

Abstract
A network of claudin strands creates continuous cell–cell contacts to form the intercellular tight junction barrier; a second protein, occludin, is associated along these strands. The physiological barrier remains stable despite protein turnover, which involves removal and replacement of claudins both in the steady state and during junction remodeling. Here we use a pulse–block–pulse labeling protocol with fluorescent ligands to label SNAP/CLIP-tags fused to claudins and occludin to identify their spatial trafficking pathways and kinetics in Madin–Darby canine kidney monolayers. We find that claudins are first delivered to the lateral membrane and, over time, enter the junction strand network from the basal side; this is followed by slow replacement of older claudins in the strands. In contrast, even at early times, newly synthesized occludin is found throughout the network. Taking the results together with our previous documentation of the mechanism for claudin strand assembly in a fibroblast model, we speculate that newly synthesized claudins are added at strand breaks and free ends; these are most common in the basalmost edge of the junction. In contrast, occludin can be added directly within the strand network. We further demonstrate that claudin trafficking and half-life depend on carboxy-terminal sequences and that different claudins compete for tight junction localization.
INTRODUCTION
Tight junctions form the selective paracellular barrier between epithelial cells required for directional transepithelial absorption and secretion. Claudins (cldns), a family of 26 small integral membrane proteins (Liu et al., 2016), are the principal components of this seal. Cldns polymerize to form a network of strands in each cell and pair with cldn strands on adjacent cells to create an angstrom-level size- and charge-selective solute barrier (Van Itallie and Anderson, 2006). This anastomosing meshwork of cldn strands is the structural hallmark of the tight junction when visualized by freeze-fracture electron microscopy (FFEM; Furuse et al., 1998). Localization of cldn-based strands to the tight junction is dependent on their interaction with members of the zonula occludens (ZO) scaffolding protein family, ZO-1/2/3 (Umeda et al., 2006). The amino acid sequences of most cldns end in PDZ-binding motifs that can bind to the first PDZ domains of ZO proteins; this scaffolding interaction is required for efficient strand formation and placement (Itoh et al., 1999). Recent x-ray crystallographic structures suggest how cldns interact across cells in trans and form antiparallel double polymer strands in cis (Suzuki et al., 2014, 2015; Zhao et al., 2018); however, these models do not provide insight into how new cldns might be incorporated into continuous tight-junction strands. By analogy with other polymeric protein systems such as actin-based microfilaments and tubulin-based microtubules or packing of connexins at gap junction plaques, it seems likely that claudins add at exposed free ends of strands and not within the polymer.
Although in most cases tight-junction strand networks are limited to the very apical end of the lateral membrane, within this restricted location there is some polarity in the strand organization, with apical strands being more continuous and with free strand ends and strand breaks mostly occurring along basal strands. This is particularly evident in intestinal crypt cells and in mitotic cells (Tice et al., 1979; Mora-Galindo, 1986), although also readily found in freeze-fracture images of tight junctions in Madin–Darby canine kidney (MDCK) cells (e.g., Figure 1, arrows) and in other epithelial cells and tissues. In addition, a cldn-binding domain of an intestinal toxin resulted in disaggregation of cldn strands preferentially from the basalmost strands, suggesting that these were more dynamic or unstable and thus more accessible than apical strands (Sonoda et al., 1999). Using SNAP-tagged cldns expressed in fibroblasts, we recently found that strands break and reform and the site of addition of new cldns is preferentially at the free ends of broken strands (Van Itallie et al., 2017).
FIGURE 1:
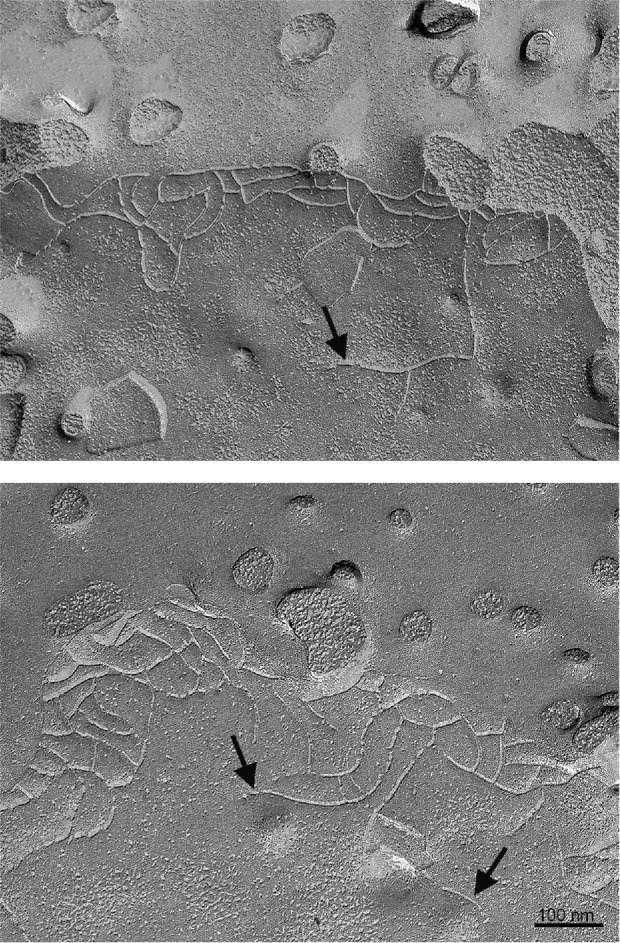
Freeze fracture electron microscopic images of MDCK II cells reveals basal free ends and strand breaks; the apical surface showing microvillar stubs is at the top of the images. Some free ends are indicated by arrows. Bar 100 nm.
Occludin (ocln), like cldns, is an integral membrane protein that interacts with ZO-1 and associates with tight-junction freeze-fracture strands in a structurally undefined way. Unlike cldns, ocln by itself cannot form strands but is instead recruited there by cldns (Furuse et al., 1998). Biochemical data suggest that interaction with cldns may actually be indirect, mediated by the ability of both proteins to bind ZO-1 (Raleigh et al., 2011). Confocal (Furuse et al., 1998) and more recently superresolution analysis (Van Itallie et al., 2017) of cldn and ocln coexpressed in fibroblasts suggest that although ocln localizes to cldn strands, its distribution is patchy rather than continuous. In addition, measurement of fluorescence recovery after photobleaching (FRAP) of ocln and cldns in epithelial cells shows fast turnover for ocln, but relatively much slower recovery for cldns (Shen et al., 2008; Van Itallie et al., 2017). This difference in FRAP behavior suggests that claudins are more stably located in the tight junction and that there is not a stable, constitutive association between these two integral membrane proteins.
Although freeze-fracture strands are restricted to the apical tight junction (Tice et al., 1979), it has long been evident that many cldns (Rahner et al., 2001; Holmes et al., 2006) and a minor fraction of ocln (Sakakibara et al., 1997) are also variably distributed along the lateral membrane. For some cldns, binding partners on the lateral membrane have been identified. For example, lateral cldn7 has been shown to interact with EpCam (Kuhn et al., 2007; Wu et al., 2013), and both cldn7 and cldn11 associate with lateral integrins (Tiwari-Woodruff et al., 2001; Lu et al., 2015). These interactions have been demonstrated to have implications for cell growth, migration, and tight junction function. However, the role and organization of most lateral cldns remain unclear. One possibility is that these cldns and the small proportion of lateral ocln represent a dynamic pool equilibrating pre- or postpolymerization with apical strands. To test the possibility that some of the lateral cldns and ocln might be in the process of trafficking to the tight junction for addition to basal strands, we made cldns 2 and 4 and ocln with SNAP and CLIP fusion protein tags (Stoops et al., 2014) and used a variety of fluorescent and nonfluorescent ligands to follow their biosynthetic progress. We found that newly synthesized cldns arrived first on the lateral membrane, then concentrated at the basal pole of the tight junction, and subsequently replaced junctional cldns, consistent with the possibility that tight junction strands form and mature in a basal to apical direction. In contrast, newly synthesized ocln appeared to be targeted directly to the tight junction.
RESULTS
Different Cldns partition differently between the tight junction and lateral membrane
Although claudin-based freeze-fracture strands are focused at the apical end of the lateral membrane, many cldns are also normally localized along the lateral membranes in tissues (Holmes et al., 2006) and cultured epithelial cells (Tokuda and Furuse, 2015). For example, in MDCK II cells, cldn2 colocalizes with the tight junction marker ZO-1 (Figure 2A, top panels) and prominently in intracellular vesicles, while cldn1 (Figure 2, middle panel), cldn3, and cldn4 (Figure 2A, bottom panels) are all found with ZO-1 at tight junctions but also distributed along the lateral cell membranes. This is most evident in z-axis stacked images, where ZO-1 marks the site of the tight junctions and the cldns are variably associated both with the junction and with the lateral membrane (Figure 2B). Lateral cldns are unevenly distributed over the membrane, concentrated in what appear to be patches, raising the question of whether cldns cluster on the lateral membrane (Figure 2A, arrows). However, what appear to be patches are unlikely to represent specific structural clusters, but simply to be focally increased fluorescent signals where there are multiple infoldings in the plasma membrane (Supplemental Figure S1). Consistent with this, the immunofluorescent signal from β1-integrin is also enhanced where it colocalizes with cldn-4 (Supplemental Figure S1). Likewise, when the lateral membrane is labeled with derivatized biotin and detected with fluorescent streptavidin, that signal is also enhanced with the increased cldn 4 signal (Supplemental Figure S1). Because both signals would be expected to distribute uniformly on the lateral membrane, the enhanced signals for cldn, β1-integrin, and streptavidin conjugates most likely all reflect increased focal concentrations due to membrane interdigitations.
FIGURE 2:

Endogenous clds are variably distributed between the apical tight junction and lateral membrane in MDCK II cells. (A) Top, Maximum-intensity projections of immunofluorescence confocal colocalization of cldn2, cldn1, ZO-1, and merge (left to right panels) show that cldn1 is more laterally disposed than cldn2 or ZO-1. Bottom, Colocalization of cldn4, cldn3, ZO-1, and merge (left to right panels) reveals cldn4 and cldn3 at the lateral membrane as well as colocalized with ZO-1. Arrows in enlargement show apparent concentrations of lateral cldns likely related to lateral membrane infoldings (see Supplemental Figure S1). (B) z-Axis images as in A show ZO-1 focused at the apical junctional region and cldns colocalized with ZO-1 but also distributed along the lateral membrane.
