL/min. The injection volume was 2 μL. All analytes were monitored by MS in negative ionization mode by Multiple Reaction Monitoring (MRM). The following transitions were observed: acidomycin (m/z 218.2 [M – H]− → m/z 158.1), diclofenac (m/z 293.2 [M – H]− → m/z 249.8, internal standard). Analyte and internal standard peak areas were integrated using Analyst (version 1.6.2, AB SCIEX). Analyte peak areas were normalized to internal standard peak areas and the analyte concentrations were determined with the appropriate standard curve.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant (AI091790 to D.S.) from the National Institutes of Health and in part by the Intramural Research Program of NIAID (AI000693-25). Mass spectrometry was carried out in the Center for Mass Spectrometry and Proteomics, University of Minnesota. Isothermal titration calorimetry was carried out using an ITC-200 microcalorimeter, funded by the NIH Shared Instrumentation Grant S10-OD017982. The Bruker-AXS D8 Venture diffractometer was purchased through a grant from NSF/MRI (#1229400) and the University of Minnesota. We gratefully acknowledge Colin Manoil for supplying A. baumannii AB5075, Ryan Hunter for supplying S. aureus USA300 and P. aeruginosa PA14, and Gary Dunny for suppling E. faecalis V583. We acknowledge the use of the Integrated Genomics Operation Core at MSKCC, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. The authors acknowledge the Minnesota Supercomputing Institute (MSI) at the University of Minnesota.
ABBREVIATIONS
- ACCs
acyl-CoA carboxylases
- ACM
acidomycin
- ACP
acyl-carrier protein
- AMP2
2-amino-2-methylpropan-1,3-diol
- AUC
area under the curve
- BioA
7,8-diaminopelargonic acid synthase
- BioB
biotin synthase
- BioD
dethiobiotin synthetase
- BioF
7-keto-8-aminopelargonic acid synthase
- BirA
biotin protein ligase
- BPL
biotin protein ligase
- CL
clearance
- DAPA
7,8-diaminopelargonic acid
- DTB
dethiobiotin
- DR
drug-resistant
- DUC
dual-control
- EcBioB
E. coli biotin synthase
- FdrA
ferredoxin reductase A
- FOR
frequency of resistance
- FLD
flavodoxin
- FLDR
flavodoxin reductase
- FprA
flavoprotein reductase A
- HPLC
high performance liquid chromatography
- IC/EC
intracellular concentration/extracellular concentration
- ITC
isothermal titration calorimetry
- IV
intravenous
- KAPA
7-keto-8-aminopelargonic acid
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- mCPBA
meta-chloroperoxybenzoic acid
- MDR-TB
multidrug-resistant TB
- MIC
minimum inhibitory concentration
- MTBC
M. tuberculosis complex
- MtBioB
M. tuberculosis biotin synthase
- MtBPL
M tuberculosis biotin protein ligase
- NTM
non-tuberculosis mycobacteria
- PK
pharmacokinetics
- PO
oral
- PTSA
para-toluenesulfonic acid
- SAM
S-adenosyl-l-methionine
- SAR
structure-activity relationship
- TB
tuberculosis
- TEMPO
(2,2,6,6-tetramethylpiperidin-1-yl)oxyl
- WT
wild type
- XDR-TB
extensively drug-resistant TB
- XRD
X-ray diffraction
Footnotes
Supporting Information: The Supporting Information is available free of charge via the Internet at http://pubs.acs.org. A complete list of MIC values for M(X)DR and DS M. tuberculosis, grow inhibition concentration-response plots for MIC determination, HPLC traces for analytes, LC-MS/MS parameters and fragmentation data, raw values for accumulation assays, far-western blots for E. coli and M. tuberculosis lysates, homology model experimentals, X-ray crystallographic data for acidomycin and 1H and 13C NMR of all compounds.
The authors declare no competing financial interests.
