

Abstract
α-Synuclein (α-syn), particularly in its aggregated forms, is implicated in the pathogenesis of Parkinson's disease and other related neurological disorders. However, the normal biology of α-syn and how it relates to the aggregation of the protein are not clearly understood. Because of the lack of the signal sequence and its predominant localization in the cytosol, α-syn is generally considered exclusively an intracellular protein. Contrary to this assumption, here, we show that a small percentage of newly synthesized α-syn is rapidly secreted from cells via unconventional, endoplasmic reticulum/Golgi-independent exocytosis. Consistent with this finding, we also demonstrate that a portion of cellular α-syn is present in the lumen of vesicles. Importantly, the intravesicular α-syn is more prone to aggregation than the cytosolic protein, and aggregated forms of α-syn are also secreted from cells. Furthermore, secretion of both monomeric and aggregated α-syn is elevated in response to proteasomal and mitochondrial dysfunction, cellular defects that are associated with Parkinson's pathogenesis. Thus, intravesicular localization and secretion are part of normal life cycle of α-syn and might also contribute to pathological function of this protein.
Keywords: synuclein, Parkinson's disease, exocytosis, protein aggregation, vesicle, neurodegeneration
Introduction
Parkinson's disease (PD) is a major neurodegenerative disease of aging, pathologically characterized by selective loss of dopamine neurons in substantia nigra and aggregated protein deposits called Lewy bodies (LB) (Forno, 1996; Dauer and Przedborski, 2003). Fibrillar aggregates of α-synuclein (α-syn) constitute a major component of these intracellular inclusions and are also found in many other neurological disorders, including dementia with Lewy bodies and multiple system atrophy (Goedert, 2001). Missense and gene multiplication mutations in α-syn gene are linked to familial forms of PD (Polymeropoulos et al., 1997; Kruger et al., 1998; Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibanez et al., 2004; Zarranz et al., 2004) and result in an acceleration of pathogenic aggregation of this protein (Conway et al., 2000; Choi et al., 2004), supporting the importance of α-syn aggregation in the pathogenesis of PD. Although animal models with transgenic overexpression of α-syn do not show bona fide Parkinson's pathology, they do exhibit neuronal loss and α-syn aggregation (Feany and Bender, 2000; Masliah et al., 2000; Giasson et al., 2002; M. K. Lee et al., 2002). Moreover, viral vector-mediated expression of α-syn in substantia nigra in monkeys and rats has led to nigrostriatal degeneration and LB-like inclusion bodies (Kirik et al., 2002, 2003; Lo Bianco et al., 2002). Together, all point to the possibilities that conformational defects and aggregation of α-syn play critical roles in the pathogenesis of PD and other α-synucleinopathies.
α-Syn localizes to nerve terminals (Iwai et al., 1995) and has been implicated in synaptic transmission (Abeliovich et al., 2000; Cabin et al., 2002; Liu et al., 2004; Yavich et al., 2004). Until recently, α-syn has been considered exclusively an intracellular protein, present mostly in the cytosol with some partitioning to vesicle fractions (George et al., 1995; H.-J. Lee et al., 2002a), thus leading to an assumption that this protein has only the cell-autonomous functions. This concept was challenged recently by the findings that α-syn is present in human CSF and blood plasma at low nanomolar concentrations in both PD and normal human subjects (Borghi et al., 2000; El-Agnaf et al., 2003). Furthermore, blood level of α-syn is doubled in familial PD patients with α-syn gene locus triplication (Miller et al., 2004). However, the mechanism underlying this phenomenon and the physiological and pathological roles extracellular α-syn may play in this process are not understood. Here, we report that a portion of α-syn is present in the lumen of vesicles (V) and can be secreted through an unconventional exocytic pathway in a constitutive manner. We also found that vesicular α-syn is highly prone to aggregation, and the aggregated forms are also secreted from the cells. These findings reveal a new branch of normal life cycle of α-syn that might affect its aggregation under pathological conditions.
Materials and Methods
Reagents. All-trans retinoic acid, saponin, z-Leu-Leu-Leu-al (MG132), rotenone, proteinase K (PK), and protease inhibitor mixture were purchased from Sigma (St. Louis, MO). OPTI-PREP iodixanol reagent was purchased from Accurate Chemicals and Scientific Corp. (Westbury, NY). Brefeldin A (BFA) was purchased from Epicenter Technologies (Madison, WI). Monoclonal antibodies for α-syn (Syn-1), protein disulfide isomerase (PDI), rab4, synapsin, and synaptotagmin 1 were purchased from BD Biosciences (San Diego, CA). Ubiquitin antibody was purchased from DakoCytomation (Carpinteria, CA). Monoclonal antibody for α-syn (H3C) and polyclonal antibody for catalase were kind gifts from Dr. J. George (University of Illinois, Urbana-Champaign, IL) and Dr. T. Imanaka (Toyama Medical and Pharmaceutical University, Toyama, Japan), respectively. Gold-conjugated anti-mouse IgG antibody was obtained from Ted Pella (Redding, CA).
Cell culture and α-syn expression. Differentiation of SH-SY5Y cells and preparation of rat primary cortical neurons were performed as described previously (Lee et al., 2004). On day 5 of differentiation, SH-SY5Y cells were infected with adenoviral vector containing cDNA for human α-syn (adeno/α-syn), β-galactosidase (lacZ; adeno/lacZ), C-terminal-tagged human α-syn (adeno/α-syn-MycHis), β-syn (adeno/β-syn-MycHis), A53T (adeno/A53T-MycHis), A30P (adeno/A30P-MycHis), or empty vector at a multiplicity of infection (m.o.i.) of 20 or as indicated. The detailed procedure for infection was described previously (H.-J. Lee et al., 2002b).
Western blotting and preparation of conditioned medium and cell extract. To collect the secreted proteins, cells were cultured in fresh medium for 3 h unless otherwise indicated. The conditioned medium was centrifuged at 10,000 × g for 20 min to remove cell debris and used for Western analysis without concentration. For extraction of cellular proteins, cells were washed twice with ice-cold PBS, collected in cold extraction buffer (PBS containing 1% Triton X-100 and protease inhibitor mixture), and incubated on ice for 10 min. The total cell extracts were mixed with Laemmli sample buffer and sonicated before Western analysis. Western blot analysis was performed according to the procedure described previously (H.-J. Lee et al., 2002b).