Newly synthesized cldn2 and 4 both traffic to the lateral membrane but with differing kinetics
Lateral cldns could 1) be newly synthesized and initially trafficked to the lateral membrane before they are incorporated into the tight junction strands; 2) have been depolymerized from tight junction strands; 3) be present in an independent, stable lateral cldn pool; or 4) be distributed on the lateral membrane due to some combination of these possibilities. To discriminate among these ideas and define the trafficking pathway, we chose to make cell lines expressing SNAP-tagged cldn 2, as a representative of a mostly apically disposed cldn, and SNAP-tagged cldn4, as an example of a cldn that is also distributed along the lateral cell membranes. The SNAP system has been used previously to define biosynthetic routes for other membrane proteins (Harder et al., 2012). Because we were concerned that overexpression of exogenous cldns might influence their half-life, trafficking, and distribution, we used CRISPR/Cas9-mediated gene editing and homologous recombination from a donor plasmid (Butkevich et al., 2018) to insert SNAP tag sequences into the gene locations encoding the amino termini of endogenous cldn2 and 4 in MDCK cells (Butkevich et al., 2018). This should allow expression of the SNAP-tagged cldns to remain under the control of their endogenous promotors and at normally expressed levels. The scheme for this is shown in Figure 3A; single cells expressing putative SNAP-tagged cldns were selected after incubation with fluorescent SNAP ligand and sorted into individual wells by FACS. Following expansion, potentially correctly edited clones were characterized by PCR analysis to detect the presence of the modified cldn2 and cldn4 genes (Figure 3B). The resulting gene-edited cell lines expressing the endogenously tagged cldns are designated SNAP(e)cldn, where (e) indicates that they function under their endogenous promoters and differentiates them from overexpressing cell lines used in some complementary experiments. Immunoblot comparison of wild-type with SNAP-tag overexpressing and gene-edited lines (Figure 3C) demonstrates that in gene-edited lines, there is loss of signal at the size of unmodified cldn2 and 4 and appearance of a single immunoreactive band corresponding to the SNAP cldn fusion protein. Immunofluorescence localization of SNAP(e)cldns (Figure 3D) is similar to that of untagged cldns. Most SNAP(e)cldn2 is vesicular or colocalizes with ZO-1 (Figure 3D, top panels), while SNAP(e)cldn4 (Figure 3E, bottom panels), like the endogenous cldn4, colocalizes with ZO-1 but also appears on the lateral membrane.
FIGURE 3:

CRISPR/Cas9 gene editing was used to insert SNAP tags at the N-terminal ends of cldns2 and 4. (A) Scheme for insertion of SNAP tags into MDCK cell DNA; CRISPR/Cas9/gRNA was used to cleave DNA specifically upstream of coding regions and homology-directed recombination was used to insert SNAP tag; cells were labeled with fluorescent SNAP tag ligand and single cells sorted into multiwell dishes. (B) Positive clones were identified by PCR (position of primers indicated in A) and by immunoblot (C), where expression was compared with wild-type MDCK II cells and cells stably overexpressing SNAP-cldns (left panel probed for ocln and cldn2, right for ocln and cldn4). The endogenously tagged cldn4 fusion protein contains five extra residues between the SNAP tag and the start of cldn4, resulting in a slightly larger fusion protein. Ocln was used as a loading control. The upper magenta band in the cldn4 panel (overlapping with and slightly above ocln) is a nonspecific band seen with lots of IR-labeled secondary antibodies. (D) Maximum intensity projections of confocal images of ZO-1 colocalized JF549 SNAP-ligand labeled SNAP(e)cldn2 (top panels) and SNAP(e)cldn4 (bottom panels) shows localization like that of endogenous cldns, as do z-view images (E).
To observe cldn biosynthesis and trafficking, a pulse–block–pulse protocol was employed, sequentially labeling the SNAP tag with two ligands fluorescing at different wavelengths. Cells expressing endogenously tagged cldns were first incubated with a cell-permeant fluorescent substrate, commonly JF549 SNAP-tag ligand; this was followed by the administration of an excess of unlabeled SNAP ligand to block any SNAP(e)cldns that had escaped labeling with JF549. Cells were then washed and incubated in the absence of fluorescent ligand for various periods of time to allow synthesis of new SNAP(e)cldns. These newly synthesized cldns were then labeled with a second fluorescent SNAP-tag ligand, most often SNAP-cell 505*; this scheme is outlined in Figure 4A. The distribution of JF549-labeled SNAP(e)cldn and SNAP-cell 505*-labeled cldn thus defines the localization of “old” cldn and “new” cldn, respectively. The change in the distribution of old and new cldn for SNAP(e)cldn2 (Figure 4B, top panels) and SNAP(e)cldn4 (Figure 4B, bottom panels) reveals that over 24 h, SNAP(e)cldns turn over, but that the time course differs between the two cldns. At time 0, the old cldn in both SNAP(e)cldn2 and 4 expressing cells is localized at cell–cell contacts and to some extent in intracellular vesicles. However, 4 h after cells are blocked and allowed to synthesize new cldns, new SNAP(e)cldn2 is mostly seen in the Golgi (Figure 4A, top panels, and Supplemental Figure S2A, top panels), while new SNAP(e)cldn4 is in the Golgi and has also begun to accumulate on the lateral membrane (Figure 4B, bottom panels). Disruption of trans-Golgi trafficking with Brefeldin A blocked membrane localization of new cldn2 and 4 (Supplemental Figure S2B). By 8 h, new SNAP(e)cldn2 begins to appear at the lateral membrane, but the predominant fluorescent signal is still from the old SNAP(e)cldn2; in contrast, at the same time, new SNAP(e)cldn4 has clearly replaced much of the old cldn4. By 24 h, both old SNAP(e)cldn2 and 4 are undetectable and have been replaced by the second pulse of protein. The signal at the junction compared with the immediate perijunctional membrane was quantified at the 4- and 8-h time points by line scanning across cell contacts and by comparing plot profiles made from 549 (magenta) and 505 (green) channels; graphs comparing the relative intensities from matched images are shown in Figure 4C. Newly synthesized cldn-4 clearly arrives at the lateral membrane and enters the tight junction faster than cldn-2.
FIGURE 4:

Pulse-chase SNAP tag labeling of SNAP(e)cldn2 and 4 reveals that newly synthesized cldns are trafficked to the lateral membranes and that cldn2 is biosynthesized and/or trafficked more slowly than cldn4. (A) Scheme of SNAP tag labeling, first with JF549 SNAP-ligand to label all cldn2 or 4 (T = 0), followed by blocking for 30 min with SNAP-cell block. This is followed by incubation for various periods of time (4, 8, and 24 h) and then labeling newly synthesized cldns with SNAP-cell 505*. (B) Fluorescence of SNAP ligand labeling of SNAP(e)cldn2 (top panels) and SNAP(e)cldn4 (bottom panel) expressing MDCK II cells. Cells are imaged after labeling with JF549 SNAP ligand and then labeled and imaged with 505*at 4, 8, and 24 h after blocking. Difference in biosynthesis/trafficking is most evident at the 4-h time point. Arrows indicate vesicular colocalization of old and new SNAP(e)cldn4; arrowhead indicates vesicular structure containing only new SNAP(e)cldn4. (C) Line scan across cell contacts at 4 and 8 h reveal more accumulation of new SNAP(e)cldn4 than SNAP(e)cldn2; normalization was performed as described in Materials and Methods; n = 14 line scans.
Notably, the old cldn2 and 4 localized to vesicles appear to represent protein designated for degradation; a fraction of these vesicles colocalize with the lysosomal marker LAMP (Supplemental Figure S2C, top panels). No new cldn2 appears in this vesicular fraction until 24 h after labeling. In contrast, although most vesicular cldn4 also appears to be old cldn, a small amount of new SNAP(e)cldn4 starts to appear in vesicles by 4 h, and this is much more marked at 8 h (Supplemental Figure S2, bottom panels, white arrows); many of these vesicles are double-labeled with JF549 and SNAP-cell 505*. Thus, a newly synthesized cohort of cldn4 both enters and is removed from the junction faster than a comparable cohort of cldn2. These results suggest that the dominant pathway for cldn trafficking is from the Golgi to the lateral membrane then to the tight junction followed by endocytic removal from the junction.
SNAP-tag cldn2 has a longer half-life than does cldn4
We previously demonstrated a longer half-life for cldn2 (≥9 h) than cldn4 (∼6 h) in MDCK cells (Van Itallie et al., 2004), which is consistent with results reported above showing the longer time course of delivery of new SNAP(e)cldn2 to cell contacts than of SNAP(e) cldn4. Although SNAP tags are relatively small (19.4 kDa), its fusion creates a protein twice the normal size of endogenous cldn. To verify that the addition of the SNAP-tag did not differentially alter cldn2 or 4 turnover, we directly measured the half-lives of SNAP(e)cldn2 and 4 by labeling with JF549 SNAP ligand, washing out excess ligand, and measuring the loss of fluorescence in the SNAP(e)cldns after separation of the proteins by SDS–PAGE (Figure 5A). Analysis of the change in the level of fluorescence in SNAP(e)cldn2 and 4 gel bands (or gel locations) revealed decay rates very similar to those we had found previously (representative examples in Figure 5, B, quantified in C); in contrast, the half-life of the free SNAP protein alone stably expressed in MDCK cells was significantly longer (approximately 17 h) than that either of SNAP(e)cldn2 (9.4 h) or SNAP(e)cldn4 (6.4 h).
FIGURE 5:

Cldn2 has a longer half-life than cldn4. (A) To measure half-life of SNAP-tagged proteins, MDCK cells expressing SNAP(e)cldn2 or SNAP(e)cldn4 were incubated with JF549 SNAP-tag ligand (time 0), washed and incubated with SNAP-cell block, and then collected after 3, 5, 7, and 9 h. (B) Duplicate samples of MDCK cells expressing SNAP(e)cldn 2 (top panels), SNAP(e)cldn4 (middle panels), and MDCK cells stably expressing the SNAP-tag protein alone were collected for analysis by SDS–PAGE and in-gel fluorescence. After fluorescence analysis, gels were transferred and probed for ocln as a loading control. (C) Quantification of duplicate or triplicate experiments were used to determine approximate half-lives by linear regression analysis.
