

Abstract
Desmogleins (Dsgs) are cadherin family adhesion molecules essential for epidermal integrity. Previous studies have shown that desmogleins associate with lipid rafts, but the significance of this association was not clear. Here, we report that the desmoglein transmembrane domain (TMD) is the primary determinant of raft association. Further, we identify a novel mutation in the DSG1 TMD (G562R) that causes severe dermatitis, multiple allergies, and metabolic wasting syndrome. Molecular modeling predicts that this G-to-R mutation shortens the DSG1 TMD, and experiments directly demonstrate that this mutation compromises both lipid raft association and desmosome incorporation. Finally, cryo-electron tomography indicates that the lipid bilayer within the desmosome is ∼10% thicker than adjacent regions of the plasma membrane. These findings suggest that differences in bilayer thickness influence the organization of adhesion molecules within the epithelial plasma membrane, with cadherin TMDs recruited to the desmosome via the establishment of a specialized mesoscale lipid raft–like membrane domain.
INTRODUCTION
A characteristic feature of epithelial cells is the assembly of specialized plasma membrane domains that mediate cell adhesion, communication, and barrier function (Sumigray and Lechler, 2015; Garcia et al., 2018). Among these structures, adherens junctions and desmosomes play overlapping but distinct roles in cell adhesion, signaling, and morphogenesis (Garcia et al., 2018). Desmosomes are particularly abundant in tissues exposed to mechanical stress, including the skin and heart (Delmar and McKenna, 2010; Amagai and Stanley, 2012; Hsu et al., 2018). These adhesive complexes are characterized by highly organized and dense arrangements of desmosomal proteins that can be visualized by electron microscopy (Al-Amoudi et al., 2007; Delva et al., 2009; Harmon and Green, 2013). Considerable progress has been made in identifying protein interactions that mediate adhesion in both adherens junctions and desmosomes, as well as the associations that anchor these adhesive structures to the cytoskeleton (Holthofer et al., 2007; Troyanovsky, 2012; Nekrasova and Green, 2013; Quinlan et al., 2017). However, the physical constraints imposed by the epithelial plasma membrane that contribute to the segregation of adherens junctions and desmosomal complexes into morphologically, biochemically, and functionally distinct structures are poorly understood.
The adhesive core of the desmosome is comprised of single pass transmembrane desmosomal cadherins termed desmogleins and desmocollins (DSC) that mediate adhesion between adjacent cells (Al-Amoudi et al., 2007; Saito et al., 2012b; Harrison et al., 2016). In humans, there are four desmoglein genes (DSG1-4), along with three desmocollins (DSC1-3) (Simpson et al., 2011). The desmosomal cadherins are coupled to the intermediate filament cytoskeleton through adaptor proteins such as plakoglobin, plakophilins, and the cytolinker protein desmoplakin (DP) (Delva et al., 2009; Al-Amoudi et al., 2011; Jones et al., 2017). These interactions form an electron-dense plaque that couples the adhesive interactions of the desmosomal cadherins to the intermediate filament cytoskeleton of adjacent cells, thus conferring tissue resilience to mechanical stress (Thomason et al., 2010; Hsu et al., 2018). Loss of desmosome function results in skin (Payne et al., 2004; Hsu et al., 2018) and heart (Delmar and McKenna, 2010; Samuelov and Sprecher, 2015) diseases characterized by tissue fragility. In the skin, loss of desmosomal adhesion manifests clinically as epidermal blisters and erosions (Payne et al., 2004; Brooke et al., 2012), and in some disorders, aberrant thickening of the epidermis (Stahley and Kowalczyk, 2015; Has and Technau-Hafsi, 2016). One example of such a disease is severe dermatitis, multiple allergies, and metabolic wasting (SAM) syndrome (Samuelov et al., 2013). This disease is typically caused by null mutations in DSG1, leading to epidermal fragility and barrier defects (Cheng et al., 2016; Has et al., 2015).
We and others have previously demonstrated that desmosomal proteins associate with lipid rafts (Nava et al., 2007; Delva et al., 2008; Resnik et al., 2011; Brennan et al., 2012; Stahley et al., 2014). Lipid rafts are sphingolipid- and cholesterol- enriched membrane microdomains that introduce spatial heterogeneity into lipid bilayers (Pike, 2004, 2006; Lingwood et al., 2009; Simons and Sampaio, 2011). These domains are critical for protein trafficking, membrane organization, and signaling (Diaz-Rohrer et al., 2014a; Honigmann and Pralle, 2016; Levental and Veatch, 2016; Sezgin et al., 2017; Santos and Preta, 2018). The sphingolipids present in rafts feature long, saturated acyl chains that, along with cholesterol, contribute to the more ordered, densely packed, and thicker membrane environment characteristic of lipid rafts (Brown and London, 2000; Lingwood et al., 2009; Honigmann and Pralle, 2016). Desmogleins and other desmosomal proteins have been shown to associate with lipid raft membrane domains as determined by detergent resistance and buoyancy on sucrose gradients (Nava et al., 2007; Resnik et al., 2011; Brennan et al., 2012; Stahley et al., 2014). In addition, disruption of lipid rafts by removal of cholesterol from cellular membranes results in weakened desmosomal adhesion, suggesting that lipid rafts play a role in desmosome homeostasis (Resnik et al., 2011; Stahley et al., 2014). However, we do not know how desmosomal cadherins target to raft domains or how incorporation into raft domains impacts desmosomal cadherin function.
In the present study, we sought to determine the mechanisms by which raft association governs desmosome assembly and identify the determinants of desmoglein partitioning to rafts. Our results indicate that the transmembrane domain (TMD) of the desmogleins is critical for raft association, and that the E-cadherin TMD does not support raft targeting. Raft association appears to be essential for desmoglein function, as a novel mutation that shortens the TMD of human DSG1 abrogates lipid raft targeting, impairs desmosome association, and causes the human skin disease SAM syndrome. Cryo-electron tomography (cryo-ET) and subtomogram averaging demonstrate that the lipid bilayer within the desmosome is thicker than the adjacent plasma membrane, consistent with predictions that the lipid bilayer is thicker at raft domains compared with nonraft membranes. Thus, our results support a model in which the desmosome is a specialized type of lipid raft membrane microdomain, and the lengthy desmoglein TMD enables efficient desmosome incorporation by facilitating desmoglein partitioning into the thicker desmosomal lipid bilayer. These findings suggest that epithelial junctional complexes achieve plasma membrane domain specification not only through selective protein interactions but also through constraints imposed by the biophysical characteristics of the plasma membrane.
RESULTS
Palmitoylation of Dsg3 is not required for lipid raft association
Desmogleins and other desmosomal components associate with lipid raft membrane microdomains (Nava et al., 2007; Delva et al., 2008; Resnik et al., 2011; Stahley et al., 2014). A number of raft-associating proteins, including plakophilins, utilize palmitoylation as a membrane raft-targeting mechanism (Greaves and Chamberlain, 2007; Levental et al., 2010; Roberts et al., 2014). Palmitoylation is a reversible posttranslational modification that occurs when palmitoyltransferases add a 16-carbon fatty acid (palmitate) to cysteine residues (Munday and Lopez, 2007; Charollais and Van Der Goot, 2009). Sequence alignments (Figure 1A) reveal that desmosomal cadherins contain conserved cysteine residues at the cytoplasmic face of the TMD, and our previous studies have shown that these residues are critical for desmoglein palmitoylation (Roberts et al., 2016). We hypothesized that palmitoylation of desmogleins would mediate lipid raft association. Therefore, we mutated cysteines 640 and 642 to alanine residues in murine DSG3 and used a lentiviral expression system to generate stable A431 cell lines expressing FLAG-tagged wild-type (WT) or mutant Dsg3(CC). A431 cells are an epidermal carcinoma cell line that has been used extensively for desmosome studies due to their human origin, relatively flat morphology, and assembly of robust desmosomes (Bornslaeger et al., 1996; Palka and Green, 1997; Setzer et al., 2004; Wahl, 2005; Sobolik-Delmaire et al., 2010; Roberts et al., 2016). Mass tag labeling confirmed that the mutation of these conserved membrane-proximal cysteines eliminated Dsg3 palmitoylation (Figure 1B). Interestingly, the loss of Dsg3 palmitoylation had no discernable effect on lipid raft association as determined by Dsg3 incorporation into buoyant and detergent-resistant membranes (DRM) (Lingwood and Simons, 2007) (Figure 1C). In addition, both WT Dsg3 and Dsg3(CC) localized to cell–cell borders as assessed by widefield immunofluorescence (Figure 1D). Furthermore, Dsg3 and Dsg3(CC) exhibited similar Triton X-100 solubility, suggesting no defect in the desmosome or cytoskeletal association of Dsg3(CC) (Figure 1, E–G). Collectively, these results indicate that palmitoylation is not required for lipid raft association of desmogleins or for normal Dsg3 subcellular distribution in quiescent A431 monolayers.
FIGURE 1:

Palmitoylation is not required for Dsg3 lipid raft association. (A) Sequence alignment of the DSGs reveals a pair of highly conserved cysteine residues (red underline) at the interface between the TMD (bolded text) and the cytoplasmic domain. (B) Mass-tag labeling replaces palmitoyl moieties on cysteine residues with mPEG, causing a size shift detectable by Western blot analysis. Dsg3 is doubly palmitoylated and mutation of the membrane-proximal cysteine residues to alanine abolishes palmitoylation. (C) Lipid raft fractionation of HeLa cells expressing Dsg3-FLAG from adenoviruses reveals no defect in lipid raft targeting of the palmitoylation-null mutant. (D) Widefield images of A431 cell lines stably harboring flag-tagged constructs. Scale bar = 20 µm. (E) Western blot of Triton X-100–soluble pools and -insoluble pools from A431 cell lines stably expressing either Dsg3 or Dsg3(EcadTMD). (F) Densitometry quantification of Dsg3 in Triton-soluble and -insoluble pools from panel E. Loss of palmitoylation has no detectable effect on the solubility of Dsg3 in Triton X-100, a classic measure of desmosome and cytoskeletal association. (G) Densitometry quantification of E-cadherin distribution between Triton-soluble and -insoluble pools.