REFERENCES
- (1).WHO Global tuberculosis report; World Health Organization; Geneva, Switzerland, 2018. [Google Scholar]
- (2).Brites D; Gagneux S, Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol. Rev. 2015, 264, 6–24. DOI: 10.1111/imr.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gagneux S, Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol 2018, 16, 202–213. DOI: 10.1038/nrmicro.2018.8. [DOI] [PubMed] [Google Scholar]
- (4).Organization W. H., Treatment of tuberculosis. Guidelines. 4th ed.; WHO/HTM/TB/2009.420: 2010. [PubMed] [Google Scholar]
- (5).Wohlleben W; Mast Y; Stegmann E; Ziemert N, Antibiotic drug discovery. Microb. Biotechnol 2016, 9, 541–548. DOI: 10.1111/1751-7915.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Baltz R, Antibiotic discovery from actinomycetes: Will a renaissance follow the decline and fall? SIM News 2005, 55, 186–196. [Google Scholar]
- (7).Grundy WE; Whitman AL; Rdzok EG; Rdzok EJ; Hanes ME; Sylvester JC, Actithiazic acid. I. Microbiological studies. Antibiot. Chemother. (Northfield) 1952, 2, 399–408. [PubMed] [Google Scholar]
- (8).Sobin BA, A New Streptomyces Antibiotic. J. Am. Chem. Soc 1952, 74, 2947–2948. DOI: 10.1021/ja01131a526. [DOI] [Google Scholar]
- (9).Tejera E; Backus EJ; Dann M; Ervin CD; Shakofski AJ; Thomas SO; Bohonos N; Williams JH, Mycobacidinian antibiotic active against acid-fast organisms. Antibiot. Chemother. (Northfield) 1952, 2, 333. [PubMed] [Google Scholar]
- (10).Schenck JR; De Rose AF, Actithiazic acid. II. Isolation and characterization. Arch. Biochem. Biophys 1952, 40, 263–269. DOI: 10.1016/0003-9861(52)90110-0. [DOI] [PubMed] [Google Scholar]
- (11).Hamada Y; Kawashima M; Miyake A; Okamoto K, Studies on acidomycin. V. Anti-acidomycin factor in rabbit’s urine observed by cylinder plate method. J. Antibiot. (Tokyo) 1953, 6, 158–162. [PubMed] [Google Scholar]
- (12).Maeda K, Chemical studies on antibiotic substances. IV. A crystalline toxic substance of Streptomyces thioluteus producing aureothricin. J. Antibiot. (Tokyo) 1953, 6, 137–138. [PubMed] [Google Scholar]
- (13).Umezawa H; Oikawa K; Okami Y; Maeda K, Thiazolidone antibiotic as an antimetabolite to biotin. J. Bacteriol 1953, 66, 118–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McLamore WM; Celmer WD; Bogert VV; Pennington FC; Sobin BA; Solomons IA, Structure and Synthesis of a New, Thiazolidone Antibiotic. J. Am. Chem. Soc. 1 1953, 75, 105–108. DOI: 10.1021/ja01097a030. [DOI] [Google Scholar]
- (15).Kawashima M; Fujii S, Biochemical studies on acidomycin. II. Increase of urinary biotin excretion of acidomycin-treated rabbits. Pharm. Bull 1953, 1, 328–331. DOI: 10.1248/cpb1953.1.328. [DOI] [PubMed] [Google Scholar]
- (16).Umezawa H; Oikawa K; Maeda K; Okami Y, Anti-biotin activity of thiazolidone antibiotic. Jpn. J. Med. Sci. Biol. 1953, 6, 395–403. [DOI] [PubMed] [Google Scholar]
- (17).Stim TB; Arnwine BC; Foster JW, Further studies on potentiation of inhibitor action through determination of reversing metabolites. J. Bacteriol 1959, 77, 566–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Takayama K; Wang C; Besra GS, Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev 2005, 18, 81–101. DOI: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Eisenreich W; Dandekar T; Heesemann J; Goebel W, Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat. Rev. Microbiol 2010, 8, 401–412. DOI: 10.1038/nrmicro2351. [DOI] [PubMed] [Google Scholar]
