

Abstract
Previously, we had shown that high magnitude stretch (HMS), rather than low magnitude stretch (LMS), induced significant apoptosis of skeletal muscle C2C12 myoblasts. However, the molecular mechanism remains obscure. In this study, we found that p53 protein accumulated in the nucleus of LMS-loaded cells, whereas it translocated into mitochondria of HMS-loaded cells. Knocking down endogenous p53 by shRNA abrogated HMS-induced apoptosis. Furthermore, we demonstrated that overaccumulation of reactive oxygen species (ROS) during HMS-inactivated AKT that was activated in LMS-treated cells, which accounted for the distinct p53 subcellular localizations under HMS and LMS. Blocking ROS generation by N-acetylcysteine (NAC) or overexpressing constitutively active AKT vector (CA-AKT) inhibited HMS-incurred p53 mitochondrial translocation and promoted its nuclear targeting. Moreover, both NAC and CA-AKT significantly attenuated HMS-induced C2C12 apoptosis. Finally, we found that Ser389 phosphorylation of p53 was a downstream event of ROS-inactivated AKT pathway, which was critical to p53 mitochondrial trafficking during HMS stimuli. Transfecting p53-shRNA C2C12s with the mutant p53 (S389A) that was unable to target p53 to mitochondria underwent significantly lower apoptosis than transfection with wild-type p53. Altogether, our study uncovered that mitochondrial localization of p53, resulting from p53 Ser389 phosphorylation through ROS-inactivated AKT pathway, prompted C2C12 myoblast apoptosis during HMS stimulation.
INTRODUCTION
A retruded mandibular can severely affect a patient’s profile, due to a relatively prominent maxilla and insufficient development of the chin (Bilgic et al., 2015). A functional appliance, which generates an appropriate mechanical stretch by maintaining a retruded mandibular in normal position, could induce normal growth and adaption of neuromuscular and skeletal tissue (Ciavarella et al., 2017). Among these adaptable orofacial tissues, masticatory muscles are highly sensitive to mechanical stimuli and respond very well to different mechanical modalities. However, in contrast to the effect of proper mechanical stimuli on muscle adaption and growth (Goldspink et al., 1995; Tidball, 2005), unphysiological mechanical stimuli lead to muscle damage (Loerakker et al., 2010; Ferry et al., 2015; Naughton et al., 2017).
Muscle satellite cells, also known as myoblasts, are highly responsible for the reconstruction of skeletal muscle. Once activated, myoblasts proliferate, differentiate, and fuse into the myofibrils to maintain muscle mass and function. However, under some pathological conditions, myoblasts proceed to apoptosis, leading to impaired muscle regeneration (Song et al., 2006). Although apoptosis of skeletal myoblasts has been intensively studied under conditions such as denervation, muscular dystrophy, and aging, the effect of mechanical stretch on the apoptosis of skeletal myoblasts was still elusive.
One of our previous works has preliminarily proved that high magnitude stretch (HMS) led to a higher ratio of myoblast apoptosis than low magnitude stretch (LMS; Tan et al., 2009). We found that reactive oxygen species (ROS) were accumulated during stretch stimulation and assumed that ROS could be one of the initial responses to mechanical stimuli in skeletal muscle and is directly correlated with HMS-induced apoptosis of myoblasts.
In a search for ROS-related factors that led to induction of myoblast apoptosis under HMS, two pathways drew our attention: p53 and AKT, both of which were shown to be involved in cell apoptosis and ROS accumulation (Liu et al., 2008; Zhang et al., 2016). In addition, they were both reported to participate in intracellular mechanosignaling (Kruger and Linke, 2009; Ebata et al., 2016). Notably, even though p53 has been well studied in oncology due to its function as the “guardian of genome,” emerging roles of p53 in skeletal muscle physiology had emerged, particularly in mechanical force–induced muscular mitochondrial biogenesis (Saleem et al., 2011; Bartlett et al., 2014). Therefore, in skeletal myoblasts, the effect of mechanical stretch on p53 and its possible relation with apoptosis requires further elucidation.
In the present study, stimulating the C2C12 skeletal myoblasts with cyclic mechanical stretch of 10% (LMS) and 20% (HMS) magnitude, we showed that HMS promoted C2C12 apoptosis, whereas LMS did not. Moreover, mitochondria translocation of p53 was crucial in HMS-induced myoblast apoptosis, and we also explored the mechanism through which ROS and AKT were involved in this process. Targeting these molecules could be a new strategy in preventing muscle damage that occurs during stretch-induced muscle reconstruction.
RESULTS
Effects of LMS and HMS on skeletal myoblast viability
Previously, we subjected C2C12 myoblasts to cyclic stretch of different magnitudes (5, 10, 15, and 20%) for 24 h and demonstrated that cell viability was reversely correlated with stretching magnitudes (Tan et al., 2009). In this study, we chose two stretching magnitudes of 10 and 20% to represent LMS and HMS, respectively, and stimulated cells for 12 and 24 h. MTT assays showed that C2C12 viability did not significantly change after LMS for 12 and 24 h, whereas it did reduce remarkably under HMS in a time-dependent manner (Figure 1A). To determine whether the reduced cell viability was due to apoptosis, transferase-mediated biotinylated UTP nickend labeling (TUNEL) staining was applied to mark the apoptotic nuclei (Figure 1B). The ratio of apoptotic cells was evaluated by an apoptotic index (AI) that was calculated as the percentage of apoptotic nuclei in each field (Figure 1C). LMS led to a minor increase, whereas HMS resulted in a substantial increase, of C2C12 apoptosis (Figure 1, B and C). On the other hand, cell cycle regulator p21 was examined to see whether the reduced cell viability and increased apoptosis was related to cell cycle regulation. As can be seen in Figure 1, D and E, even though cell viability was higher in LMS-treated myoblasts than in HMS-treated cells, the expression of p21 mRNA and protein was significantly higher in the 12 h LMS group than in the other groups. Thus, these results indicated that LMS and HMS affected C2C12 viability independently of p21.
FIGURE 1:

HMS-promoted apoptosis of C2C12 myoblasts. (A) C2C12 cells were subjected to stretches of 10 and 20% magnitudes for 12 and 24 h, and cell viability was tested by MTT assay. (B) Cells were stretched with 10 and 20% magnitudes for 12 and 24 h, followed by TUNEL and DAPI staining. The apoptotic nuclei were recognized by TUNNEL staining (white arrow). Scale bar, 50 μm. (C) The apoptotic nuclei and total nuclei were counted, and apoptotic index was calculated as the percentage of apoptotic nuclei in total nuclei number per field. Three different fields are included, and data are shown as mean ± SD. (D) Real-time PCR was performed to examine the mRNA level of p21 after stretching of 10 and 20% magnitudes for 12 and 24 h. (E) Representative WB result of p21 protein level after stretching of 10 and 20% magnitudes for 12 and 24 h. The data represent the average of at least three separate experiments. Error bars indicate ± SD for triplicate. Significant differences are indicated by asterisks: *, P < 0.05 and **, P < 0.01, compared with static control cells.
Overload of ROS was involved in HMS-induced apoptosis of myoblasts
ROS are known to be responsible for muscle cell apoptosis and related muscle damage (Lee et al., 2005; Whitehead et al., 2006). Moreover, we and others had also manifested that in skeletal muscles, ROS were activated in response to mechanical stimuli (Tan et al., 2009; Prosser et al., 2013; Ward et al., 2014). Using the oxidation-sensitive probe, we measured temporal ROS production in C2C12 cells loaded by LMS and HMS for 12 and 24 h. LMS induced a mild increase of ROS in 12 and 24 h. By contrast, ROS production was intensively elevated in HMS-loaded cells for 12 and 24 h (Figure 2, A and B).
FIGURE 2:

ROS overloading was involved in HMS-induced apoptosis of myoblasts. (A) 2′,7′-Dichlorodihydrofluorescein diacetate (DCFDA) and DAPI staining of myoblasts subjected to LMS and HMS for 12 and 24 h. ROS generation was observed under the fluorescence microscope. Scale bar, 100 μm. (B) ROS levels per 5000 cells of each group were calculated by Microplate Fluorescence Reader FL600, and the data were normalized to values obtained from the control group. (C) AV/PI staining and flow cytometry analysis displayed drastic apoptosis of HMS-stretched myoblasts and attenuated apoptosis when ROS generation was suppressed by NAC. (D) Statistical analysis of the percentage of early (AV+/PI−) and late (AV−/PI+) apoptotic cells subjected to stretch. Data combined from three independent experiments are presented as mean ± SD. (E) Western blots were performed using caspase-3 primary antibody. Same samples were immunoblotted by GAPDH as the loading control. Black arrow indicates the cleaved capase-3 fragment with molecular size around 19 kDa. Significant differences are indicated by asterisks: *, P < 0.05 and **, P < 0.01, compared with static control cells.
To clarify whether the different amount of ROS accumulating during LMS or HMS stimuli incurred the distinct apoptotic ratios of C2C12 myoblasts, ROS scavenger (N-acetylcysteine [NAC]) was applied to eliminate ROS production. We had previously found that NAC could efficiently eliminate ROS production at the concentration of 100 μM (Tan et al., 2009), and our present study chose to use this concentration for the following experiments. Flow cytometry analysis (FCAS) of Annexin V binding and PI (AV/PI) staining was performed to quantitatively evaluate the apoptosis of myoblasts. AV−/PI− (lower left quadrant), AV+/PI− (lower right quadrant), AV+/PI+ (upper right quadrant), and AV−/PI+ (left upper quadrant) were labeled as living cells, early apoptotic cells, late apoptotic cells, and necrotic cells, respectively. FCAS showed that HMS stimulation for 12 and 24 h significantly increased the early and late apoptosis of C2C12 myoblasts, whereas LMS only mildly propelled the C2C12 apoptosis (Figure 2, C and D). In addition, NAC treatment significantly prohibited myoblast apoptosis under HMS stimulation, whereas LMS-induced apoptosis was barely affected by NAC (Figure 2, C and D). Consistent with the FCAS results, the cleaved caspase-3 level was also elevated in HMS-stimulated myoblasts, which was blocked by pretreatment with NAC (Figure 2E). It should be noted that even though cellular ROS in LMS- or HMS-treated cells were eliminated effectively to the same level as in the unstretched cells, LMS- or HMS-induced apoptosis was still higher than its unstretched counterpart (Figure 2, C and D). Altogether, these results indicated that LMS-induced minor apoptosis was not attributed to the minor accumulation of ROS, whereas the excessively accumulated ROS were directly linked with HMS-induced drastic apoptosis of myoblasts.
