

Abstract
Endothelin receptor A (ETAR) promotes tumorigenesis by stimulating cell proliferation, migration, and survival. However, the mechanism of ETAR in promoting tumor growth is largely unknown. In this study, we demonstrate that ETAR stimulates colon cell proliferation, migration, and tumorigenesis through the activation of YAP/TAZ, two transcription coactivators of the Hippo tumor suppressor pathway. Endothelin-1 treatment induced YAP/TAZ dephosphorylation, nuclear accumulation, and transcriptional activation in multiple colon cancer cells. ETAR stimulation acted via downstream G-protein Gαq/11 and Rho GTPase to suppress the Hippo pathway, thus leading to YAP/TAZ activation, which was required for ETAR-induced tumorigenesis. Overall, these results indicate a critical role of the YAP/TAZ axis in ETAR signaling.
Introduction
The endothelin family consists of a group of 21-amino acid peptides produced primarily in the endothelium, epithelium, and smooth muscle cells (1,2). Among the three isoforms ofendothelins, endothelin-1 (ET-1) is the most abundant and widely expressed. Extensive studies have revealed a wide range of biological functions of ET-1, including vasoconstriction, cell proliferation, and cell migration (3). In addition, ET-1 signaling has been involved in tumorigenesis in multiple tissues (4). It acts as a growth factor in several cancers, including carcinoma of the prostate, ovary, colon, cervix, breast, kidney, lung, central nervous system tumors as well as melanoma, Kaposi sarcoma, and bone metastasis (5, 6).
ET-1 signals through plasma membrane-localized endothelin receptor A (ETAR) and endothelin receptor B (ETBR), two classical G-protein-coupled receptors (GPCR). ETAR has been reported to be a primary vasoconstrictor and growth-promoting receptor, whereas ETBR inhibits cell growth and vascular constriction (1, 7). Upon activation by ET-1, ETAR contributes to cell proliferation, migration, and survival. It also promotes tumor growth and progression, angiogenesis, and metastatic spread (4, 5, 8). The importance of ETAR in cancer indicates that ETAR-specific antagonists (e.g., BQ123, atrasentan, and ZD4054) are potential cancer therapeutic drugs. In fact, ETAR activation induces the activity of tumor-related proteinases and subsequently promotes cell migration and cell invasion, which could be blocked by BQ123 (9, 10).
Consistent with a prominent role of ET-1 in tumorigenesis, several ETAR antagonists have been tested in clinical trials. Atrasentan showed significant positive effects in brief pain inventory, PSA progression, and bone markers on patients in phase I and II trials (11). In a phase III prostate cancer trial, atrasentan treatment showed improvement in the median time to disease progression in patients, although the primary end-point was not achieved (12, 13). Atrasentan is also under phase II trials for other types of cancer, including renal, ovarian, non-small cell lung, and brain cancers (11). ZD4054 (also known as zibotentan) is a specific ETAR antagonist and has been studied in a multicenter phase II, randomized, double blind, placebo-controlled trial. Patients that received ZD4054 experienced a 35% reduction in the risk of death, which has showed improved overall survival (14). ZD4054 is being evaluated in hormone-resistant prostate cancer in phase III clinical trials (15, 16). In addition, Bosentan and YM598, two nonselective antagonists for both ETAR and ETBR, have shown promising therapeutic effect on prostate cancer in clinical trials (17).
Previous studies have shown higher expression of ETAR and ET-1 in colon cancer tissue compared with normal tissue (18–20), whereas ETBR is expressed in a contrary pattern (21). In addition, ETAR expression correlates with liver metastases of colorectal cancers (20, 21). ETAR may also promote colon cancer cell survival in response to chemotherapy, such as cisplatin (20). A preclinical study demonstrated that ZD4054 efficiently suppressed colorectal cancer cell growth and progression, indicating its potential role as adjuvant therapy in colorectal cancer (22). However, the molecular mechanism of ETAR in promoting tumor cell survival, growth, or metastasis in colorectal cancer is not fully understood.
The Hippo pathway was initially identified to be key player in organ size control in Drosophila and lately found to be important in human cancer (23–26). The core components of the mammalian Hippo pathway contain a kinase cascade ofMST1/2, MAP4Ks, and LATS1/2, in which MST1/2 or MAP4Ks phosphorylate and activate LATS. This kinase cascade regulates target genes expression by inhibiting its downstream transcriptional coactivators Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ). LATS1/2 restrict the activity of YAP/TAZ by direct phosphorylation (27). Phosphorylated YAP/ TAZ are sequestered in the cytoplasm by 14–3-3 binding (28–30). Inhibition of LATS1/2 activity leads to dephosphorylation ofYAP/TAZ, which then translocate to the nucleus, bind to the TEA domain family transcription factors (TEAD1–4), and promote the expression of target genes (31, 32). Generally, the Hippo pathway acts as an important inhibitor of tissue growth and organ size by controlling cell proliferation, apoptosis, and migration in response to a variety of extracellular and intracellular signals, including GPCR signaling (33, 34).
ETAR activation by ET-1 has been reported to induce the expression of connective tissue growth factor (CTGF) and cysteine-rich protein 61 (CYR61), two well-defined target genes of the Hippo pathway (35–38), indicating that ET-1 may act upstream of the Hippo-YAP pathway. In this study, we revealed a mechanism of YAP/TAZ activation by ET-1/ETAR and its role in colorectal tumorigenesis.
Materials and Methods
Chemicals
Chemicals that were used in this study: Endothelin-1, BQ123, Tetramethylrhodamine B isothiocyanate-conjugated Phalloidin, and Latrunculin B were purchased from Sigma-Aldrich. GSK429286 and Y27632 were obtained from Selleck-chem. C3 was purchased from Cytoskeleton Inc. Phos-tag-conjugated acrylamide was purchased from Wako Chemicals. DAPI was purchased from Invitrogen.
Antibodies
Antibodies for YAP (#4912) and Phospho-YAP (S127; #4911) were purchased from Cell Signaling Technology. The Lats1 (A300–477A) and Lats2 (A300–479A) antibodies used in immunoprecipitation were from Bethyl Laboratories. TAZ (HPA007415) antibody was obtained from Sigma-Aldrich. Anti-TEF (610922) was purchased from BD Transduction Laboratories. Antibodies for ETAR (SC33535), Gαq/11 (SC392), and CTGF (SC14939) were from Santa Cruz Biotechnology. Anti-β-actin (A00702) was purchased from GenScript. Antibody for GS was from Abcam. All antibodies were used properly following manufacturer-recommended dilutions in immunoblotting and immunofluorescence staining analysis.
Plasmids
Lats2 wild-type, Lats2 K/R-mutant, myc-Rho-L63, and Rho-GDI-GFP plasmids were described in ref. 39. The shRNA oligos for TAZ (CCGGGCGTTCTTGTGACAGATTATACTCGAGTA-TAATCTGTCACAAGAACGCTTTTTG) were annealed following the protocol from Addgene and cloned into pMKO.1 vector. YAP shRNA oligos (CCGGGCCACCAAGCTAGATAAAGAAC-TCGAGTTCTTTATCTAGCTTGGTGGCTTTTTG) was purchased from Sigma-Aldrich.
