

Abstract
Inflammatory bowel disease and colonic tumors induced by Helicobacter hepaticus (Hh) infection in susceptible mouse strains are utilized to dissect the mechanisms underlying similar human diseases. In our study, infection with genotoxic cytolethal distending toxin-producing Hh in 129/SvEv Rag2−/−Il10−/− gpt delta (RagIl10gpt) mice of both sexes for 21 weeks induced significantly more severe cecal and colonic pathology compared to uninfected controls. The mutation frequencies in the infected RagIl10gpt males were 2.1-fold higher for the cecum and 1.7-fold higher for the colon than male RagIl10gpt controls. In addition, there was a 12.5-fold increase of G:C-to-T:A transversions in the colon of Hh-infected males compared to controls. In contrast, there was no statistical significance in mutation frequencies between infected female Rag2Il10gpt mice and controls. Moreover, Hh infection in RagIl10gpt males significantly up-regulated transcription of Tnfα and iNos, and decreased mRNA levels of cecal Atm compared to the infected females; there was no significant difference in mRNA levels of Il-22, Il-17A, Ifnγ and Atr between the infected males and females. Significantly higher levels of cecal and colonic iNos expression and γH2AX-positive epithelial cells (a biomarker for double-strand DNA breaks [DSB]) in Hh-infected Rag2Il10gpt males vs. Hh-infected females were noted. Finally, Hh infection and associated inflammation increased levels of intestinal mucosa-associated genotoxic colibactin-producing pks+ Escherichia coli. Elevated Tnfα and iNos responses and bacterial genotoxins, in concert with suppression of the DSB repair responses, may have promoted mutagenesis in the lower bowel mucosa of Hh-infected male RagIl10gpt mice.
Keywords: colitis, mutation promotion, iNos expression, DSBs, H. hepaticus
Introduction
Chronic inflammation and genomic mutations are two of the hallmarks of cancer.1 Colorectal cancer (CRC) is usually associated with somatic genomic mutations; colitis-associated cancer (CAC), a subtype of CRC, develops after inflammatory bowel disease (IBD), such as Crohn’s disease and ulcerative colitis.2 A growing body of evidence indicates that chronic inflammation is a significant risk factor for several major human malignancies, including CAC.3 Although the underlying mechanisms of how chronic inflammation contributes to tumorigenesis are incompletely understood, it has been proposed that chronic inflammation stimulates overproduction of reactive oxygen and nitrogen species (RONS) by inflammatory cells, which can lead to point mutations in the host genome. Such mutations are considered a critical component for tumor initiation in cells of inflamed tissues.3–5 Additionally, an increased inflammatory cell population creates a cytokine milieu favoring cell proliferation.3
Approximately 15% of malignancies worldwide can be attributed to infection.6 Helicobacter pylori infection causes gastric inflammation and is a strong risk factor for the development of gastric cancer.7 Several enterohepatic Helicobacter species (EHS) including H. cinaedi, H. fennelliae and H. pullorum have been isolated from patients with IBD.8–10 H. hepaticus, the prototype of EHS and a genetically close relative of H. pylori, has the cdt operon which encodes cytolethal distending toxin (CDT) with DNAase I-like activity. CDT is also present in multiple pathogenic gram-negative bacteria including Campylobacter jejuni, select Escherichia coli strains and Salmonella Typhi.11 H. hepaticus infection causes IBD and colon cancer in susceptible mouse strains; these infectious models have been widely used to delineate etiopathogenesis of IBD and intestinal carcinogenesis.12 Inactivation of H. hepaticus CDT attenuated H. hepaticus-induced colitis accompanied by a reduction of double-strand DNA breaks in 129/SvEv Rag2−/− mice.13 The mechanisms underlying the role of H. hepaticus in promoting intestinal tumorigenesis have been extensively characterized.12 However, whether H. hepaticus infection initiates the intestinal tumorigenesis remains poorly understood. Additionally, select strains of E. coli carry a genomic pks (polyketide synthase) island that encodes a genotoxin colibactin; this toxin induces double-strand DNA breaks (DSBs) and cell cycle arrest in mammalian cells.14 It has been documented that pks+ E. coli infection promotes intestinal carcinogenesis in A0M/Il10-deficient 129/SvEv mice and there is higher prevalence in patients with colonic cancer compared to healthy controls.14
A λ-EG10-based transgenic C57BL/6 mouse model was specifically designed to facilitate in vivo detection of genomic point mutations and large deletions.15 It has been estimated by whole genome sequencing that C57BL/6 gpt delta mice carry tandem arrays of approximately 40 copies of the bacterial guanine phosphoribosyltransferase gene (gpt) at a single site on chromosome 17.16 In this experimental in vivo system, point mutations can be detected and quantified by 6-thioguanine (6-TG) selection, whereas deletions up to 10 kb in length are characterized by selection based on sensitivity to P2 interference (Spi−).15 We previously demonstrated that H. pylori SS1 infection in C57BL/6 gpt delta mice promoted point mutation frequencies in gastric mucosa in females but not in males compared to uninfected controls, whereas no difference in large deletions was noted.5 These findings support previous studies documenting that female C57BL/6 mice are more susceptible to inflammatory response induced by Helicobacter infection.17 Recently, B6C3F1 gpt delta mice, a F1 hybrid of the C57BL/6 gpt delta and C3H mouse strains, have been utilized to characterize high resolution mutational spectra in aflatoxin B1-induced liver tumors by comparing sequences of a 6.4 kb plasmid recovered from λ-EG10 using duplex sequencing.18 Our previous studies showed that H. hepaticus infection induced intestinal tumorigenesis in 129/SvEv Rag2−/− mice without mature T and B lymphocytes due to deficiency in recombination activating 2 gene (Rag2); the inhibition of H. hepaticus-induced pathology in this mouse strain requires the adoptive transfer of Il-10 (an inflammation-suppressive cytokine)-competent regulatory T cells.4,19 In our study, we generated 129/SvEv Rag2−/−Il10−/− gpt delta (RagIl10gpt) mice to determine if H. hepaticus infection increased mutation frequency in the host genome. We focused on accumulation of point mutations in the lower bowel in RagIl10gpt male and RagIl10gpt female mice. This approach was utilized because we previously detected no differences in genomic deletion frequencies between H. pylori SS1-infected C57BL/6 gpt delta mice and their controls.5 H. hepaticus infection in RagIl10gpt mice caused colitis at 21 weeks postinoculation, whereas loss-of-function mutations in the gpt gene were used to calculate mutation frequencies in the intestinal tissues of H. hepaticus-infected and control mice. We also characterized histopathologic changes, expression of inflammatory cytokines and genes involved in DNA damage responses, epithelial iNos expression, γH2AX foci+ epithelial cells and H. hepaticus colonization levels in the intestinal mucosa. Moreover, the influence of H. hepaticus infection on colonization dynamics of indigenous pks+ E. coli in RagIl10gpt mice was examined.
Materials and Methods
Animals, bacterial strain and experimental infections
Generation of RagIl10gpt mice and the animal husbandry were detailed in the Supporting Information. Five-week-old RagIl10gpt mice of both sexes were orally gavaged every other day with three doses of 0.2 ml (~2 × 108 organisms) of H. hepaticus 3B1 (ATCC 51449) in Brucella broth. The respective control mice were gavaged orally with vehicle only. At 21 weeks postinoculation with H. hepaticus (WPI), all the mice, including an uninfected control group (7 males, 7 females) and a H. hepaticus-infected group (7 males, 8 females), were euthanized with CO2.
