

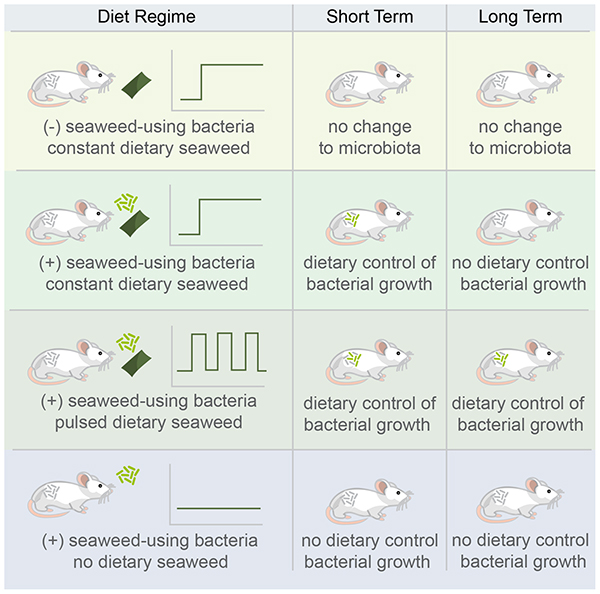
Summary
Interest in manipulating the gut microbiota to treat disease has led to a need for understanding how organisms can establish themselves when introduced into a host with an intact microbial community. Here, we employ the concept of orthogonal niche engineering: a resource typically absent from the diet, seaweed, creates a customized niche for an introduced organism. In the short term, co-introduction of this resource at 1% in the diet along with an organism with exclusive access to this resource, Bacteroides plebeius DSM 17135, enables it to colonize at a median abundance of 1%, and frequently up to 10 or more percent, both on pulsed and constant seaweed diets. In a two-month follow-up after the initial treatment period, B. plebeius stops responding to seaweed in mice initially on the constant seaweed diet, suggesting treatment regime will affect controllability. These results offer potential for diet-based intervention to introduce and control target organisms.
Graphical Abstract
INTRODUCTION
Introducing new bacteria into an intact or disturbed microbial community is one of the primary goals of microbial therapeutics. However, we have limited knowledge about the features that govern successful colonization by introduced microorganisms. Recent work has demonstrated that the presence of functional metabolic capacity beyond that of the endogenous microbiota contributed to persistence of colonization by an introduced Bifidobacterium species (Maldonado-Gómez et al., 2016). A strong predictor for engraftment of microorganisms introduced by fecal microbiota transplant (FMT) is the presence of shared organisms in the donor and recipient (Li et al., 2016; Smillie et al., 2018), likely indicating that shared functional capacity mediate colonization in this case. Further, transposon sequencing of a model gut community of Bacteroides species identified arabinoxylan as capable of modulating the abundance of a single strain (Wu et al., 2015). Given these findings, we hypothesized that we could control the colonization of an introduced organism in an intact community by providing it with exclusive access to a resource unusable by the rest of the community.
We identified a resource unlikely to be used by microorganisms in the lab-mouse gut: the red algae, Porphyra, comprising the edible seaweed nori. Nori contains complex, sulfated polysaccharides including porphyran, and unlike terrestrial plants, includes vitamin B12 (Hehemann et al., 2012), which has previously been implicated as a fitness determinant for Bacteroides thetaiotaomicron in the murine GI tract (Goodman et al., 2009). The pathways for degradation of porphyran are rare in the human gut microbiota, and almost exclusively found in metagenomic samples from individuals in populations known to eat seaweed (Pluvinage et al., 2018), primarily Japanese, who consume on average 5–10 g of seaweed daily (Hehemann et al., 2012). Horizontal gene transfer of the pathways necessary for breakdown of porphyran from a marine bacterium to B. plebeius, a gastrointestinal commensal, suggest strong selective pressures for the acquisition of this trait (Hehemann et al., 2010). This data suggests that communities without organisms capable of metabolizing certain dietary compounds (like algal polysaccharides) may be permissive to introduction of organisms capable of directly using them (like B. plebeius). Recent work demonstrated this concept in a B. ovatus strain and two other engineered strains capable of porphyran utilization (Shepherd et al., 2018).
Here, we confirmed the basic results of (Shepherd et al., 2018), and show that B. plebeius colonizes mouse guts at high levels in the presence of a preferred substrate (i.e. polysaccharides in seaweed), with no evidence of competition for this substrate from native gut bacteria. We provide a simple argument to suggest that B. plebeius competes with native microbes or the host for other nutrients to increase its biomass in the system. We conduct parametric analysis of a nonlinear dynamical model of the system to explain how apparently low levels of an exclusive resource can lead to abundant colonization of an introduced species. Enhanced colonization, however, comes at a cost to B. plebeius, with its levels depleted in the long term depending on the original seaweed treatment. We provide evidence to promote further investigation of the role of microbe-microbe and host-immune interaction in mediating these outcomes. These results provide a proof-of-principle for orthogonal niche engineering as a method for synbiotic design (i.e. controlling abundance of introduced microorganisms through manipulating resource availability) (Krumbeck et al., 2015; Panigrahi et al., 2017), but also reveal limitations to this strategy. In the context of microbial therapeutics, we show stable engraftment of a non-indigenous bacterial strain into an intact gut community in the presence of its engineered niche. However, our results also indicated that foreign microbes, when stimulated by an exclusive resource in the short term, might lose controllability in the long term.
