

Abstract
Pre-mRNA-splicing and adenosine to inosine (A-to-I) RNA-editing occur mostly cotranscriptionally. During A-to-I editing, a genomically encoded adenosine is deaminated to inosine by adenosine deaminases acting on RNA (ADARs). Editing-competent stems are frequently formed between exons and introns. Consistently, studies using reporter assays have shown that splicing efficiency can affect editing levels. Here, we use Nascent-seq and identify ∼90,000 novel A-to-I editing events in the mouse brain transcriptome. Most novel sites are located in intronic regions. Unlike previously assumed, we show that both ADAR (ADAR1) and ADARB1 (ADAR2) can edit repeat elements and regular transcripts to the same extent. We find that inhibition of splicing primarily increases editing levels at hundreds of sites, suggesting that reduced splicing efficiency extends the exposure of intronic and exonic sequences to ADAR enzymes. Lack of splicing factors NOVA1 or NOVA2 changes global editing levels, demonstrating that alternative splicing factors can modulate RNA editing. Finally, we show that intron retention rates correlate with editing levels across different brain tissues. We therefore demonstrate that splicing efficiency is a major factor controlling tissue-specific differences in editing levels.
Adenosine to inosine editing (A-to-I editing) deaminates adenosines in double-stranded RNAs leading to nucleotide differences between RNA and DNA (Licht and Jantsch 2016; Eisenberg and Levanon 2018). Inosine is primarily interpreted as guanosine by cellular machines (Basilio et al. 1962; Licht et al. 2019). Hence, adenosine deamination has a profound impact on the affected RNAs, changing their coding potential, base-pairing abilities, folding, mRNA-translation efficiency, or proteins associated with the targeted RNAs (Bass 2002; Tajaddod et al. 2015; Nishikura 2016; Licht et al. 2019).
A-to-I editing is mediated by adenosine deaminases acting on RNAs (ADARs). In mammals, two catalytically active ADAR enzymes, ADAR (ADAR1) and ADARB1 (ADAR2) are known. Both enzymes have overlapping, yet distinct substrate specificities (Bass 2002). Mice lacking Adar die at embryonic day 12.5, accompanied by liver disintegration, hematopoietic defects, and an increase in interferon signaling (Hartner et al. 2004, 2009; Wang et al. 2004). Lethality can be rescued when sensors of viral dsRNAs (Ifih1 or Mavs) are also deleted (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015). Together, this suggests a role for Adar in innate immunity, possibly helping in discriminating self from nonself RNA (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015). Adarb1 null mice die within 3 wk after birth but are rescued when a pre-edited Gria2 allele is coexpressed (Brusa et al. 1995; Higuchi et al. 2000).
ADAR and ADARB1 require double-stranded RNA structures (dsRNA) for substrate recognition and editing (Higuchi et al. 1993; Herb et al. 1996). The sequence opposing the editing site is called the editing-complementary site (ECS). For exonic editing sites with an intronic ECS, editing needs to take place before mRNA-splicing (Higuchi et al. 1993; Herb et al. 1996). Consistently, Nascent-seq in Drosophila or human cells shows that the majority of editing takes place cotranscriptionally (Rodriguez et al. 2012; Hsiao et al. 2018). The efficiency of intron-removal has a direct impact on the level of editing and less efficient splicing generally leads to increased editing at sites that depend on an intronic ECS (Licht et al. 2016). Vice versa, editing can cause changes in alternative splicing: Intronic editing in the Adarb1 transcript leads to the inclusion of a premature termination codon (Rueter et al. 1999). Moreover, splicing of the glutamate receptor is seemingly regulated by A-to-I editing (Higuchi et al. 2000; Schoft et al. 2007; Penn et al. 2013). Finally, loss of ADARs causes transcriptome-wide alternative splicing changes (Solomon et al. 2013; St Laurent et al. 2013; Mazloomian and Meyer 2015; Hsiao et al. 2018).
Editing levels increase during development and vary between tissues, a phenomenon that cannot be explained by differential expression of RNA editing enzymes alone (Wahlstedt et al. 2009; Stulić and Jantsch 2013; Huntley et al. 2016; Tan et al. 2017). For instance, editing of some but not all protein-coding sites is particularly high in human arteries (Tan et al. 2017). The possible underlying mechanisms are poorly understood but likely involve substrate-specific regulation but also global regulation at different levels (Marcucci et al. 2011; Garncarz et al. 2013; Tariq et al. 2013; Oakes et al. 2017; Tan et al. 2017).
A-to-I editing sites have been mostly identified in different human tissues (Levanon et al. 2004; Li et al. 2009b; Peng et al. 2012; Ramaswami et al. 2012, 2013; Bazak et al. 2014; Picardi et al. 2015). Most of the sites locate to noncoding regions of transcripts, especially UTRs and introns, and are deposited in various editing databases (Kiran et al. 2013; Ramaswami and Li 2014; Picardi et al. 2017). REDIportal, for instance, contains a collection of over 4.5 million human A-to-I editing sites (Picardi et al. 2017). In contrast, only ∼8000 editing sites have been deposited for mice (Ramaswami and Li 2014). Here, we used Nascent-seq to identify mouse editing sites. Moreover, by manipulating splicing, we unravel how A-to-I editing is regulated during transcript maturation and by mRNA-splicing.
Results
Nascent-seq identifies over 90,000 novel editing sites
Most mouse A-to-I editing sites known today locate to exonic regions. To explore the intronic editome, we used Nascent-seq (Menet et al. 2012; Rodriguez et al. 2012). To allow high fidelity editing-site identification, we aimed to compare wild-type and editing-null mice. To achieve this, Adarb1−/− mice were rescued by a pre-edited version of the glutamate receptor (Adarb1−/−, Gria2R/R) (Higuchi et al. 2000) and crossed with mice heterozygous for Adar (Hartner et al. 2004) carrying a homozygous Mavs deletion (Adar−/+, Mavs−/−). After crossing the heterozygous offspring, we selected for F2 mice of genotype Adarb1−/−, Gria2R/R, Adar−/+, Mavs−/− which are fully viable. Crossing of these mice to each other resulted in 25% mice carrying a homozygous deletion for Adar. These mice are smaller when compared to their heterozygous Adar−/+ littermates and have a high mortality around day 15 after birth. For editing site determination, we isolated RNA at postnatal day 14 from editing-positive mice expressing ADAR and ADARB1: Adar+/+, Mavs−/−, Adarb1+/+, Gria2R/R (termed wildtype in this study). Secondly, we isolated RNA from editing-null mice (double-knockout or dko mice) where Adar and Adarb1 have been deleted: Adar−/−, Mavs−/−, Adarb1−/−, Gria2R/R. Thirdly, we analyzed RNA from mice where only Adarb1 (Adar2) had been deleted: Adar+/+, Mavs−/−, Adarb1−/−, Gria2R/R (called Adarb1−/− mice for simplicity).
We extracted nascent RNA in triplicate from brains of wild-type and dko mice followed by depletion of polyadenylated transcripts and removal of rRNA to enrich for nascent transcripts. Nascent RNA was sequenced (125-bp, paired-end) yielding between 113 mio and 247 mio uniquely mapped reads per replicate and mapped to the mm10 RefSeq genome. Comparison with poly(A)-mRNA-seq shows an increased intronic coverage (Supplemental Fig. S1). Editing site detection was done using the RDDpred package (Kim et al. 2016). RNA-DNA differences (RDDs) were counted and plotted according to the type of difference resulting in the raw RDDpred output (Fig. 1A).
Figure 1.

