

Abstract
The centromere is located at the primary constriction of condensed chromosomes where it acts as a platform regulating chromosome segregation. The histone H3 variant CENP-A is the foundation for kinetochore formation. CENP-A directs the formation of a highly dynamic molecular neighborhood whose temporal characterization during mitosis remains a challenge due to limitations in available techniques. BioID is a method that exploits a “promiscuous” biotin ligase (BirA118R or BirA*) to identify proteins within close proximity to a fusion protein of interest. As originally described, cells expressing BirA* fusions were exposed to high biotin concentrations for 24 h during which the ligase transferred activated biotin (BioAmp) to other proteins within the immediate vicinity. The protein neighborhood could then be characterized by streptavidin-based purification and mass spectrometry. Here we describe a further development to this technique, allowing CENP-A interactors to be characterized within only a few minutes, in an in vitro reaction in lysed cells whose physiological progression is “frozen.” This approach, termed in vitro BioID (ivBioID), has the potential to study the molecular neighborhood of any structural protein whose interactions change either during the cell cycle or in response to other changes in cell physiology.
INTRODUCTION
The kinetochore is a complex multiprotein machine that directs chromosome segregation at mitosis (Santaguida and Musacchio, 2009; Ng et al., 2013; Westhorpe and Straight, 2013; Cheeseman, 2014; Hinshaw and Harrison, 2018). Kinetochore structure changes dramatically across the cell cycle from a ball of condensed chromatin containing the specialized histone H3 subtype CENP-A during interphase (Brenner et al., 1981; Moroi et al., 1981; Earnshaw et al., 1985; Shang et al., 2013) to a trilaminar plate during mitosis (Roos, 1973). To fully understand kinetochore maturation, we need not only a complete parts list, but also a dynamic map of protein neighborhoods during this remarkable structural transformation.
Following the initial discoveries of centromere proteins using autoantibodies (Moroi et al., 1980; Guldner et al., 1984; Earnshaw et al., 1985), characterization of the kinetochore proteome continued with the isolation of kinetochore subcomplexes using antibodies to CENP-A (Ando et al., 2002) and tandem affinity tags (Cheeseman and Desai, 2005; Foltz et al., 2006; Hori et al., 2008). However, such affinity-purification methods are not well suited to identify either transient low-affinity protein–protein interactions or those that occur in the context of insoluble subcellular structures such as mitotic chromosomes or kinetochores. Traditional affinity-purification methods all begin with solubilization of the target protein, a procedure that can disrupt important protein–protein interactions. This has been overcome, at least in part, by the development of methods such as EMARS (enzyme-mediated activation of radical sources; Kotani et al., 2008) and SPPLAT (selective proteomic proximity labeling assay using tyramide; Rees et al., 2015), allowing in situ proximity-based labeling of proteins in cells.
In EMARS horseradish peroxidase (HRP) is coupled to an antibody, protein ligand, or a fusion protein, for localization purposes at the plasma membrane. The labeling is performed using aryl azide-biotin or aryl azide-fluorescein. When oxidized by HRP in the presence of H2O2, these substrates bind to proteins proximal to the localized HRP. SPPLAT labeling differs only in that biotin-tyramide is used as the substrate.
Neighborhood mapping of intracellular proteins began with in vivo BioID (Roux et al., 2012; Kim and Roux, 2016; Li et al., 2017) in which a promiscuous mutant form of the bacterial biotin ligase BirA (BirAR118G, henceforth referred to as BirA*) was used to transfer biotin to nearby proteins (Choi-Rhee et al., 2004). This method was first used for mapping the neighborhood of lamin A, leading to the identification of a novel nuclear envelope protein (Roux et al., 2012). Subsequently, the method has been applied to many proteins of interest including insoluble, membrane-associated proteins in many eukaryotic cells (Beck et al., 2014; Chan et al., 2014; Firat-Karalar et al., 2014; Chen et al., 2015; Lambert et al., 2015). It has also been applied to whole plant systems (Khan et al., 2018).
APEX (proximity labeling with ascorbate peroxidase) has recently been developed as an alternative to in vivo BioID (Rhee et al., 2013). Initially APEX did not attempt to provide maps of interactors: the ascorbate peroxidase was expressed in a compartment-specific manner. To initiate labeling, cells are incubated with H2O2 and biotin-phenol (a tyramide derivative) so the reaction can occur in the same manner as described for EMARS and SPPLAT. More recently, APEX has been expressed as a fusion protein enabling it to be used in a protein-specific manner to map the neighborhoods of microproteins (Chu et al., 2017) and β-2 adrenergic receptor (Lobingier et al., 2017).
All of these methods harbor both strengths and weaknesses. The original in vivo BioID method required cells to be incubated with 50 µM biotin for 18–24 h, during which time targets are continuously biotinylated. This approach is therefore not compatible with the proteomic study of dynamic processes. In contrast, HRP-based methods offer superior temporal resolution. Cells are typically incubated with 500 µM of substrate for 30 min, before the addition of H2O2, for as little as 1 min. A concern with this approach, however, is that substrate labeling occurs wherever the fusion protein happens to be (i.e., both assembled at its ultimate destination, but also at all stages along the way to that destination following its translation), reducing its spatial resolution. Furthermore, HRP has been reported to be more prone to unfolding and loss of activity than wild-type BirA (Rees et al., 2015).
The caveats with published labeling methods and low copy numbers of kinetochore components relative to potential contaminants, led us to consider alternative approaches to study kinetochore maturation during mitosis. We noted that both wild-type (Fernandez-Suarez et al., 2008) and mutant versions of BirA (Choi-Rhee et al., 2004) had previously been shown to be functional in vitro, albeit in restricted experimental conditions. We therefore explored the possibility that BioID labeling might be achievable in lysed cells. Remarkably, this appears to work extremely well. Our new in vitro method allows essentially instantaneous labeling of protein neighborhoods from the point of view of the cell cycle, because the cells are no longer living and are “frozen” at the point of lysis. To validate the method, we use in vitro BioID (ivBioID) to report initial descriptions of the protein neighborhoods of kinetochore protein CENP-A using a CENP-A:BirA* fusion protein under the control of an inducible promoter. Here we compare changes in the CENP-A neighborhood during interphase and mitosis. We report the surprising finding that several CENP-A chaperones appear to be stably associated with both interphase and mitotic chromatin, an unexpected finding also recently reported by others (Zasadzinska et al., 2018).
RESULTS AND DISCUSSION
To identify proteins of the CENP-A neighborhood, we tagged CENP with BirAR118G (yielding BirA*-CENP-A) and generated a stable HeLa cell line in which BirA*-CENP-A was induced following doxycycline addition. Following a 24 h treatment with doxycycline plus 50 μM added biotin (Figure 1A), BirA*-CENP-A was detected at centromeres in both interphase and mitotic cells (Figure 1B, anti-myc). Labeling with fluorescent streptavidin was consistent with centromeric staining and confirmed that the BirA* was functional (Figure 1B, streptavidin 488). In addition to the anticipated centromeric biotinylation, a diffuse signal was detected throughout the nucleus, nucleoli, and mitochondria (Figure 1B).