RESULTS
Seaweed treatment does not change the composition of the mouse microbiota
We define orthogonal niche engineering as an approach involving the identification and use or engineering of an exclusive (or nearly exclusive) nutrient-species pair to introduce an organism into an intact ecosystem. This approach requires that the engineered niche (in this case through provisioning of seaweed), does not favor the growth of organisms already in a community more than the organism to be introduced (Freter, 1983). To validate that organisms in the mouse gut do not have the capacity to expand their populations significantly on seaweed-derived substrates, we introduced seaweed into mouse chow and followed the changes in the composition of the microbiota over time. We singly housed six-week old female C57BL/6 mice and randomly assigned them to two groups: (1) those receiving standard mouse chow (control) and (2) those receiving seaweed at 1% in their mouse chow (seaweed) (Fig 1A). Mice received seaweed chow continuously for 16 days, following a 32 day washout period and resumption of seaweed feeding for an additional 8 days to assess within-mouse reproducibility of the effects of seaweed feeding. Fecal samples were collected daily for the duration of the experiment, and temporally separated subsets were selected for V4 16S rDNA amplicon sequencing.
Figure 1.

Seaweed does not alter the microbiota of conventional mice. (A) Experimental design: samples were collected at the indicated time points for 16S rDNA sequencing. Green shaded region indicates period of seaweed feeding. (B) Shannon diversity over time for control animals (n = 10) and seaweed treated animals (n = 10), with green shading again indicating the period of seaweed feeding. (C) Nonmetric multidimensional scaling of the Jensen-Shannon Divergence across mice treated and untreated with seaweed and in pre- and post-time points.
We expected that seaweed treatment would not lead to compositional changes in the microbiota. We examined the change in community structure on a seaweed diet by tracking the alpha diversity over time (Figure 1B). There were no consistent changes in Shannon diversity in the seaweed-treated mice in either the initial seaweed or late seaweed treatment time points, suggesting that seaweed feeding did not coherently alter the mouse gut microbiota in a way reflected in alpha diversity. We used Jensen-Shannon Divergence and non-metric multidimensional scaling to identify whether communities became more similar after seaweed treatment. Seaweed treated communities did not cluster separately from controls, also suggesting that this treatment led to no changes observable in the community at this level (Figure 1C). Of 289 sequence variants, none showed a significant difference in abundance (Mann Whitney U test with FDR q < 0.10) before and after treatment. Additionally, we found no evidence for the presence of genes involved in porphyran breakdown based on qPCR targeting the β-porphyranase gene present in the polysaccharide utilization locus (PUL) of B. plebeius (Hehemann et al., 2012), indicating that the genetic potential to use polysaccharides present in the seaweed was absent.
Observing limited change in response to seaweed in the gut microbiome of C57BL/6 mice, we reasoned that an organism previously reported to exploit this niche would grow in its presence in the mouse gut. Further, we wanted to pick a strain that would not engraft unless given a fitness advantage. In particular, the human-derived B. plebeius DSM 17135 strain carries the genes for porphyran degradation, as do at least two other (GI commensal) isolates (a BLAST search reveals porphyran degradation genes in Bacteroides sartorii JCM 16497 and Porphyromonas bennonis JCM 16335 genomes). This pathway is carried on an integrative conjugative element, and has been repeatedly transferred between gut microbial species. The positive selection on these genes, indicated by their transmission via HGT, suggests that organisms carrying these genes will be more fit in an environment rich in porphyran.
Introduction of B. plebeius into the digestive tract of mice
To assess the feasibility of introducing B. plebeius into the GI tract of mice, we ran an experiment with four groups of outbred female Swiss mice co-housed by treatment. The treatment groups were as follows: (1) mice that were gavaged at the beginning of the experiment with 107 CFU of B. plebeius (B. plebeius only), and mice that were gavaged with B. plebeius and (2) fed seaweed continuously (seaweed) or (3) delivered pulses of seaweed with four days on and four days off (pulsed), and (4) mice that were fed seaweed but not gavaged with B. plebeius (control) (Figure 2A). We chose outbred mice in order to reduce the likelihood of observing genotype-specific response in colonization by B. plebeius. Animals were co-housed by treatment to improve exchange of B. plebeius via coprophagy, to reduce probability of stochastic extinctions, and to select for the fittest genotype across all animals, rather than a single-animal optimized genotype.
Figure 2.

B. plebeius reaches persistent high level abundance in mice consuming seaweed. (A) Experimental design: samples were collected where indicated for qPCR (n = 5 mice per group) and IgA-Sequencing; two animals per B. plebeius only, constant seaweed, and pulsed seaweed were sampled densely across until cessation of the first seaweed treatment window. Green shaded region indicates the seaweed dosing windows. (B) qPCR-based relative abundance of B. plebeius in control (n = 5), B. plebeius only (n = 5), constant seaweed (n = 5), and pulsed groups (n = 5) at indicated time points. The lower limit of detection from non-specific amplification is marked with a gray line. (C) Time series of B. plebeius in B. plebeius only (n = 2), constant seaweed (n = 2), and pulsed groups (n = 2), line colors distinguish individual animals, green shading indicates seaweed-dosing periods. See also Figure S1.
From each mouse, fecal samples were collected daily for a 35-day period, and a subset of these samples was processed for V4 16S rDNA amplicon sequencing. Follow-up samples from 2 months after cessation of the initial seaweed feeding experiment were collected to determine whether B. plebeius persisted in mice feces long after the initial treatment. During the experiment we did not note any adverse affects on the mice across the treatment groups.
B. plebeius abundantly colonizes the GI tract of mice feeding on 1% seaweed
We focused our sequencing efforts on the time series of 2 mice per treatment group, each randomly selected from the gavaged and untreated (B. plebeius only), gavaged and treated with 1% seaweed (seaweed), and gavaged and treated with pulsed 1% seaweed (pulsed) groups. In the seaweed treated groups, we expected to find enrichment for B. plebeius and dilution and eventually extinction in the absence of seaweed. In the pulsed treatment, we expected that B. plebeius levels would rise and fall to track the presence of the seaweed.