Pulse-chase experiment. Cells were infected with adeno/α-syn-MycHis. On day 3 of infection, cells were washed twice with warm DMEM (met-/cys-) and incubated in DMEM (met-/cys-)/10% dialyzed fetal bovine serum (Invitrogen, Carlsbad, CA) for 15 min. The cells were then pulse labeled in the same medium containing 0.1 mCi/ml 35S-met/cys (PerkinElmer, Boston, MA) for 30 min. After two washes with ice-cold PBS, the cells were incubated in normal growth medium for different periods before cell extraction. For BFA treatment, cells were preincubated with 0.5 μg/ml BFA or DMSO for 2 h before the labeling. BFA and DMSO were also added during the labeling and chase. For detection of aggregated α-syn, the 2 h chase at 37 or 18°C was initiated 7 h after pulse labeling in the presence of BFA. The conditioned medium and total cell extracts were prepared as described above. The total cell extracts were centrifuged at 14,000 × g for 10 min, and the supernatant was used for further characterization. For purification of α-syn-MycHis, the cell extracts and the medium were incubated with nickel-nitrilotriacetic acid (Ni-NTA) agarose beads (Qiagen, Valencia, CA) at 4°C overnight. The beads were washed with 5 mm imidazole and 10 mm imidazole four times each, and the bound proteins were eluted with 100 mm imidazole. The eluted proteins were analyzed by SDS-PAGE and autoradiography.
Microsomal vesicle preparation by flotation centrifugation. Cells grown in 10 cm dishes were washed twice in ice-cold PBS and scraped in buffer M [containing the following (in mm): 10 HEPES, pH 7.2, 10 KCl, 1 EGTA, 250 sucrose] with protease inhibitor mixture except for the sample for the PK experiment. Cells were disrupted by using N2 cell disruption bomb (Par Instrument, Moline, IL) at 450 psi for 20 min. The extract was then centrifuged at 1000 × g for 10 min to remove cell debris. The supernatant was mixed with 60% iodixanol to obtain 35% final concentration and then was layered under 1.7 ml of 30% iodixanol/buffer M in ultracentrifuge tubes. Then, 0.1 ml of 5% iodixanol/buffer M was gently layered on top and centrifuged at 200,000 × g for 2 h. The top fraction containing vesicles and the bottom fraction with cytosolic (C) proteins were obtained and used for additional analysis.
Rat brain fractionation. The fractionation procedure was based on the work of Huttner et al. (1983) and was described in detail previously (H.-J. Lee et al., 2002a).
Large dense core vesicle fractionation. The procedure was based primarily on the work of Waites et al. (2001), except that an additional step was added to remove cytosolic proteins. Cells grown in 10 cm dishes were washed twice in ice-cold PBS and scraped in buffer L [containing the following (in mm): 10 HEPES, pH 7.4, 2 EGTA, 300 sucrose] with protease inhibitor mixture. Cells were disrupted by using N2 cell disruption bomb (Par Instrument) at 450 psi for 20 min. The extract was then centrifuged at 1000 × g for 10 min to remove cell debris. To collect vesicles and remove cytosolic proteins, the supernatant was layered on the top of 1.6 m sucrose cushion and spun at 200,000 × g for 30 min. The vesicles were collected on top of the 1.6 m sucrose cushion and diluted with PBS and spun again as above. The washed vesicles were collected and diluted with PBS before loading on to a linear 0.6-1.6 m sucrose gradient. The samples were then centrifuged at 100,000 × g for 16 h using SW41Ti rotor. Fractions of 1 ml were collected from the top.
Proteinase K digestion. Proteinase K was serially diluted and added to samples to final concentrations of 0, 0.05, 0.25, and 1.25 μg/ml or as described. The samples were then incubated at 37°C for 30 min, and 5 mm PMSF was added before adding Laemmli sample buffer.
Vesicle permeabilization by saponin or sonication. For saponin treatment, saponin (3.2% stock solution in buffer M) was added to the vesicle preparations to final 0.32% with gentle vortexing (Tollefsen et al., 2001). The samples were then incubated on ice for 30 min before they were subjected to the flotation centrifugation as described above. For sonication, the vesicles were sonicated at probe amplitude of 10 for 10 s before it was subjected to the flotation centrifugation.
Electron microscopy. The sections were prepared and incubated with anti-α-syn (H3C) or anti-synapsin 1 antibody, followed by the incubation with 10 nm gold-conjugated goat anti-mouse IgG antibody as described by Gosavi et al. (2002).
Results
Secretion of α-syn through unconventional protein export pathway
To determine whether α-syn is released from cells, we transiently overexpressed human α-syn in differentiated SH-SY5Y human neuroblastoma cells using adenoviral vector and examined the release of α-syn in the culture medium. Released α-syn was detected in the culture medium as early as 2 h and accumulated over time (Fig. 1A), suggesting that α-syn is constitutively released from the cells. PD-linked mutant variants of α-syn (A53T and A30P) were also secreted from cells constitutively when expressed in SH-SY5Y cells (Fig. 1B). To demonstrate that the release of α-syn is not caused by either artifactual outcome of overexpression of foreign protein or membrane leakage of dying cells, we performed the following four sets of experiments. First, β-galactosidase protein was not detected in the medium when we overexpressed it in SH-SY5Y cells using adenoviral vector at the level comparable with the expression of α-syn (Fig. 1C). This indicates that viral vector-mediated overexpression itself is not the cause of α-syn release. Second, the amount of released α-syn in the conditioned medium correlates with the intracellular expression levels of this protein (Fig. 1D). In the same set of cultures, endogenously expressed cytosolic protein ubiquitin was not detected in the conditioned medium, indicating that α-syn release was not the result of membrane leakage caused by cell death (Fig. 1D). We also did not detect the increased release of lactate dehydrogenase, another cytosolic protein, in the culture medium under the same condition (data not shown). On the other hand, β-syn was also released from SH-SY5Y cells overexpressing this protein but to much lesser extent compared with α-syn (Fig. 1D). Third, when the cellular proteins were pulse labeled with 35S, and the secretion of labeled α-syn was followed over time in the culture medium, the amount of released α-syn reached a plateau in 2 h (Fig. 1E), again ruling out the possibility that the release of α-syn is attributable to membrane leakage, which would have led to continuous increase of extracellular α-syn. Finally, released α-syn was detected in the culture medium of rat primary cortical neurons (Fig. 1F), suggesting that neuronal endogenous α-syn is also constitutively secreted. To determine whether the release of α-syn is mediated by exocytosis, we examined the effect of low temperature, a classical blocker of vesicular exocytosis. When SH-SY5Y cells overexpressing α-syn were incubated for 3 h at either 37 or 18°C, we found that the secretion of α-syn was greatly reduced at 18°C (Fig. 1G). This suggests that α-syn is released from the cells via exocytosis in a stimulation-independent manner.