Cell culture and transfection
HCT116 cells were cultured in McCoy’s 5A medium (Sigma) supplemented with 10% FBS (Gibco) and 50 μg/mL penicillin/streptomycin. Used in our current study, HCT116, HEK293T, and ZR-75–30 cells were obtained from the ATCC in 2015, where they were characterized by Mycoplasma detection, isozyme detection, DNA fingerprinting, and cell vitality detection. SW480, HT29, SW620, SKBR3, MDA-MB-231, MCF7, and Du145 cells were purchased from cell bank of Chinese Academy of Sciences (Beijing, China) in 2014, where these cells were characterized by Mycoplasma detection, isozyme detection, DNA fingerprinting, and cell vitality detection. These cells were maintained in DMEM medium (Invitrogen) supplemented with 10% FBS (Gibco) and 50 mg/mL penicillin/streptomycin. PC3 cells were purchased in 2015 from Cell Center of the Institutes of Biomedical Sciences (IBS), Fudan University (Shanghai, China), where these cells were characterized by Mycoplasma detection, isozyme detection, DNA fingerprinting, and cell vitality detection. PC3 cells were cultured in F12 medium (Sigma) supplemented with 10% FBS (Gibco) and 50 μg/mL penicillin/streptomycin. All cell lines were cultured at 37°C with 5% CO2 and immediately stored in liquid nitrogen till use. A new frozen vial of the same batch of cells was restarted every 2 to 3 months. For serum starvation, cells were incubated in DMEM medium, McCoy’s 5A medium, or F12 medium, respectively. Cells were transfected with plasmid DNA using Lipofectamin2000 reagent (Invitrogen). siRNAs were transfected into cells following the manufacturer’s instructions for Lipofectamine RNAiMAX reagent (Invitrogen).
To generate stable YAP/TAZ knocking-down cells, pMKO-shScramble and pMKO-shTAZ retroviruses, which were produced in HEK293T cells using VSVG and GAG as packaging plasmids, were used to infect HCT116 cells. pLKO-shYAP lentivirus was produced in HEK293T cells using psPAX2 and pMD2.G as packaging plasmids. Stable pools were selected with 1 μg/mL puro-mycin (Amresco) for 5 days. These cells were subsequently infected with the pQCXIH-ETAR retroviruses and screened with 50 μg/mL hygromycin B (Amresco) for 5 days. The cells that are resistant to both puromycin and hygromycin B were maintained and used in further experiments.
Immunoblotting
Cells were lysed in SDS sample buffer and denatured by heating on 100° C for 10 minutes. Proteins were separated on 8% to 10% Bis-Tris polyacrylamide gels. Western blot images were captured by typhoon FLA 9500 (GE Healthcare) or Image Quant LAS4000 mini (GE Healthcare).
The phos-tag reagents were purchased from Wako Chemicals. The preparation of phos-tag gels containing phos-tag and MnCl2 has been done properly following the manufacturer’s instructions. On the phos-tag gels, YAP/TAZ protein can be separated into multiple bands directly dependent on the phosphorylation status.
Immunofluorescence staining
HCT116 cells were seeded in 6-well plates. Cells were washed with PBS and fixed with 4% paraformaldehyde for 30 minutes at room temperature, washed by PBS, followed by permeabilization with 0.1% Triton X-100 for 5 minutes at 4°C. Then, cells were incubated with primary antibodies overnight at 4°C after blocking with 0.5% BSA for 30 minutes at room temperature. Then, cells were washed with PBS for three times per 5 minutes. Alexa Fluor-labeled secondary antibodies were used to incubate with cells for 2 hours at room temperature. Nucleus and actin filaments were stained with DAPI and Tetramethylrhodamine B isothiocyanate-conjugated Phalloidin, respectively. Photos were taken by using an Olympus microscopy with appropriate fluorescence filters.
Kinase assay
LATS1 kinase assay was described elsewhere (39). Briefly, HCT116 cells were lysed with NP40 lysis buffer (150 mmol/L NaCl, 50 mmol/L Tris at pH 7.5, and 0.5% NP40). LATS1 was immunoprecipitated by its specific antibody. The immunoprecipitates were washed three times with lysis buffer, once with wash buffer (40 mmol/L HEPES, 200 mmol/L NaCl), and once with kinase assay buffer (30 mmol/L HEPES, 50 mmol/L potassium acetate, 5 mmol/L MgCl2). Kinase assay was performed in kinase buffer with 500 μmol/L ATP using recombinant GST-YAP as the substrate. Phosphorylation of YAP (Ser127) was detected by the phospho-Ser127-specific antibody.
RNAi
Oligonucleotides for knocking down of human Gαq, Gα11, Gαs, YAP, TAZ (WWTR1), TEAD1/3/4 and ETAR were synthetized by GenePharma company. siRNAs were transfected into cells according to the manufacturer’s instructions of lipofectamin RNAiMAX reagent (Invitrogen, 904203). A non-targeting scrambled siRNA duplex (5’-CCAUUCCGAUCCUGAUCCG-3’) was used as a negative control. The sequences of all siRNAs used in this study are shown below:
siGNAQ#2: GACACCGAGAATATCCGCTTT
siGNA11#2: GCTCAAGATCCTCTACAAGTA
siGNAS#1: GCTTGCTTAGATGTTCCAAAT
siYAP1#1: CCCAGTTAAATGTTCACCAAT
siYAP1#3: CAGGTGATACTATCAACCAAA
siWWTR1#2: CAGCCAAATCTCGTGATGAAT
siWWTR1#3: CCAGGAACAAACGTTGACTTA
siTEAD1/3/4 #1: ATGATCAACTTCATCCACAAG
siTEAD1/3/4 #2: GATCAACTTCATCCACAAGCT
siETAR#1: CATGGATTACATCGGTATTAA
siETAR#2: ACAATGACTTTGGCGTATTTC
RNA isolation, reverse transcription, and real-time PCR
RNA was isolated using TRIzol reagent following the manufacturer’s instructions (Invitrogen). With oligo-dT primers, 3 to 5 μg of RNA was used to reverse transcribed to complementary DNA (cDNA). Diluted cDNA was then proceeded for real-time PCR with gene-specific primers in the presence of SYBR Premix Ex Taq (TaKaRa) by 7500 real-time PCR system (Applied Biosystems). Using β-actin as a housekeeping control. Relative abundance of mRNA that was performed in triplicate was determined. The primer pairs used in this study were below:
β-actin-F: GCCGACAGGATGCAGAAGGAGATCA
β-actin-R: AAGCATTTGCGGTGGACGATGGA
CTGF-F: CCAATGACAACGCCTCCTG
CTGF-R: TGGTGCAGCCAGAAAGCTC
CYR61-F: AGCCTCGCATCCTATACAACC
CYR61-R: TTCTTTCACAAGGCGGCACTC
ANKRD1-F: CACTTCTAGCCCACCCTGTGA
ANKRD1-R: CCACAGGTTCCGTAATGATTT
INHBA-F: TTGCCAACCTCAAATCGTGCT
INHBA-R: CCCACACTCCTCCACGATCAT
TAZ-F: GGCTGGGAGATGACCTTCAC
TAZ-R: AGGCACTGGTGTGGAACTGAC
YAP-F: CCACAGGCAATGCGGAATATC
YAP-R: GGTGCCACTGTTAAGGAAAGG
EDNRA-F: AACTATTGCCCACAGCAGACT
EDNRA-R: CCCAGCCAGCAGCTTAAATACA
Cell migration assay
BD Falcon Cell culture chambers with 8.0-μm pores for 24-well plates were used in this assay. Serum-starved HCT116 cells were stimulated with ET-1 or vehicle for 4 hours. Cells were then suspended in serum-free DMEM and seeded into chambers at 50,000 cells per well. The bottom chamber was filled with DMEM medium supplemented with 5% FBS. After seeding for 24 hours, the membranes were fixed in 4% paraformaldehyde and stained with 0.05% crystal violet. Photos were taken by using an Olympus microscope and migrated cells were counted and quantified.
Cell proliferation
Performed in triplicate, 1 × 105 of cells were seeded into 6-well plates and cultured in McCoy’s 5A medium supplemented with 5% FBS and 50 μg/mL penicillin/streptomycin. The cell culture medium was renewed every day. Cells were trypsinized and counted daily by using Countstar IC1000.