In fact, we found that the relative abundance of B. plebeius increased by more than two orders of magnitude (p = 0.001, n = 10 (seaweed + pulsed) mice, n = 5 B. plebeius only mice, Mann Whitney U test on qPCR-based estimates of abundance) in the seaweed and pulsed groups relative to the B. plebeius only group, suggesting that seaweed treatment enriches for B. plebeius in the GI tract of these mice (Figure 2B and Figure 2C). Indeed, at some points, B. plebeius rises in abundance to nearly 50% of the whole community. As expected, we observe drops (albeit with somewhat irregular patterns) in B. plebeius abundance that corresponded to removal of seaweed from the diet, suggesting that its ability to maintain high abundance was tied to the presence of this resource (Figure 2C). Even in mice that do not receive seaweed, the non-native, human-derived, B. plebeius colonizes at low levels, but frequently falls below the limit of detection, suggesting that it has limited access to additional resources in the mouse gut to enable its persistence.
Introduced organisms influence resources availability in the gut
Observing that B. plebeius colonizes even in the absence of seaweed in the diet, we inferred that it is capable of using resources endogenous to the mouse gut. We were surprised by the frequently high abundances of B. plebeius in the gut treated with seaweed, so we constructed a quantitative model of the system to accommodate these observations and facilitate further orthogonal niche engineering strategies (see Supplementary Information). In this quantitative model, B. plebeius can grow on either seaweed as a limiting substrate or other substrates already present in the mouse gut. Increases in B. plebeius abundance increase its depletion of substrates already present in the mouse gut, and reduce the availability of these substrates for growth of other organisms. Even without the quantitative model of the system, increases of biomass on carbon substrates must accompany decreases of other substrates: biomass cannot increase on a carbon substrate without commensurate consumption of substrates containing other essential elements like nitrogen.
This modeling result predicts that B. plebeius alters the resource pool in the gut in a way that might restrict the growth of other organisms or the host. The total abundance of bacteria in the system appears to remain unchanged in the presence or absence of seaweed as measured by qPCR (see Supplementary Figure 1). Keeping this in mind, as B. plebeius increases in abundance in the system, we expect many other sequence variants to decrease, and indeed, there is an enrichment for negative correlations with B. plebeius in the seaweed-treated mice compared to the untreated mice even after correcting for differences in community size over time (121 negative correlations/193 total correlations with p < 0.01 for seaweed and pulsed groups and 1 negative correlation/22 total correlations with p < 0.01 for the B. plebeius only group, – see Methods for details). There is also a slight enrichment for negative correlations in the continuous seaweed group compared to the pulsed group.
There were few sequence variants that positively correlated with B. plebeius abundance. We hypothesized that the abundance of organisms with similar abundance and dynamic patterns might be constrained by the same limiting resource. We found that A. muciniphila significantly positively correlated with B. plebeius in two of the mice, one from the pulsed and one from the continuous seaweed diets (Figure 3A), and had no temporal relationship in the absence of seaweed. The remaining seaweed-treated mice both exhibited a decrease in A. muciniphila abundance over time, with no significant correlation to B. plebeius.
Figure 3.

Introduced commensal competes with endogenous gut commensals. (A) Scatterplot of B. plebeius and A. muciniphila abundance over time for two animals having significant correlations between these bacteria (purple = an animal from the constant seaweed group, blue = an animal from the pulsed seaweed group). (B) B. plebeius JCM 17135 and A. muciniphila DSM 26127 were cultured alone or in combination in carbon-restricted growth medium with either glucose or mucin as the primary carbon source. Bars are the standard error of the mean (n = 4 replicates/condition) (Student’s t test on log-transformed abundance data: *, p-value < 0.05, **, p-value < 0.01, ***, p-value < 0.001).
Because A. muciniphila specializes in mucin degradation (Derrien, 2004), we inferred that both organisms use mucin for growth, indicating both a resource-based niche and interaction with the mucus layer. Intriguingly, increases in A. muciniphila abundance was associated with even greater increases in B. plebeius abundance (i.e. the slope is greater than 1) (Figure 3A), suggesting that A. muciniphila may in some way facilitate the growth of B. plebeius. To test this hypothesis, we cultured B. plebeius and A. muciniphila together in the presence and absence of mucin or glucose as a limiting carbon substrate. Relative to growth on mucin alone, B. plebeius increases in abundance in co-culture with A. muciniphila. By contrast, A. muciniphila decreases in abundance in co-culture with B. plebeius, suggesting that B. plebeius grows at the expense of A. muciniphila (Figure 3B). The facilitation of A. muciniphila on B. plebeius growth is more pronounced for mucin than glucose. Such growth facilitation by and inhibition of A. muciniphila has been shown previously for other gut commensals, including B. vulgatus (Png et al., 2010).
Long-term B. plebeius persistence is reduced in mice constantly fed seaweed
By providing access to a limited amount of resource, we could explain the high level colonization of B. plebeius in the short term. We investigated whether this advantage would hold in the long term as well. We examined the presence of B. plebeius in mice two months after cessation of the initial seaweed-feeding period. We collected baseline samples and resumed 1% seaweed treatment in the groups that originally received seaweed. Surprisingly, although we observed blooms (to nearly 50% of the community) of B. plebeius on resumption of seaweed feeding in mice originally on the pulsed diet, there was no response of B. plebeius in the mice originally on the continuous diet, and the median relative abundance (1.6e-5) was just above the limit of detection for these animals (Figure 4A and Figure 4B). There was a single mouse in the pulsed seaweed group that lost B. plebeius entirely, but there was still a significant difference in the levels of B. plebeius between the constant and pulsed groups (p = 0.02, n = 5 constant mice, n = 5 pulsed mice, Mann Whitney U Test on qPCR-based estimates of total abundance) (Figure 4A), suggesting a differential effect from the initial dietary regime on the long term responsiveness of B. plebeius to seaweed amendment.
