

Abstract
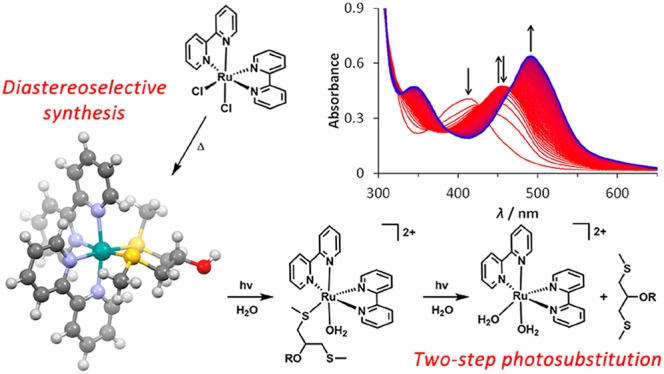
Thioethers are good ligands for photoactivatable ruthenium(II) polypyridyl complexes, as they form thermally stable complexes that are prone to ligand photosubstitution. Here, we introduce a novel symmetric chelating bis(thioether) ligand scaffold, based on 1,3-bis(methylthio)-2-propanol (4) and report the synthesis and stereochemical characterization of the series of novel ruthenium(II) polypyridyl complexes [Ru(bpy)2(L)](PF6)2 ([1]–[3](PF6)2), where L is ligand 4, its methyl ether, 1,3-bis(methylthio)-2-methoxypropane (5), or its carboxymethyl ether, 1,3-bis(methylthio)-2-(carboxymethoxy)propane (6). Coordination of ligands 4–6 to the bis(bipyridine)ruthenium center gives rise to 16 possible isomers, consisting of 8 possible Λ diastereoisomers and their Δ enantiomers. We found that the synthesis of [1]–[3](PF6)2 is diastereoselective, yielding a racemic mixture of the Λ-(S)-eq-(S)-ax-OHeq-[Ru]2+ and Δ-(R)-ax-(R)-eq-OHeq-[Ru]2+ isomers. Upon irradiation with blue light in water, [1]–[3](PF6)2 selectively substitute their bis(thioether) ligands for water molecules in a two-step photoreaction, ultimately producing [Ru(bpy)2(H2O)2]2+ as the photoproduct. The relatively stable photochemical intermediate was identified as cis-[Ru(bpy)2(κ1-L)(H2O)]2+ by mass spectrometry. Global fitting of the time evolution of the UV–vis absorption spectra of [1]–[3](PF6)2 was employed to derive the photosubstitution quantum yields (Φ443) for each of the two photochemical reaction steps separately, revealing very high quantum yields of 0.16–0.25 for the first step and lower values (0.0055–0.0093) for the second step of the photoreaction. The selective and efficient photochemical reaction makes the photocleavable bis(thioether) ligand scaffold reported here a promising candidate for use in e.g. ruthenium-based photo-activated chemotherapy.
Short abstract
Thioethers are excellent photocleavable ligands for ruthenium(II) polypyridyl complexes but may lead to the formation of several stereoisomers when they are present in bidentate ligands. Here, a chelating bis(thioether) ligand was found to coordinate to Ru(II) diastereoselectively, in spite of the four chiral centers of the resulting complex. Photosubstitution of this bis(thioether) ligand in water occurs via a selective, two-step process that involves a relatively stable mono(aqua) intermediate.
Introduction
The use of light as a trigger for the activation of metal-based anticancer agents has been actively researched over the last decades.1−5 In combination with ruthenium(II) complexes, light can be used either to drive the formation of reactive oxygen species through the sensitization of oxygen in photodynamic therapy (PDT)6−9 or to uncage photoactivatable complexes through ligand photosubstitution in photoactivated chemotherapy (PACT).10−18 This photolability can be enhanced through both steric and electronic effects.19 In our group, thioether ligands have been considered with more attention for the photocaging of bioactive ruthenium polypyridyl complexes.13,20−22 Their softness makes thioethers excellent ligands for ruthenium(II) ions, and their complexes often show good thermal stability. Under blue light irradiation, several groups have shown that thioether ligands can be selectively substituted by solvent molecules, both for monodentate ligands, e.g. 2-(methylthio)ethanol (Hmte),20,23,24 and for bidentate chelating thioether ligands.13,25−30 Examples of the latter include combinations of thioether sulfur donors with nitrogen donor atoms, e.g. 2-(methylthio)methylpyridine (mtmp),13 as well as symmetric bis(arylthioether) ligands, e.g. 1,3-bis(phenylthio)propane (bptp).26−28 The photosubstitution of some bis(thioether) ligands was previously reported to be 5–10 times more efficient than that of comparable bis(amine) ligands.26 However, these reports do not always appreciate the two-step nature of the photosubstitution of such bidentate ligands, reporting the photosubstitution quantum yields as a single number. Furthermore, the bis(thioether) ligands reported previously often have limited options for further functionalization, such as the attachment of anchoring groups, which can be very useful for the development of photoactivatable ruthenium complexes bound to inorganic surfaces or nanomaterials.
In this work, we report the coordination of the symmetric bidentate bis(thioether) ligand 1,3-bis(methylthio)-2-propanol (4) to ruthenium. We introduced an alcohol functionality in this ligand to allow for future functionalization; this substituent was added in a symmetrical position to prevent the formation of regioisomers upon metal coordination of the ligand. To exemplify these functionalization options, we also prepared the methyl- and carboxymethyl-substituted derivatives of 4, i.e. 1,3-bis(methylthio)-2-methoxypropane (5) and 1,3-bis(methylthio)-2-(carboxymethoxy)propane (6). We hence synthesized three new ruthenium polypyridyl complexes of the general formula [Ru(bpy)2(L)](PF6)2 ([1]–[3](PF6)2), where bpy = 2,2′-bipyridine and L = 4–6 (Scheme 1). We investigated the stereochemistry of the complexes using density functional theory (DFT) and NOESY NMR studies and examined both the efficiency and selectivity of the photochemistry of complexes [1]–[3](PF6)2 in aqueous solution. Our results are compared to reports by the groups of Turro and Sauvage on the related bis(thioether) chelate complexes [Ru(bpy)2(bete)](PF6)2 ([7](PF6)2, bete = 3,6-dithiaoctane), [Ru(bpy)2(bpte)](PF6)2 ([8](PF6)2, bpte = 1,2-bis(phenylthio)ethane), [Ru(phen)2(bpte)](PF6)2 ([9](PF6)2), and [Ru(phen)2(bptp)](PF6)2 ([10](PF6)2), all shown in Scheme 1.26,27 In particular, we evaluate the effects of the chelating ring size (five- vs six-membered ring), of the size and aromaticity of the thioether substituent (methyl, ethyl, or phenyl group), and of the addition of a substituent to the chelating ring (i.e., the hydroxyl or ether group in complexes [1]–[3](PF6)2) on the stereo- and photochemistry of this type of complex.
Scheme 1. Chemical Structures of New Ruthenium Polypyridyl Complexes [1]–[3](PF6)2 (Left) and Complexes [7]–[10](PF6)2, Previously Reported by the Groups of Turro (Center) and Sauvage (Right)26,27.

Experimental Section
General Considerations
Dry tetrahydrofuran (THF) was collected from a Pure-Solv MD5 dry solvent dispenser (Demaco). 1,3-Bis(methylthio)-2-propanol (4) was obtained from Alfa Aesar. All other reagents and solvents, including cis-[Ru(bpy)2Cl2], were purchased from Sigma-Aldrich and used as received. All syntheses were conducted under an oxygen-free atmosphere using standard Schlenk line techniques. Syntheses of all ruthenium complexes were performed in the absence of light. Flash column chromatography was performed on silica gel (Screening Devices BV) with a particle size of 40–64 μm and a pore size of 60 Å. TLC analysis was conducted on TLC aluminum foils with silica gel matrix (Supelco, silica gel 60, art. no. 56524) with detection by UV absorption (254 nm) or basic KMnO4 spray. Size exclusion column chromatography was performed in acetone using Sephadex LH20, loaded into a chromatography column (i.d. = 3–4 cm, l ≈ 60 cm).