Xenograft
Male BALB/c nude mice at the age of 4 to 5 weeks were purchased from SLAC Laboratory. The HCT116 stable cells were suspended in PBS and injected subcutaneously at 9 × 106 cells per mouse. Mice were sacrificed 3 weeks after injection. Weight and volume of tumors were measured. The animal experiments were performed according to the approved protocols by Animal Welfare Committee of Shanghai Medical College, Fudan University.
Results
ET-1 activates YAP/TAZ in colon cancer cell lines
To explore the role of YAP/TAZ in mediating ET-1 signaling, we first tested whether ET-1 could activate YAP/TAZ in multiple cancer cell lines. ET-1 treatment in HCT116, a colon cancer cell line, led to the dephosphorylation of YAP/TAZ in a time- and dose-dependent manner (Fig. 1A and B), as determined by immunoblotting with a phospho-YAP antibody (S127) and increased migration of YAP/TAZ on phos-tag gels. Similar results were observed in SW480 and HT29 cells, two other colon cancer cell lines (Fig. 1C; Supplementary Fig. S1A). ET-1 also increased TAZ protein levels because the stability of TAZ is potently regulated by its phosphorylation. However, ET-1 did not affect YAP/ TAZ phosphorylation in SW620 colon cancer cells (Supplementary Fig. S1B). Interestingly, the mRNA expression of ETAR in SW620 is much lower than that in other colon cell lines (Supplementary Fig. S1C). These data indicate that ET-1 induces YAP/TAZ dephosphorylation and this response depends on ETAR expression. In contrast to colon cancer cells, we observed inconspicuous change in YAP/TAZ phosphorylation in response to ET-1 treatment in several other cancer cell lines, including prostate cancer and breast cancer cells (Supplementary Fig. S2), suggesting that ET-1 regulation on YAP/TAZ activity is cell type specific.
Figure 1.
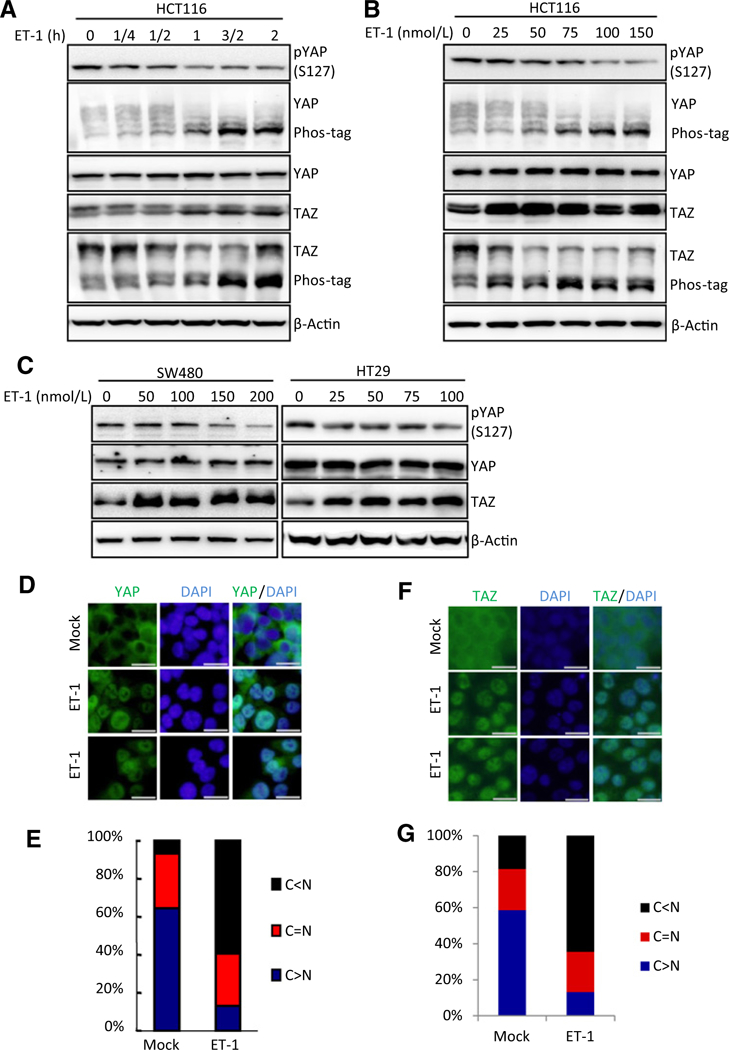
ET-1 activates YAP/TAZ in colon cancer cell lines. A and B, ET-1 activates YAP/TAZ in HCT116 cells. ET-1 induces the dephosphorylation of YAP/TAZ in time- and dose- dependent manners. HCT116 cells were serum starved for 14 hours and stimulated with ET-1 for the duration (A) or dose (B) indicated. Immunoblotting was performed with indicated antibodies. Phos-tag gels were used for assessment of YAP and TAZ phosphorylation status. C, ET-1 activates YAP/TAZ in SW480 and HT29 cells. The two cell lines were serum starved for 14 hours and were treated with ET-1 for the dose indicated for 1 hour. Immunoblotting was performed with indicated antibodies. D, ET-1 promotes YAP translocation from cytoplasm to nucleus. HCT116 cells were serum starved for 14 hours and stimulated with 100 nmol/L of ET-1 for 1 hour. Endogenous YAP(green) and nuclei (blue) were stained with specific antibody and DAPI, respectively; scale bar, 20 μm. E, Quantifications of YAP subcellular localization from at least 100 randomly selected cells in D. C, cytoplasm; N, nucleus. F, ET-1 induces TAZ translocation from cytoplasm to nucleus. HCT116 cells were serum starved for 14 hours and stimulated with 100 nmol/L of ET-1 for 1 hour. HCT116 cells were serum-starved for 14 hours and stimulated with 100 nmol/L of ET-1 for 1 hour. Endogenous TAZ (green) and nuclei (blue) were stained with specific antibody and DAPI, respectively; scale bar, 20 μm. G, Quantifications of TAZ subcellular localization from at least 100 randomly selected cells in F. C, cytoplasm; N, nucleus.
Dephosphorylation of YAP/TAZ leads to their translocation from cytoplasm to nucleus to activate target genes expression. We thus detected the localization of YAP and TAZ in response to ET-1 treatment. Consistent with YAP/TAZ dephosphorylation, ET-1 significantly induced YAP and TAZ translocation from cytoplasm to nucleus (Fig. 1D–G). Collectively, these results show that ET-1 induces activation of YAP/TAZ in ETAR-expressing colon cancer cells.
YAP/TAZ and TEADs mediate the physiologic functions of ET-1 in target gene induction
Previous studies have revealed that the activated YAP/TAZbind to the TEAD transcription factors to promote expression of target genes (31, 32). We tested the expression of YAP/TAZ target genes in response to ET-1. ET-1 treatment significantly induced the expression of CTGF, CYR61, ANKRD1, and INHBA (Fig. 2A). Knocking down of both YAP and TAZ abolished the ET-1 -induced expression of these genes (Fig. 2B and C), indicating ET-1’s effect on these genes was mediated by YAP/TAZ. Similarly, knocking down of the YAP/TAZ target transcription factor TEAD1/3/4 also abolished CTGF expression upon ET-1 stimulation (Fig. 2D), further confirming the critical role of YAP/TAZ-TEAD transcription activator in the induction of ET-1-inducible genes.
Figure 2.