- (20).Woong Park S; Klotzsche M; Wilson DJ; Boshoff HI; Eoh H; Manjunatha U; Blumenthal A; Rhee K; Barry CE III; Aldrich CC; Ehrt S; Schnappinger D, Evaluating the Sensitivity of Mycobacterium tuberculosis to Biotin Deprivation Using Regulated Gene Expression. PLoS Pathog. 2011, 7, e1002264 DOI: 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Salaemae W; Azhar A; Booker G; Polyak S, Biotin biosynthesis in Mycobacterium tuberculosis: physiology, biochemistry and molecular intervention. Protein Cell 2011, 2, 691–695. DOI: 10.1007/s13238-011-1100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lazar N; Fay A; Nandakumar M; Boyle KE; Xavier J; Rhee K; Glickman MS, Control of biotin biosynthesis in mycobacteria by a pyruvate carboxylase dependent metabolic signal. Mol. Microbiol 2017, 106, 1018–1031. DOI: 10.1111/mmi.13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bansal-Mutalik R; Nikaido H, Mycobacterial outer membrane is a lipid bilayer and the inner membrane is unusually rich in diacyl phosphatidylinositol dimannosides. Proc. Natl. Acad. Sci. USA 2014, 111, 4958–4963. DOI: 10.1073/pnas.1403078111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Park SW; Klotzsche M; Wilson DJ; Boshoff HI; Eoh H; Manjunatha U; Blumenthal A; Rhee K; Barry CE; Aldrich CC; Ehrt S; Schnappinger D, Evaluating the Sensitivity of Mycobacterium tuberculosis to Biotin Deprivation Using Regulated Gene Expression. PloS Pathog. 2011, 7 DOI: 10.1371/journal.ppat.1002264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tiwari D; Park SW; Essawy MM; Dawadi S; Mason A; Nandakumar M; Zimmerman M; Mina M; Ho HP; Engelhart CA; Ioerger T; Sacchettini JC; Rhee K; Ehrt S; Aldrich CC; Dartois V; Schnappinger D, Targeting protein biotinylation enhances tuberculosis chemotherapy. Science Translational Medicine 2018, 10 DOI: 10.1126/scitranslmed.aal1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Clark RK Jr.; Schenck JR, Actithiazic acid. III. The synthesis of DL-actithiazic acid, derivatives and homologs. Arch. Biochem. Biophys 1952, 40, 270–276. DOI: 10.1016/0003-9861(52)90111-2. [DOI] [PubMed] [Google Scholar]
- (27).Miyake A; Morimoto A; Kinoshita T, Studies on Antibiotics. I. : Acidomycin. (1). : Isolation and Chemical Structure. Pharm. Bull 1953, 1, 84–88. DOI: 10.1248/cpb1953.1.84. [DOI] [Google Scholar]
- (28).Latli B; Eriksson M; Hrapchak M; Busacca CA; Senanayake CH, A potent IkappaB kinase-beta inhibitor labeled with carbon-14 and deuterium. J. Labelled Comp. Radiopharm 2016, 59, 300–304. DOI: 10.1002/jlcr.3398. [DOI] [PubMed] [Google Scholar]
- (29).Wu ML; Aziz DB; Dartois V; Dick T, NTM drug discovery: status, gaps and the way forward. Drug Discov. Today 2018. DOI: 10.1016/j.drudis.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kawashima M; Miyake A; Hemmi T; Fujii S, Biochemical studies on acidomycin. III. Antibiotin activity of acidomycin and its related compounds. Pharm. Bull 1956, 4, 53–55. [DOI] [PubMed] [Google Scholar]
- (31).Eisenberg MA; Hsiung SC, Mode of action of the biotin antimetabolites actithiazic acid and alpha-methyldethiobiotin. Antimicrob. Agents. Chemother 1982, 21, 5–10. DOI: 10.1128/AAC.21.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Klotzsche M; Ehrt S; Schnappinger D, Improved tetracycline repressors for gene silencing in mycobacteria. Nucleic Acids Res. 2009, 37, 1778–1788. DOI: 10.1093/nar/gkp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Magwamba CC; Rukseree K; Palittapongarnpim P, Cloning, expression and characterization of histidine-tagged biotin synthase of Mycobacterium tuberculosis. Tuberculosis 2016, 98, 42–49. DOI: 10.1016/j.tube.2016.02.006. [DOI] [PubMed] [Google Scholar]