Figure 4.

Long-term persistence of B. plebeius depends on initial diet regimen. (A) qPCR-based relative abundance of B. plebeius in constant (n = 5) and pulsed (n = 5) seaweed groups at indicated time point after 1% seaweed amendment. The lower limit of detection from non-specific amplification is marked with a gray line. (B) Relative abundance of B. plebeius over time in late time points on resumption of seaweed treatment after washout in B. plebeius only (n = 3), constant (n = 3), and pulsed seaweed groups (n = 3).
B. plebeius shows increased IgA-binding when abundant after seaweed treatment
Recalling the apparent relationship of B. plebeius to A. muciniphila, and knowing A. muciniphila is heavily IgA-targeted in humans and mice (Palm et al., 2014), we wondered whether IgA-targeting, and thus immune activation against B. plebeius might be related to its disappearance from some of the mice, by inhibiting its ability to uptake seaweed polysaccharides through steric blocking or related mechanisms that prevent biofilm formation (Moor et al., 2017). Equally, IgA-targeting may be a mechanism for retaining bacteria while simultaneously controlling their growth (Donaldson et al., 2018; McLoughlin et al., 2016).
With these considerations in mind, we evaluated whether B. plebeius was detected and targeted by IgA in the gut. Only in the case that B. plebeius generates a specific response by the immune system would we expect increased binding of B. plebeius relative to the rest of the microbiota, as polyreactive IgA tends not to preferentially bind Bacteroidetes (Bunker et al., 2017). Such a difference in IgA binding might mediate control of B. plebeius abundance in the constant seaweed group at late time points, and potentially explain loss of B. plebeius. To test this hypothesis, we flow-sorted and sequenced fecal bacteria bound by IgA as described previously (Palm et al., 2014). At the late time points, B. plebeius was too low in abundance (< 1e-5) to detect in mice in the continuous seaweed treatment group. We focused our IgA-Seq efforts on late time point samples from mice originally on the pulsed and B. plebeius only groups, for which B. plebeius was detectable by qPCR. We failed to detect a signal of differential IgA-binding of B. plebeius in the B. plebeius only mice (Supplementary Figure 2), suggesting that IgA may not be an important control mechanism for B. plebeius in these animals. However, in the pulsed mice that still had B. plebeius, there is a clear enrichment for this organism when it is present in the highly IgA-bound fraction (Supplementary Figure 2). In fact, it is more IgA-bound than any other member of the microbiota in one of these mice, suggesting that the binding was highly specific to this organism. We interpret this to mean that IgA might be an important host-side control mechanism on this organism in the presence of dietary seaweed.
Discussion
Here, we provide a proof-of-concept of diet-based orthogonal niche engineering, whereby an organism is introduced with a tailored resource to establish an orthogonal niche in an intact community. Previous studies of the influence of diet on the gut microbiota have primarily focused on the alterations in the bacterial community and functional or metabolic shifts in response to dietary perturbations (Carmody et al., 2015; David et al., 2013, 2014; Desai et al., 2016; Turnbaugh et al., 2006, 2009). It has been documented that Bacteroidetes in particular display a hysteretic response (i.e. memory of past dietary exposures dampens future responses) to dietary changes (Carmody et al., 2015) that may affect their future ability to respond to substrates in the diet. We provide additional evidence that previous dietary exposures affect the ability of organisms to respond to future diets.
Similar results to those found in the present paper were recently reported (Shepherd et al., 2018), confirming and complementing those in this manuscript. Shepherd and colleagues identified a B. ovatus strain capable of utilizing porphyran through the same PUL as B. plebeius, and engineered two other strains (B. thetaiotaomicron and B. stercoris) with the same capacity. In line with our results, they successfully introduce these three porphyran users into distinct, intact microbiota in Swiss Webster mice both with seaweed in the case of B. ovatus, and purified porphyran in water in all other experiments. They find that porphyran alone does not alter the composition of the gut microbiota in Swiss Webster mice, which mirrors what we found with seaweed in C57BL/6 mice. It is likely that differences in host genetics or the endogenous microbiota across mouse lines could cause differences in the response to dietary amendments. However, the composition of the gut microbiota in both Swiss Webster (Shepherd et al., 2018) and C57BL/6 mice (this study) is not altered by the addition of porphyran or seaweed to the diet in the absence of known porphyran users.
Shepherd and colleagues show that B. ovatus can colonize the colonic crypts in the presence, but not in the absence of porphyran, allowing for co-existence of isogenic strains through apparent niche partitioning. We suspect this mechanism may extend to our system with B. plebeius, and may contribute to the differences in responsiveness of B. plebeius at late time points depending on diet. One possibility is that populations of B. plebeius from the constantly fed seaweed group may be confined to the crypts, and so less responsive to seaweed administration because of reduced access. Recent evidence suggests that crypt colonization for B. fragilis is dependent on IgA (Donaldson et al., 2018), and we present evidence that B. plebeius is heavily IgA-bound in seaweed-treated mice, which can confine bacterial growth through enchainment and aggregation of dividing cells (Moor et al., 2017). The differences in long term responses merit further consideration as they suggest controllability of introduced organisms will depend on the history of dietary exposures, as has been observed before for the response of Bacteroides to dietary fluctuations (Carmody et al., 2015).