Nascent-seq identifies almost 100,000 mouse editing sites. (A) Nascent-RNA was prepared from brains of wild-type mice and subjected to Illumina sequencing (n = 3). After mapping to the mouse genome (mm10), potential RNA-DNA differences (RDDs) were determined using RDDpred (Raw RDDpred). To improve the identification of true RDDs, we removed all RDDs that either could not be mapped unambiguously to one strand, only occurred in one replica, or were also detected in an editing-deficient mouse line (Adar−/−, Adarb1−/−; A−/−, Ab1−/−). Thereby, we enriched for A-to-G RDDs indicative of A-to-I editing (wt stranded). (B) For validation, we used Sanger sequencing. Twenty-two editing sites were tested, and representative cDNA plus corresponding gDNA sequencing traces are shown (all traces are shown in Supplemental Fig. S2).
The majority of RDDs were A-to-G/T-to-C mismatches indicating A-to-I editing followed by C-to-T/G-to-A mismatches, suggesting C-to-U editing. All other transitions were found in relatively low numbers, demonstrating the reliability of the pipeline. Still, to further improve the quality, we removed all sites that were also detected in the double-knockout (Adar−/−, Adarb1−/−) or that could not be unambiguously aligned to one strand. This resulted in the wild type-only set (“wt/stranded”). Here, we almost exclusively observed A-to-G mismatches, suggesting enrichment for true A-to-I editing events. Using Sanger sequencing, we validated 21 out of 22 editing sites; i.e., 95% (Fig. 1B; Supplemental Fig. S2; Supplemental Tables S1, S2). As expected, we did not detect any A-to-G peak in the Adar−/−, Adarb1−/− set. This supports the notion that ADAR and ADARB1 are the only active editing enzymes. Subsequently, only A-to-G transitions exclusively detected in the wild type were considered for further analysis. Thereby, we identified almost 100,000 A-to-I editing sites, which we submitted to the REDIportal (http://srv00.recas.ba.infn.it/atlas/search_mm.html) (Picardi et al. 2017).
Nascent editing sites primarily locate to introns and are associated with repeat elements
Compared to previous studies, we identified over 90,000 novel editing sites (Supplemental Fig. S3; Supplemental Table S3). Of 97,416 identified editing sites, approximately 50,000 are not edited in the Adarb1−/− mice, suggesting that they are primarily edited by ADARB1 (Fig. 2A), and ∼47,000 sites can be edited by ADAR. To estimate the overall ADAR and ADARB1 “editing activity”, we compared the editing level averaged across all sites in wild-type and Adarb1−/− mice (Fig. 2B).
Figure 2.

Both ADAR- and ADARB1-mediated editing is primarily associated with intronic regions and repeat elements. (A,B) Adarb1 deletion leads to an ∼60% reduction in editing activity. (A) The total number of editing sites is shown separately for wild type and Adarb1−/− (sites edited by ADAR). (B) The average editing level of all identified editing sites is plotted for wild type and Adarb1−/− (overall ADAR editing activity). Error bars = standard deviation. n = 3. (C) The genomic annotation for all sites (dark blue), ADAR sites (blue), and ADARB1 sites (light blue) is given (Exon, Intron, UTR, [n.a.] not annotated/intergenic). In addition, for the subgroups Exon, Intron, and UTR, the predicted effect of editing is given using Ensembl's variant effect predictor (VEP). VEP terms: intron variant (intron); noncoding transcript variant (noncoding); noncoding transcript exon variant (noncoding exon); NMD transcript variant (NMD); regulatory region (regulatory); 5′ or 3′ UTR variant (5′ or 3′ UTR); missense variant (missense); synonymous variant (synonymous). (D) The percentage of editing sites associated with a particular repeat as identified by RepeatMasker is shown. Colors as in C. (E,F) Sequences enriched close to editing sites not associated with repeat elements (upper panel) or associated with repeat elements (lower panel) depicted separately for (E) ADAR or (F) ADARB1. The height of the nucleotide indicates either the degree of overrepresentation (above the line) or the degree of underrepresentation (below the line).
The editing level in Adarb1−/− mice is more than threefold lower compared to wild type, suggesting that ADARB1 is the dominant editase in the mouse brain. Eighty-six percent of the editing sites map to intronic regions (Fig. 2C). The increased intronic coverage also fostered the identification of many lowly edited sites (Supplemental Fig. S4). Using the Ensembl variant effect predictor (VEP), we predicted the effect of editing events separately for exonic, intronic, and UTR sites (Fig. 2C; McLaren et al. 2016). The majority of exonic sites, for instance, are predicted to impact noncoding transcripts, followed by regulatory regions and missense (nonsynonymous) variants. Intronic editing sites frequently locate to noncoding transcripts and transcripts predicted to be NMD-targets. It should be noted that “noncoding transcript” also covers transcript variants that overlay with protein-coding genes. The majority of UTR editing occurs in 3′ UTRs. Some predictions appear to clash with the overlying genomic annotation (e.g., 2.2% of the exonic variants are predicted to have an effect on introns). This can be explained by multiple VEP annotations for one genomic site (see Methods). The proportions of genomic locations and effects predicted by VEP did not change when plotted separately for ADAR and ADARB1 sites. The majority of editing sites are associated with repeat elements, in particular, short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs), and long terminal repeats (LTRs) (Fig. 2D). Both ADAR and ADARB1 exhibit similar preferences toward different classes of repeat elements. Only SINEs are slightly enriched for ADARB1 editing, whereas LINEs are enriched for ADAR-mediated editing. Subsequently, we analyzed the nucleotides enriched or underrepresented around editing sites separately for ADAR and ADARB1 and split these into repeat-associated and nonrepeat-associated sites (Fig. 2E,F). The sequence-motif strongly differs with respect to repeat status but exhibits only small differences for ADAR and ADARB1 sites. This suggests that substrate preferences for both editases differ only slightly. However, based on the VEP prediction and repeat status, this only moderately impacts the overall preference for specific transcript features.
Editing-complementary sequences (ECSs) oppose editing sites and form a dsRNA with the editing region. To identify ECSs regions, we calculated the energetically most favorable hybridization site between the region ±15 nt around all editing sites identified in the Nascent-seq data and the extended surrounding region of ±2500 nts around the editing sites. Thereby, we predicted ∼50,000 ECSs (Supplemental Fig. S5A; Supplemental Table S3). To test the quality and significance of the predicted pairing, several assays were performed. First, we deduced an empirical P-value by comparing hybridization energy of the predicted ECSs to the energies calculated for 1000 input sequences with shuffled dinucleotides. An ECS was accepted at a P-value ≤0.001. As a control, we repeated the analysis with randomly selected adenosines having the same genomic features (e.g., “intronic” plus “repeat”). In contrast to true editing sites, randomly selected genomic regions did not allow identification of ECSs of similar quality (Supplemental Fig. S5B). Next, to independently confirm the predictions, we analyzed the sequence spanning the editing site to the predicted ECS using the RNA folding prediction tool RNAfold and the structure visualization tool forna (Lorenz et al. 2011; Kerpedjiev et al. 2015). The structures predicted for all 10 randomly selected transcripts again identified the proposed ECS (Supplemental Data Set S1). Lastly, for experimental validation, we cloned the genomic DNA coding for 10 editing sites with or without the corresponding predicted ECS into pcDNA3.1− (Supplemental Data Set S1, Supplemental Table S4). Following cotransfection with a plasmid expressing FLAG-rADAR2, RT-PCR, and Sanger sequencing, we validated 10 out of 10 ECS predictions (Supplemental Fig. S6; Supplemental Table S5; Supplemental Methods).