Necropsy and histopathology
At necropsy, cecum including the ileo-ceco-colic junction and colon (proximal, mid and distal) were collected for routine histological processing and sectioning. Hematoxylin and eosin of intestine were examined by a veterinary pathologist (SM) blinded to sample identity. The lower bowel tissues were graded with scores of ascending 1–4 for inflammation, edema, epithelial defects, crypt atrophy, hyperplasia and dysplasia as previously described.4 An intestinal histologic activity index (HAI) was generated by combining all categorical lesion scores for a particular intestinal segment.4
DNA isolation and in vitro packaging
Genomic DNA was extracted from cecal and colonic tissues using RecoverEase DNA Isolation Kit (Stratagene, San Diego, CA) following the manufacturer’s recommendations. λ-EG10 phages were packaged in vitro from genomic DNA using the Transpack Packaging Extract Kit (Stratagene).
gpt assay and sequencing analysis
The 6-TG selection assay was performed as previously described with minor modifications.5,15 The modifications were described in the Supporting Information. Mutations were classified as transitions, transversions, deletions, insertions or complex (multiple changes). Duplicate mutations at the same site within an individual tissue, considered clonal expansion of sibling mutations, were excluded. Mutation frequencies (MF) were obtained from 22 cecal tissues (control group: 5 males, 4 females; H. hepaticus-infected group: 7 males, 6 females) and 27 colonic tissues (control group: 7 males, 5 females; H. hepaticus-infected group: 7 males, 8 females), which were expressed as gpt mutants per 106 CmR E. coli colonies.
qPCR for H. hepaticus and pks+ E. coli
qPCR assays for quantifying H. hepaticus and pks+ E. coli were performed in the 7,500 Fast Real-Time PCR system (Life Technologies, Carlsbad, CA). Genome copy numbers of H. hepaticus or pks + E. coli were expressed per microgram of murine chromosomal DNA, which was measured by qPCR using a mammalian 18S rRNA gene-based primer and probe mixture (Thermo Fisher Scientific, Waltham, MA) as described previously.13 Additional information is described in the Supporting Information.
qPCR analyses on intestinal cytokines
Total RNA from murine cecal tissues was prepared using Trizol Reagents, and cDNA from tissue mRNA (2 μg) was reverse-transcribed using the High Capacity cDNA Archive kit (Life Technologies, Foster City, CA). mRNA expression of murine genes involved in innate immunity and oncogenesis, including Ifnγ, Tnfα, iNos, Il-17A, Il-22, Atm and Atr, was measured using primers and probes from Life Technologies and expressed as fold change in reference to sham-dosed control mice. Additional information is described in detail in the Supporting Information Methods and Results.
Immunohistochemistry
Formalin-fixed sections of cecum and colon (4 μm thickness) were processed and stained with rabbit antimouse iNos antibody (Abcam, Cat#: ab15323) or rabbit anti-γH2AX monoclonal antibody (Cell Signaling, Cat#: 9718) as previously described.13,20 Additional information is described in the Supporting Information Methods and Results.
Statistical analysis
All statistical analyses were performed using the Prism 5 software Package (GraphPad, San Diego, CA. Values of p < 0.05 were considered significant. More information on statistical analyses is described in the Supporting Information Methods and Results.
Results
Helicobacter hepaticus-infected RagIl10gpt mice developed more severe intestinal pathology of all categorical features compared to their sham controls
The ileo-ceco-colic (cecum/ICC) junction, cecum and proximal/transverse/distal colons of RagIl10gpt mice were assessed for various histopathological criteria, including inflammation, edema, epithelial defects, atrophy, hyperplasia and dysplasia, and the HAI scores for the individual mice were generated. At 21 WPI, the scores of HAI in all the different segments evaluated were significantly higher in H. hepaticus-infected RagI10gpt mice vs uninfected controls (Fig. 1, p < 0.05 or less). This was manifested by moderate histiocytic and granulocytic predominant inflammation, mild epithelial defects including surface tethering and crypt degeneration/abscess, mild mucosal and submucosal edema, minimal crypt loss/atrophy, mild to moderate epithelial hyperplasia and mild to moderate dysplasia characterized by loss of epithelial mucin, cytological atypia and architectural abnormalities. There was no significant difference in the HAI scores between H. hepaticus-infected males and females (Fig. 1, p > 0.5).
Figure 1.
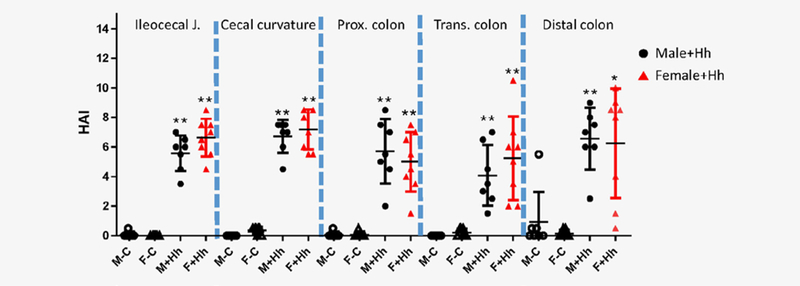
HAI scores in the lower bowel of RagIl10gpt mice at 21 WPI. Bars represent means ± standard deviation of the data of the respective groups (the group of control males, H. hepaticus-infected males or control females: n = 7; the group of infected female group: n = 8). The HAI scores were an aggregate of inflammation, epithelial defects, crypt atrophy, hyperplasia and dysplasia. All H. hepaticus-infected mice had higher HAI core of all the evaluated intestinal segments than those in uninfected controls. However, the HAI scores were comparable between the infected males and females. *represents statistical significance p values when compared to the uninfected controls: *<0.05, **<0.01.
Helicobacter hepaticus infection significantly increased mutagenic potency in the large intestine of RagIl10gpt males but not females
Rag2−/−ll0−/− mice experimentally infected with H. hepaticus, develop colonic carcinoma.4 We sought to characterize whether H. hepaticus infection in Rag2ll0gpt promotes mutagenic potency. The frequency of gpt point mutations in cecal and colonic DNA at 21 WPI was determined by sequencing all mutants selected based on 6-TG resistance. To rule out possible confounding effects of clonal expansion, all recovered mutants were sequenced. Any mutation replicating another at the same site within an individual sample was excluded from the subsequent frequency calculation. After this adjustment, a total of 243 gpt-mutated sequences were used to calculate mutation frequencies. The gpt mutation frequencies in the H. hepaticus-infected males were significantly higher in both the ceca (12.13 ± 7.97 × 10−6, p = 0.036) and colon (12.73 ± 3.68 X 10−6; p = 0.026) compared to those in control counterparts (5.57 ± 6.7 × 10−6 for the ceca, 8.07 ± 6.63 × 10−6 for the colons; Fig. 2a). In contrast, there was no significant difference in both cecal and colonic mutation frequencies between infected females and their controls. These results indicated that H. hepaticus-induced promotion of large intestine genomic point mutations was male-biased.
Figure 2.
