

Abstract
Postaxial polydactyly (PAP) is a common limb malformation that often leads to cosmetic and functional complications. Molecular evaluation of polydactyly can serve as a tool to elucidate genetic and signaling pathways that regulate limb development, specifically, the anterior-posterior specification of the limb. To date, only five genes have been identified for nonsyndromic PAP: FAM92A, GLI1, GLI3, IQCE and ZNF141. In this study, two Pakistani multiplex consanguineous families with autosomal recessive nonsyndromic PAP were clinically and molecularly evaluated. From both pedigrees, a DNA sample from an affected member underwent exome sequencing. In each family, we identified a segregating frameshift (c.591dupA [p.(Q198Tfs*21)]) and nonsense variant (c.2173A > T [p.(K725*)]) in KIAA0825 (also known as C5orf36). Although KIAA0825 encodes a protein of unknown function, it has been demonstrated that its murine ortholog is expressed during limb development. Our data contribute to the establishment of a catalog of genes important in limb patterning, which can aid in diagnosis and obtaining a better understanding of the biology of polydactyly.
Introduction
Genetically driven molecular pathways involved in limb development epitomize a complicated process that is still not fully understood. Genetic evaluation of hereditary limb malformations allows the identification of genes associated with limb defects, thereby providing valuable clues to the physiologic role of these genes without prior information on function (Grzeschik 2002; Barham and Clarke 2008). Massively parallel sequencing has proven to be a powerful approach to identify novel genes associated with rare monogenic disorders, including limb malformations (Ng et al. 2010; Parla et al. 2011; Kalsoom et al. 2013; Spielmann et al. 2016).
Postaxial polydactyly (PAP) is a common congenital limb malformation characterized by extra digit(s) at the ulnar (little finger) or fibular (5th toe) side of the hands and/or feet (Schwabe and Mundlos 2004; Umm-e-Kalsoom et al. 2012). PAP results from defective patterning of the anterior-posterior axis of the developing limb bud. It can occur as an isolated trait (nonsyndromic PAP) or as a part of a syndrome (Biesecker 2011).
A Pakistani clinical and descriptive genetic study reported that 52% of nonsyndromic polydactylies are postaxial (Malik et al. 2014). Several studies reported that males are affected twice as often as females (Castilla et al. 1973; Malik et al. 2014). Non-syndromic PAP (OMIM: 174200), can be broadly divided into two types: PAP-A, characterized by a well-developed extra fifth digit, and PAP-B showing an incompletely developed extra digit (pedunculated postminimus) attached to the fifth finger/toe (Schwabe and Mundlos 2004; Umm-e-Kalsoom et al. 2012; Kalsoom et al. 2013; Verma and El-Harouni 2015a). In Pakistan PAP-A is more prevalent than PAP-B with 64.8% of cases presenting with PAP-A (Malik et al. 2014). Non-syndromic PAP segregates with either an autosomal dominant or recessive model of inheritance with variable expression (Umm-e-Kalsoom et al. 2012; Radhakrishna et al. 1999). Four autosomal dominant loci (PAP-A1-A4) and five autosomal recessive (PAP-A5-A9) loci have been reported (Umm-e-Kalsoom et al. 2012; Kalsoom et al. 2013). Thus far five PAP genes have been identified : GLI3 [PAP-A1 (OMIM: 174200) (Al-Qattan 2012)]; ZNF141 [PAP-A6 (OMIM: 615226) (Kalsoom et al. 2013)]; IQCE [PAP-A7 (OMIM: 617642) (Umair et al. 2017)]; GLI1 [PAP-A8 (OMIM: 618123) (Palencia-Campos et al. 2017)] and FAM92A [PAP-A9 (OMIM: 618219) (Schrauwen et al. 2018)].
In this study, two Pakistani multiplex consanguineous families with autosomal recessive PAP were evaluated. In both families, homozygous KIAA0825 loss of function variants were identified which likely underlie PAP etiology.