Figure 1.
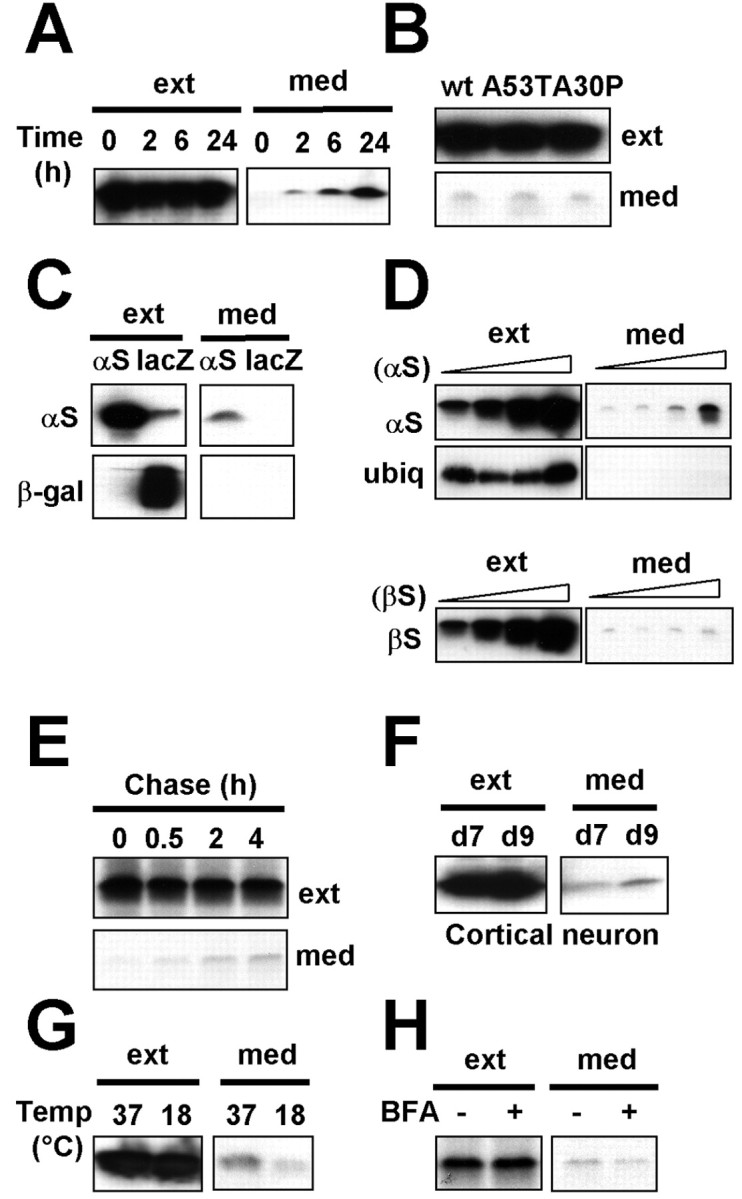
α-Syn is secreted from cells via exocytosis. A, Release of α-syn from differentiated SH-SY5Y cells. The conditioned medium (med) and the whole-cell extracts (ext) were collected at the indicated times. B, Release of α-syn mutants from differentiated SH-SY5Y cells. wt, Wild type. C, Overexpression of β-galactosidase does not cause its release. The conditioned medium and the cell extracts were collected from differentiated SH-SY5Y cells overexpressing either α-synorlacZ (β-gal). D, Secretion of α-syn correlates with its expression levels. Different doses of recombinant adenoviruses, adeno/α-syn and adeno/β-syn (1.25, 5, 20, 80 m.o.i.), were expressed in differentiated SH-SY5Y cells, and the levels of intracellular (ext) and released (med) proteins were analyzed. Released proteins were collected for 3 h. Note that ubiquitin (ubiq) was not released from the cells overexpressing α-syn. E, Pulse-chase experiment of α-syn secretion. SH-SY5Y cells expressing α-syn-MycHis were metabolically labeled with 35S-met/cys for 30 min and chased for indicated periods, and α-syn-MycHis was purified from the conditioned medium and the cell extract using Ni-NTA agarose beads. F, Secretion of endogenous α-syn from primary cortical neurons. The medium and the cell extracts were collected at day 7 (d7) or day 9 (d9) after 48 h of incubation in fresh medium. G, α-Syn secretion is blocked by low-temperature incubation. SH-SY5Y cells expressing α-syn were incubated at either 37 or 18°C for 3 h. H, Secretion of α-syn is insensitive to BFA. SH-SY5Y cells expressing α-syn-MycHis were pretreated with BFA for 2 h and metabolically labeled with 35S-met/cys for 30 min and chased for 2 h in the presence of BFA. αS, α-Syn; βS, β-syn.
These results were unexpected, because α-syn lacks any kind of signal sequence and is primarily known to be localized in the cytosol. However, the secretion of proteins that lack signal sequence has been observed for more than a decade (Nickel, 2003). Although the underlying mechanism of this unconventional export remains elusive, some of these proteins seem to use nonclassical vesicular exocytosis, because their release is resistant to BFA (Rubartelli et al., 1990, 1992; Elliott and O'Hare, 1997; Lecellier et al., 2002), a classical inhibitor of endoplasmic reticulum (ER)/Golgi-dependent protein secretion. To determine whether α-syn secretion is mediated by classical ER-Golgi pathway, newly synthesized proteins in SH-SY5Y cells expressing α-syn were pulse labeled with 35S, and secretion of labeled α-syn was analyzed in the presence and absence of BFA. BFA treatment drastically reduced protein release from cells, confirming that the classical export pathway was affected (data not shown). In contrast, as shown in Figure 1H, disruption of the classical export pathway with BFA did not block the release of α-syn. Thus, α-syn seems to use a secretory mechanism independent of ER/Golgi-related classical vesicular transport.