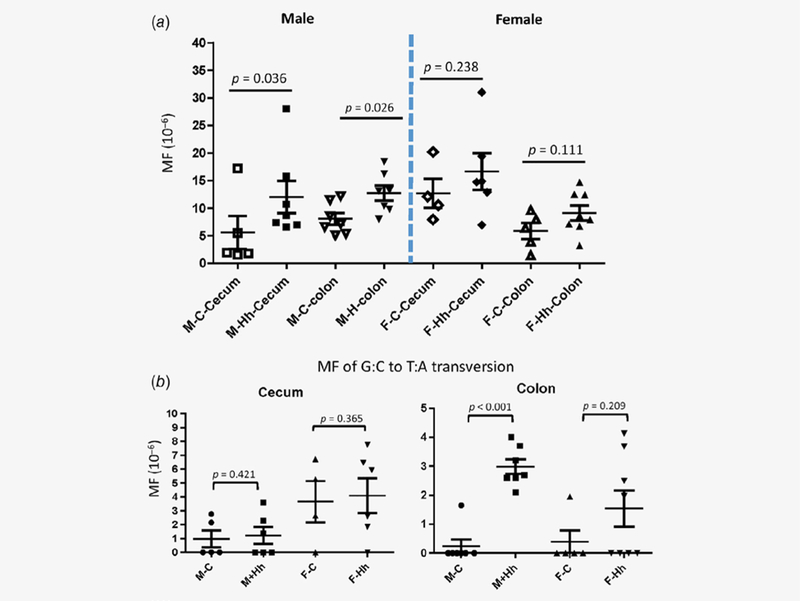
Mutation frequencies (MFs) in H. hepaticus-infected RagIl10gpt mice and uninfected controls at 21 WPI. MFs were determined using the gpt assay as described in the Materials and Methods. (a) The overall MFs. H. hepticus infection significantly increased MFs in the cecum (p = 0.036) and in the colon (p = 0.026) in males but not in females vs. their respective controls. (b) MFs of G:C-to-T:A transversion. The MF of colonic G:C-to-T:A transversion increased by 12.5-fold in the infected males over the uninfected controls. The data are presented as mean ± standard error.
Helicobacter hepaticus infection increased frequency of transversion mutations in the lower bowel of RagIl10gpt males compared to uninfected controls
For RagIl10gpt males, point mutations represented 72–88% of the total mutations detected (Supporting Information Fig. S1A). H. hepaticus infection increased the frequency of transversions (63.2% for cecum, 42.6% for colon) associated with a decrease in the frequency of transitions (24% for cecum and 29.7 for colon) when compared to the frequency of transversions (39.6% for cecum, 23.2% for colon) and the frequency of transitions (34.6% for cecum and 51.3% for colon) in uninfected males, respectively. For RagIl10gpt females, point mutations represented 83–95% of the total mutations (Supporting Information Fig. S1B). In the cecum, H. hepaticus infection slightly increased the frequency of transversion mutations (51.4%) associated with a decrease of the frequency of transition mutations (37.8%) compared to those (46.9% for transversions and 47.9% for transitions) in the controls; in the colon, H. hepaticus infection did not significantly alter the overall spectrum of mutations when compared to the uninfected females (Supporting Information Fig. S1B).
To further identify which type(s) of genomic mutations were significantly elevated, the overall MFs in the lower bowel of H. hepaticus-infected RagIl10gpt males, the means of MFs for each type of mutations in the individual groups were calculated and summarized in Table 1. The mean of the MFs of G:C to T: A (G>T) transversions in the H. hepaticus-infected RagIl10gpt males increased by 12.5-fold in the colon when compared to that in the uninfected controls (Table 1, Fig. 2b, 2p = 0.0007). The nearly fourfold elevation of the MF of G>T was also detected in the colons of H. hepaticus-infected RagIl10gpt females vs. their controls but without statistical significance (Fig. 2b, 2p = 0.173). In contrast, there were no statistical differences in the MFs of G>T in the ceca of both H. hepaticus-infected RagIl10gpt males and females compared to their respective controls (Fig. 2b). It is also worth noting that the average MF of A:T to C:G trans-versions was higher by eightfold in the ceca of H. hepaticus-infected RagIl10gpt males over the uninfected controls but did not reach statistical significance (Table 1).
Table 1.
H. hepaticus infection elevates the mutation frequency (×10−6) of G>T transversion in the colon of male mice
| Rag2−/−Il10−/− gpt129 mice | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cecum | Colon | |||||||
| M-Control | M-Hh | F-Control | F-Hh | M-Control | M-Hh | F-Control | F-Hh | |
| Effect | (n = 5) | (n = 7) | (n = 4) | (n = 6) | (n = 7) | (n = 7) | (n = 5) | (n = 8) |
| Transition | ||||||||
| G:C to A:T | 0.89 (1.13) | 2.31 (1.91) | 4.36 (2.37) | 4.94 (4.34) | 3.15 (2.66) | 2.9 (3.16) | 2.92 (1.94) | 4.55 (1.68) |
| A:T to G:C | 1.04 (1.86) | 0.61 (0.97) | 1.73 (2.17) | 1.29 (3.16) | 0.98 (1.62) | 0.83 (1.13) | 0.48 (1.07) | 1.44 (1.73) |
| Transversion | ||||||||
| G:C to T:A | 0.98 (1.36) | 1.24 (1.37) | 3.66 (2.97) | 4.10 (3.05) | 0.24 (0.58) | 3.00 (0.67)1 | 0.39 (0.97) | 1.54 (1.77) |
| G:C to C:G | 0.61 (0.94) | 0.29 (0.51) | 0.56 (1.12) | 2.15 (1.88) | 0.43 (0.95) | 0.35 (0.93) | 0.00 | 0.25 (0.70) |
| A:T to T:A | 0.00 | 0.92 (1.65) | 1.73 (2.17) | 1.07 (2.21) | 0.57 (0.83) | 1.85 (1.87) | 1.08 (1.74) | 0.22 (0.62) |
| A:T to C:G | 0.62 (0.95) | 5.22 (10.37) | 0.00 | 1.17 (1.70) | 0.64 (1.43) | 0.15 (0.41) | 0.00 | 0.48 (0.94) |
| Deletion | ||||||||
| 1 bp deletion | 1.44 (2.82) | 1.44 (1.74) | 0.66 (1.32) | 0.65 (1.58) | 2.0 (2.9) | 2.37 (2.25) | 0.51 (1.14) | 0.87 (1.73) |
| ≥2 bp deletion | 0.00 | 0.00 | 0.00 | 0.00 | 0.34 (0.76) | 0.18 (0.46) | 0.48 (1.07) | 0.00 |
| Insertion | 0.00 | 0.00 | 0.00 | 1.14 (1.79) | 0.00 | 0.00 | 0.00 | 0.00 |
| Complex mutation | 0.00 | 0.10 (0.27) | 0.00 | 0.145 (0.35) | 0.00 | 0.93 (1.25) | 0.00 | 0.22 (0.63) |
| Total | 5.57 (6.71) | 12.13 (7.97)2 | 12.7 (5.29) | 16.65 (8.11) | 8.07 (6.63) | 12.73 (3.68)3 | 5.86 (3.26) | 9.15 (3.78) |
Data are means (SD) of mutation frequencies of the individual mutation type in each group.
p < 0.001 vs. male controls.
p = 0.036 vs. male controls.
p = 0.026 vs. male controls.
Helicobacter hepaticus infection significantly elevated transcription of Tnfα and iNos and increased epithelial iNos expression in RagIl10gpt males compared to RagIl10gpt females
We previously demonstrated that several proinflammatory mediators such as nitric oxide (NO), Tnfα, Il-17 and Il-22 play an important role in inducing intestinal inflammation and cancer in H. hepaticus-infected 129/SvEv Rag2−/− or 129/SvEv Rag2−/−~Il10−/− mice.4,20 To explore the possible signaling pathways underlying the male-biased promotion of mutation frequency in RagIl10gpt mice, we measured and compared mRNA levels of cecal Il-17A, Il-22, Tnfα, iNos and Ifnγ among the groups. The mRNA levels of all these genes were significantly higher in infected mice of both sexes compared to their sham controls (Fig. 3). Infected males expressed significantly higher mRNA levels of cecal Tnfα and iNos when compared to infected females, whereas there were no significant differences in mRNA levels of cecal Il-17A, Il-22 and Ifnα between H. hepaticus-infected males vs. females.
Figure 3.