Materials and methods
Ethical approval and sampling
The study was approved by institutional review boards (IRBs) at Balochistan University of Information Technology, Engineering and Management Sciences, Quetta, Pakistan (IRB# 00010538); Baylor College of Medicine and Affiliated Hospitals, Houston, USA (IRB# H-41049); and Quaid-i-Azam University, Islamabad, Pakistan (IRB# QAU-155).
The nonsyndromic PAP families are both from Pakistan, BD204 was ascertained from the Upper Dir district of Khyber Pakhtonkhwa (Northern Pakistan) and PD-23 from the city of Mirpur Khas, Sindh Province (Southern Pakistan). For family BD204, DNA samples were obtained from two affected (V:1 and V:2) and two unaffected (IV:2 and V:3) members (Fig. 1A) and for family PD-23, from three affected (IV:2, VI:1, and VI:5) and eight unaffected (IV:4, IV:5, V:3, V:4, VI:2, VI:3, VI:4, and VI:7) members (Fig. 2A).
Fig. 1.

A Pedigree drawing of family BD204 segregating autosomal recessive postaxial polydactyly type A and B. Males are denoted by squares while females are display as circles. Family members affected with PAP are denoted by a filled in symbol and unaffected members by a clear one. Consanguineous unions are delineated by double lines. A diagonal line through a square or circle represents a deceased family member. DNA samples were obtained from individuals for which a genotype is shown. A star (*) indicates availability of exome sequence data. B Photographs and radiographs for affected members of family BD204. Individual V:1 displays PAP-A of both feet (a) and hands exhibiting an extra digit articulated with the fifth metacarpal along with mild clinodactyly in the fifth digit of both hands (b, c). For individual V:2, the right and left foot present with PAP-A (d, e), and the right hand presents with PAP-B that is characterized by a rudimentary tissue tag, extending from fifth finger (f). The left hand of V:2 presents with PAP-A, displaying a sixth finger outspread from the fifth metacarpal (g). V:2 also presents with mild along with mild clinodactyly in the fifth digit of both hands (f, g). Radiographic features of the feet of patient (V:2) reveal a diphalangeal extra digit articulated with fifth metatarsal in the right foot (h). A diphalangeal extra digit and a bifurcated and broad metatarsal is visible in the left foot (i). Radiographs of hands of affected individual (V:2) show PAP-A in left hand (k). The extra finger occurs in the form of a discrete digit having two phalanges (k). Bony abnormalities can be excluded in the right hand (j)
Fig. 2.

A Pedigree drawing of family PD-23 segregating autosomal recessive postaxial polydactyly type A. Family members affected with PAP are denoted by a filled in symbol and unaffected members by a clear one. Consanguineous unions are delineated by double lines. A diagonal line through a square or circle represents a deceased family member. DNA samples were obtained from individuals for which a genotype is shown. A star (*) indicates availability of exome sequence data. B Photographs and radiographs for family PD-23. Individual IV:2 displays PAP-A of both feet (a) and hands (b). For individual VI:1 the left foot displays postaxial hexadactyly with a well-developed diphalangeal additional toe while the right foot is clinically and radiologically normal (c, e). The hands of individual VI:1 do not show any clinical or radiological abnormality (d, f). Individual VI:5 displays unilateral PAP-A of the left hand with an extra digit arising from the fifth metacarpal, mild camptodactyly in the fifth digit of both hands. as well as PAP-A of the right foot (g, h)
DNA extraction, genotyping, quality control and homozygosity mapping
Genomic DNA was extracted from peripheral blood samples using commercially available kits. For BD204, whole-genome genotyping of 550K single nucleotide polymorphism (SNP) markers was performed using the Infinium® HumanCoreExome Chip (Illumina, USA) on genomic DNA samples from two affected (V:1 and V:2) and one unaffected (V:3) family members. The genotype data underwent quality control using MERLIN (Abecasis et al. 2002) to detect occurrences of double recombination events over short genetic distances which could be due to genotyping errors, and PEDCHECK (O’Connell and Weeks 1998) to identify Mendelian inconsistencies. Analysis of the genotype data were performed using homozygosity mapping (HomozygosityMapper) (Seelow and Schuelke 2012).