Stretch-induced elevation of p53 protein level was partially related to ROS production
Because p53 has been reported to be closely related to ROS homeostasis (Liu et al., 2008), we assumed that p53 expression should be changed in response to ROS generation during mechanical stretch. Real-time PCR results displayed that the p53 mRNA level was slightly raised upon LMS stimuli, whereas it was significantly enhanced in HMS-treated cells, regardless of pretreating with or without NAC (Figure 3A). Western blot (WB) results showed that the protein level of p53 was also enhanced to various degrees by LMS and HMS (Figure 3, B and C). When ROS production was blocked by NAC, the HMS-induced elevation of p53 protein was significantly attenuated, whereas it did not change in the LMS-treated group (Figure 3, B and C). These results demonstrated that p53 protein was stabilized in C2C12 myoblasts subjected to LMS or HMS, partially depending on the generation of ROS. However, the p53 protein level was not always relevant to the ROS amount, which can be seen when comparing Figure 2B and Figure 3C.
FIGURE 3:
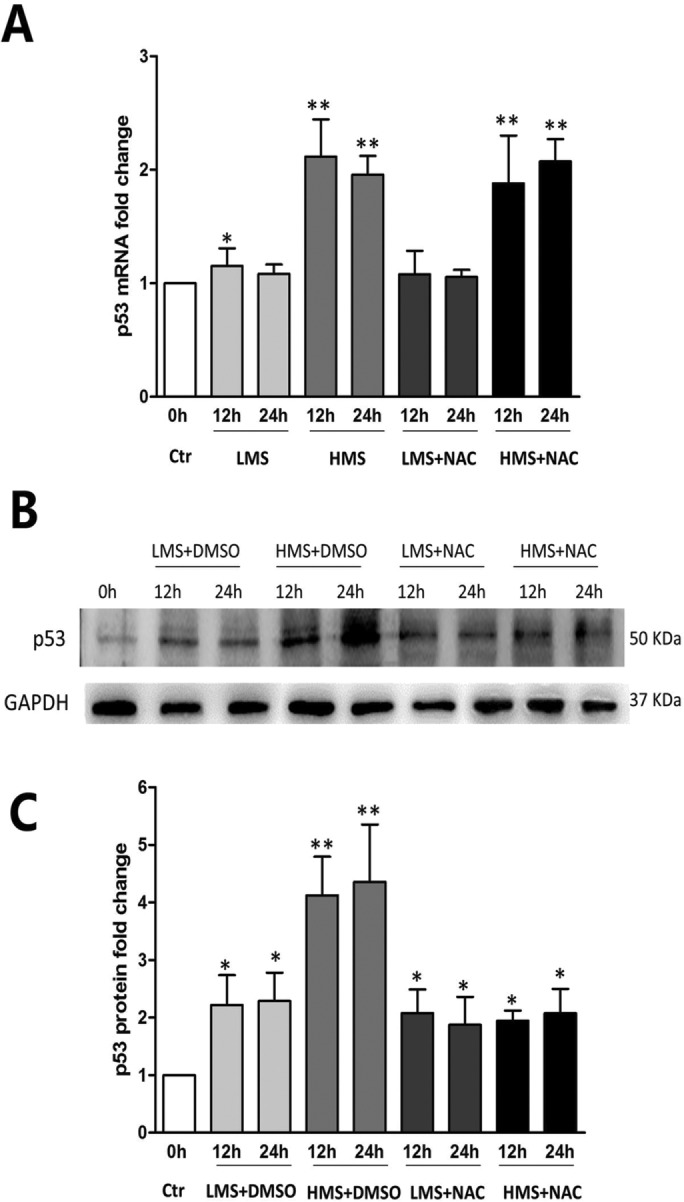
Stretch-induced elevation of p53 protein level was partially related to ROS production. (A) Real-time PCR displayed the p53 mRNA level of myoblasts under LMS or HMS stimuli with or without NAC pretreatment. (B) Representative WB result of p53 protein level in myoblasts under LMS or HMS stimuli with or without NAC pretreatment. (C) Densitometric analysis of p53 protein expressions. Data were analyzed by one-way ANOVA and are presented as mean ± SD. Significant differences are shown by *, P < 0.05 and **, P < 0.01, compared with static control cells.
p53 participated in HMS-induced apoptosis of myoblasts
In myoblasts subjected to HMS, the discrepancy between the higher apoptotic ratio in 24 h than in 12 h and similar p53 expression in these two groups led us to doubt the involvement of p53 in regulating C2C12 apoptosis (Figures 2D and 3C). To study the above hypothesis, we knocked down endogenous p53 expression through RNA interference (RNAi). Myoblasts were infected with lentivirus containing either shRNA targeting p53 or scrambled shRNA. p53 shRNA vector greatly reduced the p53 mRNA level by ∼80% (Figure 4A, top panel), and to a lesser extent, the p53 protein level (Figure 4A, bottom panel). Green fluorescent signal confirmed the infection efficiency of Sh-p53 lentivirus (Figure 4B).
FIGURE 4:

p53 participated in HMS-induced apoptosis of myoblasts. (A) C2C12 myoblasts were transfected with lentivirus vector containing p53 shRNA or scrambled shRNA. After transfection (48 h), cells were collected for real-time PCR and WB analysis to detect the efficiency of p53 silencing. Scrambled shRNA was used as negative control for both real-time PCR and WB results. (B) One week of 2 mg/ml puromycin selection generated the stable p53-knockdown myoblasts (left, light microscopy picture of Sh-p53 cells; middle, fluorescence microscopy picture of the same field; right, merge of light microscopy picture and fluorescence microscopy picture in the same field). Scale bar, 50 μm. (C) Effect of LMS or HMS on protein levels of p53 and cleaved caspase-3 in p53 shRNA or scrambled shRNA infected myoblasts. Same samples were immunoblotted by GAPDH as the loading control. Black arrow indicates the cleaved capase-3 fragment with molecular size around 19 kDa. (D) Densitometric analysis of p53 protein expressions in p53 shRNA or scrambled shRNA infected myoblasts under LMS or HMS stimulation. (E) AV/PI staining and flow cytometry analysis of apoptosis in p53 shRNA or scrambled shRNA infected myoblasts under LMS or HMS stimulation. (F) Statistical analysis of the percentage of early (AV+/PI−) and late (AV−/PI+) apoptotic cells in each group. Data were analyzed with Student’s t test for the p53 silencing group comparing to its negative control in each condition. Significant differences are shown by *, P < 0.05 and **, P < 0.01, compared with scrambled shRNA infected cells.
When subjected to either LMS or HMS for 12 and 24 h, myoblasts (Sh-p53) displayed a reduced level of p53 protein in comparison to the counterparts overexpressing scrambled shRNA (Sh-NC), showing efficient down-regulation of p53 protein under stretching stimulation (Figure 4, C and D). Intriguingly, p53 knocking down had barely any effect on LMS-induced apoptosis of C2C12 myoblasts, in either the 12 h or the 24 h loading group (Figure 4, E and F). However, when subjected to HMS for 12 and 24 h, the Sh-p53 transfected cells had alleviated apoptosis (Figure 4, E and F), which was reassured by caspase-3 immunoblot (Figure 4C). Collectively, these results demonstrated that although p53 was dispensable for LMS-induced apoptosis of C2C12 myoblasts, it did play a vital role in HMS-induced apoptosis of myoblasts, though not absolutely related to its total protein level.
The effect of stretch-generated ROS on subcellular localization of p53 in myoblasts
It has been well known that p53 functions are regulated, in part, through subcellular localization, in addition to its transcription, translation, and protein stability (O’Brate and Giannakakou, 2003). Thus, we explored p53 subcellular localization in myoblasts under different stretching stimulation, aiming to testify whether LMS or HMS distinctly influenced p53 subcellular localization. Immunofluorescence results showed that in LMS-loaded cells, p53 protein located primarily in the nucleus (Figure 5A). By contrast, p53 translocated from nucleus to cytoplasm in 12 h HMS-loaded cells, whereas it almost exclusively existed in cytoplasm in 24 h HMS-loaded cells (Figure 5A). In addition, cell fractionation experiments were applied to verify p53 protein level in nucleus and cytoplasm (Figure 5, B and C).
FIGURE 5:

The effect of ROS on subcellular localization of p53 in myoblasts under LMS or HMS stimuli. (A) Cells were loaded under either HMS or LMS for 12 and 24 h, with or without NAC pretreatment. After this, cells were fixed and permeabilized (see Materials and Methods) and then incubated with p53 antibody. After labeling, cells were extensively washed and localization of p53 was detected by immunofluorescence of anti-mouse IgG-FITC. The figure shows a representative immunostaining analysis after counterstaining with DAPI. (B) Protein samples from the above groups were separated for nuclear and cytoplasmic fractions, and WB experiments were applied using p53 antibody to detect p53 subcellular localization. H3 histone and GAPDH were used as loading controls of nuclear and cytoplasmic fractions. (C) Densitometric analysis of p53 protein expressions in nuclear or cytoplasmic samples. Data were analyzed by one-way ANOVA and are presented as mean ± SD. Significant differences are shown by *, P < 0.05 ; ***, P < 0.001 (nuclear sample); and #, p < 0.05 (cytoplasmic sample), compared with static control cells.
Next, we determined to see whether the cytoplasmic redistribution of p53 was related to ROS generated during stretch. Both immunofluorescence and WB results showed that in LMS-loaded cells pretreated with NAC, the subcellular mobilization of p53 remained unchanged (Figure 5, A–C). By contrast, in HMS-loaded cells pretreated with NAC, the diminished cytoplasmic p53 level was accompanied by the raised nuclear p53 level (Figure 5, A–C). Collectively, these data proved that p53 nuclear-to-cytoplasmic trafficking could be ascribed to ROS that were overproduced during intensive stretch loading.
HMS-inactivated AKT prompted p53 nuclear-mitochondrial redistribution and apoptosis in myoblasts
In addition to the function of nuclear p53 that induces apoptosis through transactivating proapoptotic target genes, mitochondria have also been recognized as another site where p53 promotes apoptosis via transcription-independent pathways (Speidel, 2010). Thus, to discern whether the cytoplasmic portion of p53 accumulated in the mitochondria in HMS-induced myoblasts, we conducted immunoblot and immunostaining by using purified mitochondrial fractions and MitoTracker Red CMXRos, respectively. Results from immunostaining demonstrated that cytoplasmic p53 molecules colocalized with mitochondria in HMS-loaded myoblasts (Figure 6A), which was further confirmed by immunoblot results of purified p53 subcellular fractions (Figure 6B).