All NMR spectra were recorded on a Bruker AV-300, AV-400, or AV-500 spectrometer. Chemical shifts (δ) are indicated in ppm relative to TMS or the solvent peak. Atom numbering for NMR attribution is shown in the Supporting Information (Schemes S1 and S2). Mass spectra were recorded by using a MSQ Plus Spectrometer fitted with a Dionex automatic sample injection system. High-resolution mass spectra were recorded by direct injection (2 μL of 1 μM solution in MeOH or acetonitrile and 0.1% formic acid) in a mass spectrometer (Thermo Finnigan LTQ Orbitrap) equipped with an electrospray (250 °C) with resolution R = 60000 at m/z 400 (mass range m/z 150–2000) and dioctyl phthalate (m/z 391.28428) as a lock mass. The high-resolution mass spectrometer was calibrated prior to measurements with a calibration mixture (Thermo Finnigan).
Ligand Synthesis
1,3-Bis(methylthio)-2-methoxypropane (5)
Dry and deoxygenated THF (20 mL) was placed under a dinitrogen atmosphere in a round-bottom flask containing 1,3-bis(methylthio)-2-propanol (0.56 g, 0.50 mL, 3.70 mmol), followed by the addition of solid NaH (296 mg, 7.40 mmol, 60% dispersion in mineral oil). The resulting suspension was stirred at room temperature for 20 min to allow complete deprotonation of the alcohol. Afterward the reaction mixture was cooled to 0 °C, and iodomethane (0.63 g, 0.28 mL, 4.44 mmol) was added dropwise. The resulting suspension was stirred at room temperature for 24 h and then quenched with saturated aqueous NH4Cl (5 mL) to yield a light yellow solution. The solvent was removed in vacuo, and the residue was dissolved in H2O (40 mL) and extracted with DCM (3 × 40 mL). The organic layers were combined, washed with brine, dried over MgSO4, and concentrated by rotary evaporation. Separation of the product (Rf = 0.7) and unreacted starting compound (Rf = 0.9) was performed by column chromatography (SiO2, petroleum ether 40/60/EtOAc (4/1)), ultimately resulting in 404 mg of compound 5 as a colorless oil (2.43 mmol, 66%). 1H NMR (400 MHz, δ in CDCl3): 3.50 (p, J = 5.7 Hz, 1H, H3), 3.42 (s, 3H, H6), 2.74 (ddd, J = 21.4, 13.6, 5.7 Hz, 4H, H2 + H4), 2.16 (s, 6H, H1 + H5). 13C NMR (101 MHz, δ in CDCl3): 80.6 (C3), 57.5 (C6), 37.2 (C2 + C4), 16.8 (C1 + C5). ESI-MS in CH3OH m/z exptl (calcd): 205.0 (205.0, [M + K]+). 1H NMR data match the literature data.31
1,3-Bis(methylthio)-2-(carboxymethoxy)propane (6)
Dry and deoxygenated THF (10 mL) was placed under a nitrogen atmosphere in a round-bottom flask containing 1,3-bis(methylthio)-2-propanol (0.25 g, 0.22 mL, 1.64 mmol), followed by the addition of solid NaH (328 mg, 8.20 mmol, 60% dispersion in mineral oil) and potassium iodide (22 mg, 0.133 mmol). The resulting suspension was stirred at room temperature for 20 min to allow complete deprotonation of the alcohol. Afterward the reaction mixture was cooled to 0 °C, and a solution of bromoacetic acid (342 mg, 2.46 mmol) in dry THF (2 mL) was added dropwise. The resulting suspension was heated to reflux, stirred for 22 h, and subsequently cooled to 0 °C and quenched with water (10 mL) to yield a light yellow solution. The solvent was removed in vacuo, and the residue was dissolved in H2O (30 mL) and washed with EtOAc (3 × 30 mL). The aqueous layer was acidified to pH ∼2 with 1 M HCl, followed by extraction with EtOAc (3 × 50 mL). The organic layers were combined, washed with brine, dried over MgSO4, and concentrated by rotary evaporation. Acetic acid impurities were removed from the crude product by coevaporation with toluene (3 × 50 mL), to obtain compound 6 as a colorless oil (327 mg, 1.55 mmol, 95%). 1H NMR (300 MHz, δ in CDCl3): 9.23 (s, 1H, −COOH), 4.28 (s, 2H, H6), 3.64 (p, J = 5.9 Hz, 1H, H3), 2.76 (ddd, J = 19.3, 14.2, 5.5 Hz, 4H, H2 + H4), 2.15 (s, 6H, H1 + H5). 13C NMR (75 MHz, δ in CDCl3): 173.4 (C7), 79.8 (C3), 67.5 (C6), 37.9 (C2 + C4), 16.6 (C1 + C5). HR-MS in CH3OH m/z exptl (calcd): 233.0285 (233.0384, [M + Na]+).
Ruthenium Complex Synthesis
[Ru(bpy)2(4)](PF6)2 ([1](PF6)2)
A mixture of 1,3-bis(methylthio)-2-propanol (4, 78 mg, 0.51 mmol) and cis-Ru(bpy)2Cl2 (50 mg, 0.103 mmol) was placed in a 25 mL round-bottom flask, under an N2 atmosphere. A deoxygenated mixture of EtOH and H2O (1/1 v/v, 10 mL) was added, and the reaction mixture was refluxed in the dark for 1.5 h. The resulting orange solution was cooled to room temperature, and EtOH was removed in vacuo. Water (10 mL) was added to the residue, before washing with Et2O (3 × 15 mL). A saturated aqueous KPF6 solution (∼5 mL) was then added to the aqueous layer, and the resulting orange suspension was extracted with DCM (6 × 20 mL). The combined organic layers were washed once with half saturated aqueous KPF6 and then dried by rotary evaporation. Any excess KPF6 was removed by size exclusion chromatography in acetone, and after drying overnight under high vacuum, complex [1](PF6)2 was obtained as an orange powder (50 mg, 0.058 mmol, 57%). TLC: Rf = 0.2 (SiO2, acetone/H2O/saturated aqueous KPF6 (16/4/1)). 1H NMR (500 MHz, δ in acetone-d6): 9.87 (d, J = 5.1 Hz, 1H, HA6), 9.63 (d, J = 5.6 Hz, 1H, HD6), 8.88 (t, J = 8.1 Hz, 2H, HD3 + HA3), 8.74 (dd, J = 8.2, 3.5 Hz, 2H, HB3 + HC3), 8.47 (tdd, J = 7.9, 2.9, 1.4 Hz, 2H, HD4 + HA4), 8.18 (td, J = 7.9, 1.5 Hz, 2H, HB4 + HC4), 8.08 (dddd, J = 11.2, 7.4, 5.7, 1.4 Hz, 2H, HD5 + HA5), 7.84 (d, J = 5.8 Hz, 1H, HB6), 7.79 (d, J = 5.3 Hz, 1H, HC6), 7.52 (tdd, J = 7.2, 5.6, 1.3 Hz, 2H, HB5 + HC5), 5.33 (d, J = 4.4 Hz, 1H, – OH), 4.87 (br s, 1H, H3), 3.41 (dd, J = 13.5, 3.1 Hz, 1H, H4,eq), 3.30 (dd, J = 13.1, 6.3 Hz, 1H, H2,ax), 3.01 (dd, J = 13.1, 2.1 Hz, 1H, H2,eq), 2.99–2.93 (m, 1H, H4,ax), 1.59 (s, 3H, H5), 1.36 (s, 3H, H1); 13C NMR (101 MHz, δ in acetone-d6): 158.8, 158.7, 157.6, 157.5 (all Cq), 154.6 (CD6), 154.4 (CA6), 152.2 (CC6), 152.1 (CB6), 140.0 (CA4 + CB4 + CC4 + CD4), 129.7, 129.1 (CA5 + CD5), 128.9, 128.8 (CB5 + CC5), 126.0, 125.9 (CA3 + CD3), 125.3, 125.2 (CB3 + CC3), 67.0 (C3), 41.2 (C2), 39.5 (C4), 18.0 (C5), 16.1 (C1). HR-MS in CH3CN m/z exptl (calcd): 303.5503 (303.5504, [M – 2PF6 + CH3CN]2+), 565.0662 (565.0669, [M – 2PF6 – H]+). UV–vis: λmax (ε in M–1 cm–1) in H2O: 413 nm (5.13 × 103). Anal. Calcd for C25H28F12N4OP2RuS2·H2O: C, 34.37; H, 3.46; N, 6.41. Found: C, 34.94; H, 3.61; N, 6.36.