The TMD of desmogleins mediates lipid raft association
In addition to palmitoylation, emerging evidence indicates that lipid raft association of membrane spanning proteins is also regulated by the physiochemical properties of the TMD (Lorent et al., 2017). In particular, TMD length is a critical determinant for targeting to lipid rafts (Scheiffele et al., 1997; Garcia-Garcia et al., 2007; Diaz-Rohrer et al., 2014b; Lorent et al., 2017). Sequence alignments (Figure 1A) indicate that the TMDs of the desmogleins, which associate with rafts, are considerably longer (24 amino acids) than the corresponding TMDs of classical cadherins, such as E-cadherin (21 amino acids) and VE-cadherin (20 amino acids), which exhibit minimal raft association (Stahley et al., 2014). Recent studies indicate that the free energy of raft association can be calculated based on TMD length, surface area, and palmitoylation (Lorent et al., 2017). These parameters predict efficient WT Dsg1 raft partitioning (∆Graft = 0.17), with markedly lower raft affinity for the TMD of E-cadherin (∆Graft = 0.30) (Table 1). To directly test whether the TMD is the principal motif conferring lipid raft association on the desmoglein family of proteins, we generated a chimeric cadherin in which the Dsg3 TMD was replaced with the E-cadherin TMD (Dsg3[EcadTMD]). Lentiviral transduction was used to generate stable A431 cell lines expressing either WT Dsg3-FLAG or Dsg3(EcadTMD)-FLAG. Sucrose gradient fractionations demonstrated that the Dsg3(EcadTMD) chimera was virtually excluded from DRM fractions when compared with WT Dsg3 (Figure 2, A and B). Immunofluorescence localization indicated that Dsg3(EcadTMD) localized to cell–cell contact sites. However, Triton X-100 extraction showed decreased insoluble pool partitioning as assessed by both immunofluorescence (Figure 2C) and Western blot analysis (Figure 2, D–F), suggesting decreased Dsg3(EcadTMD) association with cytoskeletal elements relative to WT Dsg3. Expression of the Dsg3(EcadTMD) mutant caused no apparent changes in endogenous E-cadherin distribution (Figure 2, A, D–F). To determine whether the Dsg3 TMD is sufficient to confer lipid raft targeting, we constructed interleukin 2 receptor (IL2R) α chain-Dsg3 chimeric proteins comprising the IL2R extracellular domain coupled to the Dsg3 cytoplasmic tail with either the IL2R TMD or the Dsg3 TMD (Figure 2G; Saito et al., 2012a). The IL2R-Dsg3 chimera harboring the Dsg3 TMD partitioned to DRM fractions, whereas the chimera containing the IL2R TMD did not partition with DRM fractions. Collectively, these studies indicate that the Dsg3 TMD is the primary determinant of Dsg3 raft association.
TABLE 1:
Summary of TMDs.
| Construct | TMD sequence | Amino acid number | Length (nm) | ΔGraft |
|---|---|---|---|---|
| WT Dsg1 | FGPAGIGLLIMGFLVLGLVPFLMI | 24 | 3.6 | 0.166 |
| Dsg1 (G578R) | FGPAGIGLLIMGFLVLR | 16 | 2.4 | 0.387 |
| E-cadherin | LGILGGILALLILILLLLLFL | 21 | 3.15 | 0.304 |
FIGURE 2:

The Dsg3 TMD is necessary for lipid raft association. (A) Sucrose gradient fractionation of A431 cells stably expressing murine Dsg3 (WT or EcadTMD mutant). Replacing the Dsg3 TMD with the shorter E-cadherin TMD abolishes lipid raft targeting. (B) Densitometry quantification of WT Dsg3 and Dsg3(EcadTMD) in DRM fractions shown in A. (C) Dsg3(EcadTMD) is more susceptible than Dsg3 WT to pre-extraction in Triton X-100 prior to fixation and immunofluorescence localization. Scale bar = 20 µm. (D) Western blot analysis indicates Dsg3(EcadTMD) is more soluble in Triton X-100 than in Dsg3 WT. (E) Quantification of Dsg3 in Triton-soluble or -insoluble pools from D. (F) Quantification of E-cadherin in Triton-soluble and -insoluble pools in cells expressing either WT Dsg3 or Dsg3(EcadTMD) mutant. (G) Lipid raft fractionation of FLAG-tagged IL2R-Dsg3 chimeras expressed in HeLa cells using an adenoviral delivery system. Inclusion of the lengthy Dsg3 TMD in the chimera (right panel) confers lipid raft targeting. *p < 0.05.
To test whether the TMD of other desmoglein family members also functions in raft association, similar experiments were conducted in the context of Dsg1. Dsg1 WT and a Dsg1(EcadTMD) chimera were generated. Both proteins were tagged with a carboxyl terminal green fluorescent protein (GFP) and stably expressed in A431 cell lines as described above. Similar to the Dsg3(EcadTMD) chimera, the Dsg1(EcadTMD) chimera showed a marked decrease in association with DRM fractions as determined by sucrose gradient fractionation (Figure 3, A and B). Additionally, Dsg1(EcadTMD) was partially excluded from Triton-insoluble fractions of cell lysates (Figure 3, C–E), similar to the results seen with Dsg3(EcadTMD) (Figure 2, D–F). Finally, both the WT Dsg1 and Dsg1(EcadTMD) demonstrated border staining characteristic of desmogleins (Figure 3F). Together, these results demonstrate a central role for the TMDs of the DSG family in lipid raft association.
FIGURE 3:

The Dsg1 TMD is critical for lipid raft association. (A) Western blot of Triton X-100 extracts and sucrose gradient fractionations of A431 cells stably expressing murine WT Dsg1 or Dsg1(EcadTMD) chimera. (B) Quantification from densitometry analysis of the percentage of total Dsg1 in the DRM fractions of sucrose gradient fractionations. (C) Differential detergent extraction and Western blot analysis indicate that Dsg1(EcadTMD) is more soluble in Triton X-100 than WT Dsg1. (D) Quantification of Dsg1 Western blots shown in C. (E) Quantification of E-cadherin Western blots shown in C. (F) Widefield images of A431 cells expressing either GFP-tagged WT Dsg1 or Dsg1(EcadTMD). Both constructs localized to cell–cell borders and no abnormal localization was observed for the Dsg1(EcadTMD) chimera. Scale bar = 20 µm. **p < 0.001.
A mutation in the TMD of DSG1 causes SAM syndrome
Loss of DSG1 function is associated with a number of autoimmune, infectious, and genetic diseases (Payne et al., 2004; Samuelov and Sprecher, 2015; Stahley and Kowalczyk, 2015). One recently discovered desmosome-associated disease is SAM syndrome (Samuelov et al., 2013). Most instances of SAM syndrome are caused by homozygous functional null mutations in the desmosomal cadherin desmoglein 1 (DSG1) (Has et al., 2015; Cheng et al., 2016). Here, we report a novel and dominantly inherited heterozygous DSG1 missense mutation within the DSG1 TMD (Figure 4). The probands presented with ichthyosiform erythrokeratoderma, diffuse palmoplantar keratosis, and multiple allergies (Figure 4A). Proband III-2 suffered metabolic wasting and died of status asthmaticus and recurrent infections. Hemotoxylin and eosin staining of skin biopsied from the proband revealed compact hyperkeratosis with parakeratosis, frequent detachment of the entire stratum corneum, and dissociation of individual corneocytes (Figure 4B). Although these findings indicate an adhesion defect, we observed minimal alterations in desmosome ultrastructure when patient epidermis was examined by electron microscopy (Figure 4C). These clinical and genetic observations led us to diagnose the patient with SAM syndrome. Unlike previously reported instances of DSG1 mutations in SAM syndrome (Samuelov et al., 2013; Has et al., 2015; Cheng et al., 2016; Danescu et al., 2017), this patient harbored a novel missense mutation in DSG1 which introduces a hydrophilic arginine residue (p.G562R) into the otherwise hydrophobic TMD of DSG1 (Figure 4, D–F). Subsequent to our characterization of this initial family, a second unrelated individual was identified with a G562R heterozygous mutation previously reported as a case of erythrokeratoderma variabilis (Ishida-Yamamoto et al., 2000). The parents of this patient lacked this mutation and were disease free. Together, these observations demonstrate that a heterozygous G562R mutation in the DSG1 TMD causes a human skin disease best characterized clinically as SAM syndrome.