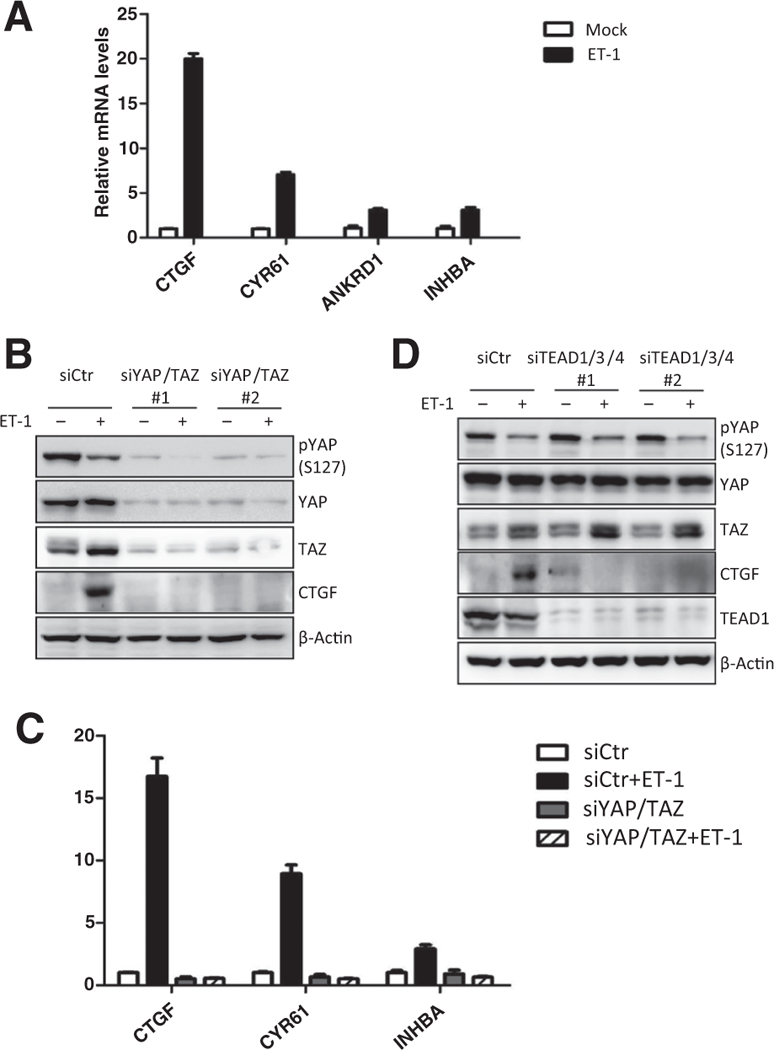
YAP/TAZ and TEAD mediate the physiologic functions of ET-1 in target gene induction. A, ET-1 induces expression of YAP/TAZ target genes. HCT116 cells were serum starved for 14 hours and stimulated with 100 nmol/L of ET-1 for 3 hours. The mRNA levels of CTGF, CYR61, ANKRD1,and INHBA were quantified by real-time PCR. Data, mean ± SD. B, YAP/TAZ are required for ET-1 to induce downstream gene expression. HCT116 cells were transfected with control or YAP/TAZ siRNAs. After 48 hours, HCT116 cells were serum starved for 14 hours and stimulated with 100 nmol/L of ET-1. Protein levels of CTGF and the knockdown efficiency of YAP/TAZ were determined by immunoblotting. C, YAP/TAZ are required for ET-1 to induce downstream gene expression. HCT116 cells were transfected with control or YAP/TAZ siRNAs. After 48 hours, HCT116 cells were serum starved for 14 hours and stimulated with 100 nmol/L of ET-1 for 3 hours. The mRNA levels of indicated target genes were measured by real-time PCR. Data, mean ± SD. D, TEAD is indispensable for ET-1 to induce target gene expression. HCT116 cells were transfected with control or TEAD1/3/4 siRNAs. Serum-starved cells were treated with 100 nmol/L of ET-1. Protein levels of CTGF and the knockdown efficiency of TEAD1 were determined by immunoblotting.
ET-1 activates YAP/TAZ through ETAR and Gαq/11
ET-1 signals through ETAR to promote cell proliferation, migration, and survival. We have found that the response of YAP/TAZ to ET-1 correlates with ETAR expression level (Supplementary Fig. S1C). To test whether ET-1 activates YAP/TAZ depends on ETAR, we used siRNA to knock down ETAR and found that ET-1 treatment could not induce YAP/TAZ dephosphorylation in the ETAR knockdown cells (Fig. 3A). We also pretreated cell with ETAR selective antagonist BQ123 (9, 10, 40) and observed similar effect that ETAR inhibition prevented YAP/TAZ activation by ET-1 (Fig. 3B). Moreover, BQ123 also blocked YAP nuclear translocation in response to ET-1 (Fig. 3C and D), demonstrating that ET-1 activates YAP/TAZ through ETAR.
Figure 3.
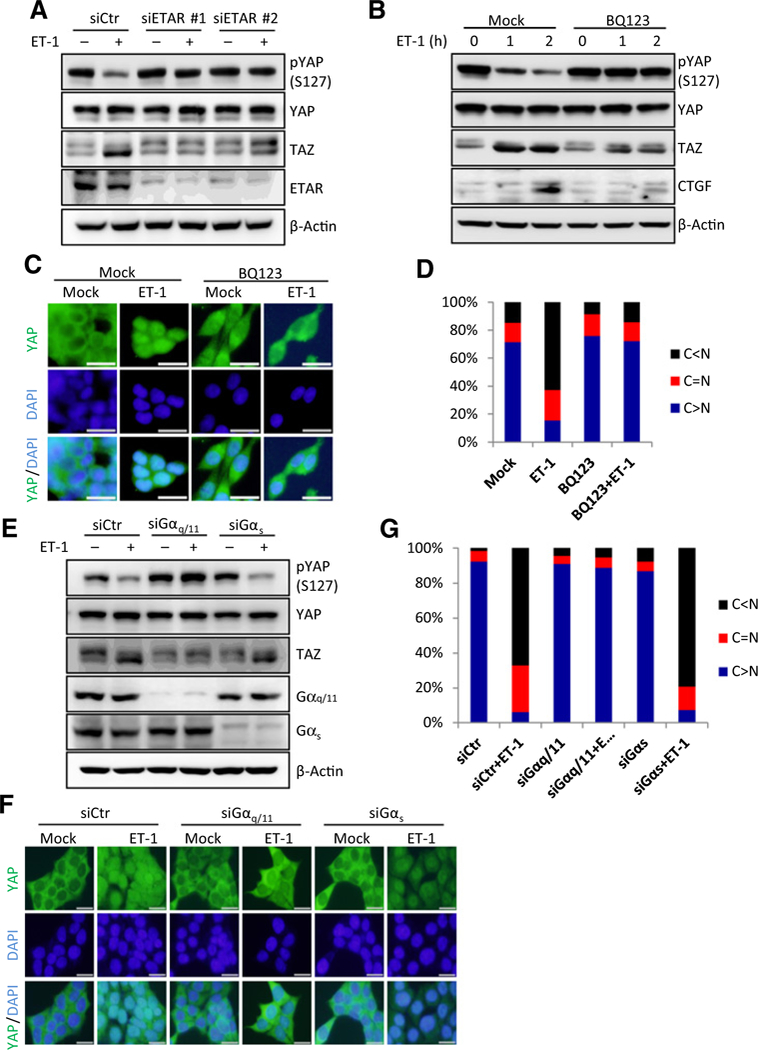
ET-1 stimulation activates YAP/TAZ through ETAR and Gαq/11. A, ET-1 activates YAP/TAZ through ETAR. HCT116 cells were transfected with control or ETAR siRNAs. Serum-starved cells were treated with 100 nmol/L of ET-1 for 1 hour. YAP/TAZ and knockdown efficiency of ETAR were determined by immunoblotting. B, BQ123 blocks ET-1-induced YAP/TAZ activation. Serum-starved HCT116 cells were pretreated with 2 μmol/L of BQ123 for 4 hours, and then were treated with 100 nmol/L of ET-1 for indicated time. Protein levels of CTGF and YAP/TAZ were determined by immunoblotting. C, BQ123 blocks YAP nuclear localization induced by ET-1 stimulation. HCT116 cells were pretreated with 2 μmol/L of BQ123 for 4 hours. Serum-starved cells were treated with 100 nmol/L of ET-1 for 1 hour. Immunofluorescence staining was performed for endogenous YAP (green) and nuclei (DAPI, blue); scale bar, 20 μm. D, Quantifications of YAP subcellular localization from at least 100 randomly selected cells in C. C, cytoplasm; N, nucleus. E, ET-1 signals through Gαq/11 to induce YAP/TAZ activation. HCT116 cells were transiently transfected with control, Gαq/11, or Gαs siRNAs. Serum-starved cells were treated with 100 nmol/L of ET-1 for 1 hour. YAP/TAZ and knockdown efficiency of Gαq/11 and Gαs were determined by immunoblotting. F, ET-1 stimulation induces YAP/TAZ nuclear localization through Gαq/11. HCT116 cells were transiently transfected with control, Gαq/11 or Gαs siRNAs. Serum-starved cells were treated with 100 nmol/L of ET-1. Endogenous YAP (green) and nuclei (blue) were stained with specific antibody and DAPI, respectively; scale bar, 20 μm. G, Quantifications of YAP subcellular localization from at least 100 randomly selected cells in F. C, cytoplasm; N, nucleus.