FIGURE 6:

Mitochondrial translocation of p53 in myoblasts under HMS stimuli. (A) Cells were loaded under HMS for 12 and 24 h, and Mito-Tracker Red CMXRos staining was used as a marker of mitochondria before regular immunofluorescence staining. The pictures were representative results displaying p53 nuclear export and mitochondrial localization after 12 and 24 h HMS stimuli. The 0 h group was used as the control group, with the same exposure time being used for the 12 and 24 h groups. The right pictures adjacent to the left ones are magnified pictures of the white rectangular area in the left pictures. (B) WB results further confirmed p53 mitochondrial translocation in myoblasts under HMS stimuli. H3 histone, GAPDH, and Cox IV were used as markers of cytoplasmic, nuclear, and mitochondrial fractions, respectively.
AKT, also known as protein kinase B (PKB), is an important regulator of p53 function and apoptosis (Abraham and O’Neill, 2014). In addition, AKT was reported to be able to differently control p53 expression and subcellular localization in response to various cellular stimuli (Pohnke et al., 2004; Saito et al., 2005; Yang et al., 2006). First, we examined AKT activity in myoblasts upon stretching stimuli. WB results showed that phospho-AKT (p-AKT) in LMS-treated cells was elevated, in contrast to unstretched cells (Figure 7A). Strikingly, when myoblasts were subjected to HMS, p-AKT expression was significantly lower than its basal level (Figure 7A). To further testify whether decreased AKT activity was directly linked with p53 mitochondrial translocation in HMS-loaded cells, HA-tagged constitutively active AKT vector (CA-AKT) was transiently overexpressed in myoblasts before being subjected to HMS to sustain the p-AKT level, whereas HA-tagged wild-type AKT plasmid (WT-AKT) was used as the negative control. Immunostaining by HA antibody was conducted to observe the transfecting efficiency of WT-AKT and CA-AKT plasmids (Figure 7B). Immunoblots of the AKT and HA tags confirmed the efficient transfection of these plasmids, whereas an immunoblot of p-AKT demonstrated that transfecting CA-AKT effectively raised AKT activity in both unstretched and HMS-stretched myoblasts (Figure 7C).
FIGURE 7:

HMS-inactivated AKT prompted p53 nuclear-mitochondrial redistribution and apoptosis. (A) WB results showed that AKT was phosphorylated in LMS-treated C2C12 cells and was dephosphorylated in HMS-treated cells. (B) Transfection efficiency of WT-AKT and CA-AKT plasmids were evaluated by immunostaining with HA antibody and observing green signal under the fluorescence microscope. (C) Representative WB results confirmed that transfecting both WT-AKT and CA-AKT vectors effectively raised the AKT level in both unstretched and HMS-stretched myoblasts, whereas only WT-AKT vector increased the p-AKT level. (D) Immunofluorescence results proved that C2C12 cells overexpressing CA-AKT resulted in p53 nuclear import subjected to HMS stimulation, comparing to the counterparts overexpressing WT-AKT that displayed p53 mitochondrial localization under HMS. The right pictures adjacent to the left ones were magnified pictures of the white rectangular area in the left pictures. (E) WB results further confirmed the diminished p53 level in mitochondrial fraction and raised the p53 level in the nuclear fraction in HMS-loaded C2C12 cells transfected by the CA-AKT vector. H3 histone, GAPDH, and Cox IV were used as markers of cytoplasmic, nuclear, and mitochondrial fractions, respectively. (F) Dose-dependent reduction of mitochondrial p53 and elevation of nonmitochondrial p53 in HMS-loaded C2C12 cells by transfection of CA-AKT vector. GAPDH and Cox IV were used as markers of nonmitochondrial and mitochondrial fractions, respectively. (G) AV/PI staining and flow cytometry analysis of apoptosis in HMS-treated C2C12 cells that were transfected with either WT-AKT vector or CA-AKT vector. (H) Statistical analysis of the percentage of early (AV+/PI−) and late (AV−/PI+) apoptotic cells in each group. Data were analyzed with Student’s t test for the CA-AKT group comparing to its negative control WT-AKT group in 12 and 24 h HMS-loaded cells. (I) Western blots were performed using caspase-3 primary antibody. Same samples were immunoblotted by GAPDH as the loading control. Black arrow indicates the cleaved capase-3 fragment with molecular size around 19 kDa. Significant differences are indicated by asterisks: *, P < 0.05 and **, P < 0.01, comparing WT-AKT with CA-AKT in 12 and 24 h loading groups.
The total p53 protein level was unaffected when transfecting CA-AKT or WT-AKT (Figure 7C). Both immunofluorescence and immunoblotting results testified that HMS-induced nuclear-mitochondrial shuttling of p53 was impeded in cells overexpressing CA-AKT, in contrast to the cells overexpressing WT-AKT (Figure 7, D and E). Moreover, when the p-AKT level was increased by transfecting the CA-AKT vector in a dose-dependent manner, the ratio of mitochondrial p53 (M-p53)/nonmitochondrial p53 (NM-p53) in myoblasts subjected to HMS for 24 h was gradually decreased (Figure 7F). Notably, FCAS of AV/PI staining and caspase-3 immunoblot results showed that HMS-induced myoblast apoptosis was drastically attenuated by transfecting CA-AKT (Figure 7, G–I). Therefore, these results corroborated the fact that in C2C12 myoblasts, HMS-inactivated AKT incurred drastic apoptosis, accompanied by p53 nuclear bypassing and mitochondrial redistribution.
ROS exquisitely regulated PI3K/AKT activity in myoblasts during stretching stimuli
Considering that AKT was reported to be redox sensitive in skeletal myoblasts (Kosmidou et al., 2001; Orzechowski et al., 2005; Yen et al., 2012), and ROS were differently generated by LMS or HMS stimuli, we speculated that the distinct effects of LMS and HMS on AKT activity might be attributed to the different generation of ROS. To elucidate the effect of ROS on AKT activation in stretched myoblasts, cells were pretreated with NAC before LMS or HMS stimulation. As can be seen in Figure 8, A and B, when the accumulation of ROS was completely blocked by NAC, LMS-induced AKT phosphorylation was nearly abolished; accordingly, HMS-dephosphorylated AKT was also partially rescued, comparing to the basal AKT activity in unstretched cells. Thus, it seemed that minor or physiological elevation of ROS in LMS-treated cells promoted AKT activation, whereas drastic accumulation of ROS during HMS stimuli dephosphorylated AKT to an extent even lower than basal p-AKT in unstretched cells.
FIGURE 8:
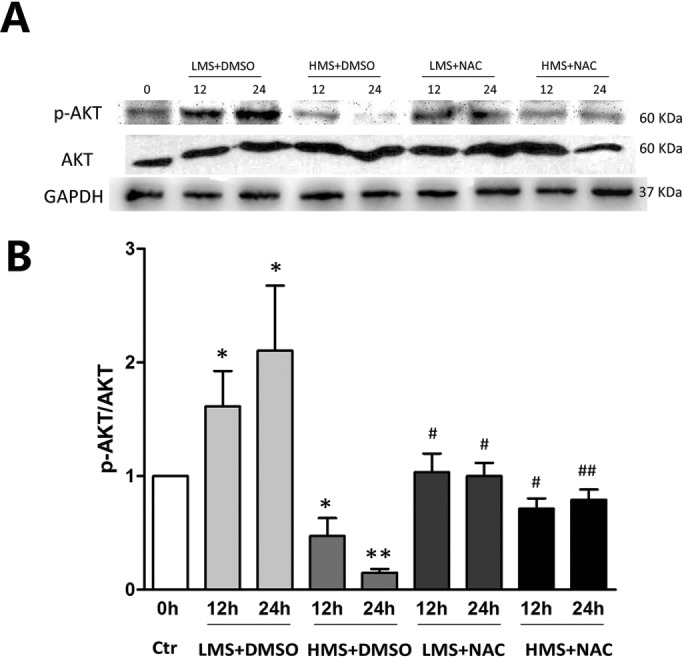
Stretching magnitude–dependent regulation of AKT by ROS. (A) Cells were stretched by LMS or HMS for 12 and 24 h, with or without pretreatment with NAC. Phospho-AKT (Ser473) level and total-AKT level were detected by WB. The same samples were immunoblotted by GAPDH as the loading control. (B) Densitometric analysis of the ratio of p-AKT/AKT. Data were analyzed by one-way ANOVA and are presented as mean ± SD. Significant differences are shown by *, P < 0.05 and **, P < 0.01, compared with static control cells, while #, P < 0.05 and ##, P < 0.01 show the significant differences between the stretching alone group and the stretching combined with NAC pretreatment group.
Phosphorylation of p53 Ser389 was required for HMS-induced p53 mitochondrial translocation and myoblast apoptosis
Posttranslational regulation, especially phosphorylation, of p53 plays an important role in determining p53 subcellular localization. Castrogiovanni et al. (2018) provided direct evidence that mutant p53 S392A was unable to mobilize to mitochondria in the human p53-null H1299 cell line under genotoxic stress. Because Ser389 of mouse p53 is homologous with Ser392 of human p53, we decided to express mutant p53 S389A plasmid in p53-shRNA C2C12 myoblasts before HMS stimulation to impede mitochondrial trafficking of p53. First of all, phosphorylation of p53 on Ser389 (p-S389) was observed in HMS-stressed myoblasts, which was not obvious in LMS-stressed cells (Figure 9A). Furthermore, in HMS-stretched cells, p-S389 of p53 existed in mitochondria and cytoplasm fractions, with no p-S389 detected in the nuclear portion (Figure 9B). To explore whether ROS-AKT signaling was involved in phosphorylation of p53 Ser389, NAC scavenger and CA-AKT plasmid were used to intervene with ROS accumulation or AKT dephosphorylation during HMS stimuli, respectively. As can be seen in Figure 9C, both NAC and CA-AKT negatively affected the level of p-S389, with NAC having a more pronounced effect than CA-AKT. These results confirmed that ROS accumulation and subsequent AKT dephosphorylation acted as upstream events of p53 Ser389 phosphorylation in HMS-treated myoblasts.