FIGURE 4:

DSG1 TMD mutation causes SAM syndrome. (A) Individual III-2 displays feet covered with hyperkeratotic yellowish papules and plaques, and ichthyosiform erythroderma with severe itch occurs over much of his body. (B) Hematoxylin and eosin staining of III-2′s skin biopsy reveals acantholytic lesions in the upper layers of the epidermis. Scale bar = 100 μm. (C) Electron micrographs of epidermal sections from the proband indicate relatively normal desmosome morphology. Scale bar = 200 nm. (D) Pedigree of affected individuals and near relatives. Inheritance determined by genomic DNA sequencing. (E) Genomic DNA sequencing of white blood cells reveals these SAM syndrome patients have a heterozygous point mutation, c.1684G>A (black arrow) in DSG1. The adjacent splice site is unaffected. (F) Schematic showing the location of the SAM-causing G-to-R substitution (red) within the TMD (blue). (G, H) Widefield microscopy of DSG1 immunofluorescence in human skin biopsies reveals both DSG1 down-regulation and inappropriate clustering at cell borders in SAM syndrome patient epidermis. SC, stratum corneum; SG, stratum granulosum; SS, stratum spinosum; SB, stratum basale. The SC/SG boundary is demarcated by a dashed line. Down-regulation of DSG1 is observed in the SG and SS. (I–K) DP is slightly down-regulated in patient skin, and DSG3 is up-regulated. Scale bar = 20 µm. (L, M) SIM indicates reduced desmosomal DSG1 in SAM syndrome patient tissue in the stratum spinosum and granulosum. Scale bar = 5 µm. *p < 0.01, **p < 0.001.
To determine how the G562R mutation impacted DSG1 organization in patient skin, biopsies from the proband were processed for immunofluorescence microscopy. DSG1 levels were markedly reduced (∼40%) in the spinous and granular layers of patient epidermis (Figure 4, G and H), and DSG1 localized in cytoplasmic puncta and aberrant clusters at cell–cell borders. Interestingly, DSG1 staining in patient stratum corneum was markedly increased, perhaps reflecting increased antibody penetration. DP levels were slightly reduced in patient epidermis, whereas DSG3 levels were markedly increased (Figure 4, I–K). To further investigate alterations in DSG1 distribution, we performed structured illumination microscopy (SIM) on patient and control epidermis. DSG1 fluorescence intensity within patient and control desmosomes was comparable in basal keratinocytes, where DSG1 expression is low and other DSG isoforms (DSG2, DSG3) are expressed. However, DSG1 fluorescence intensity in patient desmosomes was significantly reduced (Figure 4, L and M) in the spinous and granular layers where DSG1 is prominently expressed. Thus, although morphologically normal desmosomes could be observed by electron microscopy (Figure 4C), these desmosomes apparently lack sufficient DSG1 levels to support normal epidermal cohesion.
To investigate the mechanism by which the DSG1(G562R) mutation causes SAM syndrome, GFP-tagged murine WT Dsg1α and a mutant harboring the equivalent G-to-R substitution, Dsg1(G578R), were expressed in A431 epithelial cells. Widefield fluorescence imaging revealed that both WT Dsg1 and Dsg1(G578R) were present at cell-to-cell borders (Figure 5A). Interestingly, the Dsg1(G578R) mutant also exhibited a prominent perinuclear staining pattern. There were no obvious differences in DP localization in the two cell lines (Figure 5A), and plakoglobin generally colocalized with both cell–cell border and perinuclear pools of WT Dsg1 and Dsg1(G578R) (Figure 5B). To determine whether the Dsg1(G578R) mutant was defective in desmosome targeting, SIM was performed and Dsg1 fluorescence intensity was measured at cell borders both within and outside of individual desmosomes to control for possible variations in Dsg1 levels at different cell–cell contact sites. We observed that Dsg1(G578R) displayed a decreased association with desmosomes when compared with WT Dsg1 (Figure 5, C and D). Furthermore, parallel bands of GFP fluorescence signal overlapping with DP were routinely observed for WT Dsg1-GFP but not for Dsg1(G578R)-GFP (Figure 5, C and E). Finally, WT Dsg1 efficiently entered a detergent-resistant pool, consistent with incorporation into insoluble desmosome and cytoskeletal-associated complexes, whereas mutated Dsg1(G578R) remained predominantly soluble (Figure 5, F–H). Together, these findings indicate that the G-to-R TMD mutation reduces DSG1 incorporation into desmosomes in both cultured cells and patient epidermis.
FIGURE 5:

SAM-causing DSG1 mutation causes defects in junction targeting. (A) Widefield immunofluorescence micrographs of A431 cell lines stably expressing either WT Dsg1-GFP or Dsg1(G578R)-GFP reveal broadly similar distribution of DP and (B) colocalization between DSG1 and plakoglobin (PG). Scale bar = 20 µm. (C) Superresolution micrographs of A431 stable cell lines acquired using SIM reveal defects in Dsg1(G578R) desmosome targeting. Scale bar = 5 µm. (D) Desmosomes are identified by SIM imaging as regions of parallel DP immunofluorescence staining resembling railroad tracks. Quantification of Dsg1 found within DP railroad tracks compared with border Dsg1 outside of railroad tracks. (E) Quantification of railroad track appearance observed for WT or mutant Dsg1.GFP. (F–H) The G578R mutation increases solubility of the mutant in Triton X-100 as determined by Western blot analysis. E-cadherin distribution in Triton-soluble and -insoluble pools is not altered in A431 cell lines expressing the Dsg1 mutant. *p < 0.05.
In addition to being deficient in desmosome targeting, we also observed that DSG1 was present in cytoplasmic puncta in SAM syndrome patient epidermis (Figure 4G) and that Dsg1(G578R) was concentrated in perinuclear compartments in A431 cell lines (Figure 5, A and B). To determine whether the Dsg1(G578R) mutant exhibited membrane trafficking defects, cell surface proteins were biotinylated and pulse-chase experiments were conducted to measure Dsg1 turnover rates. These experiments revealed no difference in the rate of Dsg1(G578R) turnover from the plasma membrane compared with WT Dsg1 (Figure 6, A and B). We also confirmed that Dsg1(G578R) is inserted into the plasma membrane in the correct orientation by demonstrating that the c-terminal GFP tag is accessible to antibody detection only after cells have been permeabilized with Triton X-100 (Supplemental Figure S1A). Furthermore, biotinylation experiments detected no significant difference in the ratio of surface to total Dsg for WT Dsg1 or Dsg1(G578R) at steady state (Supplemental Figure S1, B and C).
FIGURE 6:

SAM-causing DSG1 mutation delays trafficking to the plasma membrane. (A) Pulse-chase experiments were performed to determine the rate of turnover of Dsg1 from the plasma membrane in A431 cells expressing murine Dsg1-GFP. Cell surface proteins were biotinylated at t = 0, washed, and incubated at 37°C for various amounts of time. Cell lysates were collected after the indicated times. Biotinylated proteins were captured using streptavidin beads and processed for Western blot analysis. (B) Quantification using densitometry revealed no significant differences in the rate of turnover of Dsg1 vs. Dsg1(G578R). (C) Dsg1(G578R) is trafficked to the plasma membrane substantially more slowly than WT. Cell surface proteins were cleaved using trypsin at t = 0. Trypsin was removed and cells were then incubated for the indicated times. The amount of newly delivered surface Dsg1 was assayed via biotin labeling followed by capture using streptavidin beads and subsequent Western blot analysis. (D) Quantification using densitometry indicates Dsg1(G578R) recovers more slowly than WT Dsg1. (E) Densitometry analysis of the Dsg1 no-trypsin condition in (C) as a ratio-to-densitometry analysis of the E-cadherin no-trypsin condition in C reveals comparable surface levels of WT Dsg1 and Dsg1(G578R). (F) Cells were cultured in low-calcium medium to cause cadherin removal from cell–cell borders and accumulation in intracellular compartments (Low Ca2+). Some cells were then switched back to normal calcium to allow for junction assembly (3 h high). Dsg1(G578R) displays increased colocalization with the Golgi apparatus protein GM130 under both conditions. Scale bar = 20 µm. (G) Quantification of colocalization of Dsg1 and GM130. *p < 0.001.
To measure rates of delivery to the plasma membrane, cell surface proteins were cleaved using trypsin, and the rate of Dsg1 recovery at the cell surface was monitored by biotinylation (Figure 6, C and D). Prior to trypsinization, Dsg1(G578R) cell surface levels were similar to WT Dsg1 (Figure 6E), again indicating that steady state cell surface levels of the mutant were comparable to WT Dsg1. However, whereas the surface pool of WT Dsg1 recovered within 3–6 h after trypsinization, Dsg1(G578R) exhibited delayed plasma membrane recovery (Figure 6D). To determine whether Dsg1(G578R) was being retained in secretory compartments, A431 cell lines were grown in low-calcium medium overnight to internalize all cadherins and subsequently switched to high-calcium medium to allow Dsg1 to traffic out to cell–cell borders. These experiments revealed that Dsg1(G578R) was retained in GM130-labeled compartments (Figure 6, F and G), indicating that the G-to-R mutation causes retention of Dsg1 in the Golgi apparatus, delaying its trafficking through the secretory pathway.
Disease-causing mutation abrogates lipid raft association of Dsg1
On the basis of our findings that the TMD of the desmogleins is critical for lipid raft association, we hypothesized that the G-to-R mutation observed in SAM syndrome patients would prevent Dsg1 from partitioning to lipid rafts. Calculations based on parameters which predict free energy of raft association from TMD sequences (Lorent et al., 2017) revealed that introduction of the SAM-causing G-to-R mutation into the DSG1 TMD dramatically alters the energetics of raft association (Table 1). Consistent with these calculations, sucrose gradient fractionations revealed that Dsg1(G578R) was virtually absent from lipid raft (DRM) fractions (Figure 7, A and B). Interestingly, we observed a notable reduction in plakoglobin association with DRM in cell lines expressing Dsg1(G578R), suggesting that the DSG1 mutant also recruited plakoglobin out of raft domains. To further test the ability of WT Dsg1 and Dsg1(G578R) to associate with lipid rafts, these proteins were transiently expressed in rat basophilic leukemia cells and giant plasma membrane vesicles (GPMV) were chemically isolated (Levental and Levental, 2015a,b). Nonraft plasma membrane domains were labeled with FAST-DiO (F-DiO), a dialkylcarbocyanine dye. WT Dsg1 efficiently partitioned into areas of plasma membrane vesicles lacking F-DiO, indicating partitioning to the liquid-ordered raft domain (Figure 7, C and D). In contrast, Dsg1(G578R) was almost entirely cosegregated with F-DiO and excluded from the liquid-ordered plasma membrane domain, indicating minimal raft affinity. Together, these findings reveal that the G-to-R TMD mutation reduces cell surface DSG1 association with lipid rafts.