[Ru(bpy)2(5)](PF6)2 ([2](PF6)2)
Complex [2](PF6)2 was synthesized using the method described for [1](PF6)2, using a mixture of 5 (85 mg, 0.516 mmol) and cis-Ru(bpy)2Cl2 (25 mg, 0.052 mmol) in a mixture of EtOH and H2O (1/1 v/v, 6 mL). The complex was obtained as a light orange powder in 69% yield (31 mg, 0.036 mmol). TLC: Rf = 0.2 (SiO2, acetone/H2O/saturated aqueous KPF6 (16/4/1)). 1H NMR (300 MHz, δ in acetone-d6): 9.81 (d, J = 5.1 Hz, 1H, HA6), 9.58 (d, J = 5.3 Hz, 1H, HD6), 8.88 (t, J = 7.5 Hz, 2H, HD3 + HA3), 8.74 (dd, J = 8.2, 4.0 Hz, 2H, HB3 + HC3), 8.46 (t, J = 7.9 Hz, 2H, HD4 + HA4), 8.23–8.04 (m, 4H, HB4 + HC4 + HD5 + HA5), 7.84 (d, J = 6.2 Hz, 1H, HB6), 7.79 (d, J = 5.8 Hz, 1H, HC6), 7.51 (tdd, J = 7.4, 5.6, 1.3 Hz, 2H, HB5 + HC5), 4.48 (br s, 1H, H3), 3.54 (s, 3H, H6), 3.53–3.44 (m, 2H, H4,eq + H2,ax), 3.16 (dd, J = 13.7, 5.6 Hz, 1H, H4,ax), 2.97 (dd, J = 13.2, 1.6 Hz, 1H, H2,eq), 1.63 (s, 3H, H5), 1.34 (s, 3H, H1). 13C NMR (75 MHz, δ in acetone-d6): 158.8, 158.6, 157.6, 157.5 (all Cq), 155.0 (CD6), 154.1 (CA6), 152.3 (CC6), 152.1 (CB6), 140.0, 140.0, 140.0, 140.0 (CA4 + CB4 + CC4 + CD4), 129.8, 129.0 (CA5 + CD5), 128.9, 128.8 (CB5 + CC5), 126.0, 125.9 (CA3 + CD3), 125.3, 125.2 (CB3 + CC3), 75.8 (C3), 57.2 (C6), 37.6 (C2), 36.9 (C4), 18.2 (C5), 15.8 (C1). HR-MS in CH3CN m/z exptl (calcd): 310.5584 (310.5583, [M – 2PF6 + CH3CN]2+). UV–vis: λmax (ε in M–1 cm–1) in H2O: 412 nm (4.04 × 103). Anal. Calcd for C26H30F12N4OP2RuS2·4H2O·0.5(CH3)2CO: C, 34.03; H 4.26; N, 5.77. Found: C, 34.00; H, 4.47; N, 5.98.
[Ru(bpy)2(6)](PF6)2 ([3](PF6)2)
Complex [3](PF6)2 was synthesized using the method described for [1](PF6)2, using a mixture of 6 (48 mg, 0.228 mmol) and cis-Ru(bpy)2Cl2 (52 mg, 0.107 mmol) in a mixture of EtOH and H2O (1/1 v/v, 10 mL). The complex was obtained as a light orange powder in 55% yield (54 mg, 0.059 mmol). TLC: Rf = 0.2 (SiO2, acetone/H2O/saturated aqueous KPF6 (16/4/1)). 1H NMR (400 MHz, δ in acetone-d6): 9.91 (d, J = 5.6 Hz, 1H, HA6), 9.55 (d, J = 5.2 Hz, 1H, HD6), 8.86 (dd, J = 14.6, 8.2 Hz, 2H, HD3 + HA3), 8.73 (t, J = 7.8 Hz, 2H, HB3 + HC3), 8.46 (qd, J = 8.0, 1.5 Hz, 2H, HD4 + HA4), 8.17 (tt, J = 7.9, 1.5 Hz, 2H, HB4 + HC4), 8.06 (ddd, J = 7.4, 5.7, 1.4 Hz, 2H, HD5 + HA5), 7.84 (dd, J = 5.7, 0.8 Hz, 1H, HB6), 7.78 (dd, J = 5.7, 0.8 Hz, 1H, HC6), 7.52 (dddd, J = 8.8, 7.2, 5.6, 1.3 Hz, 2H, HB5 + HC5), 4.77 (s, 1H, H3), 4.48 (d, J = 16.5 Hz, 1H, H6), 4.35 (d, J = 16.5 Hz, 1H, H6), 3.59 (dd, J = 13.2, 6.3 Hz, 1H, H2,ax), 3.52 (dd, J = 14.1, 2.7 Hz, 1H, H4,eq), 3.26 (dd, J = 14.0, 4.8 Hz, 1H, H4,ax), 2.98 (dd, J = 13.2, 1.6 Hz, 1H, H2,eq), 1.68 (s, 3H, H5), 1.35 (s, 3H, H1). 13C NMR (101 MHz, δ in acetone-d6): 171.3 (C7), 158.7, 158.6, 157.6, 157.5 (all Cq), 154.9 (CD6), 154.8 (CA6), 152.3 (CC6), 152.0 (CB6), 140.0, 140.0, 140.0, 139.9 (CA4 + CB4 + CC4 + CD4), 129.9, 129.2 (CA5 + CD5), 128.9, 128.8 (CB5 + CC5), 126.0, 125.8 (CA3 + CD3), 125.3, 125.2 (CB3 + CC3), 75.0 (C3), 66.7 (C6), 37.7 (C2), 36.9 (C4), 18.4 (C5), 15.9 (C1). HR-MS in CH3CN m/z exptl (calcd): 312.0410 (312.0396, [M – 2PF6]2+); UV–vis: λmax (ε in M–1 cm–1) in H2O: 412 nm (5.18 × 103). Anal. Calcd for C27H30F12N4O3P2RuS2·H2O: C, 34.81; H, 3.46; N, 6.01. Found: C, 34.91; H, 3.87; N, 5.89.
Density Functional Theory
Structure minimizations were performed using density functional theory (DFT) as implemented in the ADF software package from SCM (version 2017). The structures of the 16 possible Λ stereoisomers of [1](PF6)2, consisting of eight isomers with a chairlike metallacycle (i.e., R and S conformation for both sulfur atoms and the −OH substituent) and eight isomers with a boatlike metallacycle, were optimized in water using the conductor-like screening model (COSMO)32 to simulate the effect of solvation. The BLYP functional,33,34 combined with a TZP basis set (valence triple-ζ plus 1 polarization function) and a small frozen core for all atoms including ruthenium,35 was employed in all calculations. All boatlike structures were found to convert to chairlike structures during the structure optimization process and are thus not shown.
Photosubstitution Quantum Yields of [1]–[3](PF6)2 under Blue Light Irradiation
UV–vis experiments on the ruthenium complexes were performed on a Cary 50 Varian spectrometer equipped with a Cary Single Cell Peltier for temperature control (T = 298 K) and stirring. For the irradiation, a LED light source was used (λ = 443 nm, fwhm = 11 nm) the photon flux of which was determined by ferrioxalate actinometry (see Table S1 in the Supporting Information). Experiments were performed in 1.0 × 1.0 cm fluorescence cuvettes (QS-111, Hellma Analytics) containing 3.00 mL of solution. A stock solution of the desired complex was prepared using demineralized water, which was then diluted to the desired working concentration (Table S1) and placed in the cuvette. Irradiations were carried out under an N2 atmosphere after deoxygenation for 10 min by gentle bubbling of N2 through the sample, and the sample was kept under an inert atmosphere during the experiment by a gentle flow of N2 over the top of the cuvette. A UV–vis absorption spectrum was measured every 6 s during the experiment. Data were analyzed using Microsoft Excel 2010. The quantum yields of the photosubstitution reactions (Φ443) were calculated by fitting the time evolution of the UV–vis absorption spectra of the irradiated solution using the Glotaran software package (see the Supporting Information for a full description).36 Mass spectrometry was performed after the irradiation experiments to identify the photoproducts.
Photoirradiation Monitored by 1H NMR Spectroscopy
Deoxygenated D2O (0.6 mL) was placed in an NMR tube containing [1](PF6)2 (1 mg) under an N2 atmosphere, resulting in an orange solution (2 mM). The tube was irradiated at room temperature using a LOT 1000 W xenon arc lamp equipped with an IR short-pass filter and a 400 nm long-pass filter. The progress of the photoreaction was monitored by 1H NMR at several time points until the steady state was reached (at 60 min irradiation).