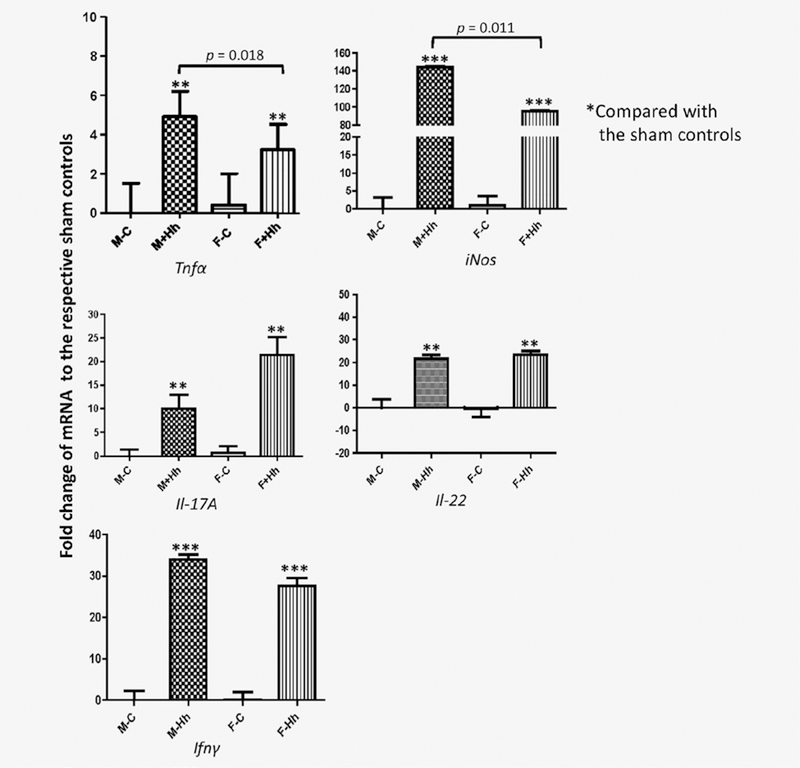
Relative mRNA levels of select cecal genes contributing to inflammation and carcinogenesis in RagIl10gpt mice. Significant transcriptional differences were found for all five genes tested in H. heapticus-infected mice irrespective of sex when compared to uninfected controls. Importantly, mRNA levels of cecal Tnfα and iNos in the infected males were significantly higher than those in the infected females. For the comparison of mRNA, the target mRNA was normalized to that of the “house-keeping” gene Gapdh. Numbers on the y-axis represent mean fold change of the individual mRNA levels in reference to the control group (defined as 0). Bars, standard deviations. *represents p values when compared to the sham, *<0.05, **<0.01, ***<0.001.
To correlate mRNA levels of iNos with epithelial iNos expression in the lower bowel of RagIl10gpt mice, we quantitatively examined levels of epithelial iNos expression in the ceca and the colon using IHC (Fig. 4). Expression of cecal and colonic iNos in the uninfected controls of both sexes was minimal and sporadic along the epithelium, whereas much stronger fluorescence signal and overall distribution on the epithelial surface and crypts were detected in H. hepaticus-infected mice (Figs. 4a and 4b). In addition, the index of epithelial iNos expression in the ceca and colon were significantly higher in H. hepaticus-infected RagIl10gpt males than those in H. hepaticus-infected RagIl10gpt females, indicating that H. hepaticus infection increased iNos expression in the lower bowel of the males compared to the females (Fig. 4c, 4p = 0.0197 and 0.0037 for the cecum and colon, respectively).
Figure 4.
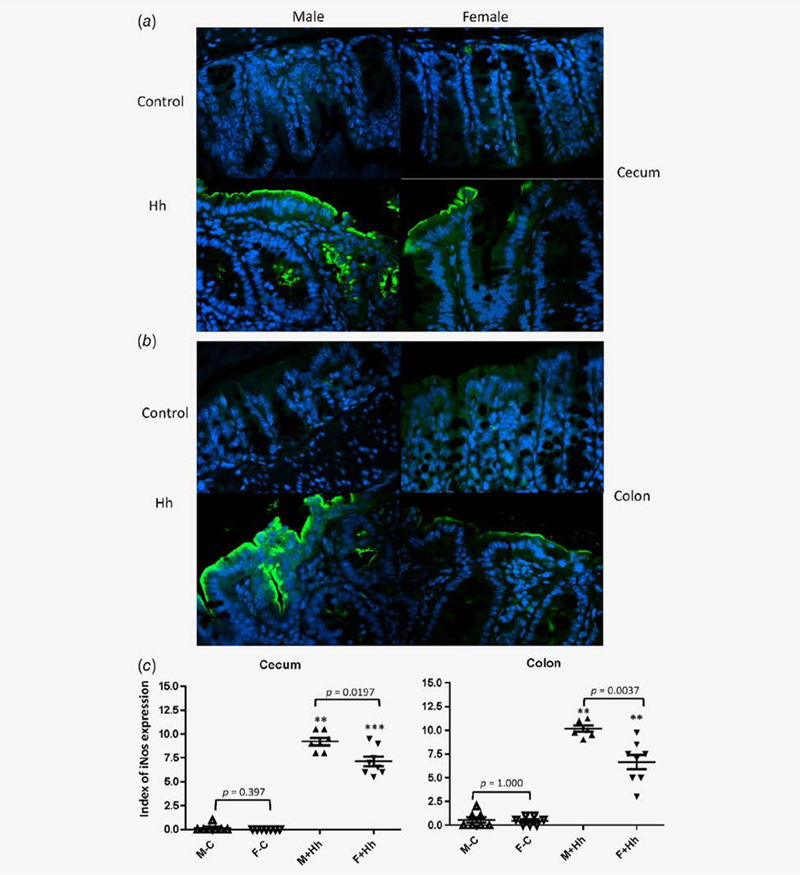
Epithelial iNos expression was significantly elevated in the lower bowel of H. hepaticus-infected RagIl10gpt males vs. H. hepaticus- infected RagIl10gpt females. The representative iNos staining images: the ceca (a) and colons (b). (c) Index of iNos epithelial cells in the lower bowel of RagIl10gpt mice. H. hepaticus infection significantly elevated iNos expression compared to the uninfected controls. *represents p values when compared to the sham controls: **<0.01, ***<0.001.
Helicobacter hepaticus infection significantly elevated epithelial γ-H2AX foci+ cells in concert with transcriptional downregulation of Atm in the lower bowel of RagIl10gpt males compared to RagIl10gpt females
It has been documented that H. hepaticus infection in Rag2-deficient mice stimulates iNos expression, which is considered an important mechanism for promoting DNA double-strand breaks (DSBs) and intestinal carcinogenesis.4,20 We sought to assess patterns of γ-H2AX foci positivity, the most sensitive marker for detecting DSBs, and the subsequent DNA damage repair response (DDR) in the lower bowel epithelial cells of RagIl10gpt mice. Uninfected RagIl10gpt controls contained only a few γ-H2AX foci+ epithelial cells, which were sporadically distributed in the cecal and colonic mucosa (Fig. 5a and 5b). In contrast, γ-H2AX foci+ epithelial cells in H. hepaticus-infected RagIl10gpt mice of both sexes had stronger fluorescence intensity, broad epithelial distribution, and were frequently clustered in the crypts compared to the uninfected controls (Fig. 5a and 5b). There was a significantly higher index of γ-H2AX foci+ epithelial cells in the lower bowel of the H. hepaticus-infected RagIl10gpt males compared to H. hepaticus-infected RagIl10gpt females (Fig. 5c, 5p = 0.0061 and 0.0014 for the cecum and colon, respectively).
Figure 5.