Additionally, Shepherd and colleagues show a correlation between B. ovatus abundance and porphyran concentration in the water and manage this control on both fiber free and fiber-rich diets. Our system administered seaweed in a fiber-containing chow and not porphyran in water. Further, our conventional mice start with more complex communities than the conventional, restricted flora mice used by Shepherd and colleagues. In all sampled mice that were originally on the seaweed diet, we observed noisy fluctuations in the abundance of B. plebeius, particularly in the pulsed diet, suggesting a strong dependence on this resource. These results contrast to the relatively tight control over introduced microbial abundance seen by Shepherd and colleagues, and may relate to the experimental differences outlined above.
We show that B. plebeius can inhibit the growth of other members of the community, particularly A. muciniphila, a primary mucin degrader, which has important implications for the engineering of this system. We use a conceptual and quantitative model to suggest that introduced organisms are not metabolically isolated from the existing community, but interact with it in ways that require consumption of resources in addition to their exclusive substrate. Indeed, we show that increases in B. plebeius during seaweed treatment do not associate with increases in total community size, meaning that other organisms will necessarily be negatively impacted. Similarly, Shepherd and colleagues show an increase in total community size largely from an increase in the population of B. ovatus when treating with porphyran on fiber-free, but not on fiber-rich diets. These results along with our own suggest that community size on fiber-containing diets is constrained by different resources than fiber-free diets, consistent with work from others on this topic (Desai et al., 2016).
The issue of resource competition will be more acute in the case of porphyran, which does not provide a source of nitrogen for growth. If total biomass of an introduced organism increases on a carbon-only substrate, nitrogen must also be incorporated to support this biomass increase, and necessarily affects the pool of available resources either to the rest of the microbial community or to the host. Important pathogens, including C. difficile and S. enterica have been shown to used mucin-derived substrates when metabolic networks among the native microbiota are disrupted, allowing them to expand to significant abundances within the community (Ng et al., 2013). For dysbiotic communities, this consequence may be considered a desirable feature, as the introduced organism could siphon resources from microorganisms that would otherwise exacerbate disease.
These results provide additional proof for the concept of orthogonal niche engineering as an approach to introducing bacteria into complex communities. The convergence on porphyran by us and another independent group (Shepherd et al., 2018) as a means to control introduced microbial abundance might question the extensibility of this strategy to other organism-substrate pairs, if there are few other combinations that exhibit these properties. Shepherd and colleagues manage to transfer this pathway to naïve organisms, but this strategy requires introduction of 34 genes. The evidence by Maldonado-Gómez and colleagues (Maldonado-Gómez et al., 2016), and our work suggests that synthetic strategies may not be necessary if it is possible to identify organism-substrate pairs that function in this way independent of community context. For example, through functional screens of human gut bacteria, it may be possible to reveal the nutritional preferences of specific strains to identify such pairs (Tramontano et al., 2018). Of course, a major limitation of this approach is ensuring that these strain-nutrient pairs will succeed independent of community context. As an alternative approach, synthetic nutrient-organism pairs (for example, engineering an organism to metabolize sucralose) may fulfill these design criteria. We caution that all strategies in this vein will be contingent on the universal requirement for nitrogen and other limiting resources in the gut. Rational synbiotic design will benefit from considerations of the interaction of introduced prebiotics and probiotics with other biotic and abiotic factors. In this system, there is very clear positive selection for growth on seaweed given that the genes were transferred from a marine bacterium to a gut commensal bacterium. It will be important to consider how off-target effects through species-species, species-diet, and species-host interactions can alter these responses.
Experimental Procedures
Animals
Six-week old female C57BL/6 wild type and outbred Swiss Webster mice (Taconic, Germantown, NY) were housed and handled in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facilities using techniques and diets including Bacteroides plebeius as specifically approved by Massachusetts Institute of Technology’s Committee on Animal Care (CAC) (MIT CAC protocol # 0912-090-15 and 0909-090-18). The MIT CAC (IACUC) specifically approved the studies as well as the housing and handling of these animals. Mice were euthanized using carbon dioxide at the end of the experiment.
B. plebeius culture
B. plebeius DSM 17135 was obtained from the DSMZ and cultured as specified. Briefly, colonies were obtained by the streak method on PYG (Modified) Agar incubated at 37°C in a Coy Anaerobic Chamber (Grass Lake, MI) for up to 48 hours. After the appearance of colonies, single colonies were inoculated in PYG (Modified) medium and incubated overnight prior to gavage or nucleic acid extraction. 25% glycerol stocks of B. plebeius were made and stored at −80°C and streaked onto fresh medium for revival and colony picking.
A. muciniphila culture
A. muciniphila DSM 26127 was obtained from the DSMZ and cultured as specified. Briefly, colonies were obtained by the streak method on BHI + 0.5% (w/v) porcine type II gastric mucin (Sigma M2738) purified as described previously (Derrien, 2004) agar, and incubated at 37°C in a Coy Anaerobic Chamber (Grass Lake, MI) for up to 48 hours. After the appearance of colonies, single colonies were inoculated into liquid medium (BHI + 0.5% (w/v) mucin), and incubated overnight before use in co-culture experiments.
Co-culture Experiments
Overnight cultures of B. plebeius and A. muciniphila grown in PYG (modified) and BHI + 0.5% (w/v) mucin, respectively, were inoculated at a starting density of 106 CFUs alone or in combination into medium 75 (Hoskins and Boulding, 1981) with either porcine type II gastric mucin (Sigma M2738) or glucose as the limiting carbon source at 0.5% (w/v). After 48 hours of growth, colony forming units were enumerated by plate counts on selective media (BHI + 0.5% (w/v) mucin) or PYG (modified) for A. muciniphila and B. plebeius, respectively).