The many intronic editing sites identified by Nascent-seq suggest that the majority of editing happens cotranscriptionally as seen before (Rodriguez et al. 2012; Hsiao et al. 2018). When we grouped editing sites according to the ratio of editing site coverage/ ECS coverage (number of reads mapping to the editing site/number of reads mapping to the ECS), we saw that editing sites with a lower ratio generally have a higher editing level and vice versa, suggesting that the co-occurrence of the ECS within the same transcript is a major determinant of editing levels. Such co-occurrence is likely modulated by transcription speed and processing efficiency (Fig. 3). This effect was particularly strong for editing sites within repeat elements (Supplemental Fig. S7).
Figure 3.
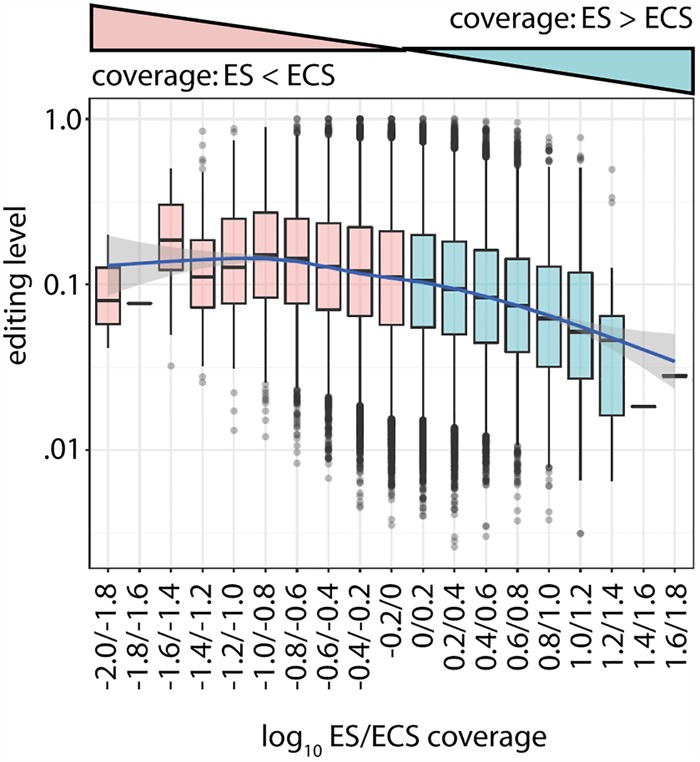
The persistence of the ECS increases editing levels. Box plot showing binned editing sites according to their log10 ES/ECS coverage (red: log10 ES/ECS coverage < 0 → ES saturated with ECS; blue: log10 ES/ECS coverage > 0 → ES deprived of ECS) and the respective editing level (left side).
Editing levels are cotranscriptionally modified by the efficiency of splicing
By studying a selected set of exonic editing sites, we previously showed that splicing efficiency is a major factor controlling the level of editing (Licht et al. 2016). To determine the impact of splicing on editing on a transcriptome-wide scale, we used the splicing inhibitor meayamycin (Gao et al. 2013). Meayamycin inhibits SF3b, a part of spliceosomal U2 snRNP at low nM concentrations. Meayamycin was used on short term cultures of two primary cell types. Firstly, we used bone marrow of 6-wk-old mice and inhibited splicing using a concentration of 5 nM meayamycin for 6 h. Secondly, we treated primary neuronal cultures established from brains of mice at embryonic day e11.5 with 15 nM meayamycin for 6 h.
For isolation of poly(A)-selected RNA and cDNA library preparation, 5 + 5 or 6 + 6 (untreated plus treated) biological replicates of primary neurons and bone marrow, respectively, were used. Libraries were sequenced in a paired-end 125-bp mode, and reads were mapped to the mouse genome (mm10). Each replicate yielded between 63 mio and 279 mio uniquely mapped reads.
Splicing inhibition works in both systems as evidenced by the larger proportion of reads mapping to intronic regions in the meayamycin-treated compared to control cells (Fig. 4A). Next, we quantified the number of reads at known editing sites (our Nascent-seq data plus known sites from the RADAR and DARNED databases [Kiran et al. 2013; Ramaswami and Li 2014]). We defined the editing level as the number of reads supporting editing divided by the total number of reads. We analyzed the bone marrow and the primary neuronal replicates (treated or untreated) separately and only considered editing sites that were supported by a minimum of five reads in each replicate of either the primary neuronal cultures or the bone marrow samples. Upon meayamycin treatment, 571 and 943 sites were found differentially edited (P-value < 0.05) in bone marrow and neuronal cultures, respectively (Fig. 4B; Supplemental Table S6). Editing generally increased upon treatment, most likely because splicing inhibition leads to a prolonged persistence of editing-competent dsRNA-structures formed within introns or between exons and introns (Fig. 4C). Consistently, the effect of splicing inhibition on editing is most pronounced for intronic and internal exonic sequences. In contrast, editing sites in UTRs are only modestly affected in the primary neuronal samples. Editing levels in bone marrow UTRs even decrease. A possible explanation could be that ADAR enzymes accumulate at intronic sequences and are depleted from UTRs under treatment conditions. This idea is supported by the analysis of the mean editing levels in meayamycin- and DMSO-treated cells (Fig. 4D). The basal editing levels (DMSO conditions) are lower in bone marrow (about 5%) than in primary neurons (about 10%). Under treatment conditions, the lower and thus potentially limiting editing activity in bone marrow may cause a shift of ADAR activity from UTR and not annotated sites to intronic and exonic sites. To further analyze this, we applied more stringent filtering using a cut-off of 10 reads minimum coverage and split editing sites into repeat-associated and nonrepeat-associated sites (Supplemental Fig. S8). Under these conditions, a similar shift in editing levels was observed, albeit the changes were stronger for repeat-associated sites. The position of the ECS played only a minor role, but the increase in editing was particularly strong for exonic sites in the bone marrow when the ECS was located in an intron, consistent with the notion that, in this scenario, the impact of splicing on editing levels should be particularly high (Supplemental Fig. S9). We validated the NGS results using Sanger sequencing with a validation rate of 83% (Fig. 4E; Supplemental Fig. S10). In sum, reduced splicing efficiency is associated with an increase in editing levels.
Figure 4.

Reduced splicing efficiency globally increases exonic and intronic editing. (A) Either bone marrow cells or primary neurons were treated with the splicing inhibitor meayamcyin (MEA) or vehicle control (DMSO). RNA was isolated after treatment, and poly(A)-selected RNA was subjected to RNA-seq. The relative intronic coverage over editing sites after treatment with MEA is shown. Bone marrow: n = 6, primary neurons: n = 5, error bars = SEM. (B) Bar plot displaying the overall number of significantly changed editing sites for bone marrow and primary neurons. (C) Box plots showing the log2 fold change (log2FC) for editing levels in untreated (DMSO) and treated (MEA) primary cells (bone marrow: left panel, primary neurons: right panel) separated into different genic locations (exonic, intronic, UTR, [n.a.] not annotated, i.e., intergenic). Dots represent single editing sites. Significantly changed sites are highlighted in red (P-value < 0.05). (D) Mean editing levels in bone marrow (left panel) or primary neurons (right panel) for grouped editing sites (intron, exon, UTR, n.a.) under DMSO conditions (blue dots) or meayamycin treatment (red dots). The blue (DMSO) or red (meayamycin) line is drawn at the mean editing level of all sites. Gray arrows indicate the shift in mean editing levels upon meayamycin treatment. (E) Validation of changed editing levels by Sanger sequencing. The editing site is marked by an arrow. The change in editing determined by RNA-seq is given below the chromatograms (+ or − indicates an increase or decrease upon treatment as determined by RNA-seq).