A reference sample of cis-[Ru(bpy)2(H2O)2](CF3SO3)2 (cis-[14](CF3SO3)2) in D2O was prepared by the addition of a drop of triflic acid to a suspension of [Ru(bpy)2(CO3)] in D2O in the absence of light. The latter was prepared following a literature procedure.37
Singlet Oxygen Generation and Phosphorescence Quantum Yield of [1]–[3](PF6)2
The singlet oxygen generation and phosphorescence quantum yields of [1]–[3](PF6)2 were determined by relative methods, using [Ru(bpy)3]Cl2 as the standard. A full description is provided in the Supporting Information.
Results and Discussion
Synthesis
Ligands 5 and 6 were obtained in good yields from the commercially available ligand 4 through deprotonation of the alcohol with sodium hydride, followed by nucleophilic substitution using iodomethane or bromoacetic acid as the electrophile, respectively (Scheme 2). Coordination of ligands 4–6 to the ruthenium center was achieved by refluxing an excess of the ligand (2–10 equiv) with cis-[Ru(bpy)2Cl2] in an ethanol/water mixture. Replacement of the two coordinating chlorides by ligands 4–6 was typically completed within 1.5 h, as shown by the color change of the solution from purple to orange. After anion exchange with KPF6, complexes [1]–[3](PF6)2 were obtained in 55–69% yield as orange solids. The complexes were all isolated as their bis(hexafluoridophosphate) salt, as confirmed by elemental analysis. The workup of compound [3](PF6)2 was performed under acidic conditions (pH ∼2) to ensure protonation of the carboxylic acid in the final solid product. All three complexes were soluble in water, despite their apolar counteranions. Coordination of the bis(thioether) ligand was clearly demonstrated by 1H NMR by a splitting of the signal of the thiomethyl groups, e.g. from a singlet at 2.16 ppm for ligand 5 in CDCl3 to two singlets at 1.63 and 1.34 ppm for complex [2](PF6)2 in acetone-d6. Further characterization of the complexes was performed using high-resolution mass spectrometry and elemental analysis.
Scheme 2. Synthesis of Ruthenium Complexes [1]–[3](PF6)2.

Conditions: (a) NaH, iodomethane in THF, 0 °C to room temperature, 24 h, 66%; (b) NaH, KI, bromoacetic acid in THF, 0 °C to reflux, 22 h, 95%; (c) (i) cis-[Ru(bpy)2Cl2] in EtOH/H2O (1/1 v/v), reflux, 1.5 h, (ii) KPF6, 57% ([1](PF6)2), 69% ([2](PF6)2), 55% ([3](PF6)2). Compounds [1]–[3](PF6)2 were obtained as racemic Λ/Δ mixtures.
As we used a racemic sample of cis-[Ru(bpy)2Cl2] for the synthesis of [1]–[3](PF6)2, we obtained racemic mixtures of the Λ and Δ enantiomers for each complex. An additional stereochemical complication is caused by the six-membered ring formed by the coordination of ligands 4–6, which induces four more sources of isomerism: the configuration (R or S) of the two sulfur atoms, the configuration of the carbon atom attached to the hydroxyl or ether group, leading to either an axial or equatorial −OR substituent, and the inversion of the six-membered metallacycle, which transforms all axial substituents on the ring into equatorial ones (see Scheme 3). With five stereogenic centers, we would expect 32 possible isomers, i.e. 16 Λ diastereoisomers and their respective Δ enantiomers. However, due to the plane of symmetry in ligands 4–6, inversion of the six-membered ring leads to the formation of one of the other diastereoisomers: e.g., ring inversion of Λ-a-[Ru]2+ (see Scheme 3) leads to the formation of Λ-h-[Ru]2+. Thus, we concluded that there are eight possible Λ diastereoisomers in total, shown in Scheme 3, all with their respective Δ enantiomers. It should be noted that the determination of enantiomer relationships is nontrivial for these complexes. For example, whereas the mirror image of Λ-a-[1]2+ is as expected Δ-a-[1]2+, the enantiomer of Λ-b-[1]2+ is Δ-c-[1]2+ because the two nonequivalent sulfur atoms in diastereoisomer b exchange with each other upon mirroring into c. A similar exchange occurs with isomers f and g. The full list of enantiomeric pairs is shown as Table 1 for convenience. According to 1D and 2D 1H NMR, which showed only a single set of 16 aromatic proton signals originating from the bipyridine ligands, all 3 complexes were obtained as a racemic mixture of a single diastereomer.
Scheme 3. Possible Stereoisomers of Complexes [1]–[3]2+, Resulting from the Inversion of either the Configuration of One of the Sulfur Atoms or the Configuration of the Carbon Atom Attached to the Hydroxyl or Ether Group.

Table 1. Enantiomer Relationships for Complexes [1]–[3]2+a.
| Λ | Λ-a-[Ru]2+ | Λ-b-[Ru]2+ | Λ-c-[Ru]2+ | Λ-d-[Ru]2+ | Λ-e-[Ru]2+ | Λ-f-[Ru]2+ | Λ-g-[Ru]2+ | Λ-h-[Ru]2+ |
| Δ | Δ-a-[Ru]2+ | Δ-c-[Ru]2+ | Δ-b-[Ru]2+ | Δ-d-[Ru]2+ | Δ-e-[Ru]2+ | Δ-g-[Ru]2+ | Δ-f-[Ru]2+ | Δ-h-[Ru]2+ |
Structural Characterization by NMR and DFT
In order to gather insight into which one of the eight diastereoisomers of [1](PF6)2 was obtained, we performed a computational study of the stability of each of these isomers in aqueous solution using DFT, employing the COSMO32 model to simulate solvent effects. We minimized the structures of the eight Λ diastereoisomers of [1]2+ shown in Scheme 3, where the six-membered ring is in a chair conformation, as well as the eight possible diastereoisomers with the six-membered ring in a boat configuration. The diastereoisomers in a boat configuration either relaxed to one of the chair configurations shown above or resulted in a twisted-boat configuration with a high energy. Thus, we concluded that a boat configuration is energetically strongly disfavored for the six-membered metallacycle in [1]2+ and that the product obtained must be in a chair configuration. The optimized structures, their structural distortion parameters, and their respective energies in water are given in Table 2, Table S2, and Figure S1. Four of the possible geometries, i.e. Λ-b-[1]2+, Λ-d-[1]2+, Λ-g-[1]2+, and Λ-h-[1]2+, were significantly higher in energy, in comparison to the other four. All of these geometries have one of the sulfur atoms in an (R)-ax orientation that leads to a steric clash of the thiomethyl group with one of the bipyridine ligands (Figure S1). Diastereoisomer Λ-(S)-eq-(S)-ax-OHeq-[1]2+ (Λ-f-[1]2+ in Scheme 3) was found to be the lowest in energy, 3.7 kJ mol–1 lower than the diastereoisomer that is second lowest, Λ-(S)-eq-(S)-ax-OHax-[1]2+ (Λ-c-[1]2+), obtained by inversion of the configuration of the carbon atom bearing the alcohol substituent. Two more diastereoisomers have relatively low energies, namely Λ-(S)-eq-(R)-eq-OHax-[1]2+ (Λ-a-[1]2+), and Λ-(S)-eq-(R)-eq-OHeq-[1]2+ (Λ-e-[1]2+), where both thiomethyl groups are found in equatorial positions. The small energy differences of ∼4 kJ mol–1 between these isomers is not enough to exclude any of these four structures purely on the basis of their computed energies.
Table 2. Absolute and Relative Energies in Water (COSMO) of the Λ Diastereoisomers of [1]2+, Optimized by DFT.