FIGURE 9:

Phosphorylation of p53 Ser389 was required for HMS-indued p53 mitochondrial translocation and myoblast apoptosis. (A) Cells were stretched by LMS or HMS for 12 and 24 h, and p53 phosphorylation on Ser389 was detected by WB. (B) Confirmation of phospho-p53 (Ser389) localization in HMS-loaded C2C12 cells for 12 and 24 h. H3 histone, GAPDH, and Cox IV were used as markers of cytoplasmic, nuclear, and mitochondrial fractions, respectively. (C) HMS-promoted phosphorylation of p53(Ser389) was dependent on the generation of ROS and subsequent dephosphorylation of AKT. Pretreatment with NAC or transfecting with CA-AKT vector efficiently blocked p53(Ser389) phosphorylation under HMS stimuli. (D) Transfection of p53-knockdown C2C12 cells with wild-type p53 vector or mutant p53 (S389A) vector partially rescued the p53 protein level. (E) Mutant p53 (S389A) could not be phosphorylated under 24 h–HMS stimuli, comparing to wild-type p53 in Sh-p53–infected C2C12 cells. (F) Mutant p53 (S389A) could not translocate into mitochondria under 24 h–HMS stimuli, comparing to wild-type p53. M and NM represent mitochondrial and nonmitochondrial portions, respectively. (G) After stimulated by HMS for 24 h, both mutant p53 (S389A) and wild-type p53 transfected cells were subjected to subcellular fraction assay, and the WB result confirmed that mutant p53 (S389A) was mainly accumulated in nonmitochondrial cytoplasm, comparing to wt-p53 that localized in mitochondria. M, C, and N represent mitochondrial, cytoplasmic, and nuclear portions, respectively. (H) AV/PI staining and flow cytometry analysis showed that transfecting wt-p53 vector could reverse the inhibited apoptosis in p53-knockdown C2C12 cells, whereas transfecting mt-p53 (S389A) failed to accomplish a similar effect. (I) Statistical analysis of the percentage of early (AV+/PI−) and late (AV−/PI+) apoptotic cells in each group. Data were analyzed by one-way ANOVA and are presented as mean ± SD. *, P < 0.05 and **, P < 0.01 represent the significant difference among the HMS-treated groups of C2C12 with distinct p53 expression. #, P < 0.05 shows the significant difference between static and stretched cells. (J) Representive WB results of caspase-3 cleavage in each group. Same samples were immunoblotted by GAPDH as the loading control. Black arrow indicates the cleaved capase-3 fragment with molecular size around 19 kDa.
Next, we transfected the Sh-p53 C2C12 myoblasts with either wt-p53 or mutant p53 (S389A) plasmids in order to see whether the nonphosphorylatable mutant S389A-p53 impaired p53 translocation to the mitochondria and induction of myoblast apoptosis under HMS. Transfection of either wt-p53 or S389A-p53 plasmid partially restored the total p53 protein level (Figure 9D). After being subjected to 24 h of HMS stimulation, total p53 protein levels in either wt-p53 or S389A-p53 transfected Sh-p53 C2C12 were as similar as normal myoblasts, except for the empty-vector transfected Sh-p53 cells (Figure 9E). However, as we expected, the p-S389 p53 level in S389A-p53 transfected Sh-p53 cells was significantly lower than its wt-p53 transfected counterparts (Figure 9E). To further testify whether phosphorylation of p53 on Ser389 was essential to its HMS-induced mitochondrial trafficking, subcellular fractions immunoblotting was conducted. As can be seen in Figure 9F, transfecting Sh-p53 myoblasts with wt-p53 plasmid showed p53 mitochondrial targeting after 24 h of HMS, which was partially abrogated by p53 Ser389 mutation to Ala389 (Figure 9F). Interestingly, although S389A mutation inhibited p53 mitochondrial localization under HMS, it did not restrain p53 in the nucleus; instead, S389A mutation seemed to confine p53 in the nonmitochondrial cytoplasm in HMS-stimulated myoblasts (Figure 9G).
Finally, FCAS of apoptosis demonstrated that transfection with wt-p53 plasmid prominently increased the apoptotic ratio of HMS-stimulated Sh-p53 C2C12 cells (Figure 9, H and I), which previously showed decreased apoptotic ratio without wt-p53 transfection (Figure 4E). Notably, transfection with S389A-p53 failed to restore the HMS-incurred apoptosis as wt-p53 did (Figure 9, H and I). Immunoblotting of cleaved capsase-3 displayed similar trends (Figure 9J). Taken together, these results demonstrated that ROS/AKT–up-regulated Ser389 phosphorylation of p53 was crucial to HMS-induced p53 mitochondrial targeting and resultant myoblast apoptosis.
20% stretch-induced ROS led to mitochondrial translocation of p53 and apoptosis in L6 myoblasts
To testify whether L6 and C2C12 skeletal myoblasts have similar mechanical properties, we also tested the effect of stretch on L6 myoblasts using different magnitudes. MTT assays showed that after 24 h stimulation, L6 proliferation was promoted by 10% stretch and inhibited by 20% stretch, while it was unchanged by 5 and 15% stretch (Supplemental Figure 1A). Notably, L6 apoptosis was significantly elevated by 20% stretch, whereas it did not change by 5, 10, and 15% stretch (Supplemental Figure 1, B and C). In addition, ROS was overproduced in L6 myoblasts under 20% stretching stimuli (Supplemental Figure 1, D and E). L6 apoptosis was partially prohibited when ROS was eliminated by NAC (Supplemental Figure 1, F and G). To further examine the AKT and p53 responses to 20% stretch-induced ROS in L6 myoblasts, WB and immunofluorescence were performed. As can be seen in Supplemental Figure 1H, the p53 level was increased and the p-AKT level was decreased in 20% stretch-stimulated L6 myoblasts, whereas ROS elimination reversed these processes. Furthermore, p53 protein accumulated mainly in mitochondria and partially in cytoplasm and nuclei by 20% stretch (Supplemental Figure 1, I and J). In contrast, NAC pretreatment led to p53 mitochondria–cytoplasm shuttling in 20% stretch-stimulated L6 myoblasts (Supplemental Figure 1, I and J). All together, these results demonstrated that L6 myoblasts seemed to display similar cellular responses to stretching stimuli, which was inconsistent with the studies by Ma et al. (2017) and Fu et al. (2018).
DISCUSSION
Skeletal muscles are constantly exposed to mechanical stimuli, such as gravity and physical exercises, which contributes to their growth and reconstruction. However, mechanical overloading (e.g., high-force contractions) incurs muscle damage rather than regeneration under some circumstances (Loerakker et al., 2010; Ferry et al., 2015; Naughton et al., 2017). Our present study compared the distinct effects of HMS and LMS on C2C12 myoblasts, and testified that HMS, rather than LMS, inactivated AKT pathway due to ROS overgeneration, which further contributed to p53 mitochondrial translocation and C2C12 apoptosis.
Stretching magnitude–dependent generation of ROS in myoblasts
Skeletal myoblasts produce transient fluxes of ROS in response to diverse stimuli (Reid et al., 1992; Zuo et al., 2000; Zuo and Clanton, 2005; Falk et al., 2006; Martins et al., 2008; Allen et al., 2010). In fact, ROS participate in a cascade of events leading to muscle regeneration and repair within physiological concentrations. However, local persistence of ROS delays the beneficial processes by oxidatively damaging differentiating myoblasts and myotubes (Gomez-Cabrera et al., 2015). Our previous study subjected C2C12 myoblasts to different stretches (magnitudes of 5, 10, 15, and 20%), and demonstrated that 15 and 20% stretches reduced cell viability because of elevated apoptosis, whereas 5 and 10% stretches did not (Tan et al., 2009). The present study further confirmed the contribution of ROS overproduction to HMS-induced C1C12 myoblast apoptosis by using ROS scavenger NAC, consistent with some other murine cell types such as SH-SY5Y cell and ATII cell that were rescued from stretch-induced apoptosis by inhibition of ROS (Kuhn et al., 2017; Xu et al., 2017).
The different responses of C2C12 to different ROS production have already been testified to by Yu et al. (2019). Interestingly in our study, inhibition of ROS caused little difference in the apoptotic ratio of LMS-treated myoblasts (Figure 2). Considering the mounting in vivo evidence testifying to the favorable effects of exercise-generated ROS on muscle functions and training-induced adaptions, it is logical to speculate that myoblasts under LMS stimuli produced a low level of ROS that possibly promoted myoblast proliferation or differentiation, rather than apoptosis. In fact, lots of research has already demonstrated that mechanical stretch is a positive stimulus for in vitro myogenic differentiation. Particularly, almost all of these studies applied the stretching magnitude of 10% roughly (Zhang et al., 2004; Kurokawa et al., 2007; Zhan et al., 2007; Chang et al., 2016; Andersen et al., 2018), while 20% stretching, on the other hand, was proved to be detrimental for myogenic differentiation (Akimoto et al., 2001).
Nuclear migration of p53 in LMS-loaded myoblasts
In 1996, Soddu et al. (1996) first demonstrated the indispensable role of p53 in myogenic differentiation. Thus, it is reasonable to conjecture that p53 nuclear localization in LMS-treated myoblasts might be related to the process. One chief consideration is that the nuclear localized p53 could be a hint of its function of transactivating genes involved in cell cycle arrest and DNA damage repair, which are critical during myogenic differentiation (Porrello et al., 2000; Latella et al., 2004; Fayzullina and Martin, 2016). In this case, the fact that p53 silence had no effect on LMS-triggered apoptosis was well consistent with the notion that p53 is not required for the differentiation associated apoptosis of skeletal myoblasts (Cerone et al., 2000). However, in LMS-loaded cells, ROS scavenger NAC had little effect on p53 protein expression and subcellular localization (Figures 3B and 5B). Thus, we assumed that ROS up-regulation and p53 nuclear localization might independently contribute to myogenic differentiation under LMS stimulation, though they were known to be closely related to each other (Liu et al., 2008).