FIGURE 7:

SAM-causing DSG1 mutation abolishes lipid raft association. (A) Sucrose gradient fractionation and Western blot analysis of A431 cell lines stably expressing WT and mutant Dsg1. (B) Quantification of results in A indicates SAM-causing mutation abolishes Dsg1 partitioning to DRM (lipid raft fractions). (C) Representative images of GPMVs isolated from rat basophilic leukemia cells expressing GFP-tagged WT Dsg1 or Dsg1(G578R). Unsaturated lipid marker F-DiO to visualize the nonraft phase. (D) Normalized line scans of Dsg1 fluorescence intensity were measured through peaks corresponding to Dsg1 intensity in raft and nonraft membrane, respectively. Background-subtracted ratios of these two intensities yield raft partition coefficients, Kp,raft. Data are shown as mean ± SEM from three independent experiments. *p < 0.05, **p < 0.001.
The lipid bilayer within desmosomes is thicker than nondesmosomal membranes
The results above illustrate a critical role for the DSG TMD in the association of this family of cadherins with lipid raft membrane microdomains and for its crucial role in epidermal homeostasis. To understand how the physiochemical properties of the desmoglein TMD confer raft and desmosome targeting, structural models of the TMDs of WT Dsg1, the Dsg1(G578R) SAM mutant, and E-cadherin were generated by the Robetta structure prediction server (Kim et al., 2004). The modeling predicts that the SAM-causing G-to-R mutation interrupts the Dsg1 TMD helix and significantly shortens the run of helical hydrophobic residues (Table 1 and Figure 8A), potentially deforming the lipid bilayer as phospholipids position to maintain energetically favorable interactions between hydrophilic and hydrophobic amino acid residues (Nezil and Bloom, 1992; de Jesus and Allen, 2013). These findings are consistent with the notion that the SAM-causing mutant disrupts lipid raft association by shortening the DSG1 TMD, thereby increasing the energy cost of entering the thicker lipid bilayer characteristic of lipid raft domains (Lorent et al., 2017).
FIGURE 8:

The desmosome bilayer is thicker than adjacent bilayers. (A) Structural models of the DSG1 WT, DSG1 SAM mutant, and E-cadherin TMDs acquired using the Robetta prediction server and depicted in schematized lipid bilayers. The length of each TMD is shown in nm and is based on TMD amino-acid number. (B) Representative slice from a cryo-ET showing a desmosome (D) with characteristic intracellular plaque attached to intermediate filaments. Directly adjacent to the desmosome, membrane remnants can be seen (DA). Other nondesmosomal (ND) regions of the plasma membrane embedded in a thin layer of ice are also visible. Scale bar = 100 nm. (C) Projections of the average of all subvolumes from the most significant class for desmosome adjacent and desmosome bilayers. (D) Schematic showing the thickness of the desmosome bilayer compared with desmosome-adjacent bilayers. The lipid bilayer within desmosomal membranes is thicker (4.5 ± 0.4 nm) when compared with membranes adjacent to desmosomes (4.0 ± 0.3 nm) or from nondesmosomal membranes (4.0 ± 0.3 nm), **p < 0.001. Intensity plots are shown superimposed to subtomogram average projections for desmosome and desmosome-adjacent membranes. (E) Summary table depicting the TMD lengths and the measured phospho-head group-to-head group distance (Dhh) as shown in D. Also shown is the estimated distance between phosphate residues (Dpp), which corresponds to the hydrophobic interior of the bilayer. This hydrophobic region of the bilayer was estimated by subtracting the predicted polar head group size (1 nm) (Lewis and Engelman, 1983) from the measured Dhh shown in D.
Experiments in model membranes suggest that the high cholesterol content (Nezil and Bloom, 1992; Kucerka et al., 2008) and long, saturated acyl chains (Lewis and Engelman, 1983; Niemela et al., 2007) present in lipid raft domains contribute to significant thickening of raft phospholipid bilayers relative to nonraft regions of the membrane. A prediction derived from such observations, and from the experimentally demonstrated presence of desmosomal proteins in rafts, is that the lipid bilayer within desmosomes in cells or tissues would be thicker than nondesmosomal regions of the plasma membrane, thereby accommodating the lengthy desmoglein TMD. To test this possibility, cryo-ET and subtomogram averaging were performed on mouse liver samples enriched in the plasma membrane fraction (Figure 8B). The thickness of lipid bilayers measured in the subtomogram averages within desmosomal and nondesmosomal regions of the plasma membrane was then determined. This analysis revealed that desmosomal bilayers were 10% thicker (4.5 ± 0.4 nm) than regions immediately adjacent to the desmosome (4.0 ± 0.3 nm, p = 2.2E-122) or at arbitrary regions of membrane visible within the tomograms (4.0 ± 0.3 nm, p = 6.4E-104) (Figure 8C). Together, these findings suggest that desmosomes represent a highly specialized plasma membrane domain that is characterized by lipid raft-associated proteins and a thickened phospholipid bilayer characteristic of lipid raft–like model membranes.
DISCUSSION
Lipid rafts have emerged as important membrane microdomains that regulate membrane organization, endocytosis, and signaling (Pike, 2006; Lingwood et al., 2009; Honigmann and Pralle, 2016; Levental and Veatch, 2016; Sezgin et al., 2017). Desmosomal proteins have been shown to associate with lipid rafts in a variety of epithelial cell types (Nava et al., 2007; Delva et al., 2008; Resnik et al., 2011; Brennan et al., 2012; Stahley et al., 2014), but the mechanisms and physiological relevance of this association are poorly understood. Here, we report that the TMDs of the desmogleins are the key determinants for targeting these cadherins to lipid rafts. A mutation within the DSG1 TMD that shortens this domain abrogates both lipid raft partitioning and desmosome association and leads to the human skin disease SAM syndrome. Cryo-ET reveals that the lipid bilayer within the desmosome is markedly thicker than the adjacent lipid bilayer, thereby favoring incorporation of the longer desmoglein TMDs into this plasma membrane domain. Collectively, our results suggest that desmosomes are a specialized mesoscale lipid raft–like membrane domain.
Essential functions for desmogleins have been exposed by human diseases in which desmogleins are targeted by autoantibodies, infectious agents, or genetic mutation (Amagai and Stanley, 2012; Stahley and Kowalczyk, 2015). DSG1 is the primary desmoglein expressed in the outermost layers of the epidermis, and DSG1 loss of function mutations leads to at least two different types of epidermal disorders. Haploinsufficiency of DSG1 causes palmoplantar keratoderma (Lovgren et al., 2017), whereas complete loss of DSG1 leads to SAM syndrome (Samuelov et al., 2013; Has et al., 2015; Cheng et al., 2016). Most individuals afflicted with SAM syndrome succumb to chronic infection in early childhood (Samuelov et al., 2013). Here, we report two separate instances of SAM syndrome, one inherited and one sporadic, caused by a glycine to arginine substitution (G562) within the hydrophobic TMD (Figure 4). Arginine residues play an important role in terminating TMDs and establishing TMD orientation within the lipid bilayer (Parks and Lamb, 1993; Reddy et al., 2014), consistent with molecular modeling indicating that the disease-causing glycine to arginine substitution shortens the DSG1 TMD (Figure 8).
Our data indicate that shortening of the DSG1 TMD by insertion of an arginine residue disrupts DSG1 function in SAM syndrome patients by preventing lipid raft association. TMD length correlates positively with raft association (Lorent et al., 2017), and our structural predictions and molecular modeling predict that desmoglein TMDs confer raft association (Table 1 and Figure 8A). This notion is consistent with our findings using both classical DRM fractionation experiments (Figures 3 and 4) and direct observations of partitioning of cell surface Dsg1 into liquid-ordered plasma membrane domains (Figure 7). In addition to shortening the Dsg1 TMD, insertion of an arginine residue into the TMD could also impact local protein and lipid packing within the desmosomal membrane domain. This could occur as polar residues, such as arginine, are thought to “snorkel” out of the hydrophobic bilayer to interact with the hydrophilic, aqueous environment, whereas hydrophobic TMD residues retain contact with the acyl chains of the lipids (Schow et al., 2011). Such an arrangement within the bilayer could increase the effective surface area of the TMD and decrease packing of lipids and proteins, thereby altering Dsg1 association with lipid raft and desmosomal components.
Interestingly, we observed defects in the rate of mutant Dsg1 delivery to the plasma membrane after trypsinization (Figure 6). In addition, plakoglobin colocalized with DSG1(G578R) pools that were retained in the Golgi apparatus (Figure 5B), and we observed a reduction in plakoglobin association with DRM fractions in cells expressing the DSG1(G578R) mutant (Figure 7). Nonetheless, Dsg1(G578R) is inserted into membranes in the correct orientation, and steady state plasma membrane levels of the mutant are similar to WT Dsg1 (Supplemental Figure S1). Surprisingly, palmitoylation does not appear to be required for desmoglein raft association (Figure 1 and Roberts et al., 2016), although it does impact desmoglein dynamics at the plasma membrane (Roberts et al., 2016). Recently, the desmosomal component plakophilin also was shown to be palmitoylated (Roberts et al., 2014), further demonstrating an important role for this reversible posttranslational modification in regulating desmosome assembly dynamics. Further studies will be needed to assess how palmitoylation is utilized in combination with other physiochemical properties of the desmoglein TMD to modulate the trafficking and adhesive properties of these unique cadherins.