Whole exome sequencing and Sanger sequencing
For family BD204 and PD-23 genomic DNA samples from one affected family member (BD204: V:2; PD-23: VI:1) was selected for exome sequencing (Figs. 1A, 2A).
Exome library preparation was performed using the Roche/Nimblegen SeqCap EZ v2.0 (~ 36.5 MB) for family BD204 and the Agilent SureSelect Exome V6 Capture Library kit for family PD-23. Barcoded libraries were pooled and sequencing was performed on an Illumina HiSeq with an average on-target coverage of 54× for BD204 and 87× for PD-23. Reads were aligned to GRCh37/Hg19 using Burrows-Wheeler Aligner (BWA-MEM) (Li and Durbin 2009) for family BD204 and using Bowtie2 (Langmead et al. 2018) and SAMTools (Li et al. 2009) for family PD-23. Duplicate removal, indel-realignment, quality recalibration, and variant detection and calling were performed using Picard and Genome Analysis Toolkit (GATK) (McKenna et al. 2010).
For family BD204, variants were annotated using ANNOVAR (Wang et al. 2010) and selected based on the following criteria: exonic and splicing variants (± 12 bp); population specific minor allele frequency (MAF) < 0.005 in the genome aggregation database (gnomAD) database (Lek et al. 2016); a scaled C-score of ≥ 10 in the combined annotation dependent depletion (CADD) database for single nucleotide variants (SNVs) (Kircher et al. 2014) and bioinformatics predictions of the variants impact. Variants located in the linkage/homozygosity region were given priority, but other genomic areas were also considered. Sanger sequencing was used to verify segregation of candidate variants within family BD204.
For data analysis of family PD-23, the Varbank pipeline v.2.3 and filter interfaces were applied to detect protein changes, affected donor and acceptor splice sites, and overlaps with known variants. Acceptor and donor splice-site mutations were analyzed using a maximum entropy model (Yeo and Burge 2004). Based on the same filtering parameters used for family BD204, variants were selected to be tested for segregation. Sanger sequencing was also used to verify segregation of candidate variants. Mutation nomenclature of KIAA0825 is based on transcript NM_001145678.1.
In silico expression analysis
To evaluate the expression of KIAA0825 (2210408IRik) in the developing mouse limb, RNA-seq expression data were downloaded and further analyzed from datasets GSE78345, GSE78372, GSE78405, GSE78465 and GSE82919 from the Gene Expression Omnibus (GEO) database (Edgar et al. 2002). Bernstein and colleagues have studied total RNA extracted from developing limbs at stages E11.5–E15.5 in C57BL/6 mouse embryos (B6NCrl substrain; mixed sex). Data were generated as part of the Encyclopedia of DNA Elements (ENCODE) project (ENCODE Project Consortium 2012; Edgar et al. 2002). Non-strand specific RNA-seq data, containing the TPM values (Transcripts per Kilobase Million), were downloaded and further analyzed with R3.1.2. Two technical replicates were generated from each embryonic stage, and the averaged expression was plotted using ggplot2.
Results
Clinical features
Pedigrees BD204 and PD-23 are consanguineous and PAP segregates with an autosomal recessive mode of inheritance (Figs. 1A, 2A). For family BD204, nonsyndromic PAP-A was present in two affected individuals (V:1 and V:2) and PAP-B was also observed in individual V:2 (Fig. 1B). Affected individuals presented bilateral, well-developed duplication of fifth digit in hands and feet, compatible with PAP-A (Fig. 1B: a–g). However, the right hand of the affected individual V:2 had a postaxial rudimentary tag that is a feature of PAP-B (Fig. 1B: f). X-rays of affected individual (V:2) revealed a diphalangeal sixth finger of the left hand while no additional bony structure was observed in the right hand (Fig. 1B: h–k). The fifth metatarsal has a larger diameter whereas the diameter of third and fourth metatarsals is smaller in individual V:2. Additionally both affected individuals V:1 and V:2 showed mild clinodactyly in the fifth digit of both hands (Fig. 1B: b, c, f, g). Radiographs showed a diphalangeal sixth digit for both feet with one digit extending from a bifurcated fifth metatarsal for the left foot (Fig. 1B: h, i). Affected individuals had normal height, teeth, nails, skin and hair without any associated craniofacial or neurological abnormalities.