Mitochondrial translocation of p53 in HMS-loaded myoblasts
In contrast to the small effect of knocking down p53 on LMS-induced apoptosis of C2C12 cells, HMS-induced apoptosis was significantly alleviated by p53 knockdown, indicating that mechanical overloading (HMS) triggered the functional switch of p53 to a proapoptotic factor in C2C12 cells. Our study further confirmed the correlation between p53 mitochondrial localization and HMS-induced myoblast apoptosis. Evidence is as follows: 1) there were more cytoplasmic p53 in the 24 h HMS loading group than in the 12 h loading group, concordant with more apoptosis in the former; 2) ROS scavenger NAC efficiently blocked p53 cytoplasmic trafficking in both 12 and 24 h HMS loading groups, while apoptosis was also substantially inhibited in these two groups; 3) immunoblotting and immunostaining results manifested the cytoplasmic p53 mostly accumulated in mitochondria; 4) overexpressing CA-AKT promoted the mitochondria-nuclear shuttling of p53, accompanied by the concomitant reduction in HMS-induced apoptosis; and 5) transfecting p53-knockdown C2C12 cells with mutant p53(S389A) not only restrained p53 from migrating to mitochondria, but also partially protected these cells from HMS-induced apoptosis. Although direct proof showing mitochondrial p53 incurred apoptosis was lacking in our study, the attenuated apoptosis resulting from impaired p53 mitochondrial migration indicated that p53 participated in HMS-triggered apoptosis via a transcription-independent manner (Speidel, 2010). To date, there is no research reporting the mechanoresponses of p53, such as subcellular localization, in skeletal myoblasts. Still, a couple of in vivo studies focusing on the effects of exercise on skeletal muscles had verified p53 nuclear or mitochondrial localization in these tissues (Siu and Alway, 2005; Saleem and Hood, 2013; Safdar et al., 2016; Tachtsis et al., 2016). These authors all agreed with the role of p53 in regulating mitochondrial biogenesis, mitochondrial DNA content, and damage repair, although their conclusions were not always consistent and were even conflicting in some cases, which could be ascribed to variable factors (species, age, exercise protocol, muscle type, etc.). Notably, comparing to these nonapoptotic functions of p53 mitochondrial accumulation in exercised skeletal muscles, our present study reported that mitochondria-localized p53 provoked severe apoptosis in HMS-treated myoblasts. This discrepancy could be attributed to two major aspects: 1) Active contraction of skeletal muscles is totally different from passive stretching of them. Thus, stretching of masticatory muscles by a functional appliance and exercise of them by chewing might represent distinct muscular biology. 2) Nonmitotic skeletal myonuclei are the major component of cell population in skeletal muscles. Thus, most of those in vivo findings were reflected by the changes in the skeletal myonuclei. In contrast, C2C12 myoblasts could be regarded as myogenic precursor cells, which constitutes a relatively small portion compared with the total myonuclei. Nevertheless, both these in vivo studies and our present study pointed out the multifunction of p53 in muscle biology.
Furthermore, our study corroborated that ROS-inactivated AKT promoted p53 mitochondrial translocation. First, AKT phosphorylation was negatively correlated with ROS during HMS stimuli (Figure 8). This was consistent with some studies that described xenobiotics-induced ROS-dephosphorylated AKT in C2C12 cells, whereas pretreatment with an antioxidant, such as NAC, restored AKT phosphorylation (Yen et al., 2012; Rahman et al., 2014). Intriguingly, other researchers stated that AKT was involved in activation of antioxidants, and inhibition of AKT led to increased ROS and C2C12 apoptosis (Kang et al., 2015; Ding et al., 2017). Overall, the complex relations between AKT and ROS in C2C12 myoblasts could be exquisitely regulated by different cellular stimuli, which can be seen in our present study that displayed opposite correlation between AKT and ROS by merely treating C2C12 cells with different magnitudes of stretch.
Second, we uncovered that p53 Ser389 phosphorylation promoted p53 to translocate into mitochondria, and ROS-dephosphorylated AKT was necessary but not sufficient to phosphorylate p53 Ser389. This was supported by the following facts: 1) overexpressing mutant p53 (S389A) constrained p53 to the nonmitochondrial cytoplasm; 2) overexpression of CA-AKT inhibited p53 Ser389 phosphorylation; and 3) NAC was shown to be more effective than CA-AKT in hindering p53 Ser389 phosphorylation. Interestingly, there were p-p53 Ser389 existing in the cytoplasm and mitochondria, but not in nuclei, which indicated that p53 Ser389 could not be phosphorylated in the nuclei. In addition, p53 was shuttled out of nuclei in WT-AKT transfected cells, whereas CA-AKT confined p53 to nuclei. Thus, these results led to the assumption that cytoplasmic sequestration of p53 by ROS-inactivated AKT is a prerequisite for its phosphorylation at Ser389. With respect to the kinases that might phosphorylate p53 Ser389 in cytoplasm, p38 was the most attractive candidate, not only because our group had participated in another research and testified p38 activation in stretched C2C12 cells (Ji et al., 2010), but also that several studies had already reported direct evidence of p38-dependent phosphorylation of p53 on Ser389 (murine; Huang et al., 1999; Keller et al., 1999) or its homology Ser392 (human; Horikawa-Miura et al., 2007; Li et al., 2007; Panickar et al., 2009; Flores-Lopez et al., 2013; Dziegielewska et al., 2014). Because p38 is a well-known downstream target of ROS, it is reasonable that AKT dephosphorylation shuttled p53 into cytoplasm, which enabled its phosphorylation at Ser389 and localization to mitochondria by p38 (Wan et al., 2017). Notably, the negative correlation between p38 and AKT had been demonstrated in a couple of experiments (Gratton et al., 2001; Kim et al., 2008; Cui et al., 2016), especially with one study conducting in vitro stretching of alveolar epithelial cells (Peng et al., 2010). Collectively, it might be worth exploring the cross-talk between p38 and AKT in our HMS-stimulated C2C12 cells to clarify whether AKT dephosphorylation and p38 phosphorylation were interconnected in phosphorylating p53 Ser389.
However, there were some discrepancies regarding p53 nuclear or mitochondrial translocation under AKT dephosphorylation. For example, some researchers displayed p53 mitochondrial trafficking by AKT dephosphorylation (Yang et al., 2006; Hu et al., 2012; Zheng et al., 2014; Wang et al., 2015), whereas others verified that dephosphorylated AKT prompted p53 nuclear localization (Pohnke et al., 2004; Saito et al., 2005; Pardossi-Piquard et al., 2009; Wang et al., 2017). Interestingly in our study, LMS-induced p53 nuclear import was proved to be irrelevant to AKT phosphorylation, whereas HMS-induced p53 nuclear export was dependent upon AKT dephosphorylation. Classically, MDM2 is well known for its ability to carry p53 out of nuclei for degradation. Many other factors, such as c-Abl, p14ARF, and CBP/p300, were reported to regulate MDM2 activity, thus affecting p53 subcellular localization (O’Brate and Giannakakou, 2003). In addition, Ser15 phosphorylation of p53 by some kinases like Chk1/2 and JNK was equally important with MDM2 in orchestrating p53 nuclear transport (Appella and Anderson, 2001). Thus, we assumed that AKT was not the only factor contributing to p53 nuclear-cytoplasmic shuttling in response to stretching stimuli, and those factors above might cooperate with AKT in modulating p53 subcellular localization under mechanical stretch.
In summary, C2C12 under LMS generated low levels of ROS, accompanied by p53 nuclear localization and AKT phosphorylation, but failed to display significant apoptosis; HMS caused severe apoptosis of C2C12, which was attributed to the overaccumulation of ROS that dephosphorylated AKT, which promoted p53 phosphorylation at Ser389 and mitochondrial translocation (Figure 10).
FIGURE 10:

An illustrative view of the different fates of C2C12 myoblasts under LMS or HMS stimulation. Left panel, LMS-induced mild elevation of ROS, resulting in the activation of AKT in C2C12 myoblasts. In addition, p53 was found in the nuclei of LMS-loaded C2C12 cells. Some studies demonstrated that lower magnitude cyclic stretch could promote myogenic differentiation, even though whether those changes in our study were related to C2C12 differentiation had not been explored. Right panel, HMS caused drastic apoptosis of C2C12 cells, which could be ascribed to the massive accumulation of ROS that dephosphorylated AKT. Inactivation of AKT in HMS-loaded C2C12 cells was shown to be necessary for p53 nuclear export in our study, a step required for its further phosphorylation on Ser389 that might involve p38 MAPK. Phospho-p53 (Ser389) translocated to mitochondria of C2C12 cells and induced their apoptosis under HMS stimuli.
MATERIALS AND METHODS
Reagents
The 3-(4,5-methylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) kit (#C0009) and 4’,6-diamidino-2-phenylindole (DAPI) (#C1006) were purchased from Beyotime Biotechnology (Shanghai, China). The In Situ Cell Death Detection Kit, TMR red (#12156792910) was purchased from Roche. Fluorescein isothiocyanate (FITC) conjugated annexin V and propidium iodide was purchased from ThermoFisher (#V13242; Waltham, MA). Oxidation-sensitive probe 2′,7′-dichlorodihydrofluorescein diacetate was purchased from Molecular Probes (#C6827; Eugene, OR). The PrimeScript RT Reagent Kit (#RR037A) and TB Green Fast qPCR Mix (#RR430A) were purchased from Takara Biomedical Technology (Shiga, Japan). Antibodies against p53 (Cell Signaling Technology; #2524), phospho-p53 (Ser392; Cell Signaling Technology; #9281), AKT (Cell Signaling Technology; #4685), phospho-AKT (Ser473; Cell Signaling Technology; #4060), caspase-3 (Cell Signaling Technology; #9662) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against GAPDH (Elabscience; #E-AB-20032), Cox IV (Elabscience; #E-AB-22002), H3 histone (Elabscience; #E-AB-14124) were purchased from Elabscience (Wuhan, China). Horseradish peroxidase (HRP)-labeled goat anti-mouse second antibody (#A0216) and HRP-labeled goat anti-rabbit second antibody (#A0208) were purchased from Beyotime (Shanghai, China). FITC-labeled goat anti-mouse second antibody (#F0257) and ROS scavenger N-acetyl-l-cysteine (NAC) (#A9165) were purchased from Sigma (St. Louis, MO). P53 shRNA and scrambled shRNA lentiviral vectors, CA-AKT, HA-AKT, wt-p53 and mt-p53 (S389A) plasmids were all purchased from Genechem Biological Company (Shanghai, China).
Cell culture
C2C12 and L6 skeletal muscle myoblasts (American Type Culture Collection) were cultured in growth medium (DMEM supplemented with 10% fetal bovine serum [FBS], 2 mM l-glutamine, 1% penicillin/streptomycin, and nonessential amino acids) at 37°C with 5% CO2. The medium was changed every 2 d. Cell passages 4–6 were used in our study.