Alternative splicing factors are a potential novel class of editing regulators
Next, we determined whether alternative splicing factors can affect editing levels. To this end, we analyzed RNA-seq data from cortices of Nova1−/− and Nova2−/− knockout mice (Saito et al. 2016). Both NOVA1 and NOVA2 are brain-specific alternative splicing factors (Ule et al. 2006). After mapping the reads to the mouse genome, we again called editing levels at all known sites, requiring a minimum of five reads in each replicate. Using these criteria, we detected 15,103 sites that were edited in any of the genotypes (Supplemental Table S7). In Nova1−/− and Nova2−/− mice, 385 and 520 editing sites, respectively, were differentially edited as compared to wild type (P-value < 0.05) (Fig. 5A). Depending on the location of the NOVA-binding motif in the pre-mRNA, NOVA proteins can act as silencers or enhancers of splicing (Ule et al. 2006). Consequently, the overall level of editing was not shifted, suggesting that NOVA proteins rather modulate editing in a target-specific way, unlike in meayamycin-treated cells (Fig. 5B). To test this idea further, we next analyzed if the change in editing levels for individual sites (where we had identified an ECS) corresponded to the change in splicing efficiency. As a measure of splicing efficiency we determined the intron-specific read-coverage (more coverage = less efficient splicing; less coverage = more efficient splicing). To achieve this, we used DEXSeq (Anders et al. 2012) and applied a P-value of 0.1 (Fig. 5C). As expected, when the splicing efficiency was reduced as compared to wild-type mice, editing levels increased (and vice versa). Still, this general trend cannot be observed for all editing sites, suggesting that NOVA1 and NOVA2 also indirectly influence editing levels. Moreover, consistent with a binary role of NOVA1 and NOVA2 in alternative splicing, we did not observe a general increase in the rate of intron retention in the Nova1−/− or Nova2−/− knockout mice (Supplemental Fig. S11).
Figure 5.

The alternative splicing factors NOVA1 and NOVA2 modulate editing levels. (A) Re-analysis of publicly available RNA-seq data from the cortices of six wild-type and three Nova1−/− and three Nova2−/− mice (Saito et al. 2016). The reads have been mapped to the mouse genome (mm10), and the editing level of known editing sites was determined. The total number of significantly changed editing sites is given (P-value < 0.05). (B) Editing levels in cortices of wild-type and either Nova1−/− (left panel) or Nova2−/− (right panel) knockout mice were determined, and the change in editing levels was plotted. Dots represent single editing sites. Significantly changed sites are highlighted in red (P-value < 0.05). A separate box plot is given for different genic locations (exonic, intronic, UTR, [n.a.] not annotated/intergenic). (C) Box plot showing the log2 fold change of editing levels for editing sites with up-regulated splicing efficiency (increase) or down-regulated splicing efficiency (decrease) for wild-type versus Nova1−/− (green) or wild-type versus Nova2−/− (blue) mice. Splicing efficiency is determined by a decrease (up-regulated splicing efficiency) or increase in intron-specific coverage (down-regulated splicing efficiency) as determined by DEXSeq (P-value < 0.1).
Splicing controls tissue-specific editing levels
Alternative splicing and intron retention levels vary between tissues and are important for defining tissue-specificity (Braunschweig et al. 2014; Baralle and Giudice 2017). Alternative splicing is enriched in the brain and regulated by brain-specific splicing factors including NOVA1 or NOVA2 (Raj and Blencowe 2015). We therefore reasoned that splicing efficiency may contribute to observed differences in editing between tissues. The impact of splicing on editing is particularly high when the editing site is coordinated with an intronic ECS (Licht et al. 2016). Most editing targets with an intronic ECS like GRIA2 or HTR2C are primarily expressed in different brain regions (Li et al. 2009b; Heraud-Farlow et al. 2017). Therefore, we collected all RNA-seq data for 13 different brain regions available from the Genotype-Tissue Expression (GTEx) Consortium (The GTEx Consortium 2013). From these, we calculated the editing level for all exonic human editing sites available through the RADAR database that are conserved between human and either chimp, rhesus, or mouse (Ramaswami and Li 2014). As a measure of splicing efficiency, we calculated the level of intron retention by dividing the exonic by intronic read coverage. Next, we removed all editing sites where no editing was detected or no reads mapped to the intron. Moreover, we required both editing levels and splicing efficiency values to be calculated for at least 10 tissues supported by a minimum of two samples, leaving a total of 45 editing sites. Subsequently, editing sites were classified according to the location of the ECS as either intronic or exonic. We defined an ECS as intronic when the editing-competent duplex was formed between the edited exon and the downstream or upstream intron, whereas an exonic ECS contains an editing-competent dsRNA-stem solely within the edited exon (Fig. 6A,B). If the location of the ECS was not known, we identified the ECS using RNA secondary structure prediction tools (Lorenz et al. 2011; Kerpedjiev et al. 2015). Subsequently, we compared our structures to published predictions (if available) and found that all of them match (Supplemental Table S8). In sum, we classified 16 sites as intronic and 29 sites as exonic (Supplemental Table S8; Supplemental Data Set S2).
Figure 6.

Splicing controls tissue-specific A-to-I editing for intron-dependent sites. (A,B) Exonic editing sites either depend on an (A) intronic editing complementary site (intronic ECS) or (B) exonic ECS. Exons are shown as blue bars, introns as thin lines. The editing site (A → I) is depicted. (C,D) Representative graphs for the correlation between average editing level and average exon/intron coverage at the downstream intron (as a measure of splicing efficiency) are shown for sites with a (C) intronic ECS like FLNB and GRIK2 or (D) exonic ECS like GABRA3 across different brain regions (RNA-seq data from GTEx). (E,F) The correlation (r = Pearson correlation coefficient) between average editing levels and average exon/intron ratio for conserved editing sites (Coordinates: hg19; [Chr.] chromosome; [Str.] strand) is plotted when the read coverage allowed calculation of editing levels and exon/intron ratio for at least 10 tissues (n = number of tissues) with minimum of two samples. Color code ranges from blue = strong negative correlation over white = no correlation to red = positive correlation. The significance is indicated by one or two asterisks: (*) P-value < 0.05, (**) P-value < 0.01.
The expected negative correlation between editing levels and splicing efficiency was observed for editing sites with an intronic ECS like FLNB or GRIK2 (Fig. 6C). In contrast, no correlation was observed at sites with an exonic ECS like GABRA3 (Fig. 6D). Other editing sites with an intronic or exonic ECS exhibit a similar trend (Fig. 6E,F). This clearly indicates that editing levels in a particular tissue largely depend on splicing efficiency when the ECS is formed with the intron and identifies tissue-specific splicing as a major factor controlling editing.
Discussion
In the mouse, only about 8000 A-to-I editing sites are known and cataloged (Ramaswami and Li 2014). Still, mice are an indispensable model system to study editing—for instance, to delineate the role of ADAR editing in innate immunity (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015). Using mice devoid in all active ADAR enzymes, we performed Nascent-seq to obtain a comprehensive set of nearly 100,000 A-to-I editing sites, most of which are novel. To our knowledge, this is the first study where a complete editing-null mouse has been used as a negative control to filter against false-positive editing sites.
ADARB2 (ADAR3) is a third member of the ADAR proteins (Chen et al. 2000). While the presence of a deaminase domain suggests that ADARB2 may have deaminating activity, no editing activity could be detected so far (Chen et al. 2000; Schneider et al. 2014). Still, it cannot be excluded that ADARB2 acts on unknown substrates. Here, we show on a transcriptome-wide scale that mice without both, ADAR and ADARB1, lack any significant mRNA-deamination activity, indicating that ADARB2 is not an active deaminase (see Fig. 1B). Transitions detected in the double-knockout mouse mostly reflect C-to-U editing but may also indicate RDDs that emerge due to unknown biological activity shortly after transcription (Wang et al. 2014).