Intravesicular localization of α-syn
Although majority of cellular α-syn is present in the cytosol, a small portion is also present in membranous vesicle fractions in tissue and cell homogenates (George et al., 1995; H.-J. Lee et al., 2002a). Given that this protein is released from the cells via exocytosis, we tested whether the vesicle-associated α-syn is present in the lumen of the vesicles. First, we determined the topology of α-syn in vesicles by examining the sensitivity to a nonspecific protease, PK; proteins exposed on the outer surface of vesicles would be digested by PK. Microsomal vesicles were isolated from SH-SY5Y cells that overexpress α-syn and incubated with different concentrations of PK. α-Syn in the vesicular fraction is protected from PK digestion, whereas cytosolic α-syn is readily digested under the same condition (Fig. 2A). The protection against PK digestion was completely abolished when vesicular membranes were solubilized with detergent (Fig. 2A, +Tx), suggesting that the protection is caused by the encapsulation by membrane. Like the wild-type protein, both A53T and A30P mutant variants were protected from PK digestion in vesicle fractions (Fig. 2B). PK resistance is a common property of lumenal proteins, such as PDI and catalase, whereas the proteins that are exposed on the surface of vesicles, such as synaptotagmin 1 (an integral membrane protein) and rab4 (a peripheral membrane protein), are sensitive to PK (Fig. 2C). This is not an artifact caused by overexpression of protein, because overexpression of unrelated cytosolic protein (β-galactosidase) did not result in PK-resistant vesicular protein (Fig. 2D), and endogenously expressed neuronal α-syn also showed PK resistance in vesicle fraction (Fig. 2E). Similarly, α-syn is resistant to PK digestion in both microsomal vesicles (P3) and crude synaptic vesicles (LP2) isolated from rat cerebrum homogenates at PK concentrations that readily digest cytosolic (S3) proteins (Fig. 2F). Again, synaptotagmin 1 and synapsin 1 (a peripheral membrane protein) were digested under the same condition. These results suggest that vesicular α-syn is located in the lumen of vesicles.
Figure 2.

Vesicular α-syn is not accessible to protease digestion. A, PK resistance of vesicular α-syn. V and C prepared from differentiated SH-SY5Y cells were incubated with different concentrations of PK. Note that detergent-mediated solubilization of membrane (1% Triton X-100) eliminates PK resistance of vesicular α-syn. B, PK resistance of mutant α-syn in vesicles. Microsomal vesicles were prepared from differentiated SH-SY5Y cells overexpressing wild type (wt) or mutants and incubated with different concentrations of PK (0, 0.1 and 1 μg/ml). C, PK digestion of other vesicular proteins. Microsomal vesicles from SH-SY5Y cells were incubated with PK. Concentrations of PK: 0, 0.1 and 1 μg/ml. D, PK sensitivity of β-galactosidase (β-gal). V and C from SH-SY5Y cells overexpressing β-galactosidase were treated with 0, 0.1, and 1 μg/ml PK. E, PK sensitivity of neuronal endogenous α-syn. Vesicular and cytosolic fractions were isolated from rat cortical neurons and treated with PK. F, PK sensitivity of brain α-syn. Microsomal vesicles (P3), crude synaptic vesicles (LP2), and cytosol (S3) were prepared from rat brain homogenate and subjected to PK digestion. +Tx, With 1% Triton X-100; -Tx, without Triton X-100; αS, α-syn; cat, catalase; ST, synaptotagmin 1; SN, synapsin 1.
To further demonstrate this, we reasoned that, under the condition that causes permeabilization of vesicular membranes without complete solubilization, lumenal proteins would be released from the vesicles, whereas both integral and peripheral membrane proteins would remain bound to the vesicles. This condition can be achieved with low concentrations of saponin, a detergent frequently used to permeabilize cellular membranes (Tollefsen et al., 2001). The vesicles from SH-SY5Y cells (Fig. 3B) and rat brain (Fig. 3C) were permeabilized with saponin, and the membrane-bound and the released proteins were separated by a flotation centrifugation (Fig. 3A). After the saponin-mediated membrane permeabilization, PDI and catalase, lumenal proteins, were released from the vesicles, whereas both peripheral (synapsin 1) and integral (synaptotagmin 1) membrane proteins remained bound to the “ghost vesicles,” indicating that the permeabilization was successfully achieved without disrupting the interaction between vesicular membranes and the associated proteins (Fig. 3B,C). Under this condition, most of vesicular α-syn was released from the vesicles, supporting the idea that α-syn is present in vesicle lumen. To further support our interpretation, we repeated this experiment after permeabilizing the vesicular membranes using sonication, which permeabilizes membranes by entirely different mechanism from saponin treatment. Sonication caused a partial permeabilization of vesicles and thus resulted in a partial release of α-syn and PDI (Fig. 3D), but no release of synaptotagmin 1 was observed. Thus, after permeabilization of vesicles, α-syn behaved like a lumenal protein.
Figure 3.

Vesicular α-syn is released from vesicles after membrane permeabilization. A, Diagram of the lumenal protein release experiment. Vesicles were prepared from differentiated SH-SY5Y cells overexpressing α-syn or rat brain tissues and were permeabilized with saponin treatment or sonication. Saponin treatment and sonication produced ghost vesicles without lumenal components. The ghost vesicles and released proteins were separated with the flotation centrifugation. B, Lumenal protein release from the SH-SY5Y vesicles after the saponin-mediated permeabilization. C, Lumenal protein release from the rat brain P3 vesicles after the saponin-mediated permeabilization. D, Protein release from the SH-SY5Y vesicles after the sonication-mediated permeabilization. Sap, Saponin; αS, α-syn; cat, catalase; ST, synaptotagmin 1; SN, synapsin 1.
Finally, localization of α-syn in vesicle lumen was confirmed by electron microscopy with immunogold labeling. Vesicles were prepared by flotation centrifugation to ensure that heavy nonvesicular material is not included in the samples. As shown in Figure 4, α-syn immunoreactivities were found in the lumen of vesicles isolated from rat brain (Fig. 4A) and SH-SY5Y cells (Fig. 4B). On the other hand, antibodies against synapsin 1 decorated the outer surface of the brain LP2 vesicles (Fig. 4C), suggesting that the immunogold labels faithfully represented the localization of the proteins in these vesicles. Together, these results demonstrate that vesicle-associated α-syn is mainly a lumenal protein.
Figure 4.

Immunoelectron microscopy of the brain vesicles and neuroblastoma vesicles. Rat brain P3 vesicles (A), SH-SY5Y vesicles (B), and rat brain crude synaptic vesicles (C) were prepared by the flotation centrifugation and examined after immunogold labeling for α-syn (A, B) and synapsin 1 (C). Note that 10 nm of gold particles label α-syn in the lumen of electron-dense vesicles and synapsin-1 on the surface of vesicles. Scale bars, 100 nm.