Application of cyclic stretch
The cells were stretched as described in our previous study. In brief, C2C12 myoblasts were seeded at a density of 3 × 105/ml on Bioflex Collagen Type I–coated six-well plates (Flexcell International, Burlington, NC) and grown under nonstretch conditions until the cells had reached 60% confluence. The cells were then exposed to cyclic stretch involving 1 s of stretch alternating with 1 s of relaxation, using a computer-controlled vacuum stretch apparatus (FX-4000T Tension Plus System; Flexcell International). The magnitude of HMS was defined as 20% deformation, whereas the magnitude of LMS was defined as 10% deformation. Cells were harvested after exposure to cyclic stretch for 12 and 24 h, respectively. Control cells were cultured on same plates without mechanical strain to avoid variations based on the attachment stratum.
MTT assay
Because all the stretch procedures were done in six-well flexible-bottom culture plates, we harvested the cells through trypsin digestion at the end of the experiments and aliquot (1%) from each treatment was replated to a 96-well plate for measuring the viable cell number by MTT assay. For the MTT assay, MTT was added (100 µl/well) to each well of the 96-well plate and incubated in 37°C for 4 h. Formazan products were solubilized with dimethyl sulfoxide, and the optical density was measured at 490 nm.
TUNEL staining
After C2C12 cells were subjected to LMS or HMS for 12 and 24 h, they were washed with phosphate-buffered saline (PBS). Then, the terminal deoxynucleotidyl TUNEL assay was performed for adherent cells with the In Situ Cell Death Detection Kit, TMR red (Roche), according to the manufacturer’s instructions. After the TUNEL reaction, nuclei were counterstained with DAPI according to the manufacturer’s instructions. Samples were analyzed under a fluorescence microscope, Nikon ECLIPSE E400, using a 520–550 or 330–385 nm excitation filter, and a 580 or 420 nm band pass filter for TUNEL and DAPI assays, respectively. Images of four microscopic fields at 100-fold magnification were captured randomly, and the TUNEL-positive cells and DAPI-positive nuclei were counted manually to calculate the percentage of apoptotic cells to total cells. Data are presented as the percentage of three independent experiments with the mean value.
Annexin V binding and propidium iodide staining
Annexin V labeling and fluorescence activated cell sorting (FACS) analysis were performed in accordance with manufacturer’s instructions. Briefly, the cells were harvested, stained with FITC conjugated AV/PI using the Apoptosis Detection Kit (R&D Systems, Minneapolis, MN) after being continuously stretched for 12, 24, and 48 h, and analyzed by flow cytometry (Epics XL; Beckman-Coulter, Krefeld, Germany). FCAS of AV/PI staining was used to quantify viable, early apoptotic, late apoptotic, and necrotic cells. Each data point represents n = 4 cell isolations with triplicate measurements. The untreated population was used to define the levels of apoptosis and number of dead cells under basal conditions. Each experiment was performed in triplicate.
Measurement of ROS
ROS measurement was done as described previously in our study (Tan et al., 2009). Brifely, cells were harvested and washed twice with PBS. Then cells were divided into two parts equally: one for cell number count and the other for ROS measurement. For ROS measurement, cells were incubated for 30 min at 37°C in the dark with the oxidation-sensitive probe 2′,7′-dichlorodihydrofluorescein diacetate. After two washes with PBS, PBS was added and fluorescence from the plates were read in a Microplate Fluorescence Reader FL600 (Bio-Tek) using a wavelength of 486 nm for excitation and 530 nm for emission. ROS levels per 5000 cells of each group were calculated. Unstained cells were applied as background, and the data were normalized to values obtained from the control group.
Quantitative real-time reverse transcription PCR
The total RNA from cultured cells under different experimental conditions was extracted using the Trizol reagent (Invitrogen). After RNA isolation, 500 ng RNA was used for cDNA synthesis using the PrimeScript RT Reagent Kit (Takara). Real-time PCR was performed using Roche Applied Science Light Cycler 480 in a reaction volume of 20 ml per tube containing 2.0 ml cDNA and 10 ml TB Green Fast qPCR Mix (Takara). Following a hot start, the samples were denatured at 95°C for 5 s, and the primer was annealed at 60°C for 40 cycles of 30 s. The mRNA levels relative to that of housekeeping gene GAPDH were calculated by the 2−ΔΔCt method. The primers used for the PCR amplifications are as follows: GAPDH forward: TGAAGGTCGGTGTGAACGG; reverse: GTGAGT GGAGTCATACTGGAA, size 150 base pairs. P53 forward: CCTCAGCATCTTATCCGAGTGG; reverse: TGGATGGTGGTACAGTCAGA, size 128 base pairs.
Western blot
The cells were harvested in RIPA lysis buffer (10 mM Tris, 5 mM EDTA, 0.25% Triton X-100, 300 mM NaCI, 1 mM phenylmethanesulfonyl fluoride [PMSF], and 1 × proteinase inhibitor) as soon as specific experiments were done. Lysates were centrifuged at 12,000 rpm for 10 min at 4°C. Equal aliquots of protein samples were separated by SDS–PAGE using 10–20% Tris-glycine gels and then were electrotransferred onto polyvinylidene fluoride membranes (Millipore, Bedford, MA). Membranes were blocked with 5% lipid-free milk in Tris-buffered saline with 0.1% Tween (TBST) for 1 h at room temperature, followed by incubation with primary antibodies overnight at 4°C. The membrane was washed three times with TBST for 10 min each time and exposed to the HRP-conjugated secondary antibody (1:5000; Santa Cruz Biotechnology) for 2 h at room temperature. The results were observed by the enhanced chemiluminescence detection assay. The intensity of each blot was observed by a computer imaging system and analyzed using Quantity One software after normalization to corresponding loading controls. The primary antibodies used were as follows: anti-p53 (1:1000), anti–phospho-p53 (Ser392; 1:1000), anti–caspase 3 (1:1000), anti-AKT (1:1000), anti–phospho-AKT (Ser473; 1:1000), anti-GAPDH (1:1000), anti–Cox IV (1:1000), and anti–H3 histone (1:500).
Immunofluorescence staining
For immunofluorescence staining, cells grown on Bioflex six-well plates were stretched and then were incubated with 200 nM MitoTracker Red CMXRos (Molecular Probes, Eugene, OR) in culture medium for 30 min. Medium was removed, and the cells were fixed with 4% formaldehyde containing 0.1% glutaraldehyde for 15 min at 37°C. After being rinsed with cold PBS (pH 7.4), the cells were permeabilized using 0.1% Triton X-100 (Fisher) for 10 min and then incubated with 1% bovine serum albumin for 1 h at room temperature. Antibodies against p53 (1:2000 dilution) were added, and the fixed cells were incubated with antibodies at 4°C overnight followed by incubation with anti-mouse immunoglobulin G (IgG)-FITC (Sigma; 1:150 dilution) for 1 h. After removal of antibodies, cells were rinsed with PBS and mounted with 90% glycerol. DAPI staining was conducted right after washing with PBS. Fluorescence was immediately observed using a Nikon ECLIPSE E400 microscope.
Subcellular fractionation
To separate cytoplasmic and nuclear fractions, the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime; #P0028) was used according to the manufacturer’s instructions. To obtain cytoplasmic, nuclear, and mitochondrial fractions synchronously, we applied the method reported by Dimauro et al. (2012). Briefly, cells were pelleted by centrifugation at 400 × g for 5 min and were resuspended in 300–500 μl of STM buffer and homogenized for 1 min on ice. The homogenate was maintained on ice for 30 min, vortexed at maximum speed for 15 s, and then centrifuged at 800 × g for 15 min. The pellet was labeled as P0 and kept on ice; the supernatant was labeled as S0 and used for subsequent isolation of mitochondrial and cytosolic fractions. P0 (containing nuclei and debris) was resuspended in 300–500 μl STM buffer, vortexed at maximum speed for 15 s, and then centrifuged at 500 × g for 15 min. The pellet was resuspended in 200–500 μl NET buffer and vortexed at maximum speed for 15 s and incubated on ice for 30 min. The nuclei were lysed with 10–20 passages and sonicated at high setting for 10–15 s with 30 s pauses. The lysate was centrifuged at 9000 × g for 30 min (at 4°C), and the resultant supernatant was the final “nuclear fraction.” Cytosolic and mitochondrial fractions were extracted from S0 by centrifugation at 11,000 × g for 10 min, and the supernatant (containing cytosol and microsomal fractions) was precipitated in cold 100% acetone at −20°C for at least 1 h followed by centrifugation at 12,000 × g for 5 min. The pellet was then resuspended in 100–300 μl STM buffer and labeled as “cytosolic fraction.” The pellet obtained above was again resuspended in 100–200 μl STM buffer and centrifuged at 11,000 × g for 10 min. Once centrifuged, the mitochondrial pellet was resuspended in 50–100 μl SOL buffer by sonication on ice at high setting for 5–10 s with 30 s pauses and labeled as “mitochondrial fraction.” Validation of the purity of the subcellular fractions was determined by examining “house-keeper” protein markers GAPDH (cytoplasmic), Cox IV (mitochondrial), and H3 histone (nuclear) by standard WB analysis.
Lentiviral vector–mediated p53 shRNA infection
The mouse p53 short hairpin RNA (shRNA) plasmid (target/sense sequences 5′-CG(T/C)GCCATGGCCATCTACAAG-3′; Mukhopadhyay et al., 2009) was constructed by Genechem Biological Company and inserted along with the green fluorescent reporter protein gene. The lentiviral vectors containing either p53 shRNA plasmid or noneffective scrambled plasmid were provided by Genechem Biological Company. The lentivirus infection experiments were conducted as below. First, a preliminary experiment was conducted to obtain the optimal value of the multiplicity of infection (MOI). Myoblasts were cultured overnight to reach 40–60% confluence and were transfected by lentivirus at MOI of 20, 40, 60, 80, and 100. After infection (48 h), the transfection efficiency was evaluated using a fluorescence microscope (Nikon, Japan). In the formal experiment, the appropriate amounts of lentivirus were added according to the appropriate MOI values (targeting shRNA vector: MOI = 80 and nontargeting shRNA vector: MOI = 60). To increase the transfection efficiency, 20% FBS and 2 mg/ml puromycin were added to select positively infected cells. This selection process lasted for 1 wk.