FIGURE 1:

Testing in vivo BioID using CENP-A. (A) Flowchart describing how HeLa tet-on cells, stable for the conditional expression of myc-BirA*CENP-A (BirA*–CENP-A), were cultured in media supplemented with doxycycline and 50 µm biotin for 24 h before processing for either microscopy or mass spectrometry. (B) Cells fixed and processed for immunofluorescence. The panels show representative examples of cells in interphase and mitosis probed with streptavidin 488 (green), anti-myc (red), and stained with DAPI (blue). (C) Immunoblot analysis of affinity-purified material from control (parental HeLa cell line) or BirA*–CENP-A expressing cells. Blots were probed with streptavidin 800 IR LI-COR labels. (D–F) HeLa cells were seeded into CLEM dishes and cell culture media was supplemented with doxycycline and 50 μm biotin for 24 h. Cells were fixed, permeabilized, and probed with Alexa Fluor 488 dye–labeled colloidal gold, conjugated to streptavidin (Molecular Probes), and imaged using LM to identify mitotic cells of interest. Cells were embedded in resin, sectioned, and imaged by transmission electron microscopy (TEM). (D) Low-magnification images showing the metaphase plate of mitotic cell chosen for CLEM. Images are DAPI (blue), regions of biotinylation (green), and the same cell reidentified by TEM. (E) Two progressive zooms of the boxed region shown in D, shown as LM merge (left), TEM (middle), and LM-TEM merge (overlay, right). Bottom panels (Zoom 2) show 9× magnifications of the white box in D. Top panels (Zoom 1) are a 3× magnification intermediate of part of the white box in D. Contrast-rich areas are visible corresponding to centromeres. The scale bar shows 100 nm (top) and 20 nm (bottom). (F) Pixel density analysis. Line scans (five to six per kinetochore—indicated in panel Fi, top) were taken through kinetochores, originating in the centromere and terminating in the cytoplasm, among kinetochore microtubules. The bottom left panel is an enlargement of the white box in Fi, showing a representative line scan passing through the kinetochore and terminating in the cytoplasm. The bottom right panel shows gold particles only, spotted onto a carbon film for comparison. Pixel densities were compiled and represented as a histogram (Fii). Predicted subcellular positions are noted beneath the histogram. Ncell = 2. Nkinetochore = 12. Bar = 20 nm. (G) Summary of the MS data returned for in vivo BirA*–CENP-A. Numbers in brackets represent the ranked position of CENP-A within the MS data, based on protein score.
To determine the distance traveled by activated biotin before derivatizing a target protein we measured the dispersion of biotin from BirA*-CENP-A using correlative light and electron microscopy (CLEM). CENP-A is an excellent substrate for this because it is organized as a linear structure in the inner kinetochore plate. To determine the range of distances traveled by activated biotin following its release from BirA*-CENP-A, we performed the in vivo BioID protocol, then stained cells with Alexa Fluor 488 streptavidin conjugated to 5 nm colloidal gold. We then measured the distribution of gold around kinetochores (Figure 1, D–F). Overview images confirm that a mitotic cell of interest was identified and followed from LM (light microscopy) imaging through to EM (electron microscopy; Figure 1D). Higher-magnification images confirmed that individual chromosomes and their centromeres could also be followed (Figure 1E, Zoom 1). Further examination of the images confirmed that centromeres visualized by LM correlated accurately with electron dense gold particles decorating linear “plates” in electron micrographs (Figure 1E, Zoom 2). The distribution of colloidal gold particles was measured using line scans, taken perpendicular to the long axis of the kinetochore plates (Figure 1Fi, red bars). High-magnification images confirmed that “in situ” gold particles (Figure 1Fi, bottom left panel) resembled free gold particles spotted onto Formvar-coated grids (Figure 1Fi, bottom right panel). The line-scan density profile revealed a remarkably narrow distribution of gold particles, with an overall span of ∼20 nm (Figure 1Fii). This correlates remarkably well with the thickness of kinetochore plates measured by thin-section electron microscopy (Rieder, 1982) and with previous estimates of the distances traveled by activated biotin (10–20 nm; Roux et al., 2012; 10 nm; Kim et al., 2014).
Because the half width of the gold distribution was only ∼10 nm, this experiment suggests that the activated biotin travels only a very limited distance before derivatization of a target protein. We would expect a sharp cutoff to this distribution outside the chromosome, because any labeled cytosolic proteins would presumably diffuse away from the site of labeling. In contrast, chromatin is relatively immobile, so we would have expected the labeling in subjacent chromatin to reflect the distance traveled by activated biotin. The fact that relatively little gold was found in the chromatin flanking the inner surface of the kinetochore strongly supports the assumption that BioID does indeed measure protein neighborhoods at the molecular level.
To characterize the specificity of the in vivo BioID method, cells expressing BirA*-CENP-A were subjected to the original BioID protocol (Materials and Methods) and then lysed and biotinylated proteins enriched by binding to streptavidin beads (Figure 1C). The affinity-purified material was then analyzed by mass spectrometry. In control experiments, cells not expressing BirA*-CENP-A were grown in the presence of 50 µm biotin, and affinity purifications performed in parallel. This in vivo analysis returned more than 500 hits, with CENP-A itself ranked at position 298, based on peptide score (Figure 1G and Supplemental Table 1). After subtracting proteins identified in the control pull downs, this experiment yielded a list of more than 300 proteins (Figure 1G and Supplemental Table 2) with CENP-A now ranked at position 109.
Although there were likely genuine proteins from the CENP-A neighborhood on this list, no other centromere proteins were detected. Furthermore, given that CENP-A itself was ranked so far down the list, we concluded that attempting to identify other members of the CENP-A neighborhood by this approach would be unlikely to succeed. It was also evident that this approach (which requires labeling for 18–24 h in the presence of high biotin concentrations) could never allow us to discriminate between cell cycle–dependent interacting partners of CENP-A.
To address these problems of labeling specificity and temporal resolution, the method needed to be improved in two key areas: 1) BirA*-specific labeling of target proteins over much shorter reaction times and 2) better discrimination between specific and nonspecific biotinylation targets.
Indeed, the intracellular pool of free biotin is an issue with the published in vivo BioID protocol, as growing cells for less than 18–24 h in medium supplemented with 50 µM biotin yielded low levels of BirA* labeling of target proteins (Roux et al., 2012). At physiological levels (e.g., the concentration of biotin in fetal bovine serum [FBS]), the uptake of biotin into cells is relatively slow (Dakshinamurti and Chalifour, 1981; Chalifour and Dakshinamurti, 1982), and although increasing the biotin concentration can increase its uptake (Chalifour and Dakshinamurti, 1982; Robinson et al., 1983), this improves only until transporter saturation is reached (Prasad et al., 1998; Daberkow et al., 2003).
We speculated that one way to solve this problem and achieve rapid labeling of substrates with low backgrounds would be to conduct the biotinylation reaction in vitro, using permeabilized cells. To test whether such an in vitro system would be feasible, we designed a two-step protocol combining mild permeabilization of cells with incubation in a biotinylation reaction buffer (Figure 2A). Strikingly, we found that BirA* was active under these conditions (Figure 2, C–E), yielding biotinylation signals comparable to those obtained with a 24 h incubation in vivo (Figure 2B).
FIGURE 2:

Rapid in vitro biotinylation (ivBioID) of CENP-A. (A) Flowchart of ivBioID method. (B) Immunofluorescence of in vivo prepared BirA*–CENP-A cells. In panels B and E, 24 h+ represents an in vivo BioID sample extracted with 0.1% Triton X-100 before fixation. Panels t = 0 represent a sample with no biotin incubation. (C) Summary of results of cells prepared using the protocol described in A, under a variety of detergent extraction conditions. (D) Immunofluorescence of ivBioID–CENP-A prepared cells. Cells were permeabilized with 0.1% Triton X-100 extraction for 2 min. Several biotinylation buffer incubation time points were tested. Cells were fixed and processed for IF as standard and labeled with streptavidin 488 (green) or anti-myc (red). In panels D and E, 15 min* represents cells incubated with biotin buffer lacking ATP. Bar = 5 μm; Zoom bar = 1 μm. (E) Bar graph showing quantification of centromeric immunofluorescence from samples prepared as for B and D. Bars show the mean fluorescence of streptavidin 488 normalized to myc fluorescence. Ncell = 20. Ncentromere = 200.
The in vitro biotinylation worked efficiently and reproducibly but was sensitive to the conditions used to permeabilize the cells. Extraction of cells with SDS at concentrations of >0.01% appeared to abolish the reaction, and at 0.01% SDS, only occasional weak labeling was observed (Figure 2C). Of the conditions tested, brief extraction of cells with Triton X-100 at concentrations up to 0.1% appeared to be the most suitable for preserving BirA* functionality in the in vitro reaction (Figure 2C), and this concentration was used for all subsequent experiments.