Cldns are incorporated into tight-junction strand networks at the basal side
It remains unknown how individual cldns are added into the continuous cldn polymer network at the tight junction. Individual molecules might be added throughout the network, possibly after exocytic delivery directly into the tight junction network membrane. Alternatively, cldns might be incorporated from a pool on the lateral membrane, a pathway strongly supported by the observations described above. We next asked whether we could visually document the entry site of newly synthesized fluorescent cldns using confocal microscopy.
z-Axis analyses (Figure 6) of the localization of old and new cldn2 reveals that at 0 h, old cldn2 (magenta, Figure 6A) is concentrated apically in tight junctions (top panel, top row). Over the next 24 h, this old cldn2 is replaced by new cldn2 (green, top panel, next three rows). Notably, the new cldn2 appears somewhat on the lateral membrane, but also concentrates just basal to the old cldn2 (for example, arrow, 8 h time point), suggesting that new cldn adds to the basal side from the lateral pool. Old cldn4, although also concentrated at apical junctions, is more strongly distributed along the lateral membrane and occasionally evident basally (Figure 6B, top panels). New (green) cldn4 clearly adds to the whole lateral membrane, but by 8 h, like cldn2, it is concentrated just below the apicalmost old cldn4 signal. By 24 h, both new cldn2 and 4 have fully replaced old cldns. Together, these results are consistent with the idea that both newly synthesized SNAP(e)cldn2 and 4 are added to the tight junction from the basal side, but also reiterate that the delivery kinetics of the two cldns differs, with cldn2 being slower than cldn4. Quantification of localization of old and new cldns from z-views at 4 and 8 h (Figure 6C) shows diffuse lateral distribution of new cldns at 4 h, but a distinct peak of localization of new cldns at 8 h that is slightly but consistently concentrated basal to the peak of old cldn localization. Notably, there is no evidence of mixing of old cldn with new cldn on lateral membrane at 8 h, suggesting that there is not a major contribution to lateral cldns either from depolymerization of the tight junction cldns or from vesicular removal from the tight junction strands and recycling to the lateral membrane. Instead, it seems likely that new lateral cldn moves up into the tight junction or is recycled directly from the lateral plasma membrane and degraded.
FIGURE 6:

Time course of z-axis analysis of SNAP(e)cldn2 (top panels) and SNAP(e)cldn4 (bottom panels) reveals that only new cldns replace old cldn by first trafficking first to the lateral membrane. Multiple isolated z-stack images of MDCK II cell lateral membranes show position of old cldn2 (A) and 4 (B) (0 h, magenta, top panels) that are gradually replaced with new (green) at 4, 8, and 24 h. Arrows indicate examples of overlap (white) between old, apical cldn (magenta) and newly synthesized cldn (green). (C) Line scans (N = 14) from the apical to the basal direction along the lateral cell membrane starting at the apicalmost fluorescent signal and extending basally for 4 µm show the apical shift of new cldn localization (top panels, cldn2; bottom panels, cldn4.
We had previously demonstrated that expression of cldns in a fibroblast model system results in the formation of large cell-to-cell strand patches and that newly synthesized cldns concentrate at free ends or breaks in the strands (Van Itallie et al., 2017). Because free strand ends are common at the basal ends of genuine epithelial cell tight junctions (for example, Figure 1), it seems likely that the observed concentration of new cldns at the basal end of the tight junction in MDCK cells means that this is the favored normal entry site for polymerization of new cldns into the tight junction strands and network.
Cldn4 trafficking efficiency to the plasma membrane and protein half-life are regulated by carboxyl-terminal sequences
Some previous studies (Thanabalasuriar et al., 2013; Yin et al., 2017) have found that the YV motif at the carboxyl (C-) terminal end of some cldns is required for proper biosynthetic trafficking to the plasma membrane. We therefore asked whether removal of the C-terminal three residues, the minimal PDZ-binding motif, from the tail of cldn4 might influence its trafficking or half-life. This would suggest a required interaction with a PDZ domain or other protein that interacts with these sequences. To test this, wild-type and truncated (-3) SNAP- and CLIP-tagged cldn4 were stably expressed in MDCK II cells; these lines are referred to as SNAP cldns, without the (e) designation. RTqPCR of the exogenous SNAP cldns showed that they were expressed at levels equivalent to endogenous cldns (Supplemental Figure S3). Removal of the three carboxyl (C-) terminal amino acid residues of cldn4 did not affect its steady-state localization but did slow the rate of appearance at the lateral membrane. This was detected in two different ways. Full-length CLIP-labeled cldn4 (CLIP cldn4) and SNAP-labeled cldn4 lacking these last three residues (SNAP cldn4(-3)) were stably coexpressed in MDCK II cells. For the first set of experiments, these cells were labeled with either JF549 CLIP ligand to detect old full-length cldn4, blocked and then subsequently labeled with CLIP505* to detect new cldn4 (Figure 7A left panels), or, in parallel, with JF549 SNAP 549 followed by SNAP505* to detect old and new cldn4(-3) (Figure 7A, right panels). Although the distribution of both old CLIP cldn4 and old SNAP cldn4(-3) (Figure 7A, top panels, 549 only) is very similar, new CLIP cldn4 (such as SNAP(e)cldn4) is localized to the plasma membrane by 3 h and more obviously at 6 h, while new SNAP cldn4(-3) shows little plasma membrane localization even after 6 h (Figure 7A, bottom panels). This delay in membrane localization for SNAP cldn4(-3) relative to CLIP cldn4 was quantified in a second set of experiments by comparing localization in the same cells after blocking the labeling of old cldns with both SNAP and CLIP cell block, and then using JF549 CLIP ligand and SNAP505* to label new cldns (Figure 7, B, quantified by line scanning in C).
FIGURE 7:

Cldn4 C-terminal amino acids are required for efficient biosynthesis and/or trafficking to the plasma membrane. (A) MDCK II cells stably coexpressing CLIP cldn4 and SNAP cldn4(-3) were labeled first only for CLIP cldn4 localization with either JF549 CLIP-ligand, followed by CLIP-cell block and CLIP-cell 505* (left panels) and second, only for SNAP cldn4(-3) using JF549 SNAP-ligand, SNAP-cell block, and SNAP-cell 505* and fixed at 0, 3, and 6 h (right panels). Fluorescence analysis shows clear lateral membrane staining of new 505*-labeled cldn at 3 h only for full-length cldn4. (B) When SNAP- and CLIP-tagged cldns in coexpressing cells are labeled in the same cell with JF549 CLIP cell ligand and SNAP-cell 505* before and after blocking, old cldns localize identically, independent of the presence of the three C-terminal amino acids, but new CLIP cell cldn appears at cell contacts before SNAP cell cldn4(-3). This finding is quantified by line scanning in C. (D) Comparison of decay in fluorescent JF549-labeled CLIP cldn4 and SNAP-cell 505* SNAP cldn4(-3) in the same cells reveals that SNAP cldn4(-3) is more stable than full-length cldn4; these results are quantified in E. (F) Coexpression of GFP-LNXp80 and SNAP cldn4 reveals colocalization in intracellular vesicles.
Along with slower appearance at the membrane, the half-life of cldn4(-3) was almost twice that of the full-length cldn4 (Figure 7, D and E). This suggests that the C-terminal amino acids may be also important in regulation of normal protein degradation. This is consistent with the finding from the Furuse laboratory that the E3 ubiquitin ligase LNX1p80 binds to the C-terminal domain of cldns and that this is involved in cldn degradation (Takahashi et al., 2009); this is a PDZ domain–mediated interaction. In support of this possibility, we found full-length SNAP cldn4 partially colocalized in intracellular vesicles with transiently expressed GFP-LNX1p80 (Figure 7F).
To test whether this difference in half-life between full length and SNAP cldn4(-3) were also due to differences in binding to (or scaffolding by) ZO-1 or ZO-2, we analyzed SNAP cldn4 biosynthetic trafficking and half-life in ZO-1/ZO-2 double knockdown cells (Z1/Z2 KD; Fanning et al., 2012). Both trafficking and half-life were very similar in the two cell backgrounds (Supplemental Figure S4), suggesting that the minimal amount of remaining ZO-1/2 in the dKDs (<5%; Fanning et al., 2012) is sufficient for normal localization and trafficking.
Together, these results suggest that beyond the cldn-PDZ binding motif interaction with ZO proteins, other proteins interacting with C-terminal residues, potentially components of the COPII sorting machinery (Yin et al., 2017) and the ubiquitination pathway (Takahashi et al., 2009), are likely to be important regulators of trafficking and half-life.
Junction remodeling during cell migration stimulates membrane localization of newly synthesized cldn4 but not cldn2
The above experiments were all performed in stable, confluent monolayers. To test whether junctional appearance of new cldns might be stimulated by membrane remodeling, we used a cell migration model to investigate cldn trafficking during the transition from stationary to migratory phenotype. MDCK II cells expressing SNAP(e)cldn2 and SNAP(e)cldn4 were plated in culture dishes inside oblong silicon inserts. Once confluent, cells were labeled with JF549 SNAP ligand to detect old cldns and were washed, and inserts were removed to stimulate cell migration at the new free edges (Figure 8). After 3.5 h, cells were labeled with SNAP-cell 505* to identify new cldns. MDCK cells expressing SNAP(e)cldn2 showed some detectable synthesis of new cldn throughout the entire cell field, but the level of new cldn was no different in the midfield, where cells were not migrating, from those at the new migrating edge (Figure 8, A, quantified in B). In contrast, there was clear enhancement of labeling of new SNAP(e)cldn4 at the edge of the migrating monolayer (arrows in Figure 8C) compared with cells in the middle of the monolayer (Figure 8, C, quantified in D). These results suggest that cldn4 biosynthesis and/or trafficking is rapidly upregulated in response to junction remodeling. In contrast, cldn2 expression and localization are little altered in acute remodeling. This relative difference correlates with the faster entry and removal of cldn4 from junctions in stable monolayers compared with cldn2.
FIGURE 8:

Migrating cells accumulate newly synthesized cldn4 but not cldn2 at cell contacts. (A) MDCK II cells expressing SNAP(e)cldn2 were plated in chambers with silicon inserts. When cells reached confluence, they were incubated with JF549 SNAP-cell ligand, washed, and blocked. Gaskets were removed (wound) and cells allowed to migrate into the resulting cell-free area. SNAP-cell 505* was added 3.5 h later. Fluorescence imaging of both the middle of the cell field (top image) and at the edge (bottom image) revealed roughly equivalent amounts of SNAP cell 549 signal and lower, but again equivalent amounts of cell contact new 505*-labeled SNAP(e)cldn2; this distribution was quantified by line scanning in B. In contrast, the same protocol applied to SNAP(e)cldn4-expressing cells (C) revealed more localization of new 505*-labeled SNAP(e)cldn4 near the free edge (lower image) than in midfield (upper image); this distribution was quantified by line scanning in D.