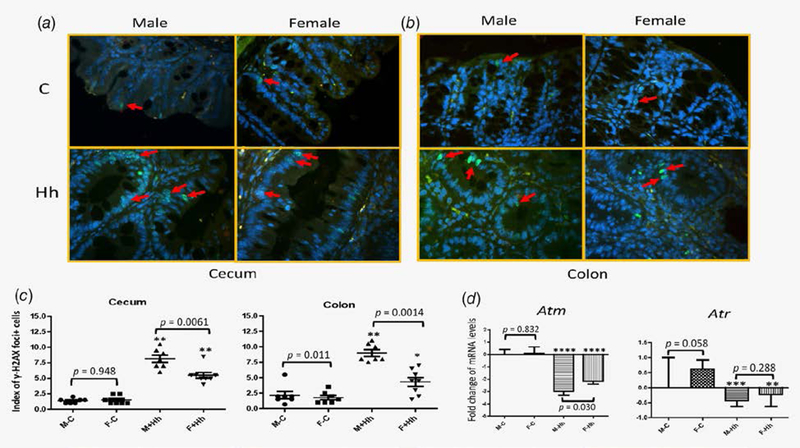
Levels of γH2AX foci+ epithelial cells and transcription of Amt were significantly increased in the lower bowel of H. hepaticus-infected RagIl10gpt males vs. H. hepaticus-infected RagIl10gpt females. The representative γH2AX staining images: the ceca (a) and colons (b). Representative γH2AX foci+ epithelial cells are denoted by red arrows. (c) Index of γH2AX foci+ epithelial cells. H. hepaticus infection significantly elevated the degrees of γH2AX foci+ epithelial cells compared to the uninfected controls. (d) Relative mRNA levels of cecal Atm and Atr. The mRNA levels of Atm and Atr were significantly increased in H. heapticus-infected mice irrespective of sex when compared to uninfected controls. The target mRNA was normalized to that of the “house-keeping” gene Gapdh. Numbers on the y-axis represent mean fold change of the individual mRNA levels in reference to the control group (defined as 0). Bars, standard deviations. *represents p values when compared to the sham controls: *<0.05, **<0.01, ***<0.001, ****<0.0001.
Ataxia-telangiectasia-mutated (ATM) and ATR (ATM- and RAD3-related) are serine/threonine kinases that belong to the superfamily of phosphatidylinositol 3-kinase-related kinases.21 ATM and ATR are recruited and activated by DNA DSBs and DNA single-strand breaks (SSBs), respectively, and then initiate activation of DNA damage repair responses via phosphorylation of tumor suppressors such as p53, CHK1 and CHK2.21 To shed light on the possible mechanisms underlying the differences in levels of γ-H2AX foci+ epithelial cells in the lower bowel between H. hepaticus-infected RagIl10gpt mice and the controls or between H. hepaticus-infected males and females, we measured mRNA levels of cecal Atm and Atr using qPCR (Fig. 5d). H. hepaticus infection significantly suppressed transcription of both Atm and Atr in RagIl10gpt mice of both sexes compared to the uninfected controls (Fig. 5d). In addition, there were significantly lower mRNA levels of Atm in H. hepaticus-infected RagIl10gpt males compared to H. hepaticus-infected RagIl10gpt females (p = 0.03), whereas mRNA levels of Atr were comparable between these two groups (p = 0.288; Fig. 5d).
Helicobacter hepaticus colonization levels were comparable between males and females
To test whether disparity of mutation frequencies, epithelial iNos expression and indices of γ-H2AX foci+ epithelial cells between RagIl10gpt females and males resulted from different H. hepaticus colonization levels, H. hepaticus colonization levels in the cecum and colon of infected RagIl10gpt mice were quantified using qPCR. There was a significantly higher H. hepaticus colonization level in the cecum compared to that in the colon (Supporting Information Fig. S2), However, H. hepaticus colonization levels were comparable between infected males and females. This result demonstrated that the difference in mutation frequencies, epithelial iNos expression and the development of γ-H2AX foci+ epithelial cells between H. hepaticus-infected RagIl10 gpt females and males were not ascribed to bacterial colonization levels.
Helicobacter hepaticus infection significantly increased the population of cecal, colonic and fecal pks+ E. coli
Select strains of E. coli produce colibactin, which is encoded by the pks island and can induce DNA double-strand breaks (DSBs) in eukaryotic cells.14 Given that our recent study demonstrated that pks+ E. coli strains are prevalent in several commercial mouse strains,22 we investigated whether H. hepaticus infection influences growth of pks+ E. coli. Because there were no significant differences in pks+ E. coli levels between sexes, mice were grouped as infected and controls for comparative analyses. At 21 WPI, H. hepaticus infection significantly increased cecal (p = 0.013), colonic (p = 0.0007) and fecal pks+ E. coli abundance (p < 0.0001) compared to that in uninfected controls (Fig. 6).
Figure 6.
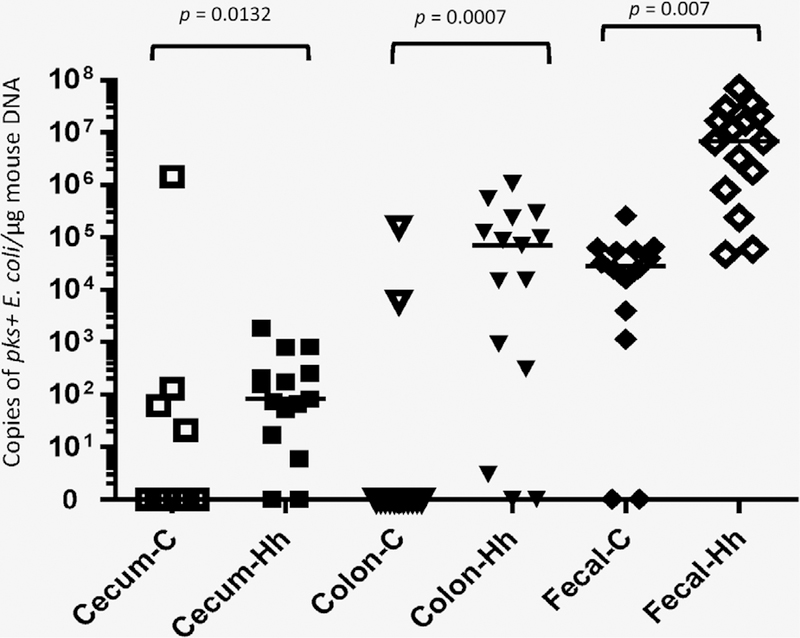
Influence of H. hepaticus infection on the levels and prevalence of pks+ E. coli in the lower bowel and feces at 21 WPI. H. hepaticus infection significantly elevated the level of fecal pks+ E. coli and also increased the prevalence of mucosa-associated pks+ E. coli in the lower bowel of the infected RagIl10gpt mice. The numbers on the y-axis represent the copies of the H. hepaticus genome expressed per μg murine DNA in the samples.
Discussion
Helicobacter hepaticus infection induces IBD and intestinal tumors in susceptible mouse strains such as Rag2- and Il-10-deficient mice.12 The host immune responses and intestinal pathological features caused by H. hepaticus infection recapitulate several features of IBD and intestinal tumorigenesis in humans and thereby have been utilized to dissect etiopathogeneses of these human diseases.12 In our study, we demonstrated that H. hepaticus infection significantly increased genomic MF in the lower bowel of male RagIl10gpt mice but not female RagIl10gpt mice. We have proposed a working model to summarize our findings depicting possible pathways leading to the male-biased promotion of genomic mutation frequencies induced by H. hepaticus infection in RagIl10gpt mice (Supporting Information Fig. S3). Increased mutation frequencies in the H. hepaticus-infected males were not due to levels of H. hepaticus colonization, but rather more likely mediated by enhancing the Tnfα-iNos signaling in RagIl10gpt males vs. females. Elevated iNos expression can enhance DSBs, as indicated by higher levels of γH2AX foci+ epithelial cells, and also directly induce point genomic mutations in the lower bowel of H. hepaticus-infected RagIl10gpt males compared to H. hepaticus-infected RagIl10gpt females.20,23 More DSBs in the H. hepaticus-infected males may also in part be due to stronger suppression of Atm transcription. Increased DSBs can lead to genomic instability and point substitution mutations.24,25 These enhanced genotoxic effects could further promote intestinal tumorigenesis in the H. hepaticus-infected RagIl10gpt males. In addition, H. hepaticus infection increased colonization levels of mucosa-associated pks+ E. coli, which produces genotoxic colibactin, suggesting that H. hepaticus infection not only caused intestinal pathology by itself but also promoted the pathogenic potential of naturally colonized pks+ E. coli.