We measured the efficiency of in vitro biotinylation (Figure 2D) by quantifying the fluorescence levels of streptavidin-Alexa 488 at centromeres following differing incubation periods with biotinylation buffer (Figure 2E). In control experiments, using the established in vivo BioID protocol, BirA*-CENP-A produced robust levels of biotinylation at centromeres (Figure 2, B and E). Our in vitro protocol, which we refer to as in vitro BioID (ivBioID), was less efficient, but yielded clear centromeric biotinylation after as little as 2 min of incubation with biotinylation buffer. Only minor differences were observed between biotinylation buffer incubations of 2, 5, and 10 min; however, longer incubations did result in greater levels of biotinylation (Figure 2, D and E). In control experiments, removing either biotin or ATP from the reaction buffer resulted in almost undetectable levels of biotinylation (Figure 2, D and E).
Clear visual evidence for the increased specificity of the in vitro biotinylation came from examining the results of in vitro biotinylation on mitotic chromosome spreads. Cells expressing BirA*-CENP-A were arrested in mitosis with nocodazole, hypotonically swollen, and centrifuged onto glass slides. The ivBioID reaction was then performed, with labeling for 20 min. This procedure gave highly specific biotin labeling of the centromeres of mitotic chromosomes (Figure 3, A–C). In contrast, when cells were processed for the conventional in vivo BioID protocol and similar chromosome spreads were prepared, the biotin labeling was seen not only at centromeres, but also all along the chromosome arms (Figure 3, D–F). It is therefore clear that the ivBioID protocol can yield a more selective labeling of the neighborhood of the target protein.
FIGURE 3:
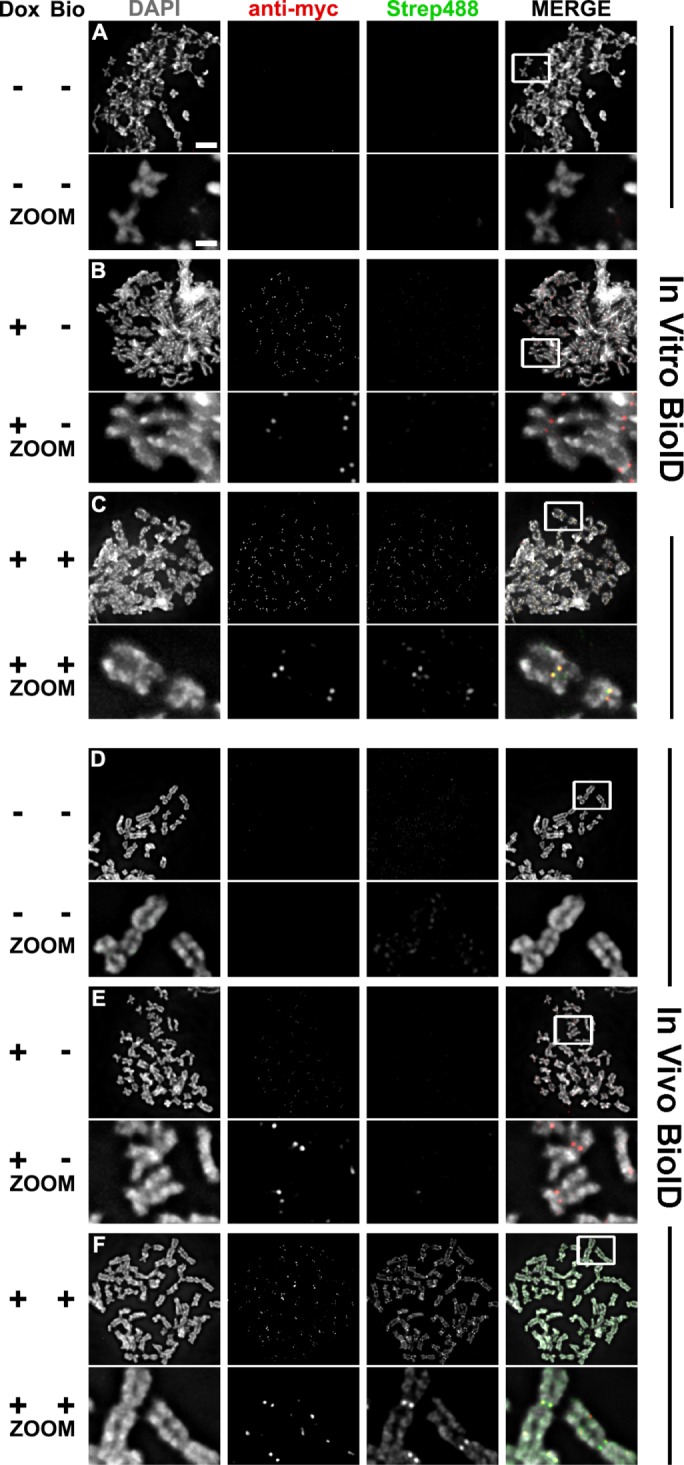
Chromosome spreads from both in vivo and in vitro prepared samples. Qualitative samples were prepared for comparison of in vivo and in vitro BioID samples. Experimental conditions include cells not expressing BirA*–CENP-A and not treated with biotin (A and D); cells expressing BirA*–CENP-A but not treated with biotin (B and E); cells expressing BirA*–CENP-A and treated with biotin (C and F). All samples were fixed and probed with DAPI, anti-myc (red), and streptavidin 488 (green). Zooms are of the white boxed region in “MERGE.” Bar = 5 and 1 μm.
Biotinylation with sub–cell cycle resolution
Having obtained evidence that ivBioID allows rapid biotinylation of substrates by BirA*, we next investigated whether this approach would also provide a more selective method for mapping protein neighborhoods. Our ultimate goal was to ask whether ivBioID could detect differences in the CENP-A neighborhood across the cell cycle. Indeed, immunofluorescence experiments confirmed that mitotic cells can also be labeled efficiently by the ivBioID protocol (Supplemental Figure 1).
For mass spectrometry analysis, we conducted an ivBioID reaction on larger amounts of interphase and mitotic cells, affinity purified the biotinylated material using streptavidin beads, and analyzed the bound proteins by mass spectrometry (Figure 4A). Coomassie staining of the streptavidin pull downs revealed that samples prepared after ivBioID of mitotic cells contained less material than those obtained from interphase cells (Figure 4B). Streptavidin labeling also suggested that levels of recovered protein were lower from mitotic than from interphase cells.
FIGURE 4:

Production of samples for ivBioID analysis of CENP-A. (A) Comparison of original in vivo BioID (top) and in vitro BioID (bottom) methodologies showing addition of the permeabilization step and post lysis biotinylation. (B) Coomassie blue stain gel of samples submitted for MS showing controls and hit samples. (C) Western blot of samples submitted for MS showing controls and hit samples. Biotinylation can be seen in the bottom panel, and anti-myc–BirA*–CENP-A can be seen in the top panel.
Mass spectrometry analysis returned a total of 659 proteins from three repeats following ivBioID of interphase cells and 506 proteins from three repeats from ivBioID of mitotic cells. In control experiments, 484 and 369 proteins were recovered from interphase or mitotic cells not expressing the BirA* fusion protein (-doxycycline) and 455 and 355 proteins were recovered from interphase or mitotic reactions performed in the absence of biotin. To identify the most specific ivBioID–CENP-A interactors we firstly background-subtracted proteins identified in control experiments; secondly, only considered hits present in at least two of the three repeats; and lastly, removed all proteins whose abundance score in the mass spectrometer fell below a minimum confidence threshold. The final list returned 20 and 9 proteins for the interphase and mitotic samples, respectively (Figure 5A). Importantly, both lists contained CENP-A, CENP-B, and CENP-C.