In SH-SY5Y cells, it has been suggested that two types of secretory vesicles exist, small synaptic-like vesicles (SSLVs) and the large dense core vesicles (LDCVs) (Goodall et al., 1997). LDCVs can be followed with a specific marker, secretogranin II, and distinguished from lighter SSLVs with synaptophysin, which is present only in light vesicles. Subcellular fractionation indicates that the distribution of α-syn correlates with secretogranin II but not with synaptophysin (Fig. 5A). In addition, with electron microscopy of LDCV-enriched fractions, α-syn immunolabels were frequently found in electron-dense vesicles similar to the LDCVs (Fig. 5B). These results suggest that α-syn is enriched in LDCVs. The presence of α-syn in the lumen of LDCVs is consistent with our finding that this protein can be released from cells via exocytosis.
Figure 5.
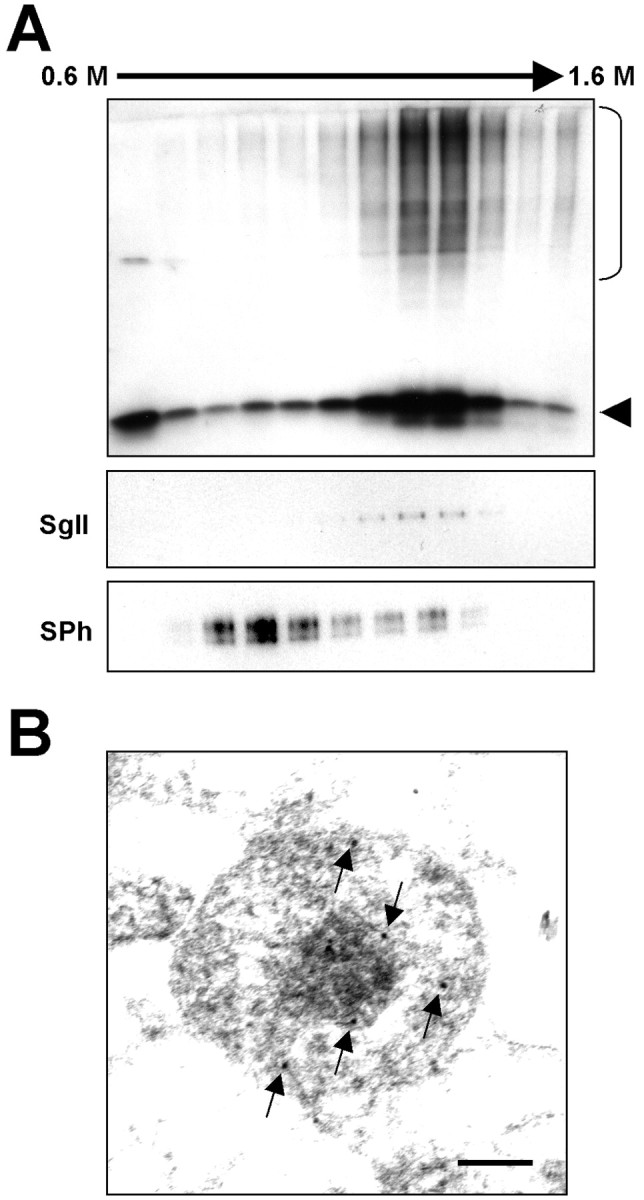
α-Syn in LDCVs. A, Subcellular fractionation of LDCVs. Cell extract from differentiated SH-SY5Y cells overexpressing α-syn was subjected to a linear sucrose gradient centrifugation, and fractions were collected from the top. High-molecular-weight aggregates are marked with a bracket on the right, and monomeric α-syn is marked with an arrowhead. SgII, Secretogranin II; SPh, synaptophysin. B, Immunoelectron microscopy of LDCV fractions (fractions 8 and 9). Arrows indicate 10 nm of gold particles labeling α-syn. Scale bar, 100 nm.
Aggregation of intravesicular α-syn
To characterize the aggregation properties of vesicular α-syn, we incubated brain P3, LP2, and S3 fractions at room temperature and compared the rate at which the high-molecular-weight aggregates were formed. We found that aggregates formed more rapidly in the P3 and LP2 vesicle fractions than in the cytosolic fraction (Fig. 6A). Vesicles from SH-SY5Y cells also showed rapid α-syn aggregation compared with the cytosol (data not shown). These vesicular aggregates were formed from the intravesicular α-syn rather than the trace amounts of cytosolic contaminants or vesicle-surface-bound α-syn, because the release of lumenal α-syn completely abolished de novo aggregation (Fig. 6B,C). We then asked whether this rapid vesicular α-syn aggregation also occurs in intact cells. To test this, SH-SY5Y cells overexpressing α-syn were cultured for different periods, and at each time point, α-syn aggregation was analyzed in the vesicle and cytosolic fractions. We found that the aggregation was faster and more robust in the vesicles than in the cytosol (Fig. 6D). Together, these results suggest that α-syn aggregation is favored in the vesicle lumen compared with the cytosol.
Figure 6.

Aggregation of the vesicular α-syn. A, Rate of α-syn aggregation in vesicles and cytosol. Rat brain fractions were incubated at room temperature for 1 or 2 d. High-molecular-weight aggregates are marked with a bracket on the right, and monomeric α-syn is marked with an arrowhead. B, C, Lumenal α-syn is responsible for vesicular α-syn aggregation. Vesicles were prepared from SH-SY5Y cells overexpressing α-syn and treated with either a buffer with (+) or without (-) saponin. Treated vesicles were isolated once again with flotation centrifugation and incubated for 2 d.α-Syn aggregation was analyzed by Western blotting (C). The asterisk indicates an unrelated protein that cross-reacts to Syn-1 antibody (Perrin et al., 2003). D, Time course of α-syn aggregation in V and C in intact cells. Sap, Saponin; RT, room temperature.