ET-1 activates several trimeric G proteins, including Gαq/11 and Gαs, to initiate intracellular signaling pathways (1). To determine which Ga proteins are involved in YAP/TAZ regulation, Gαq/11 or Gαs was silenced by siRNAs in HCT116 cells. Knocking down of Gαq/11 strongly blocked YAP/TAZ dephosphorylation in response to ET-1, while knockdown of Gαs had little effect on ET-1 -induced YAP/TAZ dephosphorylation (Fig. 3E). Consistently, ET-1 induced YAP nuclear accumulation in cells transfected with control or Gαs siRNA, but not Gαq/11 siRNA (Fig. 3F and G). Taken together, we conclude that ET-1 signals through Gαq/11 to activate YAP/TAZ.
ET-1 acts through LATS, Rho/ROCK, and actin cytoskeleton to activate YAP/TAZ
LATS1/2 are the major responsible upstream kinases for YAP/ TAZ phosphorylation and nuclear-cytoplasmic shuttling. To investigate whether LATS1/2 is involved in YAP/TAZ regulation by ET-1, we measured the kinase activity of LATS1 immuno-purified from control or ET-1-treated cells. As shown in Fig. 4A, ET-1 treatment markedly reduced LATS1 kinase activity, which was coincident with YAP/TAZ dephosphorylation. Consistently, overexpression of wild type LATS2, but not the kinase-dead mutant ofLATS2, completely blocked the effect of ET-1 on YAP/TAZ phosphorylation (Fig. 4B). These data suggest that ET-1 decreases YAP/TAZ phosphorylation mainly by inhibiting the LATS kinase activity.
Figure 4.
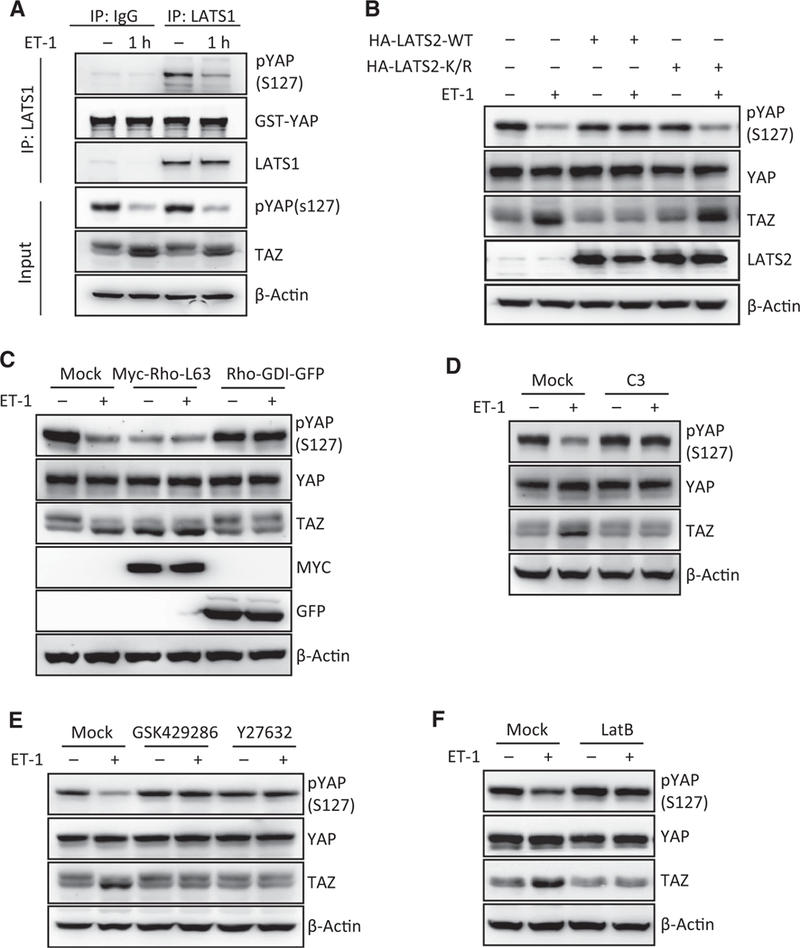
ET-1 acts through LATS, Rho/ROCK, and actin cytoskeleton to activate YAP/TAZ. A, ET-1 treatment decreases LATS1 activity. HCT116 cells were serum-starved for 14 hours and stimulated with 100 nmol/L of ET-1 for 1 hour. LATS1 was immunoprecipitated and in vitro kinase assay was performed using GST-YAP as a substrate. Phosphorylation of GST-YAP by LATS1 was determined by phospho-YAP antibody. B, Ectopic expression of LATS2 blocks ET-1-induced YAP/TAZ activation. HCT116 cells were transiently transfected with control, LATS2 wild-type (WT), or kinase dead mutant (K/R). Serum-starved cells were treated with 100 nmol/L of ET-1 for 1 hour. Phosphorylationand protein levels of YAP/TAZ were determined by immunoblotting. C, Rho GTPase is involved in YAP/TAZ activation by ET-1. HCT116 cells were transiently transfected with control, Myc-Rho-L63, or Rho-GDI-GFP mutant. Serum-starved cells were treated with 100 nmol/L of ET-1 for 1 hour. Phosphorylation and protein levels of YAP/TAZ were determined by immunoblotting. D, Inactivation of Rho GTPase prevents YAP/TAZ dephosphorylation by ET-1 stimulation. Serum-starved HCT116 cells were pretreated with C3 for 4 hours, followed by treatment with 100 nmol/L of ET-1 for 1 hour. Phosphorylation and protein levels of YAP/TAZ were determined by immunoblotting. E, ROCK is required for ETAR-induced YAP/TAZ activation. Serum-starved HCT116 cells were pretreated with GSK429286 (0.5 μmol/L) or Y27632 (1 μmol/L) for 4 hours, followed by treatment with 100 nmol/L of ET-1 for 1 hour. Phosphorylation and protein levels of YAP/TAZ were determined by immunoblotting. F, Disruption of actin cytoskeleton blocks ET-1-induced YAP/TAZ activation. Serum-starved HCT116 cells were pretreated with latrunculin B (Lat B; 1 mg/mL) for 20 minutes, followed by treatment with 100 nmol/L of ET-1 for 1 hour. Phosphorylation and protein levels of YAP/TAZ were determined by immunoblotting.