Epidemiological data suggest that patients with IBD have an increased risk for colon cancer.26 It has been proposed that dysbiosis in intestinal microbial compositions and pathogenic bacterial infections are key factors to trigger inflammation in IBD and may contribute to the development of colon cancer.27 Persistent chronic inflammation can lead to excessive production of RONS by inflammatory cells, which also is in part due to iNos-mediated generation of NO being increased in epithelial cells.20 RONS can induce DNA damage via elevation of DNA adducts such as ethno-adducts, 7,8-dihydro-8-oxoguanine (8-oxoG) and other mutagenic precursors in vitro and in vivo.28,29 Our previous studies indicated that H. hepaticus infection in Rag2−/− mice induced progression of intestinal pathology from inflammation, dysplasia to carcinoma during the duration of infection, and this process is mediated in part by elevated production of NO.4,20 The increased production of NO as evidenced by elevation of urinary nitrate excretion was correlated with H. hepaticus-induced overexpression of Tnfα and iNos primarily in macrophages and lower bowel epithelial cells in concert with neutrophils.4,13,20 Consistent with these previous findings in H. hepaticus-infected Rag2−/− mice, there were significantly higher levels of epithelial iNos expression in the cecum and colon as well as the upregulation of Tnfα and iNos, Il-17A, Il-22 and Ifnγ in H. hepaticus-infected RagIl10gpt mice of both sexes compared to their uninfected controls. Importantly, significant promotion of MF was detected only in the lower bowel of H. hepaticus-infected RagIl10 gpt males but not in H. hepaticus-infected RagIl10gpt females when compared to their respective controls. The male-biased mutation promotion was likely driven by the sufficiently elevated NO production, given that there were higher levels of iNos in the ceca and colon in concert with the transcriptional upregulation of cecal Tnfα and iNos in the H. hepaticus infected RagIl10gpt males compared to the infected RagIl10gpt females. Comparable severity of intestinal pathology between the H. hepaticus-infected males vs. females at 21 WPI suggests a longer duration (>21 WPI) of H. hepaticus infection in this mouse strain is needed to differentiate the differences in the lower bowel pathology between males and females. This premise is supported by our previous study showing that wild-type (WT) H. hepaticus infection in Rag2-deficient mice induced higher levels of cecal Tnfα and γH2AX, but similar severity of intestinal pathology compared to CDT-deficient H. hepaticus mutant infection at 10 WPI; more severe preneoplastic lesions in the lower bowel were noted in the WT H. hepaticus-infected mice compared to the H. hepaticus mutant-infected mice at 20 WPI.13
We demonstrated that cecal and colonic DBSs, indicated by the presence of histone variant γH2AX+ foci (a sensitive and reliable biomarker for DSBs), were also increased in the H. hepaticus infected RagIl10gpt males compared to the infected RagIl10gpt females. This difference was unlikely attributable solely to H. hepaticus CDT activity, since H. hepaticus colonization levels were comparable between RagIl10gpt males and females. Given that infection with the cdtB-defective H. hepaticus mutant induced less epithelial DSBs in concert with a decreased severity of dysplastic lesions compared to CDT-producing H. hepaticus in the lower bowel of Rag2-deficient mice.13 It is plausible to speculate that deficiency in CDT activity could attenuate the ability of H. hepaticus to promote lower bowel MFs in Rag2Il10gpt males. The elevated expression of iNos in the lower bowel of H. hepaticus-infected RagIl10gpt males vs. H. hepaticus-infected RagIl10gpt females at least in part led to the increased levels of DSBs in the H. hepaticus-infected RagIl10gpt males. This premise is in line with the previous finding that Il22-mediated reduction of iNos expression in the lower bowel of H. hepaticus-infected Rag2-defienct mice significantly inhibited the development of DSBs.20 In addition, H. hepaticus infection in RagIl10gpt mice of both sexes significantly decreased mRNA levels of cecal Atm and Atr compared to their controls; Atm and Atr play a pivotal role in repairing DSBs and SSBs, respectively.21 Consistent with this finding, transcriptional downregulation of Atm and Atr was noted in the large bowel of H. hepaticus-infected Rag2-deficient mice at 20 WPI.30 These data suggest that H. hepaticus-induced DNA damage could result partially from suppression of the host DNA damage repair pathways. Lower mRNA levels of cecal Atm in H. hepaticus-infected RagIl10gpt males vs. H. hepaticus-infected RagIl10gpt females may also contribute to their elevated DSBs. Furthermore, Hartung et al. reported that the H. pylori Type IV secretion system-dependent induction of DBSs is mediated by the nucleotide excision repair endonucleases XPF and XPG; this process also requires the activation and subsequent target gene expression of the NF-κB pathway.31 Whether such a mechanism in promoting H. pylori-induced DSBs is operable in H. hepaticus-associated DSBs warrants further investigation. It is generally accepted that unrepaired DBSs in eukaryotic cells can result in genomic instability by generating deletion mutations, extensive loss of heterozygosity, genomic rearrangements and chromosomal translocations with tumorigenic potential.24 Recent studies have shown that DSB-induced DNA replication can also lead to the formation of base pair substitution mutations.25 We postulate that the increased DBSs in H. hepaticus-infected RagIl10gpt males vs. H. hepaticus-infected RagIl10gpt females may have a role in the male-biased promotion of MFs that are predominantly comprised of substitution mutations.
Our data demonstrated that H. hepaticus-induced promotion of MF only occurred significantly in RagIl10gpt males but not in RagIl10gpt females when compared to their respective controls. This finding is consistent with our previous work showing that H. hepaticus infection induced more profound cancer in the lower bowel of Rag2-deficient male mice receiving Il10-deficient regulatory T cells compared to their female counterparts.19 In addition, the male predominant hepatocellular carcinoma was induced by H. hepaticus infection in immune-competent inbred AJ/Cr mice.32 Furthermore, epidemiological data indicate that there is a higher incidence of colorectal cancer in men (1 in 24) than women (1 in 48).33 Sex-based disparity of the colorectal cancer rate in humans suggests that sex hormones have a protective role in the development of this disease, which is supported by preclinical trials with hormone (estrogen) replacement therapy.34 The protective effect of estrogen on H. pylon-induced gastric carcinogenesis in INS-GAS mice has also been reported.35 Although the precise mechanisms underlying the protective effects of estrogen on the development of colon cancer are not completely understood, it has been proposed that estrogen exerts the intracellular effects via two pathways, genomic via the interaction of estrogen receptors (ER) with DNA for regulating gene transcription and nongenomic via the interaction of ER directly with cellular signaling pathway such as protein kinase K, NO and MARK.34 Despite the observation that there was no statistical difference in H. hepaticus-induced intestinal pathology between males and females, pathological effects resulting from a higher mutation rate in males could develop in a longitudinal study such as at 8 months postinfection when a high incidence of intestinal carcinomas in H. hepaticus-infected 129/SvEv Rag2−/− mice are noted.19
Our data showed that H. hepaticus infection in RagIl10gpt males significantly increased transversion mutations, particularly G>T conversions, by 12.5-fold in the colon compared to uninfected controls. In addition, the average MF of G>T was also increased by nearly 4-fold in the colons of H. hepaticus-infected RagIl10gpt females vs. their controls but without statistical significance due to the fact that 4 out of 8 H. hepaticus-infected females did not have colonic G>T mutations, suggesting that elevation of this type of point mutation in the females is also modulated by other yet identified factors in addition to H. hepaticus infection. Several factors have been reported to contribute formation of G>T transversions. An increase of iNOS expression is positively correlated with high prevalence of the G>T trans-versions in human colon tumors.36 Exposure of cultured cells to NO˙, O2− and H2O2 by cocultivation with activated macrophages or to NO delivered by the reactor system leads predominantly to the development of G>T transversions.37 Additionally, G>T can also result from DNA adducts produced by oxidative stress or lipid peroxidation such as 8-oxodG, a major product of oxidative damage to DNA, which causes this transversion by mispairing of the oxidized guanine with adenine during replication.38 This type of transversion was also increased, as one of two H. pylori SS1 infection-associated point mutations, in infected C57BL/6 gpt delta female mice and C57BL/6 Big Blue transgenic male mice infected with H. pylori SS1.5,39 Given that the G>T transversions are promoted by both gastric helicobacters and EHS in different mouse models of infection, in one an adaptive immunity-deficient RagIl10gpt mice for H. hepaticus and in another immune-competent H. pylori-infected B6 mice, we speculate that the G>T transversion may represent a Helicobacter-associated signature point mutation. This hypothesis is supported by the fact that approximately ~50% of orthologs are shared among the genomes of H. pylori and H. hepaticus ATCC 51449 (3B1).40 Interestingly, G>T transversions, a major type of substitution mutations in mutational signature 18 in human cancers, are also predominantly identified in MUTYH-mutated colorectal samples; MUTYH is a front line DNA repair defense against 8-oxoG.41,42 In addition, the COSMIC database (https://cancer.sanger.ac.uk/cosmic/signatures) for human cancer genomic studies associates G>T mutations with oxidative stress in gastric and colon cancers.