Seaweed Diet Experiment
C57BL/6 mice were used in the initial studies in Figure 1 in which 10 mice were fed with a custom chow diet (Bio-Serv, Flemington NJ) containing 1% raw seaweed nori (Izumi Brand) and 10 mice had a standard control diet (Product# F3156, AN-93G, Bio-Serv, Flemington NJ). Animals were co-housed for 6 days after arrival in the MIT animal facilities, and singly housed after separation into the seaweed treatment and control groups. Fresh fecal samples were obtained within an hour daily for all animals in all groups. Fecal samples were collected into anaerobic 25% glycerol containing 0.1% cysteine, and transferred immediately to dry ice before being stored at −80°C prior to nucleic acid extraction.
B. plebeius Gavage Experiment
For the B. plebeius gavage experiments, Swiss Webster mice were co-housed for 6 days after arrival in MIT animal facilities, and 5 mice per group were co-housed by treatment on initiation of the experiment. Fecal samples were collected for each animal for three days prior to the initiation of the experimental protocol. At day 0, mice in the B. plebeius gavage groups were gavaged only once with approximately 107 CFUs of B. plebeius DSM 17135 culture in 250 μl volumes of PYG (Modified) media, and control groups were gavaged only once with 250 μl sterile PYG (Modified) media. All groups treated with seaweed received a custom chow diet containing 1% seaweed nori at the initiation of experiments. All fecal samples were collected as described for the initial seaweed diet experiment.
Nucleic Acid Extraction
DNA from fecal samples and bacterial cultures was extracted using the MoBio High Throughput (HTP) PowerSoil Isolation Kit (MoBio Laboratories, Inc., now Qiagen) with minor modifications. Briefly, samples were homogenized with bead-beating and then 50 μl Proteinase K (Qiagen) added and samples were incubated in a 65°C water bath for 10 minutes. Samples were then incubated at 95°C for 10 minutes to deactivate the protease. All other steps remained the same.
16S Library Preparation and Sequencing
Libraries for paired-end Illumina sequencing were constructed using a two-step 16S rRNA PCR amplicon approach as described previously with minor modifications (Preheim et al., 2013). The first-step primers (PE16S_V4_U515_F, 5′ ACACG ACGCT CTTCC GATCT YRYRG TGCCA GCMGC CGCGG TAA-3′; PE16S_V4_E786_R, 5′-CGGCA TTCCT GCTGA ACCGC TCTTC CGATC TGGAC TACHV GGGTW TCTAA T 3′) contain primers U515F and E786R targeting the V4 region of the 16S rRNA gene, as described previously (Preheim et al., 2013). Additionally, a complexity region in the forward primer (5′-YRYR-3′) was added to help the image-processing software used to detect distinct clusters during Illumina next-generation sequencing. A second-step priming site is also present in both the forward (5′-ACACG ACGCT CTTCC GATCT-3′) and reverse (5′-CGGCA TTCCT GCTGA ACCGC TCTTC CGATC T-3′) first-step primers. The second-step primers incorporate the Illumina adapter sequences and a 9-bp barcode for library recognition (PE-III-PCR-F, 5′-AATGA TACGG CGACC ACCGA GATCT ACACT CTTTC CCTAC ACGAC GCTCT TCCGA TCT 3′; PE-III-PCR-001–096, 5′-CAAGC AGAAG ACGGC ATACG AGATN NNNNN NNNCG GTCTC GGCAT TCCTG CTGAA CCGCT CTTCC GATCT 3′, where N indicates the presence of a unique barcode.
Real-time qPCR before the first-step PCR was done to ensure uniform amplification and avoid overcycling all templates. Both real-time and first-step PCRs were done similarly to the manufacturer’s protocol for Phusion polymerase (New England BioLabs, Ipswich, MA). For qPCR, reactions were assembled into 20 μL reaction volumes containing the following: DNA-free H2O, 8.9 μL, HF buffer, 4 μL, dNTPs 0.4 μL, PE16S_V4_U515_F (3 μM), 2 μL, PE16S_V4_E786_R (3 μM) 2 μL, BSA (20 mg/mL), 0.5 μL, EvaGreen (20X), 1 μL, Phusion, 0.2 μL, and template DNA, 1 μL. Reactions were cycled for 40 cycles with the following conditions: 98° C for 2 min (initial denaturation), 40 cycles of 98 C for 30 s (denaturation), 52° C for 30 s (annealing), and 72° C for 30s (extension). Samples were diluted based on qPCR amplification to the level of the most dilute sample, and amplified to the maximum number of cycles needed for PCR amplification of the most dilute sample (18 cycles, maximally, with no more than 8 cycles of second step PCR). For first step PCR, reactions were scaled (EvaGreen dye excluded, water increased) and divided into three 25-μl replicate reactions during both first- and second-step cycling reactions and cleaned after the first-and second-step using Agencourt AMPure XP-PCR purification (Beckman Coulter, Brea, CA) according to manufacturer instructions. Second-step PCR contained the following: DNA-free H2O, 10.65 μL, HF buffer, 5 μL, dNTPs 0.5 μL, PE-III-PCR-F (3 μM), 3.3 μL, PE-III-PCR-XXX (3 μM) 3.3 μL, Phusion, 0.25 μL, and first-step PCR DNA, 2 μL. Reactions were cycled for 10 cycles with the following conditions: 98° C for 30 s (initial denaturation), 10 cycles of 98° C for 30 s (denaturation), 83° C for 30 s (annealing), and 72° C for 30s (extension). Following second-step clean-up, product quality was verified by DNA gel electrophoresis and sample DNA concentrations determined using Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific). The libraries were multiplexed together and sequenced using the paired-end with 250-bp paired end reads approach on the MiSeq Illumina sequencing machine at the BioMicro Center (Massachusetts Institute of Technology, Cambridge, MA).