Rho GTPase and actin cytoskeleton organization have been demonstrated to be key mediators of mechanical cues, GPCR ligands, and cell attachment in regulating the Hippo pathway (41–43). We therefore tested whether Rho GTPase is involved in YAP/TAZ activation by ET-1. Consistent with previous findings, overexpression of Rho-L63, a constitutively active mutant of Rho, significantly activated YAP/TAZ in the absence of ET-1 treatment (Fig. 4C). On the other hand, expression of Rho GDP dissociation inhibitor (Rho-GDI), which binds to Rho GTPase and inhibits GTPase cycling, completely blocked ET-1-induced YAP/TAZ dephosphorylation (Fig. 4C). Similarly, pre-treatment of cells with botulinum toxin C3, a specific inhibitor of Rho GTPases, also blocked YAP/TAZ activation by ET-1 (Fig. 4D). We also found that pretreatment with GSK429286 and Y27632, two inhibitors of Rho-associated protein kinase (ROCK), also strongly suppressed ETAR-induced YAP/TAZ dephosphorylation (Fig. 4E) and nuclear translocation (Supplementary Fig. S3A).
The Rho/ROCK pathway potently modulates actin cytoskeleton dynamics, particularly stress fiber formation, in response to various GPCR agonists (39). We therefore examined whether cytoskeletal reorganization contributes to YAP/TAZ activation in response to ET-1 stimulation. Treatment with latrunculin B (Lat B), an F-actin destabilizing reagent, blocked ET-1-induced YAP/TAZ dephosphorylation (Fig. 4F) and nuclear accumulation (Supplementary Fig. S3B). Taken together, the above data support a model that ET-1 acts through Rho/ROCK and actin cytoskeleton to regulate the Hippo pathway, leading to YAP/TAZ activation.
ET-1 promotes cell migration, proliferation, and tumor growth through YAP/TAZ
It has been reported that ETAR plays a role in the growth and progression of colorectal cancer (18, 20, 44). YAP/TAZ are also known to promote cell proliferation and migration (39, 45). Because YAP/TAZ activities are significantly elevated upon ET-1 treatment in colon cancer cells, we investigated the role of YAP/TAZ in ET-1-regulated cellular functions. Consistent with previously reported observations (3, 44), ET-1 treatment promoted the migration of HCT116 cells as determined by the Transwell cell migration assay. Notably, this effect was largely abolished by YAP/TAZ knockdown (Fig. 5A and B), suggesting an essential role of YAP/TAZ in mediating the ET-1-induced colon cancer cell migration. We then tested the role of ETAR-YAP/TAZ signaling in cell proliferation. Several stable cell lines, including ETAR overexpression, YAP/TAZ knockdown, and combination of ETAR overexpression and YAP/TAZ knockdown, were established in HCT116 cells (Fig. 5C). Our results showed that ectopic expression ofETAR enhanced cell proliferation, whereas knockdown of YAP/TAZ significantly blocked the cell growth-promoting effect of ETAR (Fig. 5D). These observations indicate that YAP/TAZ play a critical role in ETAR-induced cell growth.
Figure 5.
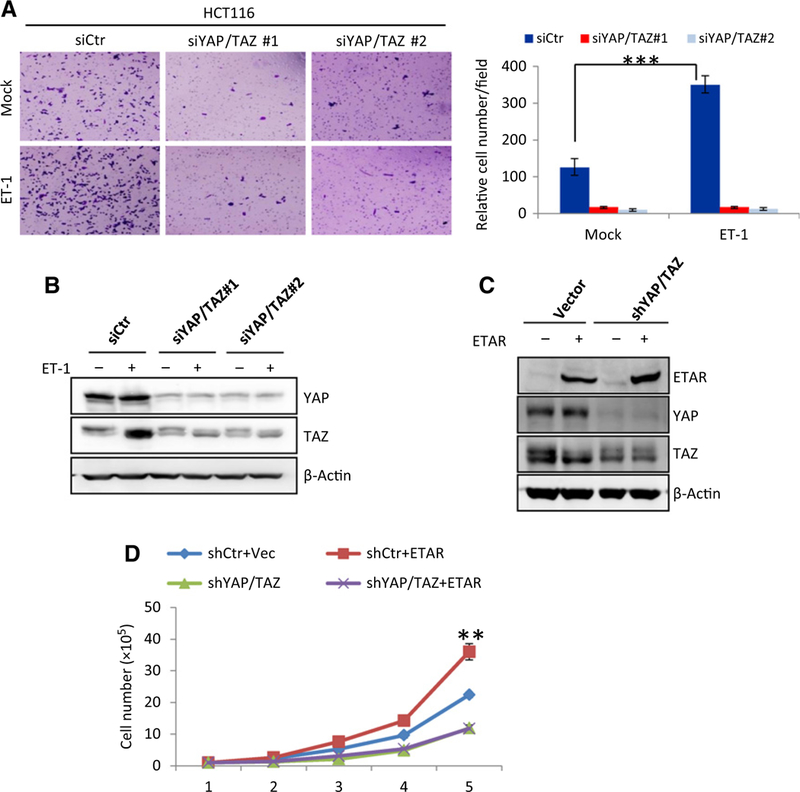
ETAR promotes cell migration and cell proliferation through YAP/TAZ. A, Loss of YAP/TAZ impairs ETAR-induced cell migration. HCT116 cells were transfected with siRNAs targeting YAP and TAZ. After 48 hours, cells were serum starved for 14 hours and stimulated with ET-1 for 4 hours. Cell migration was determined by Transwell cell migration assay as described in Materials and Methods. Cells were stained by crystal violet (left) and quantified (right). The P value of <0.001 was calculated by Student t test and shown. Data, mean ± SD. B, Verification of YAP/TAZ knockdown in HCT116 cells. Cells were transfected and treated in parallel as that in A. Protein levels of YAP/TAZ was determined by immunoblotting. C, Establishment of HCT116 stable cells with ETAR overexpression, YAP/TAZ knockdown, or the combination of both. Expressions of ETAR and YAP/TAZ were determined by immunoblotting. D, YAP/TAZ mediate ETAR-induced cell proliferation. Growth of HCT116 stable cells described in C was determined by cell growth assay. Stable cells were cultured in triplicate in McCoy’s 5A medium supplemented with 5% FBS. Cells were counted daily by Countstar IC1000. The asterisks labeled at day 5 indicate P value was 0.006 and 0.004 between groups of shCtr+ETAR and shCtr+Vec or shYAP/TAZ+ETAR, respectively. The P value of < 0.01 indicates significant difference. The P value was calculated by paired Student t test and is shown. Error bars represent cell numbers ± SD from triplicate experiments.
To gain more pathologic insight of ET-1 signaling in colon tumorigenesis, we analyzed the expression level of ETAR in various cancer types in The Cancer Genome Atlas (TCGA) database (Supplementary Fig. S4A). Renal clear cell carcinoma, thymoma, and esophageal carcinomas show higherexpression levels of ETAR than their corresponding normal tissues. Among the tissues analyzed, colon cancer samples show a significant upregulation of ETAR expression compared with normal colon tissues (Fig. 6A). The TCGA data are consistent with our results that most colon cancer cell lines express high levels of ETAR mRNA (Supplementary Fig. S1C). In contrast, no significance difference of ETAR expression is observed between breast cancer or liver cancer samples and their corresponding normal tissue samples, while prostate cancer samples show a decrease of ETAR expression compared with normal tissues (Fig. 6A). Thus, the expression of ETAR appears to be cancer type dependent. To further elucidate the function of YAP/TAZ in mediating ET-1 signaling in tumor growth, we used a xenograft model to investigate whether YAP/TAZ is required for ET-1-induced colon tumor growth. Injection of HCT116 cells with ETAR overexpression into nude mice generated tumors that were bigger than tumors from the control cell injection, indicating a tumor-promoting function of ETAR (Fig. 6B and C). Notably, knocking down of YAP/TAZ in ETAR-overexpressed cells dramatically suppressed tumor growth (Fig. 6B and C). In summary, our observations support a model that YAP/TAZ act as downstream targets of ET-1 and play an important role in mediating the tumor-promoting function of ET-1.