Nascent-seq performed on brain tissue lacking only Adarb1 showed an approximately threefold drop in editing in Adarb1−/− mice. This is consistent with previous findings showing higher expression of ADARB1 in the mouse brain as compared to ADAR (Heraud-Farlow et al. 2017). Apparently, ADAR can only partially compensate for the lack of ADARB1 editing activity. The analysis of ADAR- and ADARB1-mediated editing in the human GTEx data suggested that ADAR primarily edits repetitive sequences whereas ADARB1 is the primary enzyme targeting nonrepetitive coding sequences (Tan et al. 2017). In contrast, we show here that both ADAR and ADARB1 act on repeat elements. As we focus on the mouse brain, where ADARB1 is highly expressed, we cannot exclude that ADAR is primarily acting on repeats outside the nervous system. A potential drawback of our Nascent-seq approach is the lack of Nascent-seq data for the Adar−/− genotype. Therefore, we cannot clearly distinguish editing sites that could be edited by either ADAR or ADARB1 with equal efficiency. Such sites would be falsely identified as exclusive ADAR sites in our study. Also, if ADAR and ADARB1 needed to form heterodimers at particular sites, such sites would be falsely identified as exclusive ADARB1 sites. Thus, while we are confident that most sites are properly assigned as substrates of one of the two ADAR enzymes, our analysis clearly underestimates the number of sites where ADARB1 could act.
Using a mutational approach, we had shown previously that the efficiency of splicing is a key mechanism to control the level of editing at several selected sites (Licht et al. 2016). Here, using transcriptome-wide analysis, we can show that inhibition of splicing leads to a general increase of editing in intron-containing transcripts and mRNAs. The increase in editing upon splicing inhibition is particularly strong for exonic editing sites with adjacent intronic ECSs. Editing in UTRs does not follow a general trend, as a decrease was observed in bone marrow whereas UTR-editing in primary neurons was not shifted. Differences in basal editing activity between both primary cell systems may explain this. While the mean editing level in bone marrow is ∼5%, the level in primary neurons is almost twice as high. Moreover, ADARB1 is the primary editase in brain, while in bone marrow only ADAR is expressed at significant levels (Heraud-Farlow et al. 2017).
Consistent with the role of splicing on editing, altered editing patterns were detected in RNA-seq data from Nova1−/− or Nova2−/− knockout mice. NOVA1 and NOVA2 can either enhance splicing or silence splicing (Ule et al. 2006). Consistently, both increased and decreased editing levels were observed in Nova1−/− or Nova2−/− knockout mice. For most sites an increase in editing was accompanied with a drop in splicing efficiency and vice versa. Still, as this was not observed at all editing sites, NOVA1 and NOVA2 may also indirectly influence editing, for instance, by competing with other RNA-binding proteins. The splicing factor SRSF9 was found to repress ADARB1-mediated editing of brain-specific sites (Huang et al. 2018; Shanmugam et al. 2018; Quinones-Valdez et al. 2019). Moreover, a recent survey for the impact of RNA-binding proteins on editing levels revealed that alternative splicing factors have a great impact on editing levels (Quinones-Valdez et al. 2019). Together with our data, this suggests that alternative splicing factors are important editing regulatory factors that fine-tune editing levels similarly as established modulators of editing like AIMP2 and PIN1, or the E3 ligase WWP2 (Marcucci et al. 2011; Tan et al. 2017). Alteration of ADAR levels or its catalysis rate would obviously affect all substrates in one cell or tissue to the same extent. However, it is known that editing levels can vary in a substrate-specific manner between tissues. This is most clearly exemplified for the two RNAs encoding the paralogous proteins filamin A (FLNA) and filamin B (FLNB). While editing of the Flna transcript is highest in the cardiovascular system and the large intestine, editing of Flnb is low (Stulić and Jantsch 2013). Instead, Flnb editing levels are highest in the cartilaginous tissue and muscle (Czermak et al. 2018). Therefore, substrate-specific regulation must occur in these cases. RNA-binding proteins may compete with ADARs in a substrate-specific manner (Garncarz et al. 2013; Tariq et al. 2013; Quinones-Valdez et al. 2019). Our finding that splicing efficiency is a major factor in regulating editing levels may explain substrate-specific and tissue-specific regulation of editing levels. However, not all edited substrates follow the same trend. For instance, the editing level of the Gria2 Q/R editing site does not correlate with reduced splicing efficiency. In contrast to other editing sites, editing of the Gria2 Q/R site is close to 100% and required to support mammalian life (Higuchi et al. 2000). Seemingly, only edited Gria2 pre-mRNA can be spliced efficiently to release the mature transcript (Higuchi et al. 2000; Schoft et al. 2007; Penn et al. 2013). This suggests a specific regulation of Gria2 Q/R site editing and splicing that does not follow the general trend described here.
In sum, our study highlights the importance of cotranscriptional coordination between RNA editing and mRNA splicing and shows that splicing efficiency and splicing factors are major factors for the regulation of editing levels.
Methods
Isolation of nascent RNA and library preparation
Mavs+/− mice were acquired from Jackson Laboratory and crossed with Adar+/− and subsequently with Adarb1−/−, Gria2R/R (stock #008634, allele: Mavstm1Zjc). For Nascent-seq, mice of desired genotypes were sacrificed at p14, and nascent-RNA was isolated from brain tissue (Menet et al. 2012). After treatment with DNase I (Thermo Fisher Scientific), contaminating polyadenylated RNA was removed using the NEBNext poly(A) Isolation Module (New England Biolabs). Ribosomal RNA was removed using Ribo-Zero (Illumina). cDNA libraries were generated from 1 µg of nascent-RNA using NEBNext Ultra Directional RNA Library Prep kit for Illumina (New England Biolabs) and sequenced in paired-end mode with 125-bp read length on a HiSeq 2500 (Illumina).
Splicing inhibition in bone marrow or primary neuronal cells
Bone marrow cultures were established as described (Kroeger et al. 2009; Licht et al. 2016). Bone marrow from one mouse was split and either treated with 5 nM meayamycin for 6 h or DMSO as vehicle control (Gao et al. 2013). Primary neuronal cultures were established from mouse embryos at e11.5. Each embryo isolate was split into two wells of a six-well dish coated with poly-D-lysine and cultured as established previously (Licht et al. 2016). One well was treated with 15 nM meayamycin for 6 h, while the corresponding well was treated with DMSO as control. RNA was isolated using TriFast (VWR Peqlab) and DNase I treated (Thermo Fisher Scientific). Libraries were generated as described above but from polyadenylated RNA isolated using the NEBNext poly(A) mRNA Magnetic Isolation Module (New England Biolabs).