Our data indicated that there were differential MFs between the cecum and the colon or between sexes. Notably, H. hepaticus infection significantly increased the MFs of G>T transversion in the colon but not in the cecum of males; instead, H. hepaticus infection elevated the average level of A>C transversion in the cecum of the H. hepaticus-infected males vs. the control males by eightfold. The A>C transversions can be induced by 8-oxo-7,−8-dihydro-GTP which is formed from oxidization of dGTP and also represents one of four mutational signatures in colorectal cancer in humans.37,41 In addition, the average G>T MF was higher in the cecum than that in the colon in the control females as well as that in the cecum of the control males. Such differences are likely influenced by multiple factors since these MFs are widely distributed among the individual mice of the same group. Variations between cecal and colonic MFs of the same sex could be at least partially attributed by differential roles of key mediators involving H. hepaticus-induced inflammation and subsequent carcinogenesis in cecum vs. colon. Morrison et al. reported that neutralization of proinflammatory IL-17A exacerbated H. hepaticus-induced cecal, but not colonic, pathology in anti-Il-10R-treated C57BL/6 mice, whereas depletion of proinflammatory Il-22 attenuated H. hepaticus-induced colonic, but not cecal, pathology.43 In addition, differences in the microbial compositions along the gastrointestinal tract of mice such as C57BL/6 mice, including cecum and colon,44 could also contribute to the inconsistency in MFs or type-specific MFs between the cecum and the colon of RagIl10gpt mice. Furthermore, sex-biased MFs may result from distinct effects of male and female hormones capable of modulating the host immune responses and also the gastrointestinal microbiomes.45
It has been documented that H. hepaticus infection perturbed the mucosa-associated microbial community structure in the lower bowel of C57BL/6 mice by decreasing microbial evenness as evidenced using a terminal restriction fragment length polymorphism assay.46 In addition, elevation of the iNos-mediated nitrate respiration pathway provides a growth advantage to E. coli in the inflamed intestine of DSS-treated C57BL/6 mice.47 Furthermore, the population of pks+ E. coli in the colonic bio-films of patients with familial adenomatous polyposis and ApcMinΔ716/+ mice is enriched by concurrent colonization of Bacteroides fragilis.48 In the current study, H. hepaticus infection increased levels of fecal pks+ E. coli, presumably due to inflamed large bowel of H. hepaticus-infected RagIl10gpt mice. Infection with pks+ E. coli strains compared to their pks-deficient isogenic mutants increased DSBs in epithelial cells as evidenced by elevation of γ-H2AX foci+ and promoted colonic tumorigenesis in AOM/Il10-deficient 129/SvEv and AOM/DSS/C57BL/6 mice.14,49 In addition, pks+ E. coli produces genotoxic colibactin which induces DBSs and genomic instability in mammalian cells.14 Furthermore, a recent study reported that colibactin produced by pks+ E. coli can generate interstrand crosslinks and DNA adducts by alkylating the host DNA in HeLa cells and in germ-free C57BL/6J mice, providing mechanistic insights into the possible role of bacterial genotoxin colibactin in the development of colorectal cancer.50 Thus, the H. hepaticus-promoted increase of intestinal pks+ E. coli colonization could have synergistic tumorigenic potential with H. hepaticus in RagIl10gpt mice. Given that there were no differences in colonization of pks+ E. coli between H. hepaticus-infected RagIl10 gpt males and females, pks+ E. coli was not solely responsible for the male-biased promotion of the MF in this mouse model.
Our finding that H. hepaticus infection promoted an elevated MF in the lower bowel of RagIl10gpt mice in a male-biased manner highlights the mutagenic potential of H. hepaticus infection and highlights a putative role of sex hormones in regulating this effect. Predominant G>T transversions induced by infection with H. hepaticus, which is also the same mutational pattern noted with infection of gastric H. pylori, could arguably represent a mutation biomarker for inflammation-induced infection caused by Helicobacter spp. in mouse models. This effect may have clinical relevance since the G>T transversions are identified as prominent mutation signature in MUTYH-mutated colorectal cases and is also associated with gastric tumors, 79% of which is attributable to H. pylori infection.6,41 An increase of mucosa-associated pks+ E. coli by H. hepaticus-associated inflammation raises the concern that the common intestinal colonization of colibactin-producing E. coli in laboratory mice used to study IBD and colon cancer could potentially confound experimental results.22
Supplementary Material
What’s new?
Infection with Helicobacter hepaticus is associated with diseases of the liver and biliary tract and is a suspected risk factor for colon cancer in humans. How H. hepaticus infection potentially contributes to intestinal tumorigenesis, however, is not fully known. Here, in 129/SvEv Rag2−/−Il10−/− gpt delta mice, H. hepaticus infection with cytolethal distending toxin activity induced a male-biased increase in transversion mutation frequency in the lower bowel. This increase was associated with elevations in epithelial iNos expression and number of γH2AX foci-positive cells, biomarkers for double-strand DNA breaks. H. hepaticus-associated inflammation further increased bowel colonization by indigenous colibactin (genotoxin)-producing E. coli.
Acknowledgements
We would like to thank Alyssa T. Pappa and Parisa Zarringhalam for assistance in the preparation and submission of article, Lenzie Cheaney and Christian Kaufman for technical assistance related to mice breeding and necropsy. Our study was supported in part by NIH grants R01–0D01141, T32–0D010978 (to JGF), P30-ES002109 and P01-CA26731 (to JE and JGF) and R01-CA080024 (to JME).
Grant sponsor: National Institutes of Health;
Grant numbers: P30-ES002109, R01-OD01141, T32-OD010978, P01-CA26731, R01-CA080024
Abbreviations:
- ATM
ataxia telangiectasia-mutated
- ATR
ATM- and RAD3-related
- CDT
cytolethal distending toxin
- CRC
colorectal cancer
- CAC
colitis-associated cancer
- DSB
double-strand DNA breaks
- ER
estrogen receptors
- IBD
inflammatory bowel disease
- MF
mutation frequency
- NO
nitric oxide
- pks
polyketide synthase
- RagIl10gpt
129/SvEv Rag2−/−Il10−/− gpt delta
- RONS
reactive oxygen and nitrogen species
- WPI
weeks postinoculation
- WT
wild-type
Footnotes
Additional Supporting Information may be found in the online version of this article.