16S rDNA Sequence Data Processing and Quality Control
Paired-end reads were joined with PEAR (Zhang et al., 2014) using default settings. After read joining, the complexity region between the adapters and the primer along with the primer sequence and adapters were removed. Sequences were processed batchwise using the DADA2 (Callahan et al., 2016) pipeline in R, trimming sequences to 240 bp long after quality filtering (quality trim Q10) with maximum expected errors set to 1. A final sequence variant table combining all sequencing data was generated using DADA2. Sequence variants were classified using RDP (Maidak et al., 1996; Wang et al., 2007). The resulting count tables were used as input for analysis within R.
qPCR
qPCR was carried out as described in the 16S rDNA Library Preparation and Sequencing section. For quantification of B. plebeius, primers were designed to target the beta-porphyranase A gene (BACPLE_01693) in the porphyran degradation PUL: GH86 F: 5’-TCGAA TGTCA CAAAG CGTTC-3’ and GH86 R: 5’- ATGGA CGGGA CATTC TGTTC-3’. For direct quantification of B. plebeius abundance, a nucleic acid standard curve was prepared using 10-fold dilutions of nucleic acids extracted from B. plebeius overnight cultures quantified by NanoDrop after RNAse treatment. Mann Whitney U test was used to identify differences in B. plebeius abundance between treatment and control groups.
IgA Sorting
Pre-weighed frozen fecal samples in glycerol were thawed at 4°C and then homogenized using a handheld homogenizer and pestles (Kimble Chase Kontes) at a final dilution in sterile PBS of 100 mg per ml. Samples were processed as described previously (Palm et al., 2014). After homogenization, samples were centrifuged at 50 × g at 4°C for 15 minutes, then washed three times in 1 ml PBS/1% BSA at 8000 × g for 5 minutes each. The pre-sort fraction was collected as 20 μL after resuspension prior to the final wash and stored at −80°C. The cell pellet was resuspended in 25 μL of 20% Normal Rat Serum (Jackson Immunoresearch) in PBS/1%BSA and incubated for 20 minutes on ice. After incubation, 25 μL 1:12.5 α-mouse- IgA-PE (EBioscience, clone mA-6E1) was added to each sample and samples incubated on ice for 30 minutes. Samples were washed three times in 1 ml PBS/1% BSA as above, and finally resuspended in PBS/1% BSA and transferred to blue filter cap tubes (VWR 21008–948) for flow sorting. An average of 500,000 cells from the IgA-positive and IgA-negative bacteria were sorted in triplicate into sterile microcentrifuge tubes on the BD FACSAria II at the MIT Koch Institute Flow Cytometry Core (Cambridge, MA). Samples were centrifuged, supernatant removed, and stored at −80°C until nucleic acid extraction using the DNeasy UltraClean Microbial Kit (Qiagen).
16S rDNA Data Analysis
A DADA2 sequence variant table including all experiments was imported into R and analyzed using phyloseq (McMurdie and Holmes, 2013) and custom R scripts.
Spearman correlations between B. plebeius and all other sequence variants in the time series were determined for each of the two mice across three groups. The number of sequence variants with negative or positive correlations and unadjusted p-values less than 0.01 were determined for each group. The Fisher Exact test was used to determine whether there were differences in the proportion of positive and negative correlations between the groups for each pairwise comparison of groups.
IgA Data Analysis
We used DESeq2 to detect differences in abundance between IgA-bound and IgA-unbound fractions (McMurdie and Holmes, 2014). Using DESeq2 as applied to microbiome count data, OTUs that had an adjusted p-value of < 0.05 (Wald Test with Benjamini-Hochberg correction) between the IgA+ and IgA- fractions were considered significantly differentially abundant between the fractions.
Model Construction and Analysis
See Supplementary Information for the simulated chemostat model for bacterial growth. Code for model simulations were implemented in MATLAB R2017B.
Statistical Analysis
All statistical analyses were performed using the R statistical computing package (https://cran.r-project.org/), except statistics for comparison of colony forming units (CFU) for B. plebeius and A. muciniphila (Figure 3B) and total community size comparisons with qPCR (Supplementary Figure 1), which were performed in Excel. Statistically significant differences are shown with asterisks as follows: *p < 0.05, ** p < 0.01, ***p < 0.001. Number of animals (n) used for individual experiments and details of the statistical tests used are indicated in the figure legends. When quantities were expected to follow a normal distribution (as the log of total cell counts), we compared groups using a t test. Otherwise, a non-parametric (Mann-Whitney) test was used. For calculating correlations, we use Spearman’s rho to account for nonlinear relationships between two variables. IgA data was analyzed using DESeq2 and the Wald Test (p < 0.05) to identify significantly differentially abundant sequence variants.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jan Hendrik Hehemann, Fatima Hussain, Tami Lieberman, Mathieu Groussin, Avihu Yona, and the members of the Alm and Polz Lab for extensive discussion and experimental advice. We thank Rachel Soble for technical contributions. We thank the MIT BioMicro Center for sequencing service. We thank the Koch Institute for Flow Cytometry.
Funding was provided by the Broad Institute BN10 Training Grants. SM Kearney was funded by an NSF Graduate Research Fellowship.