Cldn2 returns to cell contacts more slowly than other cldns after calcium switch
Junction remodeling associated with cell migration could alter cldn biosynthesis, turnover, and/or delivery of new cldn to cell contacts. In the well-characterized calcium switch assay (Gonzalez-Mariscal et al., 1985, 1990), cell junctions disassemble in the absence of calcium and then reassemble when calcium is restored; junction reformation is independent of new protein synthesis (Gonzalez-Mariscal et al., 1985). To test whether there was a difference in the timing of membrane appearance of cldn2 and 4 in this assay of reassembly, filter-grown MDCK cells were incubated overnight in the absence of calcium; this treatment resulted in the loss of junctional cldn1, 2, 4, occludin, and ZO-1 (compare top [Control] and middle panels [time 0 after 15 h without calcium], Figure 9A). Six h after restoration of normal calcium levels, cldn1, cldn4, occludin, and ZO-1 have largely returned to their locations at cell–cell contacts; in contrast, cldn2 at this time is mostly intracellular and only weakly associated with cell contacts. By 24 h, all cldns, ocln, and ZO-1 are concentrated at cell–cell contacts. The early time difference (6 h) in localization is quantified by comparing line scans across cell–cell contact sites for cldn2 and cldn4 (Figure 9B). As others have found (Stankewich et al., 1996), the recovery of barrier function in the calcium switch assay, as measured by transepithelial electrical resistance (TER), is accompanied by an overshoot that returns to a steady state level by 24 h (Figure 9C). The low TER that normally characterizes MDCK II cells is largely due to expression of cldn2 in this cell line (Tokuda et al., 2010); it thus seems possible that the delay in the localization of this pore-forming cldn2 compared with electrically tighter cldn1, 3, and 4 may contribute to the overshoot seen in the recovery of TER at early times after calcium replacement.
FIGURE 9:

Cldn2 relocalizes to cell contacts after calcium switch assay more slowly than does cldn1 or cldn4. (A) MDCK II cells cultured on semipermeable membranes were incubated in low-calcium media for 15 h; calcium was added back, and cells were collected and fixed for immunofluorescence at various times after readdition. TER was measured in parallel. (A) Immunofluorescence analysis—(left panel) cldn2, cldn1, and ZO-1 and (right panel) cldn4, ocln, and ZO-1 in untreated monolayers (top images), after 15 h in low calcium (second row), 6 h after calcium readdition (third row), and 24 h after calcium readdition—reveals that by 6 h of calcium return, most the cldn1, cldn4, ocln, and ZO-1 but not cldn2 has returned to cell contacts; the difference in cell-contact localization between cldn2 and 4 is quantified in B. (C) TER measurements show that measurable TER is lost in low calcium but overshoots control levels by more than 10-fold before returning to control values at 24 h.
Cldn4 is stabilized in the absence of cldn2
Of the cldns examined (1–4) in MDCK II cells, cldn2 is the most concentrated at the apical tight junction. Recent structural analysis of the interaction between the first PDZ domain of ZO-1 and the C-terminal domain of cldn2 revealed the presence of a second interacting site between cldn2 and ZO-1 beyond the canonical PDZ binding motif (Nomme et al., 2015). Interaction at this second site, a tyrosine six residues from the C-terminus, stabilizes the cldn2-ZO-1 binding interaction and is absent in cldn1, which was found to have weaker binding to ZO-1 (Nomme et al., 2015); cldn4 also lacks a tyrosine at position 6. In addition, Tokuda and Furuse (2015) demonstrated that knockout of cldn2 in MDCK II cells resulted in an increased concentration at tight junctions in the plasma membrane localizations of cldns 1, 3, 4 and 7, suggesting that the presence of cldn2 might partially exclude tight-junction localization of the other cldns. We thus asked whether removal of cldn2 might also alter trafficking and/or the half-life of cldn4. Indeed, in cldn2 MDCK II knockout cells, SNAP cldn4 was substantially stabilized relative to expression of the same construct in wild-type MDCK II cells (Figure 10). This was evident not only from comparing SNAP cldn4 half-lives in the two cell lines (Figure 10A), but also in the delayed appearance of new SNAP cldn4 in knockout cells compared with control cells (Figure 10, B, quantified in C). In addition, as noted by Tokuda and Furuse (2015), cldn4 was somewhat more focused at the tight junction than for control cells (Figure 10D) and Z-stacks (Figure 10E) quantified by z-axis scans (Figure 10F). Together, these results suggest that cldn2 normally has a dominant effect on strand organization in MDCK II cells and its presence partially excludes other cldns spatially and temporally from the tight junction.
FIGURE 10:

SNAP cldn4 expressed in cldn2 KO MDCK II cells has a longer half-life than SNAP cldn4 in wild-type (WT) cells. (A) MDCK II control and cldn2 KO cells stably expressing SNAP cldn4 were incubated with JF549 SNAP-cell ligand, washed, blocked, and collected for SDS–PAGE. Fluorescence analysis of duplicate samples (top panels) and quantification (B) give half-live values for SNAP cldn4 in WT cells of 6.9 h and >24 h in cldn2 KO cells. Subsequent immunoblot for ocln is included as a loading control and for cldn2 to verify KO cell lines. (C) Confocal fluorescence analysis of WT and KO cells expressing SNAP cldn4 and labeled with JF549 SNAP-cell ligand, blocked, incubated for 8 h and then labeled with SNAP-cell 505* shows mixed old and new cldn4 at the lateral membrane in WT cells, but little new cldn4 in cldn2 KO cells. (D) Localization of SNAP-cell 505*-labeled new cldn is compared in WT and cldn2 KO cells by line scanning analysis (n = 14). (E) z-Axis view of SNAP cldn4 expression in WT (top panels) and cldn2 KO cells (bottom panels) reveals brighter junctional concentration of SNAP cldn4 in KO cells compared with WT. (F) z-Axis scans of SNAP cldn4 expressed in WT and cldn2 KO cells (n = 19).
Newly synthesized ocln appears first throughout tight junctions
To test whether newly synthesized ocln followed the same trafficking pattern we observed for cldns, we stably expressed SNAP-tagged ocln in MDCK II cells and employed a similar pulse–block–pulse labeling protocol. We found that after 3 h, newly synthesized SNAP ocln (Figure 11A, green) was partially intracellular, likely in Golgi, but unlike cldns, new SNAP-tagged ocln also concentrated sharply with old SNAP ocln (magenta). Z-stack images of new and old SNAP ocln (Figure 11B) show that, in contrast to what we observed for SNAP/CLIP cldns, there was colocalization of a small amount of new SNAP ocln in the middle of the magenta signal for old tight junction SNAP ocln (middle panels). This was quantified by scanning along the Z-axis (Figure 11C); these data suggest that unlike cldns, newly synthesized ocln colocalizes with older ocln. To more directly compare the trafficking of newly synthesized SNAP ocln with that for cldn4, we stably coexpressed CLIP cldn4 and SNAP ocln and compared the localization of both newly synthesized proteins at early time points. At steady state (Figure 11, D, top panels, quantified in E, left panel), both SNAP ocln (green) and CLIP cldn4 (magenta) are concentrated at apical cell contacts, with variable lateral membrane distribution. After 3 h of synthesis, new SNAP ocln (green) is sharply focused at cell junctions, while as previously shown, new CLIP cldn4 is distributed all along the lateral cell membrane but not concentrated at apical cell contacts (Figure 11, D, middle panels, quantified in E, middle panel). By 6 h, new SNAP ocln continues to concentrate at junctions, but is now also visible on the lateral membrane, while CLIP cldn4 is finally beginning to concentrate at tight junctions (Figure 11, D, bottom panels, quantified in E, right panel). These results suggest that in contrast to the path for cldn trafficking, occludin is predominantly delivered directly to the tight junction and subsequently can move to the lateral membrane, either by diffusion in the membrane or by vesicular recycling. This is consistent with much published evidence for occludin removal and reinsertion on the membrane (Morimoto et al., 2005; Fletcher et al., 2014).
FIGURE 11:

Pulse-chase SNAP tag labeling of SNAP ocln and 4 reveals that newly synthesized ocln appears at apical cell contacts. (A) Maximum-intensity projections of MDCK cells expressing SNAP ocln labeled for 0, 3, and 7 h reveals that new (green) ocln is partially intracellular but also sharply focused with old (magenta) tight junction ocln even at 3 h. (B) z-Stack images show colocalization of a small amount of new ocln with old ocln (white dots) even at 3 h (middle panels) and more dramatically at 7 h (bottom panels) with minor lateral ocln. (C) z-Axis line scans revealed colocalization of new and old ocln along the lateral membrane (N = 14). (D) Stable coexpression of CLIP cldn4 (magenta) and SNAP ocln (green) shows the early appearance of sharply focused newly synthesized ocln but not cldn (middle panels). (E) Line scans of images as in D indicate the more discrete localization of ocln than of cldn4 at 3 and 6 h (n = 12–14 scans).
DISCUSSION
Although there are numerous studies examining vesicular recycling at tight junctions of cldn (Dukes et al., 2012; Capaldo et al., 2014; Ikari et al., 2014; Lu et al., 2014; Marunaka et al., 2017) and ocln (Hopkins et al., 2003; Ivanov et al., 2004; Morimoto et al., 2005; Shen and Turner, 2005; Shen et al., 2008; Fletcher et al., 2014), there is little information about their biosynthetic trafficking pathways. In this study, we inserted SNAP and CLIP tags into the endogenous cldn2 and cldn4 genes in MDCKII cells to permit visualization of cldn trafficking pathways and showed for the first time that newly synthesized cldns are predominantly delivered first to the lateral membrane and then to the tight junction, where they enter at the basal end of the strand network. In contrast, newly synthesized SNAP ocln appears to be added directly to tight junctions. We also observe significant differences in the kinetics of membrane delivery between cldn2 and cldn4, with cldn2 having a slower delivery and longer half-life. In addition, we find that KO of cldn2 changes both the membrane targeting kinetics and half-life of cldn4, suggesting interdependence among cldn species affecting expression, localization, and turnover.
SNAP and CLIP tagging of proteins has been used previously to study trafficking of membrane proteins (Stoops et al., 2014). This system has several useful advantages for studying protein trafficking. First, the tag/label size is small (∼20 kDa) compared with antibody/fluorescent secondary antibody size (150 kDa primary antibody plus 50 kDa for fluorescent Fab fragments), allowing excellent resolution in fluorescence microscopy. Second, the availability of two versions (SNAP and CLIP) allows coexpression and thus comparison of the trafficking of two different proteins in the same cell. Third, a variety of stable and bright fluorescent and nonfluorescent ligands allow for sequential labeling. Finally, a recent report described the advantages of use of an endogenous SNAP–tagged fusion protein for superresolution microscopy (Butkevich et al., 2018). We also made endogenous SNAP–tagged cldn2 and 4 fusion proteins; however, we found no clear difference in trafficking of endogenously tagged and moderately overexpressed SNAP-tagged cldns.