For family PD-23, four individuals are affected with non-syndromic PAP-A of variable degree (Fig. 2A). Individual IV:2 has PAP-A with well-developed additional fingers and toes on both hands and feet (Fig. 2B: a, b). The hands of individual VI:1 do not show any clinical or radiological abnormality (Fig. 2B: d, f), however, his left foot displays postaxial hexadactyly with a well-developed diphalangeal additional toe (Fig. 2B: c, e). The corresponding metatarsal bone is distally broadened. The right foot of individual VI:1 is clinically and radiologically normal (Fig. 2B: c, e). Affected individual VI:5 displays unilateral PAP-A of the left hand with an extra digit arising from the fifth metacarpal along with mild camptodactyly in the fifth digit of both hands as well as PAP-A of the right foot (Fig. 2B: g, h). Family member IV:3 who was reported to be affected with PAP is deceased and no additional phenotype data could be obtained.
Homozygosity mapping
For family BD204, whole genome SNP array data revealed three homozygous regions in BD204 affected family members: 1p31.1-p21.1 (29.23 Mb); 3q13.31-q22.1 (16.52 Mb); and 5q14.3-q15 (10.34 Mb).
Exome and Sanger sequence data
For family BD204 and PD-23, no exonic/splice site variants within the known nonsyndromic PAP genes with an allele frequency < 0.05 in the gnomAD exome and genome databases were found. When considering the entire exome, for family BD204, six homozygous variants of interest were identified with two of these homozygous variants lying within regions of homozygosity. Segregation of the variants was tested using DNA samples from four BD204 family members (Fig. 1A and Supplementary Table 1). In family PD-23, one homozygous variant of note was detected and segregation was tested (Fig. 2A and Supplementary Table 1).
The rare variants detected in the exome sequence data were tested for segregation with the PAP using Sanger sequencing. For pedigree BD204 two variants segregated with PAP: c.761G > A [p.(R254Q)] within RPF1 and c.591dupA [p.(Q198Tfs*21)] within KIAA0825 that lay in homozygous regions 1p31.1-p21.1 and 5q14.3-q15, respectively (Supplementary Table 1). The homozygous variant RPF1 c.761G > A [p.(R254Q)] has a CADD C-score of 33 and is rare with a gnomAD exome MAF = 8.13 × 10−6, with only two heterozygous individuals reported in the Non-Finnish European population (MAF = 1.79 × 10−5). This variant is unlikely to be causative for PAP based on the RPF1 protein function and its implication in Wilms tumor susceptibility-5 (OMIM: 601583). The second homozygous variant c.591dupA [p.(Q198Tfs*21)] within KIAA0825 (C5orf36) segregated in family BD204 and was not observed in gnomAD exomes or genomes.
Aside from family BD204, a second homozygous variant c.2173A > T [p.(K725*)] within KIAA0825 segregated in family PD-23. This variant is rare with a MAF = 6.75 × 10−6 in gnomAD exomes, only one heterozygous allele was observed in the gnomAD Latino population (MAF = 4.32 × 10−5) and none were detected among 22,170 South Asian chromosomes.