| isomer | absolute energy in water/Hartree | relative energy (ΔE) in water/kJ mol–1 |
|---|---|---|
| Λ-a-[1]2+ | –13.05674 | 4.1 |
| Λ-b-[1]2+ | –13.05197 | 16.7 |
| Λ-c-[1]2+ | –13.05690 | 3.7 |
| Λ-d-[1]2+ | –13.05133 | 18.4 |
| Λ-e-[1]2+ | –13.05688 | 3.8 |
| Λ-f-[1]2+ | –13.05832 | 0.0 |
| Λ-g-[1]2+ | –13.05310 | 13.7 |
| Λ-h-[1]2+ | –13.05317 | 13.5 |
As the DFT calculations did not provide a conclusive answer, we turned to 1H NMR spectroscopy. The stereochemistry of the carbon atom bearing the alcohol (C3 in Figure 1) could be found from the 3J coupling constants of the protons on the adjacent carbon atom (C2). The large difference between the 3J coupling constant of the axial (3J = 6.3 Hz) and equatorial protons (3J = 2.1 Hz) suggests that the proton on C3 is positioned axially, and thus the −OH group has to be equatorial. NOESY NMR spectroscopy further confirmed the axial position of this proton (H3) by an off-diagonal correlation with the D6 proton of the bpy ligand (Figure S2). As the alcohol group is equatorial, the number of possible Λ isomers of [1]2+ in solution was reduced to two, i.e. Λ-(S)-eq-(S)-ax-OHeq-[1]2+ (Λ-f-[1]2+) and Λ-(S)-eq-(R)-eq-OHeq-[1]2+ (Λ-e-[1]2+), which differ from each other by a single inversion of sulfur chirality. In order to assess whether this thiomethyl group (C1 in Figure 1) was axial or equatorial, we examined the off-diagonal NOESY correlations of this group (Figure S3). We found a correlation of these protons to the A6 proton of the bpy ligand, over a distance of 3.29 Å versus 5.00 Å for the equatorial and axial cases, respectively. This suggested that the thiomethyl group is oriented equatorially. However, the protons on C1 also show an off-diagonal correlation to the axial proton on C3, a proton that is significantly closer if the thiomethyl group is oriented axially (3.36 Å versus 4.98 Å). Finally, a weak correlation was found to the C3 proton on the bipyridine ring, which is closer to thiomethyl group C1 in the axial conformation (5.19 Å versus 6.22 Å). All in all, this convinced us that this thiomethyl group is at least predominantly oriented axially, yet an equilibrium between its axial and equatorial positions in solution could not be fully excluded. Thus, our NMR studies suggest that [1]2+ is predominantly a racemic mixture of Λ-(S)-eq-(S)-ax-OHeq-[1]2+ (Λ-f-[1]2+, Figure 1) and Δ-(R)-ax-(R)-eq-OHeq-[1]2+ (Δ-g-[1]2+). This is also the enantiomeric pair that was found to be most stable in our DFT studies (Table 2), suggesting that the formation of the complex is under thermodynamic control. Substitution of the alcohol does not affect the stereochemistry of the complexes, as complexes [2](PF6)2 and [3](PF6)2 were also found to form as a racemic mixture of the Λ-f-[Ru]2+ and Δ-g-[Ru]2+ enantiomers, where [Ru]2+ is [2]2+ or [3]2+.
Figure 1.
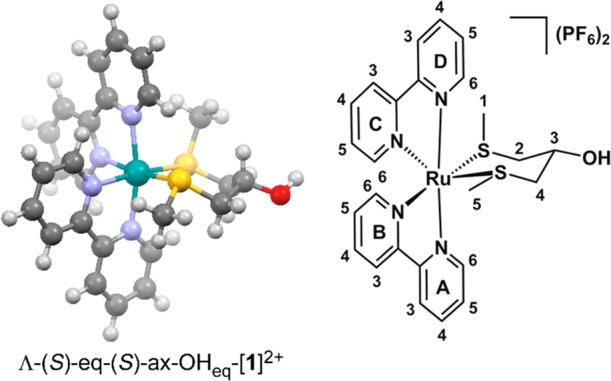
Structure of the most stable Λ diastereoisomer of [1]2+, Λ-(S)-eq-(S)-ax-OHeq-[1]2+ (Λ-f-[1]2+), optimized by DFT (BLYP/TZP) in water (COSMO), with a schematic drawing showing the atom numbering used in the text.
In recent work from our group we have shown that the bond angle variance σ2 can be used as a structural distortion parameter to quantify the steric hindrance induced by thiomethyl groups in ruthenium polypyridyl complexes that bear no straining pyridyl ligands.25,38 In the case of complex [1]2+, we observed an increase in the σ2 value by at least 25 upon the introduction of an (R)-ax sulfur atom in the Λ diastereoisomers, in comparison to their corresponding (S)-eq isomer (e.g., σ2 = 59.4 and 87.5 for Λ-a-[1]2+ and Λ-b-[1]2+, respectively, see Table S2). This increase correlates well with the energies calculated by DFT (Table 2), which show an increase by 10–15 kJ mol–1 for this inversion of the sulfur configuration. Interestingly, we could not find a direct correlation between the σ2 value and the DFT energy for the conformation of the second sulfur atom. Inversion from Λ-(S)-ax to Λ-(R)-eq for the C1 thiomethyl group led to an increase in the σ2 value of ∼17 but resulted in virtually no increase in DFT-calculated energy. This phenomenon could be explained by the fact that the calculation of the σ2 value does not take into account the intraligand interactions within bis(thioether) ligand 4. Although the Λ-(S)-ax conformation is favorable for relieving the octahedral strain on the ruthenium center, it does lead to unfavorable 1,3-diaxial interactions with the H3 proton on ligand 4, making the total energetic effect negligible. Logically, we observed no effect of orientation of the alcohol group on the σ2 value, since this does not affect the octahedral strain on the ruthenium center.
The synthesis of the related complexes [9](PF6)2 and [10](PF6)2 was also reported to be diastereoselective by Sauvage et al., who reported the same stereochemistry for the sulfur atoms as we found for [1]–[3](PF6)2.27 However, in their crystal structure the six-membered ring in [10](PF6)2 is found in a half-chair conformation, perhaps made possible by the lack of substitution at the C3 position. Overall, we can conclude that the configuration of the sulfur atoms is not influenced by the size of the ring, nor by the type of substituents on the sulfur atoms (methyl groups in [1]–[3](PF6)2 and phenyl groups in [9]- and [10](PF6)2) or by substituents on the chelating ring. However, the introduction of substituents at the C3 position on the ring does seem to force the ring into a chair conformation.
Photochemistry
All three complexes form yellow solutions in water, showing a 1MLCT absorption band around 412 nm, with molar absorption coefficients of 4.0–5.2 × 103 M–1 cm–1 (Table 3), typical for ruthenium(II) polypyridyl complexes containing two thioether donor ligands.26 Essentially no phosphorescence was observed upon irradiation of the complexes with blue light in deuterated methanol (Figure S4A), with phosphorescence quantum yields ΦP lower than 2.0 × 10–4. The complexes also appeared to be very poor singlet oxygen sensitizers (ΦΔ ≤ 0.008, Figure S4B), as expected from their photosubstitution properties (vide infra).
Table 3. Lowest-Energy Absorption Maxima (λmax), Molar Absorption Coefficients at λmax (εmax) and 443 nm (ε443), Photosubstitution Quantum Yields (Φ443) and Photosubstitution Reactivities (ξ443 = Φ443 × ε443) at 298 K in H2O, Singlet Oxygen Quantum Yield (ΦΔ), and Phosphorescence Quantum Yield (ΦP) at 293 K in MeOD for Complexes [1]–[3](PF6)2 and Photochemical Intermediates [11]–[13](PF6)2.
| complex | λmax/nm (εmax/103 M–1 cm–1) | ε443/103 M–1 cm–1 | Φ443 | ξ443 | ΦΔ | ΦP (λem/nm) |
|---|---|---|---|---|---|---|
| [1](PF6)2 | 413 (5.13) | 2.95 | 0.24 | 704 | 0.008 | 2.0 × 10–4 (624) |
| [11](PF6)2 | 453 (7.02) | 6.68 | 0.0079 | 53 | ||
| [2](PF6)2 | 412 (4.04) | 2.29 | 0.25 | 578 | 0.007 | 1.4 × 10–4 (620) |
| [12](PF6)2 | 456 (5.52) | 5.04 | 0.0093 | 47 | ||
| [3](PF6)2 | 412 (5.18) | 2.92 | 0.16 | 474 | <0.005 | 6 × 10–5 (620) |
| [13](PF6)2 | 456 (6.77) | 6.19 | 0.0055 | 34 |
In the absence of light, complexes [1]–[3](PF6)2 were found to be stable in water (Figure S5). However, all three compounds are photoreactive under blue light irradiation in water. We monitored the photoreactions of [1]–[3](PF6)2 with UV–vis absorption spectroscopy and mass spectrometry. Upon irradiation of a solution of [1](PF6)2 with a blue LED (λ = 443 ± 11 nm), we observed a two-step bathochromic shift in the 1MLCT absorbance band of the solution (Figure 2 and Figure S6). First, the absorption maximum shifted from 413 to 453 nm, accompanied by three isosbestic points at 319, 364, and 426 nm (Figure 2A). This first reaction was completed within 5 min under the irradiation conditions used (photon flux qp = 2.65 × 10–8 mol of photons s–1), at which point the absorption maximum started to shift toward longer wavelengths again. This second reaction, in which the absorption maximum changed from 453 to 491 nm, showed isosbestic points at 314, 330, 389, and 466 nm and was significantly slower than the first photoreaction (Figure 2B). Completion of this second reaction took 1 h, at which point a steady state was reached. Mass spectrometry of the reaction mixture after irradiation (Figure S7) showed a peak at m/z 247.9, corresponding to [Ru(bpy)2(CH3CN)2]2+ (calcd m/z 248.0), formed inside the mass spectrometer from the original photoproduct [Ru(bpy)2(OH2)2]2+ ([14]2+). No signals were observed that match to photoproducts resulting from expulsion of one of the bpy ligands. This result indicates that, upon blue light irradiation of [1]2+ in water, the bis(thioether) chelate 4 is selectively substituted by two water molecules.