FIGURE 5:
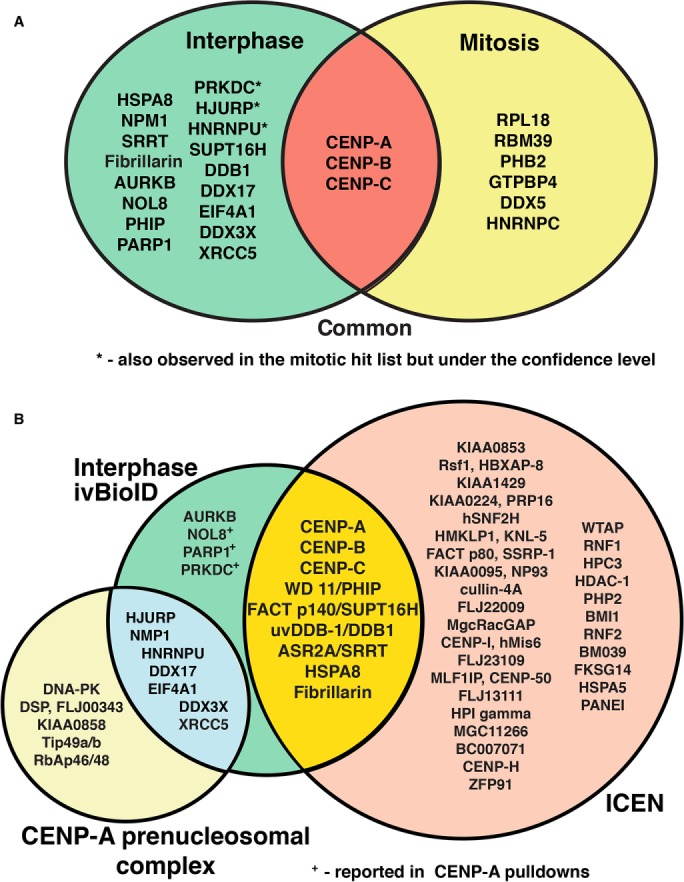
Analysis of the CENP-A neighborhood in interphase and mitosis using ivBioID. (A) Venn diagram showing the final filtered hit lists for interphase (green) and mitotic (yellow) samples and how they overlap with one another. Interphase hits labeled with * were also observed in the mitotic hit list but were under the confidence level. The overlap in red shows those proteins seen in both interphase and mitotic lists. (B) Proteins observed in the interphase ivBioID final hit list (green) show overlap with known CENP-A interacting proteins. The bulk of the proteins detected by ivBioID correspond to proteins previously found in the ICEN complex (yellow overlap; Ando et al., 2002; Obuse et al., 2004; Izuta et al., 2006) and the CENP-A prenucleosomal complex (blue overlap; Foltz et al., 2006). The other proteins not detected in our analysis but found in the original descriptions of the ICEN complex (orange) and CENP-A prenucleosomal complex (pale yellow) are also shown. Hits labeled with + were previously found in pull downs using either CENP-A or themselves as the target.
Validation of the CENP-A neighborhood in interphase and mitosis
Combining the CENP-A data from both interphase and mitotic cells over the three experiments yielded a comprehensive list of proteins proximal to CENP-A as determined by this method (Figure 5A). Importantly, most of these proteins had been previously shown in other studies to be associated with CENP-C in chromatin or in its preassembly complexes.
From the list of CENP-A interphase interactors, two groups of proteins became apparent (Figure 5B). The first is found in the interphase centromere complex (ICEN)—the first published set of CENP-A–associated proteins, which were identified by the Yoda lab. The ICEN complex was a cohort of 40 proteins purified from HeLa cells following micrococcal nuclease digestion of chromatin and immunoprecipitation with a monoclonal antibody to CENP-A (Ando et al., 2002; Obuse et al., 2004; Izuta et al., 2006). Remarkably, nine of the 20 interphase ivBioID hits are found in the list of ICEN proteins, including CENP-A itself. The other eight proteins are CENP-B, CENP-C, PHIP, SUPT16H, DDB1, SRRT, HSPA8, and FIB.
The appearance of CENP-C was expected, because this protein has been shown to bind directly to CENP-A (Dunleavy et al., 2009; Foltz et al., 2009; Carroll et al., 2010; Guo et al., 2017). Although CENP-C is not strictly dependent on CENP-A for localization to kinetochores in Drosophila (Erhardt et al., 2008) or in chicken DT40 cells (Samejima et al., 2015), it does appear to require CENP-A for kinetochore localization in human cells and in Caenorhabditis elegans (Oegema et al., 2001; Liu et al., 2006). Similarly, the interaction between CENP-B and CENP-A nucleosomes at the centromere is very well characterized, although the role of CENP-B remains under intensive study (Fachinetti et al., 2015; Hoffmann et al., 2016). The role of the other ICEN proteins at centromeres remains to be determined.
A second group of six proteins observed in the list of interphase ivBioID hits is found in the CENP-A prenucleosomal complex. These are HJURP, NPM1, DDX17, EIF4A1, XRCC5, and DDX3X. This complex is involved in CENP-A deposition. Indeed, HJURP is the chaperone that inserts CENP-A into chromatin. The CENP-A prenucleosomal complex presumably exists in both interphase and mitosis so that cells are ready for CENP-A deposition at centromeres specifically in a window during mitotic exit when CDK1 activity drops (Jansen et al., 2007; Silva et al., 2012). It was surprising that this complex was detected by ivBioID, because the protocol involves a detergent permeabilization step that would be expected to wash away soluble proteins and protein complexes. Our analysis thus suggests that the CENP-A prenucleosomal complex must be relatively stably engaged either at centromeres or at other sites on chromatin both in interphase and during mitosis. This unexpected observation has recently been confirmed by others (Zasadzinska et al., 2018).
Of the four remaining proteins found by interphase ivBioID with CENP-A, three were previously reported in immunoprecipitation studies for interactions with CENP-A. NOL8, PARP1, and PRKDC were either found by using CENP-A or themselves as the bait protein (Saxena et al., 2002; Shuaib et al., 2010; De Antoni et al., 2012; Hein et al., 2015). Remarkably, the only interphase “hit” protein not previously recovered in a complex with CENP-A is Aurora B kinase. However, Aurora B and Aurora A can both phosphorylate CENP-A on serine 7 (Zeitlin et al., 2001; Kunitoku et al., 2003; Boeckmann et al., 2013). This phosphorylation has been reported to have an unexpected role in regulating cytokinesis (Zeitlin et al., 2001) and also to be involved in localization of the CPC to centromeres (Kunitoku et al., 2003). Surprisingly, in our study, biotinylated Aurora B was observed in the absence of its three auxiliary CPC subunits INCENP, Survivin, and Borealin.
The list of mitotic proteins in the CENP-A neighborhood was much shorter. Aside from CENP-B and CENP-C we recovered two members of the prenucleosomal complex (DDX5 and HNRNPC). Again, it was unexpected and interesting that two members of the CENP-A prenucleosomal complex would be associated with mitotic CENP-A. Among the others, PBH2 was reported to be involved in kinetochore assembly and protecting sister chromatid cohesion (Takata et al., 2007; Lee et al., 2011), but has not been reported to be linked with CENP-A. The other three proteins, RPL8 (a ribosomal protein), RBM39 (a splicing factor), and GTPBP4 (a nucleolar GTP-binding protein), do not have obvious links to kinetochores or CENP-A and determination of their link to CENP-A requires further investigation.
Gene ontology analysis of hits
To better understand the CENP-A neighborhood during interphase and mitosis, gene ontology analysis was carried out on the two hit lists. The biological functions of the proteins in both of these lists (Figure 6) show a large crossover between the functions of the proteins found in both interphase and mitosis, the largest of these groups being kinetochore assembly. This was expected, particularly when considering that a greater percentage of the mitotic hits are seen to be involved in this process. Also prominent were other chromatin-related categories, including nucleosome organization, ATP-dependent chromatin remodeling, sister chromatid cohesion, and chromosome organization.