Two homozygous variants were detected within KIAA0825, a frameshift variant, c.591dupA [p.(Q198Tfs*21)] (located in the 5q14.3-q15 region of homozygosity) and a nonsense variant c.2173A > T [p.(K725*)] that segregated within families BD204 and PD-23, respectively. The amplified KIAA0825 sequence was obtained using forward (F1) and reverse (R1) primers (F1: 5′-cccatcaaccctaagtggaa-3′ and R1: 5′-tcaccattgaagatg-gagctaa-3′) within family BD204 and using forward (F2) and reverse (R2) primers (F2: 5′-ggcactgaattgcatcgaga-3′ and R2: 5′-acagagctcaccatccctaaag-3’) within family PD-23 (Supplementary Fig. 1). The Sanger sequence chromatograms are displayed in Supplementary Fig. 1. The variants were deposited in the ClinVar databased under accession numbers SCV000864221 and SCV000864222 (https://www.ncbi.nlm.nih.gov/clinvar/).
To estimate the frequency of KIAA0825 mutations in PAP, we queried exome sequences of 40 affected individuals from 31 unrelated Pakistani families and did not detect any additional potentially pathogenic variants. Taken together, these results suggest that KIAA0825 variants are likely a rare cause of autosomal recessive PAP in Pakistan.
KIAA0825 ortholog expression in the developing limb
In silico query of the ENCODE data (RNA-seq) showed that the closest mouse ortholog of KIAA0825, 2210408IRik, is expressed in the developing mouse limb from E11.5 through E15.5 compared to other known polydactyly genes (Fig. 3).
Fig. 3.
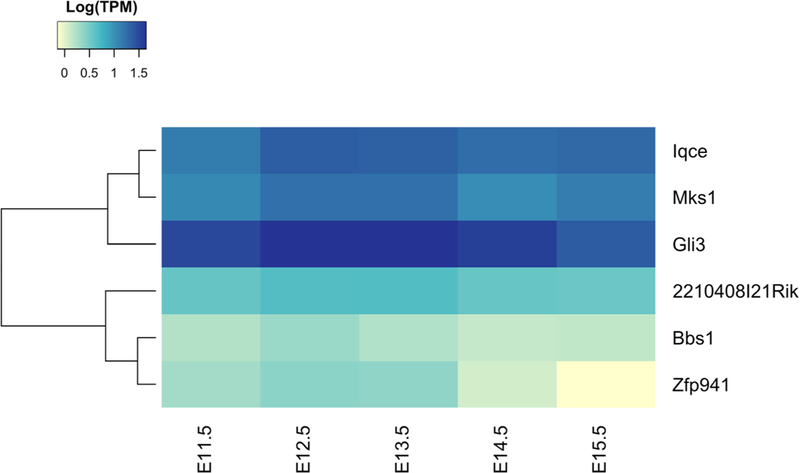
Heatmap representation of RNA-seq data queried from ENCODE of polydactyly genes in the developing mouse limb from E11.5 through E15.5. The 2210408IRik gene, the closest mouse KIAA0825 ortholog, has a steady expression level in the developing limb. Here, it is plotted together with other non- syndromic i.e., Zfp941 (ZNF141 ortholog), Iqce, Gli3 and syndromic polydactyly genes i.e., Bbs1, Mks1. TPM transcripts per kilobase million
Discussion
Human limb development is a complex process that is spatially and temporally regulated by a complex cascade of molecular pathways. The growth of the limb bud occurs in a three-dimensional axis: proximal to distal, dorsal to ventral, and anterior to posterior (radial to ulnar) (Barham and Clarke 2008). Dysregulation in anterior-posterior patterning of the developing limb may result in polydactyly that can occur as an isolated limb malformation or as part of a pleiotropic developmental anomaly syndrome (Biesecker 2011; Verma and El-Harouni 2015). Genes more specifically related to the anterior-to-posterior axis development are strongly related to the molecular basis of polydactyly (Yang and Niswander 1995; Loomis et al. 1998; Verma and El-Harouni 2015). Polydactyly can be present on the radial side (preaxial), ulnar side (postaxial; PAP) or involve non-border (central) digits (mesoaxial). PAP is the most common of these three polydactyly types with an estimated incidence of one in ~ 110 live births in Africans (Pompe van Meerder-voort 1976) and one in ~ 1340 live births in Europeans (Watson and Hennrikus 1997).