Figure 2.
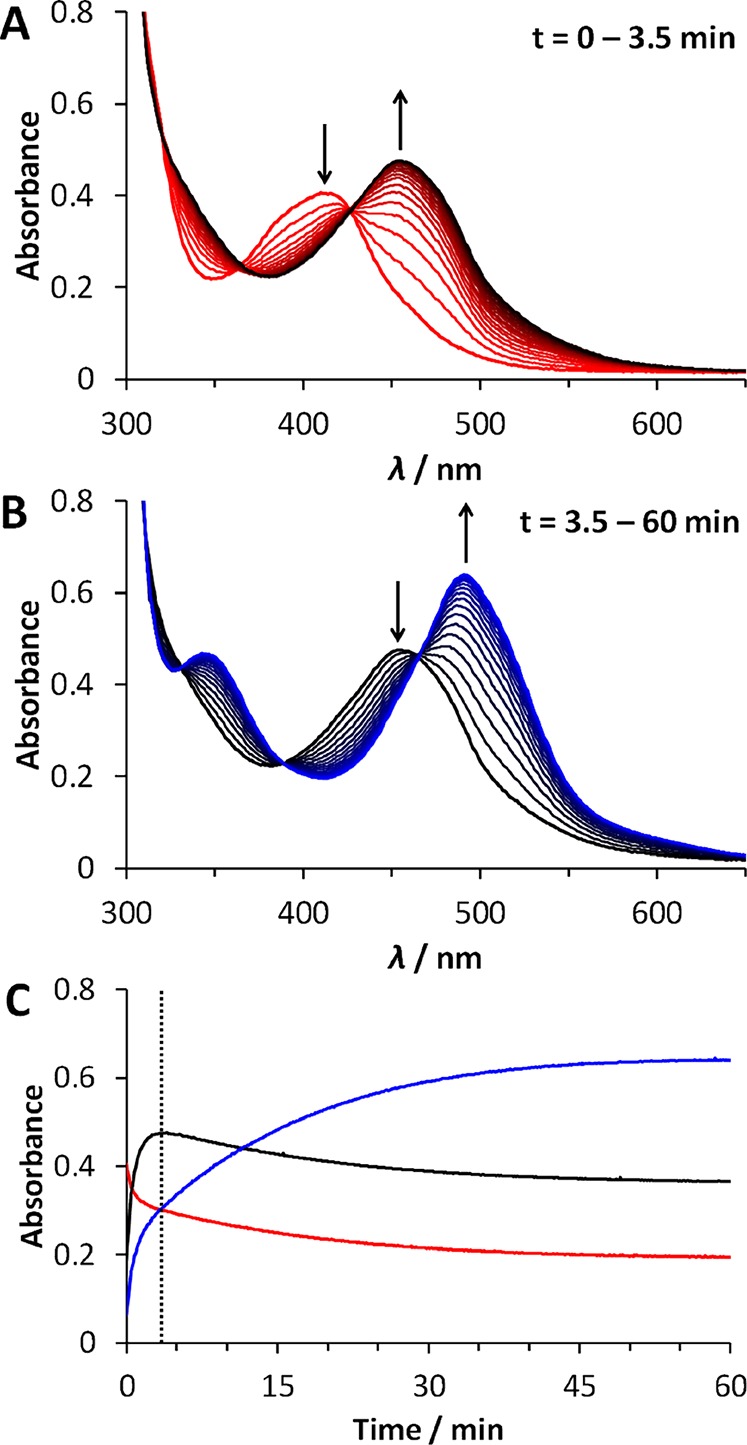
Evolution in time of the absorption spectra of a solution of [1](PF6)2 in H2O (72 μM) upon irradiation at 298 K with a 443 nm LED (qp = 2.65 × 10–8 mol of photons s–1) under N2, for t = 0–3.5 min (A, Δt = 12 s) and t = 3.5–60 min (B, Δt = 3.2 min), and the time evolution of the absorbance (C) at 413 nm (red), 453 nm (black), and 491 nm (blue) during the first 60 min of irradiation. The vertical dashed line (t = 3.5 min) indicates the completion of the first photosubstitution reaction.
The intermediate species in the photoreaction was identified by mass spectrometry, by measuring a sample after the first 5 min of irradiation (Figure S8). This sample showed the peak for the photoproduct, as well as a peak for the starting compound [1]2+ at m/z 282.7 (calcd m/z 283.0), and another signal at m/z 303.1, identified as [Ru(bpy)2(4)(CH3CN)]2+ (calcd m/z 303.6), formed inside the mass spectrometer from the original photochemical intermediate [Ru(bpy)2(4)(H2O)]2+. We hypothesized that the intermediate, which is reasonably stable, is most likely six-coordinate, with ligand 4 bound in a monodentate fashion, and the second thioether group is replaced by water: i.e., [Ru(bpy)2(κ1-4)(H2O)]2+ ([11]2+). Overall, under blue light irradiation [1](PF6)2 undergoes a two-step consecutive photochemical substitution of the bis(thioether) ligand, passing through the rather stable mono(aqua) intermediate [11]2+ (Scheme 4). This two-step photoreactivity is reminiscent of the photoreactivity observed for ruthenium polypyridyl complexes bearing two photocleavable monodentate ligands, such as cis-[Ru(bpy)2(py)2]2+ (py = pyridine),39−41 and has also been observed for the photodissociation of the ligand bete (3,6-dithiaoctane) in [7]2+ 26 or mtmp (2-(methylthio)methyl-2-pyridine) in [Ru(bpy)2(mtmp)]2+.13
Scheme 4. Two-Step Photosubstitution Reactions Observed upon Blue Light Irradiation of Solutions of [1]2+–[3]2+ in H2O.

The identity of the final products of the photoreaction was confirmed by 1H NMR spectroscopy (Figure 3). White light irradiation of a sample of [1](PF6)2 in D2O in an NMR tube resulted in the formation, at the photostationary state, of a mixture of the free ligand 4 and of the complex cations cis-[Ru(bpy)2(H2O)2]2+ (cis-[14]2+) and trans-[Ru(bpy)2(H2O)2]2+ (trans-[14]2+). Although our experimental data do not allow us to exclude direct formation of trans-[14]2+ from the photochemical intermediate [11]2+, it is most likely formed through photoisomerization of cis-[14]2+ to its trans isomer, as reported previously.37,42 As both the cis and trans isomers undergo photoisomerization, a photostationary state is obtained at the end of the irradiation experiment. Since the quantum yields for these cis–trans isomerization reactions of [14]2+ are relatively high (Φ450 = 0.023–0.045 in 0.5 M H2SO4) in comparison to the photosubstitution of [11]2+ (Table 3), we did not observe these reactions separately (whether by NMR or by UV–vis absorption spectroscopy) but they occur concomitantly. In addition, NMR experiments under light irradiation were not helpful in identifying the structure of [11]2+, as this intermediate may exist as several highly unsymmetrical isomers, the peaks of which overlap with those of the reagent or products (Figure 3).
Figure 3.

Evolution of the aromatic (A, δ 9.9–6.9 ppm) and aliphatic regions (B, δ 4.1–1.15 ppm) of the 1H NMR spectrum of a solution of [1](PF6)2 (2.0 mM) in D2O upon irradiation with a Xe lamp (λirr = 400–700 nm) for 60 min. Labeled signals correspond to the free ligand 4 (diamonds), cis-[14]2+ (circles), and trans-[14]2+ (squares).