- (34).Berkovitch F; Nicolet Y; Wan JT; Jarrett JT; Drennan CL, Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 2004, 303, 76–79. DOI: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Broach RB; Jarrett JT, Role of the [2Fe-2S]2+ cluster in biotin synthase: mutagenesis of the atypical metal ligand arginine 260. Biochemistry 2006, 45, 14166–14174. DOI: 10.1021/bi061576p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Farrar CE; Jarrett JT, Protein residues that control the reaction trajectory in S-adenosylmethionine radical enzymes: mutagenesis of asparagine 153 and aspartate 155 in Escherichia coli biotin synthase. Biochemistry 2009, 48, 2448–2458. DOI: 10.1021/bi8022569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Farrar CE; Siu KK; Howell PL; Jarrett JT, Biotin synthase exhibits burst kinetics and multiple turnovers in the absence of inhibition by products and product-related biomolecules. Biochemistry 2010, 49, 9985–9996. DOI: 10.1021/bi101023c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Fugate CJ; Jarrett JT, Biotin synthase: insights into radical-mediated carbon-sulfur bond formation. Biochim. Biophys. Acta 2012, 1824, 1213–1222. DOI: 10.1016/j.bbapap.2012.01.010. [DOI] [PubMed] [Google Scholar]
- (39).Fugate CJ; Stich TA; Kim EG; Myers WK; Britt RD; Jarrett JT, 9-Mercaptodethiobiotin is generated as a ligand to the [2Fe-2S]+ cluster during the reaction catalyzed by biotin synthase from Escherichia coli. J. Am. Chem. Soc 2012, 134, 9042–9045. DOI: 10.1021/ja3012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Reyda MR; Dippold R; Dotson ME; Jarrett JT, Loss of iron-sulfur clusters from biotin synthase as a result of catalysis promotes unfolding and degradation. Arch. Biochem. Biophys 2008, 471, 32–41. DOI: 10.1016/j.abb.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Reyda MR; Fugate CJ; Jarrett JT, A complex between biotin synthase and the iron-sulfur cluster assembly chaperone HscA that enhances in vivo cluster assembly. Biochemistry 2009, 48, 10782–10792. DOI: 10.1021/bi901393t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Taylor AM; Farrar CE; Jarrett JT, 9-Mercaptodethiobiotin is formed as a competent catalytic intermediate by Escherichia coli biotin synthase. Biochemistry 2008, 47, 9309–9317. DOI: 10.1021/bi801035b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Taylor AM; Stoll S; Britt RD; Jarrett JT, Reduction of the [2Fe-2S] cluster accompanies formation of the intermediate 9-mercaptodethiobiotin in Escherichia coli biotin synthase. Biochemistry 2011, 50, 7953–7963. DOI: 10.1021/bi201042r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ugulava NB; Frederick KK; Jarrett JT, Control of adenosylmethionine-dependent radical generation in biotin synthase: a kinetic and thermodynamic analysis of substrate binding to active and inactive forms of BioB. Biochemistry 2003, 42, 2708–2719. DOI: 10.1021/bi0261084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ugulava NB; Gibney BR; Jarrett JT, Iron-sulfur cluster interconversions in biotin synthase: dissociation and reassociation of iron during conversion of [2Fe-2S] to [4Fe-4S] clusters. Biochemistry 2000, 39, 5206–5214. DOI: 10.1021/bi9926227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ugulava NB; Gibney BR; Jarrett JT, Biotin synthase contains two distinct iron-sulfur cluster binding sites: chemical and spectroelectrochemical analysis of iron-sulfur cluster interconversions. Biochemistry 2001, 40, 8343–8351. DOI: 10.1021/bi0104625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ugulava NB; Sacanell CJ; Jarrett JT, Spectroscopic changes during a single turnover of biotin synthase: destruction of a [2Fe-2S] cluster accompanies sulfur insertion. Biochemistry 2001, 40, 8352–8358. DOI: 10.1021/bi010463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ugulava NB; Surerus KK; Jarrett JT, Evidence from Mossbauer spectroscopy for distinct [2Fe-2S](2+) and [4Fe-4S](2+) cluster binding sites in biotin synthase from Escherichia coli. J. Am. Chem. Soc 2002, 124, 9050–9051. DOI: 10.1021/ja027004j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Cramer JD; Jarrett JT, Purification, Characterization, and Biochemical Assays of Biotin Synthase From Escherichia coli. Methods Enzymol 2018, 606, 363–388. DOI: 10.1016/bs.mie.2018.06.003. [DOI] [PubMed] [Google Scholar]