A prediction based on our finding that the desmoglein TMD is responsible for partitioning to lipid rafts is that the lipid bilayer within desmosomes should be thicker than the surrounding nondesmosomal membrane. Indeed, cryo-ET revealed that the lipid bilayer within the desmosome is substantially thicker than nondesmosomal regions of the plasma membrane (Figure 8, B–D). These observations indicate that it would be energetically costly for the DSG1 G-to-R SAM mutant to enter the thicker bilayer present in desmosomes due to hydrophobic mismatch between phospholipids and Dsg1 TMD amino acid residues (Marsh, 2008; de Jesus and Allen, 2013). Therefore, it is likely that shortening of the TMD in the SAM mutant and failure to enter the thicker lipid bilayer domain of the desmosome represent a central pathophysiological mechanism of this disease-causing mutation. Indeed, we observed that the DSG1 G-to-R mutant is deficient in entering desmosomes both in patient epidermis (Figure 4) and when expressed in cultured epithelial cell lines (Figure 5). We also find that Dsg3 and Dsg1 polypeptides harboring the shorter E-cad TMD are unable to associate with lipid rafts and behave similarly to full length E-cadherin (Figures 2 and 3). The predicted E-cadherin TMD is 21 amino acids, compared with the 24 amino acid TMD of desmogleins (Table 1). Although these chimeras do not effectively enter lipid rafts as assessed by DRM fractionation assays, we do find that these Dsg(EcadTMD) chimeras can associate with desmosomes as assessed by SIM (not shown). It is likely that for these chimeras, protein–protein interactions mediated by the desmoglein cytoplasmic and extracellular domains can partially overcome the energy cost of incorporating into the thicker bilayers present in the desmosome. In addition, mismatch of TMD length and hydrophobic thickness of the bilayer can be accommodated by changes in TMD tilt within the membrane and by local bilayer deformation (Nezil and Bloom, 1992; de Jesus and Allen, 2013). In contrast, the predicted 16 amino acid TMD of the Dsg1(G578R) mutant is significantly shorter than the TMD of both desmogleins and E-cadherin, and therefore its entry into desmosomal membranes is apparently energetically prohibitive.
Together, our observations support a model in which adherens junctions and desmosomes assemble into distinct plasma membrane microdomains based not only on protein interactions but also due to the biophysical nature of the epithelial plasma membrane and the TMD characteristics of different cadherin subfamilies (Figure 9, A and B). Interestingly, early studies of desmosomal composition found that these junctions are enriched in sphingolipids and cholesterol, key components of what are now referred to as lipid rafts (Skerrow and Matoltsy, 1974; Drochmans et al., 1978). In addition, most of the major desmosomal proteins are palmitoylated, including the desmosomal cadherins and plakophilins, whereas adherens junction components lack this modification (Roberts et al., 2014, 2016). Given the key role for palmitoylation in lipid raft association, these findings further suggest that affinities for different lipid domains of the plasma membrane are central features that distinguish adherens junction and desmosomal proteins. Further studies will be needed to determine the precise structural and functional characteristics of different cadherin TMDs and how they selectively dictate lipid raft association. In addition, it will be important to discern how TMD characteristics are used in conjunction with lipid modifications such as palmitoylation to sort desmosome and adherens junction components into distinct plasma membrane domains with unique morphologies and functions. These features appear to be of fundamental importance for skin physiology, as our findings reveal that a mutation altering the structure of the desmoglein TMD is a novel pathomechanism of a desmosomal disease. This work also raises the possibility that other human disorders may result from alterations in lipid raft association or raft homeostasis. Indeed, loss of lipid raft targeting may be an underappreciated pathomechanism in human diseases that were previously conceived as generalized protein trafficking defects.
FIGURE 9:
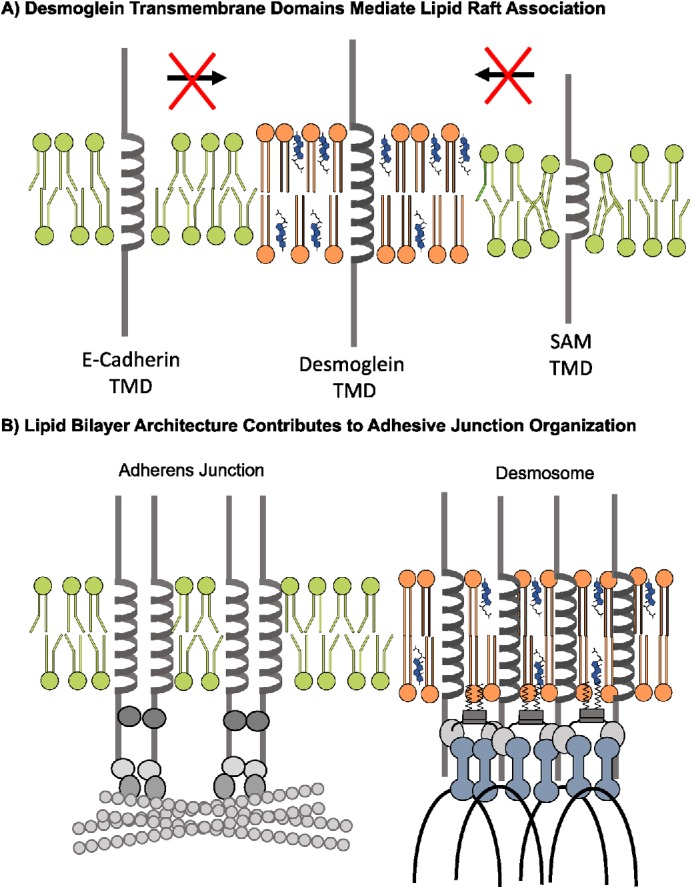
Model. (A) The extended DSG TMD facilitates lipid raft association. In contrast, the entry of E-cadherin and the Dsg1 SAM mutant into lipid rafts is unfavorable due to hydrophobic mismatch between the cadherin TMD and the phospholipid headgroups of the lipid bilayer. (B) Desmosomal proteins enter lipid raft domains through TMD affinities for raft–like membrane domains and palmitoylation of desmosomal cadherins and plaque proteins. In contrast, adherens junction components lack these raft-targeting features, resulting in exclusion of adherens junction components from lipid raft membrane domains. Thus, the biophysical properties of the bilayer associated with the desmosome promote spatial segregation of adherens junctions and desmosomes.
MATERIALS AND METHODS
Subjects
All affected and healthy family members or their legal guardians provided written and informed consent in accordance with the guidelines of the Institutional Review Board of Keio University and Emory University School of Medicine in adherence to the Helsinki guidelines. The investigators were not blinded to the allocation during experiments and outcome assessment.
Mutation analysis
Whole-exome sequencing was performed using genomic DNA isolated from the probands (II-2 and III-2) and their parents (I-1, I-2, and II-1). Whole-exome sequencing libraries were constructed using SureSelect Human All Exon V5 (Agilent) and sequenced by HiSeq2500 (Illumina). Sequencing reads were mapped to a human reference genome sequence (hs37d5) by BWA software (0.7.12-r1039). The mapped reads were realigned and variation sites were detected by GATK-3.30 software. The detected variation sites were annotated by SnpEff/SnpSift 4.1d software. Because the phenotype appeared in the proband II-2 (delivered from healthy parents) and transmitted to the proband III-2 (Figure 4D), we searched for genetic variations that de novo mutated in the proband II-2 and transmitted to the proband III-2. Only one variation was identified to fulfill the criteria, which was c.1684G>A (p.G562R) of DSG1, coding for the desmosomal cadherin desmoglein 1. Sanger sequencing confirmed the mutation was identified in the probands but not from other healthy family members (Figure 4, D and E). The mutation had not been identified in cohort studies (Genomes Project et al., 2015; Nagasaki et al., 2015; Sudmant et al., 2015; Higasa et al., 2016). The whole-exome sequencing of the probands II-2 and III-2 revealed no other variations in the exons and exon-intron boundaries of DSG1.
Immunohistochemistry and electron microscopy of patient samples
Biopsies were embedded in optimum cutting temperature solution and stored at –80°C. Prior to immunostaining, 5-µm cryosections were prepared on glass microscope slides. Primary and secondary antibodies are described below. Sections were sealed using mounting medium (ProLong Gold; ThermoFisher Scientific) and a coverslip. For electron microscopic studies, the biopsied sample was fixed in an ice-cold 2% glutaraldehyde/60 mM HEPES (pH 7.4) buffer followed by fixation with 1% osmium tetroxide, staining with 1% uranyl acetate, and embedding in Epon812. Ultrathin sections were stained with 1.5% uranyl acetate and Reynolds lead citrate and examined with an electron microscope (JEM-1010; JEOL) at the accelerating voltage of 80 kV.
Construction of mutants
Constructs were cloned using PCR and mutagenesis by the Cloning Division within Emory Integrated Genomics Core or purchased through Cyagen VectorBuilder services.
Structural predictions
Sequences for TMDs were analyzed using the Robetta structure prediction server (Kim et al., 2004).