Secretion of aggregated α-syn
Next, we asked whether aggregates are also secreted from cells. To test this, we examined the secretion of α-syn aggregates from SH-SY5Y cells expressing different levels of α-syn. At low levels of α-syn expression, only monomeric α-syn was detected in both cell extracts and conditioned medium. However, higher levels of α-syn expression led to its aggregation in cells and also the secretion of the aggregates (Fig. 7A). The release of the aggregates is also mediated by exocytosis rather than membrane damage, because the incubation at a low temperature blocked the secretion of pulse-labeled α-syn aggregates as well as the monomer (Fig. 7B). It has been shown that specific cellular defects, such as proteasomal dysfunction and mitochondrial deficits, also caused α-syn aggregation (Ciechanover and Brundin, 2003; Lee, 2003). Therefore, we asked whether these abnormal conditions would increase the secretion of α-syn aggregates. To induce proteasomal dysfunction, we cultured the cells in the presence of MG132, a proteasomal inhibitor, and the aggregation and secretion of α-syn were analyzed. Consistent with previous studies, proteasomal inhibition led to the increase in intracellular α-syn aggregation. Under this condition, increased levels of both aggregated and monomeric α-syn were found in the conditioned medium (Fig. 7C). Ubiquitin, on the other hand, was not detected in MG132-treated cells, indicating that the increased α-syn release was not attributable to the leakage of cytosolic proteins from dying cells. Addition of rotenone, a mitochondrial complex I inhibitor that induces intracellular α-syn aggregation, also led to an increase in the levels of extracellular α-syn aggregates (Fig. 7D). Collectively, these data provide evidence that cells release aggregated forms of α-syn as well as the native form, and specific cellular impairment causing α-syn aggregation leads to the elevated levels of extracellular α-syn aggregates.
Figure 7.

Secretion of aggregated α-syn. A, Secretion of α-syn (αS) aggregates parallels with intracellular aggregate formation. SH-SY5Y cells were infected with adeno/α-syn at increasing m.o.i. (0, 2.5, 5, 10, 20). Secreted proteins were collected for 16 h. High-molecular-weight aggregates are marked with a bracket on the right, and monomeric α-syn is marked with an arrowhead. The whitening in the Western data of medium (60 kDa area) is attributable to an abundant presence of serum albumin. B, Secretion of α-syn aggregates is blocked by low-temperature incubation. SH-SY5Y cells expressing α-syn-MycHis were metabolically labeled with 35S-met/cys for 2 h, and the fresh medium was added for 7 h and the medium was changed again and chased for another 2 h at either 37 or 18°C to monitor secretion of aggregates. C, Secretion of α-syn in response to MG132. Cells were treated with 10 μm MG132 for 24 h to induce aggregation, and the medium and the cell extract were collected. D, Secretion of α-syn from rotenone (Rot)-treated cells. Cells were treated with rotenone (0, 3, 10, and 30 nm) for 2 d to induce aggregation. ext, Whole-cell extract; med, conditioned medium; ubiq, ubiquitin.
Discussion
We show that a portion of α-syn is constitutively released from neuronal cells to the culture medium via exocytosis as a part of normal life cycle of this protein rather than an experimental artifact that might occur because of membrane leakage from dying cells. This conclusion is based on the facts that (1) the release of other cytosolic proteins (e.g., endogenous lactate dehydrogenase, overexpressed β-galactosidase, and endogenous ubiquitin) was not detected in the culture medium, (2) the pulse-chase experiment showed that the release of labeled α-syn reached a plateau after 2 h, although there was plenty of labeled α-syn still existed in the cytoplasm, and (3) the release of α-syn was inhibited by low temperature, a classical blocker of vesicular exocytosis. In addition, our finding of intravesicular α-syn supports the conclusion that α-syn is released from cells via exocytosis. Importantly, α-syn was detected in human CSF and blood plasma in both PD and normal human subjects (Borghi et al., 2000; El-Agnaf et al., 2003). Our current studies suggest that intravesicular localization and secretion of α-syn might be the underlying basis for the generation of the extracellular α-syn in humans.
It has been shown that α-syn binds artificial liposomes containing phospholipids with acidic head groups (Davidson et al., 1998; Jo et al., 2000), lipid droplets (Cole et al., 2002), and lipid rafts (Fortin et al., 2004). It was also demonstrated that the binding to lipid is accompanied by the acquisition of extended helical structure (Bussell and Eliezer, 2003; Chandra et al., 2003; Jao et al., 2004). Although the interaction of α-syn to certain liposomes appears to be stable (Davidson et al., 1998), its interaction with brain-derived and cell line-derived vesicles has been difficult to detect in cell-free conditions, probably as a result of rapid dissociation (Cole et al., 2002; Ding et al., 2002). The fact that α-syn binding to lipid membranes was stabilized by chemical cross-linking (Cole et al., 2002) supports the dynamic nature of the interaction between α-syn and vesicular surface. We showed that the portion of α-syn that stably cofractionated with vesicles from brain tissues and neuronal cells is in fact localized in the lumen of the vesicles rather than bound to the surface. Therefore, in cells, α-syn might be localized in vesicles in two different ways. Some are bound to the cytosolic surface of vesicles, where the interaction is controlled by dynamic equilibrium with the free cytosolic α-syn. The others enter into the lumen of vesicles, and at least some of the intravesicular α-syn appears to be secreted via exocytosis. It is not clear how α-syn translocates into vesicles, nor is it known whether binding to vesicle surface contributes to entry into vesicles in any capacity. Investigations into these problems are likely to enhance our understanding on normal biology of α-syn. In agreement with its interaction with lipid membranes and localization in vesicles, α-syn has been implicated in lipid metabolism and vesicle trafficking (Scherzer et al., 2003; Willingham et al., 2003). It remains to be seen to what extent intravesicular α-syn plays a role in lipid and vesicle-related function of this protein.
α-Syn lacks the signal sequence and is indeed secreted via an unconventional, ER/Golgi-independent protein export pathway. Similar unconventional export has been observed for the secretion of certain cytokines and angiogenic growth factors, such as interleukin-1α and -1β and fibroblast growth factor-1 and -2 (Nickel, 2003). Interestingly, there is an increasing body of evidence that the export of classical cytosolic proteins is a common phenomenon, because other leader-less proteins, such as galectins, thioredoxin, heat shock proteins, and certain cytoskeletal proteins, are also secreted from cells through unconventional export pathways (Nickel, 2003; Fevrier and Raposo, 2004). Some of these pathways involve vesicular intake of the protein before secretion, whereas others are vesicle independent. Although the physiological function of unconventional secretion is not fully understood, there are reports suggesting that some of these secreted proteins are involved in intercellular communication. A recent study showed that when purified recombinant α-syn was added to the culture medium, the extracellular α-syn was internalized into cells in a rab5a-dependent manner (Sung et al., 2001). In light of this report as well as our current findings, we speculate that the secreted forms of α-syn might enter into the neighboring cells and perform as yet unidentified functions. Alternatively, vesicular intake and secretion might represent a novel mechanism by which cells expel damaged or misfolded α-syn. In support of this, secretion of α-syn was elevated under the conditions that would increase the levels of misfolded α-syn, such as inhibition of proteasomal or mitochondrial function. These possibilities still remain speculative, but they should be testable using our tissue culture models.