- (50).Segel IH, Enzyme kinetics: Behavior and analysis of rapid equilibrium and steady-state enzyme systems. Wiley-Interscience: 1976; p 957. DOI: doi: 10.1002/kin.550080117. [DOI] [Google Scholar]
- (51).Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ, Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299 DOI: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Sarathy J; Dartois V; Dick T; Gengenbacher M, Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 1648–1653. DOI: 10.1128/AAC.02202-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kubitschek HE; Friske JA, Determination of bacterial cell volume with the Coulter Counter. J. Bacteriol 1986, 168, 1466–1467. DOI: 10.1128/jb.168.3.1466-1467.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Piddock LJ; Ricci V, Accumulation of five fluoroquinolones by Mycobacterium tuberculosis H37Rv. J. Antimicrob. Chemother 2001, 48, 787–791. DOI: 10.1093/jac/48.6.787. [DOI] [PubMed] [Google Scholar]
- (55).Danilchanka O; Mailaender C; Niederweis M, Identification of a novel multidrug efflux pump of Mycobacterium tuberculosis. Antimicrob. Agents Chemother 2008, 52, 2503–2511. DOI: 10.1128/AAC.00298-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Duckworth BP; Geders TW; Tiwari D; Boshoff HI; Sibbald PA; Barry CE III; Schnappinger D; Finzel BC; Aldrich CC, Bisubstrate Adenylation Inhibitors of Biotin Protein Ligase from Mycobacterium tuberculosis. Chem. Biol 2011, 18, 1432–1441. DOI: 10.1016/j.chembiol.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).David SE; Timmins P; Conway BR, Impact of the counterion on the solubility and physicochemical properties of salts of carboxylic acid drugs. Drug Dev. Ind. Pharm 2012, 38, 93–103. DOI: 10.3109/03639045.2011.592530. [DOI] [PubMed] [Google Scholar]
- (58).Ogata K; Izumi Y; Tani Y, Control Action of Actithiazic Acid on the Biosynthesis of Biotin-vitamers by Microorganisms. Agric. Biol. Chem 1970, 34, 1872–1874. DOI: 10.1080/00021369.1970.10859865. [DOI] [Google Scholar]
- (59).Ogata K; Izumi Y; Tani Y, The Controlling Action of Actithiazic Acid on the Biosynthesis of Biotin-vitamers by Various Microorganisms. Agric. Biol. Chem 1973, 37, 1079–1085. DOI: 10.1271/bbb1961.37.1079. [DOI] [Google Scholar]
- (60).Ifuku O; Kishimoto J; Haze S; Yanagi M; Fukushima S, Conversion of dethiobiotin to biotin in cell-free extracts of Escherichia coli. Biosci. Biotechnol. Biochem 1992, 56, 1780–1785. DOI: 10.1271/bbb.56.1780. [DOI] [PubMed] [Google Scholar]
- (61).Duin EC; Lafferty ME; Crouse BR; Allen RM; Sanyal I; Flint DH; Johnson MK, [2Fe-2S] to [4Fe-4S] cluster conversion in Escherichia coli biotin synthase. Biochemistry 1997, 36, 11811–11820. DOI: 10.1021/bi9706430. [DOI] [PubMed] [Google Scholar]
- (62).Ollagnier-de-Choudens S; Mulliez E; Fontecave M, The PLP-dependent biotin synthase from Escherichia coli: mechanistic studies. FEBS Lett. 2002, 532, 465–468. DOI: 10.1016/S0014-5793(02)03733-X [DOI] [PubMed] [Google Scholar]