Cell line generation, culture, and reagents
A431 cells were cultured in DMEM (Corning 10-013-CV) with 10% fetal bovine serum (FBS; Hyclone SH30071.03) and 1% penicillin/streptamycin (Corning 30-004-CI). Cells were stably infected with lentiviruses expressing the various murine desmoglein constructs. Blasticidin (5 µg/ml) was used to select for infected cells. No clonal isolation was performed. Cell lines expressing WT and mutant DSG1-GFP were subjected to fluorescence-activated cell sorting in order to obtain populations with roughly equal DSG1-GFP expression levels. For experiments utilizing a calcium switch, low-calcium medium was prepared as described previously (Wilson, 2014): no calcium DMEM (Life Technologies/Molecular Probes 21068028), 10% FBS, calcium chelating BT Chelex 100 resin (Bio-Rad 143-2832), and 1% penicillin/streptamycin.
Immunofluorescence
A431 cells were cultured to ∼70% confluence on glass coverslips. In experiments in which preextraction is explicitly used, cells were treated with phosphate-buffered saline (PBS)+ containing 0.2% Triton X-100 and 300 mM sucrose on ice for 1 min prior to fixation. Cells were fixed in 3.7% paraformaldehyde in PBS+ on ice for 10 min. Cells were permeabilized in PBS+ containing 0.1% Triton X-100 and 3% bovine serum albumin (BSA) for 10 min. Nonspecific antibody binding was prevented with a blocking step in PBS+ containing 3% BSA and 0.05% Triton X-100. Primary and secondary antibodies (listed below) were diluted into blocking solution. For rinse buffer, we used PBS+ containing 0.2% BSA and 0.05% Triton X-100. Cells were mounted to glass microscope slides using prolong gold mounting medium (described above).
Antibodies
Antibodies were mouse anti-DSG3 and AK15 described previously (Tsunoda et al., 2003); rabbit anti-calnexin (Enzo Life Sciences ADI-SPA-860); mouse anti-DP1/2 (Fitzgerald 10R-D108AX); rabbit anti-DP NW6 was a kind gift from Kathleen Green (Northwestern University); mouse anti-Dsg1-P124 (Progen 651111); mouse anti-plakoglobin (gamma catenin) (BD TransLabs 610253); mouse anti-E-cadherin (BD Biosciences 610252); mouse anti-flotillin 1 (BD 610820); mouse anti-flotillin 2 (BD 610383); rabbit anti-GFP Life A11122); rabbit anti-FLAG (Bethyl A190-102A). Secondary antibodies conjugated to Alexa Fluors were purchased from Invitrogen. Horseradish peroxidase–conjugated secondary antibodies were purchased from Bio-Rad.
Image acquisition and processing
Widefield fluorescence microscopy was performed using a DMRXA2 microscope (Leica, Wetzler, Germany) equipped with a 100×/1.40 NA oil immersion objective and narrow band pass filters. Images were acquired with an ORCA digital camera (Hamamatsu Photonics, Bridgewater, NJ) and processed using Fiji ImageJ. Superresolution microscopy was performed using a Nikon N-SIM system on an Eclipse Ti-E microscope system equipped with a 100×/1.49 NA oil immersion objective, 488- and 561-nm solid-state lasers in three-dimensional (3D) SIM mode. Images were captured using an EM charge-coupled device camera (DU-897; Andor Technology) and reconstructed using NIS-Elements software with the N-SIM module (version 3.22; Nikon). All microscopy was performed at room temperature. Widefield microscopy results are representative of two independent replicates with at least 10 cells each, whereas SIM results are representative of at least 50 desmosomes per condition.
Desmosome targeting analysis using SIM
To quantify desmosome targeting in cultured cells, Dsg1.GFP fluorescence was measured within regions of interest (ROI) drawn around DP railroad track staining at cell–cell borders. DP was detected using the Fitzgerald antibody as described above. This Dsg1.GFP fluorescence intensity was compared with adjacent ROI at regions of cell borders lacking desmosomes. For both WT and mutant Dsg1, targeting to desmosomes was measured as a fold enrichment of Dsg1.GFP fluorescence in desmosomes compared with nondesmosomal regions. For SAM syndrome patient and control tissue, desmosomal ROIs were defined using DP (via NW6 antibody labeling, see above) railroad tracks and DSG1 fluorescence was measured therein.
Triton solubility/insolubility
A431 cells were cultured until confluent in six-well tissue culture plates. Cells were washed twice with ice-cold PBS. The Triton-soluble pool was isolated by incubating cells with Triton buffer (1% Triton X-100, 10 mM Tris, pH 7.5, 140 mM NaCl, 5 mM EDTA, 2 mM egtazic acid [EGTA], with protease inhibitor) for 10 min on ice. Lysate was then centrifuged at 16,000 × g for 10 min at 4°C to pellet Triton-insoluble fraction. Triton-soluble supernatant was collected and mixed 1:1 with 2× Laemmli sample buffer containing 5% β-mercaptoethanol. The Triton-insoluble pellet was resuspended in 2× Laemmli sample buffer (Bio-Rad 161-0737) containing 5% β-mercaptoethanol. All samples were heated to 95°C for 10 min and vortexed for 30 s halfway through, prior to being run on a gel for Western blotting.
Isolation of DRM
DRM were isolated as described previously (Lingwood and Simons, 2007). Briefly, cells were cultured in 25-cm2 flasks (two per gradient) and washed with PBS+. Cells were collected by scraping in TNE buffer supplemented with protease inhibitors (Roche) and pelleted by centrifugation at 0.4 × g at 4°C for 5 min (5415R; Eppendorf). Cells were resuspended in TNE buffer and homogenized using a 25-gauge needle. TNE buffer containing Triton X-100 was added (final concentration of 1%) and cells were incubated on ice for 30 min. Four hundred microliters of detergent extract was mixed with 800 µl of 56% sucrose in TNE and placed at the bottom of a centrifuge tube. Volumes (1.9 ml) of 35 and 5% sucrose were layered on top of the sample. Following an 18-h centrifugation at 4°C (44,000 rpm, SW55 rotor, Beckman Optima LE-80 K Ultracentrifuge), 420-µl fractions (1–11, remaining volume combined to make up fraction 12) were removed from top to bottom of the gradient and stored at −20°C until processed for Western blot analysis. Flotillin-1 and Flotillin-2 were used as raft markers, and calnexin was used as a nonraft marker. Unless otherwise stated, all films shown are representative for at least three independent experiments.
GPMV isolation and partitioning measurements
GPMVs were isolated and imaged as described (Levental et al., 2009; Sezgin et al., 2012). Before GPMV isolation, cell membranes were stained with 5 µg/ml of F-DiO (Invitrogen), a fluorescent lipidic dye that strongly partitions to disordered phases because of double bonds in its fatty anchors (Levental et al., 2011).
Biotin labeling in pulse-chase experiments
For Dsg1 cleavage and recovery experiments, cells were grown to confluence in 35-mm cell culture plates (Corning 430165). Cells were trypsinized using TrypLE (Life Technologies 12605-010) for ∼8 min and suspended. After the indicated refractory period, surface proteins were biotinylated. For experiments monitoring protein turnover from the plasma membrane, surface proteins were biotinylated before the indicated period. Biotinylation was achieved using PBS+ containing 0.5 mg/ml EX-Link Sulfo-NHS-SS-Biotin (Thermo Scientific 21331) for 30 min at 37°C. Unbound biotin was quenched in PBS+ containing 50 mM NH4Cl for 1 min. Cells were lysed in RIPA (PBS+ containing 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate, 10 mM Tris-HCl, 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, and protease inhibitor cocktail [Roche 11836170001]), scraped to transfer from culture plate to an Eppendorf tube, and incubated for 10 min on ice. Lysate was cleared via centrifugation at 16,000 × g at 4°C for 10 min. Biotinylated protein was captured on streptavidin-coated beads during overnight incubation at 4°C. Beads were collected via centrifugation at 2500 × g at 4°C for 1 min. Protein was released from beads using Laemmli buffer containing 5% β-mercaptoethanol for 5 min at 95°C.
Mass-tagging of palmitoylated proteins
For mass-tag labeling, we followed the procedure described by Percher et al. (2016). Lysates from A431 cells expressing the indicated constructs were prepared in TEA buffer (50 mM triethanolamine; pH7.3, 150 mM NaCl, and 5 mM EDTA) containing 4% SDS. Two hundred micrograms of total cellular protein was treated with a final concentration of 10 mM neutralized TCEP for 30 min with end-over-end rotation. NEM was added to a final concentration of 25 mM and rocking continued for 2 h. NEM was removed by three rounds of chloroform/methanol/H2O precipitation. The final pellet was resuspended in TEA buffer containing 0.2% Triton X-100. Samples were treated with 0.75 M NH2OH hydroxylamine (+HA) or without (–HA) and incubated at room temperature for 1 h. Excess HA was removed with one round of chloroform/methanol/H2O precipitation and the pellet was resuspended in TEA buffer containing 0.2% Triton X-100 supplemented with 1 mM mPEG-Mal (10 kDa; Sigma). Samples were incubated with rocking for 2 h, and reactions were terminated by one round of chloroform/methanol/H2O precipitation. The final pellet was suspended in 1× Laemmli sample buffer and resolved by SDS–PAGE.
Isolation, freezing, and imaging of desmosomes
To isolate desmosomes from mouse liver, a method based on the protocol of Tsukita and Tsukita (1989) was used, in which a desmosomal fraction was obtained by sucrose density gradient centrifugation, followed by NP-40 detergent treatment. The fraction should contain only bile canaliculi-derived plasma membranes, as the homogenization and centrifugation steps are designed to free the preparation from contaminating cell fragments and nuclear membranes due to their higher densities (Neville, 1960; Song et al., 1969; Tsukita and Tsukita, 1989). The desmosomal fraction was immediately plunge-frozen on holey carbon grids, which were subsequently inserted into the column of a FEI Titan Krios at liquid nitrogen temperatures. Tilt series of the sample (+60 to –60°) were recorded and subsequently reconstructed into 3D tomograms. Subtomogram averaging was performed as described previously (Forster and Hegerl, 2007).