FIGURE 6:

GO enrichment analysis of interphase and mitotic ivBioID hit lists. GO analysis using the hit lists obtained from the MS reveals enrichment for a subset of biological processes and cellular components. Both sets of samples were run through the g-Profiler toolkit (Reimand et al., 2016) and the significant terms (P < 0.01) for each sample were recorded (Supplemental Table 3). We calculate log2 enrichment for each GO term using the fraction of these terms per sample against the ratio of these terms in the database used by g-Profiler. (A) Biological processes in which the proteins found in the hit lists are enriched. (B) Cellular process in which the proteins found in the hit lists are enriched.
Looking more generally at the cellular components in which the proteins fall (Figure 6B), all our proteins fell into only four categories: nuclear pericentric heterochromatin, granular components, centromeric protein, and nuclear lumen. All but the granular components are nuclear/chromatin specific. The association with the granular component of the nucleolus is less obvious. Indeed, some of these proteins, which are extremely abundant in cells, could be contaminants.
We can conclude from this analysis that the ivBioID method generates lists of enriched proteins that are relevant for CENP-A–related activities. Thus, the method appears to have high specificity and to be capable of detecting relatively nonabundant components of insoluble cellular structures.
Perspective
The ivBioID method reveals information about the CENP-A neighborhood that differs from interaction maps reported by classical methods such as two hybrid screens and tandem affinity pull-down screens. Thus, ivBioID provides an additional, orthogonal method for the exploration of protein relationships. The method revealed a majority of proteins that were previously known to interact directly with CENP-A, including CENP-C, CENP-B, and HJURP; it also offered insights into the changes in the CENP-A environment at interphase and mitosis. Interestingly, despite the stability of CENP-A nucleosomes in vivo (Bodor et al., 2013), its environment is characterized by dynamic chromatin, as shown by the presence of proteins specific for chromatin remodeling as well as CENP-A nucleosome assembly.
Because the ivBioID method works with cells that have been harvested and permeabilized and are no longer physiologically active, it offers what is effectively “instantaneous” time resolution of cellular processes. Thus, if cultures can be synchronized or sorted, this method should offer a useful approach to determining differences in protein neighborhoods in cells grown under normal conditions without stresses imposed by exposure to extended and high concentrations of biotin or transient shock with H2O2. Furthermore, because the bulk of soluble proteins are removed by the detergent extraction, the method exhibits a remarkably low background, provided that the goal is to look at relatively stable structural interactions.
Since these experiments were performed, a new method for in vivo biotin labeling of protein neighborhoods known as “TurboID” has been published (Branon et al., 2018). The authors used directed evolution to produce a highly mutated BirA that can biotinylate target proteins in vivo in HEK 293T cells in as little as 10 min and also in living Drosophila melanogaster and C. elegans. TurboID clearly addresses a number of the difficulties with previous in vivo BioID methods, and provides a powerful complement to the in vitro method described here. TurboID provides much longer lists of potential neighbors (which can be a strength or a weakness) and should be able to detect weak or transient interactions. In contrast, ivBioID provides lower background, much shorter hit lists, can analyze changes over even finer time intervals (e.g., a minute or even less in highly time-sensitive processes like mitosis), and will presumably detect primarily stable interactions (which may be easier to validate than weak or transient interactions). Furthermore, it should be useful in any cell type or organism, independent of the biotin transport or endogenous pools. Together, classic BioID (Roux et al., 2012) and these new methods are opening up powerful new approaches to analyzing protein proximities in cells.
MATERIALS AND METHODS
Cell culture
HeLa cells were maintained in DMEM containing 10% FBS and 100U/ml penicillin/streptomycin at 37°C and 5% CO2.
For preparation of stable cell lines, CENP-A was amplified by PCR from HeLa cDNA and cloned into pcDNA3.1-mycBioID vector (Addgene) using Not1 and Afl2 restriction sites. Myc-BirA(R118G)–CENP-A was amplified by PCR from pcDNA3.1-mycBioID–CENP-A vector and cloned into a tet-on 3G inducible expression vector (Clontech). Tet-On 3G vectors were cotransfected, with a linear resistance gene, into HeLa cells expressing the tetracycline-regulated transactivator (Clontech), using GeneJuice (Novagen). Stable cell lines were enriched using antibiotic selection for 7–14 d. Surviving clones were expanded and screened for the stable, inducible expression of Myc-BirA(R118G)–CENP-A (otherwise known as BirA*–CENP-A) by immunofluorescence microscopy. Expression of the fusion protein was observed in >90% of cells in the selected culture.
Indirect immunofluorescence microscopy
Cells were fixed in 4% paraformaldehyde (PFA) and processed as previously described (Booth et al., 2014). The primary antibodies or fluorescent labels used were as follows: anti–α-tubulin antibody (B512; Sigma); anti-myc Alexa Fluor 555 conjugate (Millipore); streptavidin Alexa Fluor 488 conjugate (Life Technologies); and anti–CENP-C. Three-dimensional data sets were acquired using a cooled CCD camera (CH350; Photometrics) on a wide-field microscope (DeltaVision Spectris; Applied Precision) with a NA 1.4 Plan Apochromat lens. The data sets were deconvolved with softWoRx (Applied Precision). Three-dimensional data sets were converted to Quick Projections in softWoRx, exported as TIFF files, and imported into Adobe Photoshop or Adobe Illustrator for final presentation.
CLEM
The CLEM processing method was adapted from previously established protocols (Booth et al., 2011, 2013, 2014). Briefly, HeLa cells conditionally expressing BirA*–CENP-A were seeded onto glass-bottomed gridded dishes (MatTek Corporation) in medium supplemented with biotin (50 µM) and doxycycline (1 µg/ml). Following a 24 h expression period, cells were fixed using a mixture of glutaraldehyde and paraformaldehyde for 1 h. Next, cells were permeabilized, blocked in 3% bovine serum albumin, and probed with Alexa Fluor 488 dye–labeled colloidal gold, conjugated to streptavidin (Molecular Probes), for 1 h. Samples were washed (in phosphate-buffered saline [PBS] containing Hoechst) and cells of interest identified using a wide-field epifluorescence microscope (DeltaVision RT; Applied Precision). Cells with centromere labeling were located and their position mapped using transmitted light to visualize reference coordinates. Cells were then postfixed for 20 min in 1.5% PFA, before osmication (0.5% osmium tetroxide in PBS), dehydrated (using a graded series of ethanol), and embedded in resin. Once cured, ultrasmall resin blocks (50 µm2) were fine trimmed and serial sections (85 nm thickness) taken at areas corresponding to previously chosen coordinate positions. Cells were visualized with a Phillips CM120 BioTwin transmission electron microscope (FEI) and micrographs acquired using a Gatan Orius CCD camera (Gatan).
The appropriate Z position correlative light/EM images of a cell were concatenated using ImageJ and then analyzed and overlaid using Photoshop Elements 6 (Adobe).
Centromeric gold labeling and pixel density analysis
Electron micrographs containing mitotic chromosomes were selected for analysis of gold particles at the centromere. The pixel density of a 1 µm region of interest (ROI) was measured using the raw data from the “plot profile” function of ImageJ. For unbiased consistency, the 1 µm ROI always started within the chromosome body and finished in the cytoplasm, with the halfway point lying at the region most dense with gold label. Approximately 150 individual pixel density measurements were taken within each 1 µm region. The data were plotted as a line-scan profile, with each data point representing the mean of 6 × 1 µm regions of interest per centromere.