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 2.Lasry A, Zinger A, Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat Immunol 2016;17:230–40. [DOI] [PubMed] [Google Scholar]
- 3.Terzic J, Grivennikov S, Karin E, et al. Inflammation and colon cancer. Gastroenterology 2010;138: 2101–2114 e5. [DOI] [PubMed] [Google Scholar]
- 4.Erdman SE, Rao VP, Poutahidis T, et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci USA 2009;106:1027–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheh A, Lee CW, Masumura K, et al. Mutagenic potency of Helicobacter pylori in the gastric mucosa of mice is determined by sex and duration of infection. Proc Natl Acad Sci USA 2010;107:15217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plummer M, de Martel C, Vignat J, et al. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health 2016;4:e609–16. [DOI] [PubMed] [Google Scholar]
- 7.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest 2007;117:60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bohr UR, Glasbrenner B, Primus A, et al. Identification of enterohepatic Helicobacter species in patients suffering from inflammatory bowel disease. J Clin Microbiol 2004;42:2766–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomson JM, Hansen R, Berry SH, et al. Enterohepatic Helicobacter in ulcerative colitis: potential pathogenic entities? PLoS One 2011;6:e17184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chai JN, Peng Y, Rengarajan S, et al. Helicobacter species are potent drivers of colonic T cell responses in homeostasis and inflammation. Sci Immunol 2017;2:eaal5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ge Z, Schauer DB, Fox JG. In vivo virulence properties of bacterial cytolethal-distending toxin. Cell Microbiol 2008;10:1599–607. [DOI] [PubMed] [Google Scholar]
- 12.Fox JG, Ge Z, Whary MT, et al. Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol 2011;4:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ge Z, Feng Y, Ge L, et al. Helicobacter hepaticus cytolethal distending toxin promotes intestinal carcinogenesis in 129Rag2-deficient mice. Cell Microbiol 2017;19:e12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arthur JC, Perez-Chanona E, Muhlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012;338:120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nohmi T, Katoh M, Suzuki H, et al. A new transgenic mouse mutagenesis test system using Spi- and 6-thioguanine selections. Environ Mol Mutagen 1996;28:465–70. [DOI] [PubMed] [Google Scholar]
- 16.Masumura K, Sakamoto Y, Kumita W, et al. Genomic integration of lambda EG10 transgene in gpt delta transgenic rodents. Genes Environ 2015;37:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Court M, Robinson PA, Dixon MF, et al. The effect of gender on Helicobacter felis-mediated gastritis, epithelial cell proliferation, and apoptosis in the mouse model. J Pathol 2003;201:303–11. [DOI] [PubMed] [Google Scholar]
- 18.Chawanthayatham S, Valentine CC III, Fedeles BI, et al. Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc Natl Acad Sci USA 2017;114:E3101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erdman SE, Rao VP, Poutahidis T, et al. CD4(+) CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res 2003;63:6042–50. [PubMed] [Google Scholar]
- 20.Wang C, Gong G, Sheh A, et al. Interleukin-22 drives nitric oxide-dependent DNA damage and dysplasia in a murine model of colitis-associated cancer. Mucosal Immunol 2017;10:1504–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell 2017;66:801–17. [DOI] [PubMed] [Google Scholar]
- 22.Garcia A, Mannion A, Feng Y, et al. Cytotoxic Escherichia coli strains encoding colibactin colonize laboratory mice. Microbes Infect 2016;18: 777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim MY, Wogan GN. Mutagenesis of the supF gene of pSP189 replicating in AD293 cells cocultivated with activated macrophages: roles of nitric oxide and reactive oxygen species. Chem Res Toxicol 2006;19:1483–91. [DOI] [PubMed] [Google Scholar]
- 24.Sakofsky CJ, Malkova A. Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit Rev Biochem Mol Biol 2017;52: 395–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saini N, Ramakrishnan S, Elango R, et al. Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 2013; 502:389–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Triantafillidis JK, Nasioulas G, Kosmidis PA. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res 2009;29:2727–37. [PubMed] [Google Scholar]
- 27.Lane ER, Zisman TL, Suskind DL. The microbiota in inflammatory bowel disease: current and therapeutic insights. J Inflamm Res 2017;10:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meira LB, Bugni JM, Green SL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest 2008;118:2516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawanishi S, Ohnishi S, Ma N, et al. Nitrative and oxidative DNA damage in infection-related carcinogenesis in relation to cancer stem cells. Genes Environ 2016;38:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mangerich A, Knutson CG, Parry NM, et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci USA 2012;109:E1820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartung ML, Gruber DC, Koch KN, et al. H. pylori-induced DNA Strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-kappaB target gene expression. Cell Rep 2015;13:70–9. [DOI] [PubMed] [Google Scholar]
- 32.Fox JG, Dewhirst FE, Tully JG, et al. Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J Clin Microbiol 1994;32:1238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and National Cancer Incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol 2017;3:524–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barzi A, Lenz AM, Labonte MJ, et al. Molecular pathways: estrogen pathway in colorectal cancer. Clin Cancer Res 2013;19:5842–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohtani M, Ge Z, Garcia A, et al. 17 beta-estradiol suppresses Helicobacter pylori-induced gastric pathology in male hypergastrinemic INS-GAS mice. Carcinogenesis 2011;32:1244–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ambs S, Bennett WP, Merriam WG, et al. Relationship between p53 mutations and inducible nitric oxide synthase expression in human colorectal cancer. J Natl Cancer Inst 1999;91:86–8. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki T, Kamiya H. Mutations induced by 8-hydroxyguanine (8-oxo-7,8-dihydroguanine), a representative oxidized base, in mammalian cells. Genes Environ 2017;39:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wood ML, Esteve A, Morningstar ML, et al. Genetic effects of oxidative DNA damage: comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine in Escherichia coli. Nucleic Acids Res 1992;20:6023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Touati E, Michel V, Thiberge JM, et al. Chronic Helicobacter pylori infections induce gastric mutations in mice. Gastroenterology 2003;124: 1408–19. [DOI] [PubMed] [Google Scholar]
- 40.Suerbaum S, Josenhans C, Sterzenbach T, et al. The complete genome sequence of the carcinogenic bacterium Helicobacter hepaticus. Proc Natl Acad Sci USA 2003;100:7901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pilati C, Shinde J, Alexandrov LB, et al. Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J Pathol 2017;242:10–5. [DOI] [PubMed] [Google Scholar]
- 43.Morrison PJ, Ballantyne SJ, Macdonald SJ, et al. Differential requirements for IL-17A and IL-22 in cecal versus colonic inflammation induced by Helicobacter hepaticus. Am J Pathol 2015;185:3290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gu S, Chen D, Zhang JN, et al. Bacterial community mapping of the mouse gastrointestinal tract. PLoS One 2013;8:e74957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Org E, Mehrabian M, Parks BW, et al. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016;7:313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuehl CJ, Wood HD, Marsh TL, et al. Colonization of the cecal mucosa by Helicobacter hepaticus impacts the diversity of the indigenous microbiota. Infect Immun 2005;73:6952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winter SE, Winter MG, Xavier MN, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 2013;339:708–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dejea CM, Fathi P, Craig JM, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018;359:592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cougnoux A, Delmas J, Gibold L, et al. Small-molecule inhibitors prevent the genotoxic and protumoural effects induced by colibactin-producing bacteria. Gut 2016;65:278–85. [DOI] [PubMed] [Google Scholar]
- 50.Wilson MR, Jiang Y, Villalta PW, et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019;363:eaar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.