Plasmids and transfection
The expression vectors (pcDNA3) encoding wild-type Akt tagged with HA, the constitutively active Akt tagged with HA, wild-type p53, and mutant p53 S389A were all constructed by the Genechem Biological Company. These plasmids were transfected using the LipofectAmine method (Life Technologies) according to the manufacturer’s protocol. In brief, C2C12 cells were plated in 24-well plates overnight and incubated in a mixture of each individual plasmid (2 μg per well) and LipofectAmine (1:5 wt/wt) in growth medium for 6 h. After incubation for 18 h, growth medium was discarded and fresh medium was added, followed by the stretching experiments.
Statistics
All experiments were performed at least three times. Independent experiments were performed in duplicate. The results are presented as mean ± SD. Data were analyzed with Student’s t test for two-group comparison and analysis of variance (ANOVA) for a more-than-two-group comparison. P < 0.05 was defined as statistically significant.
Supplementary Material
Acknowledgments
We gratefully acknowledge the technical support from Central Laboratory members of Affiliated Hospital of Qingdao University. This study was supported by the National Natural Science Foundation of China (NSFC, No.11702154) and the China Postdoctoral Science Foundation (CPSF, No. 2018M642621).
Abbreviations used:
- CA-AKT
constitutively active AKT
- FACS
fluorescence activated cell sorting
- HMS
high magnitude stretch
- LMS
low magnitude stretch
- NAC
N-acetylcysteine
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT-AKT
wild-type AKT
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-12-0770) on March 13, 2019.
REFERENCES
- Abraham AG, O’Neill E. (2014). PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc Trans , 798–803. [DOI] [PubMed] [Google Scholar]
- Akimoto T, Ushida T, Miyaki S, Tateishi T, Fukubayashi T. (2001). Mechanical stretch is a down-regulatory signal for differentiation of C2C12. Mater Sci Eng C , 75–78. [Google Scholar]
- Allen DG, Gervasio OL, Yeung EW, Whitehead NP. (2010). Calcium and the damage pathways in muscular dystrophy. Can J Physiol Pharmacol , 83–91. [DOI] [PubMed] [Google Scholar]
- Andersen JI, Pennisi CP, Fink T, Zachar V. (2018). Focal adhesion kinase activation is necessary for stretch-induced alignment and enhanced differentiation of myogenic precursor cells. Tissue Eng Part A , 631–640. [DOI] [PubMed] [Google Scholar]
- Appella E, Anderson CW. (2001). Post-translational modifications and activation of p53 by genotoxic stresses. Eur J Biochem , 2764–2772. [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Close GL, Drust B, Morton JP. (2014). The emerging role of p53 in exercise metabolism. Sports Med , 303–309. [DOI] [PubMed] [Google Scholar]
- Bilgic F, Gelgor IE, Celebi AA. (2015). Malocclusion prevalence and orthodontic treatment need in central Anatolian adolescents compared to European and other nations’ adolescents. Dental Press J Orthod , 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrogiovanni C, Waterschoot B, De Backer O, Dumont P. (2018). Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis. Cell Death Differ , 190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerone MA, Marchetti A, Bossi G, Blandino G, Sacchi A, Soddu S. (2000). p53 is involved in the differentiation but not in the differentiation-associated apoptosis of myoblasts. Cell Death Differ , 506–508. [DOI] [PubMed] [Google Scholar]
- Chang YJ, Chen YJ, Huang CW, Fan SC, Huang BM, Chang WT, Tsai YS, Su FC, Wu CC. (2016). Cyclic stretch facilitates myogenesis in C2C12 myoblasts and rescues thiazolidinedione-inhibited myotube formation. Front Bioeng Biotechnol , 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciavarella D, Laurenziello M, Guida L, Montaruli G, Gallo C, Tepedino M, Lo Muzio L. (2017). Dentoskeletal modifications in Class II deep bite malocclusion treatment with anterior bite plane functional appliance. J Clin Exp Dent , e1029–e1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Lu P, Song G, Liu Q, Zhu D, Liu X. (2016). Involvement of PI3K/Akt, ERK and p38 signaling pathways in emodin-mediated extrinsic and intrinsic human hepatoblastoma cell apoptosis. Food Chem Toxicol , 26–37. [DOI] [PubMed] [Google Scholar]
- Dimauro I, Pearson T, Caporossi D, Jackson MJ. (2012). A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes , 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Chen X, Li W, Fu Z, Shi J. (2017). Genistein protects genioglossus myoblast against hypoxia-induced injury through PI3K-Akt and ERK MAPK pathways. Sci Rep , 5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziegielewska B, Brautigan DL, Larner JM, Dziegielewski J. (2014). T-type Ca2+ channel inhibition induces p53-dependent cell growth arrest and apoptosis through activation of p38-MAPK in colon cancer cells. Mol Cancer Res , 348–358. [DOI] [PubMed] [Google Scholar]
- Ebata T, Hirata H, Kawauchi K. (2016). Functions of the tumor suppressors p53 and Rb in actin cytoskeleton remodeling. Biomed Res Int , 9231057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk DJ, Deruisseau KC, Van Gammeren DL, Deering MA, Kavazis AN, Powers SK. (2006). Mechanical ventilation promotes redox status alterations in the diaphragm. J Appl Physiol (1985) , 1017–1024. [DOI] [PubMed] [Google Scholar]
- Fayzullina S, Martin LJ. (2016). DNA damage response and DNA repair in skeletal myocytes from a mouse model of spinal muscular atrophy. J Neuropathol Exp Neurol , 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry A, Parlakian A, Joanne P, Fraysse B, Mgrditchian T, Roy P, Furling D, Butler-Browne G, Agbulut O. (2015). Mechanical overloading increases maximal force and reduces fragility in hind limb skeletal muscle from Mdx mouse. Am J Pathol , 2012–2024. [DOI] [PubMed] [Google Scholar]
- Flores-López LA, Díaz-Flores M, García-Macedo R, Ávalos-Rodríguez A, Vergara-Onofre M, Cruz M, Contreras-Ramos A, Konigsberg M, Ortega-Camarillo C. (2013). High glucose induces mitochondrial p53 phosphorylation by p38 MAPK in pancreatic RINm5F cells. Mol Biol Rep , 4947–4958. [DOI] [PubMed] [Google Scholar]
- Fu S, Yin L, Lin X, Lu J, Wang X. (2018). Effects of cyclic mechanical stretch on the proliferation of L6 myoblasts and its mechanisms: PI3K/Akt and MAPK signal pathways regulated by IGF-1 receptor. Int J Mol Sci , 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldspink DF, Cox VM, Smith SK, Eaves LA, Osbaldeston NJ, Lee DM, Mantle D. (1995). Muscle growth in response to mechanical stimuli. Am J Physiol (2 Pt 1), E288–E297. [DOI] [PubMed] [Google Scholar]
- Gomez-Cabrera MC, Salvador-Pascual A, Cabo H, Ferrando B, Vina J. (2015). Redox modulation of mitochondriogenesis in exercise. Does antioxidant supplementation blunt the benefits of exercise training? Free Radic Biol Med , 37–46. [DOI] [PubMed] [Google Scholar]
- Gratton JP, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC. (2001). Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. J Biol Chem , 30359–30365. [DOI] [PubMed] [Google Scholar]
- Horikawa-Miura M, Matsuda N, Yoshida M, Okumura Y, Mori T, Watanabe M. (2007). The greater lethality of UVB radiation to cultured human cells is associated with the specific activation of a DNA damage-independent signaling pathway. Radiat Res , 655–662. [DOI] [PubMed] [Google Scholar]
- Hu W, Wang F, Tang J, Liu X, Yuan Z, Nie C, Wei Y. (2012). Proapoptotic protein Smac mediates apoptosis in cisplatin-resistant ovarian cancer cells when treated with the anti-tumor agent AT101. J Biol Chem , 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Ma WY, Maxiner A, Sun Y, Dong Z. (1999). p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. J Biol Chem , 12229–12235. [DOI] [PubMed] [Google Scholar]
- Ji G, Liu D, Liu J, Gao H, Yuan X, Shen G. (2010). p38 mitogen-activated protein kinase up-regulates NF-κB transcriptional activation through RelA phosphorylation during stretch-induced myogenesis. Biochem Biophys Res Commun , 547–551. [DOI] [PubMed] [Google Scholar]
- Kang JS, Choi IW, Han MH, Kim GY, Hong SH, Park C, Hwang HJ, Kim CM, Kim BW, Choi YH. (2015). The cytoprotective effects of 7,8-dihydroxyflavone against oxidative stress are mediated by the upregulation of Nrf2-dependent HO-1 expression through the activation of the PI3K/Akt and ERK pathways in C2C12 myoblasts. Int J Mol Med , 501–510. [DOI] [PubMed] [Google Scholar]
- Keller D, Zeng X, Li X, Kapoor M, Iordanov MS, Taya Y, Lozano G, Magun B, Lu H. (1999). The p38MAPK inhibitor SB203580 alleviates ultraviolet-induced phosphorylation at serine 389 but not serine 15 and activation of p53. Biochem Biophys Res Commun , 464–471. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Oh JE, Kim SW, Chun YJ, Kim MY. (2008). Ceramide induces p38 MAPK-dependent apoptosis and Bax translocation via inhibition of Akt in HL-60 cells. Cancer Lett , 88–95. [DOI] [PubMed] [Google Scholar]
- Kosmidou I, Xagorari A, Roussos C, Papapetropoulos A. (2001). Reactive oxygen species stimulate VEGF production from C(2)C(12) skeletal myotubes through a PI3K/Akt pathway. Am J Physiol Lung Cell Mol Physiol , L585–L592. [DOI] [PubMed] [Google Scholar]
- Kruger M, Linke WA. (2009). Titin-based mechanical signalling in normal and failing myocardium. J Mol Cell Cardiol , 490–498. [DOI] [PubMed] [Google Scholar]
- Kuhn H, Nieuwenhuijsen H, Karthe B, Wirtz H. (2017). Stretch-induced apoptosis in rat alveolar epithelial cells is mediated by the intrinsic mitochondrial pathway. Exp Lung Res , 49–56. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Abe S, Sakiyama K, Takeda T, Ide Y, Ishigami K. (2007). Effects of stretching stimulation with different rates on the expression of MyHC mRNA in mouse cultured myoblasts. Biomed Res , 25–31. [DOI] [PubMed] [Google Scholar]