In vivo BioID method
This was based on the method described by Roux et al. (2012). Cells were seeded into 4 × 10 cm dishes and incubated for 24 h in complete media supplemented with 1 μg/ml doxycycline and 50 μM biotin. Cells were trypsinized, centrifuged, and resuspended in 1 ml lysis buffer at 25°C (50 mM Tris, pH 7.4, 500 mM NaCl, 0.4% SDS, 5 mM EDTA, 1 mM dithiothreitol [DTT], and 1× complete protease inhibitor [Roche]), and sonicated. Triton X-100 was added to 2% final concentration. After further sonication, an equal volume of 4°C 50 mM Tris (pH 7.4) was added before additional sonication (subsequent steps at 4°C) and centrifugation at 16,000 × g. Supernatants were incubated with 100 μl Dynabeads (MyOne steptavadin C1; Invitrogen) for 1 h at RT. Beads were collected and washed twice for 8 min at 25°C (all subsequent steps at 25°C) in 1 ml wash buffer 1 (2% SDS in dH2O). This was repeated once with wash buffer 2 (0.1% deoxycholate, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA, and 50 mM HEPES, pH 7.5), once with wash buffer 3 (250 mM LiCl, 0.5% NP-40, 0.5% deoxycholate, 1 mM EDTA, and 10 mM Tris, pH 8.1), and twice with wash buffer 4 (50 mM Tris, pH 7.4, and 50 mM NaCl). Ten percent of the sample was reserved for Western blot analysis. Bound proteins were eluted from the magnetic beads with 50 μl of Laemmli SDS sample buffer saturated with biotin at 98°C. Eluent (90%) was submitted for mass spectrometry analysis.
In vitro BioID method
Cells were seeded into 10 cm dishes for asynchronous cells and 15 cm dishes for mitotic cells and incubated for 24 h in complete medium supplemented with 1 μg/ml doxycycline. For mitotic samples cells were treated with nocodozole (100 ng/ml) for 13 h. Mitotic cells were enriched via mitotic shake off. Asynchronous cells were collected via trypsinization. Cells were centrifuged and resuspended in 0.1% Triton X-100 in PBS for 1.5 min at 37°C. Samples were centrifuged and washed once in warmed PBS before incubation in biotinylation buffer (40 mM Tris:HCl, pH 8.0, 5 mM MgCl2, 3 mM ATP, 100 µM KCl, 50 µM biotin) for 15 min at 37°C. Cells were centrifuged and washed once in warmed PBS, before resuspension in 100 μl solubilization buffer at 25°C (50 mM triethanolamine:HCl, pH 7.4, 100 mM NaCl, 2 mM K-EDTA, pH 7.4, 0.4% SDS, 2 mM β-mercaptoethanol, 0.1 mM phenylmethylsulfonyl fluoride, 0.1% trasylol, 1 µg/ml chymostatin, leupeptin, antipain, pepstatin [CLAP]), and sonicated at 4°C. Samples were then centrifuged at 16,000 × g. Supernatants were transferred to clean tubes before addition of 400 µl RIPA buffer (50 mM Tris:HCl, pH 8.0, 150 mM NaCl, 1% Na deoxycholate, 0.1% SDS, 1× cOmplete ULTRA protease inhibitor; Roche) and subsequent incubation with 100 μl Dynabeads (MyOne steptavadin C1; Invitrogen) for 18 h at 4°C. Beads were collected and washed twice for 5 min at 25°C rotating (all subsequent steps at 25°C) in 1 ml PBS containing 500 mM NaCl. Subsequently two washes were performed in 1 ml PBS containing 250 mM NaCl, followed by two washes in 1 ml PBS and a final wash in 1 ml mass spectrometry grade H2O. Bound proteins were eluted from the magnetic beads with 50 μl of Laemmli SDS sample buffer saturated with biotin at 95°C. Ten percent of the sample was reserved for Western blot analysis. Ninety percent of the eluent was submitted for mass spectrometry analysis.
Immunoblotting analysis
For immunoblotting, 10% of the total eluent, purified for mass spectrometry, was loaded onto polyacrylamide gels. SDS–PAGE and immunoblotting were performed following standard procedures. The antibodies or fluorescent labels used were as follows: anti-myc 9E10 (Abcam); IRDye 800CW streptavidin (LI-COR); anti-mouse IRDye 680CW (LI-COR).
Mass spectrometry analysis
Protein samples were run on gels (NuPAGE Novex 4–12% Bis-Tris gel; Life Technologies, UK), in NuPAGE buffer (MES) and visualized using Imperial protein stain (Life Technologies, UK). The stained gel bands were excised and destained with 50 mM ammonium bicarbonate (Sigma Aldrich, UK) and 100% (vol/vol) acetonitrile (Sigma Aldrich, UK), and proteins were digested with trypsin, as previously described (Shevchenko et al., 1996). Briefly, proteins were reduced in 10 mM DTT (Sigma Aldrich, UK) for 30 min at 37°C and alkylated in 55 mM iodoacetamide (Sigma Aldrich, UK) for 20 min at ambient temperature in the dark. They were then digested overnight at 37°C with 13 ng μl−1 trypsin (Pierce, UK).
Following digestion, samples were diluted with equal volumes of 0.1% trifluoroacetic acid (TFA) and spun onto StageTips as described by Rappsilber et al. (2003). Peptides were eluted in 40 μl of 80% acetonitrile in 0.1% TFA and concentrated down to 1 μl by vacuum centrifugation (Concentrator 5301; Eppendorf, UK). Samples were then prepared for liquid chromatography–mass spectrometry (LC-MS)/MS analysis by diluting them to 6 μl with 0.1% TFA. LC-MS analyses were performed on a Q Exactive mass spectrometer (Thermo Fisher Scientific, UK) coupled online, to an Ultimate 3000 RSLCnano System (Dionex; Thermo Fisher Scientific, UK). Peptides were separated on a 50 cm EASY-Spray column (Thermo Fisher Scientific, UK) assembled in an EASY-Spray source (Thermo Fisher Scientific, UK) and operated at 50°C. Mobile phase A consisted of 0.1% formic acid in water, whereas mobile phase B consisted of 80% acetonitrile and 0.1% formic acid. Peptides were loaded onto the column at a constant flow rate of 0.3 μl min−1 and eluted at a flow rate of 0.2 μl min−1 according to the following gradient: 2–40% mobile phase B in 120 min, then to 95% in 11 min. Fourier transform mass spectrometry spectra were recorded at 70,000 resolution (scan range 350–1400 m/z) and the 10 most intense peaks with charge ≥2 of the MS scan were selected with an isolation window of 2.0 Thomson for MS2 (filling 1.0E6 ions for MS scan, 5.0E4 ions for MS2, maximum fill time 60 ms, dynamic exclusion for 50 s). Fragmentation was performed by employing HCD (Olsen et al., 2007) with normalized collision energy of 27.
The MaxQuant software platform (Cox and Mann, 2008) version 1.5.2.8 was used to process raw files and searches were conducted against Homo sapiens complete/reference proteome set of Uniprot database (released in March 2017), using the Andromeda search engine (Cox et al., 2011). For the first search, peptide tolerance was set to 20 ppm, whereas for the main search peptide tolerance was set to 4.5 ppm. Isotope mass tolerance was 2 ppm and the maximum charge was 7. A maximum of two missed cleavages was allowed. Carbamidomethylation of cysteine was set as a fixed modification. Oxidation of methionine and acetylation of the N-terminal were set as variable modifications. Peptide and protein identifications were filtered to 1% false discovery rate.
Filtering and analysis of hits
The following approach was used to reduce the background and obtain hits lists. First, the three repeats were combined, yielding one list for each experimental condition. Next those lists were filtered, keeping only proteins for which at least two independent peptides had been identified. Subsequently, proteins found in either of the control lists—1) cells not expressing BirA*–CENP-A and 2) cells that had not been exposed to biotin—were subtracted from the corresponding master list of interphase or mitotic hits. The data were further filtered by removing proteins that were not observed in at least two of the repeats. Finally, a confidence level based on peptide score (a comparison of the observed spectrum for each peptide with the theoretically calculated spectrum for that peptide; Cox and Mann, 2008) was applied to the interphase and mitotic hit lists. Proteins with a score below 1 were removed, thereby yielding the final hit lists for both conditions.
Bioinformatics and gene ontology analysis
Final hit lists from both conditions were run through the g-Profiler toolkit (Reimand et al., 2016) and the significant terms (P < 0.01) for each sample were recorded in the output. This was then used to calculate log2 enrichment for each gene ontology (GO) term using the fraction of these terms per sample against the ratio of these terms in the database used by g-Profiler.