- (63).Ollagnier-de Choudens S; Sanakis Y; Hewitson KS; Roach P; Munck E; Fontecave M, Reductive cleavage of S-adenosylmethionine by biotin synthase from Escherichia coli. J. Biol. Chem 2002, 277, 13449–13454. DOI: 10.1074/jbc.M111324200. [DOI] [PubMed] [Google Scholar]
- (64).Cosper MM; Jameson GN; Hernandez HL; Krebs C; Huynh BH; Johnson MK, Characterization of the cofactor composition of Escherichia coli biotin synthase. Biochemistry 2004, 43, 2007–2021. DOI: 10.1021/bi0356653. [DOI] [PubMed] [Google Scholar]
- (65).Tse Sum Bui B; Lotierzo M; Escalettes F; Florentin D; Marquet A, Further investigation on the turnover of Escherichia coli biotin synthase with dethiobiotin and 9-mercaptodethiobiotin as substrates. Biochemistry 2004, 43, 16432–16441. DOI: 10.1021/bi048040t. [DOI] [PubMed] [Google Scholar]
- (66).Jarrett JT, Biotin Synthase: Enzyme or Reactant? Chem. Biol 2005, 12, 409–410. DOI: 10.1016/j.chembiol.2005.04.003. [DOI] [PubMed] [Google Scholar]
- (67).Wan JT; Jarrett JT, Electron acceptor specificity of ferredoxin (flavodoxin):NADP+ oxidoreductase from Escherichia coli. Arch Biochem Biophys 2002, 406, 116–126. DOI: 10.1016/S0003-9861(02)00421-6. [DOI] [PubMed] [Google Scholar]
- (68).Lu Y; Qiao F; Li Y; Sang XH; Li CR; Jiang JD; Yang XY; You XF, Recombinant expression and biochemical characterization of Mycobacterium tuberculosis 3Fe-4S ferredoxin Rv1786. Appl. Microbiol. Biotechnol 2017, 101, 7201–7212. DOI: 10.1007/s00253-017-8454-7. [DOI] [PubMed] [Google Scholar]
- (69).Choi-Rhee E; Cronan JE, A nucleosidase required for in vivo function of the S-adenosyl-L-methionine radical enzyme, biotin synthase. Chem. Biol 2005, 12, 589–593. DOI: 10.1016/j.chembiol.2005.04.012. [DOI] [PubMed] [Google Scholar]
- (70).Boissier F; Bardou F; Guillet V; Uttenweiler-Joseph S; Daffe M; Quemard A; Mourey L, Further insight into S-adenosylmethionine-dependent methyltransferases: structural characterization of Hma, an enzyme essential for the biosynthesis of oxygenated mycolic acids in Mycobacterium tuberculosis. The Journal of biological chemistry 2006, 281, 4434–4445. DOI: 10.1074/jbc.M510250200. [DOI] [PubMed] [Google Scholar]
- (71).Berney M; Berney-Meyer L; Wong KW; Chen B; Chen M; Kim J; Wang J; Harris D; Parkhill J; Chan J; Wang F; Jacobs WR Jr., Essential roles of methionine and S-adenosylmethionine in the autarkic lifestyle of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2015, 112, 10008–10013. DOI: 10.1073/pnas.1513033112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Howe MD; Kordus SL; Cole MS; Bauman AA; Aldrich CC; Baughn AD; Minato Y, Methionine antagonizes para-aminosalicylic acid activity via affecting folate precursor biosynthesis pathway in Mycobacterium tuberculosis. bioRxiv 2018. DOI: 10.1101/319038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Hedgecock LW, Antagonism of the inhibitory action of aminosalicylic acid on Mycobacterium tuberculosis by methionine, biotin and certain fatty acids, amino acids, and purines. J. Bacteriol 1956, 72, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Palmer LD; Downs DM, The thiamine biosynthetic enzyme ThiC catalyzes multiple turnovers and is inhibited by S-adenosylmethionine (AdoMet) metabolites. J. Biol. Chem. 2013, 288, 30693–30699. DOI: 10.1074/jbc.M113.500280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Tipton PA; Cleland WW, Mechanisms of decarboxylation of carboxybiotin. J. Am. Chem. Soc 1988, 110, 5866–5869. DOI: 10.1021/ja00225a043. [DOI] [Google Scholar]
- (76).Jarrett JT, Biotin synthase: A role for iron-sulfur clusters in the radical-mediated generation of carbon-sulfur bonds. 2014; p 107–131. DOI: 10.1515/9783110480436-009. [DOI] [Google Scholar]