Figure 6.
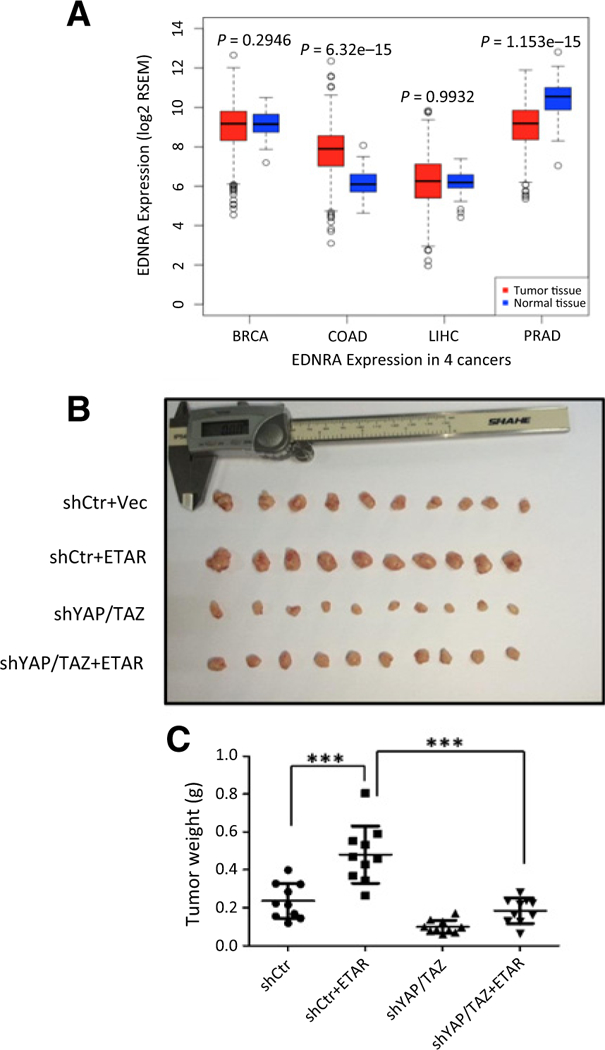
ETAR promotes xenograft tumor growth through YAP/TAZ. A, ETAR is overexpressed in colon carcinoma samples. BRCA, breast carcinoma; COAD, colon adenocarcinoma; LIHC, liver hepatocellular carcinoma; PRAD, prostate adenocarcinoma. Log2 mRNA expression from RNA Seq V2 RSEM was downloaded from TCGA data portal shown by log2 RSEM. The P value was calculated by Wilcoxon test. The corresponding P value is shown. B, YAP/TAZ are required for ETAR to promote tumor growth in a xenograft mouse model. Xenograft was performed in male nude mice using cells described in Fig. 5C. Three weeks after injection, mice were sacrificed and tumors were separated and photographed. C, Tumor weight was measured for indicated groups. The P value of <0.001 indicates significant difference. P value was calculated by Student t test and is shown.
Discussion
Previous studies have reported a role of ET-1 in the growth and progression of a wide rage of cancers, such as prostate, ovarian, renal, pulmonary, colorectal, cervical, breast carcinoma, brain tumors, melanoma, Kaposi sarcoma, as well as tumor metastases (7, 9, 46–48). ET-1 executes its biological functions mainly through ETAR, which has been proved to promote tumor growth and progression. In this study, we demonstrate the Hippo path-way serves as an important downstream module of ET-1 signaling, particularly in cell growth and tumorigenesis. We also uncover a mechanism of ET-1 in promoting colon cancer through activating the YAP/TAZ transcription coactivators. Activated ETAR couples to Gαq/11 to trigger the activation of Rho GTPase and ROCK kinase, which modulate actin cytoskeleton to inhibit LATS1/2 kinases, resulting in YAP/TAZ dephosphorylation and activation. Activated YAP/TAZ induce expressions of downstream target genes and contribute to cell proliferation and migration. ETAR overexpression, which is elevated in colon cancer samples (Fig. 6A), promotes tumor growth in vivo, whereas YAP/TAZ knockdown largely suppresses this effect. Our study reinforces the notion that ET-1-ETAR-YAP/TAZ axis is important for colon cancer, and thus indicating YAP/TAZ as potential therapeutic targets for cancers with high ETAR expression.
Endothelin was first identified as a vasconstrictor for its elevated expression in several forms of pulmonary vascular diseases (49, 50). It is involved in the pathogeneis of hypertension and vascular diseases. Endothelin receptor antagonism has emerged as an important therapeutic strategy in pulmonary arterial hypertension (PAH). Of all endothelin peptides, ET-1 appears to play the most prominent role in vascular control. Intriguingly, YAP/TAZ are also implicated in other pathophy-sological conditions, such as tissue regeneration after injury. It has been demonstrated that YAP/TAZ are activated in vascular smooth muscle cells (VSMC) by the thromboxane A2 receptor, which is important for VSMC proliferation and migration (51). Other groups have discovered YAP/TAZ function as a bridge between dysregulation of vascular stiffness and cellular metabolism (52). On the basis of our findings, one may speculate that the YAP/TAZ signaling axis also takes part in ET-1 functions in vascular physiology and diseases.
The importance of the ET axis in cancer cell growth and migration naturally leads to a notion of ETAR antagonism being a potential antitumor therapy. Several ETAR antagonists, such as atrasentan and ZD4054, have been broadly investigated in clinical trials, and the results support a potential role of ET-1 signaling in promoting cancer. We propose that high expression levels of ETAR function, at least in part, through activating YAP/ TAZ to promote tumor growth. Thus, our findings suggest that targeting the Hippo pathway components, particularly YAP/TAZ, might be a possible approach for treating high ETAR-expressing cancer. Notably, a small molecule verteporfin (trade name Visudyne) has been shown to efficiently inhibit YAP-dependent tumor growth (53, 54). Thus, verteporfin could be tested for colon cancers with high expression of ETAR as a proof of concept. As ETAR antagonists have shown varied effects in a number of cancer types, further knowledge of the mechanisms and molecular aspects of ETAR signaling in cell growth promotion is essential to understand the therapeutic value of endothelin receptor antagonism.
Supplementary Material
Acknowledgments
We thank Professor Qun-Ying Lei, Dan Ye, and the members of the Fudan MCB laboratory for support throughout the course of this study.