Footnotes
ACCESSION NUMBERS
Data from this study have been deposited in the NCBI Short-Read Archive (SRA) and 16S rDNA gene sequences and metadata can be found under PRJNA479657 (SRP151989). The R scripts, metadata, and accompanying sequence variant table to analyze the 16S rDNA gene data can be found online at the following link: https://github.com/microbetrainer/Seaweed. Sequence variant tables and metadata are additionally available on Mendeley DOI: 10.17632/86xzh5c85p.1.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, Jabri B, Antonopoulos DA, Wilson PC, and Bendelac A (2017). Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, Gerber GK, Luevano JM, Gatti DM, Somes L, Svenson KL, and Turnbaugh PJ (2015). Diet Dominates Host Genotype in Shaping the Murine Gut Microbiota. Cell Host Microbe 17, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2013). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, Erdman SE, and Alm EJ (2014). Host lifestyle affects human microbiota on daily timescales. Genome Biol 15, R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien M (2004). Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol 54, 1469–1476. [DOI] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 167, 1339–1353.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, Conner ME, Earl AM, Knight R, Bjorkman PJ, et al. (2018). Gut microbiota utilize immunoglobulin A for mucosal colonization. Science eaaq0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freter R (1983). Mechanisms that control the microflora in the large intestine. Hum. Intest. Microflora Health Dis 33–54. [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying Genetic Determinants Needed to Establish a Human Gut Symbiont in Its Habitat. Cell Host Microbe 6, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehemann J-H, Correc G, Barbeyron T, Helbert W, Czjzek M, and Michel G (2010). Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908. [DOI] [PubMed] [Google Scholar]
- Hehemann J-H, Kelly AG, Pudlo NA, Martens EC, and Boraston AB (2012). Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc. Natl. Acad. Sci 109, 19786–19791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins LC, and Boulding ET (1981). Mucin Degradation in Human Colon Ecosystems. J. Clin. Invest 67, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumbeck JA, Maldonado-Gomez MX, Martínez I, Frese SA, Burkey TE, Rasineni K, Ramer-Tait AE, Harris EN, Hutkins RW, and Walter J (2015). In Vivo Selection To Identify Bacterial Strains with Enhanced Ecological Performance in Synbiotic Applications. Appl. Environ. Microbiol 81, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SS, Zhu A, Benes V, Costea PI, Hercog R, Hildebrand F, Huerta-Cepas J, Nieuwdorp M, Salojärvi J, Voigt AY, et al. (2016). Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 352, 586. [DOI] [PubMed] [Google Scholar]
- Maidak BL, Olsen GJ, Larsen N, Overbeek R, McCaughey MJ, and Woese CR (1996). The ribosomal database project (RDP). Nucleic Acids Res 24, 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Gómez MX, Martínez I, Bottacini F, O’Callaghan A, Ventura M, van Sinderen D, Hillmann B, Vangay P, Knights D, Hutkins RW, et al. (2016). Stable Engraftment of Bifidobacterium longum AH1206 in the Human Gut Depends on Individualized Features of the Resident Microbiome. Cell Host Microbe 20, 515–526. [DOI] [PubMed] [Google Scholar]
- McLoughlin K, Schluter J, Rakoff-Nahoum S, Smith AL, and Foster KR (2016). Host Selection of Microbiota via Differential Adhesion. Cell Host Microbe 19, 550–559. [DOI] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10, e1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moor K, Diard M, Sellin ME, Felmy B, Wotzka SY, Toska A, Bakkeren E, Arnoldini M, Bansept F, Co AD, et al. (2017). High-avidity IgA protects the intestine by enchaining growing bacteria. Nature 544, 498. [DOI] [PubMed] [Google Scholar]
- Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, et al. (2013). Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, De Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, and Zhang W (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi P, Parida S, Nanda NC, Satpathy R, Pradhan L, Chandel DS, Baccaglini L, Mohapatra A, Mohapatra SS, Misra PR, et al. (2017). A randomized synbiotic trial to prevent sepsis among infants in rural India. Nature 548, 407. [DOI] [PubMed] [Google Scholar]
- Pluvinage B, Grondin JM, Amundsen C, Klassen L, Moote PE, Xiao Y, Thomas D, Pudlo NA, Anele A, Martens EC, et al. (2018). Molecular basis of an agarose metabolic pathway acquired by a human intestinal symbiont. Nat. Commun 9, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Png CW, Lindén SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, and Florin TH (2010). Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol 105, 2420–2428. [DOI] [PubMed] [Google Scholar]
- Preheim SP, Perrotta AR, Martin-Platero AM, Gupta A, and Alm EJ (2013). Distribution-Based Clustering: Using Ecology To Refine the Operational Taxonomic Unit. Appl. Environ. Microbiol 79, 6593–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, and Sonnenburg JL (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, Sauk J, Gevers D, Friedman J, Sung J, Youngster I, Hohmann EL, Staley C, Khoruts A, Sadowsky MJ, et al. (2018). Strain Tracking Reveals the Determinants of Bacterial Engraftment in the Human Gut Following Fecal Microbiota Transplantation. Cell Host Microbe 23, 229–240.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano M, Andrejev S, Pruteanu M, Klünemann M, Kuhn M, Galardini M, Jouhten P, Zelezniak A, Zeller G, Bork P, et al. (2018). Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat. Microbiol 3, 514–522. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, and Gordon JI (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1131. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, and Gordon JI (2009). The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med 1, 6ra14–6ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, McNulty NP, Rodionov DA, Khoroshkin MS, Griffin NW, Cheng J, Latreille P, Kerstetter RA, Terrapon N, Henrissat B, et al. (2015). Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science 350, aac5992–aac5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T, and Stamatakis A (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.