- (77).Brown DG; May-Dracka TL; Gagnon MM; Tommasi R, Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J. Med. Chem 2014, 57, 10144–10161. DOI: 10.1021/jm501552x. [DOI] [PubMed] [Google Scholar]
- (78).Silver LL, A Gestalt approach to Gram-negative entry. Bioorg. Med. Chem 2016, 24, 6379–6389. DOI: 10.1016/j.bmc.2016.06.044. [DOI] [PubMed] [Google Scholar]
- (79).Six DA; Krucker T; Leeds JA, Advances and challenges in bacterial compound accumulation assays for drug discovery. Curr. Opin. Chem. Biol 2018, 44, 9–15. DOI: 10.1016/j.cbpa.2018.05.005. [DOI] [PubMed] [Google Scholar]
- (80).Iyer R; Ye Z; Ferrari A; Duncan L; Tanudra MA; Tsao H; Wang T; Gao H; Brummel CL; Erwin AL, Evaluating LC-MS/MS To Measure Accumulation of Compounds within Bacteria. ACS Infect. Dis 2018. DOI: 10.1021/acsinfecdis.8b00083. [DOI] [PubMed] [Google Scholar]
- (81).Dhyse FG; Hertz R, The effects of actithiazic acid on egg white-induced biotin deficiency and upon the microbial formation of biotin vitamers in the rat. Arch. Biochem. Biophys 1958, 74, 7–16. DOI: 10.1016/0003-9861(58)90194-2. [DOI] [PubMed] [Google Scholar]
- (82).Hwang K, Actithiazic acid. IV. Pharmacological studies. Antibiot. Chemother. (Northfield) 1952, 2, 453–459. [PubMed] [Google Scholar]
- (83).Kim JH; O’Brien KM; Sharma R; Boshoff HI; Rehren G; Chakraborty S; Wallach JB; Monteleone M; Wilson DJ; Aldrich CC; Barry CE 3rd; Rhee KY; Ehrt S; Schnappinger D, A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad. Sci. USA 2013, 110, 19095–19100. DOI: 10.1073/pnas.1315860110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Iwahara S; Kikuchi M; Tochikura T; Ogata K, Some Properties of Avidin-uncombinable Unknown Biotin-Vitamer Produced by Bacillus sp. and its Role in Biosynthesis of Desthiobiotin. Agric. Biol. Chem 1966, 30, 304–306. DOI: 10.1080/00021369.1966.10858596. [DOI] [Google Scholar]
- (85).Miyake A, Studies on Antibiotics. II. : Acidomycin. (2). : Antitubercular Activity of Compounds Related to Acidomycin. Pharm. Bull 1953, 1, 89–93. DOI: 10.1248/cpb1953.1.89. [DOI] [Google Scholar]
- (86).Pennington FC; Celmer WD; McLamore WM; Bogert VV; Solomons IA, Microbiologically Active 4-Thiazolidones. J. Am. Chem. Soc. 1 1953, 75, 109–114. DOI: 10.1021/ja01097a031. [DOI] [Google Scholar]
- (87).Kai K; Takeuchi J; Kataoka T; Yokoyama M; Watanabe N, Structure–activity relationship study of flowering-inducer FN against Lemna paucicostata. Tetrahedron 2008, 64, 6760–6769. DOI: 10.1016/j.tet.2008.04.115. [DOI] [Google Scholar]
- (88).Song T; Park Y; Shamputa IC; Seo S; Lee SY; Jeon HS; Choi H; Lee M; Glynne RJ; Barnes SW; Walker JR; Batalov S; Yusim K; Feng S; Tung CS; Theiler J; Via LE; Boshoff HI; Murakami KS; Korber B; Barry CE 3rd; Cho SN, Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the beta’ subunit of RNA polymerase. Mol. Microbiol 2014, 91, 1106–1119. DOI: 10.1111/mmi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Minato Y; Dawadi S; Kordus SL; Sivanandam A; Aldrich CC; Baughn AD, Mutual potentiation drives synergy between trimethoprim and sulfamethoxazole. Nat. Commun 2018, 9, 1003 DOI: 10.1038/s41467-018-03447-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Lee JE; Cornell KA; Riscoe MK; Howell PL, Expression, purification, crystallization and preliminary X-ray analysis of Escherichia coli 5’-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Acta. Crystallogr. D Biol. Crystallogr 2001, 57, 150–152. DOI: 10.1107/S0907444900014669. [DOI] [PubMed] [Google Scholar]
- (91).Schnappinger D; O’Brien KM; Ehrt S Construction of conditional knockdown mutants in mycobacteria. Methods Mol. Biol 2015, 1285, 151–175. DOI: 10.1007/978-1-4939-2450-9_9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.