Membrane thickness measurements
For the thickness of the desmosomal membranes, 1768 selected positions (derived from three desmosomes in two tomograms) with visible cadherins were selected. As a comparison (control), 668 randomly selected positions at membranes adjacent to desmosomes (derived from three membranes in three tomograms) and 515 randomly selected positions from arbitrary membranes in the tomograms (derived from two membranes in one tomogram) were selected. Each position was cross-correlated with multiple references of a simplified membrane model of the two leaflets (dark lines representing phospholipid head groups and are included in the measurements) with various bilayer distances (3.08-, 3.52-, 3.96-, 4.4-, 4.84-, and 5.28-nm spacing) using subtomogram averaging routines with limited rotational freedom (±30° in 5° steps for all three Euler angles) after rough prealignment using the overall membrane orientation. The reference with the highest cross-correlation score then provides the bilayer spacing of each single subvolume.
Statistics
Error bars represent standard error of the mean. Significance was determined using a Student’s t test (two-tailed, heteroscedastic) and p values have been indicated. Statistical analysis of immunofluorescence results was conducted on at least two independent experiments with 10 images per condition per replicate. Statistical analysis of Western blotting was conducted on results from three independent experiments.
Supplementary Material
Acknowledgments
We thank Kathleen Green and members of the Kowalczyk lab for comments and insights during the preparation of this article. We also thank Joseph Lorent for help generating the TMD models. This work was supported by grants (R01AR048266, R01AR048266-13S1, and R01AR050501 to A.P.K.; LOEWE Dynamem to A.S.F.) and fellowships (F31AR066476 and T32GM008367 to J.D.L.) from the National Institutes of Health (NIH) and by the Practical Research Project for Rare/Intractable Diseases (16ek0109067h0003 to Y.M. and 16ek0109151h0002 to A.K.) from the Japan Agency for Medical Research and Development. I.L. is funded by the NIH/National Institute of General Medical Sciences (GM114282, GM124072, GM120351), the Volkswagen Foundation (grant 93091), and the Human Frontiers Science Program (RGP0059/2019). Additional support was provided by core facilities at Emory University, including the Integrated Cellular Imaging Core, the Emory Flow Cytometry Core, and the Cloning Division within Emory Integrated Genomics Core.
Abbreviations used:
- cryo-ET
cryo-electron tomography
- DP
desmoplakin
- DRM
detergent-resistant membrane
- DSC
desmocollins
- Dsg
desmoglein
- Ecad
epithelial cadherin
- FBS
fetal bovine serum
- F-DiO
FAST-DiO
- GFP
green fluorescent protein
- GPMV
giant plasma membrane vesicle
- HA
hydroxylamine
- IL2R
interleukin 2 receptor
- PBS
phosphate-buffered saline
- ROI
regions of interest
- SAM
severe dermatitis, multiple allergies, and metabolic wasting syndrome
- SIM
structured illumination microscopy
- TMD
transmembrane domain
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-10-0649) on April 3, 2019.
REFERENCES
- Al-Amoudi A, Castano-Diez D, Devos DP, Russell RB, Johnson GT, Frangakis AS. (2011). The three-dimensional molecular structure of the desmosomal plaque. Proc Natl Acad Sci USA , 6480–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Amoudi A, Diez DC, Betts MJ, Frangakis AS. (2007). The molecular architecture of cadherins in native epidermal desmosomes. Nature , 832–837. [DOI] [PubMed] [Google Scholar]
- Amagai M, Stanley JR. (2012). Desmoglein as a target in skin disease and beyond. J Invest Dermatol , 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornslaeger EA, Corcoran CM, Stappenbeck TS, Green KJ. (1996). Breaking the connection: displacement of the desmosomal plaque protein desmoplakin from cell-cell interfaces disrupts anchorage of intermediate filament bundles and alters intercellular junction assembly. J Cell Biol , 985–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan D, Peltonen S, Dowling A, Medhat W, Green KJ, Wahl JK, 3rd, Del Galdo F, Mahoney MG. (2012). A role for caveolin-1 in desmoglein binding and desmosome dynamics. Oncogene , 1636–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke MA, Nitoiu D, Kelsell DP. (2012). Cell-cell connectivity: desmosomes and disease. J Pathol , 158–171. [DOI] [PubMed] [Google Scholar]
- Brown DA, London E. (2000). Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem , 17221–17224. [DOI] [PubMed] [Google Scholar]
- Charollais J, Van Der Goot FG. (2009). Palmitoylation of membrane proteins (Review). Mol Membr Biol , 55–66. [DOI] [PubMed] [Google Scholar]
- Cheng R, Yan M, Ni C, Zhang J, Li M, Yao Z. (2016). Report of Chinese family with severe dermatitis, multiple allergies and metabolic wasting syndrome caused by novel homozygous desmoglein-1 gene mutation. J Dermatol , 1201–1204. [DOI] [PubMed] [Google Scholar]
- Danescu S, Leppert J, Cosgarea R, Zurac S, Pop S, Baican A, Has C. (2017). Compound heterozygosity for dominant and recessive DSG1 mutations in a patient with atypical SAM syndrome (severe dermatitis, multiple allergies, metabolic wasting). J Eur Acad Dermatol Venereol , e144–e146. [DOI] [PubMed] [Google Scholar]
- de Jesus AJ, Allen TW. (2013). The determinants of hydrophobic mismatch response for transmembrane helices. Biochim Biophys Acta , 851–863. [DOI] [PubMed] [Google Scholar]
- Delmar M, McKenna WJ. (2010). The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res , 700–714. [DOI] [PubMed] [Google Scholar]
- Delva E, Jennings JM, Calkins CC, Kottke MD, Faundez V, Kowalczyk AP. (2008). Pemphigus vulgaris IgG-induced desmoglein-3 endocytosis and desmosomal disassembly are mediated by a clathrin- and dynamin-independent mechanism. J Biol Chem , 18303–18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delva E, Tucker DK, Kowalczyk AP. (2009). The desmosome. Cold Spring Harb Perspect Biol , a002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Rohrer B, Levental KR, Levental I. (2014a). Rafting through traffic: Membrane domains in cellular logistics. Biochim Biophys Acta , 3003–3013. [DOI] [PubMed] [Google Scholar]
- Diaz-Rohrer BB, Levental KR, Simons K, Levental I. (2014b). Membrane raft association is a determinant of plasma membrane localization. Proc Natl Acad Sci USA , 8500–8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drochmans P, Freudenstein C, Wanson JC, Laurent L, Keenan TW, Stadler J, Leloup R, Franke WW. (1978). Structure and biochemical composition of desmosomes and tonofilaments isolated from calf muzzle epidermis. J Cell Biol , 427–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster F, Hegerl R. (2007). Structure determination in situ by averaging of tomograms. Methods Cell Biol , 741–767. [DOI] [PubMed] [Google Scholar]
- Garcia-Garcia E, Brown EJ, Rosales C. (2007). Transmembrane mutations to FcgammaRIIA alter its association with lipid rafts: implications for receptor signaling. J Immunol , 3048–3058. [DOI] [PubMed] [Google Scholar]
- Garcia MA, Nelson WJ, Chavez N. (2018). Cell-cell junctions organize structural and signaling networks. Cold Spring Harb Perspect Biol , a029181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. (2015). A global reference for human genetic variation. Nature , 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves J, Chamberlain LH. (2007). Palmitoylation-dependent protein sorting. J Cell Biol , 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon RM, Green KJ. (2013). Structural and functional diversity of desmosomes. Cell Commun Adhes , 171–187. [DOI] [PubMed] [Google Scholar]
- Harrison OJ, Brasch J, Lasso G, Katsamba PS, Ahlsen G, Honig B, Shapiro L. (2016). Structural basis of adhesive binding by desmocollins and desmogleins. Proc Natl Acad Sci USA , 7160–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Has C, Jakob T, He Y, Kiritsi D, Hausser I, Bruckner-Tuderman L. (2015). Loss of desmoglein 1 associated with palmoplantar keratoderma, dermatitis and multiple allergies. Br J Dermatol , 257–261. [DOI] [PubMed] [Google Scholar]
- Has C, Technau-Hafsi K. (2016). Palmoplantar keratodermas: clinical and genetic aspects. J Dtsch Dermatol Ges , 123–139; quiz 140. [DOI] [PubMed] [Google Scholar]
- Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, Doi K, Shimizu M, Nakabayashi K, Aoki Y, et al. (2016). Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet , 547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthofer B, Windoffer R, Troyanovsky S, Leube RE. (2007). Structure and function of desmosomes. Int Rev Cytol , 65–163. [DOI] [PubMed] [Google Scholar]
- Honigmann A, Pralle A. (2016). Compartmentalization of the cell membrane. J Mol Biol , 4739–4748. [DOI] [PubMed] [Google Scholar]
- Hsu CK, Lin HH, Harn HI, Hughes MW, Tang MJ, Yang CC. (2018). Mechanical forces in skin disorders. J Dermatol Sci , 232–240. [DOI] [PubMed] [Google Scholar]
- Ishida-Yamamoto A, Kelsell D, Common J, Houseman MJ, Hashimoto M, Shibaki H, Asano K, Takahashi H, Hashimoto Y, Senshu T, et al. (2000). A case of erythrokeratoderma variabilis without mutations in connexin 31. Br J Dermatol , 1283–1287. [DOI] [PubMed] [Google Scholar]
- Jones JC, Kam CY, Harmon RM, Woychek AV, Hopkinson SB, Green KJ. (2017). Intermediate filaments and the plasma membrane. Cold Spring Harb Perspect Biol , a025866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DE, Chivian D, Baker D. (2004). Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res , W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucerka N, Perlmutter JD, Pan J, Tristram-Nagle S, Katsaras J, Sachs JN. (2008). The effect of cholesterol on short- and long-chain monounsaturated lipid bilayers as determined by molecular dynamics simulations and X-ray scattering. Biophys J , 2792–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I, Byfield FJ, Chowdhury P, Gai F, Baumgart T, Janmey PA. (2009). Cholesterol-dependent phase separation in cell-derived giant plasma-membrane vesicles. Biochem J , 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I, Grzybek M, Simons K. (2011). Raft domains of variable properties and compositions in plasma membrane vesicles. Proc Natl Acad Sci USA , 11411–11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Levental I. (2015a). Giant plasma membrane vesicles: models for understanding membrane organization. Curr Top Membr , 25–57. [DOI] [PubMed] [Google Scholar]