After GLI3, ZNF141, IQCE, GLI1, and FAM92A, KIAA0825 is the sixth gene for nonsyndromic PAP. The two identified variants are very rare and co-segregate with PAP. They lead to the introduction of a premature termination codon and are predicted to be targeted by nonsense-mediated decay (NMD) in the long isoform of KIAA0825 (Hug et al. 2016). KIAA0825 has two different isoforms, a long isoform (isoform 1; NM_001145678.1; NP_001139150.1; 1275aa), and a short isoform (isoform 2; NM_173665.2; NP_775936.1; 324 aa). Both variants are likely to lead to no or limited expression of a truncated protein and a loss-of-function (LOF) of the long isoform of KIAA0825 due to NMD (isoform 1). However, the c.2173A > T variant (PD-23) does not affect the short isoform of KIAA0825, although the c.591dupA variant (BD204) is predicted to lead to the expression of a truncated KIAA0825 isoform 2, that might be partially functional or not. This might explain the milder phenotype observed in family PD-23, as the short isoform (isoform 2) of KIAA0825 is not affected in these patients. In addition, no homozygous frameshift, nonsense or splice site variants in KIAA0825 have been observed in the gnomAD database.
Homozygous 2210408I21Riktm1b(EUCOMM)Wtsi mice that were created and phenotyped by the International Mouse Phenotyping Consortium (IMPC) (Koscielny et al. 2014) have a significant decrease in bone mineral density (BMD) which was observed in both male and female mice (p = 2.02 × 10−7). The BMD mean values for 2210408I21Riktm1b(EUCOMM)Wtsi–/– males (N = 8) were 0.0428 ± 0.0011 standard deviations (SD) and for females (N = 8) 0.0439 ± 0.0025 SD compared to 0.0458 ± 0.0018 SD for male (N = 144) and 0.0478 ± 0.0018 SD for female (N = 404) controls. The 2210408IRik–/– mice (N = 16) also showed an increase of the total body fat in both males and females (p = 1.43 × 10−5) compared to controls (N = 548). A decrease in lean body mass was observed in males (p = 2.46 × 10−5) but not observed in females (p = 0.23584). Impaired glucose tolerance was also observed in 2210408IRik–/– male and female mice (p = 4.75 × 10−7). Although 2210408IRik–/– mice do not display polydactyly these data are consistent with a causative role of KIAA0825 variants for PAP.
KIAA0825 is an uncharacterized gene of unknown function. According to ENCODE, its mouse ortholog 2210408I21Rik is expressed in various tissues/organs, including in the developing limbs (Fig. 3). In mouse, the E11.5 forelimb bud reveals the first signs of a humerus and by E14.5 the most distal phalanges of the hand are formed (Martin 1990). 2210408IRik is expressed at a steady level throughout several all these time critical points. A de novo deletion of chromosome 5q14.3–15 has been associated with bilateral periventricular heterotopia, intellectual disability, and epilepsy (Cardoso et al. 2009). Interestingly, one of the three patients in this report also had postaxial polydactyly of the toes. Yet, the causative gene underlying this phenotype has not been identified, and it is unknown whether a mutation on the non-deleted chromosome was present. Overall, we provide evidence that KIAA0825 likely causes PAP in the two families under study, although additional cases are needed to confirm the causal relation of KIAA0825 to PAP.
Genes mutated in nonsyndromic and syndromic forms of polydactyly encode proteins involved in cell signaling, transcription and growth factors, proteins involved in DNA repair, structural and catalytic proteins, immunoglobulin superfamily proteins, chaperones, and gap junction proteins (Duboc and Logan 2009; Goetz and Anderson 2010; Biesecker 2011; Verma and El-Harouni 2015). However, the genetic machinery involved in limb development and anterior-posterior patterning is not fully understood. Recent technologies such as massively parallel sequencing are powerful tools to accelerate the process of gene identification for rare disorders (Ng et al. 2010; Parla et al. 2011). Hence, it is anticipated that the list of identified polydactyly genes will be extended in the next few years ultimately yielding a more complete catalog of genes involved in polydactyly phenotypes.