Recently, it has been shown that α-syn is localized in the plasma membrane of yeast cells (Outeiro and Lindquist, 2003; Dixon et al., 2005), and the plasma membrane localization is affected by the mutations in the conventional secretory pathway (Dixon et al., 2005). However, it is unclear whether and to what extent this yeast study is related to our current findings. There are no reports on the enrichment of α-syn in the plasma membrane of mammalian cells and tissues. In addition, the intravesicular localization and secretion of α-syn in our study do not involve stable membrane association, whereas the yeast study likely represents the association of α-syn to the plasma membrane on the cytosolic surface. Nevertheless, we cannot rule out the possibility that the classical secretory pathway indirectly contributes to the secretion of α-syn by providing vesicles and proteins that are required for the uptake of α-syn into the vesicles and subsequent secretion.
Our study shows that α-syn is highly prone to aggregation within the vesicles, and the aggregates are secreted from the cells. Furthermore, mitochondrial and proteasomal dysfunctions, conditions that are associated with intracellular α-syn aggregation and pathogenesis of PD (Ciechanover and Brundin, 2003; Lee, 2003), caused elevated secretion of α-syn aggregates. The secretion of α-syn aggregates is of potential importance in pathogenic function of this protein, because it has been reported that extracellular treatment with α-syn led to cell death, and this cytotoxic effect was dependent on its aggregation (Du et al., 2003; Kayed et al., 2003). Therefore, conformational abnormalities and aggregation of α-syn not only lead to cell-autonomous cytotoxicity but might also affect the viability of neighboring cells through the secretion of the aggregates. Thus, vesicle entry and secretion of α-syn, although a part of normal life cycle, may play an important role in neurodegeneration under pathogenic conditions.
Although the presence of monomeric α-syn in human blood plasma and CSF has been clearly demonstrated in previous studies (Borghi et al., 2000; El-Agnaf et al., 2003), these reports do not provide evidence for extracellular α-syn aggregates in human samples. We speculate that it might be because only a small group of cells release aggregated α-syn, compared with the infinitely larger number of cells that secrete the monomer. Therefore, it would be difficult to detect the aggregates using conventional Western blotting because of the dilution of the secreted material in body fluid. Advancement of sensitive detection methods for oligomeric or aggregated α-syn would help us resolve this issue. Another consideration is that in vivo, secreted α-syn aggregates could be cleared by either extracellular proteases or uptake by other cells such as microglia or neighboring neurons.
It is not clear why intravesicular α-syn is more prone to aggregation than the cytosolic protein. It is, however, interesting to note that vesicular membranes and their components create and maintain specific environments inside the vesicles that are different from each other and from the cytosol. Some of the known characteristics of certain vesicular environment, such as high calcium concentration (Lowe et al., 2004) and low pH (Hoyer et al., 2004), are known to increase the rate of α-syn aggregation. Alternatively, a spatially restricted compartment with a potentially high concentration of macromolecules might present molecular crowding effect, which has been shown to cause an accelerated aggregation of α-syn (Shtilerman et al., 2002; Uversky et al., 2002). Moreover, because the cytosol and vesicular lumen have different protein repertoire, the molecular interactions of α-syn with other proteins could differentiate the conformation and solubility of the protein in those locations. Thus, determination of the vesicular identity and the molecular interactions of α-syn within these vesicles would provide important clues for resolving this problem.
In conclusion, we propose vesicle entry and secretion as a new branch of normal biology of α-syn, which might affect the aggregation of this protein under pathogenic conditions. These findings raise a number of questions. What is the underlying mechanism by which α-syn translocates into vesicles? What makes the vesicle lumen more favored environment for α-syn aggregation? What are the physiological and pathological functions of the secreted α-syn? Resolution of these questions will enhance our understanding of the normal biology of α-syn and also provide insights into the pathogenic mechanism of synucleinopathies.
Footnotes
This work was supported by the Michael J. Fox Foundation (S.-J.L.), the Abramson Family Foundation (S.-J.L.), and the Parkinson's Disease Foundation/National Parkinson's Foundation joint grant (H.-J.L.). We thank Chris Rochet, Ted Fon, Yoon Suk Kim, Emmanuelle Laurine, and Hans Lee for stimulating discussions and comments on this manuscript, Julia George and Tsuneo Imanaka for antibodies, and Nafisa Ghori for the assistance in electron microscopy.
Correspondence should be addressed to Seung-Jae Lee, The Parkinson's Institute, 1170 Morse Avenue, Sunnyvale, CA 94089. E-mail: slee@thepi.org.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256016-09$15.00/0
References
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: 239-252. [DOI] [PubMed] [Google Scholar]
- Borghi R, Marchese R, Negro A, Marinelli L, Forloni G, Zaccheo D, Abbruzzese G, Tabaton M (2000) Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci Lett 287: 65-67. [DOI] [PubMed] [Google Scholar]
- Bussell Jr R, Eliezer D (2003) A structural and functional role for 11-mer repeats in alpha-synuclein and other exchangeable lipid binding proteins. J Mol Biol 329: 763-778. [DOI] [PubMed] [Google Scholar]
- Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, Ellis CE, Paylor R, Lu B, Nussbaum RL (2002) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J Neurosci 22: 8797-8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC (2003) A broken alpha-helix in folded alpha-synuclein. J Biol Chem 278: 15313-15318. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A (2004) alpha-Synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 364: 1167-1169. [DOI] [PubMed] [Google Scholar]
- Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett 576: 363-368. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Brundin P (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40: 427-446. [DOI] [PubMed] [Google Scholar]
- Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL (2002) Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J Biol Chem 277: 6344-6352. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee S-J, Rochet JC, Ding TT, Williamson RE, Lansbury Jr PT (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA 97: 571-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39: 889-909. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 273: 9443-9449. [DOI] [PubMed] [Google Scholar]
- Ding TT, Lee S-J, Rochet JC, Lansbury Jr PT (2002) Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 41: 10209-10217. [DOI] [PubMed] [Google Scholar]
- Dixon C, Mathias N, Zweig RM, Davis DA, Gross DS (2005) alpha-Synuclein targets the plasma membrane via the secretory pathway and induces toxicity in yeast. Genetics 170: 47-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du HN, Tang L, Luo XY, Li HT, Hu J, Zhou JW, Hu HY (2003) A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human alpha-synuclein. Biochemistry 42: 8870-8878. [DOI] [PubMed] [Google Scholar]