Cldns oligomerize both in trans and in cis to form strands observed in FFEM. Trans cldns oligomerization is stabilized by adhesive interactions across adjacent cells (Kubota et al., 1999). These trans interactions depend on both of the cldn’s two extracellular domains (Piontek et al., 2008; Koval, 2013) and appear to be strong, since trans endocytosis, that is, cldn from one cell coendocytosing with cldn from an adjacent cell, is common (Matsuda et al., 2004). Once stabilized by trans-interactions, cldns readily oligomerize in cis; together these modes of interaction result in very stable branching-strand networks (Shen et al., 2008; Van Itallie et al., 2017). We have recently shown using cldns expressed in fibroblasts that new cldns are added to free ends of cldn strands and at end-to-side junction points between two strands, where in this model system, strands are most likely to break and reanneal (Van Itallie et al., 2017). The finding that newly synthesized cldns concentrate at the basal pole of the tight junction before they replace older cldns is consistent with the possibility that the less well-organized basal tight-junction strands are the preferred site of entry for new cldns; a simplified model for this assembly is shown in Figure 12A. This mode of polymer growth at free ends is similar to the addition of actin and tubulin to the ends of microfilaments and microtubules and raises the question of whether cldn strands, like the cytoskeletal filaments, are controlled by proteins that catalyze monomer addition and removal and cap and sever the polymers. If such mechanisms occurred in the tight junction strands, they would have important implications for normal and pathological control of the paracellular barrier. This behavior is also reminiscent of how new gap junction proteins are added to the edges, rather than the inaccessible middle of gap junction plaques, but are removed from the middle (Gaietta et al., 2002). In an analogous manner, new septate-junction membrane proteins in Drosophila epithelial cells are added as new strands on one side of the junction, rather than being incorporated into preexisting strands (Babatz et al., 2018) Our findings might imply that more apical cldn strands are “older” than more basal strands; however, we suspect that although this may be the overall trend, there may also be differences in cldn isoform composition in apical and more basal strands. One dramatic example of this inhomogeneity was demonstrated in the junctions between sensory and nonsensory cells in the inner ear (Nunes et al., 2006), where cldn14 strands were apical to cldn6 strands. Although there is no evidence for this degree of subdomain organization in most epithelial cell junctions, it seems possible that variations in the cldn isoforms in their expression levels, relative affinities for the tight junction scaffolding, preferred cldn–cldn interactions, and sequences or modifications that differentially affect turnover rates are likely to contribute to compositional differences between apical and basal tight-junction strands.
FIGURE 12:

Model for addition of newly synthesized cldns and ocln to epithelial tight junctions. (A) Newly synthesized cldns (green) are added from the lateral membrane to basal strand breaks; old cldns (magenta) are turned over by endocytosis. (B) Although there is lateral ocln, newly synthesized ocln (green) appears first at apical junctional contacts; turnover of both new and old (magenta) ocln occurs by endocytosis.
The difference in trafficking between newly synthesized ocln and cldns was striking and somewhat unexpected. Although ocln was early recognized as associated with the cldn-based strands (Furuse et al., 1998), a fraction of ocln is normally present on the lateral membrane, and further, ocln trafficking has been reported to be targeted to the basolateral plasma membrane (Yaffe et al., 2012). However, in contrast to cldn, we found that newly synthesized ocln appeared to concentrate first at tight junctions and only later appeared on the lateral membrane. Given the finding that, unlike cldns, ocln is continually recycled at tight junctions (Shen et al., 2008) and the lack of evidence for its direct incorporation into cldn polymers (Suzuki et al., 2015), it is unsurprising that its mode of interaction with and thus assembly into tight junctions is fundamentally different from that of cldns. A model for ocln biosynthetic trafficking is shown in Figure 12B.
Our finding that SNAP cldn4(-3) is trafficked more slowly to the plasma membrane than full-length SNAP cldn4 might be explained by recent findings that cldn trafficking to the plasma membrane is dependent on its C-terminal interaction with coat protein complex II (COPII) (Thanabalasuriar et al., 2013; Yin et al., 2017; Huang et al., 2019). This is not a PDZ-domain interaction, but instead is based on the C-terminal YV sequence of cldns being recognized as a di-leucine-like (di-hydrophobic) sorting signal; mutation of the YV to AA disrupts interaction of this sequence with the COPII component Sec24C. COPII facilitates cldn transit from the endoplasmic reticulum to Golgi and thus its eventual appearance at cell contacts; depletion of Sec24C by siRNA resulted in decreased cldn surface localization (Yin et al., 2017). The C-terminal amino acids are also important in regulation of cldn4 stability and removal from the membrane, with one possible mechanism being interaction with the PDZ domain–containing ubiquitin ligase LNX1p80 (Takahashi et al., 2009). A similar interaction with a different PDZ domain–containing E3 ubiquitin ligase, PDZRN3 regulates cldn16 endocytosis and stability (Marunaka et al., 2017). Phosphorylation at S195 has also been implicated in regulation of cldn4 half-life in SMG-C6 cells (Cong et al., 2015); however, in the MDCK cell line used here, both the nonphosphorylatable mutant of S195, S195A, and the phosphomimetic mutant, S195E, had half-lives identical to that of the SNAP cldn4 wild type (unpublished data). Cldn4 half-life does appear to be independent of scaffolding to ZO-1, since cldn4 levels and distribution are normal in ZO-1 knockout cells (Tokuda et al., 2014) and expression of SNAP cldn4 in the ZO-1/2 dkD cells does not alter half-life. However, cldn4 can interact with ZO-2 and -3 (Itoh et al., 1999) and MUPP1 (multiple PDZ protein 1; Hamazaki et al., 2002; Lanaspa et al., 2008), so it is possible that some of these interactions may affect protein stability.
Coordination between cldn biosynthesis, trafficking, and junction assembly is not well understood. Early studies demonstrated that synthesis of tight-junction proteins could be regulated separately from tight-junction assembly. For example, MDCK cells grown in suspension culture and then plated in the presence of protein synthesis inhibitors formed functional tight junctions (Griepp et al., 1983). We found that cell migration stimulated the localization of new SNAP cldn4 to the cell contacts at cells near the migration front but not in the middle of the cell islands; this is not surprising, since increased protein synthesis associated with epithelial sheet migration was identified nearly 50 years ago (Trinkaus, 1973). However, the localization of new SNAP cldn2 was not different between cells near the leading edge and cells in the middle of the cell island. There is thus a fundamental difference between the regulation of cldn2 and cldn4 synthesis and localization. Consistent with our finding, Amoozadeh et al. (2018) recently showed that cldn2 expression is positively associated with increasing cell confluence, while the expression of other cldns is independent of cell confluence. Differences between cldn2 and cldn4 localization are also evident after calcium switch, since cldn4 (and cldn1 and cldn3) relocalized at cell contacts much more rapidly than does cldn2. This difference could be due to a difference in cldn2 trafficking, or alternatively, cldn2, unlike other tight junction proteins, might be proteolyzed during the incubation in low calcium.
As previously noted by Furuse and colleagues (Tokuda et al., 2014), we found that cldn2 knockout resulted in increased tight junction localization of cldn4; however, our finding of delayed cldn4 junctional delivery and substantially increased half-life in the cldn2 KO was unexpected. The most obvious explanation is that cldn2 is normally restricts tight junction access for cldn4, as previously suggested by Capaldo et al. (2014) and similar to what has been described for other cldn pairs (Angelow et al., 2007; Schlingmann et al., 2016). Cldn2 is normally present at much higher levels than is cldn4 in MDCK II cells (and sixfold higher levels than ZO-1; Supplemental Figure S3) and it binds the tight junction scaffold protein ZO-1 with higher affinity (Nomme et al., 2015). Thus, cldn2 knockout might free up the ZO scaffold and allow cldn4 to replace it in the tight junction strands. A preferred interaction between cldn2 and ZO-1 may explain why depletion of ZO-1 and ZO-2 has a more profound effect on cldn2 expression and localization than on cldn4 (Fanning et al., 2012) and further why cldn4 biosynthetic targeting and half-life in ZO-1/2 dKd cells is near normal. The greatly enhanced cldn4 stability may reflect a higher fraction of tight junction integration and/or that turnover normally is dependent on the presence of cldn2.
In summary, we find that cldn2 and cldn4 share a trafficking pattern but differ in incorporation kinetics, and we further find that there are major trafficking differences between these cldns and ocln. We speculate that to be efficiently incorporated into tight-junction strands, new cldns must be polymerized at strand breaks or free edges, most common in basal tight-junction strands; this would explain how continuous cldn strands could be renewed without loss of continuity of the cell-to-cell strand barriers. In contrast, ocln can associate with preformed strands and shows much faster turnover at the junction. However, association of ocln with tight-junction strands can modulate cldn behavior (Raleigh et al., 2011; Cording et al., 2013); further investigation is needed to better understand the importance of their interactions. Finally, the possibility of mechanisms that control the frequency and location of stand breaks and rate of cldn addition deserves investigation, since they would have important implication for the maintaining the barrier during normal function and pathologic states.
MATERIALS AND METHODS
Antibodies and reagents
Rat anti–ZO-1 (40.76, used at 1:500 for immunoblots, IB, and 1:100 for immunofluorescence, IF) has been described previously (Anderson et al., 1988). Occludin (13409-1-AP) rabbit polyclonal antibody was from ProteinTech (1:1000 IB, 1:800 IF). Rabbit cldn1 (71-7800, 1:1000 IB, 1:100 IF), mouse cldn2 (12H12, 1:500 IB, 1:100 IF), rabbit cldn3 (34-1700, 1:1000 IB, 1:100 IF), mouse cldn4 (3E2C1, 1:500 IB, 1:100 IF) and mouse LAMP2 (MA5-16561) were from Thermo Fisher. Rabbit E-cadherin (24E10, 1:1000 IB, 1:100 IF) was from Cell Signaling Technology. Mouse β1 integrin (MAB2253, 1:100, IF) was from EMD Millipore. Mouse GM130 (G65120, 1:100 IF) was from BD Transduction Laboratories. Antibodies were validated by recognizing bands of the predicted size on immunoblots, by immunolocalization, and by KO (cldn2, ZO-1, ocln). Species-specific secondary antibodies for immunofluorescence, including Alexa Fluor 488 (115-545-166) (1:100) Alexa Fluor 594–conjugated Affinipure F(ab’) fragment immunoglobulin G (IgG) (1:200) anti-rabbit (711-586-152) and anti-mouse (715-586-151), Alexa Fluor 647–conjugated Affinipure F(ab’) Fragment IgG (anti-rat712-006-153) (1:200) and immunoblots (IR-labeled 680 (anti-mouse 115-625-166) (1:2000) were from Jackson ImmunoResearch. IR-labeled 800CW was from Li-Cor (926-32211)(1:2000). Alexa Fluor 568 streptavidin (S11226 568)(1:800, IF) and rhodamine–phalloidin (R415) (1:50) and Alexa Fluor 488 phalloidin (A12379)(1:40) were from ThermoFisher.