Supplementary Material
Acknowledgements
The authors would like to thank the family members who participated in this study. This work was supported by funds from the Higher Education Commission (HEC), Islamabad, Pakistan (to WA). A Georg Forster fellowship was provided by the Alexander von Humboldt Foundation (to NW). Genotyping and exome sequencing for family BD204 was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant UM1 HG006493 (to DAN, MJB, and SML) and U24 HG008956. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Web Resources: ANNOVAR, http://annovar.openbioinformatics.org/. Burrows-Wheeler Aligner (BWA), http://bio-bwa.sourceforge.net/. Combined Annotation Dependent Depletion (CADD), http://cadd.gs.washington.edu/. Gene Expression Ommibus (GEO), https://www.ncbi.nlm.nih.gov/geo/. Genome Aggregation Database (gnomAD), http://gnomad.broadinstitute.org/. Genome Analysis Toolkit (GATK), https://software.broadinstitute.org/gatk/. ENCODE project, https://www.encodeproject.org/. HomozygosityMapper, http://www.homozygositymapper.org/. International Mouse Phenotyping Consortium (IMPC), http://www.mousephenotype.org. Varbank pipeline v.2.3, https://varbank.ccg.uni-koeln.de/.
Footnotes
Compliance with ethical standards
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00439-019-02000-0) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no competing interests.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlinrapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101. 10.1038/ng786 [DOI] [PubMed] [Google Scholar]
- Al-Qattan MM (2012) A novel frameshift mutation of the GLI3 gene in a family with broad thumbs with/without big toes, postaxial polydactyly and variable syndactyly of the hands/feet. Clin Genet 82:502–504. 10.1111/j.1399-0004.2012.01866.x [DOI] [PubMed] [Google Scholar]
- Barham G, Clarke NMP (2008) Genetic regulation of embryological limb development with relation to congenital limb deformity in humans. J Child Orthop 2:1–9. 10.1007/s11832-008-0076-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG (2011) Polydactyly: how many disorders and how many genes? 2010 update. Dev Dyn 240:931–942. 10.1002/dvdy.22609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso C, Boys A, Parrini E et al. (2009) Periventricular heterotopia, mental retardation, and epilepsy associated with 5q14.3-q15 deletion. Neurology 72:784–792. 10.1212/01.wnl.0000336339.08878.2d [DOI] [PubMed] [Google Scholar]
- Castilla E, Paz J, Mutchinick O et al. (1973) Polydactyly: a genetic study in South America. Am J Hum Genet 25:405–412 [PMC free article] [PubMed] [Google Scholar]
- Duboc V, Logan MP (2009) Building limb morphology through integration of signalling modules. Curr Opin Genet Dev 19:497–503. 10.1016/j.gde.2009.07.002 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. https://doi.org/101038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11:331–344. 10.1038/nrg2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzeschik K- H (2002) Human limb malformations; an approach to the molecular basis of development. Int J Dev Biol 46:983–991 [PubMed] [Google Scholar]
- Hug N, Longman D, Cáceres JF (2016) Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res 44:1483–1495. 10.1093/nar/gkw010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsoom U-, Klopocki E, Wasif N et al. (2013) Whole exome sequencing identified a novel zinc-finger gene ZNF141 associated with autosomal recessive postaxial polydactyly type A. J Med Genet 50:47–53. 10.1136/jmedgenet-2012-101219 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P et al. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscielny G, Yaikhom G, Iyer V et al. (2014) The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res 42:D802–D809. 10.1093/nar/gkt977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Wilks C, Antonescu V, Charles R (2018) Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics. 10.1093/bioinformatics/bty648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A et al. (2009) The Sequence Alignment/ Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis CA, Kimmel RA, Tong CX et al. (1998) Analysis of the genetic pathway leading to formation of ectopic apical ectodermal ridges in mouse Engrailed-1 mutant limbs. Development 125:1137–1148 [DOI] [PubMed] [Google Scholar]