- El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, Cookson MR, Hardy J, Allsop D (2003) alpha-Synuclein implicated in Parkinson's disease is present in extracellular biological fluids, including human plasma. FASEB J 17: 1945-1947. [DOI] [PubMed] [Google Scholar]
- Elliott G, O'Hare P (1997) Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 88: 223-233. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW (2000) A Drosophila model of Parkinson's disease. Nature 404: 394-398. [DOI] [PubMed] [Google Scholar]
- Fevrier B, Raposo G (2004) Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol 16: 415-421. [DOI] [PubMed] [Google Scholar]
- Forno LS (1996) Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol 55: 259-272. [DOI] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH (2004) Lipid rafts mediate the synaptic localization of α-synuclein. J Neurosci 24: 6715-6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George JM, Jin H, Woods WS, Clayton DF (1995) Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15: 361-372. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM-Y (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34: 521-533. [DOI] [PubMed] [Google Scholar]
- Goedert M (2001) alpha-Synuclein and neurodegenerative diseases. Nat Rev Neurosci 2: 492-501. [DOI] [PubMed] [Google Scholar]
- Goodall AR, Danks K, Walker JH, Ball SG, Vaughan PF (1997) Occurrence of two types of secretory vesicles in the human neuroblastoma SH-SY5Y. J Neurochem 68: 1542-1552. [DOI] [PubMed] [Google Scholar]
- Gosavi N, Lee H-J, Lee JS, Patel S, Lee S-J (2002) Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem 277: 48984-48992. [DOI] [PubMed] [Google Scholar]
- Hoyer W, Cherny D, Subramaniam V, Jovin TM (2004) Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 43: 16233-16242. [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P (1983) Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96: 1374-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet 364: 1169-1171. [DOI] [PubMed] [Google Scholar]
- Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T (1995) The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron 14: 467-475. [DOI] [PubMed] [Google Scholar]
- Jao CC, Der-Sarkissian A, Chen J, Langen R (2004) Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc Natl Acad Sci USA 101: 8331-8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE (2000) alpha-Synuclein membrane interactions and lipid specificity. J Biol Chem 275: 34328-34334. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486-489. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Bjorklund A (2002) Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J Neurosci 22: 2780-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Bjorklund A (2003) Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson's disease. Proc Natl Acad Sci USA 100: 2884-2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18: 106-108. [DOI] [PubMed] [Google Scholar]
- Lecellier CH, Vermeulen W, Bachelerie F, Giron ML, Saib A (2002) Intra- and intercellular trafficking of the foamy virus auxiliary bet protein. J Virol 76: 3388-3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-J, Choi C, Lee S-J (2002a) Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem 277: 671-678. [DOI] [PubMed] [Google Scholar]
- Lee H-J, Shin SY, Choi C, Lee YH, Lee S-J (2002b) Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J Biol Chem 277: 5411-5417. [DOI] [PubMed] [Google Scholar]
- Lee H-J, Khoshaghideh F, Patel S, Lee S-J (2004) Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 24: 1888-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL (2002) Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci USA 99: 8968-8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ (2003) alpha-Synuclein aggregation: a link between mitochondrial defects and Parkinson's disease? Antioxid Redox Signal 5: 337-348. [DOI] [PubMed] [Google Scholar]
- Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O (2004) alpha-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J 23: 4506-4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P (2002) alpha-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc Natl Acad Sci USA 99: 10813-10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe R, Pountney DL, Jensen PH, Gai WP, Voelcker NH (2004) Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci 13: 3245-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L (2000) Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287: 1265-1269. [DOI] [PubMed] [Google Scholar]
- Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB (2004) alpha-Synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62: 1835-1838. [DOI] [PubMed] [Google Scholar]
- Nickel W (2003) The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur J Biochem 270: 2109-2119. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S (2003) Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302: 1772-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Payton JE, Barnett DH, Wraight CL, Woods WS, Ye L, George JM (2003) Epitope mapping and specificity of the anti-alpha-synuclein monoclonal antibody Syn-1 in mouse brain and cultured cell lines. Neurosci Lett 349: 133-135. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045-2047. [DOI] [PubMed] [Google Scholar]
- Rubartelli A, Cozzolino F, Talio M, Sitia R (1990) A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J 9: 1503-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubartelli A, Bajetto A, Allavena G, Wollman E, Sitia R (1992) Secretion of thioredoxin by normal and neoplastic cells through a leaderless secretory pathway. J Biol Chem 267: 24161-24164. [PubMed] [Google Scholar]
- Scherzer CR, Jensen RV, Gullans SR, Feany MB (2003) Gene expression changes presage neurodegeneration in a Drosophila model of Parkinson's disease. Hum Mol Genet 12: 2457-2466. [DOI] [PubMed] [Google Scholar]
- Shtilerman MD, Ding TT, Lansbury Jr PT (2002) Molecular crowding accelerates fibrillization of alpha-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry 41: 3855-3860. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Han-son M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K (2003) alpha-Synuclein locus triplication causes Parkinson's disease. Science 302: 841. [DOI] [PubMed] [Google Scholar]
- Sung JY, Kim J, Paik SR, Park JH, Ahn YS, Chung KC (2001) Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. J Biol Chem 276: 27441-27448. [DOI] [PubMed] [Google Scholar]
- Tollefsen S, Wierod L, Skotte A, Rob JA, Helgeland L (2001) Saponin permeabilization of rough microsomes from rat liver reveals a novel pro-thrombin pool. Biochim Biophys Acta 1526: 249-256. [DOI] [PubMed] [Google Scholar]
- Uversky VN, M Cooper E, Bower KS, Li J, Fink AL (2002) Accelerated alpha-synuclein fibrillation in crowded milieu. FEBS Lett 515: 99-103. [DOI] [PubMed] [Google Scholar]
- Waites CL, Mehta A, Tan PK, Thomas G, Edwards RH, Krantz DE (2001) An acidic motif retains vesicular monoamine transporter 2 on large dense core vesicles. J Cell Biol 152: 1159-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham S, Outeiro TF, DeVit MJ, Lindquist SL, Muchowski PJ (2003) Yeast genes that enhance the toxicity of a mutant huntingtin fragment or alpha-synuclein. Science 302: 1769-1772. [DOI] [PubMed] [Google Scholar]
- Yavich L, Tanila H, Vepsalainen S, Jakala P (2004) Role of α-synuclein in presynaptic dopamine recruitment. J Neurosci 24: 11165-11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55: 164-173. [DOI] [PubMed] [Google Scholar]