Irradiation of complexes [2]2+ and [3]2+ resulted in very similar photoreactions, as shown in Figures S9 and S10. The UV–vis absorption spectra indicate formation of the same final photoproduct [14]2+, passing through the monodentate photochemical intermediates [12]2+ and [13]2+, as confirmed by mass spectrometry (Figures S11–S14). The quantum efficiencies of the two photochemical steps for each photoreaction were derived using global fitting of the time evolution of the UV–vis absorption spectra, using the Glotaran software package (Table 3 and Figures S15–S17).36 The photosubstitution quantum yields Φ443 were found to be similar across all three complexes, with Φ443 = 0.24, 0.25, and 0.16 for the first step of the photoreaction for [1]2+, [2]2+, and [3]2+, respectively. The second step of the photoreaction was characterized by photosubstitution quantum yields of 0.0079, 0.0093, and 0.0055, respectively. These quantum efficiencies are similar to those observed earlier for the second reaction step of bidentate pyridine-thioether ligands13 and slightly lower than those found for the substitution of monodentate thioether ligands.20 For the photosubstitution reactions of [7]2+ and [8]2+ in water, Turro et al. also reported a two-step mechanism, including a quick formation of a κ1-coordinated intermediate species. They reported overall quantum yields for the formation of [14]2+ (Φ400) of 0.024 and 0.022, respectively, rather than the quantum yields of the individual steps reported above for the photosubstitution reactions in [1]–[3]2+.26 This discrepancy in the kinetic models precludes direct comparison of photosubstitution efficiencies.
Conclusions
In this work, we have shown that the coordination of ligands 4–6 to the cis-Ru(bpy)2 scaffold under reflux in an EtOH/H2O mixture is diastereoselective, yielding complexes [1]–[3](PF6)2 as a racemic mixture of two enantiomers: namely, Λ-(S)-eq-(S)-ax-OHeq-[Ru]2+ and Δ-(R)-ax-(R)-eq-OHeq-[Ru]2+. DFT calculations showed this isomer to be the most energetically favorable, suggesting that under such conditions the synthesis is under thermodynamic control. As the obtained isomer was also found to have the smallest bond angle variance (σ2), we hypothesize that minimization of the steric hindrance induced by the thioether ligands is a major driving force for the formation of this isomer. As we obtained the same diastereoisomer that was reported for complexes [9](PF6)2 and [10](PF6)2, we conclude that the diastereoselectivity is not determined by the nature of the thioether substituent or by the chelate ring size. According to DFT, the substituent on the C3 carbon in [1]–[3](PF6)2 does force the chelate ring in a chair conformation, rather than a half-chair conformation as observed in the X-ray structures of [9](PF6)2 and [10](PF6)2.
All three complexes were found to be stable in the dark in aqueous solution but undergo efficient ligand substitution reactions upon irradiation with blue light. In all three cases, a selective substitution of the bis(thioether) ligand in two steps was observed, leading to the formation of the bis(aqua) complex [Ru(bpy)2(H2O)2]2+ ([14]2+). The reaction mechanism was found to be identical with that reported for complexes [7](PF6)2 and [8](PF6)2. A 30-fold difference in efficiency between the two steps of the photoreaction was observed, which, in combination with a high time resolution in the irradiation experiments, allowed us to determine the photosubstitution quantum yields for the individual steps, rather than the overall quantum yield. It also allowed us to identify the photochemical intermediate as the κ1-mono(thioether), mono(aqua) complex by mass spectrometry. Substitution of the alcohol group by a methoxy or carboxylate group, as in complexes [2](PF6)2 and [3](PF6)2, does not have an effect on the diastereoselectivity of the synthesis or on the selectivity of the photosubstitution reaction. Only small differences were observed in the efficiency of the photosubstitution reactions. Thus, functionalized bis(thioether) ligands are promising candidates for the binding of cis ruthenium-based PACT complexes to inorganic surfaces, as they can be functionalized, do not form too many isomers, and can be efficiently photocleaved.
Acknowledgments
The European Research Council is acknowledged for a Starting grant to S.B. Prof. E. Bouwman is kindly acknowledged for scientific discussion and support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.9b01669.
NMR spectra, spectroscopic details for photosubstitution, singlet oxygen generation, and phosphosrescence quantum yield measurements, mass spectra after irradiation, dark stability measurements of ruthenium complexes, and computational details, including calculated geometries and structural parameters (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bonnet S. Why develop photoactivated chemotherapy?. Dalton Trans 2018, 47 (31), 10330–10343. 10.1039/C8DT01585F. [DOI] [PubMed] [Google Scholar]
- Velema W. A.; Szymanski W.; Feringa B. L. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136 (6), 2178–2191. 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- Farrer N. J.; Salassa L.; Sadler P. J. Photoactivated chemotherapy (PACT): the potential of excited-state d-block metals in medicine. Dalton Trans 2009, 38 (48), 10690–10701. 10.1039/b917753a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai S.; Yang G.; Yang P.; He F.; Lin J.; Jin D.; Xing B. Recent advances in functional nanomaterials for light–triggered cancer therapy. Nano Today 2018, 19, 146–187. 10.1016/j.nantod.2018.02.010. [DOI] [Google Scholar]
- Mari C.; Pierroz V.; Ferrari S.; Gasser G. Combination of Ru(ii) complexes and light: new frontiers in cancer therapy. Chem. Sci. 2015, 6 (5), 2660–2686. 10.1039/C4SC03759F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann F.; Karges J.; Gasser G. Critical Overview of the Use of Ru(II) Polypyridyl Complexes as Photosensitizers in One-Photon and Two-Photon Photodynamic Therapy. Acc. Chem. Res. 2017, 50 (11), 2727–2736. 10.1021/acs.accounts.7b00180. [DOI] [PubMed] [Google Scholar]
- Hess J.; Huang H.; Kaiser A.; Pierroz V.; Blacque O.; Chao H.; Gasser G. Evaluation of the Medicinal Potential of Two Ruthenium(II) Polypyridine Complexes as One- and Two-Photon Photodynamic Therapy Photosensitizers. Chem. - Eur. J. 2017, 23 (41), 9888–9896. 10.1002/chem.201701392. [DOI] [PubMed] [Google Scholar]
- Shi G.; Monro S.; Hennigar R.; Colpitts J.; Fong J.; Kasimova K.; Yin H.; DeCoste R.; Spencer C.; Chamberlain L.; Mandel A.; Lilge L.; McFarland S. A. Ru(II) dyads derived from α-oligothiophenes: A new class of potent and versatile photosensitizers for PDT. Coord. Chem. Rev. 2015, 282–283, 127–138. 10.1016/j.ccr.2014.04.012. [DOI] [Google Scholar]
- Cloonan S. M.; Elmes R.; Erby M.; Bright S. A.; Poynton F. E.; Nolan D. E.; Quinn S. J.; Gunnlaugsson T.; Williams D. C. Detailed biological profiling of a photoactived and apoptosis inducing pdppz Ruthenium (II) polypyridyl complex in cancer cells. J. Med. Chem. 2015, 58 (11), 4494–4505. 10.1021/acs.jmedchem.5b00451. [DOI] [PubMed] [Google Scholar]
- Zayat L.; Calero C.; Alborés P.; Baraldo L.; Etchenique R. A New Strategy for Neurochemical Photodelivery: Metal-Ligand Heterolytic Cleavage. J. Am. Chem. Soc. 2003, 125 (4), 882–883. 10.1021/ja0278943. [DOI] [PubMed] [Google Scholar]
- Frasconi M.; Liu Z.; Lei J.; Wu Y.; Strekalova E.; Malin D.; Ambrogio M. W.; Chen X.; Botros Y. Y.; Cryns V. L.; Sauvage J.-P.; Stoddart J. F. Photoexpulsion of Surface-Grafted Ruthenium Complexes and Subsequent Release of Cytotoxic Cargos to Cancer Cells from Mesoporous Silica Nanoparticles. J. Am. Chem. Soc. 2013, 135 (31), 11603–11613. 10.1021/ja405058y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaoun N.; Renfrew A. K. A luminescent ruthenium(ii) complex for light-triggered drug release and live cell imaging. Chem. Commun. 2015, 51 (74), 14038–14041. 10.1039/C5CC05172J. [DOI] [PubMed] [Google Scholar]