- Malik S, Ullah S, Afzal M et al. (2014) Clinical and descriptive genetic study of polydactyly: a Pakistani experience of 313 cases. Clin Genet 85:482–486. 10.1111/cge.12217 [DOI] [PubMed] [Google Scholar]
- Martin P (1990) Tissue patterning in the developing mouse limb. Tissue patterning in the developing mouse limb. Int J Dev Biol 34:323–336 [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C et al. (2010) Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42:30–35. 10.1038/ng.499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266. 10.1086/301904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palencia-Campos A, Ullah A, Nevado J et al. (2017) GLI1 inactivation is associated with developmental phenotypes overlapping with Ellis-van Creveld syndrome. Hum Mol Genet 26:4556–4571. 10.1093/hmg/ddx335 [DOI] [PubMed] [Google Scholar]
- Parla JS, Iossifov I, Grabill I et al. (2011) A comparative analysis of exome capture. Genome Biol 12:R97 10.1186/gb-2011-12-9-r97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompe van Meerdervoort HF (1976) Congenital musculoskeletal malformation in South African Blacks: a study of incidence. S Afr Med J 50:1853–1855 [PubMed] [Google Scholar]
- Radhakrishna U, Bornholdt D, Scott HS et al. (1999) The phenotypic spectrum of GLI3 morphopathies includes autosomal dominant preaxial polydactyly type-IV and postaxial polydactyly type-A/B; No phenotype prediction from the position of GLI3 mutations. Am J Hum Genet 65:645–655. 10.1086/302557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauwen I, Giese AP, Aziz A et al. FAM92A underlies nonsyndromic postaxial polydactyly in humans and an abnormal limb and digit skeletal phenotype in mice. J Bone Miner Res. 10.1002/jbmr.3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe GC, Mundlos S (2004) Genetics of congenital hand anomalies. Handchir Mikrochir Plast Chir 36:85–97. 10.1055/s-2004-817884 [DOI] [PubMed] [Google Scholar]
- Seelow D, Schuelke M (2012) HomozygosityMapper2012—bridging the gap between homozygosity mapping and deep sequencing. Nucleic Acids Res 40:W516–W520. 10.1093/nar/gks487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielmann M, Kakar N, Tayebi N et al. (2016) Exome sequencing and CRISPR/Cas genome editing identify mutations of ZAK as a cause of limb defects in humans and mice. Genome Res 26:183–191. 10.1101/gr.199430.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umair M, Shah K, Alhaddad B et al. (2017) Exome sequencing revealed a splice site variant in the IQCE gene underlying post-axial polydactyly type A restricted to lower limb. Eur J Hum Genet 25:960–965. 10.1038/ejhg.2017.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umm-e-Kalsoom, Basit S, Kamran-ul-Hassan Naqvi S et al. (2012) Genetic mapping of an autosomal recessive postaxial polydactyly type A to chromosome 13q13.3-q21.2 and screening of the candidate genes. Hum Genet 131:415–422. 10.1007/s00439-011-1085-7 [DOI] [PubMed] [Google Scholar]
- Verma PK, El-Harouni AA (2015) Review of literature: genes related to postaxial polydactyly. Front Pediatr 3:8 10.3389/fped.2015.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:e164 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson BT, Hennrikus WL (1997) Postaxial type-B polydactyly. Prevalence and treatment. J Bone Jt Surg Am 79:65–68 [DOI] [PubMed] [Google Scholar]
- Yang Y, Niswander L (1995) Interaction between the signaling molecules WNT7a and SHH during vertebrate limb development: dorsal signals regulate anteroposterior patterning. Cell 80:939–947 [DOI] [PubMed] [Google Scholar]
- Yeo G, Burge CB (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11:377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.