Supplementary Material
Acknowledgments
This work was funded by the Wellcome Trust, of which W.C.E. is a Principal Research Fellow (Grant no. 073915). The Wellcome Trust Centre for Cell Biology is supported by core Grants no. 077707 and no. 092076.
Abbreviations used:
- APEX
ascorbate peroxidase
- BioID
Biotin ID
- BSA
bovine serum albumin
- CDK1
cyclin dependent kinase 1
- CENP-A
centromere protein A
- CLAP
chymostatin, leupeptin, antipain, pepstatin
- CLEM
correlative light electron microscopy
- CPC
chromosome passenger complex
- DAPI
4′,6-diamidino-2-phenylindole
- DTT
dithiothreitol
- EM
electron microscopy
- EMARS
enzyme-mediated activation of radical sources
- FBS
fetal bovine serum
- HRP
horseradish peroxidase
- ICEN
inner centromere complex
- ivBioID
in vitro Biotin ID
- LC–MS
liquid chromatography–mass spectrometry
- LM
light microscopy
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- PMSF
phenylmethylsulfonyl fluoride
- ROI
region of interest
- SPPLAT
selective proteomic proximity labeling assay using tyramide
- TFA
trifluoroacetic acid
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-12-0799) on March 20, 2019.
REFERENCES
- Ando S, Yang H, Nozaki N, Okazaki T, Yoda K. (2002). CENP-A, -B, and -C chromatin complex that contains the I-type α-satellite array constitutes the prekinetochore in HeLa cells. Mol Cell Biol , 2229–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck DB, Narendra V, Drury WJ, 3rd, Casey R, Jansen PW, Yuan ZF, Garcia BA, Vermeulen M, Bonasio R. (2014). In vivo proximity labeling for the detection of protein-protein and protein-RNA interactions. J Proteome Res , 6135–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor DL, Valente LP, Mata JF, Black BE, Jansen LE. (2013). Assembly in G1 phase and long-term stability are unique intrinsic features of CENP-A nucleosomes. Mol Biol Cell , 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann L, Takahashi Y, Au WC, Mishra PK, Choy JS, Dawson AR, Szeto MY, Waybright TJ, Heger C, Mcandrew C, et al. (2013). Phosphorylation of centromeric histone H3 variant regulates chromosome segregation in Saccharomyces cerevisiae. Mol Biol Cell , 2034–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth DG, Cheeseman LP, Prior IA, Royle SJ. (2013). Studying kinetochore-fiber ultrastructure using correlative light-electron microscopy. Methods Cell Biol , 327–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth DG, Hood FE, Prior IA, Royle SJ. (2011). A TACC3/ch-TOG/clathrin complex stabilises kinetochore fibres by inter-microtubule bridging. EMBO J , 906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth DG, Takagi M, Sanchez-Pulido L, Petfalski E, Vargiu G, Samejima K, Imamoto N, Ponting CP, Tollervey D, Earnshaw WC, Vagnarelli P. (2014). Ki-67 is a PP1-interacting protein that organises the mitotic chromosome periphery. Elife , e01641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, Ting AY. (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol , 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S, Pepper D, Berns MW, Tan E, Brinkley BR. (1981). Kinetochore structure, duplication, and distribution in mammalian cells: analysis by human autoantibodies from scleroderma patients. J Cell Biol , 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Milks KJ, Straight AF. (2010). Dual recognition of CENP-A nucleosomes is required for centromere assembly. J Cell Biol , 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalifour LE, Dakshinamurti K. (1982). The characterization of the uptake of avidin-biotin complex by HeLa cells. Biochim Biophys Acta , 64–69. [DOI] [PubMed] [Google Scholar]
- Chan PK, Srikumar T, Dingar D, Kalkat M, Penn LZ, Raught B. (2014). BioID data of c-MYC interacting protein partners in cultured cells and xenograft tumors. Data Brief , 76–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM. (2014). The kinetochore. Cold Spring Harb Perspect Biol , a015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A. (2005). A combined approach for the localization and tandem affinity purification of protein complexes from metazoans. Sci STKE , pl1. [DOI] [PubMed] [Google Scholar]
- Chen AL, Kim EW, Toh JY, Vashisht AA, Rashoff AQ, Van C, Huang AS, Moon AS, Bell HN, Bentolila LA, et al. (2015). Novel components of the Toxoplasma inner membrane complex revealed by BioID. MBio , e02357-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi-Rhee E, Schulman H, Cronan JE. (2004). Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci , 3043–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Q, Rathore A, Diedrich JK, Donaldson CJ, Yates JR, Saghatelian A. (2017). Identification of microprotein-protein interactions via APEX tagging. Biochemistry , 3299–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol , 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res , 1794–1805. [DOI] [PubMed] [Google Scholar]
- Daberkow RL, White BR, Cederberg RA, Griffin JB, Zempleni J. (2003). Monocarboxylate transporter 1 mediates biotin uptake in human peripheral blood mononuclear cells. J Nutr , 2703–2706. [DOI] [PubMed] [Google Scholar]
- Dakshinamurti K, Chalifour LE. (1981). The biotin requirement of HeLa cells. J Cell Physiol , 427–438. [DOI] [PubMed] [Google Scholar]
- De Antoni A, Maffini S, Knapp S, Musacchio A, Santaguida S. (2012). A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J Cell Biol , 269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunleavy EM, Roche D, Tagami H, Lacoste N, Ray-Gallet D, Nakamura Y, Daigo Y, Nakatani Y, Almouzni-Pettinotti G. (2009). HJURP is a cell-cycle-dependent maintenance and deposition factor of CENP-A at centromeres. Cell , 485–497. [DOI] [PubMed] [Google Scholar]
- Earnshaw WC, Halligan B, Cooke CA, Heck MMS, Liu LF. (1985). Topoisomerase-Ii is a structural component of mitotic chromosome scaffolds. J Cell Biol , 1706–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S, Mellone BG, Betts CM, Zhang W, Karpen GH, Straight AF. (2008). Genome-wide analysis reveals a cell cycle-dependent mechanism controlling centromere propagation. J Cell Biol , 805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fachinetti D, Han JS, Mcmahon MA, Ly P, Abdullah A, Wong AJ, Cleveland DW. (2015). DNA sequence-specific binding of CENP-B enhances the fidelity of human centromere function. Dev Cell , 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Suarez M, Chen TS, Ting AY. (2008). Protein-protein interaction detection in vitro and in cells by proximity biotinylation. J Am Chem Soc , 9251–9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firat-Karalar EN, Rauniyar N, Yates JR, 3rd, Stearns T. (2014). Proximity interactions among centrosome components identify regulators of centriole duplication. Curr Biol , 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Bailey AO, Yates JR, 3rd, Bassett EA, Wood S, Black BE, Cleveland DW. (2009). Centromere-specific assembly of CENP-A nucleosomes is mediated by HJURP. Cell , 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR, 3rd, Cleveland DW. (2006). The human CENP-A centromeric nucleosome-associated complex. Nat Cell Biol , 458–469. [DOI] [PubMed] [Google Scholar]
- Guldner HH, Lakomek HJ, Bautz FA. (1984). Human anti-centromere sera recognise a 19.5 kD non-histone chromosomal protein from HeLa cells. Clin Exp Immunol , 13–20. [PMC free article] [PubMed] [Google Scholar]
- Guo LY, Allu PK, Zandarashvili L, Mckinley KL, Sekulic N, Dawicki-Mckenna JM, Fachinetti D, Logsdon GA, Jamiolkowski RM, Cleveland DW, et al. (2017). Centromeres are maintained by fastening CENP-A to DNA and directing an arginine anchor-dependent nucleosome transition. Nat Commun , 15775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, et al. (2015). A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell , 712–723. [DOI] [PubMed] [Google Scholar]
- Hinshaw SM, Harrison SC. (2018). Kinetochore function from the bottom up. Trends Cell Biol , 22–33. [DOI] [PubMed] [Google Scholar]