SNAP-cell 505* (S9103S), CLIP-cell 505*(S9217S), SNAP-cell TMR*(S9105S), SNAP-cell Oregon Green (S9104S), SNAP-cell block (S9106S), and CLIP-cell block (S9220S) were from New England Biolabs. Janelia Farm cpJF549-SNAP and CLIP ligands were a kind gift from Luke Lavis, HHMI, Janelia Research Campus (Grimm et al., 2015). GFP-LNXp80 was kindly provided by Mikio Furuse (National Institute for Physiological Sciences, Okazaki, Japan).
Brefeldin A (B5936) was purchased from SigmaAldrich.
Generation of stable MDCK II cell lines expressing SNAP-Cldn 2 or SNAP-Cldn 4 under endogenous promotors using CRISPR/Cas9 and homology-arm directed repair for genome editing
To obtain N-terminally SNAP-tagged cldns under the control of the endogenous promoters in MDCK II cells, we designed guide RNAs (gRNAs) for cldn2 and cldn4 (Supplemental Table 1) and homology arms (HA) flanking the SNAP-tag for both cldn2 and cldn4. gRNAs were designed using the Broad Institute sgRNA design tool and were cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 (62988; Addgene) as previously described (Ran et al., 2013b). Homology arms were PCR-amplified in two steps (Supplemental Table 1) on MDCK II cell genomic template DNA (DNeasy DNA extraction kit, Qiagen, 69504), PCR products were purified by gel electrophoresis and cloned into pSNAPf vector (New England Biolabs, N9183S) and transformed into stellar competent cells (Escherichia coli HST08, Takara Bio, 636766). In-frame insertion of HA and SNAP-tags was confirmed by DNA sequencing (ACGT).
The two vectors containing the gRNA and SNAP-tags flanked by homology arms (Cldn2: gRNACldn2 and SNAP(e)Cldn2; Cldn4: gRNACldn4 and SNAP(e)Cldn4) were cotransfected using Lipofectamine 2000 (Invitrogen, 11668027) into Tet-off-MDCK II cells (BD Biosciences). Seven days posttransfection, cells were treated with SNAP-Cell 505* according to the manufacturer’s protocol, washed, trypsinized, washed again, and resuspended in FACS buffer (1% heat-inactivated FBS (Atlanta Biologicals, S10350) and 0.1% bovine serum albumen (Sigma, A7906) in Dulbecco’s phosphate-buffered saline (PBS) without Ca2+ and Mg2+, (Corning, 21-031-CV) at room temperature. Cells were filtered through a cell strainer cap (Falcon, 352340) before being sorted using a BDFACSAria cell sorter with BD FACSDiva 8.0.1 software (BD, San Jose, CA); untransfected MDCK II cells and cells stably expressing SNAP cldn4 were used as negative and positive controls. The signal of SNAP-Cell 505* was detected after excitation by a 488-nm laser line and emission was collected on a 515 ± 20–nm bandpass filter. Cells transfected with SNAP(e)Cldn2 and SNAP(e)Cldn4 that were SNAP-Cell 505*-bright were sorted as single cells into individual wells of a 96-well plate containing 100 µl/well complete cell culture medium.
After 2 wk in culture, individual clones were transferred into 24-well plates and tested by immunoblot for expression of the fusion protein and absence of the endogenous untagged cldn. DNA was extracted from confluent monolayers of both IB-positive and untransfected cells (DNeasy DNA extraction kit, Qiagen) and amplified using specific primers for Cldn2 and Cldn4 (primers: Supplemental Table 1) and PCR products verified by DNA sequencing (ACGT). The endogenously tagged cldn4 fusion protein contains five extra residues between the SNAP tag and the start of cldn4 (due to the position of the closest PAM site, resulting in a slightly larger fusion protein that the SNAP cldn4 used for transfection in some later experiments.
Cell culture and transfection
Tet-off MDCK II cells were cultured in DMEM (4.5 g/l glucose; Corning, 10-013-CV), 10% FBS, and penicillin/streptomycin (Corning, 30-002-CI). Unless otherwise described, cells were cultured for 6–10 d before experiments. In some experiments, stable cell lines overexpressing SNAP and CLIP-tagged cldns and mutants were generated by PCR, using pTRE human cldn4 (Van Itallie et al., 2001) as a template and cloning into the BamHI/XhoI sites in pSNAPf and pCLIPf (New England Biolabs, N9215S). Sequences of all primers are shown in Supplemental Table 1. SNAP ocln construct was made by PCR from EGFP ocln (Van Itallie et al., 2015). Cldn2 KOs were made using the pSpCas9(BB)-2A-Puro vector (62988; Addgene; Ran et al., 2013; gRNA sequences in Supplemental Table 1). All constructs were verified by sequencing. SNAP- and CLIP-tagged cldns and ocln, SNAP tag alone, and CRISPR/Cas9 constructs were transfected into MDCK II Tet-off cells using Lipofectamine 2000. In addition, SNAP cldn4 was transfected into cldn2 knockout cells and into ZO-1/ZO-2 double-knockdown cells (Fanning et al., 2012). To make stable lines, SNAP- and CLIP-tagged cldns and ocln and SNAP tag alone were cotransfected with pZeoSV2 (ThermoFisher, V85001) cell lines and selected with 1 mg/ml zeocin (InvivoGen, antzn-05) or with pBlast49 (InvivoGen, pbla49-mcs), selected with 10 µg/ml blastocidin (InvivoGen, antbl-05); individual clones were selected, and expression was verified by immunoblot analysis, except in the case of cell lines expressing SNAP alone. Expression in these cells was detected after clones in JF549 SNAP-ligand were and positive clones were detected by fluorescent imaging using the GE Typhoon Trio+ (GE Healthcare). Three expressing lines for each construct were saved and used for further analyses.
GFP-LNXp80 (Takahashi et al., 2009) was transiently transfected into MDCK cells stably expressing SNAP cldn4 using Lipofectamine 2000 and analyzed 48 h posttransfection for GFP localization after incubation of cells with JF549 SNAP tag ligand.
The calcium switch assay was performed as described previously (Van Itallie et al., 2015).
Latrunculin A (Lat A; ThermoFisher, L12370) was solubilized in dimethyl sulfoxide (Sigma, D2438); MDCK II cells expressing SNAP(e)cldn4 were plated at confluent density on 12-well filters (Corning 3460) and cultured for 3 d. Cells were incubated with SNAP-Cell 505* for 30 min, washed three times, and 20 min later, rinsed with Hank’s balanced salt solution plus calcium and magnesium (ThermoFisher 14025-092) and then incubated with 0.5 µM Lat A for 10 and 40 min. Cells were then rinsed, fixed with cold ethanol, and incubated with rhodamine phalloidin (ThermoFisher, R415) before being mounted with Mowiol (Calbiochem, 475904) with 1% n-propyl gallate (Sigma, 02370).
Biotinylation of basolateral membrane
MDCK cells expressing SNAP(e)cldn4 were cultured on Transwell filters for 6 d and treated with SNAP-Cell 505*, washed, and incubated with unlabeled media for 30 min. Cells were then washed three times with ice-cold Dulbecco’s PBS containing calcium and magnesium (D-PBS+; Corning, cat. no. 21-030-CM) with the pH adjusted to 8.0 and incubated with 10 mM EZ-link Sulfo-NHS-SS-Biotin (ThermoFisher, cat no. 21328) in D-PBS+ (pH 8.0) for 20 min at 4°C on a rocker; this incubation was repeated with fresh biotin for an additional 20 min. Filters washed with ice cold 25 mM Tris, pH 8.0, 150 mM NaCl to quench, washed with cold D-PBS+ at pH 8.0, fixed with ice-cold ethanol, and stained for ZO-1. The biotin label was visualized using fluorescent streptavidin Alexa fluor 568 conjugate (ThermoFisher, cat. no. S11226).
Time course with SNAP- and CLIP-tag ligands
MDCK II cells stably expressing either SNAP(e)cldn2, SNAP(e)cldn4, or SNAP- or CLIP-tagged cldns and mutants and SNAP ocln were cultured on glass coverslips or Transwell filters, incubated with SNAP-cell or CLIP-cell ligands (1 µM JF549 SNAP or CLIP ligands or SNAP-cell TMR* for 30 min in medium; time = 0), rinsed three times, and then SNAP-cell block (10 μM) was added for 20 min. Cells were again washed three times and incubated for various periods of time (2–24 h) before addition of 2.5 µM SNAP-cell 505* or CLIP-cell 505*and further incubation for 30 min. The times indicated in the figures are the times following block plus 30 min incubation with SNAP-cell 505* (but not including the final 30 min postsecond labeling). Cells were again washed and incubated for an additional 30 min before fixation with cold ethanol, rinsed, and mounted with Mowiol. In some experiments, SNAP-cell fluors were reversed. In one set of experiments, SNAP(e)cldn2 or 4 was incubated with JF549 and blocked and Brefeldin A (5 µg/ml) or vehicle was added. At 4 h postblock, cells were labeled with SNAP-cell 505* (plus Brefeldin A or vehicle), washed in the presence of Brefeldin A, and fixed for fluorescence analysis.
For analysis of protein half-life, MDCK II cells stably expressing either SNAP(e)cldn2, SNAP(e)cldn4, or SNAP- or CLIP-tagged cldns and mutants were cultured in duplicate 24-wellsor Transwell filters, incubated with SNAP-cell ligands (1 µM JF549 SNAP or CLIP ligands or SNAP-cell TMR*) for 30 min in medium (time = 0), and rinsed three times, and then SNAP-cell and/or CLIP-cell block (10 μM) was added for 20 min. Cells were again washed three times and incubated for various periods of time (2–24 h) before being rinsed and solubilized in SDS-loading buffer. Following SDS–PAGE, fluorescently labeled proteins were imaged using a GE Typhoon Trio+ variable mode imager (GE Healthcare) and signals in green and red channels quantified using 1D analysis in ImageQuant (GE Healthcare); then gels were transferred for analysis by immunoblot with ocln to confirm approximate protein loading. The fluorescence intensity measure in ImageQuant was normalized to time 0, and slopes and half-lives were calculated using GraphPad Prism.