- Cuello-Garibo J.-A.; Meijer M. S.; Bonnet S. To cage or to be caged? The cytotoxic species in ruthenium-based photoactivated chemotherapy is not always the metal. Chem. Commun. 2017, 53 (50), 6768–6771. 10.1039/C7CC03469E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistin F.; Balducci G.; Wei J.; Renfrew A. K.; Alessio E. Photolabile Ru Model Complexes with Chelating Diimine Ligands for Light-Triggered Drug Release. Eur. J. Inorg. Chem. 2018, 2018 (13), 1469–1480. 10.1002/ejic.201701392. [DOI] [Google Scholar]
- Kohler L.; Nease L.; Vo P.; Garofolo J.; Heidary D. K.; Thummel R. P.; Glazer E. C. Photochemical and Photobiological Activity of Ru(II) Homoleptic and Heteroleptic Complexes Containing Methylated Bipyridyl-type Ligands. Inorg. Chem. 2017, 56 (20), 12214–12223. 10.1021/acs.inorgchem.7b01642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus L. M.; Al-Afyouni K. F.; Turro C. New RuII Scaffold for Photoinduced Ligand Release with Red Light in the Photodynamic Therapy (PDT) Window. Chem. - Eur. J. 2018, 24 (45), 11550–11553. 10.1002/chem.201802405. [DOI] [PubMed] [Google Scholar]
- Li A.; Yadav R.; White J. K.; Herroon M. K.; Callahan B. P.; Podgorski I.; Turro C.; Scott E. E.; Kodanko J. J. Illuminating cytochrome P450 binding: Ru(ii)-caged inhibitors of CYP17A1. Chem. Commun. 2017, 53 (26), 3673–3676. 10.1039/C7CC01459G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragazzon G.; Bratsos I.; Alessio E.; Salassa L.; Habtemariam A.; McQuitty R. J.; Clarkson G. J.; Sadler P. J. Design of photoactivatable metallodrugs: Selective and rapid light-induced ligand dissociation from half-sandwich [Ru([9]aneS3)(N–N’)(py)]2+ complexes. Inorg. Chim. Acta 2012, 393 (0), 230–238. 10.1016/j.ica.2012.06.031. [DOI] [Google Scholar]
- White J. K.; Schmehl R. H.; Turro C. An overview of photosubstitution reactions of Ru(II) imine complexes and their application in photobiology and photodynamic therapy. Inorg. Chim. Acta 2017, 454, 7–20. 10.1016/j.ica.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldbach R. E.; Rodriguez-Garcia I.; van Lenthe J. H.; Siegler M. A.; Bonnet S. N-Acetylmethionine and Biotin as Photocleavable Protective Groups for Ruthenium Polypyridyl Complexes. Chem. - Eur. J. 2011, 17 (36), 9924–9929. 10.1002/chem.201101541. [DOI] [PubMed] [Google Scholar]
- Bahreman A.; Rabe M.; Kros A.; Bruylants G.; Bonnet S. Binding of a Ruthenium Complex to a Thioether Ligand Embedded in a Negatively Charged Lipid Bilayer: A Two-Step Mechanism. Chem. - Eur. J. 2014, 20 (24), 7429–7438. 10.1002/chem.201400377. [DOI] [PubMed] [Google Scholar]
- Siewert B.; Langerman M.; Pannwitz A.; Bonnet S. Synthesis and avidin binding of ruthenium complexes functionalized with a light-cleavable free biotin moiety. Eur. J. Inorg. Chem. 2018, 2018 (37), 4117–4124. 10.1002/ejic.201800644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S.; Collin J. P.; Gruber N.; Sauvage J. P.; Schofield E. R. Photochemical and thermal synthesis and characterization of polypyridine ruthenium(II) complexes containing different monodentate ligands. Dalton Trans 2003, 0 (24), 4654–4662. 10.1039/b310198c. [DOI] [Google Scholar]
- Zayat L.; Filevich O.; Baraldo L. M.; Etchenique R. Ruthenium polypyridyl phototriggers: from beginnings to perspectives. Philos. Trans. R. Soc., A 2013, 371 (1995), 20120330. 10.1098/rsta.2012.0330. [DOI] [PubMed] [Google Scholar]
- Cuello-Garibo J.-A.; James C. C.; Siegler M. A.; Bonnet S. Ruthenium-Based PACT Compounds Based on an N, S Non-Toxic Ligand: A Delicate Balance Between Photoactivation and Thermal Stability. Chem. Sq. 2017, 1, 2. 10.28954/2017.csq.12.002. [DOI] [Google Scholar]
- Garner R. N.; Joyce L. E.; Turro C. Effect of Electronic Structure on the Photoinduced Ligand Exchange of Ru(II) Polypyridine Complexes. Inorg. Chem. 2011, 50 (10), 4384–4391. 10.1021/ic102482c. [DOI] [PubMed] [Google Scholar]
- Collin J.-P.; Jouvenot D.; Koizumi M.; Sauvage J.-P. Ru(phen)2(bis-thioether)2+ complexes: Synthesis and photosubstitution reactions. Inorg. Chim. Acta 2007, 360 (3), 923–930. 10.1016/j.ica.2006.06.028. [DOI] [Google Scholar]
- Al-Rawashdeh N. A. F.; Chatterjee S.; Krause J. A.; Connick W. B. Ruthenium Bis-diimine Complexes with a Chelating Thioether Ligand: Delineating 1,10-Phenanthrolinyl and 2,2 ‘-Bipyridyl Ligand Substituent Effects. Inorg. Chem. 2014, 53 (1), 294–307. 10.1021/ic4022454. [DOI] [PubMed] [Google Scholar]
- Root M. J.; Sullivan B. P.; Meyer T. J.; Deutsch E. Thioether, thiolato, and 1,1-dithioato complexes of bis(2,2′-bipyridine)ruthenium(II) and bis(2,2′-bipyridine)osmium(II). Inorg. Chem. 1985, 24 (18), 2731–2739. 10.1021/ic00212a006. [DOI] [Google Scholar]
- Askes S. H. C.; Meijer M. S.; Bouwens T.; Landman I.; Bonnet S. Red Light Activation of Ru(II) Polypyridyl Prodrugs via Triplet-Triplet Annihilation Upconversion: Feasibility in Air and through Meat. Molecules 2016, 21 (11), 1460. 10.3390/molecules21111460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey E. J.; Erickson B. W.; Noyori R. New synthesis of. alpha., beta.-unsaturated aldehydes using 1,3-bis(methylthio)allyllithium. J. Am. Chem. Soc. 1971, 93 (7), 1724–1729. 10.1021/ja00736a027. [DOI] [Google Scholar]
- Pye C. C.; Ziegler T. An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Theor. Chem. Acc. 1999, 101 (6), 396–408. 10.1007/s002140050457. [DOI] [Google Scholar]
- Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38 (6), 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37 (2), 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Van Lenthe E.; Baerends E. J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24 (9), 1142–1156. 10.1002/jcc.10255. [DOI] [PubMed] [Google Scholar]
- Snellenburg J. J.; Laptenok S.; Seger R.; Mullen K. M.; van Stokkum I. H. M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Soft. 2012, 49 (3), 22. 10.18637/jss.v049.i03. [DOI] [Google Scholar]
- Durham B.; Wilson S. R.; Hodgson D. J.; Meyer T. J. Cis-trans photoisomerization in Ru(bpy)2(OH2)22+. Crystal structure of trans-[Ru(bpy)2(OH2)(OH)](ClO4)2. J. Am. Chem. Soc. 1980, 102 (2), 600–607. 10.1021/ja00522a026. [DOI] [Google Scholar]
- Fleet M. E. Distortion parameters for coordination polyhedra. Mineral. Mag. 1976, 40 (313), 531–533. 10.1180/minmag.1976.040.313.18. [DOI] [Google Scholar]
- Garner R. N.; Gallucci J. C.; Dunbar K. R.; Turro C. [Ru(bpy)2(5-cyanouracil)2]2+ as a Potential Light-Activated Dual-Action Therapeutic Agent. Inorg. Chem. 2011, 50 (19), 9213–9215. 10.1021/ic201615u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora A.; Denning C. A.; Heidary D. K.; Wachter E.; Nease L. A.; Ruiz J.; Glazer E. C. Ruthenium-containing P450 inhibitors for dual enzyme inhibition and DNA damage. Dalton Trans 2017, 46 (7), 2165–2173. 10.1039/C6DT04405K. [DOI] [PubMed] [Google Scholar]
- Pinnick D. V.; Durham B. Photosubstitution reactions of Ru(bpy)2XYn+ complexes. Inorg. Chem. 1984, 23 (10), 1440–1445. 10.1021/ic00178a028. [DOI] [Google Scholar]
- Gama Sauaia M. l.; Tfouni E.; Helena de Almeida Santos R.; Teresa do Prado Gambardella M.; Del Lama M. P. F. M.; Fernando Guimarães L.; Santana da Silva R. Use of HPLC in the identification of cis and trans-diaquabis(2,2’-bipyridine)ruthenium(II) complexes: crystal structure of cis-[Ru(H2O)2(bpy)2](PF6)2. Inorg. Chem. Commun. 2003, 6 (7), 864–868. 10.1016/S1387-7003(03)00131-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.