- Hoffmann S, Dumont M, Barra V, Ly P, Nechemia-Arbely Y, Mcmahon MA, Herve S, Cleveland DW, Fachinetti D. (2016). CENP-A is dispensable for mitotic centromere function after initial centromere/kinetochore assembly. Cell Rep , 2394–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Amano M, Suzuki A, Backer CB, Welburn JP, Dong Y, Mcewen BF, Shang WH, Suzuki E, Okawa K, et al. (2008). CCAN makes multiple contacts with centromeric DNA to provide distinct pathways to the outer kinetochore. Cell , 1039–1052. [DOI] [PubMed] [Google Scholar]
- Izuta H, Ikeno M, Suzuki N, Tomonaga T, Nozaki N, Obuse C, Kisu Y, Goshima N, Nomura F, Nomura N, Yoda K. (2006). Comprehensive analysis of the ICEN (interphase centromere complex) components enriched in the CENP-A chromatin of human cells. Genes Cells , 673–684. [DOI] [PubMed] [Google Scholar]
- Jansen LE, Black BE, Foltz DR, Cleveland DW. (2007). Propagation of centromeric chromatin requires exit from mitosis. J Cell Biol , 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Youn JY, Gingras AC, Subramaniam R, Desveaux D. (2018). In planta proximity dependent biotin identification (BioID). Sci Rep , 9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, Birendra KC, Zhu W, Motamedchaboki K, Doye V, Roux KJ. (2014). Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc Natl Acad Sci USA , E2453–E2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, Roux KJ. (2016). Filling the void: proximity-based labeling of proteins in living cells. Trends Cell Biol , 804–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani N, Gu J, Isaji T, Udaka K, Taniguchi N, Honke K. (2008). Biochemical visualization of cell surface molecular clustering in living cells. Proc Natl Acad Sci USA , 7405–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunitoku N, Sasayama T, Marumoto T, Zhang D, Honda S, Kobayashi O, Hatakeyama K, Ushio Y, Saya H, Hirota T. (2003). CENP-A phosphorylation by Aurora-A in prophase is required for enrichment of Aurora-B at inner centromeres and for kinetochore function. Dev Cell , 853–864. [DOI] [PubMed] [Google Scholar]
- Lambert JP, Tucholska M, Go C, Knight JD, Gingras AC. (2015). Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J Proteomics , 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Lin L, Equilibrina I, Uchiyama S, Matsunaga S, Fukui K. (2011). ASURA (PHB2) is required for kinetochore assembly and subsequent chromosome congression. Acta Histochem Cytochem , 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Li J, Wang L, Di LJ. (2017). Proximity labeling of interacting proteins: application of BioID as a discovery tool. Proteomics, , 28271636. [DOI] [PubMed] [Google Scholar]
- Liu ST, Rattner JB, Jablonski SA, Yen TJ. (2006). Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J Cell Biol , 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobingier BT, Huttenhain R, Eichel K, Miller KB, Ting AY, Von Zastrow M, Krogan NJ. (2017). An approach to spatiotemporally resolve protein interaction networks in living cells. Cell , 350–360.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroi Y, Hartman AL, Nakane PK, Tan EM. (1981). Distribution of kinetochore (centromere) antigen in mammalian cell nuclei. J Cell Biol , 254–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroi Y, Peebles C, Fritzler MJ, Steigerwald J, Tan EM. (1980). Autoantibody to centromere (kinetochore) in scleroderma sera. Proc Natl Acad Sci USA , 1627–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng TM, Lenstra TL, Duggan N, Jiang S, Ceto S, Holstege FC, Dai J, Boeke JD, Biggins S. (2013). Kinetochore function and chromosome segregation rely on critical residues in histones H3 and H4 in budding yeast. Genetics , 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obuse C, Yang H, Nozaki N, Goto S, Okazaki T, Yoda K. (2004). Proteomics analysis of the centromere complex from HeLa interphase cells: UV-damaged DNA binding protein 1 (DDB-1) is a component of the CEN-complex, while BMI-1 is transiently co-localized with the centromeric region in interphase. Genes Cells , 105–120. [DOI] [PubMed] [Google Scholar]
- Oegema K, Desai A, Rybina S, Kirkham M, Hyman AA. (2001). Functional analysis of kinetochore assembly in Caenorhabditis elegans. J Cell Biol , 1209–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. (2007). Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods , 709–712. [DOI] [PubMed] [Google Scholar]
- Prasad PD, Wang H, Kekuda R, Fujita T, Fei YJ, Devoe LD, Leibach FH, Ganapathy V. (1998). Cloning and functional expression of a cDNA encoding a mammalian sodium-dependent vitamin transporter mediating the uptake of pantothenate, biotin, and lipoate. J Biol Chem , 7501–7506. [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M. (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem , 663–670. [DOI] [PubMed] [Google Scholar]
- Rees JS, Li XW, Perrett S, Lilley KS, Jackson AP. (2015). Protein neighbors and proximity proteomics. Mol Cell Proteomics , 2848–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J. (2016). g:Profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res , W83–W89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science , 1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL. (1982). The formation, structure, and composition of the mammalian kinetochore and kinetochore fiber. Int Rev Cytol , 1–58. [DOI] [PubMed] [Google Scholar]
- Robinson BH, Oei J, Saunders M, Gravel R. (1983). [3H]biotin-labeled proteins in cultured human skin fibroblasts from patients with pyruvate carboxylase deficiency. J Biol Chem , 6660–6664. [PubMed] [Google Scholar]
- Roos UP. (1973). Light and electron microscopy of rat kangaroo cells in mitosis. II. Kinetochore structure and function. Chromosoma , 195–220. [DOI] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B. (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol , 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samejima I, Spanos C, Alves Fde L, Hori T, Perpelescu M, Zou J, Rappsilber J, Fukagawa T, Earnshaw WC. (2015). Whole-proteome genetic analysis of dependencies in assembly of a vertebrate kinetochore. J Cell Biol , 1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Musacchio A. (2009). The life and miracles of kinetochores. EMBO J , 2511–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Saffery R, Wong LH, Kalitsis P, Choo KH. (2002). Centromere proteins Cenpa, Cenpb, and Bub3 interact with poly(ADP-ribose) polymerase-1 protein and are poly(ADP-ribosyl)ated. J Biol Chem , 26921–26926. [DOI] [PubMed] [Google Scholar]
- Shang WH, Hori T, Martins NM, Toyoda A, Misu S, Monma N, Hiratani I, Maeshima K, Ikeo K, Fujiyama A, et al. (2013). Chromosome engineering allows the efficient isolation of vertebrate neocentromeres. Dev Cell , 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. (1996). Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem , 850–858. [DOI] [PubMed] [Google Scholar]
- Shuaib M, Ouararhni K, Dimitrov S, Hamiche A. (2010). HJURP binds CENP-A via a highly conserved N-terminal domain and mediates its deposition at centromeres. Proc Natl Acad Sci USA , 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Bodor DL, Stellfox ME, Martins NM, Hochegger H, Foltz DR, Jansen LE. (2012). Cdk activity couples epigenetic centromere inheritance to cell cycle progression. Dev Cell , 52–63. [DOI] [PubMed] [Google Scholar]
- Takata H, Matsunaga S, Morimoto A, Ma N, Kurihara D, ONO-Maniwa R, Nakagawa M, Azuma T, Uchiyama S, Fukui K. (2007). PHB2 protects sister-chromatid cohesion in mitosis. Curr Biol , 1356–1361. [DOI] [PubMed] [Google Scholar]
- Westhorpe FG, Straight AF. (2013). Functions of the centromere and kinetochore in chromosome segregation. Curr Opin Cell Biol , 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadzinska E, Huang J, Bailey AO, Guo LY, Lee NS, Srivastava S, Wong KA, French BT, Black BE, Foltz DR. (2018). Inheritance of CENP-A nucleosomes during DNA replication requires HJURP. Dev Cell , 348–362.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin SG, Shelby RD, Sullivan KF. (2001). CENP-A is phosphorylated by Aurora B kinase and plays an unexpected role in completion of cytokinesis. J Cell Biol , 1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.