SDS–PAGE and immunoblots
For immunoblots, cells were plated in 24-well dishes or 12-mm Transwell filters (Corning) and grown for 6–10 d. Cells were washed twice in ice-cold Dulbecco’s PBS (Corning) and lysed in 0.10–0.25 ml of 4X SDS sample buffer (250 mM Tris [Sigma, 93362], pH 6.8, 40% glycerol [Sigma, G8773], 0.52 M β-mercaptoethanol [Calbiochem, 4203]) and 8% SDS [Sigma, L3771]). Lysates were sonicated briefly to shear genomic DNA, heated to 95°C for 3 min, and resolved by SDS–PAGE. Gels were transferred to nitrocellulose membranes (Bio-Rad, 1620115) and then blocked in a solution of PBS and 10% nonfat dry milk powder (NFDM; Carnation) for 1 h. Filters were incubated for 1 h in primary antibodies diluted in a solution of PBS (Corning, 10X, 46-013-CM), 5% NFDM, and 0.1% Tween-20 (Affimetrix, T1003)(PBS-T); washed four times for 5 min each in PBS-T; and incubated for 30 min with 1:2000 dilution of the appropriate species-specific secondary antibodies coupled to IRDyes (Rockland, Gilbertsville, MD). After four more washes in PBS-T, filters were imaged with the Odyssey infrared imaging system (Licor Biosciences, Lincoln, NE).
Fluorescence microscopy
MDCK II cells were cultured on uncoated glass coverslips or on Transwell filters and fixed in ice-cold ethanol at 4°C for 30 min. Cells incubated with SNAP- and CLIP-tag ligands and not further immunostained were rinsed twice after fixation and mounted with Mowiol containing 1% n-propyl gallate. Cells to be immunostained were incubated in 2% normal goat serum in phosphate-buffered saline (PBS) for 60 min after fixation, and after blocking, incubated in primary antibodies for 60 min. After washing, samples were incubated with fluorescence-labeled secondary antibodies; in some cases, rhodamine or phalloidin 488 was added with the secondary antibodies. After washing, samples were mounted as above with Mowiol containing 1% n-propyl gallate.
Fluorescence imaging was done with a Zeiss (Thornwood, NY) 710 confocal microscope, using a 63× 1.4 NA oil objective or 20× air, with 488-, 561-, and 633-nm laser lines, or a Zeiss LSM 880 Airyscan in superresolution mode with a 63× 1.4 NA objective. Raw data were processed using Airyscan processing with “auto strength” (mean strength ± SD = 5.5 ± 1.3) in Zen Black software, version 2.3. For live-cell imaging, normal medium was supplemented with 20 mM HEPES, pH 7.4, and imaging was performed using a heated stage with cell culture media supplemented with 20 mM HEPES (Corning, 25-060-CI). In some experiments, superresolution images were taken using a GE OMX Blaze V4 ultrafast structured illumination microscope equipped with four scientific complementary metal-oxide semiconductor cameras using a 60× 1.42 NA lens using 488-, 561-, and 647-nm laser lines. Images were acquired using DeltaVision OMX software. Unless otherwise mentioned, all images are maximum-intensity projected through full cell heights.
Line scan analysis across cell junctions was performed as follows. A defined-length macro in Fiji (ImageJ, National Institutes of Health) was used to center a 5-µm line at the cell contacts; in matched measurements, this line position was saved using the region of interest (ROI) manager. Plot profiles were generated for each line and values transferred to Excel. In most cases, values were normalized to percent total fluorescence for each line profile and plotted using GraphPad Prism (GraphPad Software); graphs were generated using 12–20 plot profiles from multiple images.
Transmission electron microscopy
Cells were grown in 35-mm dishes postconfluence and then directly fixed in 2.5% glutaraldehyde and 1% paraformaldehyde in 0.12 M sodium cacodylate buffer, pH 7.4, for 20 min at room temperature and 40 min at 4°C. Cells were postfixed with 1% osmium tetroxide, stained en bloc with uranyl acetate, ethanol-dehydrated, and LX112 embedded. Chemicals were from Electron Microscopy Sciences and Ladd Research Industries. Thin cross-sections (70 nm) were cut, stained with uranyl acetate and lead citrate, and viewed with a JEM1400 electron microscope (JEOL USA) equipped with an AMT XR-111 digital camera (Advanced Microscopy Techniques).
Freeze-fracture electron microscopy
Freeze-fracture electron microscopy was carried out using established protocols (Medina et al., 2000).
Assays of paracellular barrier function
TER (World Precision Instruments) was carried out as previously described (Van Itallie et al., 2001). All measurements were performed in triplicate, and replicate experiments were performed at least twice.
Quantitative real-time PCR
MDCK II Tet-off cells expressing SNAP cldn2 and SNAP cldn4 were cultured for 6 d, and RNA was extracted using the RNeasy minikit (Qiagen, Germantown, MD). 1000 ng of RNA was used for cDNA synthesis with the Superscript VILO Master Mix (Life Technologies). cDNAs were diluted 1:10 and amplified using the Power Sybr Green Master Mix (Life Technologies). Quantitative RT-PCR (qRT-PCR) was performed using a LightCycler 96 thermocycler (Roche, Basel, Switzerland); qRT-PCR measurements were performed in triplicate, and values were normalized to ZO-1 mRNA. qPCR primers are listed in Supplemental Table 1.
Graphs, replicates, and image assembly
Graphing and statistics were performed using GraphPad Prism; each experiment was repeated at least once and except for SNAP(e)cldns, at least two independent clonal cell lines were analyzed. Graphs are presented using independent points, or in the case of plot profiles, as means ± SEM. Image preparation was done using ImageJ, and assembly was done in Photoshop.
Supplementary Material
Acknowledgments
We acknowledge Robert Hogg and Aparna Kishor (National Heart, Lung, and Blood Institute [NHLBI], National Institutes of Health [NIH], Bethesda, MD) for use of the LightCycler 96 thermocycler, Xufeng Wu for help with and use of the microscopes in the NHLBI Light Microscopy Imaging Core, Christoph Bleck and Erin Stempinski of the NHLBI Electron Microscopy Core, Christoph Rahner (University of Basel) for FFEM images, and Pradeep Dagur and J. Philip McCoy of the NHLBI Flow Cytometry core for help in cell sorting. In addition, we appreciate helpful insights from Michael J. Caplan (Yale University Medical School) about the use of Brefeldin A in MDCK cells. This research was supported by intramural funding by the Division of Intramural Research, NHLBI, NIH (ZIA HL006207).
Abbreviations used:
- Cldn
claudin
- COPII
coat protein complex II
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- FFEM
freeze-fracture electron microscopy
- KO
knockout
- MDCK
Madin–Darby canine kidney
- ocln
occluding
- PDZ
PSD95, Discs large, ZO-1 domain
- ROI
region of interest
- RTqPCR
real-time quantitative PCR
- ZO
zonula occludens.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-01-0008) on April 3, 2019.
REFERENCES
- Amoozadeh Y, Anwer S, Dan Q, Venugopal S, Shi Y, Branchard E, Liedtke E, Ailenberg M, Rotstein OD, Kapus A, Szaszi K. (2018). Cell confluence regulates claudin-2 expression: possible role for ZO-1 and Rac. Am J Physiol Cell Physiol , C366–C378. [DOI] [PubMed] [Google Scholar]
- Anderson JM, Stevenson BR, Jesaitis LA, Goodenough DA, Mooseker MS. (1988). Characterization of ZO-1, a protein component of the tight junction from mouse liver and Madin–Darby canine kidney cells. J Cell Biol , 1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelow S, Schneeberger EE, Yu AS. (2007). Claudin-8 expression in renal epithelial cells augments the paracellular barrier by replacing endogenous claudin-2. J Membr Biol , 147–159. [DOI] [PubMed] [Google Scholar]
- Babatz F, Naffin E, Klambt C. (2018). The Drosophila blood–brain barrier adapts to cell growth by unfolding of pre-existing septate junctions. Dev Cell , 697–710.e693. [DOI] [PubMed] [Google Scholar]
- Butkevich AN, Ta H, Ratz M, Stoldt S, Jakobs S, Belov VN, Hell SW. (2018). Two-color 810 nm STED nanoscopy of living cells with endogenous SNAP-tagged fusion proteins. ACS Chem Biol , 475–480. [DOI] [PubMed] [Google Scholar]
- Capaldo CT, Farkas AE, Hilgarth RS, Krug SM, Wolf MF, Benedik JK, Fromm M, Koval M, Parkos C, Nusrat A. (2014). Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol Biol Cell , 2710–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X, Zhang Y, Li J, Mei M, Ding C, Xiang RL, Zhang LW, Wang Y, Wu LL, Yu GY. (2015). Claudin-4 is required for modulation of paracellular permeability by muscarinic acetylcholine receptor in epithelial cells. J Cell Sci , 2271–2286. [DOI] [PubMed] [Google Scholar]
- Cording J, Berg J, Kading N, Bellmann C, Tscheik C, Westphal JK, Milatz S, Gunzel D, Wolburg H, Piontek J, et al. (2013). In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J Cell Sci , 554–564. [DOI] [PubMed] [Google Scholar]
- Dukes JD, Whitley P, Chalmers AD. (2012). The PIKfyve inhibitor YM201636 blocks the continuous recycling of the tight junction proteins claudin-1 and claudin-2 in MDCK cells. PLoS One , e28659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning AS, Van Itallie CM, Anderson JM. (2012). Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia. Mol Biol Cell , 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher SJ, Iqbal M, Jabbari S, Stekel D, Rappoport JZ. (2014). Analysis of occludin trafficking, demonstrating continuous endocytosis, degradation, recycling and biosynthetic secretory trafficking. PLoS One , e111176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Sasaki H, Fujimoto K, Tsukita S. (1998). A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol , 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science , 503–507. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Chavez de Ramirez B, Cereijido M. (1985). Tight junction formation in cultured epithelial cells (MDCK). J Membr Biol , 113–125. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Mariscal L, Contreras RG, Bolivar JJ, Ponce A, Chavez De Ramirez B, Cereijido M. (1990). Role of calcium in tight junction formation between epithelial cells. Am J Physiol , C978–C986. [DOI] [PubMed] [Google Scholar]
- Griepp EB, Dolan WJ, Robbins ES, Sabatini DD. (1983). Participation of plasma membrane proteins in the formation of tight junctions by cultured epithelial cells. J Cell Biol , 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods , 244–250, 243 p following 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki Y, Itoh M, Sasaki H, Furuse M, Tsukita S. (2002). Multi-PDZ domain protein 1 (MUPP1) is concentrated at tight junctions through its possible interaction with claudin-1 and junctional adhesion molecule. J Biol Chem , 455–461. [DOI] [PubMed] [Google Scholar]
- Harder JL, Whiteman EL, Pieczynski JN, Liu CJ, Margolis B. (2012). Snail destabilizes cell surface Crumbs3a. Traffic , 1170–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes JL, Van Itallie CM, Rasmussen JE, Anderson JM. (2006). Claudin profiling in the mouse during postnatal intestinal development and along the gastrointestinal tract reveals complex expression patterns. Gene Expr Patterns , 581–588. [DOI] [PubMed] [Google Scholar]
- Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. (2003). Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci , 725–742. [DOI] [PubMed] [Google Scholar]
- Huang J, Yin P, Zhang L. (2019). COPII cargo claudin-12 promotes hepatitis C virus entry. J Viral Hepat , 308–312. [DOI] [PubMed] [Google Scholar]
- Ikari A, Tonegawa C, Sanada A, Kimura T, Sakai H, Hayashi H, Hasegawa H, Yamaguchi M, Yamazaki Y, Endo S, et al. (2014). Tight junctional localization of claudin-16 is regulated by syntaxin 8 in renal tubular epithelial cells. J Biol Chem , 13112–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Furuse M, Morita K, Kubota K, Saitou M, Tsukita S. (1999). Direct binding of three tight junction-associated MAGUKs, ZO-1, ZO-2, and ZO-3, with the COOH termini of claudins. J Cell Biol , 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AI, Nusrat A, Parkos CA. (2004). Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell , 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval M. (2013). Differential pathways of claudin oligomerization and integration into tight junctions. Tissue Barriers , e24518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota K, Furuse M, Sasaki H, Sonoda N, Fujita K, Nagafuchi A, Tsukita S. (1999). Ca(2+)-independent cell-adhesion activity of claudins, a family of integral membrane proteins localized at tight junctions. Curr Biol , 1035–1038. [DOI] [PubMed] [Google Scholar]
- Kuhn S, Koch M, Nubel T, Ladwein M, Antolovic D, Klingbeil P, Hildebrand D, Moldenhauer G, Langbein L, Franke WW, et al. (2007). A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res , 553–567. [DOI] [PubMed] [Google Scholar]
- Lanaspa MA, Andres-Hernando A, Rivard CJ, Dai Y, Berl T. (2008). Hypertonic stress increases claudin-4 expression and tight junction integrity in association with MUPP1 in IMCD3 cells. Proc Natl Acad Sci USA , 15797–15802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Koval M, Ranganathan S, Fanayan S, Hancock WS, Lundberg EK, Beavis RC, Lane L, Duek P, McQuade L, et al. (2016). Systems proteomics view of the endogenous human claudin protein family. J Proteome Res , 339–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Johnson DL, Stewart L, Waite K, Elliott D, Wilson JM. (2014). Rab14 regulation of claudin-2 trafficking modulates epithelial permeability and lumen morphogenesis. Mol Biol Cell , 1744–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Kim DH, Fan J, Lu Q, Verbanac K, Ding L, Renegar R, Chen YH. (2015). A non-tight junction function of claudin-7-Interaction with integrin signaling in suppressing lung cancer cell proliferation and detachment. Mol Cancer , 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marunaka K, Furukawa C, Fujii N, Kimura T, Furuta T, Matsunaga T, Endo S, Hasegawa H, Anzai N, Yamazaki Y, et al. (2017). The RING finger- and PDZ domain-containing protein PDZRN3 controls localization of the Mg2+ regulator claudin-16 in renal tube epithelial cells. J Biol Chem , 13034–13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M, Kubo A, Furuse M, Tsukita S. (2004). A peculiar internalization of claudins, tight junction-specific adhesion molecules, during the intercellular movement of epithelial cells. J Cell Sci , 1247–1257. [DOI] [PubMed] [Google Scholar]
- Medina R, Rahner C, Mitic LL, Anderson JM, Van Itallie CM. (2000). Occludin localization at the tight junction requires the second extracellular loop. J Membr Biol , 235–247. [DOI] [PubMed] [Google Scholar]
- Mora-Galindo J. (1986). Maturation of tight junctions in guinea-pig cecal epithelium. Cell Tissue Res , 169–175. [DOI] [PubMed] [Google Scholar]
- Morimoto S, Nishimura N, Terai T, Manabe S, Yamamoto Y, Shinahara W, Miyake H, Tashiro S, Shimada M, Sasaki T. (2005). Rab13 mediates the continuous endocytic recycling of occludin to the cell surface. J Biol Chem , 2220–2228. [DOI] [PubMed] [Google Scholar]
- Nomme J, Antanasijevic A, Caffrey M, Van Itallie CM, Anderson JM, Fanning AS, Lavie A. (2015). Structural basis of a key factor regulating the affinity between the zonula occludens first PDZ domain and claudins. J Biol Chem , 16595–16606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes FD, Lopez LN, Lin HW, Davies C, Azevedo RB, Gow A, Kachar B. (2006). Distinct subdomain organization and molecular composition of a tight junction with adherens junction features. J Cell Sci , 4819–4827. [DOI] [PubMed] [Google Scholar]
- Piontek J, Winkler L, Wolburg H, Muller SL, Zuleger N, Piehl C, Wiesner B, Krause G, Blasig IE. (2008). Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J , 146–158. [DOI] [PubMed] [Google Scholar]
- Rahner C, Mitic LL, Anderson JM. (2001). Heterogeneity in expression and subcellular localization of claudins 2, 3, 4, and 5 in the rat liver, pancreas, and gut. Gastroenterology , 411–422. [DOI] [PubMed] [Google Scholar]
- Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, Wang Y, Wu L, Schneeberger EE, Shen L, Turner JR. (2011). Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol , 565–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. (2013a). Genome engineering using the CRISPR-Cas9 system. Nat Protoc , 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara A, Furuse M, Saitou M, Ando-Akatsuka Y, Tsukita S. (1997). Possible involvement of phosphorylation of occludin in tight junction formation. J Cell Biol , 1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlingmann B, Overgaard CE, Molina SA, Lynn KS, Mitchell LA, Dorsainvil White S, Mattheyses AL, Guidot DM, Capaldo CT, Koval M. (2016). Regulation of claudin/zonula occludens-1 complexes by hetero-claudin interactions. Nat Commun , 12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Turner JR. (2005). Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol Biol Cell , 3919–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Weber CR, Turner JR. (2008). The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J Cell Biol , 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda N, Furuse M, Sasaki H, Yonemura S, Katahira J, Horiguchi Y, Tsukita S. (1999). Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J Cell Biol , 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankewich MC, Francis SA, Vu QU, Schneeberger EE, Lynch RD. (1996). Alterations in cell cholesterol content modulate Ca(2+)-induced tight junction assembly by MDCK cells. Lipids , 817–828. [DOI] [PubMed] [Google Scholar]
- Stoops EH, Farr GA, Hull M, Caplan MJ. (2014). SNAP-tag to monitor trafficking of membrane proteins in polarized epithelial cells. Methods Mol Biol , 171–182. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, Dohmae N, Tsukita S, Nureki O, Fujiyoshi Y. (2014). Crystal structure of a claudin provides insight into the architecture of tight junctions. Science , 304–307. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Tani K, Tamura A, Tsukita S, Fujiyoshi Y. (2015). Model for the architecture of claudin-based paracellular ion channels through tight junctions. J Mol Biol , 291–297. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Iwamoto N, Sasaki H, Ohashi M, Oda Y, Tsukita S, Furuse M. (2009). The E3 ubiquitin ligase LNX1p80 promotes the removal of claudins from tight junctions in MDCK cells. J Cell Sci , 985–994. [DOI] [PubMed] [Google Scholar]
- Thanabalasuriar A, Kim J, Gruenheid S. (2013). The inhibition of COPII trafficking is important for intestinal epithelial tight junction disruption during enteropathogenic Escherichia coli and Citrobacter rodentium infection. Microbes Infect , 738–744. [DOI] [PubMed] [Google Scholar]
- Tice LW, Carter RL, Cahill MB. (1979). Changes in tight junctions of rat intestinal crypt cells associated with changes in their mitotic activity. Tissue Cell , 293–316. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff SK, Buznikov AG, Vu TQ, Micevych PE, Chen K, Kornblum HI, Bronstein JM. (2001). OSP/claudin-11 forms a complex with a novel member of the tetraspanin super family and beta1 integrin and regulates proliferation and migration of oligodendrocytes. J Cell Biol , 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda S, Furuse M. (2015). Claudin-2 knockout by TALEN-mediated gene targeting in MDCK cells: claudin-2 independently determines the leaky property of tight junctions in MDCK cells. PLoS One , e0119869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda S, Higashi T, Furuse M. (2014). ZO-1 knockout by TALEN-mediated gene targeting in MDCK cells: involvement of ZO-1 in the regulation of cytoskeleton and cell shape. PLoS One , e104994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuda S, Miyazaki H, Nakajima K, Yamada T, Marunaka Y. (2010). NaCl flux between apical and basolateral side recruits claudin-1 to tight junction strands and regulates paracellular transport. Biochem Biophys Res Commun , 390–396. [DOI] [PubMed] [Google Scholar]
- Trinkaus JP. (1973). Modes of cell locomotion in vivo. Ciba Found Symp , 233–249. [DOI] [PubMed] [Google Scholar]
- Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M. (2006). ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell , 741–754. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Anderson JM. (2006). Claudins and epithelial paracellular transport. Annu Rev Physiol , 403–429. [DOI] [PubMed] [Google Scholar]
- Van Itallie CM, Colegio OR, Anderson JM. (2004). The cytoplasmic tails of claudins can influence tight junction barrier properties through effects on protein stability. J Membr Biol , 29–38. [DOI] [PubMed] [Google Scholar]
- Van Itallie C, Rahner C, Anderson JM. (2001). Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J Clin Invest , 1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, Tietgens AJ, Anderson JM. (2017). Visualizing the dynamic coupling of claudin strands to the actin cytoskeleton through ZO-1. Mol Biol Cell , 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, Tietgens AJ, Krystofiak E, Kachar B, Anderson JM. (2015). A complex of ZO-1 and the BAR-domain protein TOCA-1 regulates actin assembly at the tight junction. Mol Biol Cell , 2769–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CJ, Mannan P, Lu M, Udey MC. (2013). Epithelial cell adhesion molecule (EpCAM) regulates claudin dynamics and tight junctions. J Biol Chem , 12253–12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe Y, Shepshelovitch J, Nevo-Yassaf I, Yeheskel A, Shmerling H, Kwiatek JM, Gaus K, Pasmanik-Chor M, Hirschberg K. (2012). The MARVEL transmembrane motif of occludin mediates oligomerization and targeting to the basolateral surface in epithelia. J Cell Sci , 3545–3556. [DOI] [PubMed] [Google Scholar]
- Yin P, Li Y, Zhang L. (2017). Sec24C-dependent transport of claudin-1 regulates hepatitis C virus entry. J Virol , 10.1128/JVI.00629-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Krystofiak ES, Ballesteros A, Cui R, Van Itallie CM, Anderson JM, Fenollar-Ferrer C, Kachar B. (2018). Multiple claudin–claudin cis interfaces are required for tight junction strand formation and inherent flexibility. Commun Biol , 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.