Nascent-seq data analysis
Sequenced reads were quality-trimmed and adapter-clipped using Trimmomatic (version 0.33) (Bolger et al. 2014) with default parameters. Quality was monitored using FastQC (version 0.11.3). Reads were mapped against the mouse reference genome (assembly mm10) using STAR (version 2.5.2b). RDDpred (Kim et al. 2016) was utilized for the detection of editing sites. MES-sites (Mapping Error-prone Sites; negative reference set) are provided for mm10 (http://epigenomics.snu.ac.kr/RDDpred/prior_data/Mouse.MES.txt.gz). Annotated editing sites from the databases DARNED (Kiran et al. 2013) and RADAR (Ramaswami and Li 2014) served as positive reference sites. DARNED and RADAR sites were converted to the mm10 coordinates using liftOver (Kuhn et al. 2013). Only RNA-DNA differences with a score ≥0.98 were considered. Subsequently, all sites overlapping with an annotated SNP, based on the variant annotation provided by the Mouse Genome Project (version 5) were removed (Keane et al. 2011). ES candidates exhibiting edited reads in at least two out of three WT samples, but in none of the dko samples, were classified as “WT only”. Conversely, sites with edited reads in at least two dko but none in WT samples were classified as “DKO only”. Editing sites were filtered and classified according to their strand topology. To this end, mapped paired-read files were split into separate half-samples containing only the first or second read in each pair. For each potential editing site, each half-sample was queried for observed substitution types considering the deriving strand. A substitution event was considered if it was observed in more than one read in more than two half-samples. We, and others before us (Levin et al. 2010), observed library-specific systematic error in the strand assignment of the reads (antisense shadow). Therefore, for sites with a relative antisense signal less than four times the global deduced antisense shadow, the signal from the less productive strand was ignored. To deduce the global antisense shadow, we applied the same strategy as described in Amman et al. (2018) for each chromosome and merged the single values by forming the average weighted by the chromosome length. The observed antisense shadows range between 0.34% and 1.74%. Integrating all this information, candidate editing sites were assigned a specific stranded substitution event or removed due to ambiguity (indicative of sequencing/mapping artifacts) or observation of corresponding substitutions on the opposing strand (indicative of a genomic variant). Finally, only unambiguous A → G events that occurred in the WT samples only were considered as true editing events. For all editing sites, the editing level was calculated using bam-readcounts (https://github.com/genome/bam-readcount). For follow-up analysis, editing sites supported by less than five reads were omitted. For ADARB1 knockout samples, editing sites were treated as edited if edited reads could be observed in more than one (out of three) samples or had a cumulative editing level greater than 1%. The obtained editing sites were characterized with respect to their genomic context (GENCODE basic annotation; release 17). Each ES was assigned to be either located in an UTR, exon, intron, or intergenic. If more than one classification applied to one ES, the higher ranked (UTR > exon > intron > intergenic) was selected. Subsequently, the potential impact of the observed A → G substitution was interrogated using Ensembl's variant predictor tool (VEP). If multiple effects for one substitution were predicted, only one variant was reported according to the following ranking: missense_variant > synonymous_variant > stop_lost > stop_ retained_variant > splice_acceptor_variant > splice_donor_variant > splice_region_variant > TF_binding_site_variant > regulatory_ region_variant > mature_miRNA_variant > 5_prime_UTR_variant > 3_prime_UTR_variant > non_coding_transcript_exon_variant > non_coding_transcript_variant > NMD_transcript_variant > intron_ variant > downstream_gene_variant > upstream_gene_variant > intergenic_variant. For a description of the variants see: www.ensembl.org/info/genome/variation/prediction/predicted_data.html#consequences.
To characterize the repeat status of an ES, RepeatMasker was used (Smit et al. 2013–2015) (www.repeatmasker.org; mm10; Repeat Library 20140131).
To calculate hybridization energies between an editing site and a potential editing complementary site (ECS), the program RNAplex was used (Tafer et al. 2011). To determine the read coverage of editing sites that are supported by at least five reads in all samples and their respective ECS, BEDTools multicov was used (Quinlan and Hall 2010).
To test for sequence motifs, all sites were separated according to their repeat association and their ADAR, ADARB1 preference. For each subset, a region ±5 nucleotides around the editing site was analyzed using pLogo generator (https://plogo.uconn.edu/) (O'Shea et al. 2013). To construct a sequence background that reflects the underlying base composition as closely, for each editing site a random 100-nt sequence stretch randomly drawn from within the surrounding ±200 bases around the editing site was used.
Validation of editing events using Sanger sequencing
For validation of editing events, Sanger sequencing on cDNA libraries and corresponding genomic DNA from the same individual was used. Editing sites with at least 10% editing (Deffit et al. 2017) were validated, as low levels of editing are difficult to discern in Sanger sequencing chromatograms (Eggington et al. 2011). Similarly, for the validation of the splicing inhibition data, only editing sites with a minimum of 20% difference in editing were considered for validation. The validation was done on previously generated cDNA libraries. One-half microliter of the library (or genomic DNA) was amplified in a 25-µL reaction using OneTaq Quick-Load 2× Master Mix with Standard Buffer (New England Biolabs) using the following PCR protocol: Initial denaturation: 1 min at 94°C, followed by 35 cycles of 94°C (30 sec), 58°C (30 sec), 68°C (30 sec), and a final elongation for 5 min. PCR products were separated using gel electrophoresis and purified by gel-elution. Sanger sequencing was done using the eluted PCR products and the reverse primer (oligonucleotide sequences: Supplemental Table S1). Geneious v11 (Biomatters) was used to analyze Sanger chromatograms. The percentage of editing is defined as the height of the G peak divided by the sum of the A + G peaks (in the case of the reverse primer: the height of the T peak divided by the sum of the T + C peaks).
Analysis of the splicing inhibition and Nova1/2 knockout cortices
Mapping and editing level detection was done as described above. Editing sites which are not supported by at least five reads in all samples were omitted. Differentially edited sites were determined applying a t-test with the Welch approximation for the degrees of freedom on the log10 values of the observed levels. Sites with an editing level of zero were set to 0.001 before applying the log10. Unmapped reads for Nova1 and Nova2 knockout mice (cortex) and corresponding wild-type controls were downloaded from the Gene Expression Omnibus (GEO) repository (GSE69711). Reads were adapter-trimmed using Trimmomatic (Bolger et al. 2014) and subsequently mapped to the mouse genome (mm10) using Bowtie 2 (Langmead and Salzberg 2012) with sensitive parameters (-L 20, -N 1). Finally, the output was processed using SAMtools to generate sorted and indexed BAM files (Li et al. 2009a). The level of editing for previously identified editing sites (Nascent-seq) was determined by counting the percent of G-reads (+strand) or C-reads (−strand) using a custom script relying on SAMtools. Differential editing was calculated with the same parameters as described for the meayamycin treatment.
To determine the effect of NOVA1 or NOVA2 deletion on splicing efficiency for individual introns, DEXSeq (Anders et al. 2012) was used. The original DEXSeq workflow was designed to test for differential exon usage between different conditions. To include also intron retention events, we modified the first step of the analysis pipeline in which a nonoverlapping exon reference annotation is produced from the gene annotation with DEXSeq's own dexseq_prepare_annotation.py Python script. We forced the script to include also intron regions by providing for each annotated transcript (i.e., exon chain) additionally a complementary intron chain. In this step, the parameter “–aggregate no” was set to prevent genes sharing exons from being merged into aggregated genes. Furthermore, transcript parts shorter than 10 bases were removed. From there, the standard DEXSeq workflow was used of counting reads and testing for differential transcript part usage (in the original pipeline called “exon usage”) between the WT and the respective mutant. Eventually, this information was linked with the information on differential editing levels between the two conditions for all editing sites that exhibit significant differential editing (P-value < 0.1). The distribution of the log2FC of editing levels was plotted separately for editing sites located in an up- or down-regulated transcript part, according to the DEXSeq analysis.
GTEx data
GTEx data were downloaded from the NCBI database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap; accession number phs000424.v6.p1). For each RNA editing site, genomic coordinates of the exon including the editing event as well as coordinates of the flanking intron were extracted by using RefSeq annotations (downloaded from UCSC) and custom scripts. Such coordinates were provided in input (in SAF format) to the featureCounts tool (Liao et al. 2014) in order to compute the number of reads supporting individual exons and introns in 329 GTEx RNA-seq experiments from 13 different brain locations. RNA-seq reads were downloaded from the dbGaP database and aligned onto the human reference genome (hg19 assembly) by means of STAR (Dobin et al. 2013). Exonic and intronic read counts were used to calculate an intron retention (IR) score that takes into account the exon to intron expression ratio. Finally, for each RNA editing site, IR scores and editing levels were correlated across brain locations. RNA editing levels for GTEx brain tissues were downloaded from REDIportal database (Picardi et al. 2017). Calculations were performed using custom Python scripts. Coordinates in the REDIportal are from the hg19 assembly. Consistently, we used hg19 for GTEx data analysis. Protein-coding genes used for our analysis are well annotated in this release. Consequently, the use of hg38 does not improve the analysis.