Grant Support
This work was supported by the National Natural Science Foundation of China (no. 31570784 to H.X. Yuan; no. 81372198 and 81522033 to D. Ye). Y. Xiong is supported by grants from NIH (GM067113, CA163834). K.L. Guan is supported by grants from NIH (CA196878 and GM51586).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
K.-L. Guanhas ownership interest (includingpatents) inVivace. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Can J Physiol Pharmacol 2008;86:485–98. [DOI] [PubMed] [Google Scholar]
- 2.Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation 2000;102:2434–40. [DOI] [PubMed] [Google Scholar]
- 3.Wu MH, Chen LM, Hsu HH, Lin JA, Lin YM, Tsai FJ, et al. Endothelin-1 enhances cell migration through COX-2 up-regulation in human chondrosarcoma. Biochim Biophys Acta 2013;1830:3355–64. [DOI] [PubMed] [Google Scholar]
- 4.Grant K, Loizidou M, Taylor l. Endothelin-1: a multifunctional molecule in cancer. BrJ Cancer 2003;88:163–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bagnato A, Spinella F. Emerging role of endothelin-1 in tumor angiogen- esis. Trends Endocrinol Metab 2003;14:44–50. [DOI] [PubMed] [Google Scholar]
- 6.Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: emerging role in cancer. Nat Rev Cancer 2003;3:110–6. [DOI] [PubMed] [Google Scholar]
- 7.Bagnato A, Natali PG. Endothelin receptors as novel targets in tumor therapy. J Transl Med 2004;2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teoh JP, Park KM, Wang Y, Hu Q, Kim S, Wu G, et al. Endothelin-1/endothelin A receptor-mediated biased signaling is a new player in modulating human ovarian cancer cell tumorigenesis. Cell Signal 2014;26: 2885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosano L, Varmi M, Salani D, Di Castro V, Spinella F, Natali PG, et al. Endothelin-1 induces tumor proteinase activation and invasiveness of ovarian carcinoma cells. Cancer Res 2001;61:8340–6. [PubMed] [Google Scholar]
- 10.Rosano L, Spinella F, Di Castro V, Nicotra MR, Albini A, Natali PG, et al. Endothelin receptor blockade inhibits molecular effectors of Kaposi’s sarcoma cell invasion and tumor growth in vivo. Am J Pathol 2003;163:753–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jimeno A, Carducci M. Atrasentan: a novel and rationally designed therapeutic alternative in the management of cancer. Expert Rev Anticancer Ther 2005;5:419–27. [DOI] [PubMed] [Google Scholar]
- 12.Carducci MA, Saad F, Abrahamsson PA, Dearnaley DP, Schulman CC, North SA, et al. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer 2007;110:1959–66. [DOI] [PubMed] [Google Scholar]
- 13.Nelson JB, Love W, Chin JL, Saad F, Schulman CC, Sleep DJ, et al. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer 2008;113:2478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren R, Liu G. ZD4054: a specific endothelin A receptor antagonist with promising activity in metastatic castration-resistant prostate cancer. Expert Opin Investig Drugs 2008;17:1237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fizazi K, Higano CS, Nelson JB, Gleave M, Miller K, Morris T, et al. Phase III, randomized, placebo-controlled study of docetaxel in combination with zibotentan in patients with metastatic castration-resistant prostate cancer. J Clin Oncol 2013;31:1740–7. [DOI] [PubMed] [Google Scholar]
- 16.Miller K, Moul JW, Gleave M, Fizazi K, Nelson JB, Morris T, et al. Phase III, randomized, placebo-controlled study of once-daily oral zibotentan (ZD4054) in patients with non-metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis 2013;16:187–92. [DOI] [PubMed] [Google Scholar]
- 17.Lalich M, McNeel DG, Wilding G, Liu G. Endothelin receptor antagonists in cancer therapy. Cancer Invest 2007;25:785–94. [DOI] [PubMed] [Google Scholar]
- 18.Asham E, Shankar A, Loizidou M, Fredericks S, Miller K, Boulos PB, et al. Increased endothelin-1 in colorectal cancer and reduction of tumour growth by ET(A) receptor antagonism. Br J Cancer 2001;85:1759–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shankar A, Loizidou M, Aliev G, Fredericks S, Holt D, Boulos PB, et al. Raised endothelin 1 levels in patients with colorectal liver metastases. Br J Surg 1998;85:502–6. [DOI] [PubMed] [Google Scholar]
- 20.Nie S, Zhou J, Bai F, Jiang B, Chen J, ZhouJ. Role of endothelin A receptor in colon cancer metastasis: in vitro and in vivo evidence. Mol Carcinog 2014;53:E85–91. [DOI] [PubMed] [Google Scholar]
- 21.Ali H, Dashwood M, Dawas K, Loizidou M, Savage F, Taylor I. Endothelin receptor expression in colorectal cancer. J Cardiovasc Pharmacol 2000;36: S69–71. [DOI] [PubMed] [Google Scholar]
- 22.Haque SU, Dashwood MR, Heetun M, Shiwen X, Farooqui N, Ramesh B, et al. Efficacy of the specific endothelin a receptor antagonist zibotentan (ZD4054) in colorectal cancer: a preclinical study. Mol Cancer Thera 2013;12:1556–67. [DOI] [PubMed] [Google Scholar]
- 23.Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015;163:811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev 2013;27:355–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan D Hippo signalinginorgansize control. Genes Dev 2007;21:886–97. [DOI] [PubMed] [Google Scholar]
- 26.Zhao B, Lei QY, Guan KL. The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol 2008; 20:638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 2007;21:2747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev 2010;24:72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007;130:1120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu CY,Zha ZY,Zhou X,Zhang H, Huang W, Zhao D, et al. Thehippotumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem 2010;285: 37159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008;22:1962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem 2009;284:13355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer 2013;13:246–57. [DOI] [PubMed] [Google Scholar]
- 34.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev 2016;30:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clines GA, Mohammad KS, Bao Y, Stephens OW, Suva LJ, Shaughnessy JD Jr., et al. Dickkopf homolog 1 mediates endothelin-1-stimulated newbone formation. Mol Endocrinol 2007;21:486–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Recchia AG, Filice E, Pellegrino D, Dobrina A, Cerra MC, Maggiolini M. Endothelin-1 induces connective tissue growth factor expression in cardiomyocytes. J Mol Cell Cardiol 2009;46:352–9. [DOI] [PubMed] [Google Scholar]
- 37.Weng CM, Yu CC, Kuo ML, Chen BC, Lin CH. Endothelin-1 induces connective tissue growth factor expression in human lung fibroblasts by ETAR-dependent JNK/AP-1 pathway. Biochem Pharmacol 2014;88: 402–11. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez-Vita J, Ruiz-Ortega M, Ruperez M, Esteban V, Sanchez-Lopez E, Plaza JJ, et al. Endothelin-1, via ETA receptor and independently of transforming growth factor-beta, increases the connective tissue growth factor in vascular smooth muscle cells. Circ Res 2005;97: 125–34. [DOI] [PubMed] [Google Scholar]
- 39.Zhou X, Wang S, Wang Z, Feng X, Liu P, Lv XB, et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest 2015;125:2123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peduto Eberl L, Bovey R, Juillerat-Jeanneret L. Endothelin-receptor antagonists are proapoptotic and antiproliferative in human colon cancer cells. Br J Cancer 2003;88:788–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 2012;26:54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep 2014;15:642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179–83. [DOI] [PubMed] [Google Scholar]
- 44.Daher Z, Noel J, Claing A. Endothelin-1 promotes migration of endothelial cells through the activation of ARF6 and the regulation of FAK activity. Cell Signal 2008;20:2256–65. [DOI] [PubMed] [Google Scholar]
- 45.Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res 2008;68:2592–8. [DOI] [PubMed] [Google Scholar]
- 46.Bagnato A, Spinella F, Rosano L. The endothelin axis in cancer: the promise and the challenges of molecularly targeted therapy. Can J Physiol Pharmacol 2008;86:473–84. [DOI] [PubMed] [Google Scholar]
- 47.Grimshaw MJ. Endothelins in breast tumour cell invasion. Cancer Lett 2005;222:129–38. [DOI] [PubMed] [Google Scholar]
- 48.Egidy G, Juillerat-Jeanneret L, Jeannin JF, Korth P, Bosman FT, Pinet F. Modulation of human colontumor-stromal interactions by the endothelin system. Am J Pathol 2000;157:1863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Channick RN, Sitbon O, Barst RJ, Manes A, Rubin LJ. Endothelin receptor antagonists in pulmonary arterial hypertension. J Am Coll Cardiol 2004;43:62S–7S. [DOI] [PubMed] [Google Scholar]
- 50.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. The N Engl J Med 1993;328:1732–9. [DOI] [PubMed] [Google Scholar]
- 51.Feng X, Liu P, Zhou X, Li MT, Li FL, Wang Z, et al. Thromboxane A2 activates YAP/TAZ protein to induce vascular smooth muscle cell proliferation and migration. J Biol Chem 2016;291:18947–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, et al. Vascularstiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 2016;126:3313–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012;26:1300–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michels S, Schmidt-Erfurth U. Photodynamic therapy with verteporfin: a new treatment in ophthalmology. Semin Ophthalmol 2001;16:201–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.