- Latella L, Lukas J, Simone C, Puri PL, Bartek J. (2004). Differentiation-induced radioresistance in muscle cells. Mol Cell Biol , 6350–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Jang MH, Kim EK, Han SW, Cho SY, Kim CJ. (2005). Nitric oxide induces apoptosis in mouse C2C12 myoblast cells. J Pharmacol Sci , 369–376. [DOI] [PubMed] [Google Scholar]
- Li L, Guo L, Tao Y, Zhou S, Wang Z, Luo W, Hu D, Li Z, Xiao L, Tang M, et al (2007). Latent membrane protein 1 of Epstein-Barr virus regulates p53 phosphorylation through MAP kinases. Cancer Lett , 219–231. [DOI] [PubMed] [Google Scholar]
- Liu B, Chen Y, St Clair DK. (2008). ROS and p53: a versatile partnership. Free Radic Biol Med , 1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerakker S, Stekelenburg A, Strijkers GJ, Rijpkema JJ, Baaijens FP, Bader DL, Nicolay K, Oomens CW. (2010). Temporal effects of mechanical loading on deformation-induced damage in skeletal muscle tissue. Ann Biomed Eng , 2577–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Fu S, Lu L, Wang X. (2017). Role of androgen receptor on cyclic mechanical stretch-regulated proliferation of C2C12 myoblasts and its upstream signals: IGF-1-mediated PI3K/Akt and MAPKs pathways. Mol Cell Endocrinol , 83–93. [DOI] [PubMed] [Google Scholar]
- Martins AS, Shkryl VM, Nowycky MC, Shirokova N. (2008). Reactive oxygen species contribute to Ca2+ signals produced by osmotic stress in mouse skeletal muscle fibres. J Physiol , 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay UK, Eves R, Jia L, Mooney P, Mak AS. (2009). p53 suppresses Src-induced podosome and rosette formation and cellular invasiveness through the upregulation of caldesmon. Mol Cell Biol , 3088–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naughton M., Miller J, Slater GJ. (2017). Impact-induced muscle damage and contact-sport: aetiology, effects on neuromuscular function and recovery, and the modulating effects of adaptation and recovery strategies. Int J Sports Physiol Perform , 1–24. [DOI] [PubMed] [Google Scholar]
- O’Brate A, Giannakakou P. (2003). The importance of p53 location: nuclear or cytoplasmic zip code? Drug Resist Updat , 313–322. [DOI] [PubMed] [Google Scholar]
- Orzechowski A, Lokociejewska M, Pawlikowska P, Kruszewski M. (2005). Preincubation with sodium ascorbate potentiates insulin-dependent PKB/Akt and c-Jun phosphorylation in L6 rat myoblasts challenged with reactive oxygen/nitrogen species. Life Sci , 496–511. [DOI] [PubMed] [Google Scholar]
- Panickar KS, Jayakumar AR, Rao KV, Norenberg MD. (2009). Ammonia-induced activation of p53 in cultured astrocytes: role in cell swelling and glutamate uptake. Neurochem Int , 98–105. [DOI] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Dunys J, Giaime E, Guillot-Sestier MV, St George-Hyslop P, Checler F, Alves da Costa C. (2009). p53-dependent control of cell death by nicastrin: lack of requirement for presenilin-dependent γ-secretase complex. J Neurochem , 225–237. [DOI] [PubMed] [Google Scholar]
- Peng XQ, Damarla M, Skirball J, Nonas S, Wang XY, Han EJ, Hasan EJ, Cao X, Boueiz A, Damico R, et al (2010). Protective role of PI3-kinase/Akt/eNOS signaling in mechanical stress through inhibition of p38 mitogen-activated protein kinase in mouse lung. Acta Pharmacol Sin , 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohnke Y, Schneider-Merck T, Fahnenstich J, Kempf R, Christian M, Milde-Langosch K, Brosens JJ, Gellersen B. (2004). Wild-type p53 protein is up-regulated upon cyclic adenosine monophosphate-induced differentiation of human endometrial stromal cells. J Clin Endocrinol Metab , 5233–5244. [DOI] [PubMed] [Google Scholar]
- Porrello A, Cerone MA, Coen S, Gurtner A, Fontemaggi G, Cimino L, Piaggio G, Sacchi A, Soddu S. (2000). p53 regulates myogenesis by triggering the differentiation activity of pRb. J Cell Biol , 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser BL, Khairallah RJ, Ziman AP, Ward CW, Lederer WJ. (2013). X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca2+]i. J Mol Cell Cardiol , 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M, Mofarrahi M, Kristof AS, Nkengfac B, Harel S, Hussain SN. (2014). Reactive oxygen species regulation of autophagy in skeletal muscles. Antioxid Redox Signal , 443–459. [DOI] [PubMed] [Google Scholar]
- Reid MB, Haack KE, Franchek KM, Valberg PA, Kobzik L, West MS. (1992). Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J Appl Physiol (1985) , 1797–1804. [DOI] [PubMed] [Google Scholar]
- Safdar A, Khrapko K, Flynn JM, Saleem A, De Lisio M, Johnston AP, Kratysberg Y, Samjoo IA, Kitaoka Y, Ogborn DI, et al (2016). Exercise-induced mitochondrial p53 repairs mtDNA mutations in mutator mice. Skelet Muscle , 7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Saito A, Hayashi T, Okuno S, Nishi T, Chan PH. (2005). Modulation of p53 degradation via MDM2-mediated ubiquitylation and the ubiquitin-proteasome system during reperfusion after stroke: role of oxidative stress. J Cereb Blood Flow Metab , 267–280. [DOI] [PubMed] [Google Scholar]
- Saleem A, Carter HN, Iqbal S, Hood DA. (2011). Role of p53 within the regulatory network controlling muscle mitochondrial biogenesis. Exerc Sport Sci Rev , 199–205. [DOI] [PubMed] [Google Scholar]
- Saleem A, Hood DA. (2013). Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-Tfam-mitochondrial DNA complex in skeletal muscle. J Physiol , 3625–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu PM, Alway SE. (2005). Subcellular responses of p53 and Id2 in fast and slow skeletal muscle in response to stretch-induced overload. J Appl Physiol (1985) , 1897–1904. [DOI] [PubMed] [Google Scholar]
- Soddu S, Blandino G, Scardigli R, Coen S, Marchetti A, Rizzo MG, Bossi G, Cimino L, Crescenzi M, Sacchi A. (1996). Interference with p53 protein inhibits hematopoietic and muscle differentiation. J Cell Biol , 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Kwak HB, Lawler JM. (2006). Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle. Antioxid Redox Signal , 517–528. [DOI] [PubMed] [Google Scholar]
- Speidel D. (2010). Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol , 14–24. [DOI] [PubMed] [Google Scholar]
- Tachtsis B, Smiles WJ, Lane SC, Hawley JA, Camera DM. (2016). Acute endurance exercise induced nuclear p53 abundance in human skeletal muscle. Front Physiol , 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Kuang W, Jin Z, Jin F, Xu L, Yu Q, Kong L, Zeng G, Yuan X, Duan Y. (2009). Inhibition of NFkappaB by activated c-Jun NH2 terminal kinase 1 acts as a switch for C2C12 cell death under excessive stretch. Apoptosis , 764–770. [DOI] [PubMed] [Google Scholar]
- Tidball JG. (2005). Mechanical signal transduction in skeletal muscle growth and adaptation. J Appl Physiol (1985) , 1900–1908. [DOI] [PubMed] [Google Scholar]
- Wan L, Zhang L, Fan K, Wang J. (2017). Aloin promotes A549 cell apoptosis via the reactive oxygen speciesmitogen activated protein kinase signaling pathway and p53 phosphorylation. Mol Med Rep , 5759–5768. [DOI] [PubMed] [Google Scholar]
- Wang J, Guo W, Zhou H, Luo N, Nie C, Zhao X, Yuan Z, Liu X, Wei Y. (2015). Mitochondrial p53 phosphorylation induces Bak-mediated and caspase-independent cell death. Oncotarget , 17192–17205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Huang D, Jiang Z, Luo Y, Norris C, Zhang M, Tian X, Tang Y. (2017). Akt3 is responsible for the survival and proliferation of embryonic stem cells. Biol Open , 850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward CW, Prosser BL, Lederer WJ. (2014). Mechanical stretch-induced activation of ROS/RNS signaling in striated muscle. Antioxid Redox Signal , 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead NP, Yeung EW, Allen DG. (2006). Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol , 657–662. [DOI] [PubMed] [Google Scholar]
- Xu Z, Liu Y, Yang D, Yuan F, Ding J, Chen H, Tian H. (2017). Sesamin protects SH-SY5Y cells against mechanical stretch injury and promoting cell survival. BMC Neurosci , 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Fraser M, Moll UM, Basak A, Tsang BK. (2006). Akt-mediated cisplatin resistance in ovarian cancer: modulation of p53 action on caspase-dependent mitochondrial death pathway. Cancer Res , 3126–3136. [DOI] [PubMed] [Google Scholar]
- Yen YP, Tsai KS, Chen YW, Huang CF, Yang RS, Liu SH. (2012). Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Arch Toxicol , 923–933. [DOI] [PubMed] [Google Scholar]
- Yu T, Dohl J, Elenberg F, Chen Y, Deuster P. (2019). Curcumin induces concentration-dependent alterations in mitochondrial function through ROS in C2C12 mouse myoblasts. J Cell Physiol , 6371–6381. [DOI] [PubMed] [Google Scholar]
- Zhan M, Jin B, Chen SE, Reecy JM, Li YP. (2007). TACE release of TNF-α mediates mechanotransduction-induced activation of p38 MAPK and myogenesis. J Cell Sci (Pt 4), 692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JS, Kraus WE, Truskey GA. (2004). Stretch-induced nitric oxide modulates mechanical properties of skeletal muscle cells. Am J Physiol Cell Physiol , C292–C299. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W. (2016). ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev , 4350965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Sheng Z, Li Y, Lu H. (2014). Salidroside inhibits oxygen glucose deprivation (OGD)/re-oxygenation-induced H9c2 cell necrosis through activating of Akt-Nrf2 signaling. Biochem Biophys Res Commun , 79–85. [DOI] [PubMed] [Google Scholar]
- Zuo L, Christofi FL, Wright VP, Liu CY, Merola AJ, Berliner LJ, Clanton TL. (2000). Intra- and extracellular measurement of reactive oxygen species produced during heat stress in diaphragm muscle. Am J Physiol Cell Physiol , C1058–C1066. [DOI] [PubMed] [Google Scholar]
- Zuo L, Clanton TL. (2005). Reactive oxygen species formation in the transition to hypoxia in skeletal muscle. Am J Physiol Cell Physiol , C207–C216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.