- Levental KR, Levental I. (2015b). Isolation of giant plasma membrane vesicles for evaluation of plasma membrane structure and protein partitioning. Methods Mol Biol , 65–77. [DOI] [PubMed] [Google Scholar]
- Levental I, Lingwood D, Grzybek M, Coskun U, Simons K. (2010). Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci USA , 22050–22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I, Veatch SL. (2016). The continuing mystery of lipid rafts. J Mol Biol , 4749–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BA, Engelman DM. (1983). Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J Mol Biol , 211–217. [DOI] [PubMed] [Google Scholar]
- Lingwood D, Kaiser HJ, Levental I, Simons K. (2009). Lipid rafts as functional heterogeneity in cell membranes. Biochem Soc Trans , 955–960. [DOI] [PubMed] [Google Scholar]
- Lingwood D, Simons K. (2007). Detergent resistance as a tool in membrane research. Nat Protoc , 2159–2165. [DOI] [PubMed] [Google Scholar]
- Lorent JH, Diaz-Rohrer B, Lin X, Spring K, Gorfe AA, Levental KR, Levental I. (2017). Structural determinants and functional consequences of protein affinity for membrane rafts. Nat Commun , 1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovgren ML, McAleer MA, Irvine AD, Wilson NJ, Tavadia S, Schwartz ME, Cole C, Sandilands A, Smith FJD, Zamiri M. (2017). Mutations in desmoglein 1 cause diverse inherited palmoplantar keratoderma phenotypes: implications for genetic screening. Br J Dermatol , 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D. (2008). Energetics of hydrophobic matching in lipid-protein interactions. Biophys J , 3996–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munday AD, Lopez JA. (2007). Posttranslational protein palmitoylation: promoting platelet purpose. Arterioscler Thromb Vasc Biol , 1496–1499. [DOI] [PubMed] [Google Scholar]
- Nagasaki M, Yasuda J, Katsuoka F, Nariai N, Kojima K, Kawai Y, Yamaguchi-Kabata Y, Yokozawa J, Danjoh I, Saito S, et al. (2015). Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun , 8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava P, Laukoetter MG, Hopkins AM, Laur O, Gerner-Smidt K, Green KJ, Parkos CA, Nusrat A. (2007). Desmoglein-2: a novel regulator of apoptosis in the intestinal epithelium. Mol Biol Cell , 4565–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasova O, Green KJ. (2013). Desmosome assembly and dynamics. Trends Cell Biol , 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville DM., Jr (1960). The isolation of a cell membrane fraction from rat liver. J Biophys Biochem Cytol , 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezil FA, Bloom M. (1992). Combined influence of cholesterol and synthetic amphiphillic peptides upon bilayer thickness in model membranes. Biophys J , 1176–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemela PS, Ollila S, Hyvonen MT, Karttunen M, Vattulainen I. (2007). Assessing the nature of lipid raft membranes. PLoS Comput Biol , e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palka HL, Green KJ. (1997). Roles of plakoglobin end domains in desmosome assembly. J Cell Sci (pt 19), 2359–2371. [DOI] [PubMed] [Google Scholar]
- Parks GD, Lamb RA. (1993). Role of NH2-terminal positively charged residues in establishing membrane protein topology. J Biol Chem , 19101–19109. [PubMed] [Google Scholar]
- Payne AS, Hanakawa Y, Amagai M, Stanley JR. (2004). Desmosomes and disease: pemphigus and bullous impetigo. Curr Opin Cell Biol , 536–543. [DOI] [PubMed] [Google Scholar]
- Percher A, Ramakrishnan S, Thinon E, Yuan X, Yount JS, Hang HC. (2016). Mass-tag labeling reveals site-specific and endogenous levels of protein S-fatty acylation. Proc Natl Acad Sci USA , 4302–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ. (2004). Lipid rafts: heterogeneity on the high seas. Biochem J , 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ. (2006). Rafts defined: a report on the Keystone symposium on lipid rafts and cell function. J Lipid Res , 1597–1598. [DOI] [PubMed] [Google Scholar]
- Quinlan RA, Schwarz N, Windoffer R, Richardson C, Hawkins T, Broussard JA, Green KJ, Leube RE. (2017). A rim-and-spoke hypothesis to explain the biomechanical roles for cytoplasmic intermediate filament networks. J Cell Sci , 3437–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy T, Manrique S, Buyan A, Hall BA, Chetwynd A, Sansom MS. (2014). Primary and secondary dimer interfaces of the fibroblast growth factor receptor 3 transmembrane domain: characterization via multiscale molecular dynamics simulations. Biochemistry , 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnik N, Sepcic K, Plemenitas A, Windoffer R, Leube R, Veranic P. (2011). Desmosome assembly and cell-cell adhesion are membrane raft-dependent processes. J Biol Chem , 1499–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BJ, Johnson KE, McGuinn KP, Saowapa J, Svoboda RA, Mahoney MG, Johnson KR, Wahl JK., 3rd (2014). Palmitoylation of plakophilin is required for desmosome assembly. J Cell Sci , 3782–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BJ, Svoboda RA, Overmiller AM, Lewis JD, Kowalczyk AP, Mahoney MG, Johnson KR, Wahl JK, 3rd (2016). Palmitoylation of desmoglein 2 is a regulator of assembly dynamics and protein turnover. J Biol Chem , 24857–24865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, Amagai M, Kowalczyk AP. (2012a). Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One , e50696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M, Tucker DK, Kohlhorst D, Niessen CM, Kowalczyk AP. (2012b). Classical and desmosomal cadherins at a glance. J Cell Sci , 2547–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, Koetsier JL, Gat A, Goldberg I, Bergman R, et al. (2013). Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet , 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelov L, Sprecher E. (2015). Inherited desmosomal disorders. Cell Tissue Res , 457–475. [DOI] [PubMed] [Google Scholar]
- Santos AL, Preta G. (2018). Lipids in the cell: organisation regulates function. Cell Mol Life Sci , 1909–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P, Roth MG, Simons K. (1997). Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J , 5501–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schow EV, Freites JA, Cheng P, Bernsel A, von Heijne G, White SH, Tobias DJ. (2011). Arginine in membranes: the connection between molecular dynamics simulations and translocon-mediated insertion experiments. J Membr Biol , 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setzer SV, Calkins CC, Garner J, Summers S, Green KJ, Kowalczyk AP. (2004). Comparative analysis of armadillo family proteins in the regulation of a431 epithelial cell junction assembly, adhesion and migration. J Invest Dermatol , 426–433. [DOI] [PubMed] [Google Scholar]
- Sezgin E, Kaiser HJ, Baumgart T, Schwille P, Simons K, Levental I. (2012). Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat Protoc , 1042–1051. [DOI] [PubMed] [Google Scholar]
- Sezgin E, Levental I, Mayor S, Eggeling C. (2017). The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Sampaio JL. (2011). Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol , a004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson CL, Patel DM, Green KJ. (2011). Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat Rev Mol Cell Biol , 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerrow CJ, Matoltsy AG. (1974). Chemical characterization of isolated epidermal desmosomes. J Cell Biol , 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolik-Delmaire T, Reddy R, Pashaj A, Roberts BJ, Wahl JK., 3rd (2010). Plakophilin-1 localizes to the nucleus and interacts with single-stranded DNA. J Invest Dermatol , 2638–2646. [DOI] [PubMed] [Google Scholar]
- Song CS, Rubin W, Rifkind AB, Kappas A. (1969). Plasma membranes of the rat liver. Isolation and enzymatic characterization of a fraction rich in bile canaliculi. J Cell Biol , 124–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahley SN, Kowalczyk AP. (2015). Desmosomes in acquired disease. Cell Tissue Res , 439–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP. (2014). Desmosome assembly and disassembly are membrane raft-dependent. PLoS One , e87809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature , 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumigray KD, Lechler T. (2015). Cell adhesion in epidermal development and barrier formation. Curr Top Dev Biol , 383–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason HA, Scothern A, McHarg S, Garrod DR. (2010). Desmosomes: adhesive strength and signalling in health and disease. Biochem J , 419–433. [DOI] [PubMed] [Google Scholar]
- Troyanovsky S. (2012). Adherens junction assembly. Subcell Biochem , 89–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Tsukita S. (1989). Isolation of cell-to-cell adherens junctions from rat liver. J Cell Biol , 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M. (2003). Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol , 2170–2178. [DOI] [PubMed] [Google Scholar]
- Wahl JK., 3rd (2005). A role for plakophilin-1 in the initiation of desmosome assembly. J Cell Biochem , 390–403. [DOI] [PubMed] [Google Scholar]
- Wilson VG. (2014). Growth and differentiation of HaCaT keratinocytes. Methods Mol Biol , 33–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.