Data access
RNA-seq data from this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena) under accession number PRJEB27264. A-to-I editing sites identified using Nascent-seq have been submitted to REDIportal (http://srv00.recas.ba.infn.it/atlas/search_mm.html). Sanger sequencing chromatograms and Python scripts are available as Supplemental Material (Supplemental_Chromatograms.zip; Supplemental_Scripts.zip).
Supplementary Material
Acknowledgments
This work was supported by grants from the Austrian Science Foundation (FWF), grant numbers P26845 and P26882 to M.F.J. and P30505 to K.L. U.K. was supported by funding from DoktoratsKolleg RNA Biology PhD Program at Vienna BioCenter. Sequencing was performed at the VBCF NGS Unit (www.viennabiocenter.org/facilities). We thank the European Elixir infrastructure (https://www.elixir-europe.org/) and Elixir-ITA for supporting the development and maintenance of REDIportal, Interomics, and PRACE (Pra15_3924 to E.P.) for computational resources to handle and process GTEx data sets. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. We also thank Kazunori Koide (Department of Chemistry, University of Pittsburgh, PA) for meayamycin. We thank Anikó Kasztner, Robert Vilvoi, Michael Janisiw, and Peter Burg for excellent technical support, and we thank all Jantsch group members for helpful discussions and acknowledge the vibrant Vienna RNA community.
Author contributions: Conceptualization: K.L. and M.F.J.; methodology: K.L., U.K., F.A., E.P., D.M., and M.F.J.; experimental work: K.L., U.K., and P.B.; scripting and software: F.A., E.P., and D.M.; analysis: K.L., U.K., F.A., and E.P.; data curation: K.L., F.A., and E.P.; writing: K.L. and M.F.J. (with input from all other authors); funding acquisition: M.F.J.; supervision: M.F.J.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.242636.118.
Freely available online through the Genome Research Open Access option.
References
- Amman F, D'Halluin A, Antoine R, Huot L, Bibova I, Keidel K, Slupek S, Bouquet P, Coutte L, Caboche S, et al. 2018. Primary transcriptome analysis reveals importance of IS elements for the shaping of the transcriptional landscape of Bordetella pertussis. RNA Biol 15: 967–975. 10.1080/15476286.2018.1462655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W. 2012. Detecting differential usage of exons from RNA-seq data. Genome Res 22: 2008–2017. 10.1101/gr.133744.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baralle FE, Giudice J. 2017. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol 18: 437–451. 10.1038/nrm.2017.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basilio C, Wahba AJ, Lengyel P, Speyer JF, Ochoa S. 1962. Synthetic polynucleotides and the amino acid code, V. Proc Natl Acad Sci 48: 613–616. 10.1073/pnas.48.4.613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass B. 2002. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71: 817–846. 10.1146/annurev.biochem.71.110601.135501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, et al. 2014. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24: 365–376. 10.1101/gr.164749.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. 10.1101/gr.177790.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh D, Feldmeyer D, Gass P, Seeburg P, Sprengel R. 1995. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270: 1677–1680. 10.1126/science.270.5242.1677 [DOI] [PubMed] [Google Scholar]
- Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. 2000. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6: 755–767. 10.1017/S1355838200000170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermak P, Amman F, Jantsch MF, Cimatti L. 2018. Organ-wide profiling in mouse reveals high editing levels of Filamin B mRNA in the musculoskeletal system. RNA Biol 15: 877–885. 10.1080/15476286.2018.1480252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deffit SN, Yee BA, Manning AC, Rajendren S, Vadlamani P, Wheeler EC, Domissy A, Washburn MC, Yeo GW, Hundley HA. 2017. The C. elegans neural editome reveals an ADAR target mRNA required for proper chemotaxis. eLife 6: e28625 10.7554/eLife.28625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL. 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319 10.1038/ncomms1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY. 2018. A-to-I RNA editing—immune protector and transcriptome diversifier. Nat Rev Genet 19: 473–490. 10.1038/s41576-018-0006-1 [DOI] [PubMed] [Google Scholar]
- Gao Y, Vogt A, Forsyth CJ, Koide K. 2013. Comparison of splicing factor 3b inhibitors in human cells. Chembiochem 14: 49–52. 10.1002/cbic.201200558 [DOI] [PubMed] [Google Scholar]
- Garncarz W, Tariq A, Handl C, Pusch O, Jantsch MF. 2013. A high-throughput screen to identify enhancers of ADAR-mediated RNA-editing. RNA Biol 10: 192–204. 10.4161/rna.23208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2013. The Genotype-Tissue Expression (GTEx) project. Nat Genet 45: 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279: 4894–4902. 10.1074/jbc.M311347200 [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. 2009. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10: 109–115. 10.1038/ni.1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heraud-Farlow JE, Chalk AM, Linder SE, Li Q, Taylor S, White JM, Pang L, Liddicoat BJ, Gupte A, Li JB, et al. 2017. Protein recoding by ADAR1-mediated RNA editing is not essential for normal development and homeostasis. Genome Biol 18: 166 10.1186/s13059-017-1301-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herb A, Higuchi M, Sprengel R, Seeburg PH. 1996. Q/R site editing in kainate receptor GluR5 and GluR6 pre-mRNAs requires distant intronic sequences. Proc Natl Acad Sci 93: 1875–1880. 10.1073/pnas.93.5.1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Single FN, Köohler M, Sommer B, Sprengel R, Seeburg PH. 1993. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 75: 1361–1370. 10.1016/0092-8674(93)90622-W [DOI] [PubMed] [Google Scholar]
- Higuchi M, Stefan M, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81. 10.1038/35017558 [DOI] [PubMed] [Google Scholar]
- Hsiao YE, Bahn JH, Yang Y, Lin X, Tran S, Yang EW, Quinones-Valdez G, Xiao X. 2018. RNA editing in nascent RNA affects pre-mRNA splicing. Genome Res 28: 812–823. 10.1101/gr.231209.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Kapeli K, Jin W, Wong YP, Arumugam TV, Koh JH, Srimasorn S, Mallilankaraman K, Chua JJE, Yeo GW, et al. 2018. Tissue-selective restriction of RNA editing of CaV1.3 by splicing factor SRSF9. Nucleic Acids Res 46: 7323–7338. 10.1093/nar/gky348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley MA, Lou M, Goldstein LD, Lawrence M, Dijkgraaf GJ, Kaminker JS, Gentleman R. 2016. Complex regulation of ADAR-mediated RNA-editing across tissues. BMC Genomics 17: 61 10.1186/s12864-015-2291-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. 2011. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477: 289–294. 10.1038/nature10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerpedjiev P, Hammer S, Hofacker IL. 2015. Forna (force-directed RNA): simple and effective online RNA secondary structure diagrams. Bioinformatics 31: 3377–3379. 10.1093/bioinformatics/btv372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Hur B, Kim S. 2016. RDDpred: a condition-specific RNA-editing prediction model from RNA-seq data. BMC Genomics 17(Suppl 1): 5 10.1186/s12864-015-2301-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiran AM, O'Mahony JJ, Sanjeev K, Baranov PV. 2013. Darned in 2013: inclusion of model organisms and linking with Wikipedia. Nucleic Acids Res 41: D258–D261. 10.1093/nar/gks961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger K, Collins M, Ugozzoli L. 2009. The preparation of primary hematopoietic cell cultures from murine bone marrow for electroporation. J Vis Exp 23: 1026 10.3791/1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn RM, Haussler D, Kent WJ. 2013. The UCSC genome browser and associated tools. Brief Bioinform 14: 144–161. 10.1093/bib/bbs038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005. 10.1038/nbt996 [DOI] [PubMed] [Google Scholar]
- Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. 2010. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7: 709–715. 10.1038/nmeth.1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009a. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, Levanon EY, Yoon JK, Aach J, Xie B, Leproust E, Zhang K, Gao Y, Church GM. 2009b. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324: 1210–1213. 10.1126/science.1170995 [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Licht K, Jantsch MF. 2016. Rapid and dynamic transcriptome regulation by RNA editing and RNA modifications. J Cell Biol 213: 15–22. 10.1083/jcb.201511041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht K, Kapoor U, Mayrhofer E, Jantsch MF. 2016. Adenosine to inosine editing frequency controlled by splicing efficiency. Nucleic Acids Res 44: 6398–6408. 10.1093/nar/gkw325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht K, Hartl M, Amman F, Anrather D, Janisiw MP, Jantsch MF. 2019. Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res 47: 3–14. 10.1093/nar/gky1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. 10.1126/science.aac7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höoner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26 10.1186/1748-7188-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. 10.1016/j.celrep.2014.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci R, Brindle J, Paro S, Casadio A, Hempel S, Morrice N, Bisso A, Keegan LP, Del Sal G, O'Connell MA. 2011. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J 30: 4211–4222. 10.1038/emboj.2011.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazloomian A, Meyer IM. 2015. Genome-wide identification and characterization of tissue-specific RNA editing events in D. melanogaster and their potential role in regulating alternative splicing. RNA Biol 12: 1391–1401. 10.1080/15476286.2015.1107703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. 2016. The Ensembl Variant Effect Predictor. Genome Biol 17: 122 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet JS, Rodriguez J, Abruzzi KC, Rosbash M. 2012. Nascent-Seq reveals novel features of mouse circadian transcriptional regulation. eLife 1: e00011 10.7554/eLife.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17: 83–96. 10.1038/nrm.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes E, Anderson A, Cohen-Gadol A, Hundley HA. 2017. Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J Biol Chem 292: 4326–4335. 10.1074/jbc.M117.779868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea JP, Chou MF, Quader SA, Ryan JK, Church GM, Schwartz D. 2013. pLogo: a probabilistic approach to visualizing sequence motifs. Nat Methods 10: 1211–1212. 10.1038/nmeth.2646 [DOI] [PubMed] [Google Scholar]
- Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, et al. 2012. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 30: 253–260. 10.1038/nbt.2122 [DOI] [PubMed] [Google Scholar]
- Penn AC, Balik A, Greger IH. 2013. Steric antisense inhibition of AMPA receptor Q/R editing reveals tight coupling to intronic editing sites and splicing. Nucleic Acids Res 41: 1113–1123. 10.1093/nar/gks1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43: 933–944. 10.1016/j.immuni.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, Manzari C, Mastropasqua F, Aiello I, D'Erchia AM, Pesole G. 2015. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep 5: 14941 10.1038/srep14941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, D'Erchia AM, Lo Giudice C, Pesole G. 2017. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res 45: D750–D757. 10.1093/nar/gkw767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinones-Valdez G, Tran SS, Jun HI, Bahn JH, Yang EW, Zhan L, Brummer A, Wei X, Van Nostrand EL, Pratt GA, et al. 2019. Regulation of RNA editing by RNA-binding proteins in human cells. Commun Biol 2: 19 10.1038/s42003-018-0271-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, Blencowe BJ. 2015. Alternative splicing in the mammalian nervous system: recent insights into mechanisms and functional roles. Neuron 87: 14–27. 10.1016/j.neuron.2015.05.004 [DOI] [PubMed] [Google Scholar]
- Ramaswami G, Li JB. 2014. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 42: D109–D113. 10.1093/nar/gkt996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Lin W, Piskol R, Tan MH, Davis C, Li JB. 2012. Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods 9: 579–581. 10.1038/nmeth.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. 10.1038/nmeth.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Menet JS, Rosbash M. 2012. Nascent-Seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell 47: 27–37. 10.1016/j.molcel.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB. 1999. Regulation of alternative splicing by RNA editing. Nature 399: 75–80. 10.1038/19992 [DOI] [PubMed] [Google Scholar]
- Saito Y, Miranda-Rottmann S, Ruggiu M, Park CY, Fak JJ, Zhong R, Duncan JS, Fabella BA, Junge HJ, Chen Z, et al. 2016. NOVA2-mediated RNA regulation is required for axonal pathfinding during development. eLife 5: e14371 10.7554/eLife.14371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider MF, Wettengel J, Hoffmann PC, Stafforst T. 2014. Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res 42: e87 10.1093/nar/gku272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoft VK, Schopoff S, Jantsch MF. 2007. Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res 35: 3723–3732. 10.1093/nar/gkm314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam R, Zhang F, Srinivasan H, Charles Richard JL, Liu KI, Zhang X, Woo CWA, Chua ZHM, Buschdorf JP, Meaney MJ, et al. 2018. SRSF9 selectively represses ADAR2-mediated editing of brain-specific sites in primates. Nucleic Acids Res 46: 7379–7395. 10.1093/nar/gky615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. http://www.repeatmasker.org. [Google Scholar]
- Solomon O, Oren S, Safran M, Deshet-Unger N, Akiva P, Jacob-Hirsch J, Cesarkas K, Kabesa R, Amariglio N, Unger R, et al. 2013. Global regulation of alternative splicing by adenosine deaminase acting on RNA (ADAR). RNA 19: 591–604. 10.1261/rna.038042.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Laurent G, Tackett MR, Nechkin S, Shtokalo D, Antonets D, Savva YA, Maloney R, Kapranov P, Lawrence CE, Reenan RA. 2013. Genome-wide analysis of A-to-I RNA editing by single-molecule sequencing in Drosophila. Nat Struct Mol Biol 20: 1333–1339. 10.1038/nsmb.2675 [DOI] [PubMed] [Google Scholar]
- Stulić M, Jantsch MF. 2013. Spatio-temporal profiling of Filamin A RNA-editing reveals ADAR preferences and high editing levels outside neuronal tissues. RNA Biol 10: 1611–1617. 10.4161/rna.26216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafer H, Amman F, Eggenhofer F, Stadler PF, Hofacker IL. 2011. Fast accessibility-based prediction of RNA–RNA interactions. Bioinformatics 27: 1934–1940. 10.1093/bioinformatics/btr281 [DOI] [PubMed] [Google Scholar]
- Tajaddod M, Jantsch MF, Licht K. 2015. The dynamic epitranscriptome: A to I editing modulates genetic information. Chromosoma 125: 51–63. 10.1007/s00412-015-0526-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. 2017. Dynamic landscape and regulation of RNA editing in mammals. Nature 550: 249–254. 10.1038/nature24041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq A, Garncarz W, Handl C, Balik A, Pusch O, Jantsch MF. 2013. RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res 41: 2581–2593. 10.1093/nar/gks1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Stefani G, Mele A, Ruggiu M, Wang XN, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. 2006. An RNA map predicting Nova-dependent splicing regulation. Nature 444: 580–586. 10.1038/nature05304 [DOI] [PubMed] [Google Scholar]
- Wahlstedt H, Daniel C, Enstero M, Ohman M. 2009. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res 19: 978–986. 10.1101/gr.089409.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. 2004. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279: 4952–4961. 10.1074/jbc.M310162200 [DOI] [PubMed] [Google Scholar]
- Wang IX, Core LJ, Kwak H, Brady L, Bruzel A, McDaniel L, Richards AL, Wu M, Grunseich C, Lis JT, et al. 2014. RNA-DNA differences are generated in human cells within seconds after RNA exits polymerase II. Cell Rep 6: 906–915. 10.1016/j.celrep.2014.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.